[level-membership-for-neurology-category]
Chapter 50A Trauma of the Nervous System
Basic Neuroscience of Neurotrauma
Experimental Models of Traumatic Brain Injury
Severe closed-head injury produces a range of cerebral lesions that may be divided into four general categories: (1) diffuse axonal injury, (2) vascular lesions including subdural hematoma, (3) contusion, and (4) neuronal degeneration in selectively vulnerable regions. In a recent review of animal models of head injury, Cernak (2005) classified models of head injury according to the biomechanics of the injury. Fluid percussion and controlled cortical impact models are characterized as a constrained direct brain deformation injury model, whereas inertial injury models and impact acceleration models are considered unconstrained in head movement and more closely mimic human TBI. Other models have specifically targeted brain trauma that occurs in combat situations. These include high-velocity penetrating and blast injury. In an attempt to investigate the effects of mechanical deformation on specific cell types, in vitro models of stretch-induced injury have also been developed.
Acceleration Concussion
Human head injury is never as pure as an experimental model, and the total human injury condition may not be addressed adequately with a single animal model. Therefore, the use of complicated models in assessing pathomechanisms after TBI can provide valuable data (Statler et al., 2001). Many of these studies have added secondary insults such as hypotension (shock), hypoxia, or hyperthermia after the primary injury to more adequately mimic human head trauma. However, this added component to the model can result in complex data interpretation. Thus, once a particular feature of human brain injury is produced in an experimental model, the pathogenesis of the injury can be critically investigated.
Military Models
The incidence of brain injury is currently estimated to be as high as 40% to 70% of combat casualties. Advancements in protective body armor have limited the number of high-velocity impact injuries to the brain, but the use of improvised explosive devices has caused blast injury to become more prevalent. Military-relevant models have only recently been developed, and much work is needed to fully explore pathomechanisms induced by these models. Two models of significance are the penetrating ballistic brain injury model and a blast injury model using a metal tube with a small amount of plastic explosives at one end. The penetrating brain injury (PBI) model mimics a bullet wound by expanding and contracting a balloon that has been inserted into the brain (Williams et al., 2005). Injury severity and placement of the probe can be varied so that different types of bullet injuries can be studied. PBI produces robust histopathological damage and increases in intracranial pressure (ICP) and hemorrhage. In addition, sensorimotor deficits are observed in this model, along with seizure activity. Blast injury models have been developed that can mimic whole-body blast or local blast injury (Cernak and Noble-Haeusslein, 2010). This type of trauma produces cognitive deficits, with ultrastructural and oxidative damage to the hippocampus. Axonal damage is also present, as indicated by increases in phosphorylated neurofilament proteins. These military-relevant injury models have the potential to provide valuable information for managing and treating combat casualties.
In Vitro Models
Using these approaches, investigators have found that trauma induces a wide range of primary cellular alterations. Astrocytic responses include hyperplasia, hypertrophy, and increased glial fibrillary acidic protein content. Increases in intracellular calcium occur, which are blocked by specific receptor antagonists. Traumatized astrocytes also produce interleukins and neurotrophic factors. Neonatal cortical neurons that are stretched undergo delayed depolarization that depends on the activation of specific receptor populations. Combined mechanical trauma and metabolic impairment in vivo also induces N-methyl-d-aspartate receptor–dependent neuronal cell death and caspase-3–dependent apoptosis. In vitro experimental approaches provide novel data concerning intracellular signaling cascades, mechanisms underlying cellular responses to trauma, and the role of specific cell types in the pathophysiology of brain trauma (Chen et al., 2009; LaPlaca et al., 2009).
Neuronal Damage After Traumatic Brain Injury
Temporal Patterns of Neuronal Death
The neuropathological sequelae of experimental and human TBI have been well described (Bramlett and Dietrich, 2004; Povlishock and Katz, 2005). In experimental TBI, temporal patterns of neuronal damage have also been characterized. As early as 6 hours after cortical contusion injury, the contused tissue appears edematous, and pyknotic neurons are apparent at the injury site. By 8 days, a cortical cavity has developed that is surrounded by a border containing necrotic tissue, a glial scar, or both. The temporal profile of neuronal damage after parasagittal FP brain injury has also been assessed with light and electron microscopy. As early as 1 hour after impact, dark shrunken neurons indicative of irreversible damage are seen in cortical layers overlying the gliding contusion that displays BBB breakdown to protein tracers. Ultrastructural studies demonstrate that early BBB dysfunction results from mechanical damage of small venules in vulnerable regions including the external capsule. In some brain regions, focal sites of acute neuronal damage are associated with extravasated protein, whereas neuronal damage in other regions appears to occur without overt BBB breakdown. Astrocytic swelling is observed early after injury, with increased glial fibrillary acidic protein immunoreactivity apparent at later times in areas, demonstrating histopathological damage (Fig. 50A.1). In terms of neuroprotection, the acuteness of this damage limits the potential for therapeutic interventions directed against the early neuronal and glial response to TBI.
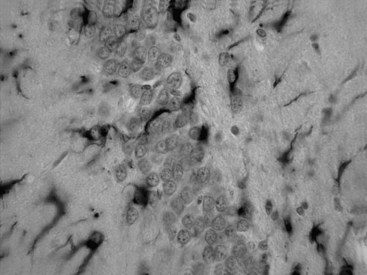
More subacute patterns of neuronal injury have been documented in various TBI models. At 3 days after moderate parasagittal FP brain injury, scattered necrotic neurons are present throughout the frontoparietal cerebral cortex remote from the impact site (Fig. 50A.2). In addition, selective neuronal damage is seen in the CA3 and CA4 hippocampal subsectors, the dentate hilus, and lateral thalamus ipsilateral to the trauma. These patterns of selective neuronal damage are associated with a well-demarcated contusion overlying the lateral external capsule. Ultrastructural changes consistent with apoptosis have been described after TBI, so delayed patterns of neuronal cell death may involve necrotic and programmed cell death processes.
Progressive Damage
Only recently has the progressive nature of the histopathological consequences of TBI been appreciated (Bramlett and Dietrich, 2007). At 2 months after moderate parasagittal FP injury, significant atrophy of the cerebral cortex, hippocampus, and thalamus is apparent in histological sections (Fig. 50A.3, A). Progressive tissue loss in the cortex and hippocampus at various times up to 1 year after lateral FP injury have been reported. Similar findings have been found in a model of controlled cortical impact injury. At 3 weeks and 1 year after injury, analysis demonstrated a significant hemispheric volume loss and expansion of the ipsilateral lateral ventricle. Thus atrophy of gray matter structures is associated with significant enlargement of the lateral ventricle.
Ventricular expansion not associated with hydrocephalus or increased ICP is felt to be a sensitive indicator of structural damage and an indirect measure of white matter atrophy. Indeed, ventricular size has been correlated with memory disturbances; patients with the highest ventricular volumes demonstrated significantly lower memory scores. Recent findings provide direct evidence of progressive white matter damage after FP brain injury (Bramlett and Dietrich, 2007). At 1 year after TBI, severe atrophy of specific tracts including the external capsule and cerebral peduncle was documented (see Fig. 50A.3, B). The importance of a progressive injury cascade after TBI in terms of other neurological conditions merits consideration. For example, if mild head trauma leads to a progressive reduction in neuronal reserve, would the person who sustained such trauma be more susceptible than others to neurodegenerative processes associated with aging? Some epidemiologic studies have indeed indicated that a history of brain trauma is a risk factor for Alzheimer disease.
Secondary and Repetitive Damage
A challenging problem faced by medical personnel responsible for the health care of amateur and professional athletes is recognizing and managing mild head injury. Returning an injured athlete to competition when the brain needs time to recover is an obvious concern. Also, a basic understanding of posttraumatic consequences that affect the vulnerability of the brain to secondary or repeated head injury remains unknown. This is a clinically important issue because TBI often is associated with respiratory suppression, resulting in secondary hypoxic insults (McHugh et al., 2007). Experimental studies have documented the detrimental consequences of secondary insults after mild to moderate TBI. The effects of secondary hypoxia on histopathological and behavioral outcome were investigated. Secondary hypoxia induced immediately after moderate FP injury resulted in significantly greater cortical and hippocampal CA1 damage and sensorimotor and cognitive deficits than were found in normoxic animals. Mild hypotension (shock) after TBI was reported to also worsen traumatic outcome. Taken together, these findings indicate the enhanced vulnerability of the posttraumatic brain to mild secondary insults.
Clinical studies indicate that patients with mild head injury may be at risk if they have a subsequent head injury (secondary impact syndrome). Recently an animal model was developed to investigate the behavioral and pathological changes associated with repetitive head injury (Weber, 2007). The consequences of single and repetitive injury induced 24 hours apart were assessed. Repetitive head injury led to greater functional impairment and structural damage than was found in the single-injury group. At the cellular level, repeated mild injury in an in vitro hippocampal cell culture model has shown elevations of neuron-specific enolase and S-100β compared to a single insult. Repetitive injury in transgenic mice that expressed mutant human Aβ precursor protein produced elevated Aβ levels and increased Aβ deposition, thus linking TBI to the mechanisms of Alzheimer disease. The known risk of developing neurodegenerative disease later in life is greater after repetitive brain trauma and makes this type of investigation extremely important.
Axonal and Dendritic Injury
Traumatic axonal injury exists as a spectrum involving widespread areas of the brain in experimental models of TBI (Buki and Povlishock, 2006). This pattern of white matter pathology can evolve from focal axonal alternations to complete transection of the axon. Reactive axonal changes using monoclonal antibodies targeted at neurofilament subunits or β-amyloid precursor protein have been characterized after FP brain injury and controlled cortical impact injury (Fig. 50A.4). Within 1 to 2 hours of injury, reactive axonal change is most conspicuous in brainstem regions, including the pontomedullary junction. This pattern of axonal damage seen in experimental models is in contrast to the human condition, in which callosal and subcortical white matter axonal damage predominates. In contrast, moderate parasagittal brain injury leads to widespread axonal damage in forebrain regions that represent reversible, irreversible, and delayed axonal perturbations. These findings may explain some of the transient and delayed functional consequences of TBI.
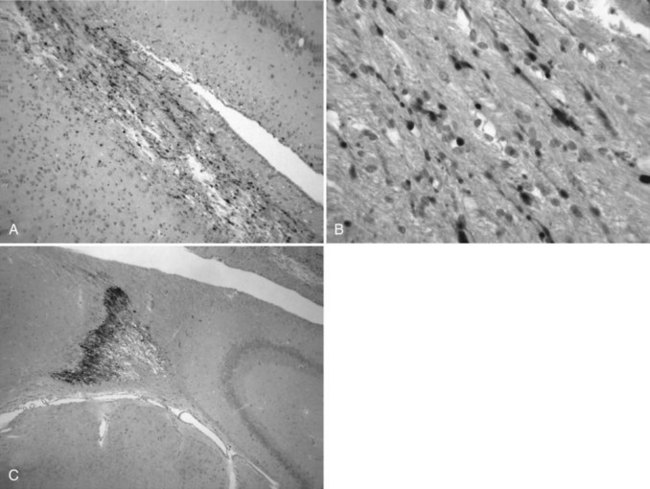
Recent evidence for neurodegeneration following severe controlled cortical impact injury in the mouse has been shown using silver staining (Hall et al., 2005). This marker identifies degenerating neurons and processes. Both hippocampal and cortical neurodegeneration is observed as late as 72 hours and continues to 7 days in select brain regions. This degeneration is associated with calpain-mediated proteolysis of cytoskeletal proteins and decreases in growth-associated protein 43 (GAP-43) expression, which may inhibit the brain’s response to elicit an attempt at plasticity to restore function.
Importance of Gender
Recent clinical and experimental data have emphasized the importance of gender on the consequences of TBI (Stein, 2007). In a study of 334 patients with TBI, female patients had a better predicted outcome at the time of discharge from an inpatient rehabilitation program. However, a meta-analysis of eight previous studies in which outcome was reported separately for men and women reported a worse outcome in women than in men. The impact of gender on TBI is an understudied area of clinical neurotrauma and should be emphasized in future trials.
In experimental models of TBI, gender also appears to influence traumatic outcome (Stein, 2007; Bramlett and Dietrich, 2001). The hemodynamic consequences of brain injury and contusion volume were significantly less in female than in male rats (Bramlett and Dietrich, 2001). In this study, ovariectomy 10 days before TBI removed the volume differences between male and female rats. Thus intact females appear to have an endogenous neuroprotective mechanism that reduces the detrimental consequences of TBI (Stein, 2007). In terms of testing neuroprotective strategies in models of TBI, gender differences must be assessed.
In this regard, in a recent randomized double-blind, placebo-controlled phase II clinical trial known as Progesterone for Traumatic Brain Injury Experimental Clinical Treatment (ProTECT), intravenous progesterone reduced the overall death rate by 50% compared to placebo. Also, there was significant improvement in the functional outcome and level of disability among patients with moderate brain injury. Based on these encouraging results, a 17-center phase III trial clinical trial is proposed (ProTECT III) that will enroll approximately 1140 subjects over a 3- to 6-year period (Wright et al., 2007). The findings from this study may provide a viable treatment option for moderate to severe TBI patients.
Basic Mechanisms of Injury
Secondary Injury Mechanisms
In head-injured patients, the extent of neurological recovery depends on the contribution of posttraumatic secondary insults (Statler et al., 2001). In the clinical setting, secondary insults include hypotension, hypoxia, hyperglycemia, anemia, sepsis, and hyperthermia. Experimental evidence indicates an increased susceptibility of the posttraumatic brain to secondary insults. For example, after midline FP brain injury, CA1 hippocampal vulnerability is enhanced with superimposed secondary ischemia. An important area of research regarding the treatment of brain injury involves the characterization of secondary injury processes, which may be targeted for intensive care management or pharmacotherapy.
Cortical spreading depression (CSD) is caused by ionic changes within tissue resulting in waves of depressed electrical activity. CSDs are not necessarily harmful to tissue unless there is damage present. It was previously thought that CSDs could only be induced experimentally; however, spontaneous self-propagating waves within damaged tissue, known as periinfarct depolarizations and similar to CSDs, have been reported (Fabricius et al., 2006). The incidence of CSD has been well described in the experimental focal ischemia literature and associated with increasing amounts of neuronal damage within the cerebral cortex. However, the presence of CSDs in the human brain-injured population had remained controversial until recently because of the inability to adequately measure the phenomenon; Strong and colleagues (2002) clearly showed the presence of CSDs following clinical TBI. Although it does not occur in all acutely injured individuals, the incidence is higher in younger patients. Such findings have resulted in a new initiative to document the occurrence of CSDs after TBI and study their relationship to outcome.
Many patients experience fever after head injury, and clinical data indicate that brain temperature may be higher than core or bladder temperature (Hayashi et al., 2004). Experimentally, posttraumatic brain hyperthermia induced artificially 24 hours after trauma increases mortality rate and aggravates histopathological outcome, including contusion size and axonal pathology. In the clinical setting, posttraumatic hyperthermia may represent a secondary injury mechanism that might negate the beneficial effects of a therapeutic agent.
A significant debilitating consequence of TBI is the development of seizures (Vespa, 2005). Some 40% to 50% of moderate to severe TBI patients develop epilepsy, and brain injuries account for 20% of symptomatic epilepsy cases (Garga and Lowenstein, 2006). Trauma-induced mechanisms of reduced seizure threshold include damage to specific neuronal populations and circuits, increased BBB damage, and aberrant mossy-fiber sprouting of dentate granule cells. Importantly, posttraumatic epilepsy has been shown to worsen traumatic outcome and aggravate cognitive problems in some clinical studies. Unfortunately, chronic seizures after brain injury are poorly controlled by available antiepileptic drugs (Temkin, 2009).
Therapeutic Interventions Directed Against Pathophysiological Processes
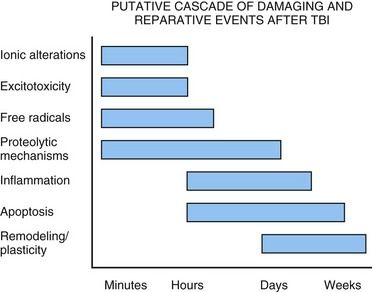
The treatment of TBI has been investigated using a variety of animal models; new therapies have been initiated, some of which have been tested in patients. Several reviews summarize the agents that have been investigated in TBI (McIntosh et al., 1998). The problem of TBI involves injury pathways that are common to other brain injuries, including cerebral ischemia. However, the pathogenesis of TBI is unique in other ways and necessitates therapeutic approaches specifically targeted at brain trauma (Fig. 50A.5). The present discussion is limited to several of the major therapeutic strategies currently being assessed experimentally and clinically (Maas et al., 2010).
Glutamate Antagonists
Excitatory amino acid neurotransmitters have been implicated in the pathophysiology of TBI. Microdialysis techniques have documented elevated levels of extracellular excitatory amino acids, whereas N-methyl-d-aspartate (NMDA) receptor antagonists, including MK-801 (dizocilpine), provide behavioral and histopathological protection against brain trauma. The role of glutamate antagonists in treating central nervous system injury has been reviewed, and clinical stroke trials of the competitive NMDA antagonist, MK-80I, were withdrawn because of harmful side effects. However, the effects of topiramate on inhibiting glutamate release have been evaluated using microdialysis in TBI patients (Alves et al., 2003). This treatment strategy shows promise in attenuating glutamate levels, but further evaluation is necessary to demonstrate clinical improvement.
Inflammation
Inflammation is a host defense mechanism initiated by injury or infection through which blood-derived leukocytes (neutrophils, monocytes and macrophages, T cells) and soluble factors (cytokines, chemokines, complement) try to restore tissue homeostasis (Morganti-Kossman et al., 2007). Although evidence supports the beneficial role of inflammatory processes in acute injury, including the production of neurotrophic factors, inflammation is also thought to contribute to the resulting neuropathology and secondary necrosis that occur after trauma. Brain trauma is associated with the production and release of proinflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6. The inhibition of cytokines such as IL-1β and TNF-α has been reported to decrease lesion size and improve behavioral outcome after TBI. Cytokine receptor antagonist (ra) drugs may also show promise in treating TBI. A report on mice treated with an IL-1ra showed a decrease in histopathological damage and lesion volume, with an improvement in behavioral recovery (Jones et al., 2005). Treatment with the potent antiinflammatory cytokine, IL-10, was reported to improve outcome as well. However, absolute TNF-α inhibition in the form of knockout mice has yielded conflicting results regarding pathological and neurological outcomes. Again, these studies emphasize the diverse actions of cytokines and both the good and bad consequences of overexpression and inhibition.
Antiapoptotic Agents
Apoptosis is a mode of cell death in both physiological and pathological processes. Evidence of apoptotic cell death has also been observed after TBI. After FP injury, apoptotic cells have been identified in the ipsilateral cortex, hippocampus, and thalamus as soon as 4 hours after injury, and immunocytochemical markers for caspase-3, -8, and -9 activation have been reported (Fig. 50A.6). The effects of FP injury on the expression of the bc1-2 protein, which regulates developmental programmed cell death, have also been investigated. Evidence of apoptosis of oligodendroglia in long tracts undergoing wallerian degeneration has been reported after spinal cord injury. Therefore, demyelination of tracts after brain or spinal cord trauma may result from apoptotic death of oligodendrocytes. Importantly, indicators of apoptotic cell death have also been observed in human tissues.
Although the molecular events leading to apoptosis are not fully understood, the family of cysteine proteases (caspases) play an active role in its pathogenesis. In reference to neuroprotection, in vitro studies have demonstrated that protease inhibitors specific to caspase-3 inhibit apoptosis. Whereas some experimental data indicate that inhibition of caspase-3 improves outcome after TBI, other studies suggest that this approach has limitations. Current emphasis is on more upstream apoptotic processes, in contrast to targeting caspase activation to attenuate damage. Calpain inhibition may target not only apoptotic cell death by regulating caspase activation but also cytoskeletal protein degradation (Raynaud and Marcilhac, 2006). More research is needed to clarify the various pathways involved in apoptotic cell death and determine which pathways may be most sensitive to therapeutic interventions. Using agents specific to apoptosis or in combination with agents that target necrosis is a potential research direction.
Therapeutic Hypothermia
Numerous studies have demonstrated that although mild to moderate hypothermia is neuroprotective in models of TBI, mild hyperthermia worsens outcome (Dietrich and Bramlett, 2010). After brain trauma, hypothermia has been shown to improve histopathological and behavioral outcomes and to influence a wide range of injury processes. Microdialysis studies report that posttraumatic hypothermia reduces the acute surge in levels of extracellular glutamate and hydroxy free radicals after injury. Posttraumatic hypothermia protects against BBB dysfunction. Hypothermia attenuates progressive cortical atrophy and subsequent ventricular enlargement. The ability of any therapeutic intervention to provide long-term protection is an important requirement for the advancement of any therapeutic strategy to the clinical setting.
As previously discussed, a significant number of patients with TBI sustain a secondary insult that may include hypotension, hypoxia, or hyperthermia. This fact has led to the use of complicated models to test novel neuroprotective agents before clinical trials. This point is important because experimental therapeutic strategies are commonly tested in simple models of brain injury as proof of concept (McIntosh et al., 1998). In this regard, posttraumatic hypothermia followed by a controlled rewarming period has been evaluated in TBI models complicated by secondary hypoxia. Taken together, these studies showed that hypothermia was protective in complicated models, but the degree of protection depended on injury severity, duration of hypothermic period, and the rewarming procedure.
The use of moderate levels of hypothermia (>32°C) also improves outcome in patient studies. Systemic hypothermia (32°-33°C) begun within 6 hours of injury (Glasgow Coma Score 4-7) resulted in no cardiac or coagulopathy-related complications, a lower seizure frequency, and more patients in the good recovery to moderate disability category. In other head trauma studies, therapeutic hypothermia attenuated intracranial hypertension but did not affect the frequency of delayed intracerebral hemorrhage. Results from a recent U.S. multicenter TBI trial failed to demonstrate a protective effect of hypothermia on traumatic outcome, but a subgroup analysis showed that patients younger than age 45 who came into the emergency room hypothermic demonstrated improvement with hypothermic treatment (Clifton et al., 2001). Obviously, more experimental and clinical studies are needed to determine what factors are most important in providing protection when using hypothermic strategies. Temperature is known to affect many pathophysiological processes after TBI, and this characteristic may be advantageous because of the multifactorial nature of trauma pathomechanisms. The cooling and rewarming periods are also important variables in determining the extent of neuroprotection. In TBI studies, prolonged periods of hypothermia (i.e., >24 hours) may therefore be necessary to protect the brain from primary and secondary injury processes. In a multicenter TBI trial from China, long-term cooling (5 days) was significantly better than short-term cooling (2 days) in terms of improved outcome in patients (Jiang et al., 2006). Because brain temperature can be elevated compared with bladder temperature in head-injured patients, normothermia or mild hypothermia should be maintained during critical postinjury periods.
Recovery of Function
Environmental Enrichment
The effect of environmental enrichment (EE) that exposes animals to a complex, highly stimulatory, and social environment has been studied in a number of TBI models. Using a midline FP injury model that produced no noticeable histopathology, EE was reported to improve cognitive function. In addition, EE has been shown to decrease overall contusion volume and improve performance in the Morris Water Maze task. The effects of EE have been suggested to be reflected in changes in dendritic arborization. Recent experimental studies have reported that animals raised with EE have larger contusion injuries and greater sensorimotor deficits after mild TBI, compared to standard laboratory cage-reared animals (Kozlowski et al., 2004). However, the sensorimotor deficit improved at a much more rapid pace in the EE animals compared to age-matched controls. Future studies combining EE with neuroprotective and reparative strategies probably will be needed to maximize improvement in outcome.
Reparative and Transplantation Strategies
After a variety of acute central nervous system injuries, there is a massive proliferation of stem or progenitor cells (Richardson et al., 2010). The identification and origin of the fate of these cells is an area of intensive investigation. In a model of FP injury, the total number of proliferating cells as identified with 5-bromo-deoxyuridine, a marker of mitotic activity, was shown to significantly increase in areas of the subventricular zone and hippocampus. In that study, proliferating cells did not express cell markers and therefore appeared not to have begun to differentiate. Targeting this endogenous proliferative response to injury may be one way to enhance recovery following TBI.
In contrast to attempts to enhance endogenous reparative events, providing new cells from exogenous sources is an alternative approach and may be necessary when neuronal loss and axonal injury are severe. Neural transplantation has been explored in TBI models (Lu et al., 2003). Fetal cortical tissue transplanted into the injury cavity improved motor function and transiently attenuated cognitive dysfunction alone and in combination with NGF infusion. Although the reestablishment of normal adult neural circuitry has not been demonstrated with fetal tissue grafts, one mechanism for improved function may be neuroprotection by release of trophic factors from the grafts (Gao et al., 2006). Recent studies have also attempted to provide cellular replacement and host-graft integration using self-renewing cell lines (Wallenquist et al., 2009). Using transplanted immortalized neural progenitor cells transduced with the mouse NGF gene to secrete NGF, improved neuromotor and cognitive function and reduced hippocampal CA3 cell death have been reported. Continued study in this exciting field may establish transplantation procedures relevant to clinical strategies to promote recovery after TBI.
Alves O.L., Doyle A.J., Clausen T., et al. Evaluation of topiramate neuroprotective effect in severe TBI using microdialysis. Ann N Y Acad Sci. 2003;993:25-34.
Bramlett H.M., Dietrich W.D. Neuropathological protection after traumatic brain injury in intact female rats versus males or ovariectomized females. J Neurotrauma. 2001;18:891-900.
Bramlett H.M., Dietrich W.D. Progressive damage after brain and spinal cord injury: pathomechanisms and treatment strategies. Prog Brain Res. 2007;161:125-141.
Bramlett H.M., Dietrich W.D. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133-150.
Buki A., Povlishock J.T. All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir (Wien). 2006;148:181-193.
Cernak I. Animal models of head trauma. NeuroRx. 2005;2:410-422.
Cernak I., Noble-Haeusslein L.J. Traumatic brain injury: an overview of pathobiology with emphasis on military populations. J Cereb Blood Flow Metab. 2010;30:255-266.
Chen Y.C., Smith D.H., Meaney D.F. In-vitro approaches for studying blast-induced traumatic brain injury. J Neurotrauma. 2009;26:861-876.
Clifton G.L., Choi S.C., Miller E.R., et al. Intercenter variance in clinical trials of head trauma—experience of the National Acute Brain Injury Study: hypothermia. J Neurosurg. 2001;95:751-755.
Dietrich W.D., Bramlett H.M. The evidence of hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics. 2010;7:43-50.
Fabricius M., Fuhr S., Bhatia R., et al. Cortical spreading depression and peri-infarct depolarization in acutely injured human cerebral cortex. Brain. 2006;129:778-790.
Gao J., Prough D.S., McAdoo D.J., et al. Transplantation of primed human fetal neural stem cells improves cognitive function in rats after traumatic brain injury. Exp Neurol. 2006;201:281-292.
Garga N., Lowenstein D.H. Posttraumatic epilepsy: a major problem in desperate need of major advances. Epilepsy Curr. 2006;6:1-5.
Hall E.D., Sullivan P.G., Gibson T.R., et al. Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma. 2005;22:252-265.
Hayashi N., Bullock R., Dietrich W.D., et al. Hypothermia for Acute Brain Damage. New York: Springer; 2004.
Jiang J.Y., Xu W., Li W.P., et al. Effect of long-term mild hypothermia or short-term mild hypothermia on outcome of patients with severe traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:771-776.
Jones N.C., Prior M.J., Burden-Teh E., et al. Antagonism of the interleukin-1 receptor following traumatic brain injury in the mouse reduces the number of nitric oxide synthase-2-positive cells and improves anatomical and functional outcome. Eur J Neurosci. 2005;22:72-78.
Kozlowski D.A., Nahed B.V., Hovda D.A., et al. Paradoxical effects of cortical impact injury on environmentally enriched rats. J Neurotrauma. 2004;21:513-519.
LaPlaca M.C., Prado G.R., Cullen D., et al. Plasma membrane damage as a marker of neuronal injury. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:1113-1116.
Lu D., Mahmood A., Chopp M. Biologic transplantation and neurotrophin-induced neuroplasticity after traumatic brain injury. J Head Trauma Rehabil. 2003;18:357-376.
Maas A.I., Roozenbeek B., Manley G.T. Clinical trials in traumatic brain injury: past experience and current developments. Neurotherapeutics. 2010;7:115-126.
McHugh G.S., Engel D.C., Butcher I, et al. Prognostic value of secondary insults in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24:287-293.
McIntosh T.K., Juhler M., Wieloch T. Novel pharmacologic strategies in the treatment of experimental traumatic brain injury. J Neurotrauma. 1998;15:731-769.
Morganti-Kossmann M.C., Satgunaseelan L., Bye N., et al. Modulation of immune response by head injury. Injury. 2007;38:1392-1400.
Povlishock J.T., Katz D.I. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20:76-94.
Raynaud F., Marcilhac A. Implication of calpain in neuronal apoptosis. FEBS J. 2006;273:3437-3443.
Richardson R.M., Singh A., Sun D., et al. Stem cell biology in traumatic brain injury: effects of injury and strategies for repair. J Neurosurg. 2010;112:1125-1138.
Statler K.D., Jenkins L.W., Dixon C.E., et al. The simple model versus the super model; translating experimental traumatic brain injury research to the bedside. J Neurotrauma. 2001;18:1195-1206.
Stein D.G. Sex differences in brain damage and recovery of function: experimental and clinical findings. Prog Brain Res. 2007;161:339-351.
Strong A.J., Fabricius M., Boutelle M.G., et al. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke. 2002;33:2738-2743.
Temkin N.R. Preventing and treating posttraumatic seizures: the human experience. Epilepsia. 2009;50(Suppl 2):10-13.
Vespa P. Continuous EEG monitoring for the detection of seizures in traumatic brain injury, infarction, and intracerebral hemorrhage: “to detect and protect.”. J Clin Neurophysiol. 2005;22:99-106.
Wallenquist U., Brännvall K., Clausen F., et al. Grafted neural progenitors migrate and form neurons after experimental traumatic brain injury. Restor Neurol Neurosci. 2009;27:323-334.
Weber J.T. Experimental models of repetitive brain injuries. Prog Brain Res. 2007;161:253-261.
Williams A.J., Hartings J.A., Lu X-C.M., et al. Characterization of a new rat model of penetrating ballistic brain injury. J Neurotrauma. 2005;22:313-331.
Wright D.W., Kellerman A.L., Hertzberg V.S., et al. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391-402.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 50A Trauma of the Nervous System
Basic Neuroscience of Neurotrauma
Experimental Models of Traumatic Brain Injury
Severe closed-head injury produces a range of cerebral lesions that may be divided into four general categories: (1) diffuse axonal injury, (2) vascular lesions including subdural hematoma, (3) contusion, and (4) neuronal degeneration in selectively vulnerable regions. In a recent review of animal models of head injury, Cernak (2005) classified models of head injury according to the biomechanics of the injury. Fluid percussion and controlled cortical impact models are characterized as a constrained direct brain deformation injury model, whereas inertial injury models and impact acceleration models are considered unconstrained in head movement and more closely mimic human TBI. Other models have specifically targeted brain trauma that occurs in combat situations. These include high-velocity penetrating and blast injury. In an attempt to investigate the effects of mechanical deformation on specific cell types, in vitro models of stretch-induced injury have also been developed.
Acceleration Concussion
Human head injury is never as pure as an experimental model, and the total human injury condition may not be addressed adequately with a single animal model. Therefore, the use of complicated models in assessing pathomechanisms after TBI can provide valuable data (Statler et al., 2001). Many of these studies have added secondary insults such as hypotension (shock), hypoxia, or hyperthermia after the primary injury to more adequately mimic human head trauma. However, this added component to the model can result in complex data interpretation. Thus, once a particular feature of human brain injury is produced in an experimental model, the pathogenesis of the injury can be critically investigated.
Military Models
The incidence of brain injury is currently estimated to be as high as 40% to 70% of combat casualties. Advancements in protective body armor have limited the number of high-velocity impact injuries to the brain, but the use of improvised explosive devices has caused blast injury to become more prevalent. Military-relevant models have only recently been developed, and much work is needed to fully explore pathomechanisms induced by these models. Two models of significance are the penetrating ballistic brain injury model and a blast injury model using a metal tube with a small amount of plastic explosives at one end. The penetrating brain injury (PBI) model mimics a bullet wound by expanding and contracting a balloon that has been inserted into the brain (Williams et al., 2005). Injury severity and placement of the probe can be varied so that different types of bullet injuries can be studied. PBI produces robust histopathological damage and increases in intracranial pressure (ICP) and hemorrhage. In addition, sensorimotor deficits are observed in this model, along with seizure activity. Blast injury models have been developed that can mimic whole-body blast or local blast injury (Cernak and Noble-Haeusslein, 2010). This type of trauma produces cognitive deficits, with ultrastructural and oxidative damage to the hippocampus. Axonal damage is also present, as indicated by increases in phosphorylated neurofilament proteins. These military-relevant injury models have the potential to provide valuable information for managing and treating combat casualties.
In Vitro Models
Using these approaches, investigators have found that trauma induces a wide range of primary cellular alterations. Astrocytic responses include hyperplasia, hypertrophy, and increased glial fibrillary acidic protein content. Increases in intracellular calcium occur, which are blocked by specific receptor antagonists. Traumatized astrocytes also produce interleukins and neurotrophic factors. Neonatal cortical neurons that are stretched undergo delayed depolarization that depends on the activation of specific receptor populations. Combined mechanical trauma and metabolic impairment in vivo also induces N-methyl-d-aspartate receptor–dependent neuronal cell death and caspase-3–dependent apoptosis. In vitro experimental approaches provide novel data concerning intracellular signaling cascades, mechanisms underlying cellular responses to trauma, and the role of specific cell types in the pathophysiology of brain trauma (Chen et al., 2009; LaPlaca et al., 2009).
Neuronal Damage After Traumatic Brain Injury
Temporal Patterns of Neuronal Death
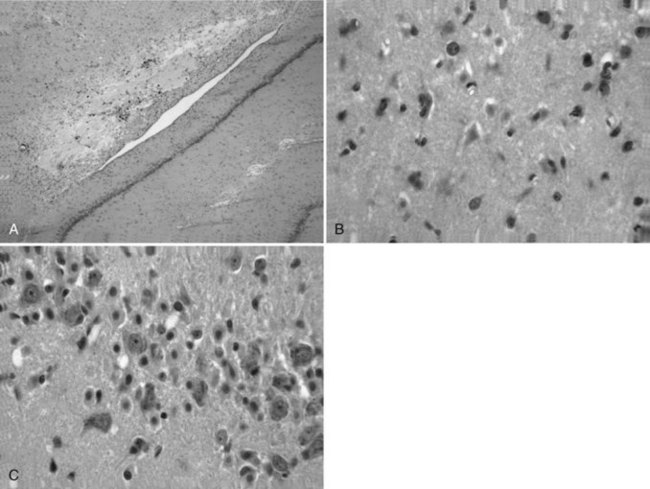
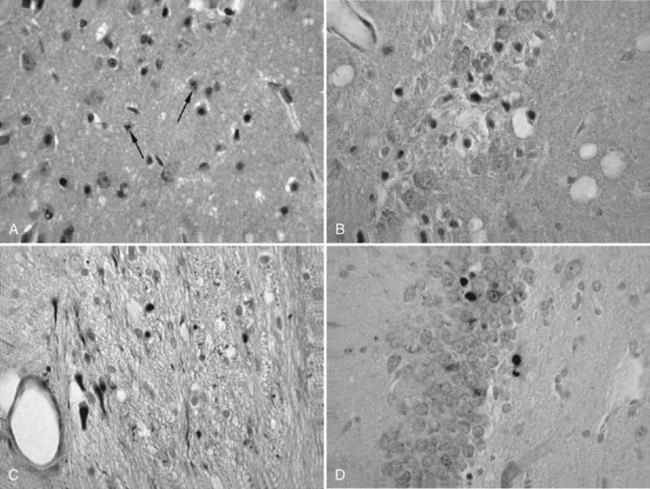
The neuropathological sequelae of experimental and human TBI have been well described (Bramlett and Dietrich, 2004; Povlishock and Katz, 2005). In experimental TBI, temporal patterns of neuronal damage have also been characterized. As early as 6 hours after cortical contusion injury, the contused tissue appears edematous, and pyknotic neurons are apparent at the injury site. By 8 days, a cortical cavity has developed that is surrounded by a border containing necrotic tissue, a glial scar, or both. The temporal profile of neuronal damage after parasagittal FP brain injury has also been assessed with light and electron microscopy. As early as 1 hour after impact, dark shrunken neurons indicative of irreversible damage are seen in cortical layers overlying the gliding contusion that displays BBB breakdown to protein tracers. Ultrastructural studies demonstrate that early BBB dysfunction results from mechanical damage of small venules in vulnerable regions including the external capsule. In some brain regions, focal sites of acute neuronal damage are associated with extravasated protein, whereas neuronal damage in other regions appears to occur without overt BBB breakdown. Astrocytic swelling is observed early after injury, with increased glial fibrillary acidic protein immunoreactivity apparent at later times in areas, demonstrating histopathological damage (Fig. 50A.1). In terms of neuroprotection, the acuteness of this damage limits the potential for therapeutic interventions directed against the early neuronal and glial response to TBI.
More subacute patterns of neuronal injury have been documented in various TBI models. At 3 days after moderate parasagittal FP brain injury, scattered necrotic neurons are present throughout the frontoparietal cerebral cortex remote from the impact site (Fig. 50A.2). In addition, selective neuronal damage is seen in the CA3 and CA4 hippocampal subsectors, the dentate hilus, and lateral thalamus ipsilateral to the trauma. These patterns of selective neuronal damage are associated with a well-demarcated contusion overlying the lateral external capsule. Ultrastructural changes consistent with apoptosis have been described after TBI, so delayed patterns of neuronal cell death may involve necrotic and programmed cell death processes.
Progressive Damage
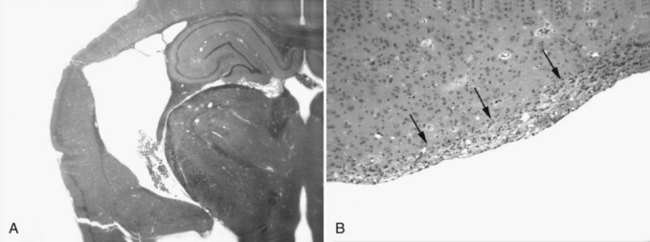
Only recently has the progressive nature of the histopathological consequences of TBI been appreciated (Bramlett and Dietrich, 2007). At 2 months after moderate parasagittal FP injury, significant atrophy of the cerebral cortex, hippocampus, and thalamus is apparent in histological sections (Fig. 50A.3, A). Progressive tissue loss in the cortex and hippocampus at various times up to 1 year after lateral FP injury have been reported. Similar findings have been found in a model of controlled cortical impact injury. At 3 weeks and 1 year after injury, analysis demonstrated a significant hemispheric volume loss and expansion of the ipsilateral lateral ventricle. Thus atrophy of gray matter structures is associated with significant enlargement of the lateral ventricle.
Ventricular expansion not associated with hydrocephalus or increased ICP is felt to be a sensitive indicator of structural damage and an indirect measure of white matter atrophy. Indeed, ventricular size has been correlated with memory disturbances; patients with the highest ventricular volumes demonstrated significantly lower memory scores. Recent findings provide direct evidence of progressive white matter damage after FP brain injury (Bramlett and Dietrich, 2007). At 1 year after TBI, severe atrophy of specific tracts including the external capsule and cerebral peduncle was documented (see Fig. 50A.3, B). The importance of a progressive injury cascade after TBI in terms of other neurological conditions merits consideration. For example, if mild head trauma leads to a progressive reduction in neuronal reserve, would the person who sustained such trauma be more susceptible than others to neurodegenerative processes associated with aging? Some epidemiologic studies have indeed indicated that a history of brain trauma is a risk factor for Alzheimer disease.
