[level-membership-for-critical-care-medicine-category]
142 Tuberculosis
 Epidemiology
Epidemiology
The World Health Organization (WHO) estimates that one-third of the world’s population is latently infected with Mycobacterium tuberculosis.1 From this pool, approximately 9 million active tuberculosis (TB) cases emerge annually, resulting in 2 million deaths and making TB the second leading cause of death by an infectious agent worldwide.2 The vast majority of TB cases (95%) occur in the developing world. Incidence rates exceed 300 cases per 100,000 persons throughout sub-Saharan Africa, the Indonesian and Philippine archipelagos, Afghanistan, Bolivia, and Peru.1,3 Areas with the most cases per year include densely populated India (2 million cases per year) and China (1.3 million cases per year).
In the United States, the TB rate continues to decline, with 3.8 new cases per 100,000 reported in 2009, the lowest rate recorded since national reporting began in 1953. Foreign-born persons and racial/ethnic minorities continue to bear a disproportionate burden of TB disease in the United States. In 2008, the TB rate within the foreign-born population in the United States was 10 times higher than in U.S.-born persons.4 TB rates among Hispanics and blacks were nearly eight times higher than among non-Hispanic whites, and rates among Asians were nearly 23 times higher than among non-Hispanic whites. Among U.S.-born racial and ethnic groups, the greatest racial disparity in TB rates was seen in the black population, who are seven times more likely to develop active TB than U.S.-born whites. Other groups at increased risk for active TB include prisoners, the homeless, and human immunodeficiency virus (HIV)-positive individuals.
The acquired immunodeficiency syndrome (AIDS) epidemic has contributed significantly to the rise in TB cases worldwide, with about 1.5 million individuals with active TB per year co-infected with HIV. HIV increases the risk of developing TB by 20.6-fold in countries where the prevalence of HIV is more than 1% in the general population.5 Co-infection with HIV contributes significantly to TB-related mortality.
 The Serious Problem of Highly Drug-Resistant Tuberculosis
The Serious Problem of Highly Drug-Resistant Tuberculosis
Drug-susceptible TB is readily curable provided adherence to medications is followed. However, drug-resistant TB requires a significantly longer course of antibiotics coupled with second-line agents that often are accompanied by difficult-to-tolerate side effects. More importantly, highly drug-resistant TB is associated with significant increase in morbidity and mortality. In the early 1990s, substantial levels of drug resistance began emerging in urban parts of the United States.6 Although the incidence of drug-resistant TB has diminished in the United States, it is increasingly problematic in many parts of the world.7
Multidrug-resistant TB (MDR-TB) is defined as resistance to two of the most powerful first-line anti-TB drugs, isoniazid (INH) and rifampin (RIF). Isolates that are resistant to multiple other combinations of anti-TB drugs but not to INH or RIF are not classified as MDR-TB. It is estimated that of the 9 million new cases of TB per year in the world, 500,000 are due to MDR-TB. Whereas drug-resistant TB is increasing at an alarming rate worldwide, particularly in India and China, prevalence in the United States decreased between 1991 and 2006 from 3.5% to 1.1%.8 MDR-TB disproportionately affects foreign-born individuals, accounting for 0.4% of TB cases occurring in U.S.-born persons and 1.3% in foreign-born individuals.9
Extensively drug-resistant tuberculosis (XDR-TB) is defined as resistance to INH, RIF, any fluoroquinolone, and to a second-line injectable (amikacin, kanamycin, or capreomycin). XDR-TB has emerged with a wide geographic distribution, including the United States, and is associated with worse treatment outcomes than MDR-TB, especially in those co-infected with HIV.7,10–15
 Tuberculosis in the Intensive Care Unit
Tuberculosis in the Intensive Care Unit Pulmonary Tuberculosis
Pulmonary Tuberculosis
Primary infection occurs following airborne implantation of tubercle bacilli into the lungs. M. tuberculosis spreads from the lungs to hilar lymph nodes, and then throughout the bloodstream (Figure 142-1). Although primary infection is usually asymptomatic in adults, it can present with fever, hilar adenopathy, lung infiltrates, pleural effusions, and even severe pulmonary disease that may mimic viral or bacterial pneumonia, which may delay the diagnosis of TB. In severely immunocompromised patients, primary TB may be aggressive and become disseminated. Pleural TB is usually a manifestation of primary TB, although it may also occur with reactivation disease. Pleural TB can present as pleuritis or empyema. Pleural biopsy specimens are more likely to yield positive cultures than pleural fluid.
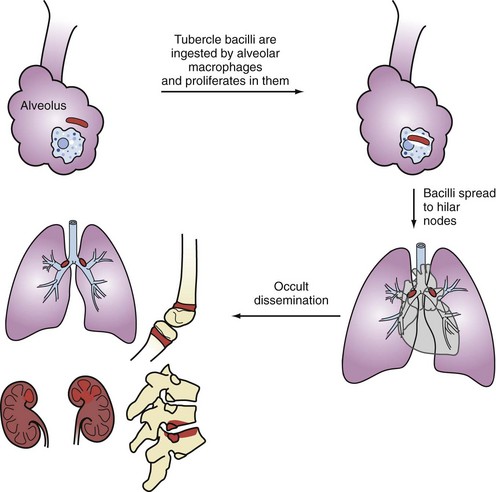
Most cases of active TB are due to reactivation of latent TB infection (LTBI). Active TB develops in about 10% of immunocompetent individuals with LTBI and tends to occur within the first 2 years of the initial infection. Typically, reactivation TB is a subacute fibrocavitary pneumonia involving the upper lobes and/or superior segments of the lower lobes. However, reactivation TB can involve any organ system and can present in a fulminant fashion with respiratory failure.16
There are some common clinical characteristics of TB patients who require ICU care. In a study of 58 ICU patients with confirmed TB, 22 (37.9%) required mechanical ventilation, and 15 (25.9%) died in the hospital.17 The factors independently associated with mortality were acute renal failure, need for mechanical ventilation, chronic pancreatitis, sepsis, acute respiratory distress syndrome (ARDS), and nosocomial pneumonia.17 Both primary and reactivation TB can cause bilateral alveolar infiltrates, hypoxic respiratory failure, and ARDS.16,18 In another study of patients hospitalized with pulmonary TB, six factors were shown to be associated with respiratory failure or death: lymphopenia, advanced age, concomitant smear-positive extrapulmonary TB, alcoholism, a high percentage of neutrophils on the peripheral white blood cell count, and lack of radiographic cavitation.19 Laboratory findings of anemia and hypoalbuminemia have been shown to be predictors for death in patients with respiratory failure due to TB.20 However, these findings are not specific to TB and commonly present in the critically ill.
Consolidation is the most frequent radiographic pattern of patients with pulmonary TB who are admitted to the ICU.21 Because this radiographic pattern is highly nonspecific, chest x-rays are often unhelpful in raising the suspicion for TB. Consolidation on initial chest radiograph has also been shown to be a strong independent risk factor for in-hospital mortality.22 One possible reason for this is a delay in the diagnosis; clinicians may be more prone to favor a diagnosis of non-tuberculous pneumonia in the absence of cavitation or miliary pattern. Another is that consolidation may be an indication of a suboptimal immune response to the infection. Pulmonary gangrene, which carries a mortality of up to 75%, can ensue when rapid progression of infiltrate causes vascular damage and death of lung tissue.23 Other life-threatening complications of pulmonary TB include hemoptysis, spontaneous pneumothoraces, bronchopleural fistulas, and empyema. Not unexpectedly, delayed recognition and treatment of nosocomial pneumonia complicating TB in patients requiring mechanical ventilation has a significant adverse effect on survival.24
Perhaps the best safeguard to prevent missing a diagnosis of pulmonary or disseminated TB in the critically ill is to maintain a high index of suspicion of it in at-risk individuals (e.g., foreign-born or immunosuppressed patients). Studies have shown that the presence of diffuse infiltrates consistent with ARDS and acute respiratory failure may cause physicians to inappropriately dismiss the diagnosis of TB.25–27 Older individuals (≥65 years) or patients with AIDS may also have delayed diagnosis of TB, due in part to atypical presentations.28,29
Hospital mortality has been reported to be 60% for patients with respiratory failure due to pulmonary TB.22 Hence, despite being a relatively rare cause of respiratory failure in ICU patients, pulmonary TB carries a poor prognosis. Early recognition of the infection is essential to reduce mortality and prevent nosocomial spread of M. tuberculosis.27
 Disseminated Tuberculosis
Disseminated Tuberculosis
Disseminated, or “miliary,” TB is more likely to occur in the very young and very old and in patients with underlying diseases such as HIV. It may result from either primary or reactivation TB. Disseminated TB typically presents subacutely with symptoms present for days to months, but it can manifest fulminantly with septic shock and multiorgan failure.30 Typical presenting signs and symptoms include fever, malaise, weight loss, dyspnea, and hypoxia.
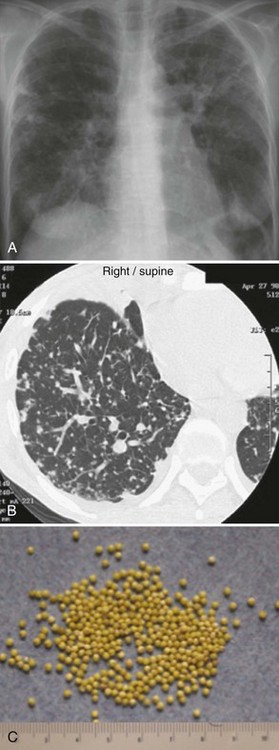
The chest radiograph (Figure 142-2, A) and computed tomography (CT) scan (see Figure 142-2, B) show a typical miliary pattern manifested by a profusion of diffuse small (<2 mm) nodules that resemble the size and uniformity of millet seeds (see Figure 142-2, C). In some cases of disseminated disease, the chest radiograph may appear normal. Virtually any organ may be involved, including the adrenals, brain, meninges, liver, pancreas, eyes, urinary tract, and skin. Bone marrow involvement by TB commonly manifests with anemia, leukemoid reaction, and thrombocytosis. The diagnosis of miliary TB can be difficult. If disseminated TB is suspected, sputum smears should be obtained even if lung disease is not apparent. Biopsy and culture of affected tissue(s), such as the bone marrow, are often required. Culture of blood, urine, and/or stool may be positive, especially in HIV-positive patients.30
 Neurologic Tuberculosis
Neurologic Tuberculosis
Tuberculous Meningitis
TB meningitis is rare in developed countries, with approximately 300 to 400 cases in the United States each year. It occurs via rupture of a subependymal tubercle that has seeded and formed during primary infection or disseminated disease. Individuals at high risk for TB meningitis include very young children with primary TB and older patients with immunodeficiency disorders such as HIV. Most patients with TB meningitis will have no known history of TB, but evidence of extrameningeal disease (e.g., pulmonary, urinary, etc.) can be found in about half of these patients.31,32 The tuberculin skin test is positive in only about 50% of patients with TB meningitis.
TB meningitis is typically a subacute disease. In one review of 58 cases, symptoms were present for 1 day to 9 months, with a median of 10 days prior to diagnosis.31 A prodromal phase of low-grade fever, malaise, headache, dizziness, vomiting, and/or personality changes may persist for 2 to 3 weeks before the patient presents for medical care. Typical findings at presentation include severe headache, altered mental status, stroke, hydrocephalus, and cranial neuropathies. These clinical features are the result of basilar meningeal fibrosis and vascular inflammation.33 Classic features of bacterial meningitis such as stiff neck and fever may be absent. When allowed to progress, coma and seizures may ensue.
The diagnosis of TB meningitis can be difficult and may be based only on clinical findings without definitive microbiological proof. Certain clinical characteristics such as longer duration of symptoms (>6 days), moderate cerebrospinal fluid (CSF) pleocytosis, and the presence of focal deficits increase the probability of TB meningitis.34,35 Characteristic CSF findings of TB meningitis include:
CSF samples should be sent for acid-fast smears, but this is associated with low sensitivity (<20%). Large volumes (10-15 mL) from several daily lumbar punctures are often needed for a microbiological diagnosis. Sensitivity is increased if four spinal taps are performed. Culture can take weeks and is also associated with low sensitivity. Stereotactic biopsy can be performed if tissue samples are needed. Mycobacterial antigens by enzyme-linked immunosorbent assay (ELISA) or radioimmunoassay have been detected in the CSF of patients with TB meningitis.36
Recent meta-analysis calculated that commercial nucleic acid amplification (NAA) assays used for the diagnosis of TB meningitis were 56% sensitive and 98% specific.37,38 Unfortunately, considerable variability in sensitivity and specificity among tests from different laboratories makes it more difficult to interpret results. Most studies conclude that commercial NAA tests can confirm TB meningitis but cannot rule it out.39 Thus a negative test neither excludes the diagnosis nor obviates the need for continued empirical therapy if the clinical suspicion is high. Comparisons of NAA and microscopy/culture using large volumes of CSF have indicated that the sensitivity of microscopy was similar to NAA for the diagnosis of TB meningitis, and repeated testing gave the highest diagnostic yield.40 The sensitivity of CSF microscopy and culture falls rapidly after the start of treatment, whereas mycobacterial DNA may remain detectable within the CSF up to a month after the start of treatment.41
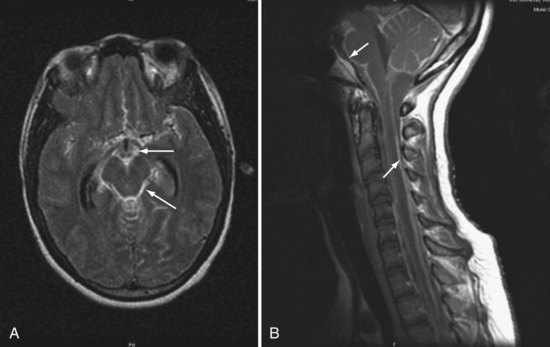
Magnetic resonance imaging (MRI) often reveals basilar meningeal enhancement (Figure 142-3) and/or hydrocephalus.32 Hypodensities due to cerebral infarcts, and ring or nodular enhancing lesions can also be seen. MRI is superior to CT for evaluating the brainstem and the extent of lesions.
The outcome of TB meningitis is improved by timely treatment. Thus empirical treatment is warranted when risk factors and clinical features are suggestive of this diagnosis, even before microbiological confirmation. Chemotherapy for TB meningitis follows the model of short-course chemotherapy for pulmonary TB—an induction phase followed by a continuation phase. But unlike pulmonary TB, the optimal drug regimen and duration of each phase of treatment are not clearly established. INH and RIF remain the most essential drugs. INH penetrates the CSF freely and has potent early bactericidal activity.42–44 RIF penetrates the CSF less well (maximum concentrations around 30% of plasma), but the high mortality from RIF-resistant TB meningitis has confirmed its central role in the treatment of CNS disease.45 INH, RIF, and pyrazinamide are considered mandatory at the beginning of TB meningitis treatment, and some centers use all three drugs for the duration of therapy.46 There are no data from controlled trials to guide choice of the fourth drug. Most authorities recommend either streptomycin or ethambutol, although neither penetrates the CSF well in the absence of inflammation, and both can produce significant adverse reactions. Therapy should be continued for 9 to 12 months.
Adjunctive corticosteroid treatment of TB meningitis has been recommended for more than 50 years, but there has been long-standing concern that corticosteroids may reduce the penetration of anti-TB drugs into the CNS.33 A recent Cochrane systematic review and meta-analysis of 7 randomized controlled trials involving 1140 participants (with 411 deaths) concluded that corticosteroids improved outcome in HIV-negative children and adults with TB meningitis, but the benefit in HIV infected individuals remains uncertain.47 The results were heavily influenced by a study performed in 545 Vietnamese adults with TB meningitis which observed that treatment with dexamethasone was associated with a significantly reduced risk of death.48 However, there was no demonstrable improvement in the combined endpoint of death or severe disability at 9-month follow-up. The survival benefit associated with corticosteroid therapy may have been due in part to a reduction in severe adverse events (9.5% versus 16%), particularly hepatitis, that necessitated changes in anti-TB drug regimens. No mortality benefit from dexamethasone was evident in 98 HIV-infected patients included in the study.48
Because there are no controlled trials comparing different corticosteroid regimens, the choice should be based on those found to be effective in published trials. One recommended regimen for adults is dexamethasone, 12 mg a day for 3 weeks, followed by gradual taper over the next 3 weeks.49 In the large study from Vietnam, patients with mild disease received intravenous (IV) dexamethasone, 0.3 mg/kg/d × 1 week, 0.2 mg/kg/d × 1 week, and then 4 weeks of tapering oral therapy.48 For patients with more severe TB meningitis, IV dexamethasone was given for 4 weeks (1 week each of 0.4 mg/kg/d, 0.3 mg/kg/d, 0.2 mg/kg/d, and 0.1 mg/kg/d), followed by 4 weeks of tapering oral dexamethasone therapy.48
Prognosis of TB meningitis largely depends on neurologic status at the time of presentation and time to treatment initiation. Most patients will die in 5 to 8 weeks if not treated. Various case series indicate a mortality rate between 7% and 65% in developed countries and up to 69% in underdeveloped areas.31,32,50 Neurologic sequelae occur in up to 50% of survivors.50 Mortality risk is highest in those with comorbidities, severe neurologic involvement on admission, rapid progression of disease, and being elderly.
Other Central Nervous System Manifestations of Tuberculosis
Other CNS manifestations of TB include brain abscesses, intracranial tuberculomas, vasculitis, radiculomyelitis, and spinal arachnoiditis. These can occur in conjunction with TB meningitis but are less likely to be seen as isolated findings in the ICU. Intracranial tuberculomas are more common among pediatric patients, especially infants, and can occur in any region of the brain. They result from hematogenous spread of TB. Tuberculous radiculomyelitis is a paradoxical reaction to the treatment of TB meningitis and may respond to corticosteroids. Signs and symptoms include subacute paraparesis, radicular pain, bladder disturbance, and paralysis.51
 Cardiovascular Tuberculosis
Cardiovascular Tuberculosis
Tuberculous Pericarditis
The diagnosis of TB pericarditis can be difficult to prove. Culture of pericardial fluid is positive in only 30% of cases, and pericardial biopsy has a yield of approximately 60%. Biopsy of the pericardium may reveal granulomatous changes consistent with TB or stains positive for acid-fast bacteria. The presence of elevated adenosine deaminase levels in the pericardial fluid has been shown to indicate TB pericarditis, but confirmation is needed.52 PCR holds promise as a more sensitive test in diagnosis of TB pericarditis.53 Many individuals are treated empirically for TB pericarditis based on clinical suspicion, positive tuberculin skin test, imaging studies, and exudative pericardial fluid with high protein and mononuclear white count. Treatment involves standard four-drug regimens as for other manifestations of TB. Prednisone, 60 mg a day, tapered over 11 weeks, is sometimes used in addition to anti-TB therapy, and has been shown to reduce the need for operative intervention.54 Pericardiectomy is sometimes necessary in the treatment of refractory or recurrent disease.
Other Cardiovascular Manifestations of Tuberculosis
In addition to the pericardium, TB may also affect the myocardium, endocardium, and epicardium (coronary arteries). These disorders are very rare. TB myocarditis occurs via direct spread from pericardium or mediastinal lymph nodes or from disseminated disease.55 Endocardial involvement may manifest as endocarditis or as mural thrombi with entrapped M. tuberculosis. TB may also affect the coronary arteries, resulting in coronary arteritis with granulomatous inflammation of the arterial wall and obliterative intimal fibrosis.56
The aorta may be affected by TB, causing aortitis, aortointestinal fistula formation, or rupture.57,58 The pathogenesis of aortitis includes septic embolization from endocarditis, seeding of a preexisting aneurysm from bacteremia, or extension from a contiguous site of infection. Signs and symptoms include fever, abdominal or back pain, and a palpable abdominal mass. Blood cultures are positive for M. tuberculosis in about 15% of cases. CT findings include air in the aortic wall, periaortic nodularity, saccular aneurysm in a noncalcified aorta, and rapidly increasing aortic diameter. Primary mycotic aneurysm of the aorta may be a sequela of chronic tuberculous aortitis.59,60
 Tuberculosis in HIV-Positive Patients
Tuberculosis in HIV-Positive Patients
HIV is the most important host risk factor for active TB.61 In many developing countries, TB is the most common opportunistic infection associated with HIV. The estimated annual risk for active TB among persons with LTBI in the general population is 12.9 per 1000 person-years. In contrast, rates of progression to active TB among HIV-infected persons with LTBI range from 35 to 162 per 1000 person-years. Because TB may be an initial manifestation of HIV infection, all patients with TB should be tested for HIV. The WHO estimates that TB causes death in 13% of persons with AIDS.62
The mechanism of increased TB susceptibility in HIV positive persons is incompletely understood. Unlike other AIDS-related opportunistic infections, CD4+ count is not always a reliable predictor of increased risk for TB disease. Alveolar macrophages (AM) are important components of an effective immune response to TB,63 and AM apoptosis represents a critical host defense mechanism that promotes M. tuberculosis elimination. In this context, one possible reason HIV increases susceptibility to TB is that HIV-infected AM have a reduced apoptotic response to M. tuberculosis compared to AM from healthy individuals.64,65
When the CD4+ count is above 350 cells/µL, pulmonary TB in AIDS patients is more likely to present with typical chest radiograph findings of upper lobe fibrocavitary disease.66 However, as the CD4+ count decreases, pulmonary TB tends to manifest with more atypical radiographic manifestations such as mediastinal adenopathy, diffuse miliary or nodular infiltrates, focal lower zone infiltrates, and lack of cavitation. Extrapulmonary TB is more common among HIV-positive patients, occurring in up to 70% of patients. Disease involving the lymph nodes is especially common. Other extrapulmonary manifestations include miliary disease, TB sepsis, and CNS disease.28 Empirical treatment may be necessary before the diagnosis is confirmed. If rapid diagnosis is needed, NAA tests can be used, although these tests are more accurate in smear-positive cases.
After initiating highly-active antiretroviral therapy (HAART) in severely immunosuppressed patients, those with subclinical or recently diagnosed TB may display a paradoxical reaction, where there is an apparent clinical worsening of TB while on appropriate anti-TB treatment.67–69 This phenomenon, also known by the more descriptive name of immune reconstitution inflammatory syndrome (IRIS), can manifest as early as 7 days after starting HAART. Signs and symptoms include fever, weight loss, and evidence of local inflammatory reactions such as lymphadenitis and worsening pulmonary disease such as increased pulmonary consolidation, nodules, and effusions. Histologically, a vigorous suppurative and necrotizing granulomatous reaction occurs, with or without caseation; cultures of infected material are almost invariably positive.
Treatment of TB in patients with HIV is similar to that in HIV-negative patients but is often complicated by drug interactions between TB medications and antiretrovirals.70 The protease inhibitors (PIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) can either induce or inhibit activity of the P450-3A (CYP3A) system. RIF can increase activity of CYP3A, leading to decreased levels of several antiretrovirals. Rifabutin is a less potent inducer of the CYP3A system and is associated with less drug-drug interactions, but dose adjustments may be needed. Despite these potential drug interactions, a RIF-based regimen should be used whenever possible. Patients with liver disease such as hepatitis C may be at increased risk for drug-induced hepatotoxicity. Another treatment issue in HIV-TB co-infection is that patients may fail to properly absorb the anti-TB drugs, which may increase the risk of treatment failure, relapses, and acquired drug resistance.71
Because of increased risk of RIF resistance, patients with HIV should not receive once weekly INH-rifapentine in the continuation phase of treatment. Twice-weekly INH-RIF or INH-rifabutin should be avoided when the CD4+ cell count is less than 100/µL. Treating drug-susceptible pulmonary TB in HIV-positive individuals for 9 months rather than the standard 6 months is associated with lower relapse rates.72,73 Recommendations regarding treatment of TB in HIV patients are frequently revised as new drugs and information become available. The following websites can assist with treatment decisions and information on drug-drug interactions:
 Tuberculosis and Immunomodulatory Therapies
Tuberculosis and Immunomodulatory Therapies
TNF-α plays a central role in the pathogenesis to various inflammatory disorders and in the pathophysiologic response to many infections. TNF-α is produced predominantly by macrophages and lymphocytes and is active both as a membrane-bound and soluble protein.74,75 In several animal models, TNF-α plays an essential part in the host defense to TB.76 One mechanism by which TNF-α potentiates host defense is by its ability to induce apoptosis of infected cells. Macrophage apoptosis helps to contain M. tuberculosis by maintaining granuloma integrity, increasing efficiency of antigen presentation, and promoting killing of intracellular M. tuberculosis.77 Administration of antibodies neutralizing TNF-α resulted in reactivation of TB in a mouse model.78 Interruption of the normal TNF-α controlled response to TB reduces apoptosis, disrupts granuloma integrity, and predisposes to disseminated infection.
TNF-α antagonists are increasingly used for the treatment of various chronic inflammatory disorders. Currently licensed TNF-α antagonists fall into two main types: monoclonal neutralizing anti-TNF-α antibodies and soluble p75 subunits of the TNF-α receptor (TNFα-R). The soluble TNFα-Rs antagonize TNF-α function by acting as decoys to bind TNF-α. Three monoclonal anti-TNF-α antibodies (infliximab, adalimumab, and certolizumab pegol) and two TNFα-Rs (etanercept and abatacept) are in clinical use. Patients treated with TNF-α blockers have a TB incidence rate of 1.17 per 1000 patient-years, 12.2 times that of the general population.79 Almost all of these cases are due to reactivation of LTBI.
Important differences have emerged among the TNF-α antagonists in regard to the risks of reactivation TB. Consistently, the excess risk is associated with infliximab and adalimumab rather than etanercept. For example, compared with etanercept, infliximab is associated with a two- to sevenfold greater risk of TB, shorter time to TB onset (17 versus 48 weeks), and a higher proportion of TB cases with disseminated or extrapulmonary disease (25 versus 10%).80,81 It is not entirely clear why the neutralizing antibodies to TNF-α put people at greater risk of reactivation TB than soluble TNF-α receptors. Possible reasons include a longer duration of action of infliximab and adalimumab and their ability to bind to membrane-bound TNF-α with greater affinity than etanercept.74 As a result, infliximab can induce death in T cells that express the membrane-bound TNF-α, whereas etanercept cannot. In addition, anti-TNF-α antibodies can inhibit T-cell activation and interferon gamma (IFN-γ) production, whereas etanercept cannot. Thus the pharmacokinetic and biological differences between the two main types of TNF-α antagonists may account for the greater susceptibility to intracellular pathogens with the use of the anti- TNF-α antibodies.74,82
Antagonists to other inflammatory cytokines are also being used in the management of patients with rheumatologic and inflammatory disorders. Interleukin 1 (IL-1) receptor antagonist (IL-1Ra) is the naturally occurring protein that prevents the action of IL-1 and IL-1β by competitively binding to IL-1R. Anakinra is a recombinant human form of IL-1Ra. In a case report, anakinra was associated with reactivation TB.83
 Diagnosis of Tuberculosis
Diagnosis of Tuberculosis
Although rapid and inexpensive, acid-fast smear microscopy is limited by its poor sensitivity (~50% sensitivity in culture-confirmed pulmonary TB cases) and suboptimal specificity (50%-80%) in settings where nontuberculous mycobacteria are commonly isolated.84–86 NAA testing has become a routine procedure in many settings, because NAA tests can reliably detect M. tuberculosis in specimens 1 or more weeks earlier than culture.85 Because of the increasing use of NAA tests and the potential impact on patient care and public health, the Centers for Disease Control and Prevention (CDC) and the Association of Public Health Laboratories (APHL) made recommendations for using NAA tests for laboratory confirmation of TB. CDC recommends that NAA testing be performed on at least one respiratory specimen from each patient in whom a diagnosis of TB is being considered but has not yet been established, and for whom the test result would alter case management or TB control activities.87,88
 Treatment of Tuberculosis
Treatment of Tuberculosis
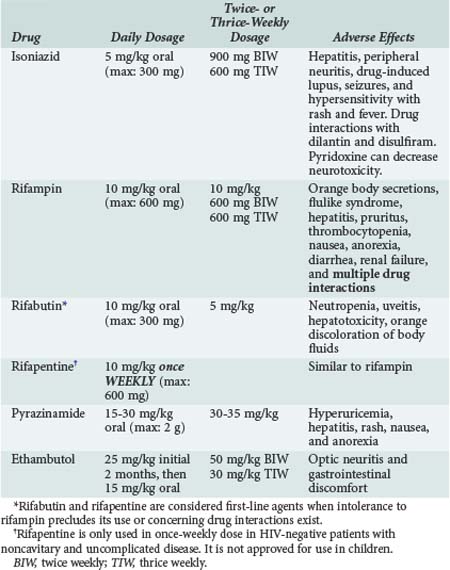
Standard treatment of adults with drug-susceptible TB is a three- or four-drug regimen for at least 6 months.89,90 The typical course of therapy for drug-susceptible disease is 2 months of INH, RIF, pyrazinamide (PZA), and ethambutol (EMB) (initial phase), followed by 4 months of INH and RIF (continuation phase) (Tables 142-1 and 142-2). A 9- to 12-month regimen is suggested for TB meningitis, for pulmonary TB that is slow to respond to therapy (e.g., those with cavitary lesions and persistent sputum culture positivity even after 2 months of an appropriate four-drug regimen), or when PZA is not used in the induction regimen. EMB can be discontinued when drug susceptibility studies show sensitivity to INH and RIF. Streptomycin (SM) can be used instead of EMB if resistance is unlikely or susceptibility is shown. The continuation phase can be daily therapy, twice-weekly therapy, or thrice-weekly therapy for drug-susceptible TB (see Table 142-1). See the HIV section for details of treating TB in HIV-positive patients. Specific guidelines including information on first- and second-line agents have been published by the CDC.91
TABLE 142-1 Current Regimens for Treatment of Drug-Susceptible Tuberculosis
| Regimen | Initial Phase | Continuation Phase |
|---|---|---|
| Daily or 5 days per week* | 8 weeks of INH, RIF, PZA, ± EMB | 18 weeks of INH and RIF |
| Intermittent† | (a) 2 weeks of daily INH, RIF, PZA, and EMB (or SM); then 6 weeks of INH, RIF, PZA, EMB BIW or TIW | 18 weeks of INH and RIF BIW |
| (b) 8 weeks of thrice-weekly INH, RIF, PZA, and EMB (or SM) | 18 weeks of INH and RIF TIW |
BIW, twice weekly; EMB, ethambutol; INH, isoniazid; PZA, pyrazinamide; RIF, rifampin; SM, streptomycin; TIW, thrice weekly.
* The daily regimen is employed when patients self-administer their drugs. There is enough redundancy that if patients miss some of their doses, the outcome will remain acceptable.
† The intermittent regimens are intended for directly-observed therapy (DOT). Regimen (a) entails a total of 62 doses and has yielded over 95% success rates for the past 22 years in Denver, Colorado.97 Regimen (b) involves 78 doses and has also resulted in success rates of approximately 95% in Hong Kong, where it is the standard regimen.98
When MDR-TB is suspected or confirmed, additional drugs that may be used include amikacin, a fluoroquinolone (levofloxacin, moxifloxacin), capreomycin, ethionamide, cycloserine, and/or para-aminosalicylic acid. Local public health departments should be contacted to meet reporting requirements and will usually be responsible for treatment monitoring. Directly observed therapy (DOT) should be implemented whenever possible. Patients with MDR-TB require DOT and longer therapy (generally 18 months of treatment after the last negative sputum culture). Surgical resection after 2 to 3 months of treatment may improve outcome.91
Parenteral therapy may be required in ICU patients and is recommended for patients with fulminant disease (Table 142-3). INH and RIF are available in parenteral forms; EMB and PZA are not. Other active medications available for IV use include the aminoglycosides, fluoroquinolones, and capreomycin. In patients with renal failure, dose adjustments are required for those taking EMB, PZA, cycloserine, an aminoglycoside, capreomycin, or a fluoroquinolone. INH and PZA should probably be withheld in the setting of severe liver failure. An expert in the treatment of TB should be consulted when treating the complicated ICU patient or those with MDR-TB.
TABLE 142-3 Selected Parenteral Medications Used in Treating Tuberculosis91
| Medication | Preparation | Initial Dosage in Adults (Maximum Dosage) |
|---|---|---|
| Isoniazid | PO, IV, IM | 5 mg/kg/d (300 mg) |
| Rifampin | PO, IV | 10 mg/kg/d (600 mg) |
| Streptomycin | IV, IM | 10-15 mg/kg/d or 750-1000 mg/d |
| Amikacin | IV, IM | Same as above |
| Kanamycin | IV, IM | Same as above |
| Capreomycin | IV, IM | Same as above |
| p-Aminosalicylic acid | PO, IV | 8-12 g/d in 2 or 3 doses |
| Levofloxacin | PO, IV | 500-1000 mg/d |
| Moxifloxacin | PO, IV | 400 mg/d |
Notes: Table shows routine daily dosing. Dosages may differ in children and in patients in intermittent therapy. Persons over age 59 should receive the lower dose for aminoglycosides (750 mg).
IM, intramuscular; IV, intravenous, PO, oral.
Corticosteroids are generally recommended in the treatment of several TB conditions, including TB meningitis and pericarditis, as discussed above.92 Their role in patients with respiratory failure due to TB and in patients with severe AIDS-associated TB has not been proven, but many have used corticosteroids for these conditions. Typical therapy includes prednisone, 40 to 80 mg per day, tapered over a few weeks.
 Risk to Healthcare Workers
Risk to Healthcare Workers
An awareness that caring for TB patients poses a risk to healthcare workers (HCWs) did not emerge until the 1950s and 1960s when studies established that M. tuberculosis infection was transmitted by the airborne route.93 However, occupational transmission received little attention until numerous outbreaks of TB and MDR-TB occurred in U.S. and European hospitals in the late 1980s and early 1990s.94 At that time, more than 20 HCWs became ill with MDR-TB, and at least 10 died.95 Hundreds of HCWs may be latently infected with MDR-TB and thus represent a relatively large reservoir of individuals at risk for future reactivation MDR-TB.
Pulmonologists are at higher risk for occupational exposure to TB compared to other medical specialists. Atypical presentations of TB can put providers at increased risk when TB is not suspected and proper precautions are not taken.96 Bronchoscopy requires close contact with patients and provokes coughing, which likely contributes to the tuberculin skin test conversion rate of 11% among pulmonary fellows.96 DMF-HEPA respirators should be used when performing bronchoscopy on patients with known or suspected TB.96
Chan ED, Strand MJ, Iseman MD. Treatment outcomes in extensively resistant tuberculosis. N Engl J Med. 2008;359:657-659.
Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TT, Nguyen TC, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351:1741-1751.
Erbes R, Oettel K, Raffenberg M, Mauch H, Schmidt-Ioanas M, Lode H. Characteristics and outcome of patients with active pulmonary tuberculosis requiring intensive care. Eur Respir J. 2006;27:1223-1228.
Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Bréban M, et al. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: the three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum. 2009;60:1884-1894.
Nahid P, Gonzalez LC, Rudoy I, de Jong BC, Unger A, Kawamura LM, et al. Treatment outcomes of patients with HIV and tuberculosis. Am J Respir Crit Care Med. 2007;175:1199-1206.
1 Raviglione MC, Snider DEJ, Kochi A. Global epidemiology of tuberculosis: morbidity and mortality of a worldwide epidemic. JAMA. 1995;273:220-226.
2 Lienhardt C, Rodrigues LC. Estimation of the impact of human immunodeficiency virus infection on tuberculosis: tuberculosis risks re-visited? Int J Tuberc Lung Dis. 1997;1:196-204.
3 WHO Report. Global Tuberculosis Control: Surveillance, Planning, Financing. Geneva: World Health Organization; 2002.
4 Centers for Disease Control and Prevention. Update: Trends in tuberculosis–United States. MMWR Morb Mortal Wkly Rep. 2009;58:249-253.
5 WHO Report. Global Tuberculosis Control 2009: Surveillance, Planning, Financing. Geneva: World Health Organization; 2009.
6 Frieden TR, Sterling T, Pablos-Mendez A, Kilburn JO, Cauthen GM, Dooley SW. The emergence of drug-resistant tuberculosis in New York City. N Engl J Med. 1993;328:521-526.
7 Chan ED, Iseman MD. Multi-drug resistant and extensively drug-resistant tuberculosis: a review. Curr Opin Infect Dis. 2008;21:587-595.
8 Centers for Disease Control and Prevention. Update: Trends in tuberculosis–United States. MMWR Morb Mortal Wkly Rep. 2008;57:281-285.
9 Cain KP, Benoit SR, Winston CA, Mac Kenzie WR. Tuberculosis among foreign-born persons in the United States. JAMA. 2008;300:405-412.
10 Gandhi NR, Shah NS, Andrews JR, Vella V, Moll AP, Scott M, et al. HIV Coinfection in multidrug- and extensively drug-resistant tuberculosis results in high early mortality. Am J Respir Crit Care Med. 2010;181:80-86.
11 Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368:1575-1580.
12 Migliori GB, Lange C, Girardi E, Centis R, Besozzi G, Kliiman K, et al. Extensively drug-resistant tuberculosis is worse than multidrug-resistant tuberculosis: Different methodology and settings, same results. Clin Infect Dis. 2008;46:958-959.
13 Chan ED, Strand MJ, Iseman MD. Treatment outcomes in extensively resistant tuberculosis. N Engl J Med. 2008;359:657-659.
14 Raviglione MC, Smith IM. XDR tuberculosis–implications for global public health. N Engl J Med. 2007;356:656-659.
15 Singh JA, Upshur R, Padayatchi N. XDR-TB in South Africa: No time for denial or complacency. PloS Med. 2007;4:e50.
16 Penner C, Roberts D, Kunimoto D, Manfreda J, Long R. Tuberculosis as a primary cause of respiratory failure requiring mechanical ventilation. Am J Respir Crit Care Med. 1995;151:867-872.
17 Erbes R, Oettel K, Raffenberg M, Mauch H, Schmidt-Ioanas M, Lode H. Characteristics and outcome of patients with active pulmonary tuberculosis requiring intensive care. Eur Respir J. 2006;27:1223-1228.
18 Dyer RA, Potgieter PO. The adult respiratory distress syndrome and bronchogenic pulmonary tuberculosis. Thorax. 1984;39:383-387.
19 Barnes PF, Leedom JM, Chan LS, Wong SF, Shah J, Vachon LA, et al. Predictors of short-term prognosis in patients with pulmonary tuberculosis. J Infect Dis. 1988;158:366-371.
20 Mehta JB, Fields CL, Byrd RP, Roy TM. Nutritional status and mortality in repsiratory failure caused by tuberculosis. Tenn Med. 1996;89:369-371.
21 Wu JY, Ku SC, Shu CC, Fan JY, Chen HY, Chen YC, et al. The role of chest radiography in the suspicion for and diagnosis of pulmonary tuberculosis in intensive care units. Int J Tuberc Lung Dis. 2009;13:1380-1386.
22 Lee PL, Jerng JS, Chang YL, Chen CF, Hsueh PR, Yu CJ, et al. Patient mortality of active pulmonary tuberculosis requiring mechanical ventilation. Eur Respir J. 2003;22:141-147.
23 Khan FA, Rehman M, Marcus P, Azueta V. Pulmonary gangrene occurring as a complication of pulmonary tuberculosis. Chest. 1980;77:76-80.
24 Lin SM, Wang TY, Liu WT, Chang CC, Lin HC, Liu CY, et al. Predictive factors for mortality among non-HIV-infected patients with pulmonary tuberculosis and respiratory failure. Int J Tuberc Lung Dis. 2009;13:335-340.
25 Chauveau P, Pellerin M, Bochereau G, Poirson B, Blin F, Kleinknecht D. Acute respiratory distress syndrome of tuberculous origin in an intensive care unit. Nouv Presse Med. 1981;10:3053-3056.
26 Heffner JE, Strange C, Sahn SA. The impact of respiratory failure on the diagnosis of tuberculosis. Arch Intern Med. 1988;148:1103-1108.
27 Leibowitz RE. Critical care and tuberculosis. Crit Care Nurs Clin North Am. 1995;7:661-666.
28 Barnes PF, Lakey DL, Burman WJ. Tuberculosis in patients with HIV infection. Infect Dis Clin North Am. 2002;16:107-126.
29 Counsell SR, Tan JS, Dittus RS. Unsuspected pulmonary tuberculosis in a community teaching hospital. Arch Intern Med. 1989;149:1274-1278.
30 Gachot B, Wolff M, Clair B, Regnier B. Severe tuberculosis in patients with human immunodeficiency virus infection. Intensive Care Med. 1990;16:487-488.
31 Kent SJ, Crowe SM, Yung A, Lucas CR, Mijch AM. Tuberculous meningitis: A 30-year review. Clin Infect Dis. 1993;17:987-994.
32 Verdon R, Chevret S, Laissy JP, Wolff M. Tuberculous meningitis in adults: Review of 48 cases. Clin Infect Dis. 1996;22:982-998.
33 Alzeer AH, FitzGerald JM. Corticosteroids and tuberculosis: risks and use as adjunct therapy. Tuberc Lung Dis. 1993;74:6-11.
34 Kumar R, Singh SN, Kohili N. A diagnostic rule for tuberculosis meningitis. Arch Dis Child. 1999;81:221-224.
35 Thwaites GE, Chau TTH, Stepniewska K, Phu NH, Chuong LV, Sinh DX, et al. Diagnosis of adult tuberculosis meningitis by use of clinical and laboratory features. Lancet. 2002;360:1287-1292.
36 Chan ED, Heifets L, Iseman MD. Immunologic diagnosis of tuberculosis: a review. Tuberc Lung Dis. 2000;80:131-140.
37 Noordhoek GT, Kolk AH, Bjune G, Catty D, Dale JW, Fine PE, et al. Sensitivity and specificity of PCR for detection of Mycobacterium tuberculosis: a blind comparison study among seven laboratories. J Clin Microbiol. 1994;32:277-284.
38 Pai M, Flores LL, Pai N, Hubbard A, Riley LW, Colford JMJ. Diagnostic accuracy of nucleic acid amplification tests for tuberculous meningitis: a systematic review and meta-analysis. Lancet Infect Dis. 2003;3:633-643.
39 Dinnes J, Deeks J, Kunst H, Gibson A, Cummins E, Waugh N, et al. A systematic review of rapid diagnostic tests for the detection of tuberculosis infection. Health Technol Assess. 2007;11:1-196.
40 Thwaites GE, Caws M, Chau TT, Dung NT, Campbell JI, Phu NH, et al. Comparison of conventional bacteriology with nucleic acid amplification (amplified Mycobacterium direct test) for diagnosis of tuberculous meningitis before and after inception of antituberculosis chemotherapy. J Clin Microbiol. 2004;42:996-1002.
41 Donald PR, Victor TC, Jordaan AM, Schoeman JF, van Helden PD. Polymerase chain reaction in the diagnosis of tuberculous meningitis. Scand J Infect Dis. 1993;25:613-617.
42 Ellard GA, Humphries MJ, Gabriel M, Teoh R. Penetration of pyrazinamide into the cerebrospinal fluid in tuberculous meningitis. BMJ. 1987;294:284-285.
43 Ellard GA, Humphries MJ, Allen BW. Cerebrospinal fluid drug concentrations and the treatment of tuberculous meningitis. Am Rev Respir Dis. 1993;148:650-655.
44 Kaojarern S, Supmonchai K, Phuapradit P, Mokkhavesa C, Krittiyanunt S. Effect of steroids on cerebrospinal fluid penetration of antituberculous drugs in tuberculous meningitis. Clin Pharmacol Ther. 1991;49:6-12.
45 Thwaites GE, Lan NT, Dung NH, Quy HT, Oanh DT, Thoa NT, et al. Effect of antituberculosis drug resistance on response to treatment and outcome in adults with tuberculous meningitis. J Infect Dis. 2005;192:79-88.
46 Humphries M. The management of tuberculous meningitis. Thorax. 1992;47:577-581.
47 Prasad K, Singh MB. Corticosteroids for managing tuberculous meningitis. Cochrane Database Syst Rev 2008;CD002244.
48 Thwaites GE, Nguyen DB, Nguyen HD, Hoang TQ, Do TT, Nguyen TC, et al. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N Engl J Med. 2004;351:1741-1751.
49 American Thoracic Society, Centers for Disease Control, and Infectious Diseases Society of America. Treatment of tuberculosis. MMWR Morb Mortal Wkly Rep. 2003;52:1-77.
50 Bidstrup C, Anderson PH, Skinhoj P, Andersen AB. Tuberculous meningitis in a country with a low incidence of tuberculosis: Still a serious disease and a diagnostic challenge. Scand J Infect Dis. 2002;34:811-814.
51 Hernandez-Albujar S, Arribas JR, Royo A, Gonzalez-Garcia JJ, Pena JM, Vazquez JJ. Tuberculous radiculomyelitis complicating tuberculous meningitis: Case report and review. Clin Infect Dis. 2000;30:915-921.
52 Voigt MD, Kalvaria I, Trey C, Berman P, Lombard C, Kirsch RE. Diagnostic value of ascites adenosine deaminase in tuberculous peritonitis. Lancet. 1989;1:751-754.
53 Rana BS, Jones RA, Simpson IA. Recurrent pericardial effusion: The value of polymerase chain reaction in the diagnosis of tuberculosis. Heart. 1999;82:246-247.
54 Dooley DP, Carpenter JL, Rademacher S. Adjunctive corticosteroid therapy for tuberculosis: A critical repraisal of the literature. Clin Infect Dis. 1997;25:872-887.
55 Dada MA, Lazarus NG, Kharsany ABM, Sturm AW. Sudden death caused by myocardial tuberculosis: Case report and review of the literature. Am J Forensic Med Pathol. 2000;21:385-388.
56 Lie JT. Coronary vasculitis: A review in the current scheme of classification of vasculitis. Arch Pathol Lab Med. 1987;111:224-233.
57 Allins AD, Wagner WH, Cossman DV, Gold RN, Hiatt JR. Tuberculous infection of the descending thoracic and abdominal aorta: Case report and literature review. Ann Vasc Surg. 1999;13:439-444.
58 Goldbaum TS, Lindsay J, Levy C, Silva CA. Tuberculous aortitis presenting with an aortoduodenal fistula: A case report. Angiology. 1986;37:519-523.
59 de Kruijf E, van Rijn AB, Koelma IA, Kuijpers TJ, van’t Wout JW. Tuberculous aortitis with an aortoduodenal fistula presenting as recurrent gastrointestinal bleeding. Clin Infect Dis. 2000;31:841-842.
60 Long R, Guzman R, Greenberg H, Safneck J, Hershfield E. Tuberculous mycotic aneurysm of the aorta: Review of published medical and surgical experience. Chest. 1999;115:522-531.
61 Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximib, a tumor necrosis factor-α neutralizing agent. N Engl J Med. 2002;345:1098-1104.
62 WHO Report. Global tuberculosis control: Surveillance, Planning, Financing. Geneva: World Health Organization; 2007.
63 Kaufmann SH. How can immunology contribute to the control of tuberculosis? Nat Rev Immunol. 2001;1:20-30.
64 Patel NR, Swan K, Li X, Tachado SD, Koziel H. Impaired M. tuberculosis-mediated apoptosis in alveolar macrophages from HIV+ persons: potential role of IL-10 and BCL-3. J Leukoc Biol. 2009;86:53-60.
65 Patel NR, Zhu J, Tachado SD, Zhang J, Wan Z, Saukkonen J, Koziel H. HIV impairs TNF-alpha mediated macrophage apoptotic response to Mycobacterium tuberculosis. J Immunol. 2007;179:6973-6980.
66 Burman WJ, Jones BE. Clinical and radiographic features of HIV-related tuberculosis. Semin Respir Infect. 2003;18:263-271.
67 Chien JW, Johnson JL. Paradoxical reactions in HIV and pulmonary TB. Chest. 1998;114:933-936.
68 Kunimoto DY, Chui L, Nobert E, Houston S. Immune mediated “HAART” attack during treatment for tuberculosis. Int J Tuberc Lung Dis. 1999;3:944-947.
69 Orlovic D, Smego RA. Paradoxical tuberculous reactions in HIV-infected patients. Int J Tuberc Lung Dis. 2001;5:370-375.
70 Centers for Disease Control. Clinical update: impact of HIV protease inhibitors on the treatment of HIV-infected tuberculosis patients with rifampin. MMWR Morb Mortal Wkly Rep. 1996;45:921-925.
71 Peloquin CA, Nitta AT, Burman WJ, Brudney KF, Miranda-Massari JR, McGuinness ME, et al. Low antituberculosis drug concentrations in patients with AIDS. Ann Pharmacother. 1996;30:919-925.
72 Swaminathan S, Narendran G, Venkatesan P, Iliayas S, Santhanakrishnan R, Menon PA, et al. Efficacy of a 6-month versus 9-month intermittent treatment regimen in HIV-infected patients with tuberculosis: a randomized clinical trial. Am J Respir Crit Care Med. 2010;181:743-751.
73 Nahid P, Gonzalez LC, Rudoy I, de Jong BC, Unger A, Kawamura LM, et al. Treatment outcomes of patients with HIV and tuberculosis. Am J Respir Crit Care Med. 2007;175:1199-1206.
74 Dinarello CA. Differences between anti-tumor necrosis factor-α monoclonal antibodies and soluble TNF receptors in host defense impairment. J Rheumatol.. 2005;32(Suppl.):40-47.
75 Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53:45-53.
76 Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, et al. Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561-572.
77 Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J.. 2009;50:1-11.
78 Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, Tsang E, et al. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun. 2001;69:1847-1855.
79 Tubach F, Salmon D, Ravaud P, Allanore Y, Goupille P, Bréban M, et al. Risk of tuberculosis is higher with anti-tumor necrosis factor monoclonal antibody therapy than with soluble tumor necrosis factor receptor therapy: The three-year prospective French Research Axed on Tolerance of Biotherapies registry. Arthritis Rheum. 2009;60:1884-1894.
80 Wallis RS, Broder M, Wong J, Lee A, Hoq L. Reactivation of latent granulomatous infections by infliximab. Clin Infect Dis. 2005;41(Suppl.):S194-S198.
81 Wallis RS, Broder M, Wong J, Beenhouwer D. Granulomatous infections due to tumor necrosis factor blockade: correction. Clin Infect Dis. 2004;39:1254-1255.
82 Saliu OY, Sofer C, Stein DS, Schwander SK, Wallis RS. Tumor-necrosis-factor blockers: differential effects on mycobacterial immunity. J Infect Dis. 2006;194:486-492.
83 Settas LD, Tsimirikas G, Vosvotekas G, Triantafyllidou E, Nicolaides P. Reactivation of pulmonary tuberculosis in a patient with rheumatoid arthritis during treatment with IL-1 receptor antagonists (anakinra). J Clin Rheumatol. 2007;13:219-220.
84 American Thoracic Society; CDC. Council of the Infectious Disease Society of America. Diagnostic standards and classification of tuberculosis in adults and children. Am J Respir Crit Care Med. 2000;161:1376-1395.
85 Moore DF, Guzman JA, Mikhail LT. Reduction in turnaround time for laboratory diagnosis of pulmonary tuberculosis by routine use of a nucleic acid amplification test. Diagn Microbiol Infect Dis. 2005;52:247-254.
86 Guerra RL, Hooper NM, Baker JF, Alborz R, Armstrong DT, Maltas G, et al. Use of the amplified Mycobacterium tuberculosis direct test in a public health laboratory: test performance and impact on clinical care. Chest. 2007;132:946-951.
87 Centers for Disease Control and Prevention. National plan for reliable tuberculosis laboratory services using a systems approach: recommendations from CDC and the Association of Public Health Laboratories Task Force on Tuberculosis Laboratory Services. MMWR Morb Mortal Wkly Rep. 2005;54:1-12.
88 Taegtmeyer M, Beeching NJ, Scott J, Seddon K, Jamieson S, Squire SB, et al. The clinical impact of nucleic acid amplification tests on the diagnosis and management of tuberculosis in a British hospital. Thorax. 2008;63:317-321.
89 Chan ED, Iseman MD. Current medical treatment for tuberculosis. BMJ. 2002;325:1282-1286.
90 Iseman MD. A Clinician’s Guide to Tuberculosis. Baltimore: Lippincott, Williams and Wilkins; 1999.
91 Centers for Disease Control and Prevention. Treatment of tuberculosis. MMWR Morb Mortal Wkly Rep. 2003;52:1-80.
92 Barnes PF, Barrows SA. Tuberculosis in the 1990s. Ann Intern Med. 1993;119:400-410.
93 Sepkowitz KA. Tuberculosis and the health care worker: a historical perspective. Ann Intern Med. 1994;120:71-79.
94 Menzies D, Fanning A, Yuan L, Fitzgerald M. Tuberculosis among health care workers. N Engl J Med. 1995;332:92-98.
95 Sepkowitz KA. AIDS, tuberculosis, and the health care worker. Clin Infect Dis. 1995;20:232-242.
96 Raucher BG. Infection control in pulmonary and critical care medicine. Sem Respir Infect. 1999;14:372-382.
97 Cohn DL, Catlin BJ, Peterson KL, Judson FN, Sbarbaro JA. A 62-dose, 6-month therapy for pulmonary and extrapulmonary tuberculosis: A twice-weekly, directly observed, and cost-effective regimen. Ann Intern Med. 1990;112:407-415.
98 Hong Kong Chest Service / British Medical Research Council controlled trial of 2, 4, and 6 months of pyrazinamide in 6-month, three-times weekly regimens for smear-positive pulmonary tuberculosis, including an assessment of a combined preparation of isoniazid, rifampin, and pyrazinamide: results at 30 months. Am Rev Respir Dis. 1991;143:700-706.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
142 Tuberculosis
Epidemiology
The World Health Organization (WHO) estimates that one-third of the world’s population is latently infected with Mycobacterium tuberculosis.1 From this pool, approximately 9 million active tuberculosis (TB) cases emerge annually, resulting in 2 million deaths and making TB the second leading cause of death by an infectious agent worldwide.2 The vast majority of TB cases (95%) occur in the developing world. Incidence rates exceed 300 cases per 100,000 persons throughout sub-Saharan Africa, the Indonesian and Philippine archipelagos, Afghanistan, Bolivia, and Peru.1,3 Areas with the most cases per year include densely populated India (2 million cases per year) and China (1.3 million cases per year).
In the United States, the TB rate continues to decline, with 3.8 new cases per 100,000 reported in 2009, the lowest rate recorded since national reporting began in 1953. Foreign-born persons and racial/ethnic minorities continue to bear a disproportionate burden of TB disease in the United States. In 2008, the TB rate within the foreign-born population in the United States was 10 times higher than in U.S.-born persons.4 TB rates among Hispanics and blacks were nearly eight times higher than among non-Hispanic whites, and rates among Asians were nearly 23 times higher than among non-Hispanic whites. Among U.S.-born racial and ethnic groups, the greatest racial disparity in TB rates was seen in the black population, who are seven times more likely to develop active TB than U.S.-born whites. Other groups at increased risk for active TB include prisoners, the homeless, and human immunodeficiency virus (HIV)-positive individuals.
The acquired immunodeficiency syndrome (AIDS) epidemic has contributed significantly to the rise in TB cases worldwide, with about 1.5 million individuals with active TB per year co-infected with HIV. HIV increases the risk of developing TB by 20.6-fold in countries where the prevalence of HIV is more than 1% in the general population.5 Co-infection with HIV contributes significantly to TB-related mortality.
The Serious Problem of Highly Drug-Resistant Tuberculosis
Drug-susceptible TB is readily curable provided adherence to medications is followed. However, drug-resistant TB requires a significantly longer course of antibiotics coupled with second-line agents that often are accompanied by difficult-to-tolerate side effects. More importantly, highly drug-resistant TB is associated with significant increase in morbidity and mortality. In the early 1990s, substantial levels of drug resistance began emerging in urban parts of the United States.6 Although the incidence of drug-resistant TB has diminished in the United States, it is increasingly problematic in many parts of the world.7
Multidrug-resistant TB (MDR-TB) is defined as resistance to two of the most powerful first-line anti-TB drugs, isoniazid (INH) and rifampin (RIF). Isolates that are resistant to multiple other combinations of anti-TB drugs but not to INH or RIF are not classified as MDR-TB. It is estimated that of the 9 million new cases of TB per year in the world, 500,000 are due to MDR-TB. Whereas drug-resistant TB is increasing at an alarming rate worldwide, particularly in India and China, prevalence in the United States decreased between 1991 and 2006 from 3.5% to 1.1%.8 MDR-TB disproportionately affects foreign-born individuals, accounting for 0.4% of TB cases occurring in U.S.-born persons and 1.3% in foreign-born individuals.9
Extensively drug-resistant tuberculosis (XDR-TB) is defined as resistance to INH, RIF, any fluoroquinolone, and to a second-line injectable (amikacin, kanamycin, or capreomycin). XDR-TB has emerged with a wide geographic distribution, including the United States, and is associated with worse treatment outcomes than MDR-TB, especially in those co-infected with HIV.7,10–15
Pulmonary Tuberculosis
Primary infection occurs following airborne implantation of tubercle bacilli into the lungs. M. tuberculosis spreads from the lungs to hilar lymph nodes, and then throughout the bloodstream (Figure 142-1). Although primary infection is usually asymptomatic in adults, it can present with fever, hilar adenopathy, lung infiltrates, pleural effusions, and even severe pulmonary disease that may mimic viral or bacterial pneumonia, which may delay the diagnosis of TB. In severely immunocompromised patients, primary TB may be aggressive and become disseminated. Pleural TB is usually a manifestation of primary TB, although it may also occur with reactivation disease. Pleural TB can present as pleuritis or empyema. Pleural biopsy specimens are more likely to yield positive cultures than pleural fluid.
Most cases of active TB are due to reactivation of latent TB infection (LTBI). Active TB develops in about 10% of immunocompetent individuals with LTBI and tends to occur within the first 2 years of the initial infection. Typically, reactivation TB is a subacute fibrocavitary pneumonia involving the upper lobes and/or superior segments of the lower lobes. However, reactivation TB can involve any organ system and can present in a fulminant fashion with respiratory failure.16
There are some common clinical characteristics of TB patients who require ICU care. In a study of 58 ICU patients with confirmed TB, 22 (37.9%) required mechanical ventilation, and 15 (25.9%) died in the hospital.17 The factors independently associated with mortality were acute renal failure, need for mechanical ventilation, chronic pancreatitis, sepsis, acute respiratory distress syndrome (ARDS), and nosocomial pneumonia.17 Both primary and reactivation TB can cause bilateral alveolar infiltrates, hypoxic respiratory failure, and ARDS.16,18 In another study of patients hospitalized with pulmonary TB, six factors were shown to be associated with respiratory failure or death: lymphopenia, advanced age, concomitant smear-positive extrapulmonary TB, alcoholism, a high percentage of neutrophils on the peripheral white blood cell count, and lack of radiographic cavitation.19 Laboratory findings of anemia and hypoalbuminemia have been shown to be predictors for death in patients with respiratory failure due to TB.20 However, these findings are not specific to TB and commonly present in the critically ill.
Consolidation is the most frequent radiographic pattern of patients with pulmonary TB who are admitted to the ICU.21 Because this radiographic pattern is highly nonspecific, chest x-rays are often unhelpful in raising the suspicion for TB. Consolidation on initial chest radiograph has also been shown to be a strong independent risk factor for in-hospital mortality.22 One possible reason for this is a delay in the diagnosis; clinicians may be more prone to favor a diagnosis of non-tuberculous pneumonia in the absence of cavitation or miliary pattern. Another is that consolidation may be an indication of a suboptimal immune response to the infection. Pulmonary gangrene, which carries a mortality of up to 75%, can ensue when rapid progression of infiltrate causes vascular damage and death of lung tissue.23 Other life-threatening complications of pulmonary TB include hemoptysis, spontaneous pneumothoraces, bronchopleural fistulas, and empyema. Not unexpectedly, delayed recognition and treatment of nosocomial pneumonia complicating TB in patients requiring mechanical ventilation has a significant adverse effect on survival.24
Perhaps the best safeguard to prevent missing a diagnosis of pulmonary or disseminated TB in the critically ill is to maintain a high index of suspicion of it in at-risk individuals (e.g., foreign-born or immunosuppressed patients). Studies have shown that the presence of diffuse infiltrates consistent with ARDS and acute respiratory failure may cause physicians to inappropriately dismiss the diagnosis of TB.25–27 Older individuals (≥65 years) or patients with AIDS may also have delayed diagnosis of TB, due in part to atypical presentations.28,29
Hospital mortality has been reported to be 60% for patients with respiratory failure due to pulmonary TB.22 Hence, despite being a relatively rare cause of respiratory failure in ICU patients, pulmonary TB carries a poor prognosis. Early recognition of the infection is essential to reduce mortality and prevent nosocomial spread of M. tuberculosis.27
Disseminated Tuberculosis
Disseminated, or “miliary,” TB is more likely to occur in the very young and very old and in patients with underlying diseases such as HIV. It may result from either primary or reactivation TB. Disseminated TB typically presents subacutely with symptoms present for days to months, but it can manifest fulminantly with septic shock and multiorgan failure.30 Typical presenting signs and symptoms include fever, malaise, weight loss, dyspnea, and hypoxia.
The chest radiograph (Figure 142-2, A) and computed tomography (CT) scan (see Figure 142-2, B) show a typical miliary pattern manifested by a profusion of diffuse small (<2 mm) nodules that resemble the size and uniformity of millet seeds (see Figure 142-2, C). In some cases of disseminated disease, the chest radiograph may appear normal. Virtually any organ may be involved, including the adrenals, brain, meninges, liver, pancreas, eyes, urinary tract, and skin. Bone marrow involvement by TB commonly manifests with anemia, leukemoid reaction, and thrombocytosis. The diagnosis of miliary TB can be difficult. If disseminated TB is suspected, sputum smears should be obtained even if lung disease is not apparent. Biopsy and culture of affected tissue(s), such as the bone marrow, are often required. Culture of blood, urine, and/or stool may be positive, especially in HIV-positive patients.30
Neurologic Tuberculosis
Tuberculous Meningitis
TB meningitis is rare in developed countries, with approximately 300 to 400 cases in the United States each year. It occurs via rupture of a subependymal tubercle that has seeded and formed during primary infection or disseminated disease. Individuals at high risk for TB meningitis include very young children with primary TB and older patients with immunodeficiency disorders such as HIV. Most patients with TB meningitis will have no known history of TB, but evidence of extrameningeal disease (e.g., pulmonary, urinary, etc.) can be found in about half of these patients.31,32 The tuberculin skin test is positive in only about 50% of patients with TB meningitis.
TB meningitis is typically a subacute disease. In one review of 58 cases, symptoms were present for 1 day to 9 months, with a median of 10 days prior to diagnosis.31 A prodromal phase of low-grade fever, malaise, headache, dizziness, vomiting, and/or personality changes may persist for 2 to 3 weeks before the patient presents for medical care. Typical findings at presentation include severe headache, altered mental status, stroke, hydrocephalus, and cranial neuropathies. These clinical features are the result of basilar meningeal fibrosis and vascular inflammation.33 Classic features of bacterial meningitis such as stiff neck and fever may be absent. When allowed to progress, coma and seizures may ensue.
The diagnosis of TB meningitis can be difficult and may be based only on clinical findings without definitive microbiological proof. Certain clinical characteristics such as longer duration of symptoms (>6 days), moderate cerebrospinal fluid (CSF) pleocytosis, and the presence of focal deficits increase the probability of TB meningitis.34,35 Characteristic CSF findings of TB meningitis include:
CSF samples should be sent for acid-fast smears, but this is associated with low sensitivity (<20%). Large volumes (10-15 mL) from several daily lumbar punctures are often needed for a microbiological diagnosis. Sensitivity is increased if four spinal taps are performed. Culture can take weeks and is also associated with low sensitivity. Stereotactic biopsy can be performed if tissue samples are needed. Mycobacterial antigens by enzyme-linked immunosorbent assay (ELISA) or radioimmunoassay have been detected in the CSF of patients with TB meningitis.36
Recent meta-analysis calculated that commercial nucleic acid amplification (NAA) assays used for the diagnosis of TB meningitis were 56% sensitive and 98% specific.37,38 Unfortunately, considerable variability in sensitivity and specificity among tests from different laboratories makes it more difficult to interpret results. Most studies conclude that commercial NAA tests can confirm TB meningitis but cannot rule it out.39 Thus a negative test neither excludes the diagnosis nor obviates the need for continued empirical therapy if the clinical suspicion is high. Comparisons of NAA and microscopy/culture using large volumes of CSF have indicated that the sensitivity of microscopy was similar to NAA for the diagnosis of TB meningitis, and repeated testing gave the highest diagnostic yield.40 The sensitivity of CSF microscopy and culture falls rapidly after the start of treatment, whereas mycobacterial DNA may remain detectable within the CSF up to a month after the start of treatment.41
Magnetic resonance imaging (MRI) often reveals basilar meningeal enhancement (Figure 142-3) and/or hydrocephalus.32 Hypodensities due to cerebral infarcts, and ring or nodular enhancing lesions can also be seen. MRI is superior to CT for evaluating the brainstem and the extent of lesions.
The outcome of TB meningitis is improved by timely treatment. Thus empirical treatment is warranted when risk factors and clinical features are suggestive of this diagnosis, even before microbiological confirmation. Chemotherapy for TB meningitis follows the model of short-course chemotherapy for pulmonary TB—an induction phase followed by a continuation phase. But unlike pulmonary TB, the optimal drug regimen and duration of each phase of treatment are not clearly established. INH and RIF remain the most essential drugs. INH penetrates the CSF freely and has potent early bactericidal activity.42–44 RIF penetrates the CSF less well (maximum concentrations around 30% of plasma), but the high mortality from RIF-resistant TB meningitis has confirmed its central role in the treatment of CNS disease.45 INH, RIF, and pyrazinamide are considered mandatory at the beginning of TB meningitis treatment, and some centers use all three drugs for the duration of therapy.46 There are no data from controlled trials to guide choice of the fourth drug. Most authorities recommend either streptomycin or ethambutol, although neither penetrates the CSF well in the absence of inflammation, and both can produce significant adverse reactions. Therapy should be continued for 9 to 12 months.
Adjunctive corticosteroid treatment of TB meningitis has been recommended for more than 50 years, but there has been long-standing concern that corticosteroids may reduce the penetration of anti-TB drugs into the CNS.33 A recent Cochrane systematic review and meta-analysis of 7 randomized controlled trials involving 1140 participants (with 411 deaths) concluded that corticosteroids improved outcome in HIV-negative children and adults with TB meningitis, but the benefit in HIV infected individuals remains uncertain.47 The results were heavily influenced by a study performed in 545 Vietnamese adults with TB meningitis which observed that treatment with dexamethasone was associated with a significantly reduced risk of death.48 However, there was no demonstrable improvement in the combined endpoint of death or severe disability at 9-month follow-up. The survival benefit associated with corticosteroid therapy may have been due in part to a reduction in severe adverse events (9.5% versus 16%), particularly hepatitis, that necessitated changes in anti-TB drug regimens. No mortality benefit from dexamethasone was evident in 98 HIV-infected patients included in the study.48
Because there are no controlled trials comparing different corticosteroid regimens, the choice should be based on those found to be effective in published trials. One recommended regimen for adults is dexamethasone, 12 mg a day for 3 weeks, followed by gradual taper over the next 3 weeks.49 In the large study from Vietnam, patients with mild disease received intravenous (IV) dexamethasone, 0.3 mg/kg/d × 1 week, 0.2 mg/kg/d × 1 week, and then 4 weeks of tapering oral therapy.48 For patients with more severe TB meningitis, IV dexamethasone was given for 4 weeks (1 week each of 0.4 mg/kg/d, 0.3 mg/kg/d, 0.2 mg/kg/d, and 0.1 mg/kg/d), followed by 4 weeks of tapering oral dexamethasone therapy.48
Prognosis of TB meningitis largely depends on neurologic status at the time of presentation and time to treatment initiation. Most patients will die in 5 to 8 weeks if not treated. Various case series indicate a mortality rate between 7% and 65% in developed countries and up to 69% in underdeveloped areas.31,32,50 Neurologic sequelae occur in up to 50% of survivors.50 Mortality risk is highest in those with comorbidities, severe neurologic involvement on admission, rapid progression of disease, and being elderly.
Other Central Nervous System Manifestations of Tuberculosis
Other CNS manifestations of TB include brain abscesses, intracranial tuberculomas, vasculitis, radiculomyelitis, and spinal arachnoiditis. These can occur in conjunction with TB meningitis but are less likely to be seen as isolated findings in the ICU. Intracranial tuberculomas are more common among pediatric patients, especially infants, and can occur in any region of the brain. They result from hematogenous spread of TB. Tuberculous radiculomyelitis is a paradoxical reaction to the treatment of TB meningitis and may respond to corticosteroids. Signs and symptoms include subacute paraparesis, radicular pain, bladder disturbance, and paralysis.51
