Congenital and Neonatal Disorders
Colonic Atresia and Stenosis
Overview: The colon is the least involved segment in intestinal atresias; colonic atresia constitutes between 1.8% and 15% of intestinal atresias, with an overall incidence of approximately 1 in 20,000 births. Colonic stenosis is very rare, with fewer than 15 cases reported in the literature. It typically consists of stricturelike stenosis, although membranous stenosis also has been reported. The anatomic descriptive classification of small intestinal atresias (see Chapter 103) also is applied to colonic atresias.1,2
Colonic atresia is associated with other colonic abnormalities, as well as extracolonic and extraintestinal abnormalities. Approximately 22% of patients also may have Hirschsprung disease or hypoganglionosis.3,4 Extraintestinal anomalies include the musculoskeletal system, heart, abdominal wall, eyes, and central nervous system.5
Etiology: The etiology of colonic atresia is thought to be similar to that of small bowel atresias, that is, related to an ischemic event in utero that leads to resorption of the involved bowel and discontinuity of the proximal and distal segments.6 More recently, mutations involving fibroblast growth factor 10 or its receptor have resulted in intestinal atresia in mice, which suggests that genetic determinants also may play a role.7
Clinical Presentation: Infants with colonic atresia present with abdominal distension, vomiting, and failure to pass meconium. Polyhydramnios is uncommon, because the atresia is sufficiently distal to allow resorption of swallowed amniotic fluid. The onset of vomiting may be delayed compared with more proximal atresias and may become feculent.5,8 Patients with colonic stenosis demonstrate similar findings proportionate to the degree of stenosis.
Imaging: Abdominal radiographs in patients with colonic atresia show distal obstruction with multiple dilated loops of bowel and abdominal distension with flank protuberance and diaphragmatic elevation. However, the proximal colon characteristically is dilated far more than the other loops of bowel (Fig. 106-1, A). This disproportionate dilatation is due to competence of the ileocecal valve, which allows intestinal contents forward into the proximal colonic segment but does not allow decompression proximally. Air-fluid levels are seen on horizontal beam radiographs, and prone images show no gas in the rectum (Fig. 106-1, B).
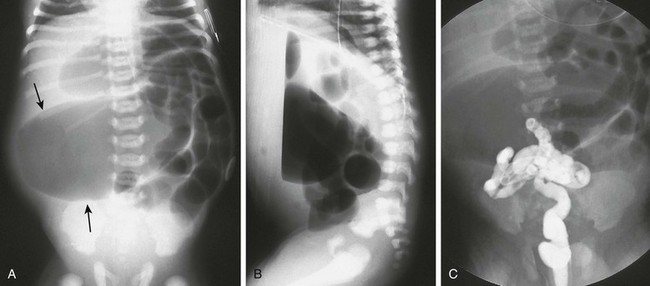
Figure 106-1 Colonic atresia.
A, A supine abdominal radiograph of a neonate with abdominal distension and bilious vomiting demonstrates protuberance of the flanks and elevation of the diaphragms. Gas-filled dilated loops of bowel are present, but one loop is dilated far more than other loops, and represents the atretic proximal colonic segment (arrows). An orogastric tube decompresses the stomach. B, A prone cross-table lateral radiograph shows a large air-fluid level located within the atretic proximal colonic segment. No gas is present in the rectum. C, A contrast enema shows a microcolon terminating blindly at the atretic segment.
A contrast enema performed on these patients demonstrates a microcolon that terminates blindly, devoid of the filling defects characteristic of a meconium ileus (Fig. 106-1, C).
Treatment: The treatment of colonic atresia is surgical. Because 15% to 20% of patients with colonic atresia also have proximal intestinal atresia, the proximal bowel should be evaluated. Hirschsprung disease should be excluded with suction biopsy before reestablishing intestinal continuity, and coexisting extraintestinal anomalies should be identified.5
Anorectal Malformations
Overview: Anorectal malformations encompass the spectrum of anal atresia and stenosis, with an incidence of approximately 1 : 5000 live births worldwide. Approximately one third of patients have isolated lesions, but two thirds of patients are affected by other abnormalities encompassing the gastrointestinal tract and multiple other systems. Approximately 95% of patients have a fistula to the urethra (males), vagina (females), or the perineum; however, 95% of patients with Down syndrome and anorectal malformation have no fistula.9
Anatomically, anorectal malformations are classified as high, intermediate, or low lesions, depending on whether the atresia lies above or below the levator sling. Some authors object to this classification because the anomaly is a spectrum, rather than three separate and distinct types. The classification system of Levitt and Peña (Table 106-1) provides both prognostic information and implications for optimal surgical management.9 These authors note that girls with high lesions with a high rectovaginal fistula who later present with a persistent/unrepaired urogenital sinus abnormality actually represent girls with a persistent cloaca, which are classified by the length of the persistent cloacal canal.10
Table 106-1
Classification of Anorectal Malformations
| Males | Females |
| Perineal fistula | Perineal fistula |
| Rectourethral fistula | Vestibular fistula |
| Bulbar | Persistent cloaca |
| Prostatic | ≤3 cm common channel |
| Recto-bladder neck fistula | >3 cm common channel |
| Imperforate anus without fistula | Imperforate anus without fistula |
| Rectal atresia | Rectal atresia |
| Complex defects | Complex defects |
From Levitt M, Peña A. Anorectal malformations. In: Coran A, et al. eds. Pediatric surgery. Philadelphia, Mosby; 2012.
Rectal atresia often is discussed among the anorectal malformations, although patients with rectal atresia have a normal anal canal and external physical findings. The rectum is atretic 1 to 2 cm above the anus.10
Etiology: Anorectal atresia with rectourethral or rectovaginal fistulas may be the result of failure of the urorectal septum to descend to the cloacal membrane and the eventual site of the perineal body; if the cloaca is too small, the hindgut may then terminate anteriorly, entering either the urethra in the male or the vagina in the female. Rectoanal atresias are thought to be related to vascular accidents, similar to the atresias that occur in the small bowel and colon. Imperforate anus is due to failure of breakdown of the anal membrane.11
No single genetic abnormality is associated with anorectal malformations. The most common chromosomal abnormalities are trisomy 21 and a microdeletion at chromosome 22q11.2, although abnormalities in multiple chromosomes have been identified.12 Approximately 15% of patients with rectovestibular or rectoperineal fistulas, which are indicative of low lesions, have a positive family history for anorectal malformations. Genetic studies in both animals and humans have implicated defects in the sonic hedgehog, Wnt5a, and Skt genes. These studies suggest that the pathogenetic mechanisms of high and low fistulas differ and that fistula formation may be due to a genetic mutation rather than obstruction.
Clinical Manifestations: Anorectal malformations are clinically apparent at birth, with the exception of patients with rectal atresia, who have a normal external appearace. Physical examination may reveal a perineal or vestibular fistula, although the possibility of such a fistula is best evaluated after 24 hours, because meconium may not appear in the perineum before this time. If fecal material appears in the urine, a rectourethral fistula may be inferred. Patients who demonstrate a “flat bottom” (i.e., the buttock crease is not visible) have poor development of pelvic musculature and a more guarded prognosis.9 Girls with a single perineal orifice have a cloacal malformation (Fig. 106-2).
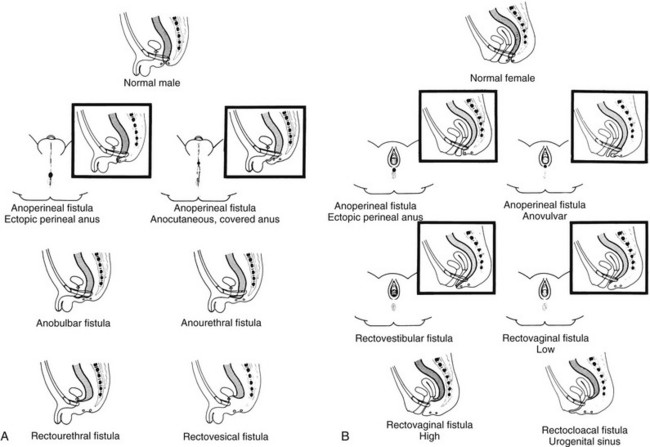
Figure 106-2 Physical examination findings in boys and girls with anorectal malformations.
A, A schematic diagram of normal and ectopic termination of the hindgut in the male. B, A schematic diagram of normal termination and common sites of ectopic termination of the hindgut in the female. (From Santulli TV. In: Mustard WT, et al, eds. Pediatric surgery. ed 2. St Louis: Mosby–Year Book; 1969.)
The best known group of anomalies associated with anorectal malformations is the VACTERL association (vertebral anomalies, anal atresia, cardiac abnormalities, esophageal atresia with or without tracheoesophageal fistula, and renal and limb abnormalities). Associated genetic syndromes include trisomy 21, trisomy 8, and fragile X syndrome.12 Cardiovascular abnormalities are seen in about one third of patients, most commonly atrial septal defect and persistent ductus arteriosus, followed by tetralogy of Fallot and ventricular septal defect.9 Vertebral anomalies occur in approximately one third of patients (Fig. 106-3, A and B), and their severity may be correlated with the complexity of the anorectal lesion. Coronal clefts often are seen in patients with imperforate anus and have been reported as being nine times more common in boys than in girls.13 Esophageal atresia with tracheoesophageal fistula is seen in approximately 10% of patients, and abnormalities affecting the duodenum (atresia or malrotation) are found in 1% to 2%. Hirschsprung disease, although reported in these patients, is rare.14 Genitourinary anomalies are common, presenting in one third to one half of cases, again correlated with increasing complexity of the defect. These anomalies range from reflux to renal dysplasia or agenesis, with cryptorchidism and hypospadias seen in male patients. Females may demonstrate müllerian abnormalities, including duplications and obstructions, particularly in the setting of cloacal abnormalities.9,10
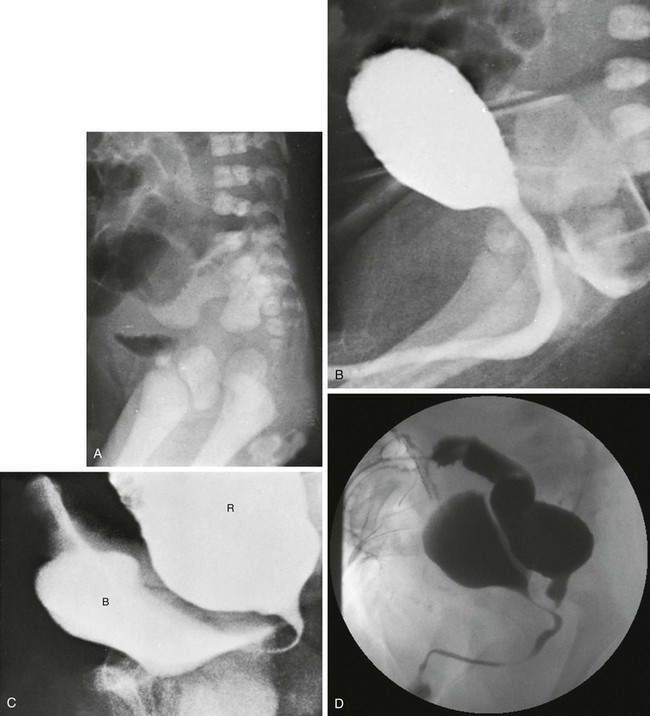
Figure 106-3 High anorectal malformation in a male neonate.
A, A lateral radiograph of the pelvis and lower abdomen the demonstrating gas in the urinary bladder. In addition, numerous lumbar coronal clefts are identified. B, A voiding cystogram demonstrates a rectoprostatic fistula. Note how the rectum bulges inferiorly below the level of the fistula. This accounts for the spuriously small distance sometimes seen between rectal gas and anal marker in such patients, although the bowel edge may lie above the puborectalis sling. C, Examination of the rectal pouch with contrast material identifies a rectovesical fistula in a male infant. D, Examination of the rectal pouch with contrast material shows the fistulous connection to the posterior urethra in a male infant. (A, From Berdon WE, et al. The radiologic evaluation of imperforate anus. An approach correlated with current surgical concepts. Radiology. 1968;90(3):466-471.)
Imaging: The goal of imaging is to elucidate associated anomalies and to determine fistulous anatomy. The sacral ratio9 quantifies the degree of sacral hypoplasia by measuring the distance between the iliac crests and the inferior border of the sacroiliac joints (A-B) and the distance between the inferior border of the iliac joints and the tip of the sacrum (C-D); the CD/AB ratio should be >0.77. An increase in the ratio to approach 1.0 indicates better sacral development and better prognosis.
Several imaging methods have been used for evaluation of the atretic anatomy in patients without a perineal fistula. The invertogram relies on visualizing the gas-filled rectum and relating its termination to pelvic bony landmarks—particularly extension below the coccyx, which would classify it as a low lesion.9 However, problems such as meconium distal to gas and movement of the rectum with infant straining and crying often render this examination inexact and problematic. Voiding cystourethrography can be useful in visualizing the fistula if contrast material flows retrograde into the atretic distal colon during voiding (Fig. 106-3, C). Voiding cystourethrography does not always work because meconium may obstruct the fistula. Instilling water-soluble contrast material into the rectal pouch after a colostomy often is successful in outlining fistulas to the bladder (Fig. 106-3, D) or urethra (Fig. 106-3, E). Occasionally, gas may outline some of the pertinent anatomy (e-Fig. 106-4).
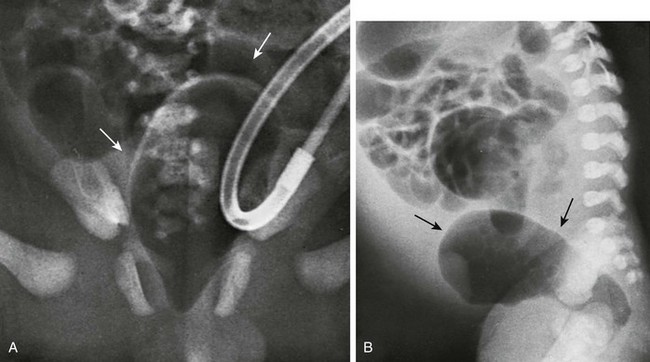
e-Figure 106-4 High anorectal malformation in a female neonate with a cloaca.
A, A pneumovagina (arrows) represents gas entering a giant vagina (arrows) above the narrow urogenital sinus. Urine also fills the vagina because of the coexisting urogenital sinus. B, A pneumovagina (arrows) in an infant with a rectovaginal and urethrovaginal connection. (From Berdon WE, et al. The radiologic evaluation of imperforate anus. An approach correlated with current surgical concepts. Radiology. 1968;90(3):466-471.)
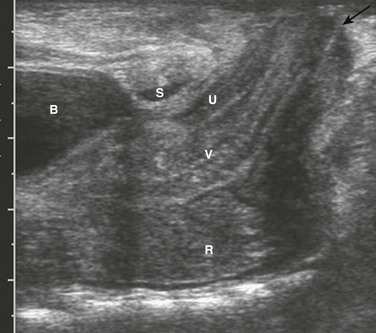
Ultrasound also is used to evaluate the distal position of the rectum (Fig. 106-5). The meconium in the pouch allows easy visualization of the distal rectum; however, changes in apparent position with straining make it somewhat challenging. Measurements of 10 mm ± 4 correlate with a low lesion, whereas measurements of 24 ± 6 tend to correlate with intermediate and high lesions.15 Ultrasound can identify the puborectalis musculature; the presence of this musculature correlates with low-type lesions, whereas its absence correlates with high-type lesions.16
Magnetic resonance imaging (MRI) also has been used to identify the internal anatomy. High field strengths, small field of view, and sequences without fat suppression, which increase the conspicuity of the muscles of the pelvic floor, are helpful in visualizing the position of the distal rectal pouch with respect to the levator mechanism.17 MRI also can be used to evaluate the rectum and levator sling after surgery (Fig. 106-6).
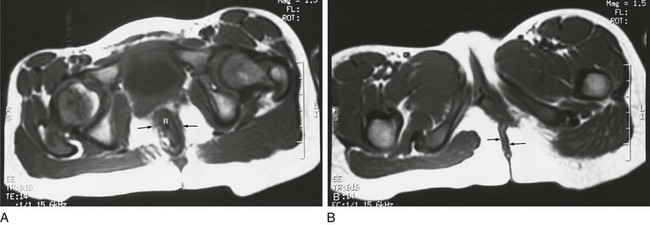
Figure 106-6 Postoperative magnetic resonance after anal atresia repair.
A, An axial T1-weighted section at the level of the pubic bone shows the rectum (R) contained within the puborectalis muscle (arrows). B, An axial T1-weighted image through the region of the pubic bone on another child after surgery shows that the rectum (R) is not enclosed by muscle on the left side. Little puborectalis muscle is seen on the left.
Patients with rectal atresia who have normal external anatomy are evaluated with contrast enema. The enema demonstrates a very short distal rectum that terminates blindly (e-Fig. 106-7).
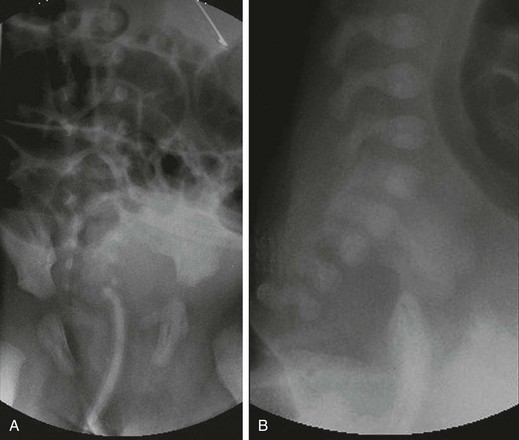
e-Figure 106-7 Rectal atresia.
A, An anteroposterior view of a neonate with distal bowel obstruction during an attempt to perform a contrast enema. The catheter is in place but would not advance further. Note the multiple dilated loops of bowel and the decompressing orogastric tube in the left upper quadrant. B, A lateral view during same procedure shows the inability of contrast material to progress further than the tip of the catheter, instead spilling onto the fluoroscopy table.
Treatment: The treatment of anorectal malformations is surgical but varies with the level and complexity of the lesion. Most boys can undergo repair with a posterior sagittal approach alone, although some with a high lesion also will require an abdominal approach to mobilize a very high rectum. In girls, 30% of cloacas need to be repaired through an abdominal approach. Babies with rectal atresia typically undergo an initial colostomy, with later anastomotic repair of the atretic rectum with the anal canal.10
Currarino Association
Overview: Although Currarino syndrome initially was described in three infants in 1981 as a triad of anorectal malformation (anorectal stenosis) with rectoperineal fistula,18 sacral anomaly (classically crescentic abnormality resembling a scimitar), and a presacral mass,19 this abnormality actually encompasses a much wider spectrum of phenotypic abnormalities and underlying genetic defects.
Etiology: Currarino syndrome is an autosomal-dominant disorder with variable penetrance; approximately 50% of the cases are sporadic. A mutation located at 7q36 encoding for the HLXB9 gene responsible for nuclear transcription factor HB9 plays a role in the dorsoventral separation of the caudal embryo; other genes related to the same differentiation pathway also may exist.20,21
Clinical Presentation: The patients typically present with constipation and may undergo anorectal dilatation. The presacral mass is often small and may not be detected unless the syndrome is suspected; an unrecognized presacral mass may present later with complications such as meningitis or development of malignancy.20–22 The spectrum of presacral masses includes teratoma, rectal duplication cyst, anterior meningocele, leiomyosarcoma, and ectopic nephroblastoma.
Other abnormalities that can be seen in the full spectrum of the syndrome include Hirschsprung disease, dysganglionosis, bladder motility abnormalities, vesicoureteral reflux, rib anomalies, urinary and gynecologic abnormalities, cord tethering and intraspinal lipomas.18,23
Imaging: Radiographs of the sacrum may reveal the typical scimitar-shaped sacral abnormality (Fig. 106-8, A); other sacral defects, typically deletion abnormalities, may be present. In a minority of patients the sacrum is normal.21
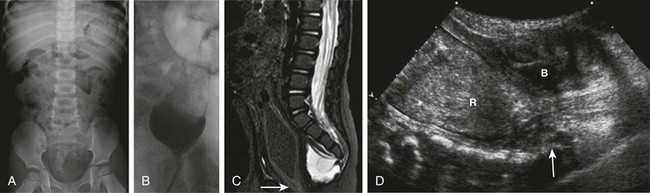
Figure 106-8 Currarino syndrome.
A, A supine abdominal radiograph in 23-month-old boy with a history of constipation demonstrates an increased amount of stool in the colon and a deletion anomaly along the left side of the sacrum, resembling a scimitar. B, A contrast enema demonstrates narrowing of the distal rectum. C, A sagittal T2-weighted lumbar spine image demonstrates a cystic retrorectal mass that proved to represent an anterior meningocele. Note the narrowing of the distal rectum (arrow). D, An anterior sagittal pelvic sonogram of a different male neonate shows a dilated proximal rectum (R) distally narrowed and a retrorectal mass (arrow) that proved to represent a mature teratoma. B, Bladder.
Those patients with anorectal stenosis may have a rectoperineal fistula or an anteriorly placed anus and present with constipation, sometimes in early adulthood. Contrast enema shows marked narrowing of the distal rectum, with excessive stool proximally (Fig. 106-8, B). If the presacral mass is of sufficient size, it will exert a visible mass effect upon the posterior wall of the distal rectum.
MRI best demonstrates the presacral masses well and provides a window into the spinal canal (Fig. 106-8, C). In young infants, the presacral mass often is visible sonographically (Fig. 106-8, D).
Hirschsprung Disease
Overview: Hirschsprung disease is the result of failure of normal bowel innervation as a result of the arrest of proximal to distal migration of vagal neural crest cells, and therefore it is considered a neurocristopathy. As a result of abnormal arrest of migration, a variable length of distal bowel lacks parasympathetic Auerbach (intermuscular) and Meissner (submucosal) plexuses and is unable to participate in normal peristalsis, resulting in failure of relaxation and functional obstruction. Pathologic evaluation also shows abnormal acetylcholinesterase staining and hypertrophied nerve fibers (e-Fig. 106-9).
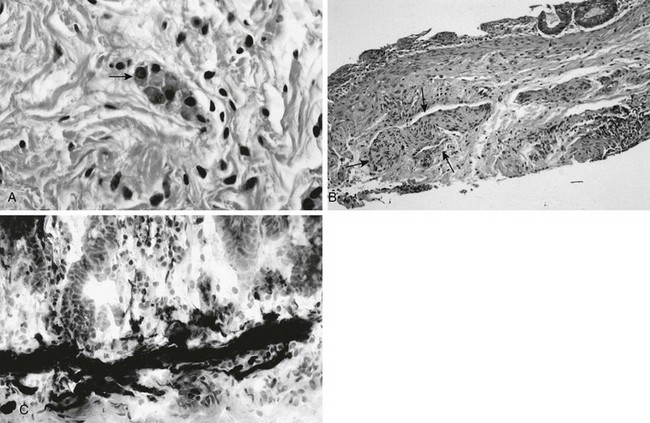
e-Figure 106-9 Pathologic findings in Hirschsprung disease.
A, In a normal newborn, a suction biopsy and hematoxylin and eosin staining reveals submucosal ganglion cells (arrow). B, Hirschsprung disease, suction biopsy shows submucosal, abnormally thick nerves (arrows) and no ganglion cells. C, Hirschsprung disease, acetylcholinesterase stain of a biopsy specimen reveals abnormal stain of twiglike nerve processes in the lamina propria. (Courtesy Dr. R. Raba, Children’s Hospital of Michigan, Detroit, MI.)
Hirschsprung disease occurs in approximately 1 per 5000 live births and is responsible for approximately 15% to 20% of cases of neonatal bowel obstruction, presenting in the newborn period in approximately 80% of cases. The incidence is less among African Americans and Asian Americans, quoted as 2.1 and 2.8 per 10,000 live births, respectively.24 The transition point between normal and abnormal bowel can occur anywhere and can extend through a variable length of the small bowel; rarely, total intestinal aganglionosis is present.24 The presence of the transition point at the rectosigmoid is termed short-segment aganglionosis and occurs in approximately 80% to 90% of cases. Males predominate in the incidence of short-segment aganglionosis (the boy : girl ratio is 4 : 1); however, male preponderance diminishes in longer segment involvement.
Hirschsprung disease is most commonly associated with Down syndrome, which is present in approximately 2% to 10% of patients with Hirschsprung disease, with a 5 : 1 boy to girl ratio.24 Other syndromes associated with Hirschsprung disease include Waardenburg, Shprintzen-Goldberg, McKusick-Kaufman, Bardet-Biedl, Currarino syndrome, and with central hypoventilation (Ondine syndrome), termed Haddad syndrome.24–26 Patients with longer segment aganglionosis are more likely to demonstrate Haddad syndrome than are those with short-segment disease.27 Between 5% and 30% of patients with Hirschsprung disease have limb, skin, central nervous system, kidney, cardiac, and other malformations.25
Etiology: The etiology of Hirschsprung disease is not precisely known. One theory is that the migrating ganglion cells do not reach the distal segment because they are fewer in number or mature prematurely. However, it also is possible that the ganglion cells reach the distal bowel but perish or are unable to proliferate because of a deficient microenvironment.28 Multiple genetic mutations have been identified, with the most common being the RET proto-oncogene located at 10q11, which is found in approximately 15% to 35% of sporadic cases and 50% of familial cases. EDNRB, which is located at 13q22, is identified in approximately 5% of cases.29,30 A positive family history is encountered in a minority of patients (<10%), but this percentage increases to nearly 25% in patients with total aganglionosis, with the degree of risk proportional to the length of the affected segment and the degree of consanguinity.31,32
Clinical Presentation: Hirschsprung disease in the neonate presents with distal obstruction, including abdominal distension and bilious vomiting. Failure to pass meconium beyond the first 24 hours is seen in up to 90% of patients.28 Rarely, perforation of the cecum or appendix can occur in neonates, typically in those with long-segment disease.33,34 In older children, Hirschsprung disease presents with constipation, abdominal distension, and vomiting, with failure to thrive in more severe cases. In late presentations, the zone of transition is typically low.28
Hirschsprung-associated enterocolitis is a major cause of morbidity and mortality in patients with Hirschsprung disease, affecting approximately 10% to 50% of patients.28,35 Clinically it can present either insidiously or abruptly with diarrhea, fever, abdominal distension, colicky abdominal pain, and hematochezia. If the underlying diagnosis is not known, the clinical situation can be confusing, because the expected clinical finding in patients with Hirschsprung disease is constipation rather than diarrhea. The pathogenesis is unknown, but implicated mechanisms include stasis and bowel dilatation, an abnormal epithelial lining, abnormal mucin production, abnormal local immune mechanisms, and viral and bacterial pathogens, notably Clostridium difficile, which are promoted by initial intestinal stasis. The incidence of enterocolitis increases with delayed diagnosis, long-segment disease, and comorbid conditions, such as those found in patients with Down syndrome.35 Postoperative risk factors include development of postoperative intestinal obstruction or development of an anastomotic leak.35 Contrast enemas generally are not indicated but can be performed carefully with water-soluble iso-osmolal contrast with surgical consultation in cases in which the diagnosis is not certain.
Imaging: In neonates, the abdominal radiographs demonstrate distal bowel obstruction (Fig. 106-10). On prone cross-table lateral radiographs, gas usually is noted in the rectum, unlike the situation with small bowel or colonic atresias; however, the caliber of the air-filled rectum is clearly smaller than that of more proximal bowel, and a zone of transition may be identifiable if it is located at the rectosigmoid junction.
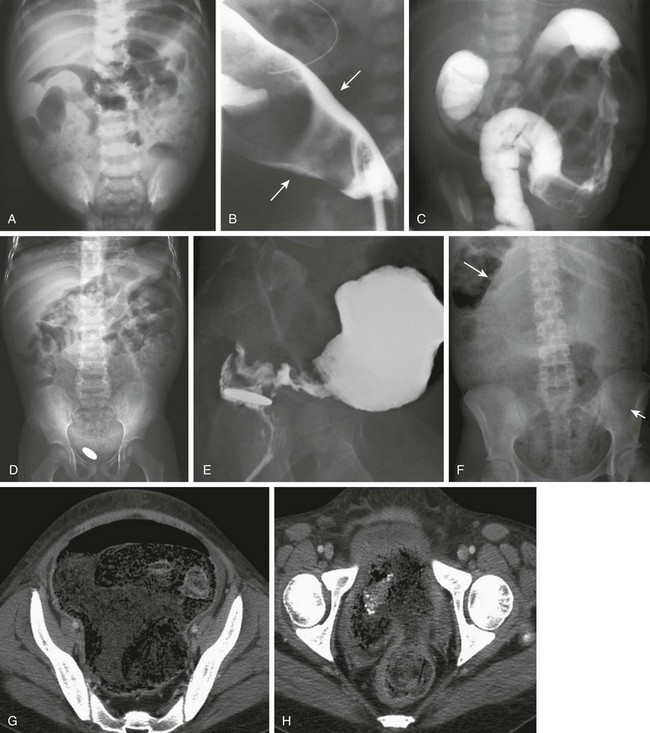
Figure 106-10 Hirschsprung disease.
A, A supine abdominal radiograph of a 2-week-old baby with abdominal distention and inability to pass stool reveals retention of stool and dilated bowel loops. B, A contrast enema in a lateral projection shows the transition zone (arrows) at the rectosigmoid junction, which was confirmed surgically. An orogastric tube is incidentally noted. C, An abdominal image during a contrast enema in a 2-day-old girl with a positive family history of Hirschsprung disease and signs of distal bowel obstruction. The examination shows an abnormal distal descending colon with an irregular wall resulting from disordered contraction and an apparent transition zone at the splenic flexure. At surgery, Hirschsprung disease with transition zone at the splenic flexure was confirmed. D, An abdominal radiograph in a 4-year-old after surgery for repair of an interrupted aortic arch; this child presented with constipation and a retained coin within the rectum for several months. The radiograph shows marked fecal dilatation of the sigmoid, which does not extend into the rectum; the rectum contains a retained coin. E, A lateral view of a contrast enema in the same patient as in D clearly shows the zone of transition and the retained coin. F, An abdominal radiograph of a 15-year-old girl with diabetes and a history of intermittent diarrhea who presented acutely with diarrhea and abdominal distension. The abdominal radiograph shows marked stool burden and colonic dilatation. The sigmoid extends into the abdomen and displaces the transverse colon cephalad (arrows). G, Computed tomography (CT) for the same patient shown in F was requested because of a clinical concern of appendicitis. CT at the upper pelvis shows a dilated sigmoid entering the pelvis. H, CT lower in pelvis shows the zone of transition.
Radiologic diagnosis is made with contrast enema, which is geared toward identification of the zone of transition (see Fig. 106-10 and e-Fig. 106-11). Routine use of enemas prior to the diagnostic procedure should be avoided, but digital examination in the older age group should not affect the radiographic findings. The study is done with a small catheter, which should be placed as close to the external sphincter as possible in neonates. Water-soluble iso-osmolal agents are preferred in neonates, because hyperosmolal media will lead to increased bowel distension; barium is not optimal if there is another cause of obstruction, such as meconium ileus, nor desirable in case of perforation. In older patients being evaluated for constipation, barium typically is used. Initial images must be obtained in the lateral projection, as soon as the contrast begins to enter the colon, and multiple fluoroscopic stored images are used to document the flow of contrast and the caliber of the colon. Imaging should not be delayed until full colonic distension is achieved because this might obliterate the zone of transition. The enema is terminated when the zone of transition is identified.
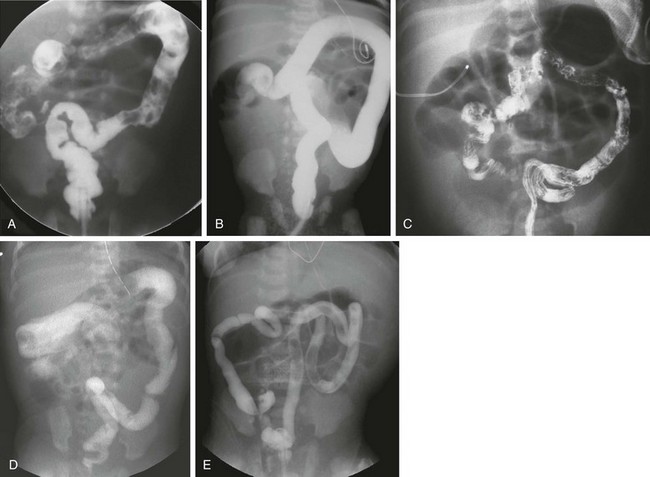
e-Figure 106-11 Hirschsprung disease—total colon and small bowel aganglionosis.
A, An anteroposterior image of a contrast enema of a neonate with distal bowel obstruction; the colon has an overall normal appearance, without evidence of a zone of transition. Total colonic aganglionosis was documented at surgery. B, An anteroposterior image of a contrast enema of a neonate with Down syndrome. The examination shows a colon of uniform caliber, although less redundant than typical. Total colonic aganglionosis was found at surgery. C, An image from a contrast enema in another neonate with total colonic aganglionosis demonstrates the appearance of a microcolon. D, An image from a contrast enema in a 6-day-old girl with distal obstruction. The examination shows a gradual caliber change, with a questionable transition point at the distal transverse colon. Total colonic aganglionosis was documented at surgery. E, An image from a contrast enema in a 1-day-old infant with distal obstruction. The examination shows a colon only slightly larger than a typical microcolon. At surgery there was aganglionosis of the entire colon and of the small bowel to the jejunum, 51 cm from the ligament of Treitz. Because of the short length of his normal bowel, the child became dependent on total parenteral nutrition.
In patients with total colonic aganglionosis, radiographic diagnosis can be problematic. The colon may have a normal appearance, or there can be a pseudotransition zone, sometimes at the splenic flexure, or a microcolon (see e-Fig. 106-11).
In addition, patients with Hirschsprung disease initially can present with meconium plug or with small left colon syndrome (Fig. 106-12); such patients should be monitored carefully, and if normal bowel function does not return, further evaluation such as suction biopsy should be considered.
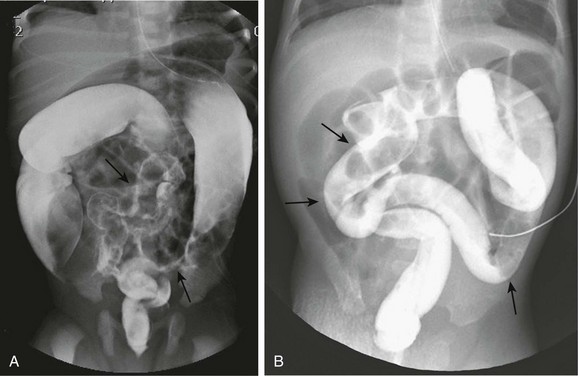
Figure 106-12 Hirschsprung disease simulating small left colon and meconium plug syndromes.
A, A spot image from a contrast enema on a 2-day-old infant with abdominal distension shows a very small caliber sigmoid and distal descending colon (arrows) with a small caliber rectum, suggesting small left colon syndrome. At surgery, Hirschsprung disease was found, with a transition zone at the splenic flexure. B, A spot image from a contrast enema in a neonate with distal bowel obstruction demonstrates a uniform caliber colon with multiple filling defects (arrows) suggesting meconium plugs. At surgery, Hirschsprung disease was identified involving the entire colon and the distal 20 cm of ileum.
A definitive diagnosis is made by characteristic biopsy findings, which show absence of ganglion cells, abnormal acetylcholinesterase staining, and hypertrophied nerve fibers (see e-Fig. 106-9). Manometric studies, with failure of relaxation of the internal sphincter with rectal distension, are helpful after the immediate neonatal period.
Abdominal radiographs of patients with Hirschsprung enterocolitis may show dilatation of bowel loops, with an irregular outline consistent with spasm, along with mucosal disruption and ulceration (Fig. 106-13, A). Contrast enemas are not indicated, but if performed, they will show ulceration and spasm (Fig. 106-13, B).
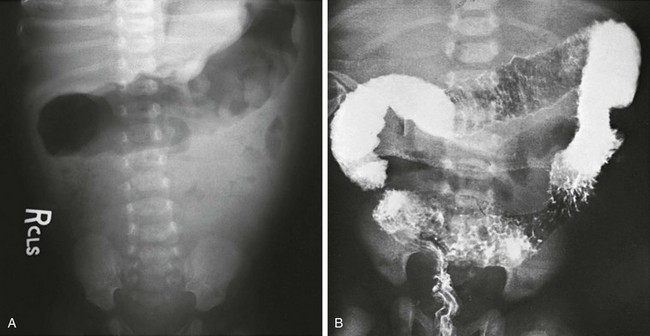
Figure 106-13 Hirschsprung enterocolitis.
A, An abdominal radiograph of a child with Hirschsprung-associated enterocolitis shows an irregular wall in the gas-filled transverse colon. B, Lethal enterocolitis in a 12-day-old boy who presented with fever, foul diarrhea, and shock. A contrast enema (performed elsewhere) shows extensive edema, ulceration, and spasm. The patient died upon arrival at the referral institution.
Treatment: Surgical repair of Hirschsprung disease is achieved by resecting the abnormal distal bowel and bringing normally innervated bowel through the sphincteric mechanism. Swenson devised and performed the first successful surgical repair; this procedure consists of resection of the aganglionic segment and anastomosis of normal bowel containing ganglion to the sphincteric mechanism. The Duhamel and Soave procedures are modifications of the Swenson procedure; they are designed to minimize risk of injury to the sphincteric mechanism. The Duhamel procedure involves leaving the anterior portion of the aganglionic rectum in place and anastomosis of ganglion-containing bowel to its posterior wall. The Soave or endorectal pull-through procedure involves resection of the mucosa and submucosa of the aganglionic bowel and pulling the normal bowel containing ganglion through the muscular rectal sleeve.
Meconium Plug, Small Left Colon, and Dysmotility of Prematurity
Overview: Meconium plug and small left colon syndromes refer to conditions of colonic dysmotility that present as distal bowel obstruction in neonates and that overlap with each other as well as with Hirschsprung disease. In patients with meconium plug syndrome, a plug of meconium is identified within the colon, which, when evacuated, has a characteristic “white head” (Fig. 106-14, A). Small left colon syndrome also may contain meconium plugs within the colon, but in addition a variable portion of the left colon (typically the descending colon and sigmoid) have a characteristically small caliber, resembling a microcolon. In these patients, the rectum typically is of normal caliber. These conditions also overlap with the failure to pass meconium and at times obstructive symptoms that may be seen in very low or extremely low birth weight infants (<1500 g and <1000 g birth weight, respectively).
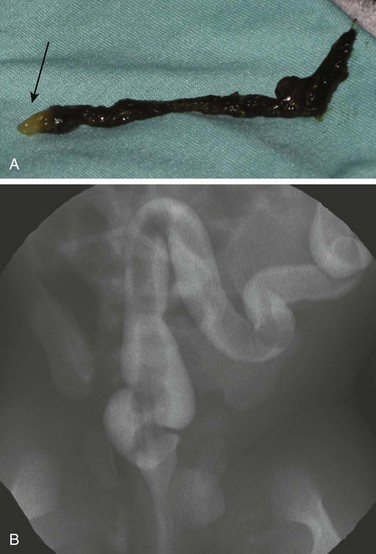
Figure 106-14 Meconium plug.
A, A meconium plug evacuated after a diagnostic contrast enema demonstrated the distinctive white tip (arrow). B, A fluorostore image from a contrast enema in a term neonate with vomiting and bowel distension demonstrates the long filling defect characteristic of meconium plug syndrome. The child was relieved of the obstruction after evacuation of the plug, without recurrence of symptoms.
Etiology: The etiology of these conditions is believed to be delayed maturation of effective peristalsis and/or abnormally increased water absorption by the colon, leading to a more tenacious meconium. Approximately 40% to 50% of small left colon syndrome cases occur in conjunction with maternal diabetes; conversely, small left colon syndrome has been cited as occurring in approximately 4.7% of infants with a history of maternal diabetes.36
Clinical Presentation: Presentation is abdominal distension and bilious vomiting, typically in a term infant. Symptoms may present in very low birth weight and extremely low birth weight patients in the first day of life or later, typically around 10 to 14 days of age, with increasing abdominal distension and failure to pass meconium.37
Meconium plug and small left colon syndromes could be the presenting findings in patients with Hirschsprung disease. It is important to monitor these patients closely after initial relief of the obstruction; if symptoms recur or do not abate, a biopsy is necessary to evaluate for Hirschsprung disease.38
Imaging: Abdominal radiographs show typical findings of distal obstruction with multiple dilated loops of bowel, although the degree of dilatation tends to be less than that seen in other causes of distal obstruction, such as ileal atresia or meconium ileus.39 As with other cases of distal bowel obstruction, a diagnostic enema should be performed with water-soluble contrast material with a near iso-osmolal concentration. In cases of meconium plug, the enema demonstrates a colon of normal caliber, typically with a long filling defect caused by the meconium plug (Fig. 106-14, B), which the patient may evacuate at the conclusion of the enema. In patients with small left colon syndrome, the examination will reveal a small caliber colon involving a variable length of sigmoid and descending colon, with a typically normal caliber rectum (Fig. 106-15).
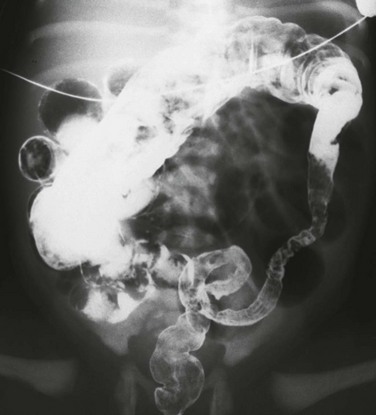
Treatment: Patients with meconium plug and small left colon syndrome typically do well after the enema examinations. A biopsy for Hirschsprung disease should be performed in patients who have a protracted recovery or a recurrence of symptoms. In premature infants with obstruction, treatment with enema also is advocated,37 although the procedure entails more risks in these fragile infants.
Chronic Intestinal Pseudoobstruction
Overview: Chronic intestsinal pseudoobstruction (CIPO) refers to a characteristic, chronically recurring, and at times massive dilatation of the intestinal tract without an identifiable mechanical cause. The term encompasses a heterogeneous group of disorders that affect the enteric nervous system (ENS) and intestinal smooth muscle, resulting in failure of normal intestinal motility. The condition can be primary (subdivided into neuropathic or myopathic) or secondary, when it occurs in association with various underlying systemic conditions. Patients with the myopathic form of primary disease tend to have involvement of other organ systems, particularly the urinary tract, and may require a vesicostomy.
Neuronal intestinal dysplasia (NID), a form of primary CIPO, is a disorder of intestinal innervation that can be diffuse or localized. NID has two subtypes.40 Type A consists of aplasia or hypoplasia of sympathetic innervation of the intestine and constitutes less than 5% of cases. Type B accounts for approximately 95% of cases and affects the parasympathetic system, with hyperplasia of the submucosal and myenteric plexus, the presence of dysplastic and ectopic ganglion cells, and increased acetylcholinesterase staining. Type B NID has been found in the proximal intestine of patients with Hirschsprung disease.41–43
Secondary disease is associated with multiple conditions, including scleroderma, dermatomyositis, muscular dystrophies, infiltrative diseases such as amyloidosis, nervous system diseases such as myotonic dystrophy, familial dysautonomia, postviral syndromes such as Epstein-Barr, cytomegalovirus, herpes zoster and rotavirus, fetal alcohol syndrome, and mitochondrial disorders.44
Etiology: The etiology of intestinal pseudoobstruction and neuronal dysplasia is not known. The ENS generates the rhythmic enteric activity and includes the interstitial cells of Cajal, neurons, and glial cells. The network of interstitial cells of Cajal has been found to be abnormal in some patients with CIPO, and delayed maturation of these cells has been reported in neonates with transient CIPO. Ganglion cells containing nitric oxide synthase, which mediates sphincteric relaxation, have been found to be increased in some patients with CIPO.44 Most cases of this disease are sporadic, but familial clusters have been reported, particularly in consanguineous families, suggesting possible genetic causes.23
Clinical Findings: Children present with periodic vomiting, diarrhea, abdominal distension, constipation, pain, and weight loss. Bacterial overgrowth can occur as a result of stasis. NID type A presents in the neonatal period with acute episodes of intestinal spasticity, diarrhea, and hematochezia. NID type B present with chronic constipation and/or pseudoobstruction, typically in the first 3 years of life.23 Patients in whom small bowel bacterial overgrowth develops as a result of intestinal stasis may exhibit diarrhea, weight loss, and macrocytic anemia.44
Multiple anomalies have been documented in patients with primary CIPO. Hirschsprung disease occurs in as many as 10% of cases.23 Related associations include multiple endocrine neoplasia (MEN) type IIb and neurofibromatosis.45 Other associations include malrotation, anorectal malformations, congenital short small bowel, and intestinal atresia, as well as extraintestinal problems such as bladder dysfunction, Down syndrome, and histiocytosis.23
Imaging: Radiographs and cross-sectional imaging demonstrate dilated loops of bowel, particularly the colon; air-fluid levels suggest obstruction but really represent severe adynamic ileus. Megacolon typically is found at contrast enema, without the diagnostic findings of Hirschsprung disease. Contrast small bowel and enema studies show poor motor activity in affected regions but otherwise are not specific. Gut dilatation is common in the colon, the stomach, and the small bowel and can assume massive proportions (e-Fig. 106-16).
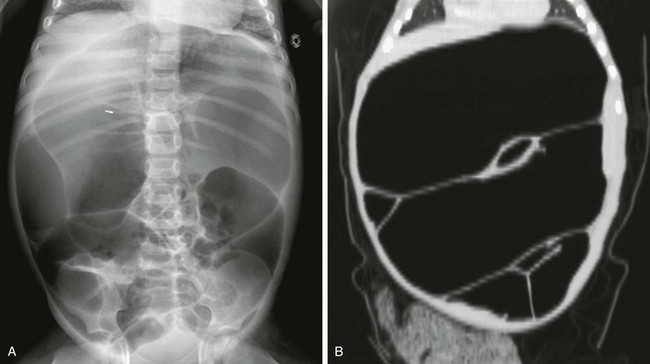
e-Figure 106-16 Pseudoobstruction.
A, An abdominal radiograph of a 5-year-old boy with myopathic type pseudoobstruction who was largely dependent on total parenteral nutrition because of prolonged episodes of feeding intolerance. During this acute presentation, the radiograph demonstrates remarkable dilatation of the colon. B, A computed tomography coronal reformat image confirms colonic dilatation; the small bowel was not dilated.
Treatment: A gastrostomy may be used for feeding (although feeding often is not tolerated) or for venting. Enteral feedings through a jejunostomy are often, but not always, tolerated. Medical management includes use of prokinetic agents and antiemetics. Management of patients with small bowel bacterial overgrowth includes vitamins, antibiotics, and possibly probiotics.44 Some patients may need intestinal transplantation or combined hepatic and intestinal transplantation if liver failure complicates total parenteral nutrition (TPN).44
Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome
Overview: Also known as Berdon syndrome, the megacystis-microcolon-intestinal hypoperistalsis syndrome, or MMIHS, was first described by Berdon and colleagues in 1976.46 Approximately 182 cases currently have been reported,41 with an approximately 2 : 1 female predominance.47 Malrotation is associated in many patients.46,47 This syndrome is considered one of the most severe forms of functional intestinal obstruction in the newborn and often is fatal.47 Without treatment, most patients succumb within the first 6 months of life.48
Etiology: The etiology of MMIHS is not certain, but case analyses suggest that at least some cases may have an autosomal-recessive inheritance pattern. Histology shows normal ganglion cells in most patients, but the number of these cells is decreased in some patients and increased in others, who show hyperganglionosis as well as giant ganglia.47 Interstitial cells of Cajal are responsible for generating the electrical slow wave activity that results in coordinated smooth muscle peristaltic activity, and abnormalities in these cells have been found in the urinary bladder49 and in the intestinal myenteric plexus50 of patients with MMIHS. In addition, abnormalities of the smooth muscle cells themselves, with evidence of thinning, vacuolar degeneration, and increased connective tissue in both the bowel and bladder, have been found in these patients.50–52
Clinical Presentation: Infants with MMIHS present with abdominal distension and bilious vomiting as a result of obstruction; abdominal distension is exacerbated by the dilated urinary bladder, and bowel sounds are decreased or absent.
Imaging: Abdominal radiographs show marked abdominal distension and a largely gasless bowel, with the markedly distended urinary bladder displacing bowel loops. Ultrasound demonstrates the markedly distended urinary bladder, typically in association with hydroureteronephrosis. Upper gastrointestinal examination demonstrates hypoperistalsis or aperistalsis of the stomach, duodenum, and small bowel. Contrast enema demonstrates a microcolon, with malrotation in more than half of the cases (e-Fig. 106-17).41
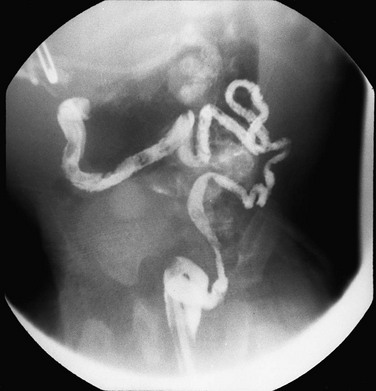
Colonic Duplications
Overview: Gastrointestinal duplications are discussed in Chapter 103; in this chapter findings that are characteristic of colonic duplications are discussed. Approximately 17% of all gastrointestinal duplications are colonic,53 and approximately 5% are rectal; they can be cystic or tubular. Cystic duplications consist of a relatively small segment of duplicated bowel that typically does not communicate with the adjacent lumen. The incidence of gastric mucosa within colonic duplications is reportedly less than in other locations, and therefore they can remain asymptomatic and be discovered incidentally later in life.
Tubular duplications typically connect with the gastrointestinal tract and often drain into the perineum via a fistula, which usually is located anterior to the rectum, or drain to a single or duplicated anus. In an extensive review of 57 cases, Yousefzadeh et al.54 defined the colon proper as that ending in a perineal anus and the duplicated colon as that ending at a perineal fistula, at a communication with the colon proper, or blindly; however, in some cases both colons end in a duplicated perineal anus or fistulae. The duplications are of variable length and can be classified as extending above or below the peritoneal reflection. If they do not extend below the peritoneal reflection and therefore do not have a perineal opening, communication with the lumen can be at both ends or at a single end. They can extend proximally to involve the cecum, appendix, and terminal ileum.
Approximately 80% of tubular colonic duplications are associated with multiple other anomalies, including renal anomalies, duplication of the genitourinary tract (e.g., uterus, bladder, urethra, or penis), and vertebral and cord abnormalities (e-Fig. 106-18). Rectal duplications are cystic structures located in the presacral space (e-Fig. 106-19).
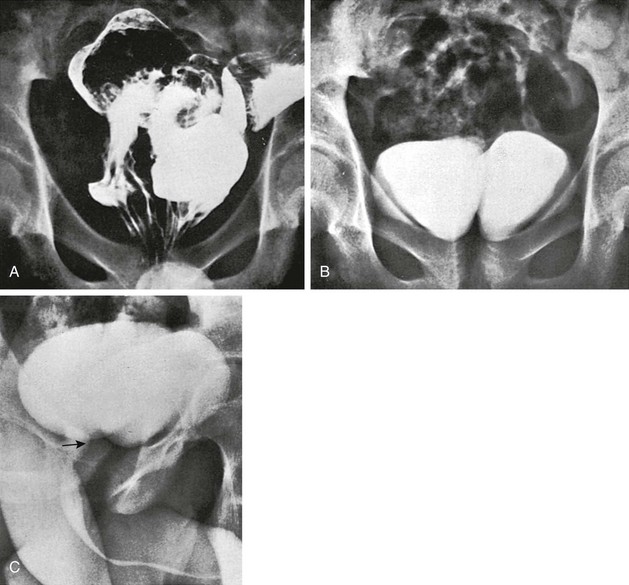
e-Figure 106-18 Duplication of the entire colon and bladder.
A, A contrast enema of the pelvis in a 15-year-old boy who had been treated in the newborn period for anal atresia demonstrates two rectums. Surgical exploration revealed duplication of the entire colon, from appendix to rectum. B, A frontal radiograph of the pelvis after excretory urography shows two bladders. Each upper urinary tract emptied into the bladder on its respective side. C, A voiding urethrogram demonstrates a single urethra (arrow) originating from the right side.
Etiology: The putative etiology of duplications was discussed in Chapter 103; the partial or abortive twinning theory invokes a split in the primitive streak during the embryonic period, with a relatively late split resulting in duplications of the colon.
Clinical Findings: Diagnosis of colonic duplication varies with the type and mode of presentation. Duplications may be found incidentally, or secondary to complications. Cystic duplications will show a mass, which may be complicated by bowel obstruction due to volvulus or intussusception. Nausea, vomiting, and abdominal pain are common presenting symptoms. If the duplication has gastric mucosa, patients may become symptomatic as a result of inflammation and ulceration. Obstruction may occur through intussusception, with the duplication cyst as a leading point, or as a result of an acutely enlarging mass, obstructing the colon even if originating in adjacent small bowel (see Fig 103-35, A through C). Finally, colonic and rectal duplications may present as a second opening in the perineum. Rectal duplications can present with constipation as well as urinary obstruction; because they are located behind the rectum, they also may mimic presacral meningoceles or cystic teratomas (e-Fig. 106-19).53,54 In girls in whom the duplication terminates in a perineal/vaginal opening, the duplication may simulate a rectovaginal fistula.
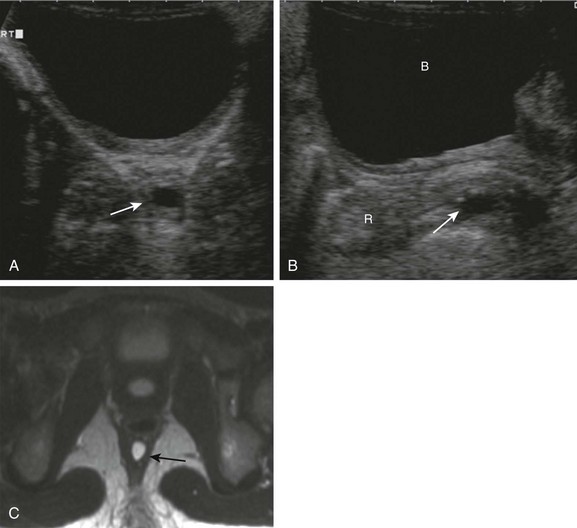
e-Figure 106-19 Rectal duplication.
A, An axial sonogram in a 6-year-old girl with abdominal pain shows a small cyst (arrow) inseparable from the rectal wall. B, A sagittal midline sonographic image demonstrates the retrorectal location of the cyst. C, An axial T2-weighted magnetic resonance image confirms the small cystic lesion (arrow) and better demonstrates its retrorectal location. B, bladder; R, rectum.
Imaging: Initial suspicion on plain films can be followed up with sonography, computed tomography (CT), or MRI. Tubular duplications with a perineal fistula are best evaluated with contrast enema via both perineal orifices; antegrade contrast studies also may be useful. CT with multiplanar reconstructions and MRI likely will be diagnostic, but their exact role in evaluation remains undefined. Rectal duplications can be identified with plain films and contrast enemas via a mass effect upon the rectum and upon the adjacent genitourinary structures if they are sufficiently large. Sonography and MRI are the best modalities to evaluate noncommunicating cysts (e-Fig. 106-19), although CT also may be diagnostic if it is performed as the initial procedure. When the duplication communicates with the perineum, introduction of contrast material is diagnostic. Investigation of other organ system anomalies should be undertaken with the appropriate imaging modality.
Necrotizing Enterocolitis
Overview: Necrotizing enterocolitis (NEC) is a termed coined in 195355 to describe an inflammatory condition affecting the gastrointestinal tract of neonates, most commonly premature infants who survive the first few days of life and are receiving enteral nutrition. Despite significant advances in neonatal intensive care, prevention of NEC remains an elusive goal. NEC is considered the most common newborn surgical emergency, and it has greater morbidity and mortality than all other surgical gastrointestinal conditions in this age group combined.56 Approximately 90% of cases of NEC occur in premature infants. The incidence is inversely proportional to gestational age, with most cases occurring in infants weighing between 500 and 750 g. NEC affects up to 11.5% of infants weighing less than 750 g and 4% of infants weighing between 1250 and 1500 g.57 Approximately 7% to 13% of cases occur in full-term infants56; risk factors include congenital heart disease and other conditions that affect splanchnic blood flow, such as maternal cocaine use and decreased prenatal umbilical blood flow.
A classification of enterocolitis severity based on clinical parameters was suggested in 1978 by Bell and colleagues,58 and with modifications, it is still used today.59,60 Stage I is sensitive but nonspecific, as it includes infants in whom NEC is suspected but not definitely diagnosed. Stage IIA is mild but definite NEC, stage IIB is NEC of moderate severity, stage IIIA refers to advanced NEC, and stage IIIB refers to NEC that has advanced to intestinal perforation.
Etiology: The most important risk factors for the development of NEC are prematurity and enteral feedings, which have been instituted in 90% of infants in whom NEC develops. Premature neonates have poorly developed intestinal motility that leads to stasis and altered ability to maintain homeostasis between proinflammatory and antiinflammatory compounds, to regulate splanchnic vasoconstriction and blood flow, and to maintain mucosal integrity. Proinflammatory compounds that are upregulated in NEC include platelet activating factor, tumor necrosis factor, and certain types of interleukins, leukotrienes, and oxygen free radicals. Accelerated apoptosis resulting from overproduction of inflammatory compounds has been shown to result in a break at the villus tip of the intestinal mucosal barrier, facilitating bacterial translocation and the inflammatory cascade.56,57,61
Clinical Findings: In Bell stage I disease, the infant exhibits relatively nonspecific symptoms, including systemic signs such as apnea, bradycardia, and temperature instability and abdominal signs consisting of abdominal distension, hemoccult positive stools, and increased gastric residuals. These signs also could be present in patients with other conditions, such as sepsis, and therefore they are sensitive but relatively nonspecific. By the time the child reaches stage IIA, clinical findings include abdominal tenderness and grossly bloody stools; by stage IIB, the child has progressed to thrombocytopenia and mild acidosis, as well as abdominal wall edema and tenderness. Infants with severe stage IIIA disease have metabolic and respiratory acidosis requiring intubation and signs of disseminated intravascular coagulation, with worsening abdominal wall erythema, edema, and induration. Patients with stage IIIB disease show clinical evidence of shock and gut perforation.
Complications of NEC in survivors relate to whether short bowel syndrome has developed after resection, with dependence on TPN and its attendant complications. Strictures occur in 9% to 36% of NEC survivors, most often in the colon (70% to 80%), with 21% of these strictures at the splenic flexure. Strictures are more common after medical management and present with failure to gain weight, rectal bleeding, or signs and symptoms of obstruction.5,56
Imaging: Abdominal radiographs have been the mainstay of imaging NEC, and the Bell criteria therefore have corresponding radiographic findings (Fig. 106-20). Findings of radiographs of infants with Bell stage I disease, similar to the physical findings, are nonspecific, demonstrating a normal bowel gas pattern or a mild ileus. Radiographic findings of stage IIA disease are more specific; an ileus pattern and focal pneumatosis can be seen. As the child’s disease progresses to stage IIB, radiographs show extensive pneumatosis with or without portal venous gas and ascites (see Fig. 106-20, A and B). Stage IIIA demonstrates more prominent ascites and persistent bowel loops, and pneumoperitoneum is the hallmark of stage IIIB disease (see Fig. 106-20, C). Periodic imaging is important and is used more frequently in patients who have more advanced disease.
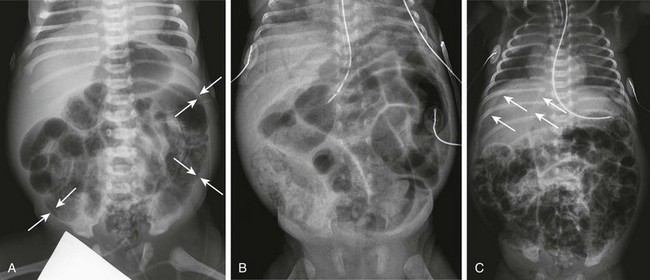
Figure 106-20 Necrotizing enterocolitis—acute.
A, Supine abdominal radiograph of a 7-day-old male premature infant presenting with abdominal distension and hematochezia demonstrates dilated loops of bowel and extensive pneumatosis. Arrows point to examples of areas in which the mucosa and submucosa are outlined as a white line bounded by luminal and subserosal gas. B, A supine abdominal radiograph of a 29-day-old premature infant progressing to acidosis and shock. In addition to marked abdominal distension, there is intramural gas and extensive portal venous gas seen over the liver. C, A supine chest and abdomen radiograph of a 10-day-old premature infant with extensive necrotizing enterocolitis progressing to perforation and free intraperitoneal air. Thin arrows point to the edge of the free air overlying the liver, and thicker arrows point to the outline of the falciform ligament. The patient is intubated and shows pulmonary abnormalities consistent with edema and atelectasis.
Intramural gas has a characteristic appearance as it wraps around the subserosa of the involved bowel loops. When the lumen is filled with gas, the mucosa and submucosa are outlined as a white line bounded by gas in the lumen centrally and subserosal intramural gas peripherally62 (see Fig. 106-20, A). When perforation is suspected, a horizontal beam radiograph is important to assess for small amounts of free air that may be difficult to detect with routine radiographs taken in the supine position.
Ultrasound has been shown to be useful and often complementary to radiography. Ultrasound can depict intramural and portal venous gas in many cases and can provide additional information about the presence, character and amount of ascites, the presence or absence of peristalsis, thickness of the bowel wall, and blood flow. Early in NEC, the bowel wall is thickened, loses the differential echogenicity of its layers, and becomes hyperemic; as the disease progresses, the bowel wall becomes thin and blood flow can no longer be detected.62
Patients in whom strictures develop may be evaluated with fluoroscopic examinations, typically contrast enema; 80% of strictures develop in the colon, and 15% develop in the terminal ileum56 (Fig. 106-21).
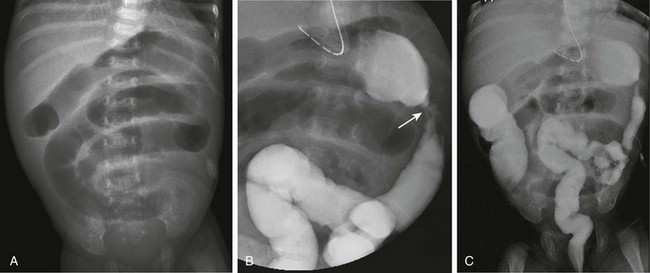
Figure 106-21 Necrotizing enterocolitis (NEC)—development of strictures.
A, An abdominal radiograph of 7-week-old premature infant with a history of NEC who presents with increasing residuals, vomiting, and abdominal distension shows dilated loops of bowel consistent with a distal, partial obstruction. B, A spot image from a contrast enema demonstrates an area of narrowing in the proximal descending colon, consistent with a stricture. C, An overhead radiograph confirms the partial obstruction at the site of the post-NEC stricture, which was confirmed surgically.
Treatment: Initial treatment of patients with NEC is medical, with fluid resuscitation, antibiotics, and TPN. If medical treatment fails, surgery may be indicated. The principal goals of surgical intervention are to remove bowel that has become gangrenous while preserving intestinal length. Optimal surgical timing, when the bowel is gangrenous but has not yet perforated, is difficult to define clinically or by imaging.56 Further, perforation is not always clinically or radiographically obvious; in fact, pneumoperitoneum in infants with surgically proven perforation is present in only approximately 63% of cases.56 Sonographic findings associated with adverse outcome include focal fluid collections and three or more additional findings: increased bowel wall echogenicity, bowel wall thickening or thinning, free fluid with internal echoes, absent bowel wall perfusion, and intramural gas.63 Additional criteria that suggest gangrene prior to perforation include abdominal wall erythema, fixed appearance of intestinal loop(s) on radiography or ultrasound, and clinical deterioration while receiving medical therapy.56,62 Patients with perforation who are deemed very poor surgical candidates may undergo initial peritoneal drainage.56,62
Cox, SG, et al. Colonic atresia: spectrum of presentation and pitfalls in management. A review of 14 cases. Pediatr Surg Int. 2005;21(10):813–818.
Emil, S, et al. Meconium obstruction in extremely low-birth-weight neonates: guidelines for diagnosis and management. J Pediatr Surg. 2004;39(5):731–737.
Epelman, M, et al. Necrotizing enterocolitis: review of state-of-the-art imaging findings with pathologic correlation. Radiographics. 2007;27(2):285–305.
Levitt, MA, Pena, A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33.
Martucciello, G, et al. Associated anomalies in intestinal neuronal dysplasia. J Pediatr Surg. 2002;37(2):219–223.
Puri, P, Shinkai, M. Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg. 2005;14(1):58–63.
Swenson, O. How the cause and cure of Hirschsprung’s disease were discovered. J Pediatr Surg. 1999;34(10):1580–1581.
References
1. Grosfeld, JL, Ballantine, TV, Shoemaker, R. Operative management of intestinal atresia and stenosis based on pathologic findings. J Pediatr Surg. 1979;14(3):368–375.
2. Martin, L, Zerella, J. Jejuno-ileal atresia: a proposed classification. J Pediatr Surg. 1976;11:399–403.
3. Cox, SG, et al. Colonic atresia: spectrum of presentation and pitfalls in management. A review of 14 cases. Pediatr Surg Int. 2005;21(10):813–818.
4. Fishman, SJ, et al. Nonfixation of an atretic colon predicts Hirschsprung’s disease. J Pediatr Surg. 2001;36(1):202–204.
5. Arca, M, Oldham, K. Atresia, stenosis and other obstructions of the colon. In: Coran A, et al, eds. Pediatric surgery. Philadelphia: Mosby, 2012.
6. Louw, J, Barnard, C. Congenital intestinal atresia observations on its origin. Lancet. 1955;266(6899):1065–1067.
7. Fairbanks, TJ, et al. A genetic mechanism for cecal atresia: The role of the Fgf10 signaling pathway. J Surg Res. 2004;120(2):201–209.
8. Dalla Vecchia, LK, et al. Intestinal atresia and stenosis: a 25-year experience with 277 cases. Arch Surg. 1998;133(5):490–496. [discussion 496-497].
9. Levitt, M, Peña, A. Anorectal malformations. In: Coran A, et al, eds. Pediatric surgery. Philadelphia: Mosby, 2012.
10. Levitt, MA, Pena, A. Anorectal malformations. Orphanet J Rare Dis. 2007;2:33.
11. Sadler, T. Digestive system. In: Langman’s medical embryology. Philadelphia: Lippincott Williams & Wilkins; 2010.
12. Herman, RS, Teitelbaum, DH. Anorectal malformations. Clin Perinatol. 2012;39(2):403–422.
13. Berdon, WE, et al. The radiologic evaluation of imperforate anus. An approach correlated with current surgical concepts. Radiology. 1968;90(3):466–471.
14. Levitt, MA, Kant, A, Pena, A. The morbidity of constipation in patients with anorectal malformations. J Pediatr Surg. 2010;45(6):1228–1233.
15. Haber, HP, et al. Transperineal sonography for determination of the type of imperforate anus. AJR Am J Roentgenol. 2007;189(6):1525–1529.
16. Han, TI, Kim, IO, Kim, WS. Imperforate anus: US determination of the type with infracoccygeal approach. Radiology. 2003;228(1):226–229.
17. Nievelstein, RA, et al. Magnetic resonance imaging in children with anorectal malformations: embryologic implications. J Pediatr Surg. 2002;37(8):1138–1145.
18. Martucciello, G, et al. Currarino syndrome: Proposal of a diagnostic and therapeutic protocol. J Pediatr Surg. 2004;39(9):1305–1311.
19. Currarino, G, Coln, D, Votteler, T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981;137(2):395–398.
20. Fleury, J, et al. Currarino syndrome as an etiology of a neonatal Escherichia coli meningitis. J Perinatol. 2007;27(9):589–591.
21. Cretolle, C, et al. Spectrum of HLXB9 gene mutations in Currarino syndrome and genotype-phenotype correlation. Hum Mutat. 2008;29(7):903–910.
22. Sen, G, et al. Familial Currarino syndrome presenting with peripheral primitive neuroectodermal tumour arising with a sacral teratoma. Pediatr Blood Cancer. 2008;50(1):172–175.
23. Martucciello, G, et al. Associated anomalies in intestinal neuronal dysplasia. J Pediatr Surg. 2002;37(2):219–223.
24. Parisi, MA. Hirschsprung disease overview. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews [Internet]. Seattle, Wash: University of Washington, Seattle, 2002. [Jul 12 [updated 2011 Nov 10]].
25. Amiel, J, Lyonnet, S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet. 2001;38(11):729–739.
26. Haddad, GG, et al. Congenital failure of automatic control of ventilation, gastrointestinal motility and heart rate. Medicine (Baltimore). 1978;57(6):517–526.
27. Bajaj, R, et al. Congenital central hypoventilation syndrome and Hirschsprung’s disease in an extremely preterm infant. Pediatrics. 2005;115(6):e737–e738.
28. Langer, JC. Hirschsprung disease. In: Coran A, et al, eds. Pediatric surgery. Philadelphia: Mosby, 2012.
29. Carrasquillo, MM, et al. Genome-wide association study and mouse model identify interaction between RET and EDNRB pathways in Hirschsprung disease. Nat Genet. 2002;32(2):237–244.
30. Heanue, TA, Pachnis, V. Prospective identification and isolation of enteric nervous system progenitors using Sox2. Stem Cells. 2011;29(1):128–140.
31. Rudolph, C, Benaroch, L. Hirschsprung disease. Pediatr Rev. 1995;16(1):5–11.
32. Badner, JA, et al. A genetic study of Hirschsprung disease. Am J Hum Genet. 1990;46(3):568–580.
33. Newman, B, Nussbaum, A, Kirkpatrick, JA, Jr. Bowel perforation in Hirschsprung’s disease. AJR Am J Roentgenol. 1987;148(6):1195–1197.
34. Newman, B, et al. Appendiceal perforation, pneumoperitoneum, and Hirschsprung’s disease. J Pediatr Surg. 1988;23(9):854–856.
35. Hackam, DJ, Filler, RM, Pearl, RH. Enterocolitis after the surgical treatment of Hirschsprung’s disease: risk factors and financial impact. J Pediatr Surg. 1998;33(6):830–833.
36. Ellis, H, Kumar, R, Kostyrka, B. Neonatal small left colon syndrome in the offspring of diabetic mothers—an analysis of 105 children. J Pediatr Surg. 2009;44(12):2343–2346.
37. Siddiqui, MM, Drewett, M, Burge, DM. Meconium obstruction of prematurity. Arch Dis Child Fetal Neonatal Ed. 2012;97(2):F147–F150.
38. Keckler, SJ, et al. Current significance of meconium plug syndrome. J Pediatr Surg. 2008;43(5):896–898.
39. Berrocal, T, et al. Congenital anomalies of the small intestine, colon, and rectum. Radiographics. 1999;19(5):1219–1236.
40. Fadda, B, et al. Neuronal intestinal dysplasia. Critical 10-years’ analysis of clinical and biopsy diagnosis. Z Kinderchir. 1983;38(5):305–311.
41. Puri, P. Intestinal dysganglionosis and other disorders of intestinal motility. In: Coran A, et al, eds. Pediatric surgery. Philadelphia: Mosby, 2012.
42. Puri, P, et al. Neuronal colonic dysplasia: an unusual association of Hirschsprung’s disease. J Pediatr Surg. 1977;12(5):681–685.
43. Berger, S, et al. Congenital malformations and perinatal morbidity associated with intestinal neuronal dysplasia. Pediatr Surg Int. 1998;13(7):474–479.
44. Rodriguez, L, Flores, A. Chronic intestinal pseudo-obstruction. In: Liacouras C, Piccoli D, Bell L, eds. Liacouras & Piccoli: pediatric gastroenterology: requisites. Philadelphia: Mosby, 2008.
45. Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 52-1991. An eight-year-old girl with recurrent abdominal distention after surgical correction of Hirschsprung’s disease. N Engl J Med. 1991;325(26):1865–1874.
46. Berdon, WE, et al. Megacystis-microcolon-intestinal hypoperistalsis syndrome: a new cause of intestinal obstruction in the newborn. Report of radiologic findings in five newborn girls. AJR Am J Roentgenol. 1976;126(5):957–964.
47. Puri, P, Shinkai, M. Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg. 2005;14(1):58–63.
48. Raofi, V, et al. Combined living-related segmental liver and bowel transplantation for megacystis-microcolon-intestinal hypoperistalsis syndrome. J Pediatr Surg. 2008;43(2):e9–e11.
49. Piaseczna Piotrowska, A, et al. Interstitial cells of Cajal in the human normal urinary bladder and in the bladder of patients with megacystis-microcolon intestinal hypoperistalsis syndrome. BJU Int. 2004;94(1):143–146.
50. Piotrowska, AP, et al. Alterations in smooth muscle contractile and cytoskeleton proteins and interstitial cells of Cajal in megacystis microcolon intestinal hypoperistalsis syndrome. J Pediatr Surg. 2003;38(5):749–755.
51. Rolle, U, et al. Megacystis-microcolon-intestinal hypoperistalsis syndrome: evidence of intestinal myopathy. Pediatr Surg Int. 2002;18(1):2–5.
52. Rolle, U, Puri, P. Structural basis of voiding dysfunction in megacystis microcolon intestinal hypoperistalsis syndrome. J Pediatr Urol. 2006;2(4):277–284.
53. Lund, D. Alimentary tract duplications. In: Coran A, et al, eds. Pediatric surgery. Philadelphia: Mosby, 2012.
54. Yousefzadeh, DK, et al. Tubular colonic duplication—review of 1876-1981 literature. Pediatr Radiol. 1983;13(2):65–71.
55. Schmid, O, Quaiser, K. Uer eine besondere schwere Verlaufende form von Enteritis beim Saugling. Oesterr Z Kinderh. 1953;8:114.
56. Sylvester, K, Liu, G, Albanese, CT. Necrotizing enterocolitis. In: Coran A, et al, eds. Pediatric surgery. Philadelphia: Mosby, 2012.
57. Wu, SF, Caplan, M, Lin, HC. Necrotizing enterocolitis: old problem with new hope. Pediatr Neonatol. 2012;53(3):158–163.
58. Bell, MJ, et al. Neonatal necrotizing enterocolitis. Therapeutic decisions based upon clinical staging. Ann Surg. 1978;187(1):1–7.
59. Walsh, MC, Kliegman, RM. Necrotizing enterocolitis: treatment based on staging criteria. Pediatr Clin North Am. 1986;33(1):179–201.
60. Kliegman, RM, Walsh, MC. Neonatal necrotizing enterocolitis: pathogenesis, classification, and spectrum of illness. Curr Probl Pediatr. 1987;17(4):213–288.
61. Posencheg, M, Pereira, G. Necrotizing enterocolitis. In: Liacouras C, Piccoli D, Bell L, eds. Liacouras & Piccoli: pediatric gastroenterology: requisites. Philadelphia: Mosby, 2008.
62. Epelman, M, et al. Necrotizing enterocolitis: review of state-of-the-art imaging findings with pathologic correlation. Radiographics. 2007;27(2):285–305.
63. Silva, CT, et al. Correlation of sonographic findings and outcome in necrotizing enterocolitis. Pediatr Radiol. 2007;37(3):274–282.