[level-membership-for-critical-care-medicine-category]
38 Traumatic Brain Injury
It is estimated that 3.2 million people are living with long-term disability related to traumatic brain injury (TBI).1 In addition to the personal toll, the direct and indirect costs of these disabilities are estimated to exceed $60 billion annually.2 Americans sustain an estimated 1.6 million TBIs each year. Approximately 290,000 require hospitalization, and 51,000 die of their injuries.3 However, the true incidence of TBI is unknown because current surveillance methodologies do not capture those treated in non-hospital settings (e.g., primary care office) or those who do not seek treatment at all.
TBI death rates in the United States fell during the 1980s. A substantial decline in motor vehicle–related fatalities was primarily responsible. At the same time, the incidence of gunshot wounds to the head rose, and in 1990, firearms surpassed motor vehicle crashes as the single largest cause of death due to TBI in some urban areas. However, in-hospital mortality rose to 8% during the last decade, presumably because of the increase in hospitalization rates of severe and moderate TBI.4
 Pathophysiology
Pathophysiology
Primary Injury
Injury to the brain is caused by external forces to the head that strain the tissue beyond its structural tolerance.5 These forces can be classified as contact or inertial.6 Contact forces typically produce focal injuries such as skull fractures, contusions, and epidural or subdural hematomas. Inertial forces result from the brain undergoing acceleration or deceleration (translational, rotational, or both) and can occur without head impact. Inertial forces can cause focal or diffuse brain injuries: pure translational acceleration leads to focal injuries such as contrecoup contusions, intracerebral hematomas, and subdural hematomas, whereas rotational or angular acceleration, common with high-speed motor vehicle crashes, usually causes diffuse injuries. Although external signs of head injury such as scalp abrasions, lacerations, and hematomas are common with blunt-force trauma, the brain can also be severely injured solely by inertial forces, without evidence of scalp or facial injuries.
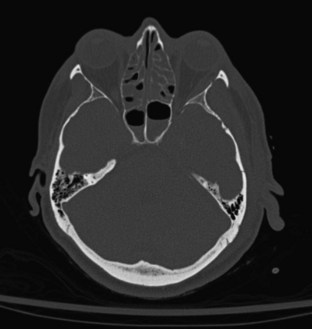
Skull fracture results from a contact force to the head that is usually severe enough to cause at least brief loss of consciousness. Linear fractures are the most common type of skull fracture and typically occur over the lateral convexities of the skull. Most often, they are nondisplaced cracks in the skull (linear fractures), but a particularly intense impact can cause a gap (diastasis) between the edges of the fracture. A depressed skull fracture, in which skull fragments are pushed into the cranial vault, usually results from blunt force by an object with a relatively small surface area, such as a hammer (Figure 38-1). The base of the skull can be fractured by severe blunt trauma to the forehead or the occiput. Basilar skull fractures are most common in the anterior skull base and often involve the cribriform plate, disrupting the olfactory nerves (Figure 38-2). Posterior basilar skull fractures may extend through the petrous bone and internal auditory canal, thereby damaging the acoustic and the facial nerves.
Figure 38-1 Right frontal depressed skull fracture caused by an assault with a hammer (axial CT scan, bone window).
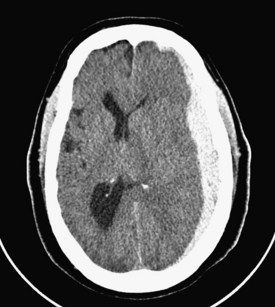
Common posttraumatic intracranial lesions are hemorrhage (epidural, subdural, and intraparenchymal), contusion, and diffuse brain injury. Subdural hematomas are seen in 20% to 25% of all comatose victims of TBI (Figure 38-3). They develop between the surface of the brain and the inner surface of the dura and are believed to result from the tearing of bridging veins over the cortical surface or from disruption of major venous sinuses or their tributaries. The hematoma typically spreads over most of the cerebral convexity; the dural reflections of the falx cerebri prevent expansion to the contralateral hemisphere. Swelling of the cerebral hemisphere is common in those with subdural hematomas, given the associated damage to underlying brain tissue. Underlying cerebral contusions were found in 67% of patients with subdural hematomas in one series.7 Subdural hematomas are classified as acute, subacute, or chronic, each having a characteristic appearance on computed tomography (CT): acute hematomas are bright white, subacute lesions are isodense with brain tissue and are therefore often overlooked, and chronic hematomas are hypodense relative to the brain.
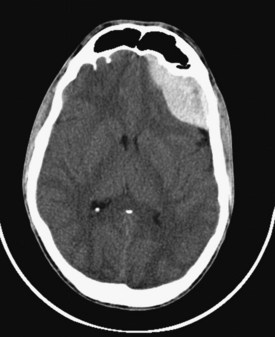
Epidural hematomas develop between the inner table of the skull and the dura, usually when the middle meningeal artery or one of its branches is torn by a skull fracture. They occur in 8% to 10% of those rendered comatose by TBI.8,9 The majority of epidural hematomas are located in the temporal or parietal regions, but they can also occur over the frontal or occipital lobes and (rarely) in the posterior fossa. They appear as hyperdense mass lesions on CT. Unlike subdural hematomas, their spread is limited by the suture lines of the skull, where the dura is very adherent. Because an epidural space normally does not exist, the clot must strip the dura from the inner table of the skull as it enlarges, resulting in its classic biconvex or lenticular shape (Figure 38-4). Epidural hematomas are uncommon in infants and toddlers, presumably because their skulls are more deformable and less likely to fracture, and in TBI victims older than 60 years, because the dura is extremely adherent to the skull.
Traumatic subarachnoid hemorrhage often results from tearing of the corticomeningeal vessels. Though common after severe TBI, subarachnoid hemorrhage does not produce a hematoma or mass effect.10 However, it may be associated with an increased risk for posttraumatic vasospasm, which may adversely affect clinical outcome.11
Contusions are heterogeneous lesions comprising punctate hemorrhage, edema, and necrosis and are often associated with other intracranial lesions. One or more contusions occur in 20% to 25% of patients with severe TBI. Because they evolve over time, contusions may not be evident on the initial CT scan or may appear as small areas of punctate hyperdensities (hemorrhages) with surrounding hypodensity (edema) (Figure 38-5). Local neuronal damage and hemorrhage lead to edema that may expand over the next 24 to 48 hours. With time, contusions may coalesce and look more like intracerebral hematomas. Depending on their size and location, they may cause significant mass effect, resulting in midline shift, subfalcine herniation, or transtentorial herniation. Contusions are most common in the inferior frontal cortex and the anterior temporal lobes,12 where the surface of the inner table of the skull is very irregular; they result from shifting of the brain over this irregular surface at the time of impact. Direct blunt-force trauma to the head can produce a contusion in the tissue underlying the point of impact (coup contusion). If the head was in motion upon collision with a rigid surface, a contusion may occur in the brain contralateral to the point of impact (contrecoup contusion).
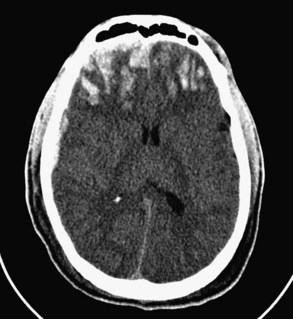
Diffuse axonal injury refers to lacerations or punctate contusions at the interface between the gray and white matter. Such punctate contusions are thought to result from the disparate densities of the gray and white matter and the consequent difference in centripetal force associated with a rotational vector of injury.13 Thus, diffuse axonal injury most often occurs after a high-speed motor vehicle crash, during which severe angular and rotational forces are applied to the head. Diffuse axonal injury was once thought to result solely from mechanical disruption at the time of impact; however, more recent research has identified cases in which the histologic footprints of diffuse axonal injury, such as fragmentation of axons and axonal swelling, do not appear until 24 to 48 hours after the incident, suggesting that some cases are a secondary manifestation of trauma.14,15 Diffuse axonal injury is present in almost half of all patients with severe TBI and in a third of those who die, and it is a common cause of persistent vegetative or minimally conscious state.
Posttraumatic intracranial lesions cause neurologic dysfunction via direct and in some cases indirect mechanisms. By destroying brain tissue, contusions and intraparenchymal hemorrhage cause deficits directly related to the function of the damaged tissue. Uncal herniation is also an important mechanism of temporary or permanent neurologic deficits.16,17 Semirigid dural reflections divide the intracranial contents into compartments. The tentorium cerebelli separates the anterior and middle cranial fossae from the posterior cranial fossa. The brainstem, specifically the midbrain, traverses an opening, the tentorial foramen, in the anterior central portion of this partition. The medial portion of the temporal lobe, the uncus, lies on both sides of the tentorial foramen. Because the most common TBIs, such as hematomas and contusions, are usually located over the lateral surfaces of the brain, and because the brain’s extreme lateral surface is the rigid skull, such lesions tend to depress the brain medially. Therefore, a subdural hematoma over the surface of the temporal lobe or a hemorrhagic contusion of the temporal lobe itself is likely to displace the medial portion of the temporal lobe (uncus) into the tentorial foramen (i.e., uncal herniation). Such displacement compresses the midbrain, which contains neurons that are part of the reticular activating system. At the base of the midbrain is the crus cerebri, which contains pyramidal fibers from the cortex, and the third cranial nerve, which exits the midbrain through the interpeduncular cistern. Midbrain compression due to uncal herniation damages the reticular activating system, causing loss of consciousness; stretches the third cranial nerve and its associated parasympathetic fibers, causing pupil dilatation and loss of the light reflex; and injures the pyramidal fibers in the crus cerebri, causing abnormal posturing responses in the contralateral arm and leg.
Intracranial hypertension is a major cause of posttraumatic neurologic morbidity and mortality.18 The intracranial pressure (ICP) is defined by the volume of CSF, blood, and brain tissue in the cranial vault. The volume of these components is dynamic, and the brain can accommodate moderate changes in any of the three. For example, the blood volume can rise or fall by as much as 30% to 40%, CSF absorption can increase to reduce the size of the ventricles by up to 90%, and brain tissue itself is compressible. Thus, the intracranial volume can gain 100 to 150 mL, equivalent to a moderate-sized subdural hematoma, without the ICP increasing significantly. When these buffering mechanisms have been exhausted, however, even a small increase in the size of a hematoma will cause a rapid rise in ICP. If appropriate treatment is delayed, the ICP may approach the mean arterial pressure (MAP), causing a hydrostatic block of blood flow to the brain and brain death. Intracranial hypertension, particularly if refractory to medical or surgical treatment, is the most common cause of death after severe TBI.
Secondary Injury
Posttraumatic ischemia initiates a cascade of metabolic events that lead to the surplus production of oxygen free radicals,19–21 excitatory amino acids,22,23 cytokines,24,25 and other inflammatory agents.26 Glutamate and aspartate are the excitatory amino acids most commonly implicated in excitotoxic injury,27 which is mediated by activation of N-methyl-D-aspartate, α-amino-3-hydroxy-5-methylisoazole-4-proprionic acid, or kainic acid receptors.23 Overactivation of these receptors causes an excessive influx of ionized calcium into the cytosol, and elevated amounts of ionized intracellular calcium play a key role in neurodegeneration after injury to the central nervous system (CNS).28,29 In addition, posttraumatic nonischemic events such as an increase in intracellular free Ca++ via receptor-gated or voltage-dependent ion channels induce the release of oxygen free radicals from mitochondria.30 Excessive levels of highly reactive oxygen free radicals cause lipid peroxidation of cell membranes, oxidation of intracellular proteins and nucleic acids, and activation of phospholipases A2 and C, which hydrolyze membrane phospholipids, thereby releasing arachidonic acid. The liberation of arachidonic acid triggers the generation of free fatty acids, leukotrienes, and thromboxane B2, all of which are associated with neurodegeneration and poor outcome after experimental TBI.31–33 Inflammatory cytokines, particularly interleukin (IL)-1, IL-6, and tumor necrosis factor, also are overproduced after TBI.34–36 In animal models, posttraumatic activation of microglia is a principal source of these cytokines.25 IL-1 and IL-6 provoke an exuberant cellular inflammatory response believed to be responsible for astrogliosis, edema, and tissue destruction.26,37
TBI also increases extracellular potassium levels,38 leading to an imbalance of intracellular and extracellular K+, disruption of the Na+/K+-ATPase cell membrane regulatory mechanisms, and subsequent cell swelling.39,40 Astrocyte swelling has been attributed to the clearance of excessive extracellular K+.41 High levels of extracellular K+ have also been implicated as the cause of widespread neuronal depolarization and spreading depression seen after experimental TBI.27,38,42 Moreover, potassium stimulates increased oxygen uptake in glial cells, potentially depriving adjacent neurons of oxygen.43,44 Severe TBI also causes a substantial decrease in extracellular magnesium (Mg++) levels, thereby impairing normal glycolysis, cellular respiration, oxidative phosphorylation, and the biosyntheses of DNA, RNA, and protein.45–47 Because Mg++ competes with Ca++ at voltage-gated cell membrane–associated Ca++ channels, reduced levels of Mg++ will result in an abnormal influx of Ca++ into the cell.
 Prehospital Care
Prehospital Care
The acutely injured brain is vulnerable to further damage from systemic hypotension, cerebral hypoperfusion, hypercarbia, hypoxemia, and elevated ICP. Preventing these physiologic insults is crucial to limiting secondary brain injury. Care of the TBI victim always should begin with evaluating and securing a patent airway and restoring normal breathing and circulation. Early endotracheal intubation usually benefits comatose patients. Securing and maintaining an airway are essential to optimal oxygenation and ventilation, and early intubation has been found to reduce mortality after severe TBI.48
The airway is usually most easily and safely secured by orotracheal intubation, a method in which most emergency medical personnel are trained and experienced. Patients with severe maxillofacial trauma may require nasotracheal intubation, but this is less desirable because it is a relatively blind procedure. The nasal passageways can be irritated, causing blood pressure (BP) and ICP to surge, and in those with severe anterior skull base fractures, the tube can inadvertently be passed into the brain. A third alternative for securing the airway is the laryngeal mask airway, an easily learned and rapidly applied device that has undergone successful field trials.49 However, it does not protect against aspiration and cannot be used to achieve high airway pressures. A surgical airway (cricothyroidotomy) should be performed only after other attempts to secure an airway have failed, and only by an experienced provider.
The patient should be sedated and pharmacologically paralyzed before intubation, because irritation of the oropharynx typically causes transient hypertension, tachycardia, increased ICP, and agitation that can interfere with the procedure. Fentanyl, a short-acting opioid agonist that produces analgesia and sedation, is the most commonly used sedative. The usual dose is 3 to 5 µg/kg body weight, administered intravenously (IV) 3 minutes before intubation. Etomidate, an alternative to opioids, provides adequate sedation and is less likely to cause hypotension. Some prefer thiopental because it is an ultra-short-acting barbiturate and is thus less likely to conceal the neurologic status when the patient reaches the trauma center; however, it is more likely than other agents to cause hypotension. Neuromuscular blocking agents commonly used for tracheal intubation include succinylcholine (1.5 mg/kg IV), which has the advantages of rapid onset, complete reliability, and very short duration of action. This last attribute is particularly important in the prehospital setting, where attempts at intubation sometimes fail. Vecuronium (0.01 mg/kg IV) is an alternative that offers the theoretical advantage of being a nondepolarizing muscle relaxant. Because it has a relatively long duration of action (1 to 2 hours), it is less forgiving of failed intubation attempts. Table 38-1 shows a recommended rapid-sequence intubation pathway.
TABLE 38-1 Recommended Rapid-Sequence Induction for Severely Head-Injured Patients
Rapid fluid resuscitation and restoration of a normal BP are critical in the prehospital setting, because hypotension has been associated with doubling of the mortality rate after severe TBI.50 The most likely cause of hypotension is hemorrhage, usually in the abdomen or chest; therefore, hypovolemia should be assumed. Lactated Ringer’s or normal saline solutions should be infused through a large-bore IV catheter as quickly as possible until normotension is achieved. Although preclinical studies suggest that hypertonic saline may be more effective than isotonic solutions for rapid volume resuscitation,51,52 results of several small clinical trials have not been convincing.53,54
In all cases of severe TBI, defined as a Glasgow Coma Scale (GCS) score of 3 to 8 and an inability to follow commands, patients should be treated as if they have a spinal fracture until an adequate examination of the spine proves otherwise. Among those who survive long enough to reach the emergency department, the likelihood of a cervical spine fracture is 2% to 7%. More troubling, however, is that an estimated 10% to 25% of all posttraumatic spinal cord injuries are iatrogenic, occurring during transport to the hospital.55 After respiratory and hemodynamic stabilization, the patient should be placed in a neutral position on a flat, hard surface. If the patient requires immediate tracheal intubation, it should be performed while another person provides in-line cervical spine immobilization. A rigid cervical spine collar should be placed as soon as possible. Next, the patient should be placed on a backboard; the cervical spine can then be further immobilized with a buttress of foam or towels placed on both sides of the head. To prevent any movement during transport, the patient should be strapped to the board in several locations.
The organization of emergency medical services and regional trauma programs has improved outcomes for victims of trauma, particularly those with severe TBI.56 A very large prospective study of the cost and outcomes associated with trauma center designation found more than a 25% reduction in in-hospital mortality for those with severe TBI who were initially treated at a level I trauma center compared to similarly injured patients treated at hospitals of similar size that were not designated trauma centers.57 This is likely because designation as a level I or II trauma center by the American College of Surgeons Committee on Trauma or a state health department ensures the availability of immediate neurosurgical care when the patient arrives. Therefore, every effort should be made to transport severely injured patients directly to a designated trauma center. Nonetheless, if an adequate airway or venous access cannot be obtained in the field, some patients may need to undergo respiratory or hemodynamic stabilization at a nearby emergency department en route to the trauma center. Once hemodynamic and airway stability is achieved, immediate transport to a designated trauma center should occur without delays for imaging or secondary surveys.
 Emergency Department Care
Emergency Department Care
Upon arrival at the trauma center, the emergency medical personnel should concisely report their prehospital assessment and management, including mechanism of injury, stabilizing maneuvers, medications given, initial vital signs, GCS score, and hemodynamic stability during transport. A thorough physical and radiographic examination to identify all life-threatening injuries should then be performed. Most trauma centers follow the Advanced Trauma Life Support protocol, a comprehensive routine that has proved successful in quickly detecting all major injuries.58 First the airway is reassessed, and the need for tracheal intubation is carefully reconsidered. For patients intubated in the field, proper placement of the tracheal tube is verified both clinically and radiographically. When the airway is secure and adequate oxygenation is confirmed using a percutaneous oxygen saturation monitor or arterial blood gas analysis, two large-bore IV catheters are inserted to provide sufficient venous access for high-volume fluid resuscitation. An isotonic saline solution is infused to continue volume replacement, which probably began at the scene. Any life-threatening injuries such as overt hemorrhage, tension pneumothorax, or cardiac tamponade should be treated immediately upon discovery. A brief neurologic examination is performed, including assessment of the GCS score (Table 38-2), pupillary size and reaction to light, and symmetry and extent of extremity movements. The head is palpated to detect fractures, lacerations, or penetrating wounds, and lacerations are probed gently to ascertain the presence of a depressed skull fracture or foreign body. Large lacerations are compressed with pressure dressings or temporarily sutured to prevent further hemorrhage. Careful inspection of the head should reveal hemotympanum, periorbital or mastoid ecchymosis, and CSF rhinorrhea or otorrhea.
| Response | Points |
|---|---|
| Speech | |
| Alert, oriented, and conversant | 5 |
| Confused, disoriented, but conversant | 4 |
| Intelligible words, not conversant | 3 |
| Unintelligible sounds | 2 |
| No verbalization, even with painful stimulus | 1 |
| Eye Opening | |
| Spontaneous | 4 |
| To verbal stimuli | 3 |
| To painful stimuli | 2 |
| None, even with painful stimuli | 1 |
| Motor | |
| Follows commands | 6 |
| Localizes painful stimulus | 5 |
| Withdraws from painful stimulus | 4 |
| Flexor posturing with central pain | 3 |
| Extensor posturing with central pain | 2 |
| No response to painful stimulus | 1 |
Data from Teasdale G, Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;2(7872):81-84.
Coagulopathy resulting from TBI is thought to occur when hypoperfusion causes activation of the protein C pathway, thereby inducing alterations in the clotting cascade.60 It is also commonly seen as a result of therapeutic anticoagulation with warfarin, especially in the geriatric population. Rapid identification and correction of coagulopathy is critical to prevent expansion of intracranial hematomas and allow surgical intervention. Fresh frozen plasma and cryoprecipitate can be used to correct the INR to 1.3 or less. Recombinant factor VIIa has been shown to decrease blood product requirements and costs associated with correction of coagulopathy.61 Other studies demonstrate more rapid correction times compared to blood product administration, although differences in outcomes have not been significant. Product expense and concern for thromboembolic complications have limited the routine use of recombinant factor VIIa.
 Definitive Treatment
Definitive Treatment
Critical to determining the severity of the brain injury and appropriate treatment are CT findings combined with a reliable postresuscitation GCS score and assessment of pupil size and reactivity. In the case of an acute subdural hematoma, for example, a patient with a moderate-sized lesion who has normal pupil size and reactivity and is able to follow commands might safely be treated nonoperatively. Conversely, surgery is unlikely to benefit an elderly patient with fixed and dilated pupils and a GCS score of 3 or 4, regardless of the CT findings. Other determining factors include size and location of the hematoma, presence and extent of an underlying contusion or brain swelling, and results of the neurologic examination. Neurologic deterioration, particularly a decline in mental status, suggests enlargement of the hematoma, and a new CT scan should be obtained promptly. Hematomas less than 10 mm thick that cause a midline shift of less than 5 mm can usually be observed, especially if they do not involve the middle cranial fossa.62 If nonoperative management is chosen for an intracranial hematoma, the patient should be monitored with frequent neurologic assessments in the intensive care unit (ICU). If the patient cannot follow commands, ICP monitoring is recommended.
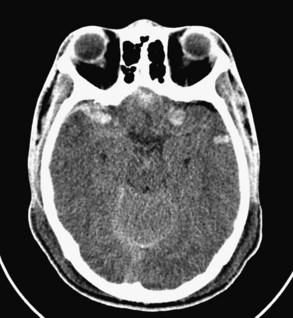
The initial signs and symptoms of contusions vary greatly, depending on their size and location and the presence of other associated lesions. A small contusion may cause only a headache or no symptoms at all. If located in an eloquent area of the brain, such as the speech or motor areas, it may cause focal neurologic symptoms. Larger contusions, especially those involving the frontal or temporal lobes, typically cause elevated ICP and coma. Patients with small or deep-seated contusions without mass effect initially can be managed nonoperatively. The contusion should be followed closely with serial CT scans, however, because there is a 20% to 30% risk that the contusion will enlarge during the next 24 to 48 hours. The ICP should be monitored if the patient cannot follow commands. As with hematomas in the middle cranial fossa, contusions of the temporal lobes should be closely watched with CT scans. A temporal contusion can enlarge to the point of uncal herniation without a significant rise in ICP, so the threshold for evacuation of these lesions should be low (Figure 38-6). Unilateral frontal or temporal lobectomies are usually well tolerated and do not cause measurable neurologic deficits, while allowing space for further brain swelling.
Physiologic Monitoring
Continuous ICP monitoring is essential for all patients who have severe TBI and abnormal CT findings, because intracranial hypertension develops in 53% to 63% of such patients.63 ICP monitoring is also recommended for comatose patients who are older than 40 years and have unilateral or bilateral motor posturing or a systolic blood pressure (SBP) less than 90 mm Hg, even if no abnormalities are seen on the initial CT scan.64 The gold standard for ICP monitors is the ventricular catheter coupled to an external strain-gauge transducer.65 It is accurate, reliable, and far less expensive than newer self-contained pressure-sensing devices. In addition, ventricular pressure is considered more reflective of global ICP than is subdural, subarachnoid, or epidural pressure. Catheters placed in these extracerebral spaces are more prone to occlusion and, owing to the effects of compartmentalization, typically record a pressure that is lower than the global ICP. Other advantages of the ventriculostomy method of ICP monitoring are that the system can be re-zeroed after insertion—not possible with most of the newer self-contained devices—and CSF can be withdrawn to treat intracranial hypertension. The overall complication rate for ventricular ICP monitoring is 7.7% (infection, 6.3%; hemorrhage, 1.4%),63 and some studies indicate that the infection rate increases significantly when a catheter remains in place for more than 5 days.66
Alternatives to the ventriculostomy technique have been developed that provide relatively accurate measurements of global ICP, are easier to insert, and may cause fewer complications. They include devices that contain a pressure-sensing transducer (either strain-gauge or fiber-optic technology) within the tip of the catheter.65 These pressure sensors provide reliable ICP measurements even if they are inserted into the white matter and are often used when a ventricular catheter is difficult to insert because of small or collapsed ventricles. The primary disadvantage is that CSF drainage is not possible. In addition, these devices can be calibrated only once, before insertion, and with some of them, measurement drift is as much as 1 to 2 mm Hg per day.
The cerebral perfusion pressure (CPP), defined as the difference between MAP and ICP, is a calculated physiologic measurement that is used to describe actual cerebral perfusion. Some have suggested that maintaining the CPP above a certain threshold is more important than any particular MAP or ICP.67
Devices that monitor the oxygen partial pressure of oxygen (PO2) of brain tissue can be used to determine whether cerebral oxygenation is adequate. These monitors continually measure the tissue PO2 in the small region of brain into which they are inserted. Studies suggest that mortality may be decreased in those undergoing oxygen directed therapy.68 Although no methods are available for continuously monitoring global CBF, transcranial Doppler insonation of the middle cerebral arteries can provide indirect information. Positron emission tomography (PET) or CBF measurements with xenon, either as a radiolabeled agent or as a CT contrast medium, can provide periodic snapshots of the blood flow.
Medical Treatment
Hypotension, defined as a MAP of less than 90 mm Hg, should be treated aggressively. Normovolemia should be restored by infusing isotonic saline as needed to achieve a central venous pressure of 7 to 12 cm H2O. Hypotonic intravenous solutions can exacerbate cerebral edema and should be avoided. If the patient is anemic, packed red blood cells should be transfused to restore the hematocrit to at least 30%. If hypotension is refractory to volume resuscitation, the patient should be given a continuous IV infusion of a vasopressor medication, with the dose titrated to raise the MAP above 90 mm Hg. Norepinephrine has been shown to be most efficacious at maintaining MAP and CPP without deleteriously affecting ICP.69
Although some advocate the used of induced hypertension to raise the CPP above 70 mm Hg, particularly if the ICP is elevated and difficult to reduce,70 others do not support this practice. A prospective, randomized clinical trial of patients with TBI compared a group whose CPP was kept above 70 mm Hg via induced hypertension with a group whose CPP was allowed to drift to 60 mm Hg.71 Six-month clinical outcomes did not differ between the two groups. Moreover, the group whose CPP was kept above 70 mm Hg required more vasopressor agents and had a significantly higher incidence of ARDS and other pulmonary complications. Others have found that the brain tissue PO2 in patients with TBI typically does not fall until the CPP drops below 60 mm Hg.72 Based on these findings, the current recommendation is to maintain a CPP above 60 mm Hg.
Intracranial hypertension is defined as sustained ICP greater than 20 mm Hg. Several clinical studies have found that mortality and morbidity increase significantly when the ICP persistently remains above this threshold.73 Based on this association and the widely accepted premise that elevated ICP can compromise cerebral perfusion and cause ischemia, the aggressive treatment of intracranial hypertension is almost uniformly endorsed. Before beginning therapy for intracranial hypertension, however, medical or physiologic conditions that can increase ICP should be considered and, if present, treated. These include seizures, fever, jugular venous outflow obstruction (e.g., poorly fitting cervical collars), and agitation.
If intracranial hypertension is refractory to sedation and neuromuscular blockade, intermittent ventricular CSF drainage is used. Intermittent rather than continuous drainage enables reliable measurement of the ICP. If these measures fail to reduce the ICP, a bolus administration of mannitol is recommended (0.25 to 1 g/kg every 3 to 4 hours as needed). This osmotic diuretic lowers ICP and increases CPP by expanding the blood volume, reducing the blood viscosity, and increasing CBF and oxygen delivery to the tissues within a few minutes of infusion. Its duration of effect averages 3 to 5 hours. Continuous infusion is less desirable than bolus infusion, because the former is more likely to lead to extravasation of the drug into brain tissue, causing a reverse osmotic gradient and increased edema and ICP.74 The serum osmolarity and sodium level should be monitored frequently during mannitol administration. The drug should be discontinued if the serum sodium level exceeds 160 mg/dL or the osmolarity exceeds 320 mOsm in order to minimize the risk of acute tubular necrosis and renal failure. The intravascular volume should also be closely monitored to prevent dehydration. Recent studies have shown that hypertonic saline may also be effective at reducing ICP. Hypertonic saline appears to create osmotic mobilization of water across the blood-brain barrier. Concentrations ranging from 3% to 23.4% have been used to decrease ICP.75
If despite these measures the ICP remains above 20 mm Hg, the ventilatory rate can be adjusted to reduce the arterial PCO2 to 30 mm Hg. Hyperventilation should be used cautiously during the first 24 to 48 hours after injury, however, because it will cause cerebral vasoconstriction at a time when CBF is already critically reduced. Evidence also suggests that even brief periods of hyperventilation can lead to secondary brain injury by causing an increase in extracellular lactate and glutamate levels.76 Prophylactic hyperventilation is always contraindicated in the absence of elevated ICP.77 If hyperventilation is used, the brain tissue PO2 or jugular venous oxygen saturation should be monitored to detect any cerebral ischemia that the treatment might cause. The risk of tissue ischemia and poor outcome may increase if the brain tissue PO2 falls below 10 mm Hg.72
If the CT scan does not reveal an intracranial mass lesion requiring surgery, the next recommended treatment for intracranial hypertension is high-dose barbiturates. Barbiturates are thought to be effective by reducing cerebral metabolic demand and blood flow, and preclinical studies suggest significant cerebral protective effects.78 Pentobarbital is the most commonly used drug for this purpose and is administered as an IV loading dose of 10 to 15 mg/kg over 1 to 2 hours, followed by a maintenance infusion of 1 to 2 mg/kg per hour. The dose can be increased until intracranial hypertension subsides or MAP begins to fall. Continuous electroencephalographic monitoring is recommended while increasing the dose until a burst suppression pattern is observed. Hypotension, the most common adverse effect of barbiturates, can usually be averted by ensuring a normal intravascular volume before administering the drug.
Only a few options remain when intracranial hypertension is recalcitrant to all these measures, and they are controversial and not uniformly embraced. Therapeutic moderate hypothermia has been used in several clinical trials over the past decade. The body temperature is lowered to 32°C to 33°C as soon as possible after injury and kept at that temperature for 24 to 48 hours using surface cooling techniques. Although some clinical trials have not found that this treatment improves neurologic outcome compared with normothermia, they have consistently shown that hypothermia significantly reduces ICP.79,80 Moreover, hypothermia does not cause significant medical complications when used for no longer than 48 hours.
Some advocate the use of decompressive craniectomies, such as large lateral or bifrontal bone flaps, with or without a generous temporal or frontal lobectomy In one study of patients with severe TBI, 6-month outcomes were similar for a group that had large decompressive craniectomies and a group that did not, even though the craniectomy group had lower initial GCS scores and more severe radiographic injuries.81 Importantly, the craniectomy group did not have a higher incidence of persistent vegetative state. Two studies reported good outcomes in 56% to 58% of patients whose refractory intracranial hypertension was treated with decompressive craniectomy as a last resort,82,83 and another study suggested that decompressive temporal lobectomy, when performed soon after injury, improves the outcome for young patients.84 However, others found that decompressive craniectomy does not improve ICP, CPP, or mortality rates.85 The decision to perform decompressive surgery should take into account the patient’s ultimate prognosis. Because age has such a profound impact on the likelihood of a meaningful recovery, these therapies are recommended only for patients who are younger than 40 years old.
Patients who have TBI, particularly those who are comatose or have significant non-CNS injuries, are at high risk for pneumonia and other infections, fever, malnutrition, seizures, deep venous thrombosis (DVT), pulmonary embolism, and other maladies endemic to the ICU. Most of these complications cause secondary brain injury and should be diagnosed and treated without delay. Fever is very common in the ICU and occurs in more than 79% of patients within the first week following injury.86 Preclinical studies have found that there is a log increase in neuronal death in ischemic brain regions for every degree of brain temperature above 39°C,87 and this effect is observed for 24 hours or more after injury.88 Clinical studies of TBI patients have shown that the brain temperature is often 1°C to 2°C higher than body temperature,89 though the effect of hyperthermia on ICP is less clear.90,91 Consequently, the body temperature should be kept below 37°C at all times, and infectious or other causes of fever should be aggressively sought and treated.
Patients who are comatose, those being maintained on neuromuscular blocking agents, and those with pelvic or long-bone fractures are at high risk for deep venous thrombosis and pulmonary embolism. They should receive early prophylaxis, which typically includes the use of lower extremity sequential compression devices as well as subcutaneous heparin or enoxaparin. The early (2 to 3 days after injury) use of minidose heparin or low-molecular-weight heparin is safe and has not been found to cause or worsen intracranial hemorrhage after TBI.92,93 DVT is prevalent in TBI patients despite early use of prophylaxis.94
Malnutrition is also common after severe TBI. The resting metabolic expenditure typically increases by 140% in a non-paralyzed patient with severe TBI.95 Branched-chain amino acids from muscle protein are used preferentially for energy metabolism, potentially compromising the effectiveness of physical therapy. Nitrogen wasting is also increased, with excretion of as much as 9 to 12 g/day. Thus, early enteral or parenteral feeding is advisable, with the aim of providing at least 140% of the daily basal metabolic caloric requirements by the third or fourth day after injury.96 A normal-sized adult patient usually needs 2000 to 3000 kcal/day. Because parenteral feeding increases the risk of infection, continuous enteral administration is preferable. For a patient expected to be in a prolonged coma, a percutaneous gastrostomy or surgical jejunostomy provides a convenient and well-tolerated route to administer tube feeding. Hyperglycemia is associated with TBI and is associated with prolonged hospital stays and increased mortality.97 Aggressive management of hyperglycemia has been shown to decrease complications and improve long-term outcome,98 but the optimal blood glucose range in patients with severe TBI remains controversial, and tight glucose control may be problematic.99,100
Posttraumatic contusions and subdural hematomas are well-known causes of generalized seizures which can precipitate secondary injury. Anticonvulsant prophylaxis, usually with phenytoin, is therefore recommended for patients with these lesions. The drug should be given for the first 7 days after injury; a prospective clinical trial found no advantage to longer prophylactic treatment.101 A common side effect of phenytoin is fever; this should be considered if infectious causes of fever have been ruled out. If a patient has seizures, especially if they are prolonged, the associated cerebral hypermetabolism will cause secondary brain injury. Seizures should thus be treated aggressively, up to and including the use of general anesthesia if necessary. Subclinical seizures may occur in up to 33% of patients in the first week following TBI.102 Therefore, EEG should be considered for those with unexplained depressed mental status, abruptly deteriorating cerebral oxygenation, or a sudden increase in ICP; however, enlarging intracranial mass lesions remain the most likely cause.
 Physical Therapy and Rehabilitation
Physical Therapy and Rehabilitation
Rehabilitation of TBI patients should begin in the ICU during the first few days after injury, in consultation with a physiatrist, and include passive range-of-motion exercises and functional splinting of the extremities. Mobilization helps prevent DVT, and studies indicate that early sitting of comatose patients may hasten the return of consciousness. Supplementing physical therapy with central neurostimulant medications is being investigated for those with more severe injuries and minimal responsiveness.103 Rehabilitation after TBI entails many other factors that are critical to optimizing outcome, but a thorough review is beyond the scope of this chapter.
 Penetrating Injuries
Penetrating Injuries Mild and Moderate Injury
Mild and Moderate Injury
Rotational, acceleration, and deceleration forces are common causes of these injuries, particularly those that result in loss of consciousness. The impact usually is not intense enough to cause intracranial hematoma, cerebral contusion, skull fracture, or brain swelling. Although a small amount of subarachnoid hemorrhage may be present, usually in the sulci over the frontal or temporal lobes, CT findings are usually normal. Abnormal magnetic resonance imaging (MRI) findings have been reported in as many as 30% of these patients, most commonly diffuse hyperdense lesions on T2-weighted images. These lesions are thought to represent focal or punctate contusions.104,105 Functional MRI often shows abnormal activation patterns, particularly if the patient has lost consciousness or is symptomatic at the time of the study.105
A patient with persistent symptoms or neurologic deficits should have a CT scan of the head and be admitted to the hospital for observation. This is particularly important for those with GCS scores of 13 or less, because the risk of an intracranial hematoma or contusion large enough to require emergent craniotomy increases as the GCS score decreases among patients whose initial GCS scores are 9 to 13. As many as 40% have CT abnormalities, and 8% require neurosurgical intervention.106
Athletes—especially those involved in contact sports such as boxing, football, soccer, wrestling, and field hockey—are at high risk for mild and moderate TBI. One report found that 47% of high school football players sustained a concussion, with 35% sustaining multiple concussions.107 Multiple concussions are much more likely to cause prolonged or permanent neurologic disability than a single concussion, particularly if they occur over a short time span. Second impact syndrome is a rare but potentially lethal problem first noted in athletes in 1973 and later implicated as the cause of sudden death in several high school football players.108
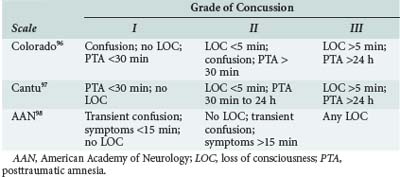
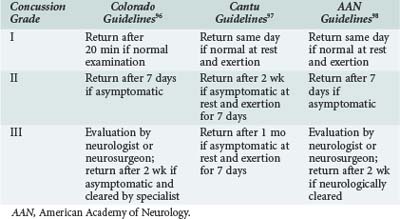
Because sports-related concussions are associated with such disabling and potentially life-threatening consequences, coaches and athletic trainers must carefully consider whether an athlete should be advised to return to play or retire from athletic competition after a concussion. Several groups have devised concussion grading scales to evaluate concussion severity and developed guidelines to determine when an athlete can safely resume play. The most widely adopted scales are those developed by Kelly and colleagues at the University of Colorado,109 Cantu,110 and the American Academy of Neurology111 (Tables 38-3 and 38-4). Most authorities recommend that athletes abstain from play for at least one season if, during that season, they sustain three or more grade I or II concussions or two grade III concussions.112 In addition, many athletic organizations at the high school, college, and professional levels have adopted neuropsychological testing as a means of objectively evaluating the cognitive and neuropsychological consequences of each concussion.113 Comparison of postinjury and preseason scores is a powerful tool for guiding return-to-play decisions.
A common sequela of mild or moderate TBI is postconcussion syndrome, a constellation of symptoms that can be disabling for weeks or even months. The most common symptoms are headache, irritability, dizziness, tinnitus, lethargy, and sleep disturbance. One or more of these symptoms develop in approximately 30% of patients 1 week after a mild or moderate TBI, but they usually subside within 3 months.114 After 1 year, only 7% of patients report residual symptoms, most commonly persistent headache. Postconcussion syndrome is best treated by a primary care physician or neuropsychologist who thoroughly understands the disorder. Cognitive testing is recommended for patients whose symptoms last more than a few weeks, because symptoms such as frustration and irritability are often linked to an inability to resume normal daily activities. Neuropsychological testing often indicates deficits in attention and concentration rather than memory. If such testing identifies specific deficits, cognitive rehabilitation is recommended.115 Persistent headaches, dizziness, and tinnitus should be treated symptomatically after a CT scan of the head establishes the absence of intracranial lesions. Posttraumatic disturbances of the ossicles of the inner ear semicircular canals can cause severe positional vertigo which can be immediately improved with canalith repositioning. In other cases, dizziness is more complex and may result from problems with the vestibular-ocular reflex, so patients with vertigo or tinnitus may benefit from evaluation by an otolaryngologist. Factors associated with an adverse long-term outcome after a concussion include old age, prolonged posttraumatic amnesia, and a below-normal premorbid intellectual capacity.114
 Prognosis
Prognosis
Several clinical and radiographic characteristics have proved useful for outcome prediction, but they must be used in concert.116 Moreover, these criteria are more reliable for predicting death or vegetative survival than for accurately predicting mild or no dysfunction and a complete return to normalcy. The most powerful outcome predictors are age, initial GCS score (particularly the motor component), pupil size and reaction to light, ICP, and the nature and extent of intracranial injuries.
Old age correlates most consistently with a poor outcome after TBI. In the Traumatic Coma Data Bank study of more than 700 patients with severe TBI, the incidence of death, persistent vegetative state, or severe disability was 92% for those older than 60 years, 86% for those older than 56 years, and 50% for younger patients.117 The older groups had a higher incidence of traumatic intracranial mass lesions, midline shift, and subarachnoid hemorrhage, and the presence of these insults correlated strongly with poor outcome. Subsequent studies confirmed the low probability of a good recovery for patients older than 60 years whose initial GCS scores are 8 or less.118
The second most important predictor of outcome is the initial postresuscitation GCS score. Among patients with severe closed head injuries in the Traumatic Coma Data Bank study, good outcomes occurred in 4.1% of those with an initial GCS score of 3, in 6.3% whose score was 4, and in 12.2% whose score was 5. Again, later clinical studies corroborated the strong direct correlation between initial GCS score and outcome.119
Unilaterally or bilaterally dilated pupils that are unreactive to light usually reflect uncal herniation and significant brainstem compression and damage, so this sign is ominous. Several large clinical studies found that patients with bilaterally fixed and dilated pupils had a greater than 90% likelihood of death or vegetative survival.120,121 Also, intracranial hypertension refractory to medication is associated with a 43% mortality rate and 0% chance of a functional outcome.122
Various studies have analyzed the effect of the type and size of posttraumatic intracranial lesions on outcome in terms of both the specific lesions and the CT-defined characteristics of their mass effect. Subdural hematomas are associated with the worst prognosis. One study found that only 26% of patients with these clots had a functional recovery.123 However, the prognosis for patients with subdural hematomas is also related to how soon after injury the clot is evacuated, with the best outcomes in those who have surgery within 2 hours.7
Epidural hematomas pose a much lower risk of mortality because, unlike subdural hematomas, they usually are not associated with underlying cerebral contusions or swelling. If left untreated, however, epidural hematomas can cause uncal herniation and death. One report noted an increase in mortality from 17% to 65% if an epidural hematoma was not evacuated within 2 hours after the onset of coma.8
The presence of traumatic subarachnoid hemorrhage is associated with a 50% greater risk of death.10 The link between traumatic subarachnoid hemorrhage and worse outcomes is controversial, however. Many believe that this condition merely indicates a more severe TBI and has no direct association with outcome.
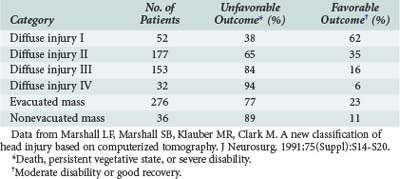
Marshall and colleagues devised a CT-based classification scheme that proved prognostically useful when applied to the patients in the Traumatic Coma Data Bank study (Tables 38-5 and 38-6).124 The classification emphasizes the mass effect of posttraumatic intracranial lesions. Not surprisingly, these investigators found the worst outcomes among patients with large intracranial mass lesions and uncal herniation.
TABLE 38-5 Computed Tomographic Classification of Traumatic Brain Injury
| Category | Definition |
|---|---|
| Diffuse injury I | No visible intracranial pathology |
| Diffuse injury II | Cisterns present, with midline shift 0 to 5 mm; no high-density lesion >25 mL |
| Diffuse injury III (swelling) | Cisterns compressed or absent, with midline shift 0 to 5 mm; no high-density lesion >25 mL |
| Diffuse injury IV (shift) | Midline shift >5 mm; no high-density lesion >25 mL |
| Evacuated mass lesion | Any lesion surgically evacuated |
| Nonevacuated mass lesion | High-density lesion >25 mL; not surgically evacuated |
Data from Marshall LF, Marshall SB, Klauber MR, Clark M. A new classification of head injury based on computerized tomography. J Neurosurg. 1991;75(Suppl):S14-S20.
The patient’s salvageability and prognosis after a penetrating injury are far clearer than for those with closed head injuries. Most victims of gunshot wounds to the head die before or shortly after hospital admission. Among 314 patients with civilian craniocerebral gunshot wounds, 92% died; 73% of them were pronounced dead at the scene of the injury, and 12% died within 3 hours of injury.125 In the Traumatic Coma Data Bank study, the mortality rate was 88% for the 151 patients with gunshot wounds to the head.126 No patient with an initial GCS score of 8 or less regained normal neurologic function, and only three recovered to the level of moderate disability, suggesting that the initial GCS score is an even more powerful predictor of outcome for these patients than for those with closed TBI. A meta-analysis of recent clinical studies examining civilian gunshot wounds to the head found that favorable outcomes (Glasgow Outcome Scale scores of 4 or 5) occurred in only 5 of 490 patients with initial GCS scores of 3 to 5.127 Mortality rates ranged from 51% to 87% for patients with scores of 8 or less. In contrast, those whose initial GCS scores were 13 to 15 all survived and had favorable outcomes. Other clinical signs associated with death or a poor outcome are fixed and dilated pupils, intracranial hypertension, and hypotension. Also, a gunshot wound is more likely to be lethal if self-inflicted.
The CT-defined extent of intracranial injury caused by the missile also has prognostic significance. Hyperdense lesions with a volume greater than 15 mL, midline shift of more than 3 mm, compressed or absent basal cisterns, subarachnoid hemorrhage, and intraventricular hemorrhage are all associated with mortality rates of 80% to 90%, as is a bullet trajectory that traverses both hemispheres, the basal ganglia, or the posterior fossa.126,128
Key Points
Chestnut RM, Marshall SB, Piek J, et al. Early and late systemic hypotension as a frequent and fundamental source of cerebral ischemia following severe brain injury in the Traumatic Coma Data Bank. Acta Neurochir Suppl (Wien). 1993;59:121-125.
Haas B, Jurkovich GJ, Wang J, Rivara FP, Mackenzie EJ, Nathens AB. Survival advantage in trauma centers: expeditious intervention or experience? J Am Coll Surg. 2009;208(1):28-36.
Narayan RK, Kishore PR, Becker DP, et al. Intracranial pressure: to monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg. 1982;56(5):650-659.
Temkin NR, Dikmen SS, Wilensky AJ, et al. A randomized, double-blind study of phenytoin for the prevention of posttraumatic seizures. N Engl J Med. 1990;323(8):497-502.
1 Zaloshnja E, Miller T, Langlois JA, Selassie AW. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J Head Trauma Rehab. 2008;23(6):394-400.
2 Finkelstein E, Corso P, Miller T. The Incidence and Economic Burden of Injuries in the United States. New York: Oxford University Press; 2006.
3 Rutland-Brown W, Langlois JA, Thomas KA, Yongli LX. Incidence of traumatic brain injury in the United States, 2003. J Head Trauma Rehab. 2006;21(6):544-548.
4 Colantonio A, Croxford R, Farooq S, Laporte A, Coyte PC. Trends in hospitalization associated with traumatic brain injury in a publicly insured population, 1992–2002. J Trauma. 2009;66(1):179-183.
5 Gennarelli TA, Meaney DF. Mechanisms of primary head injury Wilkins RH. Rengachary. SS. Neurosurgery New York: McGraw-Hill; 1996:2611-2622.
6 Graham DI, Adams JH, Nicoll JA, et al. The nature, distribution and causes of traumatic brain injury. Brain Pathol. 1995;5:397-406.
7 Seelig JM, Becker DP, Miller JD, et al. Traumatic acute subdural hematoma: Major mortality reduction in comatose patients treated within four hours. N Engl J Med. 1981;304:1511-1512.
8 Haselsberger K, Pucher R, Auer LM. Prognosis after acute subdural or epidural haemorrhage. Acta Neurochir (Wien). 1988;90:111-116.
9 Jamieson KG, Yelland JD. Extradural hematoma: Report of 167 cases. J Neurosurg. 1968;29:13-23.
10 Servadei F, Murray GD, Teasdale GM, et al. Traumatic subarachnoid hemorrhage: Demographic and clinical study of 750 patients from the European brain injury consortium survey of head injuries. Neurosurgery. 2002;50:261-267.
11 Armonda RA, Bell RS, Vo AH, Ling G, DeGraba TJ, Crandall B, et al. Wartime Traumatic Cerebral Vasospasm: Recent review of combat casualties. Neurosurgery. 2006;59:1215-1225.
12 Gurdjian ES. Cerebral contusions: Re-evaluation of the mechanism of their development. J Trauma. 1976;16:35-51.
13 Vernon DD, Woodward GA, Skjonsberg AK. Management of the patient with head injury during transport. Crit Care Clin. 1992;8:619-631.
14 Povlishock JT, Christman CW. The pathobiology of traumatically induced axonal injury in animals and humans: A review of current thoughts. J Neurotrauma. 1995;12:555-564.
15 Schweitzer JB, Dohan FCJr. Diffuse axonal injury: Windows for therapeutic intervention allowed by its pathobiology [editorial]. Virchows Arch A Pathol Anat Histopathol. 1993;423:153-156.
16 Andrews BT, Pitts LH. Functional recovery after traumatic transtentorial herniation. Neurosurgery. 1991;29:227-231.
17 Athiappan S, Muthukumar N, Srinivasan US. Influence of basal cisterns, midline shift and pathology on outcome in head injury. Ann Acad Med Singapore. 1993;22:452-455.
18 Miller JD, Becker DP, Ward JD, et al. Significance of intracranial hypertension in severe head injury. J Neurosurg. 1977;47:503-510.
19 Ikeda Y, Long DM. The molecular basis of brain injury and brain edema: The role of oxygen free radicals. Neurosurgery. 1990;27:1-11.
20 Kontos HA, Povlishock JT. Oxygen radicals in brain injury. Cent Nerv Sys Trauma. 1986;3:257-263.
21 Traystman RJ, Kirsch JR, Koehler RC. Oxygen radical mechanisms of brain injury following ischemia and reperfusion. J Appl Physiol. 1991;71:1185-1195.
22 Katayama Y, Becker DP, Tamura T, Ikezaki K. Early cellular swelling in experimental traumatic brain injury: A phenomenon mediated by excitatory amino acids. Acta Neurochir Suppl (Wien). 1990;51:271-273.
23 Nilsson P, Laursen H, Hillered L, Hansen AJ. Calcium movements in traumatic brain injury: The role of glutamate receptor-operated ion channels. J Cereb Blood Flow Metab. 1996;16:262-270.
24 Taupin V, Toulmond S, Serrano A, et al. Increase in IL-6, IL-1 and TNF levels in rat brain following traumatic lesion: Influence of pre- and post-traumatic treatment with Ro5 4864, a peripheral-type (p site) benzodiazepine ligand. J Neuroimmunol. 1993;42:177-185.
25 Woodroofe MN, Sarna GS, Wadhwa M, et al. Detection of interleukin-1 and interleukin-6 in adult rat brain, following mechanical injury, by in vivo microdialysis: Evidence of a role for microglia in cytokine production. J Neuroimmunol. 1991;33:227-236.
26 Rostworowski M, Balasingam V, Chabot S, et al. Astrogliosis in the neonatal and adult murine brain post-trauma: Elevation of inflammatory cytokines and the lack of requirement for endogenous interferon-gamma. J Neurosci. 1997;17:3664-3674.
27 Katayama Y, Becker DP, Tamura T, Hovda DA. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg. 1990;73:889-900.
28 Fineman I, Hovda DA, Smith M, et al. Concussive brain injury is associated with a prolonged accumulation of calcium: A 45Ca autoradiographic study. Brain Res. 1993;624:94-102.
29 Shapira Y, Yadid G, Cotev S, Shohami E. Accumulation of calcium in the brain following head trauma. Neurol Res. 1989;11:169-172.
30 Tymianski M, Tator CH. Normal and abnormal calcium homeostasis in neurons: A basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery. 1996;38:1176-1195.
31 DeWitt DS, Kong DL, Lyeth BG, et al. Experimental traumatic brain injury elevates brain prostaglandin E2 and thromboxane B2 levels in rats. J Neurotrauma. 1988;5:303-313.
32 Dhillon HS, Carbary T, Dose J, et al. Activation of phosphatidylinositol bisphosphate signal transduction pathway after experimental brain injury: A lipid study. Brain Res. 1995;698:100-106.
33 Kiwak KJ, Moskowitz MA, Levine L. Leukotriene production in gerbil brain after ischemic insult, subarachnoid hemorrhage, and concussive injury. J Neurosurg. 1985;62:865-869.
34 Goodman JC, Robertson CS, Grossman RG, Narayan RK. Elevation of tumor necrosis factor in head injury. J Neuroimmunol. 1990;30:213-217.
35 McClain C, Cohen D, Phillips R, et al. Increased plasma and ventricular fluid interleukin-6 levels in patients with head injury. J Lab Clin Med. 1991;118:225-231.
36 Young AB, Ott LG, Beard D, et al. The acute-phase response of the brain-injured patient. J Neurosurg. 1988;69:375-380.
37 Schoettle RJ, Kochanek PM, Magaree MJ, et al. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats. J Neurotrauma. 1990;7:207-217.
38 Takahashi H, Manaka S, Sano K. Changes in extracellular potassium concentration in cortex and brain stem during the acute phase of experimental closed head injury. J Neurosurg. 1981;55:708-717.
39 Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101-148.
40 Kimelberg HK, Biddlecome S, Bourke RS. SITS-inhibitable Cl– transport and Na+-dependent H+ production in primary astroglial cultures. Brain Res. 1979;173:111-124.
41 Kimelberg HK. Current concepts of brain edema: Review of laboratory investigations. J Neurosurg. 1995;83:1051-1059.
42 Sugaya E, Takato M, Noda Y. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J Neurophysiol. 1975;38:822-841.
43 Hertz L, Dittmann L, Mandel P. K+ induced stimulation of oxygen uptake in cultured cerebral glial cells. Brain Res. 1973;60:517-520.
44 Siesjo BD. Cell damage in the brain: A speculative synthesis. J Cereb Blood Flow Metab. 1981;1:155-185.
45 Aikawa JK. Biochemistry and physiology of magnesium [review]. World Rev Nutr Diet. 1978;28:112-142.
46 Aikawa JK. Magnesium. West J Med. 1980;133:333-334.
47 Vink R, McIntosh TK. Pharmacological and physiological effects of magnesium on experimental traumatic brain injury. Magnes Res. 1990;3:163-169.
48 Winchell RJ, Hoyt DB. Endotracheal intubation in the field improves survival in patients with severe head injury: Trauma Research and Education Foundation of San Diego. Arch Surg. 1997;132:592-597.
49 Sasada MP, Gabbott DA. The role of the laryngeal mask airway in pre-hospital care. Resuscitation. 1994;28:97-102.
50 Chesnut RM, Marshall SB, Piek J, et al. Early and late systemic hypotension as a frequent and fundamental source of cerebral ischemia following severe brain injury in the Traumatic Coma Data Bank. Acta Neurochir Suppl (Wien). 1993;59:121-125.
51 Anderson JT, Wisner DH, Sullivan PE, et al. Initial small-volume hyper-tonic resuscitation of shock and brain injury: Short- and long-term effects. J Trauma. 1997;42:592-600.
52 Doyle JA, Davis DP, Hoyt DB. The use of hypertonic saline in the treatment of traumatic brain injury [review]. J Trauma. 2001;50:367-383.
53 Shackford SR, Bourguignon PR, Wald SL, et al. Hypertonic saline resuscitation of patients with head injury: A prospective, randomized clinical trial. J Trauma. 1998;44:50-58.
54 Simma B, Burger R, Falk M, et al. A prospective, randomized, and controlled study of fluid management in children with severe head injury: Lactated Ringer’s solution versus hypertonic saline. Crit Care Med. 1998;26:1265-1270.
55 Diliberti T, Lindsey RW. Evaluation of the cervical spine in the emergency setting: Who does not need an x-ray? Orthopedics. 1992;15:179-183.
56 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Trauma systems [review]. J Neurotrauma. 2000;17:457-462.
57 Haas B, Jurkovich GJ, Wang J, Rivara FP, Mackenzie EJ, Nathens AB. Survival advantage in trauma centers: expeditious intervention or experience? J Am Coll Surg. 2009;208(1):28-36.
58 Subcommittee on Advanced Trauma Life Support of the American College of Surgeons Committee on Trauma. Advanced Trauma Life Support. Chicago: American College of Surgeons; 1993.
59 Teasdale G, Jennett B. Assessment of coma and impaired consciousness: A practical scale. Lancet. 1974;2:81-84.
60 Cohen MJ, Brohi K, Ganter MT, Manley GT, Mackensie RC, Pittet JF. Early coagulopathy after traumatic brain injury: the role of hypoperfusion and the Protein C pathway. J Trauma. 2007;63(6):1254-1262.
61 Brown CVR, Foulkrod KH, Lopez D, et al. Recombinant factor VIIa for the correction of coagulopathy before emergent craniotomy in blunt trauma patients. J Trauma. 2010;68(2):348-352.
62 Servadei F, Nasi MT, Cremonini AM, et al. Importance of a reliable admission Glasgow coma scale score for determining the need for evacuation of posttraumatic subdural hematomas: A prospective study of 65 patients. J Trauma. 1998;44:868-873.
63 Narayan RK, Kishore PR, Becker DP, et al. Intracranial pressure: To monitor or not to monitor? A review of our experience with severe head injury. J Neurosurg. 1982;56:650-659.
64 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Indications for intracranial pressure monitoring [review]. J Neurotrauma. 2007;24S:S37-S44.
65 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Recommendations for intracranial pressure monitoring technology [review]. J Neurotrauma. 2007;24S:S45-S54.
66 Clark WC, Muhlbauer MS, Lowrey R, et al. Complications of intracranial pressure monitoring in trauma patients. Neurosurgery. 1989;25:20-24.
67 Rosner MJ, Rosner SD, Johnson AH. Cerebral perfusion pressure: Management protocol and clinical results. J Neurosurg. 1995;83:949-962.
68 Stiefel M, Spiotta A, Gracias V, et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxygen. J Neurosurg. 2005;103:805-811.
69 Johnston J, Steiner L, Chatfield D, et al. Effect of cerebral perfusion pressure augmentation with dopamine and norepinephrine on global and focal brain oxygenation after traumatic brain injury. Intensive Care Med. 2004;30(5):791-797.
70 Rosner MJ, Rosner SD. Cerebral perfusion pressure management of head injury. In: Avezaat CJJ, van Eijndhoven JHM, Maas AIR, editors. Intracranial Pressure VIII. Berlin: Springer-Verlag; 1993:540-543.
71 Robertson CS, Valadka AB, Hannay HJ, et al. Prevention of secondary ischemic insults after severe head injury. Crit Care Med. 1999;27:2086-2095.
72 Kiening KL, Hartl R, Unterberg AW, et al. Brain tissue pO2-monitoring in comatose patients: Implications for therapy. Neurol Res. 1997;19:233-240.
73 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Intracranial pressure treatment threshold [review]. J Neurotrauma. 2007;24S:S55-S58.
74 Kaufman AM, Cardozo E. Aggravation of vasogenic edema by multiple dose mannitol. J Neurosurg. 1992;77:584-589.
75 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Hyperosmolar therapy [review]. J Neurotrauma. 2007;24S:S14-S20.
76 Marion DW, Puccio A, Wisniewski SR, et al. Effect of hyperventilation on extracellular concentrations of glutamate, lactate, pyruvate, and local cerebral blood flow in patients with severe traumatic brain injury. Crit Care Med. 2002;30:2619-2625.
77 Muizelaar JP, Marmarou A, Ward JD, et al. Adverse effects of prolonged hyperventilation in patients with severe head injury: A randomized clinical trial. J Neurosurg. 1991;75:731-739.
78 Roberts I. Barbiturates for acute traumatic brain injury. Cochrane Database Syst Rev 2000;(2):CD000033.
79 Schreckinger M, Marion DW. Contemporary management of traumatic intracranial hypertension: is there a role for therapeutic hypothermia? Neurocrit Care. 2009.
80 Marion D, Bullock RW. Current and future role of therapeutic hypothermia. J Neurotrauma. 2009;26(3):455-467.
81 Coplin WM, Cullen NK, Policherla PN, et al. Safety and feasibility of craniectomy with duraplasty as the initial surgical intervention for severe traumatic brain injury. J Trauma. 2001;50:1050-1059.
82 Guerra WKW, Piek J, Gaab MR. Decompressive craniectomy to treat intracranial hypertension in head injury patients. Intensive Care Med. 1999;25:1327-1329.
83 Kunze E, Meixensberger J, Janka M, et al. Decompressive craniectomy in patients with uncontrollable intracranial hypertension. Acta Neurochir Suppl (Wien). 1998;71:16-18.
84 Litofsky NS, Chin LS, Tang G, et al. The use of lobectomy in the management of severe closed-head trauma. Neurosurgery. 1994;34:628-632.
85 Munch E, Horn P, Schurer L, et al. Management of severe traumatic brain injury by decompressive craniectomy. Neurosurgery. 2000;47:315-322.
86 Thompson HJ, Kirkness CJ, Mitchell PH. Intensive care management of fever following traumatic brain injury. Intensive Crit Care Nurs. 2007;23:91-96.
87 Busto R, Dietrich WD, Globus MTT, et al. Small differences in intra-ischemic brain temperature critically determine the extent of ischemic neuronal injury. J Cereb Blood Flow Metab. 1987;7:729-738.
88 Baena RC, Busto R, Dietrich WD, et al. Hyperthermia delayed by 24 hours aggravates neuronal damage in rat hippocampus following global ischemia. Neurology. 1997;48:768-773.
89 Henker RA, Brown SD, Marion DW. Comparison of brain temperature with bladder and rectal temperatures in adults with severe head injury. Neurosurgery. 1998;42:1071-1075.
90 Rossi S, Zanier E, Mauri I, Columbo A, Stochetti N. Brain temperature, core temperature and intracranial pressure in acute cerebral damage. J Neurol Neurosurg Psychiatry. 2001;71(4):448-454.
91 McIlvoy L. The impact of hypothermia and hyperthermia on acute brain injury. AACN Clin Issues. 2005;16(4):488-500.
92 Kim J, Gearhart MM, Zurick A, et al. Preliminary report on the safety of heparin for deep venous thrombosis prophylaxis after severe head injury. J Trauma. 2002;53:38-42.
93 Norwood SH, McAuley CE, Berne JD, et al. Prospective evaluation of the safety of enoxaparin prophylaxis for venous thromboembolism in patients with intracranial hemorrhagic injuries. Arch Surg. 2002;137:696-701.
94 Reiff DA, Haricharan RN, Bullington NM, Griffin RL, McGwin GJr, Rue LW3rd. brain injury is associated with the development of deep vein thrombosis independent of pharmacological prophylaxis. J Trauma. 2009;66(5):1436-1440.
95 Young B, Ott L, Norton J, et al. Metabolic and nutritional sequelae in the non-steroid treated head injury patient. Neurosurgery. 1985;17:784-791.
96 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Nutrition [review]. J Neurotrauma. 2007;24S:S77-S82.
97 Jeremitsky E, Omert L, Dunham C, Wilberger J, Rodriguez A. The impact of hyperglycemia on patients with severe brain injury. J Trauma. 2005;58(1):47-50.
98 Van Beek J, Mushkudianai N, Steyerberg E, et al. Prognostic value of admission laboratory parameters in traumatic brain injury: Results from the IMPACT Study. J Neurotrauma. 2007;24(2):315-328.
99 Vespa P, Boonyaputthikul R, McArthur DL, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. 2006;34:850-856.
100 Oddo M, Schmidt JM, Carrera E, et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36:3233-3238.
101 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Antiseizure prophylaxis [review]. J Neurotrauma. 2007;24S:S87-S90.
102 Ronne-Engstrom E, Winkler T. Continuous EEG monitoring in patients with traumatic brain injury reveals a high incidence of epileptiform activity. Acta Neurol Scand. 2006;114(1):47-53.
103 Meythaler JM, Brunner RC, Johnson A, Novack TA. Amantadine to improve neurorecovery in traumatic brain injury—associated diffuse axonal injury: A pilot double-blind randomized trial. J Head Trauma Rehabil. 2002;17:300-313.
104 Arfanakis K, Haughton VM, Carew JD, et al. Diffusion tensor MR imaging in diffuse axonal injury. Am J Neuroradiol. 2002;23:794-802.
105 Johnston KM, Ptito A, Chankowsky J, Chen JK. New frontiers in diagnostic imaging in concussive head injury. Clin J Sport Med. 2001;11:166-175.
106 Stein SC, Ross SE. Moderate head injury: A guide to initial management. J Neurosurg. 1992;77:562-564.
107 Powell JW, Barber-Foss KD. Traumatic brain injury in high school athletes. JAMA. 1999;282:958-963.
108 Saunders RL, Harbaugh RE. The second impact in catastrophic contact-sport head trauma. JAMA. 1984;252:538-539.
109 Kelly JP, Nichols JS, Filley CM, et al. Concussion in sports: Guidelines for the prevention of catastrophic outcome. JAMA. 1991;266:2867-2869.
110 Cantu RC. Return to play guidelines after a head injury. Clin Sports Med. 1998;17:45-60.
111 Goodman D, Gaetz M. Return-to-play guidelines after concussion: The message is getting through. Clin J Sport Med. 2002;12:265.
112 Wilberger JE. Traumatic brain injury in athletes. In: Marion DW, editor. Traumatic Brain Injury. New York: Thieme Medical Publishers; 1999:165-172.
113 Maroon JC, Field M, Lovell M, et al. The evaluation of athletes with cerebral concussion. Clin Neurosurg. 2002;49:319-332.
114 Willer B, Leddy JJ. Management of concussion and post-concussion syndrome. Curr Treat Options Neurol. 2006;8(5):415-426.
115 Ben Yishay Y, Diller L. Cognitive remediation in traumatic brain injury: Update and issues. Arch Phys Med Rehabil. 1993;74:204-213.
116 MRC CRASH Trial CollaboratorsPerel P, Arango M, Clayton T, Edwards P, et al. Predicting outcome after traumatic brain injury: practical prognostic models based on large cohort of international patients. Br Med J. 2008;336:425-429.
117 Marshall LF, Gautille T, Klauber MR, et al. The outcome of severe closed head injury. J Neurosurg. 1991;75:S28-S36.
118 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Age [review]. J Neurotrauma. 2000;17:573-581.
119 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Glasgow coma scale score [review]. J Neurotrauma. 2000;17:563-571.
120 Braakman R. Early prediction of outcome in severe head injury. Acta Neurochir (Wien). 1992;116:161-163.
121 Narayan RK, Greenberg RP, Miller JD, et al. Improved confidence of outcome prediction in severe head injury. J Neurosurg. 1981;54:751-762.
122 Marmarou A, Anderson RL, Ward JD, et al. Impact of ICP instability and hypotension on outcome in patients with severe head trauma. J Neurosurg. 1991;75:S59-S66.
123 Dent DL, Croce MA, Menke PG, et al. Prognostic factors after acute subdural hematoma. J Trauma. 1995;39:36-42.
124 Marshall LF, Marshall SB, Klauber MR, Clark M. A new classification of head injury based on computerized tomography. J Neurosurg. 1991;75(Suppl):S14-S20.
125 Siccardi D, Cavaliere R, Pau A, et al. Penetrating craniocerebral missile injuries in civilians: A retrospective analysis of 314 cases. Surg Neurol. 1991;35:455-460.
126 Aldrich EF, Eisenberg HM, Saydjari C, et al. Predictors of mortality in severely head-injured patients with civilian gunshot wounds: A report from the NIH Traumatic Coma Data Bank. Surg Neurol. 1992;38:418-423.
127 [No authors listed]. Part 2. Prognosis in penetrating brain injury. J Trauma. 2001;51(2 Suppl):S44-S86.
128 Neuroimaging in the management of penetrating brain injury [review]. J Trauma. 2001;51:S-11.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
38 Traumatic Brain Injury
It is estimated that 3.2 million people are living with long-term disability related to traumatic brain injury (TBI).1 In addition to the personal toll, the direct and indirect costs of these disabilities are estimated to exceed $60 billion annually.2 Americans sustain an estimated 1.6 million TBIs each year. Approximately 290,000 require hospitalization, and 51,000 die of their injuries.3 However, the true incidence of TBI is unknown because current surveillance methodologies do not capture those treated in non-hospital settings (e.g., primary care office) or those who do not seek treatment at all.
TBI death rates in the United States fell during the 1980s. A substantial decline in motor vehicle–related fatalities was primarily responsible. At the same time, the incidence of gunshot wounds to the head rose, and in 1990, firearms surpassed motor vehicle crashes as the single largest cause of death due to TBI in some urban areas. However, in-hospital mortality rose to 8% during the last decade, presumably because of the increase in hospitalization rates of severe and moderate TBI.4
Pathophysiology
Primary Injury
Injury to the brain is caused by external forces to the head that strain the tissue beyond its structural tolerance.5 These forces can be classified as contact or inertial.6 Contact forces typically produce focal injuries such as skull fractures, contusions, and epidural or subdural hematomas. Inertial forces result from the brain undergoing acceleration or deceleration (translational, rotational, or both) and can occur without head impact. Inertial forces can cause focal or diffuse brain injuries: pure translational acceleration leads to focal injuries such as contrecoup contusions, intracerebral hematomas, and subdural hematomas, whereas rotational or angular acceleration, common with high-speed motor vehicle crashes, usually causes diffuse injuries. Although external signs of head injury such as scalp abrasions, lacerations, and hematomas are common with blunt-force trauma, the brain can also be severely injured solely by inertial forces, without evidence of scalp or facial injuries.
Skull fracture results from a contact force to the head that is usually severe enough to cause at least brief loss of consciousness. Linear fractures are the most common type of skull fracture and typically occur over the lateral convexities of the skull. Most often, they are nondisplaced cracks in the skull (linear fractures), but a particularly intense impact can cause a gap (diastasis) between the edges of the fracture. A depressed skull fracture, in which skull fragments are pushed into the cranial vault, usually results from blunt force by an object with a relatively small surface area, such as a hammer (Figure 38-1). The base of the skull can be fractured by severe blunt trauma to the forehead or the occiput. Basilar skull fractures are most common in the anterior skull base and often involve the cribriform plate, disrupting the olfactory nerves (Figure 38-2). Posterior basilar skull fractures may extend through the petrous bone and internal auditory canal, thereby damaging the acoustic and the facial nerves.
Figure 38-1 Right frontal depressed skull fracture caused by an assault with a hammer (axial CT scan, bone window).
Common posttraumatic intracranial lesions are hemorrhage (epidural, subdural, and intraparenchymal), contusion, and diffuse brain injury. Subdural hematomas are seen in 20% to 25% of all comatose victims of TBI (Figure 38-3). They develop between the surface of the brain and the inner surface of the dura and are believed to result from the tearing of bridging veins over the cortical surface or from disruption of major venous sinuses or their tributaries. The hematoma typically spreads over most of the cerebral convexity; the dural reflections of the falx cerebri prevent expansion to the contralateral hemisphere. Swelling of the cerebral hemisphere is common in those with subdural hematomas, given the associated damage to underlying brain tissue. Underlying cerebral contusions were found in 67% of patients with subdural hematomas in one series.7 Subdural hematomas are classified as acute, subacute, or chronic, each having a characteristic appearance on computed tomography (CT): acute hematomas are bright white, subacute lesions are isodense with brain tissue and are therefore often overlooked, and chronic hematomas are hypodense relative to the brain.
Epidural hematomas develop between the inner table of the skull and the dura, usually when the middle meningeal artery or one of its branches is torn by a skull fracture. They occur in 8% to 10% of those rendered comatose by TBI.8,9 The majority of epidural hematomas are located in the temporal or parietal regions, but they can also occur over the frontal or occipital lobes and (rarely) in the posterior fossa. They appear as hyperdense mass lesions on CT. Unlike subdural hematomas, their spread is limited by the suture lines of the skull, where the dura is very adherent. Because an epidural space normally does not exist, the clot must strip the dura from the inner table of the skull as it enlarges, resulting in its classic biconvex or lenticular shape (Figure 38-4). Epidural hematomas are uncommon in infants and toddlers, presumably because their skulls are more deformable and less likely to fracture, and in TBI victims older than 60 years, because the dura is extremely adherent to the skull.
Traumatic subarachnoid hemorrhage often results from tearing of the corticomeningeal vessels. Though common after severe TBI, subarachnoid hemorrhage does not produce a hematoma or mass effect.10 However, it may be associated with an increased risk for posttraumatic vasospasm, which may adversely affect clinical outcome.11
Contusions are heterogeneous lesions comprising punctate hemorrhage, edema, and necrosis and are often associated with other intracranial lesions. One or more contusions occur in 20% to 25% of patients with severe TBI. Because they evolve over time, contusions may not be evident on the initial CT scan or may appear as small areas of punctate hyperdensities (hemorrhages) with surrounding hypodensity (edema) (Figure 38-5). Local neuronal damage and hemorrhage lead to edema that may expand over the next 24 to 48 hours. With time, contusions may coalesce and look more like intracerebral hematomas. Depending on their size and location, they may cause significant mass effect, resulting in midline shift, subfalcine herniation, or transtentorial herniation. Contusions are most common in the inferior frontal cortex and the anterior temporal lobes,12 where the surface of the inner table of the skull is very irregular; they result from shifting of the brain over this irregular surface at the time of impact. Direct blunt-force trauma to the head can produce a contusion in the tissue underlying the point of impact (coup contusion). If the head was in motion upon collision with a rigid surface, a contusion may occur in the brain contralateral to the point of impact (contrecoup contusion).
Diffuse axonal injury refers to lacerations or punctate contusions at the interface between the gray and white matter. Such punctate contusions are thought to result from the disparate densities of the gray and white matter and the consequent difference in centripetal force associated with a rotational vector of injury.13 Thus, diffuse axonal injury most often occurs after a high-speed motor vehicle crash, during which severe angular and rotational forces are applied to the head. Diffuse axonal injury was once thought to result solely from mechanical disruption at the time of impact; however, more recent research has identified cases in which the histologic footprints of diffuse axonal injury, such as fragmentation of axons and axonal swelling, do not appear until 24 to 48 hours after the incident, suggesting that some cases are a secondary manifestation of trauma.14,15 Diffuse axonal injury is present in almost half of all patients with severe TBI and in a third of those who die, and it is a common cause of persistent vegetative or minimally conscious state.
Posttraumatic intracranial lesions cause neurologic dysfunction via direct and in some cases indirect mechanisms. By destroying brain tissue, contusions and intraparenchymal hemorrhage cause deficits directly related to the function of the damaged tissue. Uncal herniation is also an important mechanism of temporary or permanent neurologic deficits.16,17 Semirigid dural reflections divide the intracranial contents into compartments. The tentorium cerebelli separates the anterior and middle cranial fossae from the posterior cranial fossa. The brainstem, specifically the midbrain, traverses an opening, the tentorial foramen, in the anterior central portion of this partition. The medial portion of the temporal lobe, the uncus, lies on both sides of the tentorial foramen. Because the most common TBIs, such as hematomas and contusions, are usually located over the lateral surfaces of the brain, and because the brain’s extreme lateral surface is the rigid skull, such lesions tend to depress the brain medially. Therefore, a subdural hematoma over the surface of the temporal lobe or a hemorrhagic contusion of the temporal lobe itself is likely to displace the medial portion of the temporal lobe (uncus) into the tentorial foramen (i.e., uncal herniation). Such displacement compresses the midbrain, which contains neurons that are part of the reticular activating system. At the base of the midbrain is the crus cerebri, which contains pyramidal fibers from the cortex, and the third cranial nerve, which exits the midbrain through the interpeduncular cistern. Midbrain compression due to uncal herniation damages the reticular activating system, causing loss of consciousness; stretches the third cranial nerve and its associated parasympathetic fibers, causing pupil dilatation and loss of the light reflex; and injures the pyramidal fibers in the crus cerebri, causing abnormal posturing responses in the contralateral arm and leg.
Intracranial hypertension is a major cause of posttraumatic neurologic morbidity and mortality.18 The intracranial pressure (ICP) is defined by the volume of CSF, blood, and brain tissue in the cranial vault. The volume of these components is dynamic, and the brain can accommodate moderate changes in any of the three. For example, the blood volume can rise or fall by as much as 30% to 40%, CSF absorption can increase to reduce the size of the ventricles by up to 90%, and brain tissue itself is compressible. Thus, the intracranial volume can gain 100 to 150 mL, equivalent to a moderate-sized subdural hematoma, without the ICP increasing significantly. When these buffering mechanisms have been exhausted, however, even a small increase in the size of a hematoma will cause a rapid rise in ICP. If appropriate treatment is delayed, the ICP may approach the mean arterial pressure (MAP), causing a hydrostatic block of blood flow to the brain and brain death. Intracranial hypertension, particularly if refractory to medical or surgical treatment, is the most common cause of death after severe TBI.
Secondary Injury
Posttraumatic ischemia initiates a cascade of metabolic events that lead to the surplus production of oxygen free radicals,19–21 excitatory amino acids,22,23 cytokines,24,25 and other inflammatory agents.26 Glutamate and aspartate are the excitatory amino acids most commonly implicated in excitotoxic injury,27 which is mediated by activation of N-methyl-D-aspartate, α-amino-3-hydroxy-5-methylisoazole-4-proprionic acid, or kainic acid receptors.23 Overactivation of these receptors causes an excessive influx of ionized calcium into the cytosol, and elevated amounts of ionized intracellular calcium play a key role in neurodegeneration after injury to the central nervous system (CNS).28,29 In addition, posttraumatic nonischemic events such as an increase in intracellular free Ca++ via receptor-gated or voltage-dependent ion channels induce the release of oxygen free radicals from mitochondria.30 Excessive levels of highly reactive oxygen free radicals cause lipid peroxidation of cell membranes, oxidation of intracellular proteins and nucleic acids, and activation of phospholipases A2 and C, which hydrolyze membrane phospholipids, thereby releasing arachidonic acid. The liberation of arachidonic acid triggers the generation of free fatty acids, leukotrienes, and thromboxane B2, all of which are associated with neurodegeneration and poor outcome after experimental TBI.31–33 Inflammatory cytokines, particularly interleukin (IL)-1, IL-6, and tumor necrosis factor, also are overproduced after TBI.34–36 In animal models, posttraumatic activation of microglia is a principal source of these cytokines.25 IL-1 and IL-6 provoke an exuberant cellular inflammatory response believed to be responsible for astrogliosis, edema, and tissue destruction.26,37
TBI also increases extracellular potassium levels,38 leading to an imbalance of intracellular and extracellular K+, disruption of the Na+/K+-ATPase cell membrane regulatory mechanisms, and subsequent cell swelling.39,40 Astrocyte swelling has been attributed to the clearance of excessive extracellular K+.41 High levels of extracellular K+ have also been implicated as the cause of widespread neuronal depolarization and spreading depression seen after experimental TBI.27,38,42 Moreover, potassium stimulates increased oxygen uptake in glial cells, potentially depriving adjacent neurons of oxygen.43,44 Severe TBI also causes a substantial decrease in extracellular magnesium (Mg++) levels, thereby impairing normal glycolysis, cellular respiration, oxidative phosphorylation, and the biosyntheses of DNA, RNA, and protein.45–47 Because Mg++ competes with Ca++ at voltage-gated cell membrane–associated Ca++ channels, reduced levels of Mg++ will result in an abnormal influx of Ca++ into the cell.
Prehospital Care
The acutely injured brain is vulnerable to further damage from systemic hypotension, cerebral hypoperfusion, hypercarbia, hypoxemia, and elevated ICP. Preventing these physiologic insults is crucial to limiting secondary brain injury. Care of the TBI victim always should begin with evaluating and securing a patent airway and restoring normal breathing and circulation. Early endotracheal intubation usually benefits comatose patients. Securing and maintaining an airway are essential to optimal oxygenation and ventilation, and early intubation has been found to reduce mortality after severe TBI.48
The airway is usually most easily and safely secured by orotracheal intubation, a method in which most emergency medical personnel are trained and experienced. Patients with severe maxillofacial trauma may require nasotracheal intubation, but this is less desirable because it is a relatively blind procedure. The nasal passageways can be irritated, causing blood pressure (BP) and ICP to surge, and in those with severe anterior skull base fractures, the tube can inadvertently be passed into the brain. A third alternative for securing the airway is the laryngeal mask airway, an easily learned and rapidly applied device that has undergone successful field trials.49 However, it does not protect against aspiration and cannot be used to achieve high airway pressures. A surgical airway (cricothyroidotomy) should be performed only after other attempts to secure an airway have failed, and only by an experienced provider.
The patient should be sedated and pharmacologically paralyzed before intubation, because irritation of the oropharynx typically causes transient hypertension, tachycardia, increased ICP, and agitation that can interfere with the procedure. Fentanyl, a short-acting opioid agonist that produces analgesia and sedation, is the most commonly used sedative. The usual dose is 3 to 5 µg/kg body weight, administered intravenously (IV) 3 minutes before intubation. Etomidate, an alternative to opioids, provides adequate sedation and is less likely to cause hypotension. Some prefer thiopental because it is an ultra-short-acting barbiturate and is thus less likely to conceal the neurologic status when the patient reaches the trauma center; however, it is more likely than other agents to cause hypotension. Neuromuscular blocking agents commonly used for tracheal intubation include succinylcholine (1.5 mg/kg IV), which has the advantages of rapid onset, complete reliability, and very short duration of action. This last attribute is particularly important in the prehospital setting, where attempts at intubation sometimes fail. Vecuronium (0.01 mg/kg IV) is an alternative that offers the theoretical advantage of being a nondepolarizing muscle relaxant. Because it has a relatively long duration of action (1 to 2 hours), it is less forgiving of failed intubation attempts. Table 38-1 shows a recommended rapid-sequence intubation pathway.
TABLE 38-1 Recommended Rapid-Sequence Induction for Severely Head-Injured Patients
Rapid fluid resuscitation and restoration of a normal BP are critical in the prehospital setting, because hypotension has been associated with doubling of the mortality rate after severe TBI.50 The most likely cause of hypotension is hemorrhage, usually in the abdomen or chest; therefore, hypovolemia should be assumed. Lactated Ringer’s or normal saline solutions should be infused through a large-bore IV catheter as quickly as possible until normotension is achieved. Although preclinical studies suggest that hypertonic saline may be more effective than isotonic solutions for rapid volume resuscitation,51,52 results of several small clinical trials have not been convincing.53,54
In all cases of severe TBI, defined as a Glasgow Coma Scale (GCS) score of 3 to 8 and an inability to follow commands, patients should be treated as if they have a spinal fracture until an adequate examination of the spine proves otherwise. Among those who survive long enough to reach the emergency department, the likelihood of a cervical spine fracture is 2% to 7%. More troubling, however, is that an estimated 10% to 25% of all posttraumatic spinal cord injuries are iatrogenic, occurring during transport to the hospital.55 After respiratory and hemodynamic stabilization, the patient should be placed in a neutral position on a flat, hard surface. If the patient requires immediate tracheal intubation, it should be performed while another person provides in-line cervical spine immobilization. A rigid cervical spine collar should be placed as soon as possible. Next, the patient should be placed on a backboard; the cervical spine can then be further immobilized with a buttress of foam or towels placed on both sides of the head. To prevent any movement during transport, the patient should be strapped to the board in several locations.
The organization of emergency medical services and regional trauma programs has improved outcomes for victims of trauma, particularly those with severe TBI.56 A very large prospective study of the cost and outcomes associated with trauma center designation found more than a 25% reduction in in-hospital mortality for those with severe TBI who were initially treated at a level I trauma center compared to similarly injured patients treated at hospitals of similar size that were not designated trauma centers.57 This is likely because designation as a level I or II trauma center by the American College of Surgeons Committee on Trauma or a state health department ensures the availability of immediate neurosurgical care when the patient arrives. Therefore, every effort should be made to transport severely injured patients directly to a designated trauma center. Nonetheless, if an adequate airway or venous access cannot be obtained in the field, some patients may need to undergo respiratory or hemodynamic stabilization at a nearby emergency department en route to the trauma center. Once hemodynamic and airway stability is achieved, immediate transport to a designated trauma center should occur without delays for imaging or secondary surveys.
Emergency Department Care
Upon arrival at the trauma center, the emergency medical personnel should concisely report their prehospital assessment and management, including mechanism of injury, stabilizing maneuvers, medications given, initial vital signs, GCS score, and hemodynamic stability during transport. A thorough physical and radiographic examination to identify all life-threatening injuries should then be performed. Most trauma centers follow the Advanced Trauma Life Support protocol, a comprehensive routine that has proved successful in quickly detecting all major injuries.58 First the airway is reassessed, and the need for tracheal intubation is carefully reconsidered. For patients intubated in the field, proper placement of the tracheal tube is verified both clinically and radiographically. When the airway is secure and adequate oxygenation is confirmed using a percutaneous oxygen saturation monitor or arterial blood gas analysis, two large-bore IV catheters are inserted to provide sufficient venous access for high-volume fluid resuscitation. An isotonic saline solution is infused to continue volume replacement, which probably began at the scene. Any life-threatening injuries such as overt hemorrhage, tension pneumothorax, or cardiac tamponade should be treated immediately upon discovery. A brief neurologic examination is performed, including assessment of the GCS score (Table 38-2), pupillary size and reaction to light, and symmetry and extent of extremity movements. The head is palpated to detect fractures, lacerations, or penetrating wounds, and lacerations are probed gently to ascertain the presence of a depressed skull fracture or foreign body. Large lacerations are compressed with pressure dressings or temporarily sutured to prevent further hemorrhage. Careful inspection of the head should reveal hemotympanum, periorbital or mastoid ecchymosis, and CSF rhinorrhea or otorrhea.
| Response | Points |
|---|---|
| Speech | |
| Alert, oriented, and conversant | 5 |
| Confused, disoriented, but conversant | 4 |
| Intelligible words, not conversant | 3 |
| Unintelligible sounds | 2 |
| No verbalization, even with painful stimulus | 1 |
| Eye Opening | |
| Spontaneous | 4 |
| To verbal stimuli | 3 |
| To painful stimuli | 2 |
| None, even with painful stimuli | 1 |
| Motor | |
| Follows commands | 6 |
| Localizes painful stimulus | 5 |
| Withdraws from painful stimulus | 4 |
| Flexor posturing with central pain | 3 |
| Extensor posturing with central pain | 2 |
| No response to painful stimulus | 1 |
Data from Teasdale G, Jennett B. Assessment of coma and impaired consciousness: a practical scale. Lancet. 1974;2(7872):81-84.
Coagulopathy resulting from TBI is thought to occur when hypoperfusion causes activation of the protein C pathway, thereby inducing alterations in the clotting cascade.60 It is also commonly seen as a result of therapeutic anticoagulation with warfarin, especially in the geriatric population. Rapid identification and correction of coagulopathy is critical to prevent expansion of intracranial hematomas and allow surgical intervention. Fresh frozen plasma and cryoprecipitate can be used to correct the INR to 1.3 or less. Recombinant factor VIIa has been shown to decrease blood product requirements and costs associated with correction of coagulopathy.61 Other studies demonstrate more rapid correction times compared to blood product administration, although differences in outcomes have not been significant. Product expense and concern for thromboembolic complications have limited the routine use of recombinant factor VIIa.
Definitive Treatment
Critical to determining the severity of the brain injury and appropriate treatment are CT findings combined with a reliable postresuscitation GCS score and assessment of pupil size and reactivity. In the case of an acute subdural hematoma, for example, a patient with a moderate-sized lesion who has normal pupil size and reactivity and is able to follow commands might safely be treated nonoperatively. Conversely, surgery is unlikely to benefit an elderly patient with fixed and dilated pupils and a GCS score of 3 or 4, regardless of the CT findings. Other determining factors include size and location of the hematoma, presence and extent of an underlying contusion or brain swelling, and results of the neurologic examination. Neurologic deterioration, particularly a decline in mental status, suggests enlargement of the hematoma, and a new CT scan should be obtained promptly. Hematomas less than 10 mm thick that cause a midline shift of less than 5 mm can usually be observed, especially if they do not involve the middle cranial fossa.62 If nonoperative management is chosen for an intracranial hematoma, the patient should be monitored with frequent neurologic assessments in the intensive care unit (ICU). If the patient cannot follow commands, ICP monitoring is recommended.
[/not-level-membership-for-critical-care-medicine-category]
