73 Tumor-Targeted Radioisotope Therapy
History of Radioimmunotherapy
In the late 1940s, a method was developed for linking iodine-131 (131I) to proteins, including antibodies (Abs), without significantly altering biologic function, such as Ab immunologic specificity.1 Using this technique, Pressman and Korngold2 demonstrated in 1953 that intravenously administered Ab accumulated to a slightly greater extent in tumors than in normal tissue.3
Clinical trials involving radioimmunotherapy (RIT), or tumor-targeted systemic radiotherapy, began in the 1950s with polyclonal Abs when Beierwaltes4 administered 131I-labeled rabbit Ab to 14 patients with metastatic melanoma and achieved a pathologically documented complete response (CR) in one patient. In the 1960s, Spar and coworkers5 administered 131I-antifibrinogen polyclonal Abs to cancer patients and observed symptomatic improvement in some cases. In 1965, polyclonal Abs began to be developed against specific tumor-associated antigens (e.g., carcinoembryonic antigen). This was associated with greater Ab uptake in tumors. In the 1970s, Ettinger et al.6,7 began to treat patients with cholangiocarcinomas and hepatomas with 131I-anticarcinoembryonic antigen and 131I-antiferritin polyclonal Abs in combination with EBRT (EBRT), doxorubicin, and 5-fluorouracil chemotherapy. A 30% or greater decrease in tumor size was observed in six of nine evaluable patients. Order et al.8 pursued an aggressive combined modality approach to hepatoma, with one trial arm including radiolabeled polyclonal Abs. Because of their polyclonal nature, these tumorphilic radiolabeled Ab mixtures were heterogeneous in their pharmacokinetic and tumor binding properties.
When Kohler and Milstein published, in 1975, a technique9 for the production of monoclonal antibodies (mAbs) in which each clone selected could produce one molecular Ab species of a predefined specificity, this Nobel Prize-winning technology also provided the critical keystone for the development of RIT. With this hybridoma approach, mAbs to predefined cancer-associated antigens could be produced in gram quantities, thus revolutionizing the field. With the exception of anti-idiotypic antibodies, no mAb is absolutely specific for tumor, although tumor to the more sensitive normal tissue radiation dose ratios (therapeutic index [TI]) range from 2 : 1 to greater than 30 : 1 with tumor-targeting radiolabeled mAbs in cancer patients.10,11
MAbs have been conjugated with chemotherapeutic agents, biologic toxins, and radioisotopes in an attempt to use them to selectively target malignant cells while sparing normal tissues.12 Treatment of cancer with mAbs conjugated with chemotherapeutic agents or toxins has faced a number of obstacles, including: (1) the requirement that every malignant cell express the target antigen; (2) the development of multidrug resistance; (3) the degradation of the drug or toxin by lysosomes following endocytosis; (4) the formation of Abs against the toxin; and (5) a dose-limiting capillary leak syndrome manifested by hypoalbuminemia and edema.13,14
Tumor-targeted radioisotopes have several advantages over drugs or toxins: (1) β particles emitted by radionuclides such as iodine-131, yttrium-90 (90Y), or copper-67 (67Cu) can kill adjacent tumor cells (crossfire; Table 73-1), partially overcoming heterogeneous antigen expression and poor tumor penetration; (2) radioisotopes are less subject to multidrug resistance mechanisms; and (3) pharmacokinetic and dosimetric measurements can be obtained from scintigraphic imaging, helping to prevent unacceptable toxicity and optimize the prescribing of therapy.
Radioimmunoconjugates, however, have usually been made with the entire 150 kD mAb, resulting in large molecules that have slow blood clearance, retention in normal organs involved in Ab metabolism, and slow, incomplete tumor penetration. Combined with antigenic heterogeneity, only 0.0001% to 0.1% of the injected dose of Abs binds to each gram of tumor.15 Consequently, RIT has typically produced tumor doses of less than 20 Gy unless bone marrow support allowed larger injected doses. Challenges facing RIT, along with possible solutions,16 are presented in Table 73-2.
Table 73-2 Challenges to Improve the Therapeutic Index for Radioimmunotherapy
| Problems Facing Improvement of the Therapeutic Index | Possible Solution |
|---|---|
| Marrow toxicity | Small targeting molecules |
| Pretargeting approach | |
| Normal tissue antigen | Preinfusion of cold Ab |
| Pretargeting approach | |
| Nonspecific Fc receptor binding | Saturate receptors with cold Ab |
| Intracavitary (e.g., intraperitoneal administration) | |
| Ab fragments: F(ab′)2, svFc, diabody… | |
| Pretargeting | |
| Tumor penetration | Ab fragments: F(ab′)2, svFc, diabody… |
| Multimodality approach: chemotherapy, external beam… | |
| Pretargeting | |
| Inject Ab directly into the tumor |
Ab, Antibody; HAMA, human antimouse antibodies; mAb, monoclonal antibody.
Antibody Structure and Function
These contain variable and constant regions, with the variable regions of the heavy and light chains (VH and VL) containing the antigen-binding site (Fig. 73-1A). The major constant portion (Fc) of the Ab molecule contains a number of molecular signals that mediate biologic functions such as complement binding, hepatocyte binding, and recruitment or activation of effector cells, such as monocytes and natural killer cells. Pepsin digests of Abs result in a short divalent (F(ab′)2) antigen-binding molecule without most of the Fc fraction, and papain digestion results in an even smaller antigen-binding monovalent fragment (Fab) (see Fig. 73-1A).17,18 Genetic engineering, using the VH and VL MAb DNA, has fashioned smaller binding units, fragment (Fv), as well as the linked single chain fragments (scFvs). These may provide the building blocks for tumor-targeting molecules of the future (Fig. 73-1B).19

Many tumor-binding mAbs can produce antitumor effects: (1) complement-mediated cytotoxicity20; (2) Ab-dependent cellular cytotoxicity21,22; (3) receptor-mediated apoptosis23,24; (4) interference with growth-related receptors25–29; (5) stimulation of the humoral immune system producing a vaccine-like response30–32; or (6) the triggering of apoptosis by appropriate intracellular signal transduction. Smaller binding units (scFv) seldom have these functions, but can be linked into molecular formats developed to improve the TI, such as small high-affinity rapid-targeting radioactive molecules or larger multidentate and multifunctional pretargeted RIT drugs (see Fig. 73-1B).13
Radioisotopes
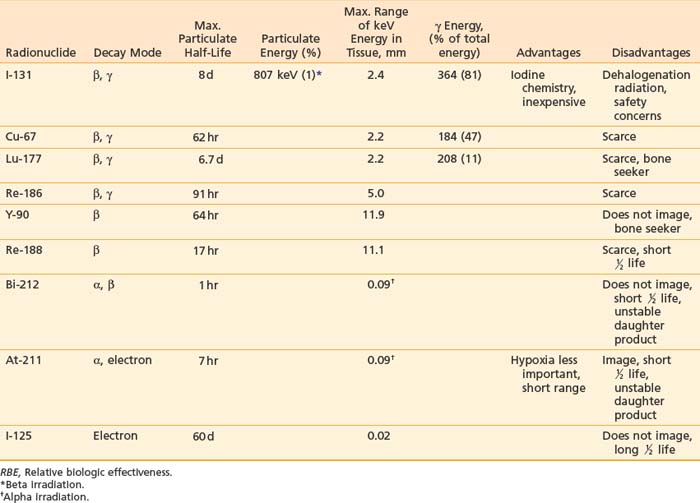
Many radioisotopes suitable for systemic tumor-targeted radionuclide therapy are presented in Table 73-1. Radioisotopes undergo decay because of the inherent instability of their nuclei. The most important types of decay for medical use lead to γ emission, β and α particles, and electron capture resulting in auger electrons. 131I has both β and γ emissions, the latter being less absorbed by the body, allowing quantitative scintigraphic imaging to determine the pharmacokinetics and calculate the dose to tumors and normal organs from the radiopharmaceutical. By contrast, β particles travel only millimeters to centimeters in tissue and deposit their energy in the vicinity of the point of decay. Beta emissions from radioisotopes such as 131I, 177Lu, 90Y, or 67Cu targeting antigen-positive tumor cells can kill nearby antigen-negative tumor cells through a “crossfire” effect, which increases the homogenicity of tumor radiation.33–36
131I has been used in many clinical trials because of its well-known radiochemistry, availability, and low cost. However, if the radioiodinated MAb is taken into some tumor cells via internalization (endocytosis), the 131I can be enzymatically removed and excreted from the tumor before it can deposit all of its particulate energy, thus, lowering the potential tumor dose. The same process, however, can result in a lower dose to the liver and kidneys since these products, small iodinated peptides and free iodine, are rapidly excreted via the kidneys. It should be noted that free iodine in the blood can be concentrated in the thyroid gland if not blocked in advance by an oral potassium iodine solution. Since the γ rays emitted by 131I have a relatively high energy (364 keV),37 practical considerations are needed to minimize the dose of radiation that is delivered to family members and health care personnel. Patients receiving high doses who are unable to be properly managed at home may need hospitalization to limit the irradiation of others.
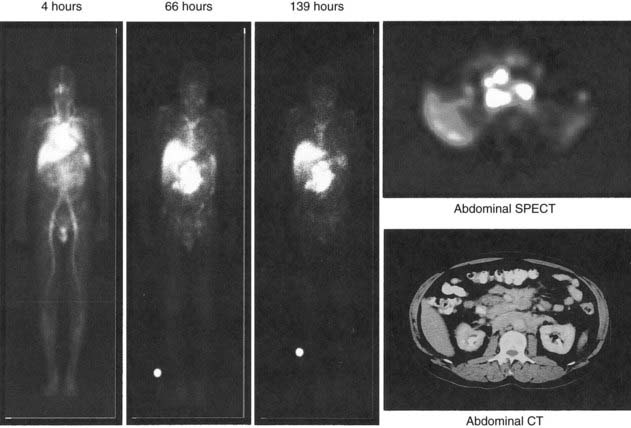
Yttrium-90, a pure β emitter, has three properties that make it an attractive choice for RIT38–40: (1) a high β energy (Eβmax = 2.29 MeV; maximum range of particulate energy in tissue = 12 mm), which enables it to kill adjacent tumor cells and thus, increase the homogenicity of tumor radiation33,34; (2) metal chemistry, which facilitates the presynthesis of storable Ab chelate conjugates that can be quickly and easily radiolabeled in RIT form when needed; and (3) a half-life (2.67 days) that is useful with intact mAbs, but may take 1 to 3 days to concentrate in tumors, compared with smaller fragments and peptides, with maximal concentration within a day. However, 90Y needs well-selected chelation chemistry since free 90Y will accumulate in cortical bone, increasing the radiation to bone marrow. Furthermore, although 90Y-labeled Abs allow easy patient management because they have no primary γ emissions, pharmacokinetics and thus, dosimetry cannot be accurately quantitated by imaging the secondary 90Y emissions. Thus, γ-emitting indium-111 (111In) in the same chelated mAb, when concurrently or sequentially administered, acts as an effective surrogate to provide pharmacokinetic information for its 90Y counterpart (Fig. 73-2).
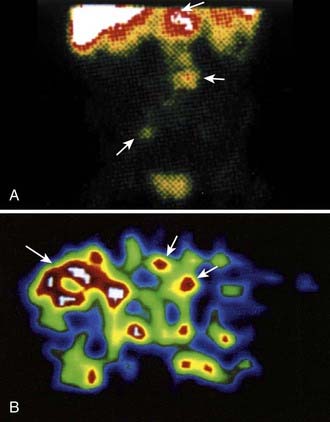
Other radioisotopes have been studied for use in RIT. Generally their availability, cost, labeling, or chemistry characteristics have outweighed the suitability of their physical characteristics for therapy. An example is 67Cu, suitable for RIT because it has 99mTc-like photons for imaging (Fig. 73-3), has a sufficiently long physical half-life for use with intact murine mAb (62 hours), emits β particles similar to 131I, and does not accumulate in bone. Preliminary studies demonstrate 67Cu may provide a better TI than 131I, based in part on its relatively long retention in tumors,41,42 resulting in 1.5- to 5-fold higher tumor doses when equivalent radioactivities (mCi) are administered.43,44 The disadvantage of 67Cu is availability and cost.
Alpha particles deposit energy over a much shorter range than β particles, typically 50 to 100 µm, targeting lesions of 1 to 2 mm diameters (micrometastasis). Thus, radioisotopes that undergo α decay, such as astatine-211 (211At) and bismuth-212 (212Bi) must be within approximately 1 to 2 cell diameters of a tumor cell to kill it.45,46 Although nearby normal cells are spared by α emitters, adjacent nontargeted tumor cells are spared as well. Therefore, these radionuclides are ideal candidates for targeting microscopic residual disease or leukemia, where β particles are relatively inefficient, depositing much of their energy outside their small tumor volume. 211At and 212Bi have been used in clinical RIT trials of leukemia and brain tumors.46,47 The short physical half-lives (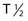 ) of these radionuclides (
) of these radionuclides (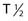 212Bi = 45.6 min;
212Bi = 45.6 min; 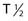 211At = 7 hr) and complex decay cascades has made routine clinical use more difficult (see Table 73-1).45,48
211At = 7 hr) and complex decay cascades has made routine clinical use more difficult (see Table 73-1).45,48
However, the development of new chelators for longer-lived α emitters (e.g., Actinium-225 (225Ac) with 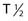 = 10 days) may make future use of RIT with α emitters logistically more feasible and widely available.49 Another promising approach for increasing the efficacy of RIT with α emitters is to link them to small engineered antitumor antibody fragments that are rapidly taken up by targeted tumor. In a preclinical model, diabody molecules were found to be effective agents for targeted radioimmunotherapy of solid tumors using 211At.50
= 10 days) may make future use of RIT with α emitters logistically more feasible and widely available.49 Another promising approach for increasing the efficacy of RIT with α emitters is to link them to small engineered antitumor antibody fragments that are rapidly taken up by targeted tumor. In a preclinical model, diabody molecules were found to be effective agents for targeted radioimmunotherapy of solid tumors using 211At.50
Chelates
Isotopes are attached to mAbs by different methods. The stability of isotope on the mAb is crucial for RIT to maximize the TI. 131I is covalently linked to the Fc portion by iodination of tyrosine residues, whereas radioisotopes of metals such as 90Y, 67Cu, 177Lu, and rhenium-188 (188Re) are more firmly attached by an intermediary molecule known as a chelate. Diethylene-triaminepentaacetic acid (DTPA) was initially used for 111In and 90Y.51 Macrocyclic bifunctional agents48 such as DOTA (1,4,7,11-tetrazacyclododecane-N,N′,N″,N′″-tetraacetic acid) or TETA (1,4,7,11-tetraazacyclotetradecane-N,N′,N″,N′″-tetraacetic acid)52 have increasingly been used because they are useful radiometals with more stable properties in vivo.52–55 For example, stability of yttrium-DOTA minimizes the loss of 90Y to the bone.55
Radiobiology
RIT differs in three important ways from conventional external beam radiotherapy (EBRT) with EBRT using linear accelerators: (1) radiation is continuously delivered to tumors at a low dose rate that initially increases with time as radiolabeled Abs accumulate in a tumor (maximal tumor dose rates approximate ≤0.40 Gy/hr for 90Y-labeled and 67Cu-labeled Abs and <0.10 Gy/hr for 131I-labeled Abs56) and then decreases because of physical decay and biologic clearance35 (compared with a high [∼150 Gy/hr]) constant dose rate intermittently delivered to a limited region of the body with (EBRT); (2) a low dose of radiation is delivered to the whole body; and (3) Abs themselves may, in some cases, exert antitumor effects through biologic mechanisms. For most tumors, one would expect the relative effectiveness of RIT to be approximately 20% less than that of dose-equivalent EBRT, since the low dose rates characteristic of RIT allow time for repair of sublethal damage.56–59 This has not necessarily been the case.
Most in vitro radiobiologic studies of dose rate effects on tumors of 0.40 Gy/hr or less have used nearly constant dose rates.57 In general, these studies have indicated that dose rates of 0.23 Gy/hr or more are required to stop the growth of malignant epithelial cells in vitro,60 although lower dose rates such as 0.09 to 0.11 Gy/hr can halt the growth of HeLa and Morris hepatoma cells61 (dose rates as low as 0.05 Gy/hr can stop the growth of lymphoma cells in vitro).62
Although radiolabeled Abs emit radiation at a low dose rate, several studies of human tumor xenografts in mice have suggested that RIT can, in certain instances, exert greater antitumor activity than dose-equivalent EBRT.63–65 A number of explanations have been proposed to account for an inverse relationship between dose rate and cell killing: (1) reoxygenation during the protracted course of irradiation delivered by RIT, increasing the radiosensitivity of tumor cells66; (2) targeting by RIT of a rapidly proliferating subpopulation of tumor cells (growth fraction) that is largely responsible for tumor doubling67; and (3) low dose rate irradiation that, in some cases, may cause tumor cells to accumulate in the radiosensitive G2/M phase of the cell cycle (G2 block) to a greater extent than conventional dose rate irradiation.68 Furthermore, the importance of sublethal damage repair mechanisms may be minimal for tumors (e.g., lymphomas that radiobiologically have a small shoulder and large α/β ratio),58 and some MAbs have biologic effects that may acutely increase tumor blood flow, elicit an inflammatory or cell mediated immune response, or sensitize tumor cells to the effects of radiation.69 In addition, low-dose-rate irradiation may induce apoptosis to a much greater extent than high-dose-rate irradiation in some tumor types (e.g., lymphoma),70,71 and this effect may be enhanced in a synergistic manner by various biologic effects of some tumor targeting mAbs.
There is evidence that apoptosis is the major response of many tumors to RIT. In experimental human lymphoma tumors (Raji) treated with 67Cu-1-2IT-BAT-Lym-1, apoptosis preceded tumor regression by 4 to 6 days. In these therapy-resistant human lymphoma tumors, apoptosis was convincingly demonstrated to be a major mechanism for the effectiveness of RIT and occurred by p53-independent mechanisms.72 Human breast carcinoma xenografts (HBT3477) have also demonstrated increased apoptotic activity independent of p53 after RIT.73 In addition to the induction of apoptosis, RIT and the associated low dose rate and relatively low dose irradiation can have a variety of important biological effects on tumors, normal tissues and the immune response.74 Potential clinical applications of the low dose rate irradiation from RIT include augmentation of antitumor responses, with possible use in conjunction with tumor vaccines, and radioadaption of normal tissues. Additional studies are needed to optimize the therapeutic utility of both bystander and low dose rate effects in order to maximize the therapeutic index of RIT.74
Linear energy transfer (LET) represents the average energy (keV) locally imparted to a medium by radiation in traversing one micrometer along its path or track.66 Because energy exchanges with matter are widely spaced with low LET radiation, a β particle has only a small probability of releasing enough energy along its track to produce DNA breaks. Approximately 200 DNA double-strand breaks per cell are required to sterilize 99% of a tumor-cell population when low LET radiation is administered.34 The cytotoxicity of low LET radiation may also be diminished by tumor hypoxia.75 In addition, low LET radiation may not be completely tumoricidal because repair of sublethal damage can occur at low dose rates.74
By using a radionuclide that emits β particles with a high energy (e.g., 90Y rather than 131I), dose rates can be increased up to tenfold because of the higher energy deposited by each disintegration of 90Y relative to 131I (see Table 73-1).76,77 High LET irradiation (α emitters) is densely ionizing along particle tracks and consequently is more efficient at producing DNA breaks than low LET radiation. The cell killing produced by high LET radiation is less dependent upon hypoxia77 since it directly produces irreparable DNA breaks, whereas low LET radiation forms highly reactive molecules that produce repairable DNA breaks and relies upon oxygen to prevent repair of these breaks.66 In conclusion, a better understanding of mechanisms and timing of RIT-induced tumor cell death may lead to the selection of combined therapies that enhance tumor responses without increased toxicities.
Dosimetry and Therapeutic Index
The concept of dosimetry is important for treatment planning and the assessment of results. Compared with dosimetry for EBRT, dosimetry for RIT is less precise and is dependent upon (1) the kinetics of uptake and clearance of radiolabeled Abs; (2) the distribution of radiolabeled Abs; and (3) the radioisotope attached to the Abs.78–80
Serial quantitative studies with γ camera imaging (see Fig. 73-2) have traditionally been used to measure radioactivity, determine pharmacokinetics, and calculate dosimetry. Magnetic resonance (MR) and computed tomography (CT) imaging provide normal tissue and tumor volumes.81,82 However, accurate measurement of the radioactivity in small, deep-seated tumors, surrounded by background radioactivity, is difficult. Several excellent overviews of quantitative imaging techniques and approaches to treatment planning in RIT have been published.80,82–85 The medical internal radiation dose (MIRD) method has traditionally been used to calculate absorbed doses of radiation to normal organs, tumor, and the whole body following the administration of radiopharmaceuticals.86 The MIRD method makes a number of assumptions, including: (1) radioactivity is uniformly distributed in that entity, and (2) nonpenetrating, (e.g., β) emissions from radionuclides are completely absorbed in a tumor or organ. These assumptions are reasonable provided that the range of β emissions does not exceed the diameter of the target, which is usually not the case. Specific absorbed fractions (S factors) for penetrating emissions have been calculated for many organs using mathematical anthropomorphic phantoms for a “standard” man, woman, child, and infant that approximate human anatomy. Actual patient organ volumes provide the best alternative.87 For radionuclides that undergo α decay (or electron capture), energy deposition needs to be considered at the cellular and subcellular level,88 which entails the use of a Monte Carlo model89 or analytical microdosimetry with Fourier transform techniques.90
The dosimetric process currently employed in many therapeutic regimens consists of the pretherapy administration of a tracer dose of RIT (or its γ-emitting surrogate) followed by serial planar conjugate view imaging using a γ camera to determine the quantitative spatial distribution of the radionuclide (see Fig. 73-3A). Three-dimensional images from γ camera tomographic scintigraphy (SPECT) provide more detailed pictorial information about deep-seated tumors, but have inherent problems with providing quantitative data (see Fig. 73-3B). Using appropriate standards and planar imaging data, cumulated activity (area under the curve [AUC]), (i.e., the amount of radioactivity integrated over time or residence time), can be calculated for organs or tumor and related to the organ or tumor volume. MIRD “S” values provide the tissue-absorbed fraction for final calculation of absorbed dose to specific organs and tumors.91
Various dosing methods for prescribing treatment doses for RIT have been used (Table 73-3).91 Most clinical trials present their results by reporting the injected activity in millicuries (mCi) or Becquerels (Bq) per body surface area (m2) or weight (kg). One mBq is equal to 2.7 × 10−2 mCi (0.027 mCi). The tumor-absorbed doses are reported either in absolute numbers (cGy or rads) or relative to the injected activity (e.g., cGy/mBq, rads/mCi). Except for intratumoral administration, the average tumor doses delivered over several days in most RIT using nonmyeloablative therapy are 7 to 30 Gy, and in myeloablative regimens, are 30 to 60 Gy.
| Dosing Methods | Example |
|---|---|
| Radiation dose (cGy)-based methods/prior dosimetry study | |
| Marrow radiation dose (nonmyeloablative) | 131I-tositumomab |
| Total body surrogate | |
| Blood/body surrogate | |
| Marrow imaging | |
| Critical (dose-limiting) organ radiation (myeloablative) | 131I-tositumomab |
| Radionuclide dose (radioactivity, gBq or mCi)-based methods | |
| Fixed total gBq (mCi) | 131I-antitenascin, 90Y-antitenascin |
| gBq (mCi) per unit body weight (kg) | 90Y-tiuxetan ibritumomab, 90Y-CC49 |
| gBq (mCi) per unit body surface area (m2) | 131I-Lym-1, 131I-hMN14 |
Adapted from DeNardo GL, Juweid ME, White CA, et al: Role of radiation dosimetry in radioimmunotherapy planning and treatment dosing, Crit Rev Oncol Hematol 39:203, 2001.
Approaches to Improve Therapeutic Index
Enhancing the TI is critical to the future success of systemic tumor-targeted radiation therapy, because dose intensification is needed to enhance the efficacy of RIT.92 Circulating radiolabeled mAb irradiates the most sensitive tissue, bone marrow. Marrow toxicity is thus the most frequent dose-limiting toxicity from non–marrow-supported RIT. In an effort to reduce marrow radiation from exposure to hours of circulating radioactive drug, small molecule radionuclide carriers, cleavable linkages of radioactivity, pretargeting, or multistep strategies have been developed that demonstrate exciting potential. These approaches have been applied to increase the TI in both NHL and solid tumors.11–13,93,94
Cleavable Linkers
Organs such as the liver or kidneys are involved in the metabolic pathway of proteins and thus concentrate radiolabeled mAb (in the liver) or small fragments (in kidney tubules). Several strategies have been described to accelerate clearance of radioactivity from these organs and thus to enhance TI.95 These include cleavable linkers, the pretargeting approach previously mentioned to lower blood and marrow dose, and Ab Fvs.
As an example, 90Y DOTA linked to m170 mAb by a peptide preferentially catabolized in the liver resulted in a one third reduction in liver concentration and thus radiation doses in prostate and breast cancer patients, compared to the more conventional DOTA-mAb linkage.96 Importantly, tumor targeting was not diminished (see Fig. 73-3). The results of this study were consistent with in vitro data, which indicated that the peptide linker of DOTA-peptide-mAb is catabolized by cathepsin B, releasing the small radioactive molecule that should be rapidly excreted.95 A variety of concepts have been used to develop linkers to lower this retention of radioactivity in the renal tubule, since small radioactive mAb fragments and peptides have little hepatic uptake and more rapid blood clearance, but usually demonstrate high localization in the kidneys. Zimmermann et al.97 evaluated the peptide-linked copper chelators CPTA-triglycyl-L-p-isothiocyanato-phenylalanine (CPTA-R1-NCS) and the DOTA-triglycyl-L-p-isocyanato-phenylalanine (DOTA-R1-NCS) coupled to F(ab′)2 Fvs. DOTA-R1-F(ab′)2 achieved the best tumor/tissue ratios. Arano et al.98–100 took advantage of lysine-specific carboxypeptidase activity in the tubular cell brush border to develop linkages that appear effective in reducing renal concentration of radiometal from radiolabeled Ab fragments and peptides.
Pretargeted RIT for Dose Intensification
This drug delivery system separates the specific localization of the initially injected larger tumor-targeting mAb from the subsequently injected smaller radioactive molecule, which is quickly bound at the tumor site or rapidly excreted in the kidney. The procedure was first described by Goodwin et al.101 and provides an alternative to increase the therapeutic ratio by selectively targeting the tumor tissue while minimizing the radiation-absorbed dose to normal tissues, particularly blood and marrow, by the excretion of unbound radioactivity. A number of pretargeting systems have been described that use different first-step conjugates94 that can be biotinylated,102 streptavidin conjugated,103 or use bifunctional Abs,104 or oligonucleotide conjugates.105 An important proof of principle pretargeting study for delivering systemic radiation therapy to metastatic cancers has been reported by Breitz et al.106 using a streptavidin/biotin strategy in a three-step pretargeting approach with an anti-CD20 mAb in patients with NHL. Tumor-to-whole body-ratios were obtained that were substantially higher than those achieved with conventional anti-CD20 RIT. In these pretargeting studies using scFv with a streptavidin conjugate, the clearing agent biotin-galactose–human serum albumin was used before injection of radiolabeled DOTA-biotin,107,108 with resultant reduction in circulating Ab by greater than 95%. Weiden et al.11 reported a three-step procedure in patients with NHL. The anti-CD20, C2B8 Ab conjugated to streptavidin (C2B8/SA), was followed by 90Y-DOTA-biotin after a clearing agent; 90Y-DOTA-biotin was tolerated at administrated doses of 29 ± 23 cGy/mCi and mean tumor to whole body ratios of 38 : 1. In six of seven patients with injected activities of 30 to 50 mg/m2 90Y, three CRs and one partial response (PR) were observed. However, high cure rates and high tumor-to-normal ratios in mice,93,109 as well as promising clinical results110 using bispecific Abs (F[ab′]2 fragment) followed by radiolabeled bivalent hapten, have also been achieved without the use of clearing agents.
RIT results in patients reported by Barbet et al.93 using bispecific Abs as the pretargeting agent and subsequent radioiodinated bivalent haptens to bind and crosslink antigens on targeted tumor cells, demonstrated useful effects of modestly increasing radioactive hapten circulation time, with greatly increased tumor retention by crosslinking of the bivalent second agent on the tumor. This bivalent radiolabeled hapten approach increased the tumor-to-blood ratio eight times over previous methods.111
Fractionated Radioimmunotherapy for Dose Intensification
Dose distribution after RIT is known to be highly inhomogeneous, due to the inability of mAb to penetrate uniformly throughout the tumor.112,113 Vascular density, inhomogeneous tumor blood flow, or interstitial pressure are among the related factors that cause an uneven tumor distribution.112,114,115 Single administration of low-dose-rate irradiation may lead to cold spots in the tumor and subsequently to tumor recurrence.116 Several strategies have been proposed to overcome this biologic problem related to mAb.117 The goal of using multiple fractions in RIT is to deliver a more homogeneous distribution of antibody, and therefore radioactivity, with higher associated cumulative radiation doses in tumor and fewer and/or less severe side effects.117,118
There is experimental and clinical evidence that fractionated RIT is effective in different tumor systems. Schlom et al. demonstrated the effect of fractionation in a human colon xenograft using 131I-B72.3 against the TAG-72 antigen. Lethal doses in mice were fractionated to assess the toxicity and the tumor growth effect. One 600-mCi dose produced a mortality rate of 60% in the mice, whereas two 300 mCi weekly doses produced 90% response rates and 10% toxic deaths.119 Dose fractionation permitted further dose escalation in this model. Three weekly doses were allowed with improved results. The same effect was also observed by Buchsbaum et al.120
Clinical experiences with multiple doses of RIT are described earlier in this chapter.121,122 Several important issues must be addressed for optimization of fractionated RIT: (1) decreased immunogenicity of mAb; (2) number of radionuclide doses; (3) dose amount per cycle; (4) interval between doses; and (5) optimal radionuclide physical half-life for a specific treatment interval.117
Clinical Studies
Lymphoma
Since 1985, promising results for RIT in NHL have been described. A summary of the main mAbs (anti-CD20, -CD22, -Lym-1) used in different clinical trials, isotopes, and commercial names are described in Table 73-4. RIT for NHL uses dose regimens that consist of either a single dose of a radionuclide chemically linked to an antibody, at either nonmyelosuppressive or myeloablative dose levels, as well as the use of multiple (fractionated) doses, consisting of dose spacing by days or weeks.
Lym-1
After developing pharmacokinetic data in patients for 131I-mAb on fragments from 1982 to 1984, DeNardo et al.123 conducted an early clinical study of 131I-Lym-1 in patients with advanced lymphoma (Fig. 73-4). Substantial responses in most of the first 10 patients124 led to expanded studies and studies by others. DeNardo et al.125 have summarized their results in patients with B-cell malignancies from the past two decades. Two consecutive trials have been reported using fractionated RIT. Thirty patients with relapsed B-cell malignancies were treated with repeated cycles (10 to 100 mCi) of 131I-Lym-1 as part of a “low-dose fractionated” phase I/II study. PR was achieved in 17 patients (57%; 13 NHL patients and four CLL patients). Advanced disease often interrupted therapy prematurely. However, 18 NHL patients who received at least 180 mCi of 131I-Lym-1, 94% responded to the therapy.121 In contrast there were no responses in patients previously treated with unconjugated Lym-1,126 which also had no dose-limiting toxicity. In a second phase II trial designed to find the maximum tolerated dose (MTD) for at least the first two of up to five doses of fractionated RIT, which binds the β subunit on HLA-DR10; 20 patients with relapsed or refractory NHL were treated in cohorts in a dose-escalating study with 131I-Lym-1 (40 to 100 mCi/m2 at 4-week intervals). The response rate was 71% for those who received at least two doses of 131I-Lym-1. Seven of the responses were complete, with a mean duration of 14 months. All three patients in the 100-mCi/m2 cohort had complete remissions. Responses were observed in all histologic grades and were more rapid and complete when higher doses of radioactivities of 131I-Lym-1 were delivered; however, myelosuppression occurred earlier and was more severe.122 In general, two or more 80-mCi/m2 intravenous infusions of 131I-Lym-1 at 4-week-intervals were well tolerated.
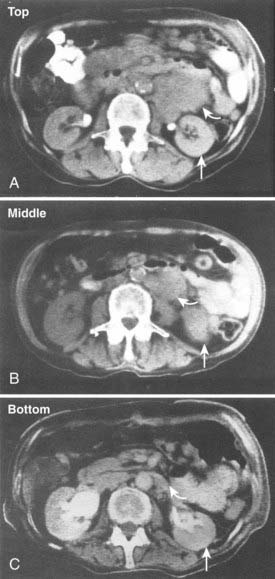
A phase I study for NHL patients with 67Cu-2IT-BAT-Lym-1 resulted in a 58% (seven of 12) overall response rate (ORR).127 For a given level of administered radioactivity, higher absorbed doses of radiation were achieved in the tumors with 67Cu-2IT-BAT-Lym-1 than 131I-Lym-1. When Lym-1 was labeled with 90Y (90Y-2IT-BAD-Lym-1), five of eight patients that failed previous chemotherapy had a PR or stabilization of NHL after RIT.128
Anti-B1 (131I-Tositumomab, Bexxar)
Several phase I/II studies assessed the efficacy and safety of 131I-tositumomab. Kaminski et al. treated 59 patients with refractory/relapsed NHL in a phase I/II single-center study.129 Fifty-three patients received individualized therapeutic doses, delivering a specified total-body radiation dose based on the patient-specific clearance rate of a preceding dosimetric dose. Unlabeled Ab was given before labeled dosimetric and therapeutic doses to improve biodistribution. Forty-two (71%) of 59 patients responded and 20 patients (34%) had a CR. Thirty-five (83%) of 42 patients with low-grade or transformed NHL responded, versus seven (41%) of 17 patients with de novo intermediate-grade NHL (P = .005). The median progression-free survival for the 42 responders was 12 months and 20.3 for those with CRs. Reversible hematologic toxicity was dose limiting. Only 10 patients (17%) had human anti-mouse Abs detected. Similar information was provided in a multicenter trial reporting a 57% response rate and 32% CR rate.130 Both studies demonstrate that a single dose of iodine 131I-tositumomab can produce frequent and durable responses in low-grade or transformed NHL, with a very acceptable toxicity profile.
A pivotal multicenter clinical trial with 131I-anti-CD20 (131I-tositumomab) in patients with low grade or transformed low grade NHL (who had failed previous chemotherapy) compared responses and duration of response in 60 patients to that of their response to their last chemotherapy regimen (LCR).131 After 131I-tositumomab, a response was observed in 39 patients (65%; with 81% response rate in patients with low grade histologies, and 39% response rate in patients with transformed low grade NHL), compared with responses in 17 patients (28%) after their last chemotherapy regimen (P < .001). The median duration of response and CR rates were 6.5 months for 131I-tositumomab compared to 3.4 months for the LCR (P < .001), and 17% CR after 131I-tositumomab (P < .001), compared to 3% after LCR. The median duration of response for CR was 6.1 months after the LCR, and had not been reached with follow-up of more than 47 months following 131I-tositumomab treatment. Another study compared the relative contribution of the antibody (tositumomab) and radionuclide (131I) to both efficacy and toxicity in a randomized two-arm open label study, with unilateral cross over of patients treated with unlabeled MAb to the RIT arm at the time of disease progression.132 On Arm A (RIT), patients received the 131I-tositumomab 450 mg unlabeled MAb with 131I (5 mCi) on 35 mg MAb on day 0. On day 0, patients on Arm B received 485 mg tositumomab (the same mg amount of total MAb as Arm A). Following imaging studies in Arm A, patients were treated on day 7 with 450 mg unlabeled MAb and 131I (dose estimated to deliver 75 Gy to the whole body) on 35 mg of MAb. On day 7, patients in Arm B received 485 mg of tositumomab. The overall response rate was 55% for Arm A and 17% for Arm B (P = .001), with CR rates of 33% and 8%, respectively (P = .012). The median duration of response was 7.6 m for Arm B and had not been reached in Arm A at the time of publication. Hematologic toxicity was more profound in Arm A, with 33% of patients experiencing grade III or IV neutropenia and thrombocytopenia, compared with 8% and 0%, respectively for Arm B.132
Other studies have also demonstrated benefit from retreatment with 131I-tositumomab, 133 as well as the use of 131I-tositumomab in rituximab failures,134 with an overall response rate of 65%, CR rate of 38%, and median progression-free survival of 24.5 months for responders. Kaminski et al.135 performed a Phase II trial of 131I-tositumomab in 76 untreated patients with newly-diagnosed, low-grade B-cell NHL. The overall response rate was 95%, with median duration of response of 71.6 months. The CR rate was 75% with the median duration of CR not reached at the time of publication. Five-year progression-free survival was 59% with a median follow-up of 61.6 months. In contrast to previously-treated patients, no supportive care was needed, and 65% of patients made a human antimouse antibody (HAMA) response, which would most likely preclude future treatment with murine or chimeric antibody, and thereby raises an important question about the optimal time for RIT with immunogenic antibodies in the natural history of NHL.
Given the efficacy and favorable toxicity profile (reversible myelosuppression and generally mild nonhematologic toxicity) in the studies reported above, recent studies have been initiated to investigate the utility of 131I-tositumomab: (1) in combination with chemotherapy in the adjuvant setting, (2) in other histologic types of NHL (e.g., diffuse large cell lymphoma [DLCL]), and (3) combination with EBRT. A phase II trial of cycloposphamide doxorubicin vineristine prednisolone (CHOP) combined with 131I-tositumomab (SWOG 9911) in newly-diagnosed patients with follicular NHL resulted in a response rate of 80% with 52% CRs.136 These encouraging results provided a compelling rationale for an ongoing phase III trial (SWOG 0016) initially comparing CHOP, CHOP + rituximab, and CHOP + 131I-tositumomab in patients with stage III, IV or bulky II newly-diagnosed follicular NHL.
2B8 (90Y-Anti-CD20, Ibritumomab, Zevalin)
Ibritumomab is the mouse parent of rituximab (anti-CD20). When radiolabeled using the chelate MX-DTPA (tiuxetan) to the mAb, it forms Zevalin. Knox et al. used cold ibritumomab before each administration in 18 patients treated with 13.5 to 50 mCi 90Y-ibritumomab. The ORR was 72% and there was a 33% CR rate. They observed that doses less than or equal to 40 mCi were nonmyeloablative.137 In a phase I/II dose escalation trial of 90Y-ibritumomab, patients received 111I-ibritumomab (for dosimetry) on day 0, followed by a therapeutic dose of 90Y-ibritumomab on day 7, in a dose-escalation fashion, starting at 0.2 mCi/kg. Both radioimmunoconjugates were preceded by two doses of the unlabeled rituximab alone. The MTD was approximately, 0.4 mCi/kg, with an estimated tumor dose of 17 Gy, and transient and generally mild hematologic toxicity. Adverse events were primarily hematologic and correlated with the baseline extent of marrow involvement with NHL and baseline platelet counts, suggesting an inverse relationship between toxicity and bone marrow reserve.138 Witzig et al. treated 68 patients, observing that the MTD was approximately 0.4 mCi/kg IDEC-Y2B8 and 0.3 mCi/kg for patients with baseline platelet counts of ≥150,000/µL, and 100,000 to 149,000/µL, respectively (see Fig. 73-2). The ORR for the intent-to-treat population (n = 51) was 67% with 26% CR. Response rates were 82% for low-grade disease (n = 34); 43% for intermediate-grade disease (n = 14); and 0% for mantle-cell disease (n = 3). Responses occurred in 41% of patients with bulky disease (≥7 cm) and 50% of the patients with splenomegaly. Time to progression for patients treated with 0.4 mCi/kg in responders and duration of response was 15.4 months and 14.4 months, respectively. Time to progression for patients with CRs treated at this dose level ranged between 28.3 and 36.4 months. One patient (2%) developed an anti-Ab response (human antichimeric Ab/human antimouse Ab).139
A prospective randomized trial was designed to evaluate the efficacy of 90Y-ibrutumomab compared to rituximab alone, as given therapeutically as a single agent (375 mg/m2 a week × 4 weeks, compared with 250 mg/m2 on days 1 and 8 in the RIT arm).140,141 A total of 143 patients were randomized in IDEC106-04. ORR rate was 80% for the 90Y-ibritumomab tiuxetan group versus 56% for the rituximab group (P = .002). CR rates were 30% and 16% in the 90Y-ibritumomab tiuxetan and rituximab groups, respectively (P = .04).141 Interestingly, there were no significant differences in duration of response or time to progression between the two study arms. As with 131I-tositumomab, nonhematologic toxicity was generally mild, and hematologic toxicity was reversible, with grade III or IV neutropenia and thrombocytopenia in 58% and 62% of patients, respectively. This clearly demonstrated additional therapeutic value in adding the isotope 90Y to the anti-CD20 mAb. 90Y-ibritumomab tiuxetan was also efficacious in patients that had previously failed treatment with rituximab (n = 57), with an overall response rate of 74%, CR rate of 15% for patients with follicular NHL (n = 54), and median time to progression in responders of 8.7 months.142 There are currently over 50 ongoing clinical trials of 90Y-ibritumomab tiuxetan either alone for the treatment of other NHL histologies, in combination therapy with a variety of chemotherapy drugs for both nonmyeloablative and myeloablative stem cell transplantation regimens, or in combination with radiation therapy. Examples of such studies, from which results are available are described below.
Encouraging results have been obtained in studies of 90Y-ibritumomab for the treatment of patients with relapsed or refractory DLCL not appropriate for stem cell transplantation143,144 and in patients with mantle cell145–147 and marginal zone lymphoma.148 In a multicenter study of 414 patients with advanced stage previously untreated nonfollicular NHL following induction chemotherapy, patients were randomized to either 90Y-ibritumomab tiuxetan or no further treatment. Progression-free survival for the adjuvant RIT arm was 37 months, and 54.6 months for those patients with a CR, compared to 13.5 and 29.9 months, respectively, for patients with no further treatment following chemotherapy.149 These are very encouraging results and demonstrate the potential utility of using RIT in an adjuvant setting. A variety of studies are currently investigating the safety and efficacy of this radiolabeled MAb combined with different chemotherapy regimens as either 1st- or 2nd-line therapy. Examples of the former include ongoing SWOG and ECOG studies.
RIT for T Cell Lymphoma and Hodgkin’s Disease
Early studies in other lymphomas included seven patients with cutaneous T-cell lymphoma who were treated with 100 to 150 mCi 131I-T101.150 Two PRs lasting 2 months and four minor responses lasting 3 weeks to 3 months were observed. Toxicity consisted of fever, pruritus, dyspnea, urticaria, neutropenia, and thrombocytopenia. All of the patients developed a HAMA response. Lenhard et al.151 treated 37 patients with progressive advanced Hodgkin’s disease with three 50-mCi cycles of 131I-antiferritin polyclonal Abs and reported a 3% CR rate and a 38% PR rate. Leukopenia and thrombocytopenia were the primary toxicities. In a subsequent study,152 12 patients with poor-prognosis Hodgkin’s disease were treated with 90Y-antiferritin polyclonal Abs followed by high-dose cyclophosphamide, carmustine, and etoposide chemotherapy and autologous bone marrow transplantation. The progression-free survival rate at 1 year was estimated to be 21%. Four patients (33%) experienced early, transplant-related mortality. More recently, 90Y-antiferritin polyclonal antibody resulted in a 70% response rate, with a median duration of response of 8 months in patients with refractory or relapsed Hodgkin’s disease, including patients who had failed previous transplant.153
RIT as a Component of Preparatory Regiments for Bone Marrow and Stem Cell Transplantation
Using a nonmarrow normal organ dosimetry-driven approach with myeloablative doses, Press et al.154,155 administered 234 to 777 mCi 131I-anti-CD20 or 131I-anti-CD37 to 19 patients with NHL. Tumor doses ranged from 10 to 91 Gy. The high-dose RIT was followed by autologous bone marrow reinfusion. A CR was achieved in 16 cases (84%), a PR was achieved in two cases (11%), and a minor response (40% tumor reduction without regrowth for 18 months) was achieved in the remaining patient (5%). The median response duration was more than 18 months for all patients.156 Nine patients had a durable complete remission without further therapy. Median survival was more than 35 months.156 Toxicity included myelosuppression, nausea, diarrhea, three serious infections, and two cases of cardiopulmonary toxicity at the MTD. Updated results from the phase I and II trials, with a median follow-up time of 42 months, showed157 a CR rate of 79% and PR rate of 7%, with an estimated overall and progression-free survival rate of 68% and 42%, respectively. Fourteen of 29 patients remained in unmaintained remissions that ranged from 27 to 87 months after RIT. Late toxicities were uncommon except for elevated thyroid-stimulating hormone levels in approximately 60% of the subjects. Two patients developed second malignancies, and there were no reported cases of myelodysplasia. A subsequent study then defined the MTD of this RIT with high-dose chemotherapy and autologous stem cell transplant (ASCT). Fifty-two patients received a trace-labeled infusion of 131I-tositumomab followed by serial quantitative γ-camera imaging and estimation of absorbed doses of radiation to tumor sites and normal organs. Twenty-five Gy to critical normal organs from 131I-tositumomab was considered the MTD that could be safely combined with 60 mg/kg etoposide and 100 mg/kg cyclophosphamide. The estimated overall survival and progression-free survival of all RIT treated patients at 2 years was 83% and 68%, respectively. These findings were favorable when compared with those in a nonrandomized control group of patients who underwent transplantation, external-beam TBI, and etoposide and cyclophosphamide therapy during the same period (overall survival of 53% and progression-free survival of 36% at 2 years), even after adjustment for confounding variables in a multivariable analysis.158
Myeloablative regimens using 131I-anti-CD20 antibodies in patients with relapsed mantle cell lymphoma after high dose chemotherapy and ASCT have yielded encouraging results, as reported by Behr et al. with 6 out of 7 patients achieving CRs and one PR, with tumor doses up to 100 Gy after 261 to 495 mCi of injected activity.159 Gopal et al.160 also reported very promising results using high-dose 131I-anti-CD20 MAb, etoposide and cyclophosphamide, and ASCT in patients with relapsed mantle cell lymphoma, with 100% survival and approximately 60% progression-free survival at 60 months. Encouraging results have also been reported in early clinical trials using 90Y-ibritumomab tiuxetan,161,162 with 2-year overall survival of 90% in one study.161
Leukemia
Chronic Lymphocytic Leukemia
Although chronic lymphocytic leukemia (CLL) is usually indolent and responsive to treatment early in its course, later stages are characterized by inexorable progression despite conventional treatment. DeNardo et al.163,164 treated five CLL patients who had failed to respond to intensive multidrug chemotherapy with 131I-Lym-1 (a murine IgG2a MAb recognizing a tumor-associated antigen that is an HLA-DR variant). CR occurred in one patient, and greater than 70% reduction of lymphadenopathy was observed in the remaining four patients, with normalization of the leukocyte counts in two of those patients. The responses, however, were of short duration. Since CLL patients have heavy marrow infiltration with malignant cells, radiation-induced thrombocytopenia from “bystander” irradiation limited the amount of 131I-Lym-1 that could be administered.
Acute Nonlymphocytic Leukemia
For acute nonlymphocytic leukemia (AML) patients who have relapsed after bone marrow transplantation or are refractory to chemotherapy, or have acute nonlymphocytic leukemia secondary to previous chemotherapy, there is no curative therapy.165 Schwartz et al.166 reported results from treating patients with myeloid leukemias with 131I-M195, a murine IgG2a mAb that binds CD33, a cell-surface glycoprotein present on committed erythroid and myelomonocytic progenitor cells, but not on the earliest pluripotent stem cells. Twenty-four patients, including seven who had failed to respond before bone marrow transplantation, received 50 to 210 mCi/m2 131I-M195 in divided doses. Twenty-three patients (96%) demonstrated decreases in peripheral blood counts, and 20 patients (83%) demonstrated fewer bone marrow blasts. Eight patients had sufficient marrow cytoreduction to proceed to bone marrow transplantation. Three of these achieved marrow remission, one of more than 6 and one of 9 months duration. Two patients in blastic phase temporarily reverted to their original myelodysplastic states. Thirty-seven percent developed HAMA, limiting retreatment. A humanized IgG1 M195 (HuM195)167 capable of inducing Ab-dependent cellular cytotoxicity22 without significant immunogenicity168 continues in clinical trials.
These studies use targeted radiotherapy to eliminate minimal residual disease after relapse or as part of the conditioning regimen before stem cell transplantation.165 A clinical trial of myeloablative therapy using 131I-antilabeled CD33 with M195 or HuM195, combined with busulfan and cyclophosphamide, and followed by infusion of HLA-compatible bone marrow, was performed in 30 patients with AML. Patients received 120 to 380 mCi of 131I, delivering up to 5 Gy to the bone marrow. Results showed that 27 of 30 patients obtained complete remissions. In one study, median survival was 4.9 months and three patients have ongoing CRs at 59 to 90 months after transplant.169 Reske et al,170 in a phase I study, added rhenium-188-labeled CD66a, b, c, e specific for normal bone marrow, in addition to the conventional conditioning with high-dose chemotherapy and 12 Gy total body irradiation. They reported a death rate of 15% after relapse. Nine of the 27 patients (33%) died from transplantation-related complications, and 14 patients (52%) were alive in complete clinical remission. These studies demonstrate the feasibility of using RIT as a part of preparatory regimens for bone marrow transplant (BMT) or stem cell transplant (SCT) to enhance leukemic cytoreduction with acceptable toxicity when used with standard high dose chemotherapy transplant protocols. Randomized studies are needed in the future to determined whether or not there is a statistically-significant benefit to this approach.
Recently, clinical trials have been performed with α particle–emitting radionuclides, which are particularly promising for the treatment of microscopic residual leukemic disease165 because of their emission properties (short range, high LET). Eighteen patients total with relapsed AML (14 patients), refractory AML (3 patients), and chronic myologenous leukemia (CML) were treated with the α emitter 213Bi (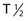 45.6 mins, 8 MeV)—HuM195 in a phase I trial and received 16 to 95 mCi in three to six fractions over 2 to 4 days. Ten of the 12 evaluable patients had reductions in peripheral blood leukemia cells, and 13 of the 18 patients had decreases in the percentages of bone marrow blasts.165 Following these studies, the first-in-man phase I trial was performed using 225Ac-HuM195 (anti-CD33) in patients with AML.171 225Ac is an isotope generator that emits four α-emitting isotopes and has a 10-day
45.6 mins, 8 MeV)—HuM195 in a phase I trial and received 16 to 95 mCi in three to six fractions over 2 to 4 days. Ten of the 12 evaluable patients had reductions in peripheral blood leukemia cells, and 13 of the 18 patients had decreases in the percentages of bone marrow blasts.165 Following these studies, the first-in-man phase I trial was performed using 225Ac-HuM195 (anti-CD33) in patients with AML.171 225Ac is an isotope generator that emits four α-emitting isotopes and has a 10-day 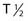 . 225Ac-immunoconjugates are 1000 × more potent than 213Bi analogues in vitro and have prolonged survival in animal models.48 In this phase I trial171, three out of six patients had elimination of peripheral blood blasts and dose-related reductions in BM involvement. The results of this phase I/feasibility study are very encouraging, and suggest that isotope generating α-emitters may have the potential to significantly enhance the therapeutic index of RIT for this clinical indication.
. 225Ac-immunoconjugates are 1000 × more potent than 213Bi analogues in vitro and have prolonged survival in animal models.48 In this phase I trial171, three out of six patients had elimination of peripheral blood blasts and dose-related reductions in BM involvement. The results of this phase I/feasibility study are very encouraging, and suggest that isotope generating α-emitters may have the potential to significantly enhance the therapeutic index of RIT for this clinical indication.
Other promising results have been obtained using 131I-anti CD45 (BC8) combined with cytoxan and TB1 as a transplant regimen for acute leukemia and MDS.172 In a phase I trial, 34 patients were treated with 76 to 612 mCi 131I-BC8. The MTD was 10.5 Gy to the liver. Seven of 25 patients with AML/MDS had a median disease-free survival of 65 months, and three of nine ALL patients were disease-free at 19, 54, and 66 m following transplant, with marrow and spleen doses of approximately 24 Gy and 50 Gy, respectively.
Solid Tumors
Malignant Melanoma
Carrasquillo et al. between 1982 and 1984, administered multiple 3 to 342 mCi infusions of 131I-labeled Fab fragments to 10 patients with malignant melanoma. There was one PR (10%) lasting 3 months. The main toxicities were neutropenia and thrombocytopenia.173 Other MAb to melanoma are now being evaluated to targets on melanoma, as this tumor presents many possible antigen targets.174,175
Glioma
There is special interest in RIT of glioma patients since higher doses may be achieved due to the intratumoral or compartmental administration of the radioimmunoconjugates.176 Using a systemic route of administration, Brady et al.177 treated 14 recurrent malignant glioma patients (seven had an anaplastic astrocytoma and seven had glioblastoma multiforme [GBM]) with multiple 7- to 50-mCi intra-arterial infusions of 125I-antiepidermal growth factor receptor mAb. One CR (7%) and two PRs (14%) of brief duration were observed. Median survival from the date of initial diagnosis was 14 months. In primary disease, 59 patients with primary high-grade gliomas of the brain (13 astrocytomas with anaplastic foci and 46 with GBM) were treated after tumor resection and postoperative radiation therapy, using multiple intravenous treatments with 125I-antiepidermal growth factor receptor. Cumulative injected doses ranged from 40 to 296 mCi with no significant life-threatening toxicities. At 1 year, 34 (58%) of the 59 patients were alive with a median overall survival of 13.5 months.178
Antitenascin mAbs provide another option for targeting brain tumors.176,179 Tenascin is present in the extracellular matrix of malignant glioma tissue. Both mAb BC-2 and mAb 81C6 have been used in clinical trials; 81C6 offers the advantage that several normal tissue-associated variants are not recognized by the mAb.176 Riva et al. treated 24 recurrent malignant glioma patients with one to three intratumoral infusions of 131I-antitenascin mAb. Seventeen patients were evaluable. Three CRs (18%) and three PRs (18%) were observed, typically lasting approximately 13 months. Median survival from the date of initial diagnosis was 16 months.
Both Riva et al.180 and Coknor and Bigner181 reported compartmental administration in recurrent and primary gliomas of the brain, using a single dose of 131I-labeled mAb 81C6. Thirty-four previously irradiated patients with recurrent malignant gliomas were treated with 131I-labeled 81C6. The MTD was 100 mCi. The median survival for patients with glioblastoma was 56 weeks and for the entire series, 60 weeks.181 A subsequent dose escalation study (42 patients) and phase II trial (33 patients) in patients with primary gliomas were performed using a combined modality approach with surgery, postoperative radiotherapy, and 131I-labeled 81C6. The MTD was 120 mCi. At higher doses delayed neurotoxicity was dose limiting.182 The median survival for all patients was 86.7, and 79.4 weeks for those with GBM. Eleven patients remained alive at a median follow-up of 93 weeks (range: 49 to 220 weeks). Nine patients (27%) developed reversible hematologic toxicity, and histologically confirmed, treatment-related neurologic toxicity occurred in five patients (15%).183 Paganelli et al102 enrolled 37 high-grade glioma patients, 20 with glioblastoma, in a controlled, open, nonrandomized study using a three-step avidin-biotin pretargeting approach to target 90Y-biotin to the tumor. All patients received surgery and radiotherapy and were disease free. Nineteen patients received adjuvant treatment with RIT. The median survival for the glioblastoma patients was 33.5 months, and all 12 glioblastoma patients randomized to conventional therapy alone (no RIT) died after a median survival from diagnosis of 8 months. In the RIT-treated grade III glioma patients, the median disease-free interval was 56 months (range: 15 to 60) and survival cannot be calculated, because only two patients within this group died.
Cheung et al. treated nine neuroblastoma patients with multiple 30- to 300-mCi infusions of 131I-3F8.184 Autologous bone marrow was reinfused into seven patients. Two PRs (22%) and two minor responses (22%) lasting several months were observed.184
Paganelli et al. treated patients with pretargeted RIT with and without temozolomide or mitoxantrone.179,185 The duration of major systemic side effects and the percent of long-term survivors with this combination therapy was particularly noteworthy, with a mean survival of 25 months in patients treated with temozolomide.179
Androgen-Independent Prostate Carcinoma
Conceptually, tumor-targeted high-dose RIT with marrow support or RIT as part of combined multimodality therapy (CMRIT), can offer effective therapy for this lethal disease. Meredith et al. used 75 mCi/m2 131I-CC49 (anti-TAG-72) mAb in 15 patients with metastatic hormone-resistant prostate cancer.186 Tumor doses ranged from 2 to 11 Gy. Six of 10 evaluable symptomatic patients had bone pain relief, but no patients met radiographic or prostate-specific antigen (PSA) criteria for an objective response. O’Donnell et al. reported 111In/90Y-m170 mAb therapy in a phase I dose escalation study in 17 patients treated with 5 to 20 mCi/m2 in similar prostate cancer patients.187 Tumor doses ranged from 2 to 20 Gy, with similar responses. Ongoing CMRIT studies with Taxol have shown more evidence of PSA and tumor responses, with the MTD not yet determined.188
Neuroendocrine Tumors
Radiolabeled receptor-binding peptides have been shown to be an important class of radiopharmaceuticals for tumor diagnosis and therapy. The high affinity of the peptide for its receptor and the internalization of the receptor-peptide complex facilitates retention of the radionuclide in receptor-expressing tumors, whereas its relatively small size facilitates rapid clearance from the blood. Peptides most successfully used for these purposes are somatostatin analogs that bind to receptors overexpressed on neuroendocrine tumors. In a clinical phase II study, targeted irradiation with the radiolabeled somatostatin analogue 90Y-DOTATOC was assessed for palliation in 41 patients with neuroendocrine gastroenteropancreatic and bronchial tumors.189 Eighty-two percent of the patients had therapy-resistant and progressive disease. Four intravenous injections (6000 mBq/m2) of 90Y-DOTATOC, administered at intervals of 6 weeks, resulted in an overall response of 24% and 36% for endocrine pancreatic tumors. CRs were found in 2% (one of 41), and PRs in 22% of patients (9 of 41). The median duration of response was not reached by 26 months. The 2-year survival time was 76% ± 16%. Excellent palliation was achieved as 83% of the patients suffering from the malignant carcinoid syndrome obtained a significant reduction of symptoms. The treatment was well tolerated.189
In another study, 30 patients with cancers expressing somatostatin receptors were treated with 90Y-DOTATOC in a dose escalation schemata with six patients per group receiving equivalent-activity doses in each of three cycles over 6 months (1.11 to 3.3 gBq per cycle). After a total dose of 3.33 gBq, one patient developed grade II renal toxicity 6 months later. Total injected activity was limited by the tolerated kidney dose, estimated to be 20 to 25 Gy. CRs or PRs occurred in 23% of patients.190
When patients with gastroenteropancreatic neuroendocrine tumors were treated with 177Lu-DOTATATE, complete or partial remissions were documented in an impressive 30% of patients and minor responses in 21% of patients, with 26% of patients with progressive disease at the start of peptide receptor radionuclide therapy (PRRP) showing stabilization.191 More than 475 patients with metastasized neuroendocrine tumor (NET) have been treated with radiolabeled somatostatin analogues. Sixty-eight of these patients have received four or more cycles of PRRT. For PRRT with Y-90-DOTA-TATE (Y-90) and/or Lu-177-DOTA-TATE (Lu-177), a fractionated, low dose protocol was used, applying three different treatment regimens: 90Y alone (n = 19), 177Lu alone (n = 8) and 90Y+177Lu (n = 41) in combination every 3 to 5 months: 90Y, 2 to 5 GBq; 177Lu, 3 to 7 GBq. Renal tubular extraction rate (TER) was monitored every 3 to 4 months using Tc-99m-MAG3. The median follow-up time in 90Y/177Lu/90Y+177Lu combination therapy was 36/22/25 months, respectively. Only two patients with long-standing hypertension (HT) and diabetes mellitus (DM) had preterminal end-stage renal disease with no need for dialysis. Thus, although PRRT appears to be nephrotoxic, risk factors need to be seriously considered, but significant toxicity can be prevented if used in fractionated low dose.192 In summary, these pharmaceuticals are very useful radioligands for somatostatin-expressing tumor therapy and doses can be administered safely with a low risk of myelotoxicity; however, the cumulative radiation dose to the kidneys always requires careful evaluation.190,193
Breast
DeNardo et al.194–196 administered multiple 60- to 70-mCi/m2 infusions of 131I-chimeric L6 (ChL6) to 10 women with metastatic breast cancer. ChL6 as part of the pre-RIT regimen was noted to have in vivo biologic activities resulting in a fall in serum complement and IL2R195 although it had had significant clinical effects relative to tumor response in the nonradioactive studies.197 In the initial 131I RIT studies, 4 of 10 patients with advanced breast cancer had a PR, lasting up to 5 months, and a minor response occurred in 2 of 10 patients. A phase I trial then explored the use of single high-dose 90Y-MX-DTPA-BrE-3 and autologous hematologic cell support.198 Nine women with heavily pretreated disease were enrolled.198 Objective PRs were noted in four of eight (50%) patients with measurable tumors.198 Eleven patients were treated with 30 to 60 mCi of 90Y-m170. Three had a PR (70% tumor reduction), three had a minor response (measurable but <70% tumor reduction), and two had stable disease for more than 1 month. Cyclosporin was administered to suppress HAMA199–201 and peripheral blood stem cells (PBSC) infusion prevented prolonged myelosuppression. Phase I dose escalation studies with 90Y-DOTA-peptide-m170 included combination therapy with Taxol with modest responses at low doses; the MTD was not reached. New progress in breast cancer therapy awaits further outcomes from trials of combined biologic active therapy.195,199
Gastrointestinal Malignancies
Intensive research of RIT has occurred in colon cancer. However, response rates in most clinical trials are below 10%, and the best reported rates are in the subgroup of patients with minimal metastatic disease. Behr et al. have treated 21 patients refractory to 5-fluorouracil with small volume metastatic disease (≤3 cm) and nine patients in an adjuvant setting after resection of liver metastases. Treatment consisted of a single injection of 60 mCi/m2 of 131I-hMN-14 (humanized anti-CEA Ab). The objective response rate in 19 assessible patients was 16%, and the ORR was 58% (partial and minor responses) with a mean duration of 9 months. Seven of nine patients with adjuvant treatment remained free of disease for up to 36 months of follow-up. Five patients with stabilized disease were retreated at the same dose level and two of four assessible patients obtained a PR.202 Liersch et al.203 recently tested whether adjuvant radioimmunotherapy (RAIT) given after resection of liver metastases (LM) of colorectal cancer can improve survival. Resection of LM from colorectal cancer is the standard of care in this setting, yet two thirds will eventually relapse, and adjuvant systemic chemotherapy has failed to improve survival.203 Twenty-three patients, following resection for LM of colorectal cancer, received a dose of 40 to 60 mCi/m2 131I-labetuzumab, a humanized monoclonal antibody against carcinoembryonic antigen. Disease-free survival, and overall survival were analyzed, and efficacy was compared retrospectively with a similar contemporaneous group of control patients (n = 19) treated at the same institution during the same period of time, but without RAIT.
At a median follow-up of 91 months, the median overall survival for RAIT patients was 58.0 months versus 31.0 months for the control group (P = .032). Corresponding survival rates (Kaplan–Meier analyses) were estimated to be 94.7% at 1 year, 78.9% at 2 years, 68.4% at 3 years, and 42.1% at 5 years with RAIT, compared to 94.7%, 68.4%, 36.8%, and 15.8%, respectively, for the controls. RAIT was beneficial independently of bilobar involvement, size and number of LM, or resection margins. Transient myelosuppression was the principal adverse effect. This is the first evidence of a promising survival advantage of adjuvant RAIT after long-term follow-up of colorectal cancer patients given salvage resection of LM.203
Other Tumors
In Stewart et al.’s phase I/II study,204–206 encouraging preliminary results were observed in ovarian cancer patients (stage IIB or greater) with no evidence of gross residual disease after cytoreductive surgery and cisplatin or carboplatin-based chemotherapy, who subsequently received a single 5- to 30-mCi intraperitoneal infusion of 90Y-labeled mAb. Long-term results from the same group included 52 patients; 31 had residual disease following chemotherapy and 21 were in complete remission. In the group of 21 patients who had achieved complete remission following surgery, conventional chemotherapy, and intraperitoneal RIT, the median survival has not been reached. The maximum follow-up is 12 years, with a survival rate of 78% at more than 10 years.207
Human Anti-Mouse Antibodies
After exposure to murine Abs, immunocompetent patients may develop HAMAs. A HAMA response usually results in rapid clearance of the therapeutic mAb from the circulation, thereby reducing tumor uptake. On very rare occasions, a HAMA response has also produced serum sickness and anaphylactoid reactions. There is considerable variability in the development of a HAMA response among patients. Despite the adverse effects of HAMA, some investigators have reported an association between improved survival after unlabeled or labeled mAb and HAMA formation, presumably secondary to immunocompetence.208 In general, there is a lower frequency of HAMA in patients who are immunosuppressed as a result of their disease and/or previous chemotherapy, such as in previously treated patients with leukemia, NHL, or Hodgkin’s disease (3% to 35%). The incidence of HAMAs increases in patients following three or more infusion of intact murine Ab.209 HAMAs can be detected in some patients as soon as 1 week after the infusion of murine Abs210 and may persist for months or years, precluding subsequent Ab infusions. Cyclosporin given orally from 2 days before until 2 weeks after the infusion of murine Abs can suppress HAMA formation.200,201 Lower levels of HAMA have been reported after fludarabine in combination with tositumomab.211 It should be noted that the cold preinfusion of Rituxan (rituximab) (anti-CD20), which results in several weeks of asymptomatic loss of circulating normal B lymphocytes, also is associated with almost no HAMA before treatment with murine mAb as radioactive Zevalin (ibritumomab).139 The amount of previous chemotherapy received before RIT may influence the development of HAMA, because the reported rates in naïve treated patients for low-grade NHL of 65%212 are reduced to 17% in refractory/relapsed disease.129
Toxicity
Overall, the acute side effects of RIT are minimal and vary with the amount of mAb given and its biologic properties. One study by Dillman et al.213 described toxic reactions that occurred in 82 cancer patients who received 0.5- to 500-mg intravenous infusions of one particular murine mAb in which approximately one fifth of the patients developed fever, chills, and diaphoresis 2 to 8 hours postinfusion. Although this is far more than usually seen, even these reactions typically lasted no more than a few hours. Acute allergic reactions were observed in fewer than 1% of the patients in such studies, but rapid infusions of a large quantity of mAbs increased the likelihood of these reactions. To reduce the risk of an allergic response, patients are commonly medicated with acetaminophen and diphenhydramine before the delivery of Abs, particularly when large quantities of intact murine IgG2a or human chimeric IgG1 Abs are given.
Myelosuppression
Myelosuppression has been the dose-limiting toxicity of almost all systemic RIT without marrow support.214 Most groups have chosen to avoid severe neutropenia and thrombocytopenia by limiting the amount of radioactivity administered per infusion. Alternatively, myeloablative doses of RIT may be delivered if PBSC215,216 or bone marrow cells217 are reinfused once radioactivity in the body has adequately cleared.
Nonmarrow Normal Organ Toxicity
Grade III and IV nonmarrow organ toxicity is very unusual, since marrow provides the major dose-limiting toxicity. However, in a dose escalation trial, with marrow transplant, Press et al.217 reported that the MTD for a single infusion of 131I-labeled mAb was defined by severe postural hypotension when 31 Gy was delivered to the lungs. Life-threatening congestive cardiomyopathy and hemorrhagic pneumonitis developed in another patient 2 months after a single infusion of 131I-labeled mAb that delivered 27 Gy to the lungs. When dose escalation is performed, hepatic toxicity has been observed in beagles.218 Although serious hepatic toxicity was not noted in systemically given RIT trials, in patients in whom high doses of radioactivity was delivered, hyperbilirubinemia and transient elevation of serum alanine aminotransferase level has been repeated.22,217
Neurologic Complications
Neurotoxic events in brain tumors have been reported with doses greater than 100 to 120 mCi of 131I-labeled mAb.181,182 At the dose of 100 mCi, the estimated radiation absorbed dose by the brain was 6 to 7 Gy,219 and at doses of 120 mCi patients developed neurologic toxicity at rates of 15%.183 Cumulative doses from previous or concurrent brain irradiation may have to be taken into account in designing such treatment plans.
Renal Complications
Renal adverse events were reported after 90Y-DOTATOC; Paganelli et al. reported renal doses as high as 11 to 25 Gy220 This is in the upper limit of tolerance after external beam irradiation.221 Moll et al.222 reported that five of seven patients with cumulative doses greater than 200 mCi/m2 developed renal failure. End-stage renal failure occurred in 3 patients within 6 months, and chronic renal failure in two patients. Renal biopsies performed in three patients showed typical signs of thrombotic microangiopathy involving glomeruli, arterioles, and small arteries. The histopathologic lesions were identical to those found after external radiotherapy, which suggests a causal relationship between 90Y-DOTATOC and renal thrombotic microangiopathy.222
Information about long-term renal effects from RIT irradiation are lacking due to the few patients receiving high renal levels and the short-term follow-up available in these advanced cancer patients. Work with bone-seeking radioactive drugs for bone pain or aggressive CMRIT for myeloma with Homium223 has suggested both radiation dose and dose rate must be considered in the renal toxicity.224
Radiosensitization and Combined Modality Rit
Since RIT is a systemically targeted form of radiation, potential radiosensitizers for use with conventional external beam irradiation may also be useful when combined with RIT. The success of using combined modality therapy consisting of chemotherapy and/or biologic response modifiers with radiotherapy in most solid tumors225,226 provides a strong rationale for combining these agents with RIT. Experimental studies with RIT have shown the potential of chemotherapeutic drugs (e.g., Taxol,227,228 taxotere,227 topotecan,229 dacarbazine,230 and gemcitabine231) and radiosensitizers such as the hypoxic cytotoxin SR 4233 (Tirapazamine),232,233 and the redox active agent Motexafin Gadolinium234 to potentiate the efficacy of RIT in animal models. As with EBRT, the timing, dose schedule, and route of administration of the RIT in relation to the administration of these adjuvant therapies will be critical.158 There is evidence from a randomized phase II trial that isotope therapy with strontium-89 increases survival in advanced carcinoma of the prostate as consolidation therapy after response to chemotherapy.235 The use of chemotherapy in combination with RIT is an active area of clinical research for the treatment of lymphoma, as described earlier in this chapter, as well as in solid tumors, with early clinical trials demonstrating acceptable toxicity with nearly full doses of both RIT and adjuvant chemotherapy,236–239 and some promising clinical results.
Several trials have studied the effect of combining involved field conventional EBRT with RIT. Msirikale et al.240 reported that patients with hepatoma receiving 6 to 9 Gy in two to three treatments before RIT had approximately threefold improvement in RIT tumor uptake. This finding is consistent with reports that low-dose EBRT increases tumor vascular permeability and Ab uptake in human breast241 and melanoma242 xenografts in nude mice. At higher radiation doses (10 to 30 Gy), external beam radiation decreased the interstitial fluid pressure, with a single dose of 30 Gy decreasing the interstitial pressure by 35%. This could have been, in part, responsible for the increased uptake of mAb following single or fractionated radiation.243 In another study, Buchegger et al.244 found acceptable toxicity in patients treated with 131I-anti-CEA monoclonal antibody and EBRT to the liver. As described, RIT and radiation therapy are also being studied in ongoing trials in patients with non-Hodgkin’s lymphoma.
Angiogenesis
Tumor vasculature is highly heterogeneous in most solid tumors, resulting in underperfused areas leading to abnormal tumor blood flow. Combined with the lack of lymph drainage in tumors, this produces an abnormal high interstitial pressure, jeopardizing the normal transport of macromolecules to the stroma and tumor cell compartment.245 Targeting neovasculature with agents that selectively increase permeability246 and normalize blood flow, thereby also increasing tumor pO2, may help to improve the efficacy of RIT. The ανβ3-integrin receptor may be used to target tumor vasculature for enhancing tumor permeability because it is highly and preferentially expressed in tumors. Antagonists of ανβ3-integrin have been shown to induce apoptosis of endothelial neovascular cells. The cyclic RGD pentapeptide is an antagonist of the ανβ3-integrin receptor and, when given 1 hour before247 In-ChL6, resulted in a 40% to 50% increase in tumor uptake compared to the control tumor uptake 24 hours after administration.247,248 The integrins ανβ3 and ανβs can also be targets for RIT, as reported by Li et al.249 in which 90Y-labeled nanoparticles were selectively targeted to these integrins in xenograft models, resulting in tumor growth delay in vivo, with histopathogical analysis of excised treated tumors demonstrating nanoparticle binding to tumor vasculature, decreased vessel density and apoptosis. Hallahan et al., using 99mTc-biapcitide (RGD peptidomimetic), showed selective binding to all neoplasms irradiated after peptide administration, compared to untreated neoplasms or tumors irradiated before administration of the RGD peptidomimetic.250 These studies demonstrate the possibility of guiding drugs selectively to tumors with the use of radiation-induced receptors.250 Gene therapy approaches have also been used in animal models to increase the level of expression of targeted tumor associated antigens or receptors, resulting in increased radiolabeled antibody or peptide uptake in tumors,251 suggesting that gene transfer techniques may also have the potential to further enhance the efficacy of RIT.
Conclusion
B-cell NHLs, which are particularly susceptible to radiation-induced apoptosis and radiobiologically have a small shoulder, large α/β ratio, and reduced sublethal damage repair capacity, have shown the best single-agent efficacy of RIT. A number of challenges face RIT for solid tumors including insufficient tumor penetration, antigenic modulation, antigenic heterogeneity, and HAMA. Many possible approaches to these challenges, including high-dose RIT, small molecules, dose fractionation, pretargeting strategies and the use of radionuclides such as 90Y, 67Cu, and α emitters, along with the development of humanized or multifunctional Abs, are under clinical investigation. High-dose therapy with bone marrow/stem cell support has already demonstrated a high tumor response rate, perhaps some curative responses, and nonmarrow normal organ toxicities that correlate with calculated dose to those organs from imaging. Although the field of RIT is relatively young, a great deal of progress has been made. RIT is ready for clinical use in lymphomas, and considerable advances have been made in the treatment of solid tumors, with the most promising results observed when RIT is integrated in multidisciplinary approaches or used in low tumor burden scenarios. In the future, the real challenge facing RIT is dose intensification, with increased tumor radiation absorbed dose while minimizing normal tissue exposure. Variations in clinical RIT choices (antibodies and isotopes), dosing schedules, and dosimetry methods emphasize the need for clinical studies in the future, with sophistication in dosimetry, treatment planning, and clinical trial methodology.252
1 Eisen HN, Keston AS. The immunologic reactivity of bovine serum albumin labeled with trace-amounts of radioactive iodine (I-131). J Immunol. 1950;63:71-80.
2 Pressman D, Korngold L. The in vivo localization of anti-Wagner-osteogenic sarcoma antibodies. Cancer. 1953;6:619-623.
3 Pressman D, Day ED, Blau M. The use of paired labeling in the determination of tumor-localizing antibodies. Cancer Res. 1957;17:845-850.
4 Beierwaltes WH. Radioiodine-labeled compounds previously or currently used for tumor localization. In: Agency IAE, editor. Proceedings of an advisory group meeting on tumor localization with radioactive agents. Panel Proceedings Series. Vienna, Austria: International Atomic Energy Agency; 1974:47-56.
5 Spar IL, Bale WF, Marrack D, Dewey WC, McCardle RJ, Harper PV. 131I-labeled antibodies to human fibrinogen. Diagnostic studies and therapeutic trials. Cancer. 1967;20:865-870.
6 Ettinger DS, Order SE, Wharam MD, Parker MK, Klein JL, Leichner PK. Phase I–II study of isotopic immunoglobulin therapy for primary liver cancer. Cancer Treat Rep. 1982;66(2):289-297.
7 Ettinger DS, Dragon LH, Klein J, Sgagias M, Order SE. Isotopic immunoglobulin in an integrated multimodal treatment program for a primary liver cancer. A case report. Cancer Treat Rep. 1979;63:131-134.
8 Order SE, Stillwagon GB, Klein JL, et al. Iodine 131 antiferritin, a new treatment modality in hepatoma: a Radiation Therapy Oncology Group study. J Clin Oncol. 1985;3:1573-1582.
9 Kohler G, Milstein C. Continuous culture of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495-497.
10 Esteban JM, Colcher D, Sugarbaker P, et al. Quantitative and qualitative aspects of radiolocalization in colon cancer patients of intravenously administered MAb B72.3. Int J Cancer. 1987;39(1):50-59.
11 Weiden PL, Breitz HB. Pretargeted radioimmunotherapy (PRIT) for treatment of non-Hodgkin’s lymphoma (NHL). Crit Rev Oncol Hematol. 2001;40(1):37-51.
12 Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer. 2001;1:118-129.
13 Goldenberg DM, Sharkey RM, Paganelli G, Barbet J, Chatal JF. Antibody pretargeting advances cancer radioimmunodetection and radioimmunotherapy. J Clin Oncol. 2006;24(5):823-834.
14 Roninson IB. The role of the MDR1 (P-glycoprotein) gene in multidrug resistance in vitro and in vivo. Biochemical Pharmacology. 1992;43:95-102.
15 Mach JP, Forni M, Ritschard J, et al. Use and limitations of radiolabeled anti-CEA antibodies and their fragments for photoscanning detection of human colorectal carcinomas. Oncodev Biol Med. 1980;1:49-69.
16 DeNardo GL, DeNardo SJ. Overview of obstacles and opportunities for radioimmunotherapy of cancer. In: Goldenberg DM, editor. Cancer therapy with radiolabeled antibodies. Boca Raton, Florida: CRC Press; 1994:141-154.
17 Larson SM, Carrasquillo JA, Krohn KA, et al. Localization of 131I-labeled p97-specific Fab fragments in human melanoma as a basis for radiotherapy. J Clin Invest. 1983;72:2101-2114.
18 Moldofsky PJ, Powe J, Mulhern CB, et al. Metastatic colon carcinoma detected with radiolabeled F(ab′)2 monocolonal antibody fragments. Radiology. 1983;149(2):549-555.
19 Albrecht H, DeNardo SJ. Recombinant antibodies: From the laboratory to the clinic. Cancer Biother Radiopharm. 2006;21(4):285-304.
20 Oettgen H, Old LJ. The history of cancer immunotherapy. Philadelphia: J.B. Lippincott; 1991.
21 Eisenthal A, Shiloni E, Rosenberg SA. Characterization of IL-2-induced murine cells which exhibit ADCC activity. Cell Immunol. 1988;115:257-272.
22 Caron PC, Co MS, Bull MK, Avdalovic NM, Queen C, Scheinberg DA. Biological and immunological features of humanized M195 (anti-CD33) monoclonal antibodies. Cancer Res. 1992;52:6761-6767.
23 Takahashi S, Maecker HT, Levy R. DNA fragmentation and cell death mediated by T cell antigen receptor/CD3 complex on a leukemia T cell line. Eur J Immunol. 1989;19:1911-1919.
24 Trauth BC, Klas C, Peters AM, et al. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301-305.
25 Sauvage CA, Mendelsohn JC, Lesley JF, Trowbridge IS. Effects of monoclonal antibodies that block transferrin receptor function on the in vivo growth of a syngeneic murine leukemia. Cancer Res. 1987;47:747-753.
26 Cuttitta F, Carney DN, Mulshine J, et al. Bombesin-like peptides can function as autocrine growth factors in human small-cell lung cancer. Nature. 1985;316:823-826.
27 Waldmann TA, Goldman CK, Bongiovanni KF, et al. Therapy of patients with human T-cell lymphotrophic virus I-induced adult T-cell leukemia with anti-Tac, a monoclonal antibody to the receptor for interleukin-2. Blood. 1988;72(5):1805-1816.
28 Trowbridge IS, Lopez F. Monoclonal antibody to transferrin receptor blocks transferrin binding and inhibits human tumor cell growth in vitro. Proc Natl Acad Sci U S A. 1982;79:1175-1179.
29 Masui H, Kawamoto T, Sato JD, Wolf B, Sato G, Mendelsohn J. Growth inhibition of human tumor cells in athymic mice by anti-epidermal growth factor receptor monoclonal antibodies. Cancer Res. 1984;44:1002-1007.
30 Fagerberg J, Frodin JE, Ragnhammar P, Steinitz M, Wigzell H, Mellstedt H. Induction of an immune network cascade in cancer patients treated with monoclonal antibodies (ab1). II. Is induction of anti-idiotype reactive T cells (T3) of importance for tumor response to MAb therapy? Cancer Immunol Immunother. 1994;38:149-159.
31 Herlyn D, Wettendorff M, Schmoll E, et al. Anti-idiotype immunization of cancer patients: modulation of the immune response. Proc Natl Acad Sci USA. 1987;84(22):8055-8059.
32 Mittelman A, Chen ZJ, Kageshita T, et al, Active specific immunotherapy in patients with melanoma. A clinical trial with mouse antiidiotypic monoclonal antibodies elicited with syngeneic anti-high-molecular-weight-melanoma-associated antigen monoclonal antibodies, [published erratum appears in J Clin Invest 1991 Feb;87(2):757]; J Clin Invest; 86. 6; 1990:2136-2144.
33 Nourigat C, Badger CC, Bernstein ID. Treatment of lymphoma with radiolabeled antibody: elimination of tumor cells lacking target antigen. J Natl Cancer Inst. 1990;82:47-50.
34 Cobb LM, Humm JL. Radioimmunotherapy of malignancy using antibody targeted radionuclides. Br J Cancer. 1986;54:863-870.
35 O’Donoghue JA. The impact of tumor cell proliferation in radioimmunotherapy. Cancer. 1994;73(3 Suppl):974-980.
36 Wheldon TE, O’Donoghue JA. The radiobiology of targeted radiotherapy. Int J Radiat Biol. 1990;58:1-21.
37 Griffiths GL. Radiochemistry of therapeutic radionuclides. In: Goldenberg DM, editor. Cancer therapy with radiolabeled antibodies. Boca Raton, FL: CRC Press; 1995:47-61.
38 Order SE, Klein JL, Leichner PK, Frincke J, Lollo C, Carlo DJ. Yttrium antiferritin—a new therapeutic radiolabeled antibody. Int J Radiat Oncol Biol Phys. 1986;12:277-281.
39 Parker BA, Vassos AB, Halpern SE, et al. Radioimmunotherapy of human B-cell lymphoma with Y-90-conjugated antiidiotype monoclonal antibody. Cancer Res [Suppl]. 1990;50(3):1022s-1028s.
40 Vriesendorp HM, Herpst JM, Germack MA, et al. Phase I–II studies of yttrium-labeled antiferritin treatment for end-stage Hodgkin’s disease, including radiation therapy oncology group 87-01. J Clin Oncol. 1991;9:918-928.
41 DeNardo SJ, DeNardo GL, Kukis DL, Mausner LF, Moody DC, Meares CF. Pharmacology of 67Cu-TETA-Lym-1 antibody in patients with B cell lymphoma [abstract]. Antibody Immunoconj Radiophar. 1991;4:36.
42 DeNardo GL, DeNardo SJ, O’Donnell RT, et al. Are radiometal-labeled antibodies better than Iodine-131-labeled antibodies: comparative pharmacokinetics and dosimetry of copper-67-, iodine-131-, and yttrium-90-labeled Lym-1 antibody in patients with non-Hogkin’s lymphoma. Clinical Lymphoma. 2000;1(2):118-126.
43 DeNardo GL, DeNardo SJ, Levy NB. Treatment of B cell malignancies with 131I-Lym-1 and mechanisms for improvement. In: Epenetos A, editor. Monoclonal antibodies 2, applications in clinical oncology. New York: Chapman & Hall Medical; 1993:355-367.
44 DeNardo GL, Kukis DL, Shen S, DeNardo DA, Meares CF, DeNardo SJ. Copper-67 versus iodine-131 labeled Lym-1 antibody: comparative pharmacokinetics and dosimetry in patients with non-Hodgkin’s lymphoma. Clin Cancer Res. 1999;5:533-541.
45 Mattes MJ. Radionuclide-antibody conjugates for single-cell cytotoxicity. Cancer. 2002;94:1215-1223.
46 Imam SK. Advancements in cancer therapy with alpha-emitters: a review. Int J Radiat Oncol Biol Phys. 2001;51:271-278.
47 Brechbiel MW. Targeted alpha-therapy: past, present, future? Dalton Trans. 2007;43:4918-4928.
48 McDevitt MR, Ma D, Lai LT, et al. Tumor therapy with targeted atomic nanogenerators. Science. 2001;294(5546):1537-1540.
49 Kennel SJ, Brechbiel MW, Milenic DE, Schlom J, Mirzadeh S. Actinium-225 conjugates of MAb CC49 and humanized delta CH2CC49. Cancer Biother Radiopharm. 2002;17(2):219-231.
50 Robinson MK, Shaller C, Garmestani K, et al. Effective treatment of established human breast tumor xenografts in immunodeficient mice with a single dose of the alpha-emitting radioisotope Astatine-211 conjugated to anti-HER/neu diabodies. Clin Cancer Res. 2008;14(3):875-882.
51 Hnatowich DJ, McGann J. DTPA-coupled proteins–procedures and precautions. Int J Rad Appl Instrum [B]. 1987;14:563-568.
52 Moi MK, DeNardo SJ, Meares CF. Stable bifunctional chelates of metals used in radiotherapy. Cancer Res. 1990;50(Suppl):789-793.
53 Stimmel JB, Stockstill ME, Kull FCJ. Yttrium-90 chelation properties of tetraazatetraacetic acid macrocycles, diethylenetriaminepentaacetic acid analogues, and a novel terpyridine acyclic chelator. Bioconjug Chem. 1995;6:219-225.
54 Harrison A, Walker CA, Parker D, et al. The in vivo release of 90Y from cyclic and acyclic ligand-antibody conjugates. Int J Rad Appl Instrum [B]. 1991;18:469-476.
55 DeNardo GL, Kroger LA, DeNardo SJ, et al. Comparative toxicity studies of yttrium-90 MX-DTPA and 2-IT-BAD conjugated monoclonal antibody (BrE-3). Cancer. 1994;73:1012-1022.
56 Dillehay LE, Williams JR. Radiobiology of dose-rate patterns achievable in radioimmunoglobulin therapy. Front Rad Ther Oncol. 1990;24:96-103.
57 Hall EJ. Radiation dose rate: a factor of importance in radiobiology and radiotherapy. Br J Radiol. 1972;45:81-97.
58 Fowler JF. Radiobiological aspects of low dose rates in radioimmunotherapy. Int J Radiat Oncol Biol Phys. 1990;18:1261-1269.
59 Shipley WU, Peacock JH, Steel GG, Stephens TC. Continuous irradiation of the Lewis lung carcinoma in vivo at clinically-used “ultra” low dose-rates. Int J Radiat Oncol Biol Phys. 1983;9:1647-1653.
60 Mitchell JB, Bedford JS, Bailey SM. Dose-rate effects on the cell cycle and survival of S3 HeLa and V79 cells. Radiat Res. 1979;79:520-536.
61 Szechter A, Schwartz G, Barsa JM. Continuous and fractionated irradiation of mammalian cells in culture—1. The effect of growth rate. Int J Radiat Oncol Biol Phys. 1978;4:991-1000.
62 Courtenay VD. Radioresistant mutants of L5178Y cells. Radiat Res. 1969;38:186-203.
63 Knox SJ, Levy R, Miller RA, et al. Determinants of the antitumor effect of radiolabeled monoclonal antibodies. Cancer Res. 1990;50:4935-4940.
64 Wessels BW, Vessella RL, Palme DF, Berkopec JM, Smith GK, Bradley EW. Radiobiological comparison of external beam irradiation and radioimmunotherapy in renal cell carcinoma xenografts. Int J Radiat Oncol Biol Phys. 1989;17:1257-1263.
65 Ning S, Trisler K, Wessels BW, Knox SJ. Radiobiologic studies of radioimmunotherapy and external beam radiotherapy in vitro and in vivo in human renal cell carcinoma xenografts. Cancer. 1997;80(12 Suppl):2519-2528.
66 Hall EJ. Radiobiology for the radiologist. Philadelphia: J.B. Lippincott Company; 1994. pp 29–43, 107–131
67 Rostock RA, Klein JL, Leichner PK, Order SE. Distribution of and physiologic factors that affect 131I-antiferritin tumor localization in experimental hepatoma. Int J Radiat Oncol Biol Phys. 1984;10:1135-1141.
68 Knox SJ, Sutherland W, Goris ML. Correlation of tumor sensitivity to low-dose-rate irradiation with G2/M-phase block and other radiobiological parameters. Radiat Res. 1993;135:24-31.
69 Sands H, Jones PL, Shah SA, Palme D, Vessella RL, Gallagher BM. Correlation of vascular permeability and blood flow with monoclonal antibody uptake by human Clouser and renal cell xenografts. Cancer Res. 1988;48:188-193.
70 Macklis RM, Lin JY, Beresford B, Atcher RW, Hines JJ, Humm JL. Cellular kinetics, dosimetry, and radiobiology of alpha-particle radioimmunotherapy: induction of apoptosis. Radiat Res. 1992;130:220-226.
71 Macklis RM, Beresford BA, Palayoor S, Sweeney S, Humm JL. Cell cycle alterations, apoptosis, and response to low-dose-rate radioimmunotherapy in lymphoma cells. Int J Radiat Oncol Biol Phys. 1993;27:643-650.
72 Kroger LA, DeNardo GL, Gumerlock PH, et al. Apoptosis related gene and protein expression in human lymphoma xenografts (Raji) after low dose rate radiation using 67Cu-2IT-BAT-Lym-1 radioimmunotherapy. Cancer Biother Radiopharm. 2001;16(3):213-225.
73 Winthrop MD, DeNardo SJ, Muenzer JT, Chi SG, Gumerlock PH. p53-independent response of a human breast cancer xenograft to radioimmunotherapy. Cancer. 1997;80(Suppl 12):2529-2537.
74 Sgouros G, Knox SJ, Joiner MC, Morgan WF, Kassis AI. MIRD continuing education: bystander and low-dose-rate effects? Are these relevant to radionuclide therapy? J Nucl Med. 2007;48:1683-1691.
75 Kurtzman SH, Russo A, Mitchell JB, et al. 212Bismuth linked to an antipancreatic carcinoma antibody: model for alpha-particle-emitter radioimmunotherapy. J Natl Cancer Inst. 1988;80(6):449-452.
76 Wessels BW, Rogus RD. Radionuclide selection and model absorbed dose calculations for radiolabeled tumor associated antibodies. Med Phys. 1984;11:638-645.
77 Brown I. Astatine-211: Its possible applications in cancer therapy. Int J Rad Appl Instrum [A]. 1986;37:789-798.
78 Buchsbaum DJ, Wessels BW. Radiolabeled antibody tumor dosimetry. In: Goldenberg DM, editor. Cancer therapy with radiolabeled antibodies. Boca Raton, Florida: CRC; 1995:21-32.
79 Buchsbaum DJ, Wessels BW. Introduction: radiolabeled antibody tumor dosimetry. Med Phys. 1993;20:499-501.
80 DeNardo GL, Raventos A, Hines HH, et al. Requirements for a treatment planning system for radioimmunotherapy. Int J Radiat Oncol Biol Phys. 1985;11:335-348.
81 Scott AM, Macapinlac HA, Divgi CR, et al. Clinical validation of SPECT and CT/MRI image registration in radiolabeled monoclonal antibody studies of colorectal carcinoma. J Nucl Med. 1994;35:1976-1984.
82 Sgouros G, Chiu S, Pentlow K, et al. Three-dimensional dosimetry for radioimmunotherapy treatment planning. J Nucl Med. 1993;34(9):1595-1601.
83 Leichner PK, Koral KF, Jaszczak RJ, Green AJ, Chen GTY, Roeske JC. An overview of imaging techniques and physical aspects of treatment planning in radioimmunotherapy. Med Phys. 1993;20(2):569-577.
84 Stabin MG. Internal dosimetry in the use of radiopharmaceuticals in therapy—science at a crossroads? Cancer Biother Radiopharm. 1999;14(2):81-89.
85 DeNardo SJ, Williams LE, Leigh BR, Wahl R. Radioimmunotherapy: Choosing an optimal dose for clinical response. Cancer. 2002;94(Suppl 4):1275-1286.
86 Watson EE, Stabin MG, Siegel JA. MIRD formulation. Review. Med Phys. 1993;20(2 Pt 2):511-514.
87 Fisher DR. Radiation dosimetry for radioimmunotherapy. Cancer. 1994;73:905-911.
88 Humm JL, Roeske JC, Fisher DR, Chen GT. Microdosimetric concepts in radioimmunotherapy. Med Phys. 1993;20:535-541.
89 Humm JL. A microdosimetric model of At-211 labeled antibodies for RIT. Int J Radiat Oncol Biol Phys. 1987;13:1767-1773.
90 Stinchcomb TG, Roeske JC. Analytic microdosimetry for radioimmunotherapeutic alpha emitters. Med Phys. 1992;19:1385-1393.
91 DeNardo GL, Juweid ME, White CA, Wiseman GA, DeNardo SJ. Role of radiation dosimetry in radioimmunotherapy planning and treatment dosing. Crit Rev Oncol Hematol. 2001;39:203-218.
92 DeNardo GL, DeNardo SJ, Balhorn R. Systemic radiotherapy can cure lymphoma: a paradigm for other malignancies? Cancer Biother Radiopharm. 23(4), 2008.
93 Barbet J, Kraeber-Bodere F, Vuillez JP, Gautherot E, Rouvier E, Chatal JF. Pretargeting with the affinity enhancement system for radioimmunotherapy. Cancer Biother Radiopharm. 1999;14(3):153-167.
94 Chinol M, Grana C, Gennari R, Cremonesi M, Geraghty JG, Paganelli G. Pretargeted radioimmunotherapy of cancer. In: Abrams PG, Fritzberg AR, editors. Radioimmunotherapy of cancer. New York: Marcel Dekker, Inc; 2000:169-193.
95 Kukis DL, Novak-Hofer I, DeNardo SJ. Cleavable linkers to enhance selectivity of antibody-targeted therapy of cancer. Cancer Biother Radiopharm. 2001;16(6):457-467.
96 DeNardo SJ, Yuan A, Richman C, et al. Therapeutic index enhancement by DOTA peptide linkage in 111In/90Y DOTA-Lym-1 and m170 MAbs in clinical trials [abstract]. J Nucl Med. 2002;43(5 Suppl):117P.
97 Zimmermann K, Gianollini S, Schubiger PA, Novak-Hofer I. A triglycine linker improves tumor uptake and biodistributions of 67-Cu-labeled anti-neuroblastoma MAb chCE7 F(ab′)2 fragments. Nucl Med Biol. 1999;26:943-950.
98 Arano Y, Fujioka Y, Akizawa H, et al. Chemical design of radiolabeled antibody fragments for low renal radioactivity levels. Cancer Res. 1999;59:128-134.
99 Arano Y. Strategies to reduce renal radioactivity levels of antibody fragements, The Quarterly Journal of Nuclear. Medicine. 1998;42(4):262-270.
100 Li L, Olafsen T, Anderson AL, Wu A, Raubitschek AA, Shively JE. Reduction of kidney uptake in radiometal labeled peptide linkers conjugated to recombinant antibody fragments. Site-specific conjugation of DOTA-peptides to a Cys-diabody. Bioconjug Chem. 2002;13:985-995.
101 Goodwin DA, Meares CF, McCall MJ, McTigue M, Chaovapong W. Pre-targeted immunoscintigraphy of murine tumors with indium-111-labeled bifunctional haptens. J Nucl Med. 1988;29:226-234.
102 Paganelli G, Magnani P, Zito F, et al. Three-step monoclonal antibody tumor targeting in carcinoembryonic antigen-positive patients. Cancer Res. 1991;51:5960-5966.
103 Sung C, van Osdol WW. Pharmacokinetic comparison of direct antibody targeting with pretargeting protocols based on streptavidin-biotin binding. J Nucl Med. 1995;36(5):867-876.
104 Le Doussal JM, Barbet J, Delaage M. Bispecific-antibody-mediated targeting of radiolabeled bivalent haptens: theoretical, experimental and clinical results. Int J Cancer. 1992;7(Suppl):58-62.
105 Bos ES, Kuijpers WH, Meesters-Winters M, et al. In vitro evaluation of DNA-DNA hybridization as a two-step approach in radioimmunotherapy of cancer. Cancer Res. 1994;54:3479-3486.
106 Breitz HB, Weiden PL, Appelbaum JW, et al. Pretargeted radioimmunotherapy (PRIT) for treatment of non-Hodgkin’s lymphoma: preliminary results. J Nucl Med. 1999;40(5):19P.
107 Breitz HB, Weiden PL, Beaumier PL, et al. Clinical optimization of pretargeted radioimmunotherapy with antibody-streptavidin conjugate and 90Y-DOTA-biotin. J Nucl Med. 2000;41(1):131-140.
108 Axworthy DB, Reno JM, Hylarides MD, et al. Cure of human carcinoma xenografts by a single dose of pretargeted yttrium-90 with negligible toxicity. Proc Natl Acad Sci U S A. 2000;97(4):1802-1807.
109 Gautherot E, Rouvier E, Daniel L, et al. Pretargeted radioimmunotherapy of human colorectal xenografts with bispecific antibody and 131I-labeled bivalent hapten. The Journal of Nuclear Medicine. 2000;41(3):480-487.
110 Weichselbaum R, Beckett MA, Vokes EE, et al. Cellular and molecular mechanisms of radioresistance. Cancer Treat Res. 1995;74:131-140.
111 Leung SO, Qu Z, Sharkey RM, et al. Pretargeting colonic tumors with engineered bispecific antibodies for improved radioimmunotherapy (RAIT). [abstract]. J Nucl Med. 2000;41:270.
112 Jain RK. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990;9:253-266.
113 Humm JL. Dosimetric aspects of radiolabeled antibodies for tumor therapy. J Nucl Med. 1986;27:1490-1497.
114 Jain RK, Baxter LT. Mechanisms of heterogenious distibution of monoclonal antibopdies and other macromolecules in turmors; significance of elevated interstitial pressure [abstract]. Cancer Res. 1988;48:7022.
115 Shockley TR, Lin K, Nagy JA, Tompkins RG, Yarmush ML, Dvorak HF. Spatial distribution of tumor-specific monoclonal antibodies in human melanoma xenografts. Cancer Res. 1992;52:367-376.
116 O’Donoghue JA. The implications of nonuniform tumor doses for radioimmunotherapy. J Nucl Med. 1999;40:1337-1341.
117 DeNardo GL, Schlom J, Buchsbaum DJ, et al. Rationales, evidence and design considerations in fractionated radioimmunotherapy. Cancer. 2002;94(4):1332-1348.
118 Withers HR, Taylor JM, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27:131-146.
119 Schlom J, Molinolo A, Simpson JF, et al. Advantage of dose fractionation in monoclonal antibody-targeted radioimmunotherapy. J Natl Cancer Inst. 1990;82:763-771.
120 Buchsbaum DJ, Khazaeli MB, Liu TP, Bright S. Fractionated radioimmunotherapy of human colon carcinoma xenografts with I-131-labeled monoclonal antibody CC49. Cancer Res. 1995;55(Suppl):5881-5887.
121 DeNardo GL, DeNardo SJ, Lamborn KR, et al. Low-dose fractionated radioimmunotherapy for B-cell malignancies using 131I-Lym-1 antibody. Cancer Biother Radiopharm. 1998;13(4):239-254.
122 DeNardo GL, DeNardo SJ, Goldstein DS, et al. Maximum tolerated dose, toxicity, and efficacy of 131I-Lym-1 antibody for fractionated radioimmunotherapy of non-Hodgkin’s lymphoma. J Clin Oncol. 1998;16(10):3246-3256.
123 DeNardo SJ, DeNardo GL, O’Grady LF, et al. Treatment of a patient with B cell lymphoma by I-131 Lym-1 monoclonal antibodies. Int J Biol Markers. 1987;2:49-53.
124 DeNardo SJ, DeNardo GL, O’Grady LF, et al. Treatment of B cell malignancies with I-131 Lym-1 monoclonal antibodies. Int J Cancer. 1988;3(Suppl 3):96-101.
125 DeNardo GL, DeNardo SJ. Treatment of B-lymphocyte malignancies with 131I-Lym-1 and 67Cu- 2IT-BAT-Lym-1 and opportunities for improvement. In: Goldenberg DM, editor. Cancer Therapy with Radiolabeled Antibodies. Boca Raton, Fl: CRC Press; 1994:217-227.
126 Hu E, Epstein AL, Naeve GS, et al. A phase 1a clinical trial of Lym-1 monoclonal antibody serotherapy in patients with refractory B cell malignancies. Hematol Oncol. 1989;7:155-166.
127 O’Donnell RT, DeNardo GL, Kukis DL, et al. A clinical trial of radioimmunotherapy with 67Cu-2IT-BAT-Lym-1 for non-Hodgkin’s lymphoma. J Nucl Med. 1999;40:2014-2020.
128 O’Donnell RT, Shen S, DeNardo SJ, et al. A phase I study of 90Y-2IT-BAD-Lym-1 in patients with non-Hodgkin’s lymphoma. Anticancer Res. 2000;20:3647-3656.
129 Kaminski MS, Estes J, Zasadny KR, et al. Radioimmunotherapy with iodine (131)I tositumomab for relapsed refractory B-cell non-Hodgkin lymphoma: update results and long-term follow-up of the University of Michigan experience. Blood. 2000;96(4):1259-1266.
130 Vose JM, Wahl RL, Saleh M, et al. Multicenter phase II study of iodine-131 tositumomab for chemotherapy-relapsed/refractory low-grade and transformed low-grade B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 2000;18(6):1316-1323.
131 Kaminski MS, Zelenetz AD, Press OW, et al. Pivotal study of iodine I 131 tositumomab for chemotherapy-refractory low-grade or transformed low-grade B-cell non-Hodgkin’s lymphomas. J Clin Oncol. 2001;19:3918-3928.
132 Davis TA, Kaminski MS, Leonard JP, et al. The radioisotope contributes significantly to the activity of radioimmunotherapy. Clin Cancer Res. 2004;10(23):7792-7798.
133 Bistron MJ, Leahy MF, Hicks RJ, et al. Repeat treatment with iodine-131-rituximab is safe and effective in patients with relapsed indolent B-cell non-Hodgkin’s lymphoma (NHL) who had previously responded to this therapy [abstract]. Blood. 2007:118.
134 Horning SJ, Younes A, Jain V, et al. Efficacy and safety of tositumomab and iodine-131 tositumomab (Bexxar) in B cell lymphoma, progressive after rituximab. J Clin Oncol. 2005;23(4):712-719.
135 Kaminski MS, Tuck M, Estes J, et al. 131I-Tositumomab therapy as initial treatment for follicular lymphoma. N Engl J Med. 2005;352(5):441-449.
136 Press OW, Unger JM, Braziel RM, et al. A phase II trial of CHOP chemotherapy followed by tositumomab/iodine I 131 tositumomab for previously untreated follicular non-Hodgkin’s lymphoma: five-year follow-up of Southwest Oncology Group Protocol S9911. Blood. 2003;102:1606-1612.
137 Knox SJ, Goris ML, Trisler K, et al. Yttrium-90-labeled anti-CD20 monoclonal antibody therapy of recurrent B-cell lymphoma. Clin Cancer Res. 1996;2:457-470.
138 Wiseman GA, White CA, Stabin M, et al. Phase I/II 90Y-Zevalin (yttrium-90 ibritumomab tiuxetan, IDEC-Y2B8) radioimmunotherapy dosimetry results in relapsed or refractory non-Hodgkin’s lymphoma. Eur J Nucl Med. 2000;27(7):766-777.
139 Witzig TE, White CA, Wiseman GA, et al. Phase I/II trial of IDEC-Y2B8 radioimmunotherapy for treatment of relapsed or refractory CD20+ B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 1999;17:3793-3803.
140 Witzig TE. Radioimmunotherapy for patients with relapsed B-cell non-Hodgkin lymphoma. Cancer Chemother Pharmacol. 2001;48(Suppl 1):S91-S95.
141 Witzig TE, Gordon LI, Cabanillas F, et al. Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 2002;20(10):2453-2463.
142 Witzig TE, Flinn IW, Gordon LI, et al. Treatment with ibritumomab tiuxetan radioimmunotherapy in patients with rituximab-refractory follicular non-Hodgkin’s lymphoma. J Clin Oncol. 2002;20(15):3262-3269.
143 Morschhauser F, Illidge TM, Huglo D, et al. Efficacy and safety of yttrium-90 ibritumomab tiuxetan in patients with relapsed or refractory diffuse large B-cell lymphoma not appropriate for autologous stem-cell transplantation. Blood. 2007;110:54.
144 Botto B, Bello M, Benevolo G, et al. Radioimmunotherapy (RIT) with 90Y-ibritumomab tiuxetan (Zevalin) for the treatment of relapsed or resistant aggressive diffuse large B-cell lymphoma (DLBCL) heavily pretreated with rituximab + chemotherapy: a GIMURELL experience. [abstract]. Blood. 2007:110.
145 Jurczak W, Giza A, Zimowska-Curylo D, et al. 90Y-Zevalin (90Y-ibritumomab tiuxetan) radioimmunotherapy (RIT) consolidation in mantle cell lymphoma (MCL) patients, not illegible for intensive therapy protocols [abstract]. Blood. 2007:110.
146 Weigert O, von Schilling C, Rummel MJ, et al. Efficacy and safety of a single-course of yttrium-90 ibritumomab tiuxetan for treatment of patients with relapsed or refractory mantle cell lymphoma after/not appropriate for autologous stem cell transplantation—final analysis of a phase II trial of the European MCL network [abstract]. Blood. 2007:110.
147 Smith MR, Zhang L, Gordon LI, et al. Phase II study of R-CHOP followed by 90Y-ibritumomab tiuxetan in untreated mantle cell lymphoma: Eastern Cooperative Oncology Group study E1499 [abstract]. Blood. 2007;110:11.
148 Vanazzi A, Ferrucci PF, Grana C, et al. Efficacy of 90Y-ibritumomab tiuxetan in marginal-zone lymphoma (MZL) [abstract]. Blood. 2007:110.
149 Zinzani PL, Tani M, Fanti S, et al. A phase 2 trial of fludarabinie and mitoxantrone chemotherapy followed by yttrium-90 ibritumomab tiuxetan for patients with previously untreated, indolent, nonfollicular, non-Hodgkin lymphoma. Cancer. 2008;112:856.
150 Rosen ST, Zimmer AM, Goldman-Leikin R, et al. Radioimmunodetection and radioimmunotherapy of cutaneous T cell lymphomas using an 131I-labeled monoclonal antibody: an Illinois Cancer Council study. J Clin Oncol. 1987;5(4):562-573.
151 Lenhard REJr, Order SE, Spunberg JJ, Asbell SO, Leibel SA. Isotopic immunoglobulin: a new systemic therapy for advanced Hodgkin’s disease. J Clin Oncol. 1985;3:1296-1300.
152 Bierman PJ, Vose JM, Leichner PK, et al. Yttrium 90-labeled antiferritin followed by high-dose chemotherapy and autologous bone marrow transplantation for poor-prognosis Hodgkin’s disease. J Clin Oncol. 1993;11:698-703.
153 Decaudin D, Levy R, Lokiec F, et al. Radioimmunotherapy (RIT) of refractory or relapsed Hodgkin’s lymphoma (HL) with 90-yttrium-labeled antiferritin antibody [abstract]. Blood. 2007:110.
154 Press OW, Eary JF, Badger CC, et al. Treatment of refractory non-Hodgkin’s lymphoma with radiolabeled MB-1 (anti-CD37) antibody. J Clin Oncol. 1989;7:1027-1038.
155 Press OW, Eary JF, Badger CC, et al. High-dose radioimmunotherapy of B cell lymphomas. Front Radiat Ther Oncol. 1990;24:204-213.
156 Press OW, Eary JF, Appelbaum FR, Bernstein ID. Treatment of relapsed B cell lymphomas with high-dose radioimmunotherapy and bone marrow transplantation. In: Goldenberg DM, editor. Cancer therapy with radiolabeled antibodies. Boca Raton, FL: CRC Press, Inc.; 1995:229-237.
157 Liu SY, Eary JF, Petersdorf SH, et al. Follow-up of relapsed B-cell lymphoma patients treated with iodine-131-labeled anti-CD20 antibody and autologous stem-cell rescue. J Clin Oncol. 1998;16(10):3270-3278.
158 Press OW, Eary JF, Gooley T, et al. A phase I/II trial of iodine-131-tositumomab (anti-CD20), etoposide, cyclophosphamide, and autologous stem cell transplantation for relapsed B-cell lymphomas. Blood. 2000;96(9):2934-2942.
159 Behr TM, Griesinger F, Riggert J, et al. High-dose myeloablative radioimmunotherapy of mantle cell non-Hodgkin lymphoma with the iodine-131-labeled chimeric anti-CD20 antibody C2B8 and autologous stem cell support. Results of a pilot study. Cancer. 2002;94(4 Suppl):1363-1372.
160 Gopal AK, Rajendran JG, Petersdorf SH, et al. High-dose chemo-radioimmunotherapy with autologous stem cell support for relapsed mantle cell lymphoma. Blood. 2002;99:3158-3162.
161 Czuczman MS, Emmanouilides C, Darif M. Treatment-related myelodysplastic syndrome and acute myelogenous leukemia in patients treated with ibritumomab tiuxetan radioimmunotherapy. J Clin Oncol. 2007;25(27):4285.
162 Vanazzi A, Ferrucci PF, Grana C, et al. High dose 90-yttrium ibritumomab tiuxetan with PBSC support in refractory-resistant NHL patients [abstract]. Blood. 2007:110.
163 DeNardo SJ, DeNardo GL, O’Grady LF, et al. Pilot studies of radioimmunotherapy of B cell lymphoma and leukemia using I-131 Lym-1 monoclonal antibody. Antibody Immunoconj Radiophar. 1988;1:17-33.
164 DeNardo GL, Lewis JP, DeNardo SJ, O’Grady LF. Effect of Lym-1 radioimmunoconjugate on refractory chronic lymphocytic leukemia. Cancer. 1994;73:1425-1432.
165 Jurcic JG. Antibody therapy for residual disease in acute myelogenous leukemia. Crit Rev Oncol Hematol. 2001;38:37-45.
166 Schwartz MA, Lovett DR, Redner A, et al. Dose-escalation trial of M195 labeled with iodine 131 for cytoreduction and marrow ablation in relapsed or refractory myeloid leukemias. J Clin Oncol. 1993;11(2):294-303.
167 Co MS, Avdalovic NM, Caron PC, Avdalovic MV, Scheinberg DA, Queen C. Chimeric and humanized antibodies with specificity for the CD33 antigen. J Immunol. 1992;148(4):1149-1154.
168 Caron PC, Jurcic JG, Scott AM, et al. A phase 1B trial of humanized monoclonal antibody M195 (anti-CD33) in myeloid leukemia: specific targeting without immunogenicity. Blood. 1994;83(7):1760-1768.
169 Burke JM, Caron PC, Papadopoulos EB, et al. Cytoreduction with iodine-anti-CD33 antibodies before bone marrow transplantation for advanced myeloid leukemia. Bone Marrow Transplant. 2003;32:549.
170 Reske SN, Bunjes D, Buchmann I, et al. Targeted bone marrow irradiation in the conditioning of high-risk leukemia prior to stem cell transplantation. Eur J Nucl Med. 2001;28(7):807-815.
171 Rosenblat TL, McDevitt MR, Pandit-Taskar N, et al. Phase I trial of the targeted alpha-particle nano-generator actinium-225 (225AC)-HuM195 (Anti-CD33) in acute myeloid leukemia (AML) [abstract]. Blood. 2007:110.
172 Matthews DC, Appelbaum FR, Eary JF, et al. Phase I study of 131I-anti-CD45-antibody plus cyclophosphamide and total body irradiation for advance acute leukemia and myelodysplastic syndrome. Blood. 1999;94:1237.
173 Carrasquillo JA, Krohn KA, Beaumier P, et al. Diagnosis of and therapy for solid tumors with radiolabeled antibodies and immune fragments. Cancer Treat Rep. 1984;68:317-328.
174 Dadachova E, Revskaya E, Sesay MA. Pre-clinical evaluation and efficacy studies of a melanin-binding IgM antibody labeled with 188Re against experimental human metastatic melanoma in nude mice. Cancer Biol Ther. 2008;7(7):1116-1127.
175 Macey DJ, DeNardo SJ, DeNardo GL, Goodnight JK, Unger MW. Uptake of indium-111-labeled monoclonal antibody ZME-018 as a function of tumor size in a patient with melanoma. Am J Physiol Imag. 1988;3:1-6.
176 Wikstrand CJ, Cokgor I, Sampson JH, Bigner DD. Monoclonal antibody therapy of human gliomas: current status and future approaches. Cancer Metastasis Rev. 1999;18(4):451-464.
177 Brady LW, Woo DV, Markoe AM, et al. Treatment of malignant gliomas with 125I-labeled monoclonal antibody against epidermal growth factor receptor. Antibody Immunoconj Radiophar. 1990;3:169.
178 Snelling L, Miyamoto CT, Bender H, et al. Epidermal growth factor receptor 425 monoclonal antibodies radiolabeled with iodine-125 in the adjuvant treatment of high-grade astrocytomas. Hybridoma. 1995;14(2):111-114.
179 Bartolomei M, Mazzetta C, Handkiewicz-Junak D, et al. Combined treatment of glioblastoma patients with locoregional pre-targeted 90Y-biotin radioimmunotherapy and temozolomide. Q J Nucl Med. 2004;48(3):220-228.
180 Riva P, Franceschi G, Riva N, Casi M, Santimaria M, Adamo M. Role of nuclear medicine in the treatment of malignant gliomas: the locoregional radioimmunotherapy approach. Eur J Nucl Med. 2000;27(5):601-609.
181 Bigner DD, Brown MT, Friedman AH, et al. Iodine-131-labeled antitenascin monoclonal antibody 81C6 treatment of patients with recurrent malignant gliomas: phase I trial results. J Clin Oncol. 1998;16(6):2202-2212.
182 Cokgor I, Akabani G, Kuan CT, et al. Phase I trial results of iodine-131-labeled antitenascin monoclonal antibody 81C6 treatment of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2000;18(22):3862-3872.
183 Reardon DA, Akabani G, Coleman RE, et al. Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol. 2002;20(5):1389-1397.
184 Cheung NKV, Yeh SDJ, Gulati S, Burch L, Kushner BH, Larson S. 131I-3F8 targeted radiotherapy of neuroblastoma (NB): a phase I clinical trial [abstract]. Proc Am Assoc Cancer Res. 1990;31:284.
185 Boiardi A, Bartolomei M, Silvani A, et al. Intratumoral delivery of mitoxantrone in association with 90-Y radioimmunotherapy (RIT) in recurrent glioblastoma. J Neuro Oncol. 2005;72:125.
186 Meredith RF, Bueschen AJ, Khazaeli MB, et al. Treatment of metastatic prostate carcinoma with radiolabeled antibody CC49. J Nucl Med. 1994;35:1017-1022.
187 O’Donnell RT, DeNardo SJ, Yuan A, et al. Radioimmunotherapy with 111In/90Y-2IT-BAD-m170 for metastatic prostate cancer. Clin Cancer Res. 2001;7:1561-1568.
188 Richman CM, DeNardo SJ, O’Donnell RT, et al. High-dose radioimmunotherapy combined with fixed, low-dose Paclitaxel in metastatic prostate and breast cancer using a MUC-1 monoclonal antibody, M170, linked to Indium-111/Yttrium-90 via a cathepsin B cleavable linker with cyclosporine to prevent human anti-mouse antibody. Clin Cancer Res. 2005;11(16):5920-5927.
189 Waldherr C, Pless M, Maecke HR, Haldemann A, Mueller-Brand J. The clinical value of [90Y-DOTA]-D-Phe1-Tyr3-octreotide (90Y-DOTATOC) in the treatment of neuroendocrine tumours: a clinical phase II study. Ann Oncol. 2001;12(7):941-945.
190 Cybulla M, Weiner SM, Otte A. End-stage renal disease after treatment with 90Y-DOTATOC. Eur J Nucl Med. 2001;28(10):1552-1554.
191 Kwekkeboom DJ, Bakker WH, Kam BL, et al. Treatment of patients with gastro-entero-pancreatic (GEP) tumours with the novel radiolabelled somatostatin analogue [177Lu-DOTA,(0) Tyr3] octreotate. Eur J Nucl Med Mol Imaging. 2003;30:417.
192 Prasad V, Baum R. Long term follow-up of tubular renal function after peptide receptor radionuclide therapy with Y-90 DOTA-TATE/Lu-177 DOTA-TATE [abstract]. J Nucl Med. 2008;49(Suppl 1):104.
193 Forrer F, Uusijarvi H, Storch D, et al. Treatment with 177Lu-DOTATOC of patients with relapse of neuroendocrine tumors after treatment with 90Y-DOTATOC. J Nucl Med. 2005;46:1310.
194 DeNardo SJ, O’Grady LF, Richman CM, et al. Radioimmunotherapy for advanced breast cancer using I-131-ChL6 antibody. Anticancer Res. 1997;17:1745-1752.
195 DeNardo SJ, Mirick GR, Kroger LA, et al. The biologic window for chimeric L6 radioimmunotherapy. Cancer. 1994;73(Suppl):1023-1032.
196 DeNardo SJ. Radioimmunotherapy in the treatment of metastatic breast cancer: an overview. In: Goldenberg DM, editor. Cancer therapy with radiolabeled antibodies. Ann Arbor, MI: CRC Press, Inc; 1995:189-201.
197 Goodman GE, Hellstrom I, Yelton DE, et al. Phase I trial of chimeric (human-mouse) monoclonal antibody L6 in patients with non-small-cell lung, colon, and breast cancer. Cancer Immunol Immunother. 1993;36:267-273.
198 Schrier DM, Stemmer SM, Johnson T, et al. High-dose 90Y Mx-diethylenetriaminepentaacetic acid (DTPA)-BrE-3 and autologous hematopoietic stem cell support (AHSCS) for the treatment of advanced breast cancer: a phase I trial. Cancer Res. 1995;55:5921s-5924s.
199 Richman CM, DeNardo SJ, O’Donnell RT, et al. Dosimetry-based therapy in metastatic breast cancer patients using 90Y monoclonal antibodies 170H.82 with autologous stem cell support and cyclosporin A. Clin Cancer Res. 1999;5(Suppl):3243s-3248s.
200 Ledermann JA, Begent RH, Massof C, Kelly AM, Adam T, Bagshawe KD. A phase I study of repeated therapy with radiolabelled antibody to carcinoembryonic antigen using intermittent or continuous administration of cyclosporin A to supress the immune response. Int J Cancer. 1991;47:659-664.
201 Weiden PL, Wolf SB, Breitz HB, et al. Human anti-mouse antibody suppression with cyclosporin A. Cancer. 1994;73(3 Suppl):1093-1097.
202 Behr TM, Liersch T, Greiner-Bechert L, et al. Radioimmunotherapy of small-volume disease of metastatic colorectal cancer. Cancer. 2002;94(4 Suppl):1373-1381.
203 Liersch T, Meller J, Bittrich M, et al. Update of carcinoembryonic antigen radioimmunotherapy with 131I-labetuzumab after salvage resection of colorectal liver metastases: comparison of outcome to a contemporaneous control group, Annals. Surg Oncol. 2007;14:2577.
204 Stewart JSW, Hird V, Snook D, et al. Intraperitoneal radioimmunotherapy of ovarian cancer: pharmacokinetics, toxicity, and efficacy of I-131 labeled monoclonal antibodies. Int J Radiat Oncol Biol Phys. 1989;16(2):405-413.
205 Stewart JS, Hird V, Snook D, et al. Intraperitoneal radioimmunotherapy for ovarian cancer: pharmacokinetics, toxicity, and efficacy of I-131 labeled monoclonal antibodies. Int J Radiat Oncol Biol Phys. 1989;16(2):405-413.
206 Hird V, Maraveyas A, Snook D, et al. Adjuvant therapy of ovarian cancer with radioactive monoclonal antibody. Br J Cancer. 1993;68:403-406.
207 Epenetos AA, Hird V, Lambert H, Mason P, Coulter C. Long term survival of patients with advanced ovarian cancer treated with intraperitoneal radioimmunotherapy. Int J Gynecol Cancer. 2000;10(S1):44-46.
208 Lamborn KR, DeNardo GL, DeNardo SJ, et al. Treatment-related parameters predicting efficacy of Lym-1 radioimmunotherapy in patients with B-lymphocytic malignancies. Clin Cancer Res. 1997;3:1253-1260.
209 Reynolds JC, Del Vecchio S, Sakahara H, et al. Anti-murine response to mouse monoclonal antibodies: clinical findings and implications. Int J Rad Appl Instrum [B]. 1989;16(2):121-125.
210 Pimm MV, Perkins AC, Armitage NC, Baldwin RW. The characteristics of blood-bourne radiolabels and the effect of anti-mouse IgG antibodies on lozalization of radiolabeled monoclonal antibody in cancer patients. J Nucl Med. 1985;26(9):1011-1023.
211 Leonard JP, Coleman M, Kostakoglu L, et al. Fludarabine monophosphate followed by Iodine I-131 tositumomab for untreated low-grade and follicular non-Hodgkin’s lymphoma (NHL) [abstract]. Blood. 1999;94(10 Suppl 1 Part 1):90a.
212 Kaminski MS, Estes J, Tuck M, et al. Iodine I131 tositumomab therapy for previously untreated follicular lymphoma [abstract]. Proc Am Soc Clin Oncol. 2000:19.
213 Dillman RO, Beauregard JC, Halpern SE, Clutter M. Toxicities and side effects associated with intravenous infusions of murine monoclonal antibodies. J Biologic Response Modifiers. 1986;5:73-84.
214 DeNardo GL, DeNardo SJ, Macey DJ, Shen S, Kroger LA. Overview of radiation myelotoxicity secondary to radioimmunotherapy using 131I-Lym-1 as a model. Cancer. 1994;73(Suppl):1038-1048.
215 Richman CM, Schuermann TC, Wun T, et al. Peripheral blood stem cell mobilization for hematopoietic support of radioimmunotherapy in patients with breast carcinoma. Cancer. 1997;80(Suppl:):2728-2732.
216 Richman CM, DeNardo SJ. Systemic radiotherapy in metastatic breast cancer using 90Y-linked monoclonal MUC-1 antibodies. Crit Rev Oncol Hematol. 2001;38(1):25-35.
217 Press OW, Eary JF, Appelbaum FR, et al. Radiolabeled-antibody therapy of B cell lymphoma with autologous bone marrow support. N Engl J Med. 1993;329(17):1219-1224.
218 Vriesendorp HM, Shao Y, Blum JE, Quadri SM, Williams JR. Fractionated intravenous administration of 90Y-labeled B72.3 GYK-DTPA immunoconjugate in beagle dogs. Nucl Med Biol. 1993;20:571-578.
219 Akabani G, Reist CJ, Cokgor I, et al. Dosimetry of 131I-labeled 81C6 monoclonal antibody administered into surgically created resection cavities in patients with malignant brain tumors. J Nucl Med. 1999;40(4):631-638.
220 Paganelli G, Zoboli S, Cremonesi M, et al. Receptor-mediated radiotherapy with 90Y-DOTA-D-Phe1-Tyr3-octreotide. Eur J Nucl Med. 2001;28(4):426-434.
221 Cassidy JR. Clinical radiation neuropathy. Int J Radiat Oncol Biol Phys. 1995;31(5):1249-1256.
222 Moll S, Nickeleit V, Mueller-Brand J, Brunner FP, Maecke HR, Milhatsch MJ. A new cause of renal thrombotic microangiopathy: yttrium 90-DOTATOC internal radiotherapy. Am J Kidney Dis. 2001;37(4):847-851.
223 Sears HF, Atkinson B, Mattis J, et al. Phase I clinical trial of monoclonal antibody in treatment of gastrointestinal tumours. Lancet. 1982;1(8275):762-765.
224 Breitz H, Wendt R, Stabin M, Bouchet L, Wessels B. Dosimetry of high dose skeletal targeted radiotherapy (STR) with 166Ho-DTMP. Cancer Biother Radiopharm. 2003;18(2):225-230.
225 O’Connell MJ, Martenson JA, Wieand HS, et al. Improving adjuvant therapy for rectal cancer by combining protracted-infusion fluorouracil with radiation therapy after curative surgery. N Engl J Med. 1994;331(8):502-507.
226 Herskovic A, Martz K, al-Sarraf M, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med. 1992;326(24):1593-1598.
227 O’Donnell RT, DeNardo SJ, Miers L, et al. Combined modality radioimmunotherapy for human prostate cancer xenografts with taxanes and 90Yttrium-DOTA-peptide-ChL6. Prostate. 2002;50(1):27-37.
228 DeNardo SJ, Kukis DL, Kroger LA, et al. Synergy of Taxol and radioimmunotherapy with yttrium-90-labeled chimeric L6 antibody: efficacy and toxicity in breast cancer xenografts. Proc Natl Acad Sci U S A. 1997;94:4000-4004.
229 Ng B, Kramer E, Liebes L, et al. Radiosensitization of tumor-targeted radioimmunotherapy with prolonged topotecan infusion in human breast cancer xenografts. Cancer Res. 2001;61(7):2996-3001.
230 Stein R, Chen S, Reed L, Richel H, Goldenberg DM. Combining radioimmunotherapy and chemotherapy for treatment of medullary thyroid carcinoma: effectiveness of dacarbazine. Cancer. 2002;94(1):51-61.
231 Cardillo TM, Blumenthal RD, Ying Z, Gold DV. Combined gemcitabine and radioimmunotherapy for the treatment of pancreatic cancer. Int J Cancer. 2002;97(3):386-392.
232 Wilder RB, McGann JK, Sutherland WR, et al. The hypoxic cytotoxin SR 4233 increases the effectiveness of radioimmunotherapy in mice with human non-Hodgkin’s lymphoma xenografts. Int J Radiat Oncol Biol Phys. 1994;28(1):119-126.
233 Wilder RB, Langmuir VK, Mendonca HL, Goris ML, Knox SJ. Local hyperthermia and SR 4233 enhance the antitumor effects of radioimmunotherapy in nude mice with human colonic adenocarcinoma xenografts. Cancer Res. 1993;53(13):3022-3027.
234 Evens AM, Spies S, Patton D, et al. A phase I/II trial of radioimmunotherapy (RIT) with 90Yttrium-ibritumomab Tiuxetan (Zevalin®; 90Y-Zevalin) Combined with the redox-active agent motexafin gadolinium (MGd): prompt and high remission rates in patients with rituximab-refractory non-hodgkin’s lymphoma (NHL) [abstract]. Blood. 118(11), 2007.
235 Tu S, Millikan RE, Mengistu B, et al. Bone-targeted therapy for advanced androgen-independent carcinoma of the prostate; a randomised phase II trial. Lancet. 2001;357:336-341.
236 Meredith RF, Alvarez RD, Partridge EE, et al. Intraperitoneal radioimmunochemotherapy of ovarian cancer: a phase I study. Cancer Biother Radiopharm. 2001;16(4):305-315.
237 Forero A, Meredith RF, Khazaeli MB, et al. Phase I study of 90Y-CC49 monoclonal antibody therapy in patients with advanced non-small cell lung cancer: effect of chelating agents and paclitaxel co-administration. Cancer Biother Radiopharm. 2005;20:467.
238 Richman CM, DeNardo SJ, O’Donnell RT, et al. Combined modality radioimmunotherapy (RIT) in metastatic prostate and breast cancer using paclitaxel and a MUC monoclonal antibody m170, linked to yttrium. J Clin Oncol. 2004;23:176.
239 Wong JY, Shibata S, Williams LE, et al. A phase I trial of 90Y-anti-carcinoembryonic antigen chimeric T84.66 radioimmunotherapy with 5-fluorouracil in patients with metastatic colorectal cancer. Clin Cancer Res. 2003;9:5842.
240 Msirikale JS, Klein JL, Schroeder J, et al. Radiation enhancement of radiolabeled antibody deposition in tumors. Int J Radiat Oncol Biol Phys. 1987;13:1839-1844.
241 Warhoe KA, DeNardo SJ, Wolkov HB, et al. Evidence for external beam irradiation enhancement of radiolabeled monoclonal antibody uptake in breast cancer. Antibody Immunoconj Radiophar. 1992;5:227-235.
242 Stickney DR, Gridley DS, Kirk GA, Slater JM. Enhancement of monoclonal antibody binding to melanoma with single dose radiation or hyperthermia. NCI Monogr. 1987;3:47-52.
243 Znati CA, Rosenstein M, Boucher Y, Epperly MW, Bloomer WD, Jain RK. Effect of radiation on interstitial fluid pressure and oxygenation in a human tumor xenograft. Cancer Res. 1996;56(5):964-968.
244 Buchegger F, Allal AS, Roth A, et al. Combined radioimmunotherapy and radiotherapy of liver metastases from colorectal cancer; a feasibility study. Anticancer Res. 2000;20(3B):1889-1996.
245 Dvorak HF, Nagy JA, Feng D, Dvorak AM. Tumor architecture and targeted delivery. In: Abrams PG, Fritzberg AR, editors. Radioimmunotherapy of cancer. New York: Marcel Dekker, Inc; 2000:107-135.
246 Burke PA, DeNardo SJ. Antiangiogenic agents and their promising potential for combined therapy. Crit Rev Oncol Hematol. 2001;39:155-171.
247 DeNardo SJ, Burke PA, Leigh BR, et al. Neovascular targeting with cyclic RGD peptide (cRGDf-ACHA) to enhance delivery of radioimmunotherapy. Cancer Biother Radiopharm. 2000;15:71-79.
248 Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL. Cilengitide targeting of avb3 integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts. Cancer Res. 2002;62:4263-4272.
249 Li L, Wartchow CA, Danthi SN, et al. A novel antiangiogenesis therapy using an integrin antagonist or anti-Flk-1 antibody coated 90Y-labeled nanoparticles [abstract]. Int J Radiat Oncol Biol Phys. 2004;58:1215-1227.
250 Hallahan DE, Qu S, Cmelak A, et al. Radiation-mediated control of drug delivery. Amer J Clin Oncol. 2000;24:473.
251 Buchsbaum DJ, Rogers BE, Khazaeli MB, et al. Targeting strategies for cancer radiotherapy. Clin Cancer Res. 1999;5:3048s-3055s.
252 Goldenberg DM, Rossi EA, Sharkey RM, McBride WJ, Chang CH. Multifunctional antibodies by the dock-and-lock method for improved cancer imaging and therapy by pretargeting. J Nucl Med. 2008;49(1):158-163.