Chapter 50B Trauma of the Nervous System
Craniocerebral Trauma
Classification
Traumatic brain injury is classified by mechanism, severity, and morphology (Table 50B.1).
Mechanism
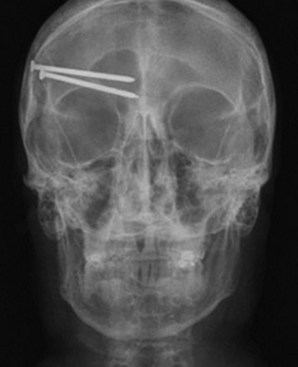
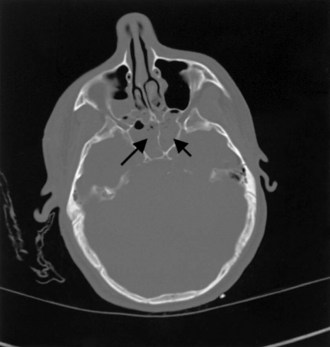
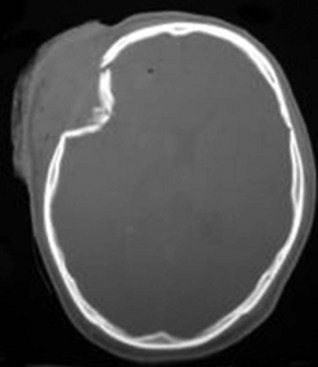
Gunshot wounds are the most prevalent penetrating head injuries (Fig. 50B.1), accounting for 35% of U.S. deaths from TBI in individuals younger than age 45. Self-inflicted injuries—for example, with power tools like nail guns (Fig. 50B.2)—are another cause of craniocerebral trauma (Testerman and Dacks, 2007), but gunshot wounds are the most lethal type of TBI, 90% of them resulting in death. They cause soft-tissue damage, often comminuted depressed skull fractures, and direct injury to the brain tissue from the missile. Beyond the laceration along the bullet path, further damage is done by shock waves, especially with high-velocity weapons. Depending on the type of penetrating injury, contamination may be a concern. However, bullet fragments are considered sterile owing to their exposure to heat from the firearm and are not routinely removed.
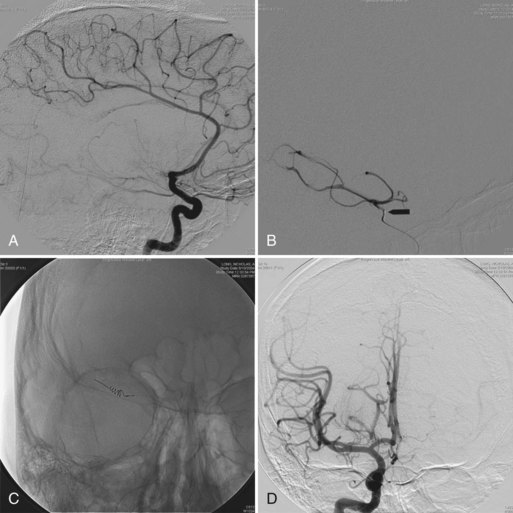
Vascular injury can occur with any type of TBI but is more common with penetrating trauma (25%–36% incidence) than with blunt injuries (<1%). Traumatic aneurysms, or pseudoaneurysms, can develop when all layers of the vessel wall rupture and surrounding cerebral tissue forms the aneurysmal wall. Such pseudoaneurysms can present as delayed subarachnoid hemorrhage (SAH) and can lead to death or severe disability. Therefore patients with penetrating injuries routinely undergo a cerebral angiogram to screen for traumatic pseudoaneurysm. A follow-up angiogram within the first months after a penetrating injury is recommended (du Trevou and van Dellen, 1992).
Blast is a rare mechanism of injury in civilian life, but it is common in combat. With ongoing militarily conflicts, blast injury has become more frequent in U.S. military personnel. American military members who returned from deployment to Iraq or Afghanistan self-reported a 12% to 20% incidence of mild TBI; explosions were recognized as a common injury mechanism (Wolf et al., 2009). Blast injuries can occur (1) as a direct result of supersonic waves of intense air (primary blast injury), (2) from being struck by objects set in motion by the blast (secondary blast injury), and (3) when a person is forcefully propelled by the blast (tertiary blast injury) (DePalma et al., 2005; Kocsis and Tessler, 2009). The brain is obviously vulnerable to secondary and tertiary blast injury, but whether primary blast forces directly injure the brain is controversial (Taber et al., 2006). The severity of blast exposure necessary to cause persistent symptoms is unclear (Hicks et al., 2010).
The severity of injury resulting from exposure to a blast can range from mild to fatal. Displacement, stretching, and shearing forces of the primary blast wave can affect the brain directly and lead to such sequelae as concussion, hemorrhage, severe edema, or diffuse axonal injury (DAI). Systemic acute air embolism from pulmonary disruption is believed to occlude the blood vessels of the brain or spinal cord and lead to stroke (Guy et al., 2000; Wolf et al., 2009). Reports from war zones suggest that brain swelling occurs within hours after a blast injury, and that the risk of mortality can be decreased by early decompressive craniectomy. Others have found that cerebral vasospasm occurred more often after a blast injury than with blunt TBI and led to a worse outcome than blunt severe TBI. Blast exposure on the mild end of the spectrum led to somatic, behavioral, psychological, and cognitive symptoms similar to postconcussion syndrome. There is well-recognized overlap between posttraumatic stress disorder and blast injury, and misdiagnosis can occur in both directions.
Injury Severity
There are different methods to stratify TBI by severity, and all are arbitrary. The most commonly used is the Glasgow Coma Scale (GCS), which was first published in 1974 by Graham Teasdale and Bryan K. Jennett, two neurosurgeons at the University of Glasgow (Teasdale and Jennett, 1974). The GCS assesses level of consciousness. It consist of three subscores: eye opening (maximal score: 4), verbal response (maximal score: 5), and motor response (maximal score: 6). Despite some shortcomings, it has excellent interrater agreement (weighted kappa >0.75) for verbal and total scores and intermediate agreement (weighted kappa 0.4-0.75) for motor and eye scores (Holdgate et al., 2006). Today it has been widely adopted by emergency medicine services and physicians to communicate the severity of injury in a consistent way. However, it is not designed to detect focal neurological deficits and is not a replacement for a thorough neurological examination.
A GCS score of 15 to 13 is considered to indicate mild TBI, and a score of 8 or below is a universally accepted definition of coma and severe head injury. Scores between 13 and 8 are classified as moderate. It is true that moderate and severe TBI are rare; mild TBI is eight times more common. Moderate and severe TBI are seen equally often. One can imagine that the GCS score shows significant ceiling effect in patients with mild TBI. Therefore, more sensitive measures exist for this purpose. The two most often used are the Cantu system (Cantu, 1996) and the American Academy of Neurology system (1997).
Despite its high incidence, no objective test to diagnose mild TBI currently exists. Only one of ten head CT scans has a positive finding in mild TBI patients. Therefore, special factors need to be considered when determining the need of a CT scan (Box 50B.1). Diagnosis is hampered by the lack of obvious injuries on computed tomography (CT) or conventional magnetic resonance imaging (MRI) and usually is based solely on clinical symptoms. Preliminary studies are investigating brain injury biomarkers to diagnose mild TBI (Dash et al., 2010; Hergenroeder et al., 2008; Kochanek et al., 2008). Although the initial progress has been promising, these markers are not yet used in the clinical setting (Springborg et al., 2009; Unden and Romner, 2009). Others have shown that an integrated approach with magnetoencephalography and diffusion tensor imaging (DTI) is more sensitive than conventional CT and MRI in detecting subtle neuronal injury in mild TBI and postconcussion syndrome (Huang et al., 2009).
Box 50B.1 Factors to Consider When Determining Need of CT in Patients with Head Injury
CT, Computed tomography; GCS, Glasgow Coma Scale; LOC, loss of consciousness.
A concussion is defined as injury to the brain caused by a hard blow or violent shaking, producing a sudden and temporary impairment of brain function such as a brief loss of consciousness or disturbance of vision and equilibrium. Concussions are equivalent to a mild TBI with negative CT findings. Their annual incidence in the United States is 128 per 100,000 population (Ropper and Gorson, 2007). The loss of consciousness is believed to result from rotational forces exerted on the upper midbrain and thalamus, impairing the function of the reticular neurons. Headache, nausea, dizziness, irritability, and impaired ability to concentrate can persist for days after the event. Persistence of these symptoms for weeks is called postconcussion syndrome and can last from 1 month to a year.
Patients with severe TBI (GCS score ≤ 8) often have injuries in at least one other organ system (Saul and Ducker, 1982). There is also a 5% incidence of associated spine fractures. About one-fourth of patients with a severe TBI undergo neurosurgical intervention. Early diagnosis of TBI improves outcome by reducing secondary injury, which can develop subsequent to the impact and includes edema, hypoxia, hypotension, ischemia, and elevated intracranial pressure (ICP) (Chesnut et al., 1993a; Chesnut et al., 1993b).
Morphology
Traumatic brain injury can also be classified by the underlying morphology and can be subdivided as injuries to the skull itself and intracranial injuries, which can be focal or diffuse (see Table 50B.1).
Skull Fractures
There are two types of skull fractures. Linear fractures traverse the full thickness of the skull from the outer to inner table, are usually fairly straight, and involve no bone displacement. The common method of injury is blunt force trauma in which the energy from the blow is transferred over a wide surface area of the skull. Fractures can be located in the calvarium or in the skull base. Skull base fractures are associated with cranial nerve (CN) palsy. Depending on their location, CN VII (temporal bone) and CN VI (clivus) are at risk; they are also prone to causing cerebrospinal fluid (CSF) leaks (Fig. 50B.3). In depressed skull fractures, the method of injury is similar to linear skull fractures, but in this case the inner and outer table are more shattered, and pieces are displaced into the brain (Fig. 50B.4). They may or may not lacerate the dura. The presence of skull fractures consistently has been associated with a higher incidence of intracranial lesions, neurological deficits, and poorer outcome (Bullock et al., 2006a). Hence, cranial fractures are important indicators of intracranial injuries, and CT scanning should be performed to rule out serious intracranial pathology in all patients with a known or suspected cranial fracture. A large study of patients with a TBI found that 71% of 850 patients with a cranial fracture had an intracranial lesion (e.g., contusion, hematoma), compared with only 46% of 533 patients without a cranial fracture (Macpherson et al., 1990).
Intracranial Injuries
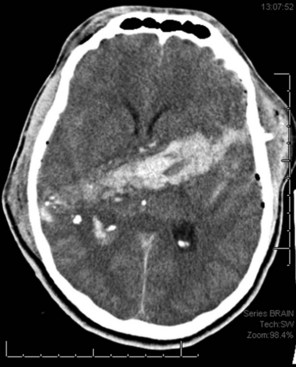
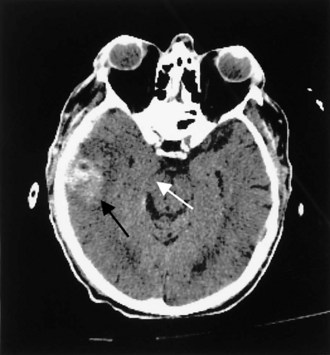
Contusions occur when the brain moves within the skull enough to collide with the skull, causing bruising of the brain parenchyma (hemorrhage and edema). The most common locations are the frontal and temporal poles, because these areas most often strike the bony surfaces of the skull base. Often, a second injury (contrecoup) is seen diagonally across the site of the direct impact (coup injury) when the accelerated brain strikes the skull on the opposite site. In contrast to concussion, contusions are visible on a head CT scan. Edema surrounding a contusion has lower signal intensity than brain on CT scan (Fig. 50B.5). Hemorrhages have higher CT signal intensity and are commonly seen as multiple bright areas of variable size. Progression of contusions is common, with delayed hemorrhage occurring over the first 24 hours in 25% of cases. However, neurosurgical intervention is rare.
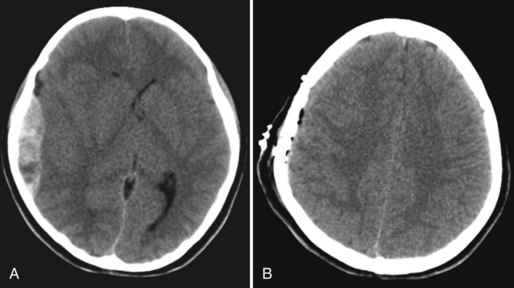
An epidural hematoma (EDH) is bleeding between the dura and the skull. The peak incidence of EDH occurs in the second decade, and the mean age of TBI victims with EDH is between 20 and 30 years of age. Traffic-related accidents, falls, and assaults account for 53% of EDH in adults; falls account for half of EDH in children. Most (70%–90%) EDH are associated with a skull fracture and bleeding from a lacerated artery or indirect bone bleeding. Arterial bleeding was identified as the source of EDH in 36% of adults in a systematic review. Rupture of the middle meningeal vein, diploic veins, or venous sinuses also can cause EDH. The most common location of EDH is temporal; because the temporal bone is thinner than the rest of the skull, it breaks more easily, often resulting in a laceration of the middle meningeal artery (Bullock et al., 2006b) (Fig. 50B.6).
Subdural hematoma (SDH) develops between the brain parenchyma and the dura (Fig. 50B.7). It occurs when the brain moves within the skull enough to tear a surface vein, also called a bridging vein, that runs from the brain surface to the dural venous sinus. The most common locations are the frontal and parietal convexities. On a CT scan, an acute SDH typically appears as a bright (hyperdense), extraaxial, crescent-shaped homogeneous collection of fluid that conforms to the cerebral surface. Unlike an EDH, its spread is not limited by suture lines; it can spread over the whole convexity, but it almost never crosses the midline. An SDH can be acute or chronic. Herein, acute SDH is defined as an SDH diagnosed within 14 days of TBI. Chronic SDH contain blood older than 14 days or blood of different ages. This accounts for their appearance. They may be of mixed density or isodense to gray matter. In these cases, it can be identified by its mass effects, including sulcal effacement, inward buckling of the gray/white interface, and presence of midline shift.
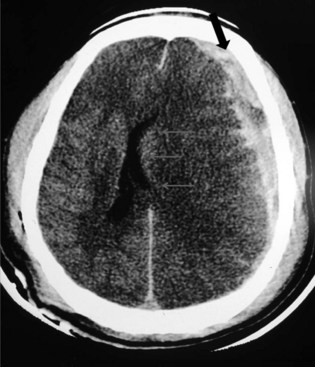
In studies conducted after the introduction of CT, the incidence of SDH was estimated to be 11% of all patients with TBI (Servadei et al., 2000). The most common injuries involving traumatic SDH are motor vehicle collisions in the younger population and falls in those older than 75 years. Between 37% and 80% of patients with acute SDH present with an initial GCS score of 8 or less, and dilated pupils are seen preoperatively in 50% of these patients. Mortality among patients who arrive at the hospital in a coma and undergo surgical evacuation is between 57% and 68%.
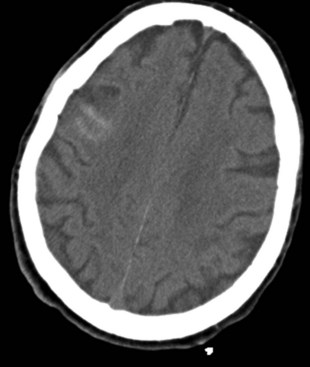
Traumatic subarachnoid hemorrhage (tSAH), seen with mild and severe TBI alike, refers to microhemorrhages into the subarachnoid space from crushed or ruptured smaller vessels. CT scans show hyperdense linear areas in superficial brain sulci (Fig. 50B.8). In contrast to aneurysmal SAH, the blood is superficial in the cortex and not present in the basal cisterns. In some instances, for example, if blood is found in the sylvian fissure, a vascular study (CT angiography, four-vessel digital subtraction angiography) is needed to rule out an aneurysm rupture. Most of the time, traumatic SAH indicates a more severe underlying brain injury and is seen in conjunction with DAI, SDH, or EDH. It has been shown that the presence of traumatic SAH is an independent factor for poor outcome. This finding could reflect a more extensive underlying injury that CT scan or conventional MRI cannot detect, or it could be attributable to a less recognized phenomenon: posttraumatic vasospasm. The incidence of posttraumatic vasospasm in patients with a mean GCS score of 7 (range 3-15) is similar to that following aneurysmal SAH (see Chapter 51C). Posttraumatic vasospasm is often overlooked, but one study found that hemodynamically significant vasospasm has been found in 44% of patients with tSAH (Oertel et al., 2005). Patients were at highest risk of developing hemodynamically significant vasospasm on day 3. Younger patients and patients with GCS scores of 8 or less were more likely to develop posttraumatic vasospasm.
Diffuse axonal injury is very common severe head injuries and is associated with significant morbidity. It is characterized clinically by rapid progression to coma in the absence of specific focal lesions. Adam classified DAI as mild, moderate, or severe (Adams et al., 1989). The duration of coma after a TBI correlates with the severity of DAI. Patients who survive the most severe form rapidly lapse into coma and remain unconscious, vegetative, or severely disabled.
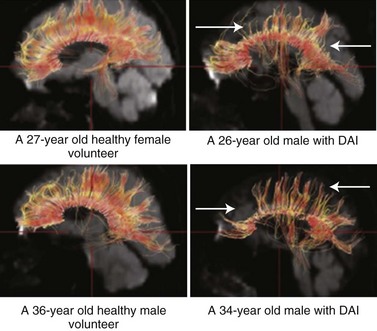
MRI is more sensitive than CT in detecting DAI. Gradient echo MRI is most sensitive to areas of hemorrhage, and fluid-attenuated inversion recovery images are best for visualizing nonhemorrhagic lesions. Diffusion tensor imaging is a new imaging technology more sensitive to DAI than conventional MRI. With DTI, the connectivity and integrity of neuronal tracts can be investigated. DTI is helpful when imaging tissue with an internal structure such as neural axons in white matter of the brain. Water molecules will then diffuse more rapidly in the direction aligned with the internal structure, and more slowly as it moves perpendicular to the preferred direction. With this information, injury to the white matter (diffusion anisotropy) can be detected and neural tracts followed through the brain (Fig. 50B.9). DTI measurements correlate with injury severity (acute GCS score) and outcome (Rankin Score at discharge) (Li and Feng, 2009; Sugiyama et al., 2007).
Pathophysiology
Following primary brain injury, a cascade of cellular and biochemical events occur in the initial minutes and extend into the subsequent days, leading to secondary neuronal degeneration (secondary brain injury) and ultimately neuronal impairment or cell death. Much of the research concerning TBI is directed at affecting the mechanisms of secondary brain injury. These mechanisms include free radical species, excitatory amino acid toxicity, hypoxia, impaired cerebrovascular autoregulation, hyperthermia, blood-brain barrier breakdown, and hypercalcemia. Hypoxia and hypoperfusion are recognized as leading contributors to secondary brain injury. A single hypotensive episode in which systolic blood pressure falls below 90 mm Hg has been associated with worse outcomes after severe TBI (Chesnut et al., 1993a). All these factors contribute to brain swelling, which leads to increased ICP, further exacerbating secondary injury.
Cerebrovascular dysregulation, often seen after TBI, is thought to contribute to secondary brain ischemia. One study found that cerebral blood flow (CBF) that was decreased (<18 mL/100 g/min) in the initial 4 to 12 hours after injury was associated with poor outcome (Piek et al., 1992). Increased CSF levels of endothelin-1 also are implicated in cerebrovascular dysregulation. Elevated CBF is often seen, especially in children and younger trauma patients. An increase in cerebral perfusion pressure (CPP) can be detrimental for such patients. Multimodal monitoring with a CBF probe, cerebral oximetry, or transcranial Doppler can help identify these patients so that elevated ICP can be treated appropriately.
After a TBI, excitotoxicity occurs upon the release of excessive amounts of excitatory amino acids such as glutamate, resulting in neuronal injury. Excitotoxic damage occurs in two phases: sodium-dependent neuronal swelling, followed by delayed calcium-dependent neuronal degeneration. These effects are mediated through activation of N-methyl-d-aspartate (NMDA) and glutamate receptors, leading to a rise in the intracellular calcium-mediated activation of proteases and lipases, which facilitates neuronal degeneration and necrotic cell death. In contrast to necrotic cell death, apoptosis or programmed cell death is marked not by swelling and dissolution of cell membranes but rather by DNA fragmentation and the formation of apoptotic cell bodies associated with neuronal shrinkage. Apoptosis is a cellular event triggered by either intrinsic mechanisms (initiated in the mitochondria) or extrinsic mechanisms (tumor necrosis factor [TNF] superfamily of cell-surface death receptors) which activate a cascade of enzymes called caspases that lead to cell termination. Apoptosis is thought to contribute to secondary neuronal injury after a TBI event. Animal studies have shown that developing neurons are more susceptible than mature neurons to excitotoxic injury, probably because more calcium is transmitted via the NMDA-mediated calcium channel in the immature brain (Geddes-Klein et al., 2006). However, although the administration of NMDA antagonists following TBI in immature rats led to decreased excitotoxic-mediated neuronal death, apoptotic cell death increased. The role of excitotoxicity and apoptosis following trauma to the developing brain warrants further investigation.
Studies of CSF support a role for inflammation after TBI. Interleukin (IL)-6, IL-8 (Minambres et al., 2003; Gopcevic et al., 2007), and soluble adhesion molecules (i.e., sICAM-1 [Otto et al., 2000]) were increased in the CSF following TBI. Whether these inflammatory mediators contribute to neurodegeneration or neuroprotection remains to be determined.
Treatment
Evaluation
Early intervention is essential in successful TBI care; as in any emergency situation, the CABs (circulation, airway, breathing) apply. Another priority of prehospital care is to optimize perfusion and oxygenation to prevent hypoxia and hypertension. Once in the emergency room, further measures can be taken if necessary to achieve adequate cardiopulmonary function. Next, a neurological examination is performed to triage patients accordingly. The GCS score is widely used to convey the severity of TBI, but a low GCS score upon arrival in the emergency room may be the result of sedation in the field. Only after the patient is weaned from sedation can injury severity be accurately assessed. Trauma patients with altered mental status, pupillary asymmetry, and flexion or extension posturing are at high risk for SDH or EDH compressing the brain and brainstem and must be evaluated with a CT scan of the head. CT is central to acute TBI diagnosis. There are two externally validated rules for when to require a CT scan after mild TBI: the Canadian CT Head Rule (CCTHR) (Stiell et al., 2001) and the New Orleans Criteria (NOC) (Haydel et al., 2000). These rules have 100% sensitivity for neurosurgical lesions and 83% to 98% sensitivity for nonoperative lesions. The CCTHR has a greater specificity and hence has led to fewer CT scans than the NOC (Levine, 2010). Neither addresses the presence of coagulopathy and anticoagulation, which others deem important risk factors for intracranial hemorrhage (Cohen et al., 2006; Ivascu et al., 2008). These criteria were updated in 2007 by the World Health Organization Taskforce on mild TBI and the Neurotraumatology Committee of the World Federation of Neurosurgical Societies (Fabbri et al., 2004) and in 2008 by the Centers for Disease Control and Prevention and the American College of Emergency Physicians. All in all, research findings agree on several indications for urgent CT scanning of the head after a minor TBI (see Table 50B.1).
Management of Diffuse Injuries
Patients with complicated mild TBI (positive CT findings) and moderate TBI are admitted to the intensive care unit (ICU) and followed with serial neurological examinations. Although intracranial complications are uncommon after a mild TBI, in 6% to 12% of patients, CT shows abnormalities (Ibanez et al., 2004; Smits et al., 2005) in the form of a contusion, tSAH, SDH, or EDH. Another CT scan obtained within 12 to 24 hours is currently the standard of care in many hospitals. However, studies of 1 million emergency department visits for mild TBI found that neurosurgical intervention was required in only 0.13% to 0.3% of these patients (Fabbri et al., 2004; Haydel et al., 2000; Jeret et al., 1993; Miller et al., 1996; Nagy et al., 1999). Over the past several years, evidence has suggested that the routine use of follow-up CT scans in patients with complicated mild TBI may not be necessary (Fabbri et al., 2008; Huynh et al., 2006; Nagy et al., 1999; Sifri et al., 2006; Velmahos et al., 2006). Most of these cases are not operative, and the patient can be discharged home after a short observation period (Thiruppathy and Muthukumar, 2004). The only exception is an EDH, which can present with a lucid interval after a brief loss of consciousness and then progress to coma and death (Levine, 2010).
Concussions are graded according to the severity of symptoms: presence or absence of loss of consciousness and time (< or > 15 minutes) for resolution of symptoms (Table 50B.2). Data to guide treatment of concussion are lacking. Clinical experts suggest a benefit from the use of mild analgesics for headache, avoidance of narcotics, and use of meclizine or promethazine and vestibular exercise for dizziness. Brief seizures may occur immediately after the initial impact and easily can be mistaken as a sign of a severe head injury.
| Grade | Cantu System | AAN System |
|---|---|---|
| 1 (Mild) | A. PTA <30 min | A. Transient confusion |
| B. No LOC | B. No LOC | |
| C. Symptoms resolved in <15 min | ||
| 2 (Moderate) | A. LOC <5 min or B. PTA >30 min |
As above, but symptoms last >15 min (still no LOC) (PTA is common) |
| 3 (Severe) | A. LOC >5 min or B. PTA >24 hrs |
Any LOC, whether brief (seconds) or prolonged |
AAN, American Association of Neurology; LOC, Loss of consciousness; PTA, posttraumatic amnesia.
Because 10% of all head injuries are sports related, return to play after concussion is an important issue. The American Academy of Neurology (1997), the Canadian Academy of Sports Medicine (2000), and several international symposia (McCrory et al., 2009) have developed guidelines for return to play after TBI, based mostly on expert opinion. Because no consensus has been reached regarding the most appropriate set of guidelines, the return-to-play decision remains in the realm of clinical judgment on an individualized basis. All guidelines agree that a symptomatic player should never return to play because of the potentially increased vulnerability of the brain to a second injury. This sensitivity may outlast the symptoms, which tend to resolve within 1 week. Evidence suggests that after a concussion, neuronal metabolic equilibrium and oxygen consumption are abnormal for several months. This suggests an initial and rapid phase of functional recovery driven by compensatory mechanisms, followed by a prolonged neuronal recovery period during which subtle deficits in brain functioning are present but not apparent to standard clinical assessment tools (Ellemberg et al., 2009). Depending on the grade of the concussion, the athlete may resume play within 1 week or 1 month (Mayers, 2008) (Table 50B.3). An objective measure of postural stability increases the sensitivity to ongoing neurological impairment in the return-to-play decision-making process and minimizes the consequences of mitigating factors such as practice effects and motivation. During recovery in the first few days after a concussion, it is important to emphasize to the athlete that physical and cognitive rest are required.
Table 50B.3 Guidelines for the Management of Sports-Related Concussion*
| Symptoms | First Concussion | Second Concussion |
|---|---|---|
| Grade 1: No loss of consciousness, transient confusion, resolution of symptoms and mental abnormalities in <15 min† | Remove from play | Allow return to play after 1 wk if there are no symptoms at rest or with exertion |
| Examine at 5-min intervals | ||
| May return to play if symptoms disappear and results of mental function examination return to normal within 15 min | ||
| Grade 2: As above, but with mental symptoms for >15 min | Remove from play and disallow play for rest of day | Allow return to play after 2-wk period of no symptoms at rest or with exertion |
| Examine for signs of intracranial lesion at sidelines and obtain further examination by a trained person on same day | Remove from play for season if imaging shows abnormality | |
| Allow return to play after 1 wk if neurologic examination normal | ||
| Grade 3: Any loss of consciousness | Perform thorough neurologic examination in hospital and obtain imaging studies when indicated | Withhold from play until symptoms have been absent for at least 1 mo |
| Assess neurologic status daily until postconcussive symptoms resolve or stabilize | ||
| Remove from play for 1 wk if loss of consciousness lasts seconds; for 2 wks if it lasts minutes; must be asymptomatic at rest and with exertion to return to play |
* These guidelines reflect consensus opinion, are not evidence-based, and are under revision. Adapted from the American Academy of Neurology guidelines.
† Testing includes orientation, repetition of digit strings, recall of word list at 0 and 5 minutes, recall of recent game events, recall of current events, pupillary symmetry, finger-to-nose and tandem gait tests, Romberg test, and provocative testing for symptoms with a 4-yd (3.5-m) sprint, 5 push-ups, and 5 knee bends.
Management of nonoperative severe TBI is directed by the evidence-based Guidelines for the Management of Severe Traumatic Brain Injury. The most recent (third) edition was published in 2007 by the Brain Trauma Foundation (Povlishock and Bullock, 2007) (Table 50B.4). With severe TBI, ICP-guided and/or CCP-guided therapy are the fundamental principles underlying all treatment. Patients with a GCS score of 8 or below qualify for ICP monitoring. The gold standard remains the external ventricular drain (EVD), which allows both ICP measurement and CSF drainage to reduce ICP. Other more expensive options are fiberoptic or microstrain gauge devices, which are placed in the brain parenchyma. These devices cannot be recalibrated during monitoring and have a negligible drift. Subarachnoid, subdural, and epidural monitors measure ICP via a fluid-coupled or pneumatic mechanism and are less accurate than an EVD.
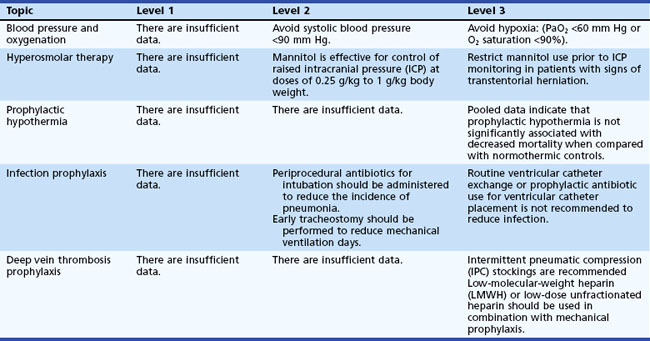
An escalating protocol should be followed to control ICP (Box 50B.2), using sedation, hyperosmolar therapy in the form of mannitol and hypertonic saline, and hyperventilation. The sequence in which these treatment options are chosen varies from hospital to hospital. Currently, at my institution, patients with increased ICP initially receive a 250-mL hypertonic 3% saline bolus and continuous 3% saline infusion if needed. Mannitol is reserved for an emergency situation such as an acutely dilated pupil and imminent brain herniation. Serum sodium can be elevated safely to 155 mEq/L and a serum osmolality of 320 mOsm/kg. Beyond those levels, renal failure can develop. If hypertonic saline administration is stopped suddenly, rebound intracranial hypertension is a risk but can be avoided by tapering the hypertonic saline solution over 48 hours.
Box 50B.2 Escalating Protocol for Treatment of Intracranial Pressure Above 20 mm Hg for 10 Minutes


























ICP that remains elevated despite maximal medical efforts is an indication for decompressive craniectomy or bilateral frontal craniectomy. Depending on the underlying pathology, one side of the skull or the frontal bone bilaterally (Kjellberg procedure) is removed to give the swollen brain more room. To date, the effectiveness of decompressive craniectomy is controversial. Findings from non-randomized trials and controlled trials with historical controls, however, suggest that decompressive craniectomy may be a useful option for intractable ICP. The results of a trial published in 2011 did not show improved functional outcome after decompressive hemicraniectomy (Cooper et al., 2011). However, the results of this trial need to be interpreted with caution because of concerning design flaws. Nonetheless, decompressive craniectomy is not without risk. One study showed a 35% rate of complications that included subdural effusion, hydrocephalus, and infection. The incidence of posttraumatic hydrocephalus is higher in patients after decompressive craniectomy. This maybe due to disturbance of CSF dynamics after decompressive craniectomy. Bilateral decompressive craniectomy is sometimes performed but is associated with an unfavorable outcome in 46% of cases (Bao et al., 2010). Some patients suffer delayed neurological deterioration, described as “syndrome of the trephined.” It is thought that that brain function is impaired by the atmospheric pressure.
Hypothermia is an alternative therapy that should be reserved for patients with persistent intracranial hypertension refractory to other medical or surgical interventions. A recent meta-analysis of all prospective TBI clinical trials found that therapeutic moderate hypothermia (32°C–34°C [89.6°C–93.2°F]) resulted in less mortality (RR 0.51; 95% CI, 0.33, 0.79) and increased likelihood of a good outcome (RR 1.91; 95% CI, 1.28, 2.85) compared with normothermia during acute care, but the risk of pneumonia increased (RR 2.37; 95% CI, 1.37, 4.10) (Peterson et al., 2008). Problems associated with hypothermia include increased bleeding risk, arrhythmias, and increased susceptibility to infection and sepsis.
The ability to limit secondary brain injury after a severe TBI relies on ensuring the delivery of an adequate supply of oxygen and metabolic substrate to the brain and to detect it as early as possible. Once the ICP rises, it may be too late to intervene, so measuring PbtO2 directly should detect compromise in oxygen delivery earlier than relying on ICP alone. Several studies have investigated the relationship between outcome and focal PbtO2, which can be measured with the Licox monitor (Stiefel et al., 2004; Narotam et al., 2006). They found that likelihood of death increased with duration of time PbtO2 was less than 15 mm Hg, prompting the question, Does oxygen-directed therapy improve outcome?, which has not been shown so far. The effect of hyperoxemia after TBI are also unclear. While mild hyperoxemia is beneficial in TBI patients, a study of 3420 patients found worse outcomes with extreme hyperoxemia (O2 487 mm Hg). The detrimental effects of hyperoxemia have been demonstrated in patients with ischemic brain injury not due to TBI. Whether the same pathophysiological process of post-reperfusion hyperoxemia is harmful in TBI remains to be determined (Davis et al., 2009).
The metabolic state of the brain can also be assessed with cerebral microdialysis, which monitors energy metabolites over extended periods but only in a small focal volume of the brain. The threshold for abnormal metabolism is evidenced by a lactate/pyruvate ratio of 40. Microdialysis in patients with severe TBI showed that tight systemic glucose control is associated with reduced cerebral extracellular glucose availability and increased prevalence of brain energy crisis (microdialysis glucose <0.7 mmol/L, with a lactate/pyruvate ratio > 40), which in turn correlates with increased mortality, so intensive insulin therapy may impair cerebral glucose metabolism after severe brain injury (Oddo et al., 2008). The combined use of microdialysis variables and PbtO2 in addition to ICP, mean arterial pressure (MAP), and CPP was found to have the best predictive accuracy for outcome after severe TBI (Low, 2009).
Hyperthermia following a severe TBI is common, potentiates secondary injury, and worsens neurological outcome. Induced normothermia (36.5°C–37.5°C) via an intravascular cooling catheter is effective prophylaxis to reduce fever burden (Puccio et al., 2009).
No evidence yet exists to guide the decision for blood transfusion (whether and when) after a severe TBI. Although intuitively it makes sense that increasing a patient’s hematocrit will lead to increased oxygen delivery to the brain, this benefit has not been clearly shown. Over recent years, evidence has emerged that blood transfusion may actually have a negative effect after TBI, even after controlling for anemia as a possible confounder (Carlson et al., 2006). Stored blood has decreased deformability and may in fact exacerbate ischemia.
Management of Focal Injuries
Between 22% and 50% of patients with EDH are comatose on admission or immediately before surgery. Additionally, 47% of patients with an EDH have a lucid interval—that is, they awaken after initial unconsciousness and then deteriorate neurologically, which may obscure the development of EDH. A patient with an EDH can go from normal neurological status to coma within minutes. Most often, the patient becomes agitated and restless and may have an episode of emesis because of the increased ICP. This is generally followed by focal neurological signs such as hemiparesis, seizures, pupillary dilation, decrease in consciousness, and in the worst-case scenario, decortication. Because of the high association between skull fractures and EDH, screening x-rays of the skull can help identify the risk for EDH. Guidelines for surgical management of acute EDH list the following criteria for surgical evacuation of an EDH (Bullock et al., 2006b):


The 2006 guidelines for surgical management of SDH give the following criteria for cases of SDH (Bullock et al., 2006c):
 An acute SDH thicker than 10 mm or causing a midline shift greater than 5 mm on CT scans should be surgically evacuated, regardless of the patient’s GCS score.
An acute SDH thicker than 10 mm or causing a midline shift greater than 5 mm on CT scans should be surgically evacuated, regardless of the patient’s GCS score.

 ICP monitoring should be performed in all comatose (GCS score ≤8) patients with an acute SDH.
ICP monitoring should be performed in all comatose (GCS score ≤8) patients with an acute SDH.
The GCS score, pupillary examination, comorbidities, CT findings, age, and ICP, influence the decision to evacuate an acute SDH. Neurological deterioration over time is common and also can influence the decision to operate. Although the time from neurological deterioration to surgery influences outcome, optimal timing of surgery is difficult to pinpoint, because most patients who undergo early surgery for SDH also have a more severe injury and worse outcome than patients undergoing delayed evacuation. The magnitude of preoperative midline shift also seems to correlate with unfavorable outcome (Nelson et al., 2010). Age is an independent predictor of outcome in patients with SDH; age older than 50 years is statistically correlated with poorer outcome. A study showed that at 3 months after SDH evacuation, patients between the ages of 18 and 30 years had 25% mortality, whereas those 50 years and older had 75% mortality (Hukkelhoven et al., 2003).
Late Complications
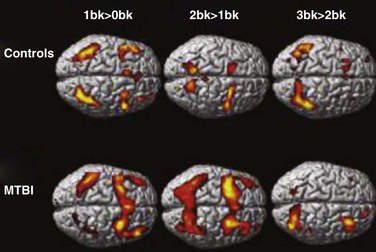
Postconcussion syndrome has an incidence of 4% to 59%. The entire spectrum of TBI severity is associated with a risk for psychiatric conditions and long-standing neurological deficits. Functional MRI has shown that working memory recruitment is different even 1 year after mild TBI, compared to healthy controls (McAllister et al., 2006) (Fig. 50B.10). Therefore, thorough neurocognitive assessment after these patients are released from acute care is imperative to offer TBI survivors the best chance for recovery. Approaches used to treat neurocognitive deficits have been cognitive and behavioral therapy with pharmacological treatment.
Outcomes
Outcome after severe TBI remains poor; approximately one-third die, and 25% survive with severe disabilities. A number of factors play an important role in predicting outcome. One study retrospectively analyzed 846 cases of severe TBI to clarify the prognostic effects of multiple factors (Jiang et al., 2002). One year after injury, GCS score, age, pupillary response and size, hypoxia, hyperthermia, and ICP were associated with outcome, indicating that prevention of hypoxia and hyperthermia and control of high ICP may be useful means to improve outcome of patients with severe head injury. A GCS score of 3 on presentation has been recognized as a poor prognostic factor. Mortality approaches 100% in the presence of bilateral fixed, dilated pupils.
Age by itself is an independent predictor of outcome. With or without surgery, outcome in the elderly is worse than in the young. Different age thresholds have been given in the literature. Depending on the statistical analysis performed, studies found worse outcomes in patients older than 39 and 65 years of age. Multiple regression analysis showed that above 39 years of age, every 10 years’ increase in age leads to a 10% increase in mortality (Kuhne et al., 2005). Even with timely and satisfactory surgery, unexplained clinical deterioration occurs in patients aged 70 years or older. For people 65 and older, falls are the leading cause of TBI-related death, accounting for 40% of all injury deaths in that age group. Falls can also cause other injuries such as hip fractures that can impede independent living and increase the risk of premature death (Cruise et al., 2006). An epidemiology study over 10 years showed that fall-related TBI increased by 126% and the related case fatality rate decreased from 32% to 18%, meaning that more elderly people are living with TBI disabilities (Fletcher et al., 2007). However, rehabilitation efforts in the elderly have often been poor because of negative attitudes (ageism) toward the recovery potential in this group. Despite their relative disadvantages, the elderly might see improved outcomes with rehabilitation programs intended for their age group.
Some CT findings can predict outcome. Accounting for the length, width, depth, and location of SDH and EDH; number, volume, and location of contusions; compression of ventricles and the basal cistern; and presence or absence of traumatic SAH, the most important CT-defined predictor of outcome is the magnitude of midline shift (Nelson et al., 2010).
Finally, the facility where patients with TBI are treated affects outcome. Patients with severe TBI treated in American College of Surgeons (ACS)-designated level 1 trauma centers have better survival rates and outcomes than those treated in ACS-designated level 2 centers (DuBose et al., 2008).
Future of Traumatic Brain Injury
An important but poorly understood feature of TBI it spatiotemporal progression or “blossoming” of a hemorrhagic contusion. Between 25% and 50% of traumatic hemorrhagic contusions and hematomas show evidence of progression during the acute postinjury period on serially obtained CT scans. The fact that it is possibly preventable makes it an attractive therapeutic target once the molecular mechanisms responsible for blossoming of hemorrhagic contusions are clarified (Simard et al., 2009).
Establishing brain injury proteins as biomarkers in TBI care could help with diagnosis, triage, management, and prognosis in the very heterogeneous population of TBI victims. Doing so also may help identify patients with TBI of similar severity so they can be allocated appropriately to TBI trials. The beta subunit of S100 protein (S100 beta) has the potential to serve as a good screening biomarker because of its high sensitivity for abnormal findings on CT scans of the head. Using S100 beta to determine whether patients with mild TBI should have a head CT was the first attempt to introduce biomarkers in clinical TBI care (Unden and Romner, 2009). An analysis showed that if blood test results require less time than imaging, and if head CT scan rates for patients with isolated mild TBI are relatively high, S100 beta will lower costs (Ruan et al., 2009). Other promissing biomakers for TBI are currently under investigation: ubiquitin C-terminal hydrolase-L1 (UCH-L1), and alpha II-spectrin breakdown products (Brophy et al., 2011; Mondello et al., 2010).
A safe and effective neuroprotective treatment for acute-stage TBI has yet to be found. The search for a “magic bullet” drug targeting a single receptor for the treatment of TBI has failed thus far for a variety of reasons. The pathophysiology of ischemic brain injury and TBI involves several mechanisms leading to neuronal injury, including excitotoxicity, free-radical damage, inflammation, necrosis, and apoptosis. In 2007, a randomized clinical trial of progesterone for acute TBI showed that intravenous administration of progesterone had no serious side effects and showed signs of benefit, especially in patients with moderate TBI (Wright et al., 2007). In 2009, a prospective, randomized, double-blind placebo-controlled clinical trial found a good safety profile of cyclosporin A infusion when given at the chosen dose of 5 mg/kg, infused over 24 hours, during the early phase after severe TBI (Mazzeo et al., 2009). Because of its ability to preserve mitochondrial integrity in experimental brain-injury models, cyclosporin A recently was proposed for human use after TBI to provide improved behavioral outcomes as well as significant histological protection.
Finally, supplemental magnesium has been considered because it positively affects many of the processes involved in secondary injury and consistently improves outcome in animal models of TBI. In 2007, however, a randomized controlled trial investigated the use of magnesium sulfate for neuroprotection after TBI and found that infusions of magnesium given to patients for 5 days beginning within 8 hours after a moderate or severe TBI were not neuroprotective and might even have a negative effect in the treatment of significant head injury (Temkin et al., 2007).
1997 Practice parameter: the management of concussion in sports (summary statement). Report of the Quality Standards Subcommittee. Neurology. 1997;48:581-585.
2000 Guidelines for assessment and management of sport-related concussion. Canadian Academy of Sport Medicine Concussion Committee. Clin J Sport Med. 2000;10:209-211.
Adams J.H., Doyle D., Ford I., et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49-59.
Bao Y.H., Liang Y.M., Gao G.Y., et al. Bilateral decompressive craniectomy for patients with malignant diffuse brain swelling after severe traumatic brain injury: a 37-case study. J Neurotrauma. 2010;27:341-347.
Brophy G.M., Mondello S., Papa L., et al. Biokinetic analysis of ubiquitin C-terminal hydrolase-L1 (UCH-L1) in severe traumatic brain injury patient biofluids. J Neurotrauma. 2011;28:861-870.
Bullock M.R., Chesnut R., Ghajar J., et al. Surgical management of depressed cranial fractures. Neurosurgery. 2006;58:S56-S60. discussion Si-iv
Bullock M.R., Chesnut R., Ghajar J., et al. Surgical management of acute epidural hematomas. Neurosurgery. 2006;58:S7-15. discussion Si-iv
Bullock M.R., Chesnut R., Ghajar J., et al. Surgical management of acute subdural hematomas. Neurosurgery. 2006;58:S16-S24. discussion Si-iv
Cantu R.C. Head injuries in sport. Br J Sports Med. 1996;30:289-296.
Carlson A.P., Schermer C.R., Lu S.W. Retrospective evaluation of anemia and transfusion in traumatic brain injury. J Trauma. 2006;61:567-571.
Chesnut R.M., Marshall L.F., Klauber M.R., et al. The role of secondary brain injury in determining outcome from severe head injury. J Trauma. 1993;34:216-222.
Chesnut R.M., Marshall S.B., Piek J., et al. Early and late systemic hypotension as a frequent and fundamental source of cerebral ischemia following severe brain injury in the Traumatic Coma Data Bank. Acta Neurochir Suppl (Wien). 1993;59:121-125.
Cohen D.B., Rinker C., Wilberger J.E. Traumatic brain injury in anticoagulated patients. J Trauma. 2006;60:553-557.
Cooper D.J., Rosenfeld J.V., Murray L., et al. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011;364:1493-1502.
Cruise C.M., Sasson N., Lee M.H. Rehabilitation outcomes in the older adult. Clin Geriatr Med. 2006;22:257-267. viii
Dash P.K., Zhao J., Hergenroeder G., et al. Biomarkers for the diagnosis, prognosis, and evaluation of treatment efficacy for traumatic brain injury. Neurotherapeutics. 2010;7:100-114.
Davis D.P., Meade W., Sise M.J., et al. Both hypoxemia and extreme hyperoxemia may be detrimental in patients with severe traumatic brain injury. J Neurotrauma. 2009;26:2217-2223.
DePalma R.G., Burris D.G., Champion H.R., et al. Blast injuries. N Engl J Med. 2005;352:1335-1342.
du Trevou M.D., van Dellen J.R. Penetrating stab wounds to the brain: the timing of angiography in patients presenting with the weapon already removed. Neurosurgery. 1992;31:905-911. discussion 911-902
DuBose J.J., Browder T., Inaba K., et al. Effect of trauma center designation on outcome in patients with severe traumatic brain injury. Arch Surg. 2008;143:1213-1217. discussion 1217
Ellemberg D., Henry L.C., Macciocchi S.N., et al. Advances in sport concussion assessment: from behavioral to brain imaging measures. J Neurotrauma. 2009;26:2365-2382.
Fabbri A., Servadei F., Marchesini G., et al. Prospective validation of a proposal for diagnosis and management of patients attending the emergency department for mild head injury. J Neurol Neurosurg Psychiatry. 2004;75:410-416.
Fabbri A., Servadei F., Marchesini G., et al. Observational approach to subjects with mild-to-moderate head injury and initial non-neurosurgical lesions. J Neurol Neurosurg Psychiatry. 2008;79:1180-1185.
Fletcher A.E., Khalid S., Mallonee S. The epidemiology of severe traumatic brain injury among persons 65 years of age and older in Oklahoma, 1992-2003. Brain Inj. 2007;21:691-699.
Geddes-Klein D.M., Serbest G., Mesfin M.N., et al. Pharmacologically induced calcium oscillations protect neurons from increases in cytosolic calcium after trauma. J Neurochem. 2006;97:462-474.
Gopcevic A., Mazul-Sunko B., Marout J., et al. Plasma interleukin-8 as a potential predictor of mortality in adult patients with severe traumatic brain injury. Tohoku J Exp Med. 2007;211:387-393.
Guy R.J., Glover M.A., Cripps N.P. Primary blast injury: pathophysiology and implications for treatment. Part III: injury to the central nervous system and the limbs. J R Nav Med Serv. 2000;86:27-31.
Haydel M.J., Preston C.A., Mills T.J., et al. Indications for computed tomography in patients with minor head injury. N Engl J Med. 2000;343:100-105.
Hergenroeder G., Redell J.B., Moore A.N., et al. Identification of serum biomarkers in brain-injured adults: potential for predicting elevated intracranial pressure. J Neurotrauma. 2008;25:79-93.
Hicks R.R., Fertig S.J., Desrocher R.E., et al. Neurological effects of blast injury. J Trauma. 2010;68:1257-1263.
Holdgate A., Ching N., Angonese L. Variability in agreement between physicians and nurses when measuring the Glasgow Coma Scale in the emergency department limits its clinical usefulness. Emerg Med Australas. 2006;18:379-384.
Huang M., Theilmann R.J., Robb A., et al. Integrated imaging approach with MEG and DTI to detect mild traumatic brain injury in military and civilian patients. J Neurotrauma. 2009;26:1213-1226.
Hukkelhoven C.W., Steyerberg E.W., Rampen A.J., et al. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99:666-673.
Huynh T., Jacobs D.G., Dix S., et al. Utility of neurosurgical consultation for mild traumatic brain injury. Am Surg. 2006;72:1162-1165. discussion 1166-1167
Ibanez J., Arikan F., Pedraza S., et al. Reliability of clinical guidelines in the detection of patients at risk following mild head injury: results of a prospective study. J Neurosurg. 2004;100:825-834.
Ivascu F.A., Howells G.A., Junn F.S., et al. Predictors of mortality in trauma patients with intracranial hemorrhage on preinjury aspirin or clopidogrel. J Trauma. 2008;65:785-788.
Jeret J.S., Mandell M., Anziska B., et al. Clinical predictors of abnormality disclosed by computed tomography after mild head trauma. Neurosurgery. 1993;32:9-15. discussion 15-16
Jiang J.Y., Gao G.Y., Li W.P., et al. Early indicators of prognosis in 846 cases of severe traumatic brain injury. J Neurotrauma. 2002;19:869-874.
Kochanek P.M., Berger R.P., Bayr H., et al. Biomarkers of primary and evolving damage in traumatic and ischemic brain injury: diagnosis, prognosis, probing mechanisms, and therapeutic decision making. Curr Opin Crit Care. 2008;14:135-141.
Kocsis J.D., Tessler A. Pathology of blast-related brain injury. J Rehabil Res Dev. 2009;46:667-672.
Kuhne C.A., Ruchholtz S., Kaiser G.M., et al. Mortality in severely injured elderly trauma patients—when does age become a risk factor? World J Surg. 2005;29:1476-1482.
Levine Z. Mild traumatic brain injury: part 1, determining the need to scan. Can Fam Physician. 2010;56:346-349.
Li X.Y., Feng D.F. Diffuse axonal injury: novel insights into detection and treatment. J Clin Neurosci. 2009;16:614-619.
Low D. Prediction of outcome utilizing both physiological and biochemical parameters in severe head injury. J Neurotrauma. 2009;26:1177-1182.
Macpherson B.C., MacPherson P., Jennett B. CT evidence of intracranial contusion and haematoma in relation to the presence, site and type of skull fracture. Clin Radiol. 1990;42:321-326.
Mayers L. Return-to-play criteria after athletic concussion: a need for revision. Arch Neurol. 2008;65:1158-1161.
Mazzeo A.T., Brophy G.M., Gilman C.B., et al. Safety and tolerability of cyclosporin A in severe traumatic brain injury patients: results from a prospective randomized trial. J Neurotrauma. 2009;26:2195-2206.
McAllister T.W., Flashman L.A., McDonald B.C., et al. Mechanisms of working memory dysfunction after mild and moderate TBI: evidence from functional MRI and neurogenetics. J Neurotrauma. 2006;23:1450-1467.
McCrory P., Meeuwisse W., Johnston K., et al. Consensus statement on concussion in sport. Third International Conference on Concussion in Sport, Zurich, November 2008. Phys Sportsmed. 2009;37:141-159.
Miller E.C., Derlet R.W., Kinser D. Minor head trauma: is computed tomography always necessary? Ann Emerg Med. 1996;27:290-294.
Minambres E., Cemborain A., Sanchez-Velasco P., et al. Correlation between transcranial interleukin-6 gradient and outcome in patients with acute brain injury. Crit Care Med. 2003;31:933-938.
Mondello S., Robicsek S.A., Gabrielli A., et al. alphaII-spectrin breakdown products (SBDPs): diagnosis and outcome in severe traumatic brain injury patients. J Neurotrauma. 2010;27:1203-1213.
Nagy K.K., Joseph K.T., Krosner S.M., et al. The utility of head computed tomography after minimal head injury. J Trauma. 1999;46:268-270.
Narotam P.K., Burjonrappa S.C., Raynor S.C., et al. Cerebral oxygenation in major pediatric trauma: its relevance to trauma severity and outcome. J Pediatr Surg. 2006;41:505-513.
Nelson D.W., Nystrom H., MacCallum R.M., et al. Extended analysis of early computed tomography scans of traumatic brain injured patients and relations to outcome. J Neurotrauma. 2010;27:51-64.
Oddo M., Schmidt J.M., Carrera E., et al. Impact of tight glycemic control on cerebral glucose metabolism after severe brain injury: a microdialysis study. Crit Care Med. 2008;36:3233-3238.
Oertel M., Boscardin W.J., Obrist W.D., et al. Posttraumatic vasospasm: the epidemiology, severity, and time course of an underestimated phenomenon: a prospective study performed in 299 patients. J Neurosurg. 2005;103:812-824.
Otto V.I., Heinzel-Pleines U.E., Gloor S.M., et al. sICAM-1 and TNF-alpha induce MIP-2 with distinct kinetics in astrocytes and brain microvascular endothelial cells. J Neurosci Res. 2000;60:733-742.
Peterson K., Carson S., Carney N. Hypothermia treatment for traumatic brain injury: a systematic review and meta-analysis. J Neurotrauma. 2008;25:62-71.
Piek J., Chesnut R.M., Marshall L.F., et al. Extracranial complications of severe head injury. J Neurosurg. 1992;77:901-907.
Povlishock J.T., Bullock M. Guidelines for the Management of Severe Head Injury: Management and Prognosis of Severe Traumatic Brain Injury, third ed. NY, New York: Brain Trauma Foundation; 2007.
Puccio A.M., Fischer M.R., Jankowitz B.T., et al. Induced normothermia attenuates intracranial hypertension and reduces fever burden after severe traumatic brain injury. Neurocrit Care. 2009;11:82-87.
Ropper A.H., Gorson K.C. Clinical practice. Concussion. N Engl J Med. 2007;356:166-172.
Ruan S., Noyes K., Bazarian J.J. The economic impact of S-100B as a pre–head CT screening test on emergency department management of adult patients with mild traumatic brain injury. J Neurotrauma. 2009;26:1655-1664.
Saul T.G., Ducker T.B. Effect of intracranial pressure monitoring and aggressive treatment on mortality in severe head injury. J Neurosurg. 1982;56:498-503.
Servadei F., Nasi M.T., Giuliani G., et al. CT prognostic factors in acute subdural haematomas: the value of the “worst” CT scan. Br J Neurosurg. 2000;14:110-116.
Sifri Z.C., Homnick A.T., Vaynman A., et al. A prospective evaluation of the value of repeat cranial computed tomography in patients with minimal head injury and an intracranial bleed. J Trauma. 2006;61:862-867.
Simard J.M., Kilbourne M., Tsymbalyuk O., et al. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma. 2009;26:2257-2267.
Smits M., Dippel D.W., de Haan G.G., et al. External validation of the Canadian CT Head Rule and the New Orleans Criteria for CT scanning in patients with minor head injury. JAMA. 2005;294:1519-1525.
Springborg J.B., Unden J., Ingebrigtsen T., et al. [Brain injury marker S100B can reduce the use of computer tomography in minor head injuries–secondary publication]. Ugeskr Laeger. 2009;171:978-981.
Stiefel M.F., Heuer G.G., Smith M.J., et al. Cerebral oxygenation following decompressive hemicraniectomy for the treatment of refractory intracranial hypertension. J Neurosurg. 2004;101:241-247.
Stiell I.G., Wells G.A., Vandemheen K., et al. The Canadian CT Head Rule for patients with minor head injury. Lancet. 2001;357:1391-1396.
Sugiyama K., Kondo T., Higano S., et al. Diffusion tensor imaging fiber tractography for evaluating diffuse axonal injury. Brain Inj. 2007;21:413-419.
Taber K.H., Warden D.L., Hurley R.A. Blast-related traumatic brain injury: what is known? J Neuropsychiatry Clin Neurosci. 2006;18:141-145.
Teasdale G., Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81-84.
Temkin N.R., Anderson G.D., Winn H.R., et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6:29-38.
Testerman G.M., Dacks L.M. Multiple self-inflicted nail gun head injury. South Med J. 2007;100:608-610.
Thiruppathy S.P., Muthukumar N. Mild head injury: revisited. Acta Neurochir (Wien). 2004;146:1075-1082. discussion 1082-1073
Unden J., Romner B. A new objective method for CT triage after minor head injury–serum S100B. Scand J Clin Lab Invest. 2009;69:13-17.
Velmahos G.C., Gervasini A., Petrovick L., et al. Routine repeat head CT for minimal head injury is unnecessary. J Trauma. 2006;60:494-499. discussion 499-501
Wolf S.J., Bebarta V.S., Bonnett C.J., et al. Blast injuries. Lancet. 2009;374:405-415.
Wright D.W., Kellermann A.L., Hertzberg V.S., et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391-402. e391-392