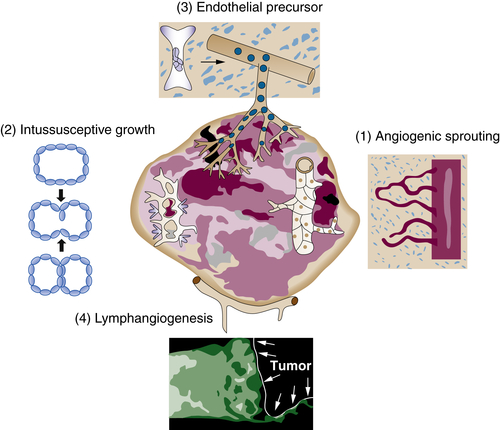
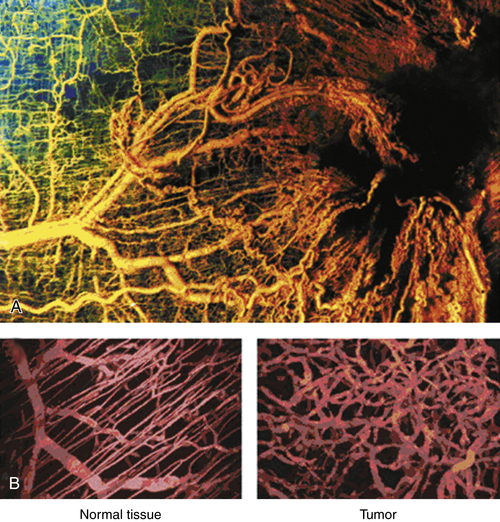
Tumor Vasculature
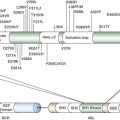
Critical Signaling Factors—Targets for Therapy
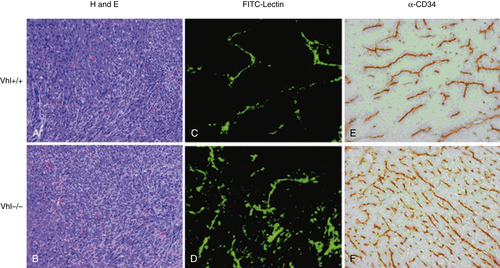
Pro-angiogenic Factors
VEGF
bFGF
Angiopoietins/Tie Receptors
PDGF
Anti-angiogenic Factors
Thrombospondin 1 (TSP-1)
Endostatin and Tumstatin
Targeting Tumor Angiogenesis in Patients
VEGF-Based Therapies
Metronomic Therapy
Remaining Challenges
1. VEGF and the quest for tumour angiogenesis factors . Nat Rev Cancer . 2002 ; 2 : 795 – 803 .
2. Inflammation and cancer: back to Virchow? Lancet . 2001 ; 357 : 539 – 545 .
3. Vascular reactions of normal and malignant tissues in vivo. III. Vascular reactions of mice to fibroblasts treated in vitro with methylcholanthrene . J Natl Cancer Inst . 1950 ; 11 : 555 – 580 .
4. Tumor angiogenesis: therapeutic implications . N Engl J Med . 1971 ; 285 : 1182 – 1186 .
5. Angiogenesis in cancer and other diseases . Nature . 2000 ; 407 : 249 – 257 .
6. Mechanisms of angiogenesis . Nature . 1997 ; 386 : 671 – 674 .
7. Endothelial cells and VEGF in vascular development . Nature . 2005 ; 438 : 937 – 945 .
8. Recent advances in vascular development . Curr Opin Hematol . 2012 ; 19 : 176 – 183 .
9. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis . Trends Mol Med . 2011 ; 17 : 347 – 362 .
10. Lymphangiogenesis and cancer . Genes Cancer . 2011 ; 2 : 1146 – 1158 .
11. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction . Blood . 2010 ; 116 : 829 – 840 .
12. Macrophage diversity enhances tumor progression and metastasis . Cell . 2010 ; 141 : 39 – 51 .
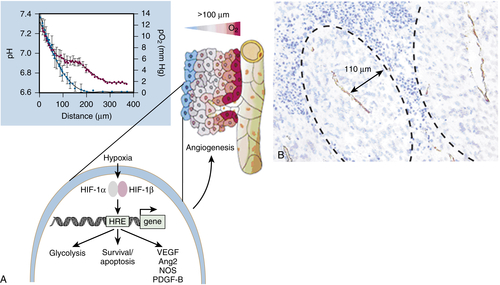
13. Exploiting tumour hypoxia in cancer treatment . Nat Rev Cancer . 2004 ; 4 : 437 – 447 .
14. VEGF as a key mediator of angiogenesis in cancer . Oncology . 2005 ; 69 ( suppl 3 ) : 4 – 10 .
15. The multifaceted circulating endothelial cell in cancer: towards marker and target identification . Nat Rev Cancer . 2006 ; 6 : 835 – 845 .
16. EPCs and pathological angiogenesis: when good cells go bad . Microvasc Res . 2010 ; 79 : 207 – 216 .
17. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells . Cell . 2006 ; 124 : 175 – 189 .
18. Contribution of endothelial progenitors and proangiogenic hematopoietic cells to vascularization of tumor and ischemic tissue . Curr Opin Hematol . 2006 ; 13 : 175 – 181 .
19. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors . Cancer Cell . 2005 ; 8 : 211 – 226 .
20. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2 . Cancer Res . 2007 ; 67 : 8429 – 8432 .
21. Tumour vascular targeting . Nat Rev Cancer . 2005 ; 5 : 436 – 446 .
22. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours . Nature . 1996 ; 379 : 88 – 91 .
23. Hypoxia-induced angiogenesis: good and evil . Genes Cancer . 2011 ; 2 : 1117 – 1133 .
24. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn’t tell us . J Natl Cancer Inst . 2002 ; 94 : 883 – 893 .
25. Angiogenesis in life, disease and medicine . Nature . 2005 ; 438 : 932 – 936 .
26. Endogenous angiogenesis inhibitors . APMIS . 2004 ; 112 : 496 – 507 .
27. Angiogenesis as a therapeutic target . Nature . 2005 ; 438 : 967 – 974 .
28. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele . Nature . 1996 ; 380 : 435 – 439 .
29. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene . Nature . 1996 ; 380 : 439 – 442 .
30. VEGF receptor signalling—in control of vascular function . Nat Rev Mol Cell Biol . 2006 ; 7 : 359 – 371 .
31. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice . Nature . 1995 ; 376 : 62 – 66 .
32. The neuropilins and their role in tumorigenesis and tumor progression . Cancer Lett . 2006 ; 231 : 1 – 11 .
33. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway . Mol Cell . 2008 ; 30 : 393 – 402 .
34. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression . Nat Rev Cancer . 2012 ; 12 : 9 – 22 .
35. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol . 2011 ; 8 : 393 – 404 .
36. Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death . Dev Biol . 1999 ; 209 : 254 – 267 .
37. The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling . Proc Natl Acad Sci U S A . 2000 ; 97 : 8386 – 8391 .
38. HIF-1 alpha is required for solid tumor formation and embryonic vascularization . EMBO J . 1998 ; 17 : 3005 – 3015 .
39. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT . Nature . 1997 ; 386 : 403 – 407 .
40. HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature . Dev Cell . 2006 ; 11 : 81 – 92 .
41. HIF-1 as a target for drug development . Nat Rev Drug Discov . 2003 ; 2 : 803 – 811 .
42. Development of novel therapeutic strategies that target HIF-1 . Expert Opin Ther Targets . 2006 ; 10 : 267 – 280 .
43. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis . Cytokine Growth Factor Rev . 2005 ; 16 : 159 – 178 .
44. Tie receptors and their angiopoietin ligands are context-dependent regulators of vascular remodeling . Exp Cell Res . 2006 ; 312 : 630 – 641 .
45. Molecular control of angiopoietin signalling . Biochem Soc Trans . 2011 ; 39 : 1592 – 1596 .
46. Insight into the physiological functions of PDGF through genetic studies in mice . Cytokine Growth Factor Rev . 2004 ; 15 : 215 – 228 .
47. PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival . Nat Cell Biol . 2005 ; 7 : 870 – 879 .
48. Inhibitors of growth factor receptors, signaling pathways and angiogenesis as therapeutic molecular agents . Cancer Metastasis Rev . 2006 ; 25 : 243 – 252 .
49. Endogenous inhibitors of angiogenesis . Cancer Res . 2005 ; 65 : 3967 – 3979 .
50. Annu Rev Med . 2006 ; 57 : 1 – 18 .
51. Thrombospondins in cancer . Cell Mol Life Sci . 2008 ; 65 : 700 – 712 .
52. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin . Proc Natl Acad Sci U S A . 1990 ; 87 : 6624 – 6628 .
53. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy . Proc Natl Acad Sci U S A . 2003 ; 100 : 12917 – 12922 .
54. Integrins and angiogenesis: a sticky business . Exp Cell Res . 2006 ; 312 : 651 – 658 .
55. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis . Cell . 1996 ; 86 : 353 – 364 .
56. Phase I study of recombinant human endostatin in patients with advanced solid tumors . J Clin Oncol . 2002 ; 20 : 3792 – 3803 .
57. Targeting angiogenesis: a review of angiogenesis inhibitors in the treatment of lung cancer . Lung Cancer . 2003 ; 42 ( suppl 1 ) : S81 – S91 .
58. Recombinant human angiostatin (rhAngiostatin) in combination with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer: a phase II study from Indiana University . Ann Oncol . 2006 ; 17 : 97 – 103 .
59. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo . Nature . 1993 ; 362 : 841 – 844 .
60. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer . N Engl J Med . 2004 ; 350 : 2335 – 2342 .
61. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer . N Engl J Med . 2003 ; 349 : 427 – 434 .
62. Age-related macular degeneration . Lancet . 2012 ; 379 : 1728 – 1738 .
63. Bevacizumab in non small cell lung cancer: development, current status and issues . Curr Med Chem . 2012 ; 19 : 961 – 971 .
64. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis . Cancer Cell . 2009 ; 15 : 220 – 231 .
65. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis . Cancer Cell . 2009 ; 15 : 232 – 239 .
66. Circulating endothelial cells as biomarkers in clinical oncology . Microvasc Res . 2010 ; 79 : 224 – 228 .
67. Chemotherapy: failure of bevacizumab in early-stage colon cancer . Nat Rev Clin Oncol . 2011 ; 8 : 10 – 11 .
68. Lessons from the adjuvant bevacizumab trial on colon cancer: what next? J Clin Oncol . 2011 ; 29 : 1 – 4 .
69. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases . Nat Rev Drug Discov . 2011 ; 10 : 417 – 427 .
70. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs . Cancer Cell . 2012 ; 21 : 82 – 91 .
71. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science . 2006 ; 312 : 1171 – 1175 .
72. Clinical translation of angiogenesis inhibitors . Nat Rev Cancer . 2002 ; 2 : 727 – 739 .
73. Harnessing preclinical mouse models to inform human clinical cancer trials . J Clin Invest . 2006 ; 116 : 847 – 852 .
74. Cancer biomarkers—an invitation to the table . Science . 2006 ; 312 : 1165 – 1168 .