CHAPTER 44 Transposition of the Great Arteries
Historically, CTGA was first described by Matthew Baillie, a Scottish pathologist, in 1797. The first corrective surgery for CTGA was the atrial switch procedure developed by Senning1 in 1959 and Mustard2 in 1964. Atrial switching effectively converts a CTGA to a CCTGA, in which the RV pumps the systemic circulation. Although this circulation is compatible with life, the systemic RV is prone to early failure. To address the shortcomings of atrial switching, Jatene and colleagues3 perfected the arterial switch procedure in 1975, in which the LV pumps the systemic circulation. It has become the preferred surgical treatment for CTGA. The Rastelli procedure4 is used when the CTGA is associated with pulmonary stenosis. CCTGA was first described by the famous Bohemian pathologist Carl von Rokitansky in 1875. Early treatments focused on correcting secondary lesions, such as ventricular septal defect (VSD). In favor of an anatomically correct circulation, Ilbawi and associates5 and others have advocated the double switch procedure, which combines the atrial switch and arterial switch procedures, or the atrial switch and Rastelli procedures. This procedure restores the LV as the systemic ventricle.
Definition and Classifications
CTGA is defined by ventriculoarterial discordance. It also can be described by the inlet-outlet features of the ventricles: the presence of subaortic infundibulum (Fig. 44-1), the absence of subpulmonary infundibulum, and mitral-pulmonary fibrous continuity (Fig. 44-2). Subaortic infundibulum implies a connection of the aorta to the infundibulum, which is a morphologic feature of the RV. The absence of subpulmonary infundibulum means that the pulmonary trunk is not connected to the RV. Because fibrous continuity of the inlet-outlet valves is a feature of the LV, a mitral-pulmonary fibrous continuity implies that the pulmonary trunk is connected to the LV.
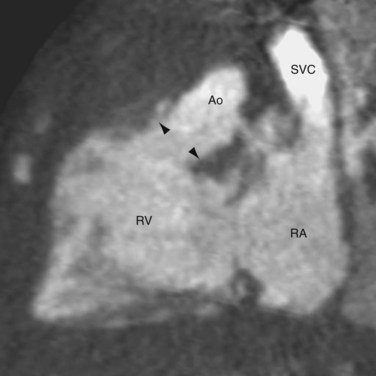
 FIGURE 44-1
FIGURE 44-1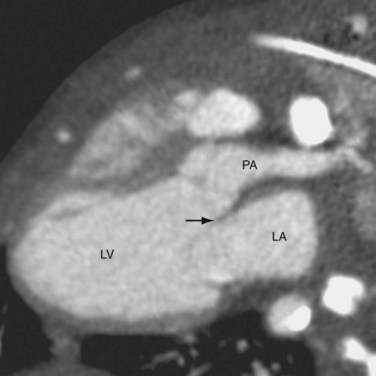
 FIGURE 44-2
FIGURE 44-2CTGA is known by a number of other names. Because CTGA comprises 90% of all transposition cases, it is sometimes referred simply as transposition of the great arteries. Other designations are simple transposition and dextrotransposition of the great arteries (D-TGA). The letter D refers to D-loop of the ventricles and not the positional relationship of the great arteries. D-loop refers to the rightward folding of the heart tube during early embryologic development. This crucial event leads to the normal positions of the LV and RV at birth (Fig. 44-3). Usually, D-TGA corresponds with the physiology of CTGA, but this is not always the case. In situs inversus, the CTGA physiology corresponds to L-loop ventricles and CCTGA physiology corresponds to D-loop ventricles. To avoid confusion, it is best to use the terms CTGA and CCTGA, which correctly describe the underlying physiology.
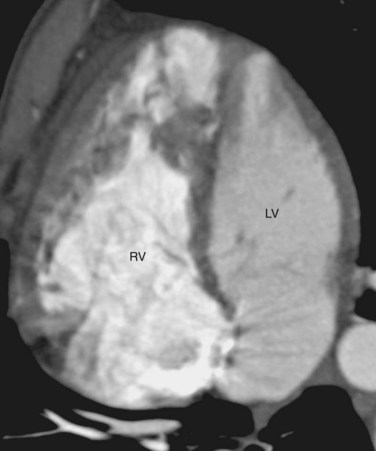
 FIGURE 44-3
FIGURE 44-3CCTGA is defined by the presence of atrioventricular and ventriculoarterial discordance. Alternative names for CCTGA are corrected transposition, double discordance, and levotransposition of the great arteries (L-TGA). In a patient with normal situs, L-TGA implies L-loop ventricles (Fig. 44-4) and atrioventricular discordance, in addition to ventriculoarterial discordance. Like D-TGA, the term L-TGA is best avoided in favor of CCTGA.
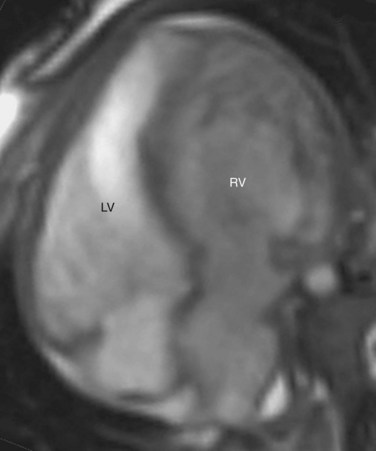
 FIGURE 44-4
FIGURE 44-4Van Praagh proposed a three-letter scheme that labels the sidedness of the atrial, ventricular, and arterial segments,6 and it has been used to categorize various anatomic manifestations of TGA. In this scheme, the first letter denotes the atrial situs, with S meaning solitus and I meaning inversus. The second letter denotes the ventricular loop, with D meaning D-loop, and L meaning L-loop. The third letter denotes the positions of the great arteries, with D meaning that the aortic root is on the right side of the pulmonary root (Fig. 44-5) and L meaning that the aortic root is on the left side of the pulmonary root (Fig. 44-6). In situs solitus and CTGA, the most common segmental anatomy is S, D, D, although S, D, L exists in a minority of cases. In situs inversus and CTGA, the most common pattern is I, L, L, which is the mirror image of S, D, D. In CCTGA, the segmental anatomy is usually S, L, L for situs solitus and I, D, D for situs inversus. Van Praagh and coworkers,7 in a pathology series of CCTGA, reported one case of S, L, D.
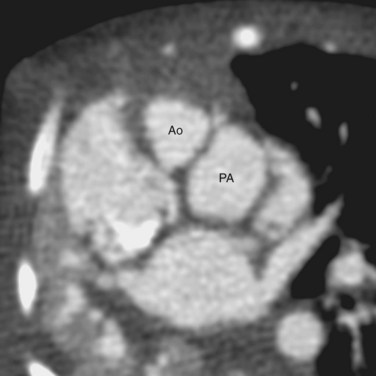
 FIGURE 44-5
FIGURE 44-5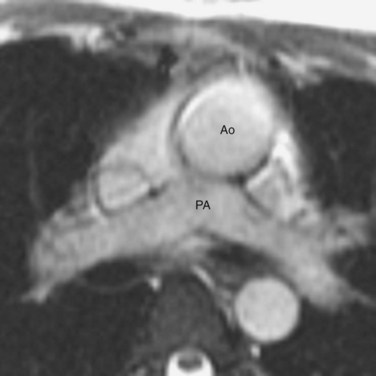
 FIGURE 44-6
FIGURE 44-6Epidemiology and Genetics
CTGA occurs in 5% to 7% of cases of congenital heart disease, with an incidence of 20 to 30/100,000 live births. It is the second most common cyanotic heart disease, behind tetralogy of Fallot, and it is the most common cyanotic heart disease that manifests in the neonatal period. There is no known racial predilection, but there is a 2 : 1 male predominance. CTGA has been reported in trisomies 18 and 21,8 although the vast majority of cases have no identified genetic defect. Chromosomal 22q11 deletion, commonly found in conotruncal anomalies, is not associated with CTGA. Furthermore, the risk of recurrence in the offspring of individuals with CTGA is very low.9 It has a greater incidence in mothers with diabetes. These observations suggest that genetics may play a less important role in the development of CTGA than environmental factors, possibly intrauterine hormonal disturbance or exposure to teratogens. CTGA is rarely associated with other extracardiac anomalies, with the possible exception of coarctation.
Etiology and Pathophysiology
Embryology
The embryology of CTGA is not well understood. The loss of the normal spiral relationship between the ascending aorta and the pulmonary trunk in CTGA suggests a problem with the process of conotruncal septation. In normal development during the fifth week of gestational age, two intertwined, spiraling flow streams in the conotruncus are thought to induce the proper spiraling septation that divides the conotruncus into the aorta and pulmonary trunk.10 The spiraling flow streams may be disturbed, leading to abnormal conotruncal septation and CTGA. This may also explain why the infundibulum septum, formed during the same process, is often abnormal in CTGA.
Associated Cardiac Anomalies
Knowledge of the coronary artery anatomy is crucial for the surgical management of CTGA. In CTGA, as in normal cardiac anatomy, the two main coronary arteries most often arise from the two aortic sinuses of Valsalva closest to, or facing, the pulmonary valve. In the most common coronary pattern for CTGA, which occurs in 67% of cases, the right coronary artery (RCA) originates from the right facing sinus and the left main coronary artery (LMCA) originates from the left facing sinus (Fig. 44-7). The LMCA then bifurcates into the left anterior descending (LAD) artery and left circumflex (LCx) artery, as expected. Less commonly, the LCx arises from the RCA in 22% of cases. Other variations include a single RCA (9.5%), a single LMCA (3%), reversed RCA and LMCA (3%), and others (Fig. 44-8).11
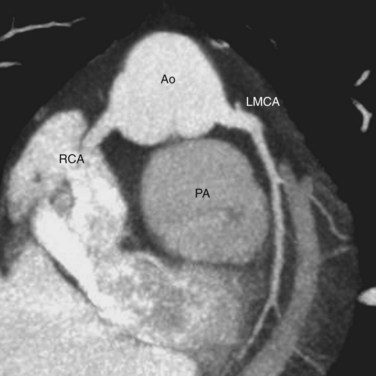
 FIGURE 44-7
FIGURE 44-7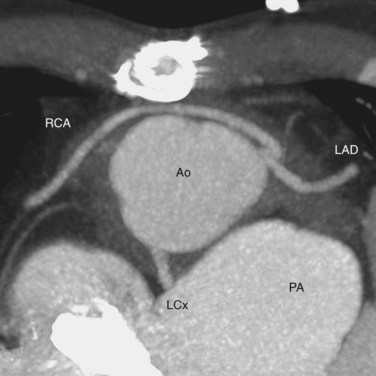
 FIGURE 44-8
FIGURE 44-8Only a small fraction (1% to 9%) of CCTGA cases are isolated without any associated anomalies.12 The most common associations are VSD and pulmonary stenosis. VSD occurs in 60% to 80% of cases (Fig. 44-9). It can be of any type and the VSD can be multiple. Pulmonary stenosis occurs in 30% to 50% of cases, and it can be valvular or subvalvular. There is a frequent association with tricuspid abnormalities, which include dysplastic leaflets, Ebstein-like displacement of the leaflets, and a tricuspid valve straddling the ventricular septum. Other less common associated anomalies are atrial septal defect (ASD), patent ductus arteriosus (PDA), pulmonary atresia, mitral valve abnormalities, coarctation, and interrupted aortic arch. CCTGA manifests with dextrocardia in 25% of cases and situs inversus in 5% of cases.
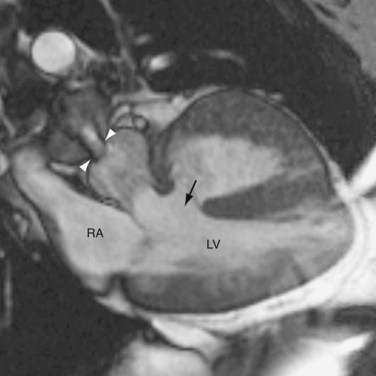
 FIGURE 44-9
FIGURE 44-9The aortic root position is typically anterior and to the left of the pulmonary root. Together with normal situs and L-loop ventricle, S, L, L is the most common segmental description for CCTGA. Variations in coronary anatomy are common, but the branching pattern of the coronary arteries generally follows the ventricles. Because the left ventricle is positioned on the right, the LMCA commonly arises from the right-facing sinus and travels to the right before branching into a LAD and a LCx. The latter lies in the right atrioventricular groove. Similarly, the RCA commonly arises from the left-facing sinus and lies in the left atrioventricular groove. Single coronary origin is also common.13
MANIFESTATIONS
Clinical Presentation
In practice, some mixing of the oxygenated and deoxygenated blood occurs through shunts between the pulmonary and systemic circulations, providing some oxygen to the systemic organs. The degree of cyanosis depends on (1) the size and direction of these shunts and (2) the degree of obstruction to the pulmonary flow. In CTGA-IVS, the most common anatomic arrangement, shunting is limited and the neonate presents with cyanosis within hours of birth. Cyanosis worsens as the PDA closes in the first few days of life. Prognosis in this scenario is poor, with a 30% survival rate at 1 month. In CTGA-VSD, the VSD is usually large and mixing occurs freely at the ventricular level. Patients may present with mild cyanosis, even after the PDA closes. As the pulmonary resistance drops in the first week of life, a large shunting from the systemic RV to pulmonary LV may occur. This can lead to congestive heart failure and later pulmonary vascular obstructive disease. The latter is the cause of pulmonary hypertension and Eisenmenger syndrome. The survival rate at 1 year is 50%, which is better than that for CTGA-IVS. In CTGA-VSD-LVOTO, the LVOT obstruction limits flow to the pulmonary circulation. The physiology in this situation is similar to that of tetralogy of Fallot. Severe obstruction would lead to diminished pulmonary flow and worsening cyanosis. Little obstruction would lead to large shunting and the same outcome as CTGA-VSD. If the obstruction is balanced just right, the pulmonary circulation may be protected from harmful shunt flow at a level of tolerable cyanosis. The survival rate for CTGA-VSD-LVOTO is 50% at 3 years, with some surviving into adulthood. Left untreated, the overall survival rates for CTGA are 70% at 1 week, 50% at 1 month, and 10% at 1 year.14
Late complications of CCTGA are caused by RV failure and conduction abnormalities. The RV is not structurally homologous to the LV, and the RV has not been evolved to carry lifelong systemic pressure load. In men, RV failure commonly occurs during the fifth and sixth decades of life. In women, it may manifest during the first pregnancy. The pathophysiology of the RV failure is not well understood. Exercise studies have shown that the cardiac reserve of the system RV is less than the normal systemic LV. The right coronary system may not provide adequate perfusion to support the hypertrophic RV, leaving the systemic RV vulnerable to ischemia. RV with exuberant hypertrophy is particularly at risk because the perfusion per unit weight of tissue is decreased. The closed tricuspid valve, which normally operates against a 20 mm Hg systolic pulmonary pressure, must now hold against a 100 mm Hg systolic systemic pressure. This, together with the dysplastic and dysmorphic tricuspid leaflets that often accompany CCTGA, produces substantial regurgitation and volume load to the RV, causing further stress to the RV (Fig. 44-10). Patients without associated cardiac anomalies generally fare better, but both groups show substantial decline in RV function between ages 40 and 50 years. The probabilities of developing RV failure in patients with and without associated anomalies are 55% and 25%, respectively, at age 40, and 90% and 65% at age 50 years.15
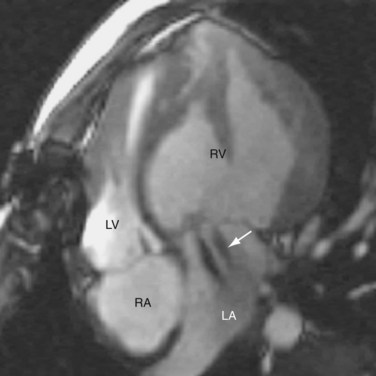
 FIGURE 44-10
FIGURE 44-10DIFFERENTIAL DIAGNOSIS
From Imaging Findings
Both CTGA and CCTGA share the configuration of parallel ascending aorta and pulmonary trunk (Fig. 44-11). Distinguishing between the two types of transpositions requires additional imaging evidence. In CTGA, the ventricles are usually in their normal positions (see Fig. 44-3) and the aortic root tends to be on the right of the pulmonary root see (Fig. 44-5). In CCTGA, the ventricular positions are reversed (see Fig. 44-4) and the aortic root tends to be on the left of the pulmonary root (see Fig. 44-6). However, as discussed earlier, these signs are not completely reliable. It is better to trace the connections between the venous returns and great arteries. In CTGA, the pulmonary veins are ultimately connected to the pulmonary artery through the left ventricle and the venae cavae are connected to the aorta through the right ventricle. In CCTGA, blood flow from the pulmonary veins reaches the aorta through the right ventricle and blood flow from the venae cavae reaches the pulmonary trunk through the left ventricle.
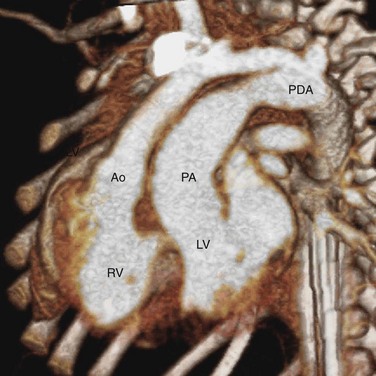
 FIGURE 44-11
FIGURE 44-11Differentiation between CTGA and double-outlet right ventricle (DORV) can be challenging, especially when the DORV has a doubly committed large VSD. In DORV, more than 50% of both the aorta and pulmonary trunk should be aligned with the right ventricle. In contrast, CTGA has the aorta aligned mostly with the right ventricle and the pulmonary trunk aligned mostly with the left ventricle. When both outlet valves are separated from the inlet valves by intervening conal muscle, or bilateral conus, this is considered a sign of DORV (Fig. 44-12). In CTGA, the mitral valve remains in fibrous continuity with the pulmonary valve, without intervening conal muscle.
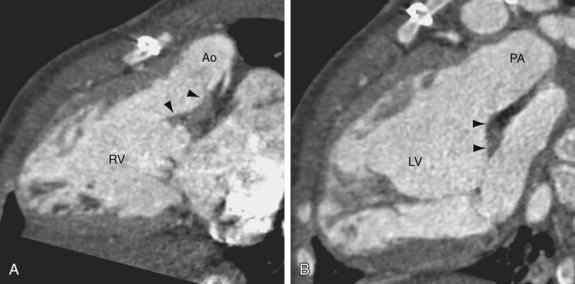
 FIGURE 44-12
FIGURE 44-12SYNOPSIS OF TREATMENT OPTIONS
Surgical and Interventional
Surgery for Complete Transposition of the Great Arteries
In CTGA patients presenting with severe cyanosis, keeping the ductus arteriosus open may not be sufficient. Creating or enlarging the ASD to promote shunting at the atrial level is a valid palliative treatment. This is now performed at the bedside with an endovascular balloon atrial septostomy under echocardiography guidance. In this procedure, first described by Rashkind and Miller in 1966,16 a balloon catheter with deflated balloon is passed from the right atrium, through the foramen ovale, to the left atrium. The balloon is inflated and forcibly retracted back to the right atrium, tearing the atrial septum to allow increased interatrial mixing. The goal is to increase systemic oxygen saturation to the 50% to 80% range. Although generally safe and widely used, balloon septostomy has been associated with an increased risk of stroke.17
The preferred surgical correction of CTGA today is the arterial switch procedure, or the Jatene procedure, named after its creator, Adib Jatene.3 Although conceptually straightforward, the arterial switch operation is technically difficult because of the need to transpose the tiny coronary arteries with the aorta. For this reason, the atrial switch procedure was developed first. Arterial switching became possible in the 1970s when a surgical technique was developed to reconnect the coronary arteries to the transposed aorta via coronary buttons. In this procedure, the ascending aorta and pulmonary trunk are transected. The coronary ostia, along with a large button of surrounding aortic wall, are excised from the aortic sinus of Valsalva. The proximal segments of the coronary arteries are freed from the surface of the heart to prevent tension or distortion after reimplantation. In a surgical manipulation called the Lecompte maneuver, the transected upper aorta is moved behind the pulmonary trunk so that the left and right main pulmonary arteries are now straddling the aorta (Fig. 44-13). The aorta is anastomosed to the proximal pulmonary trunk, creating a neoaorta. Openings are created at the appropriate positions on the neoaorta and the coronary buttons are sutured to these openings (Fig. 44-14). Finally, the transected pulmonary trunk, now located anteriorly to the aorta, is anastomosed to the proximal aorta. Any defects created by the coronary buttons are closed. If ASD and VSD are present, they are closed during the surgery.
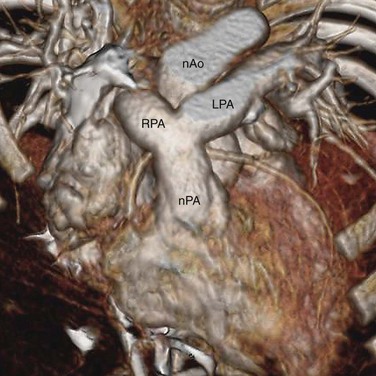
 FIGURE 44-13
FIGURE 44-13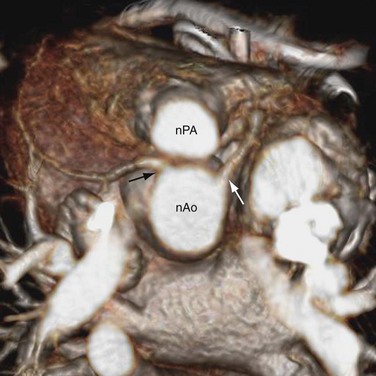
 FIGURE 44-14
FIGURE 44-14The surgical outcome of the arterial switch procedure is favorable. Depending on the series, the surgical mortality is reported to be 2% to 5%.18 Sudden death is uncommon. Late mortality related to coronary occlusion, myocardial infarction, and left ventricular failure occurs in fewer than 1%. Survival rate is 88% at 10 years and unchanged at 15 years.19 Because the coronary arteries are surgically manipulated, the long-term patency of the coronary arteries is of particular concern. In a series of 165 patients who underwent arterial switching, 7% had coronary occlusion, 5% developed more than 50% coronary stenosis, and 4% developed less than 50% coronary stenosis or coronary stretching.20 The rate for surgical intervention for coronary obstruction was 2.4% in this series. Other complications include pulmonary stenosis in 5% to 25% of patients. The obstruction most often occurs at the left main pulmonary artery as it crosses the aorta (Fig. 44-15), followed by the right main pulmonary artery and the pulmonary valve compressed between the aorta and the sternum. Aortic regurgitation is detected in 16% of patients at 5-year follow-up, but only 4% had grade 2 or above, which is considered clinically significant.
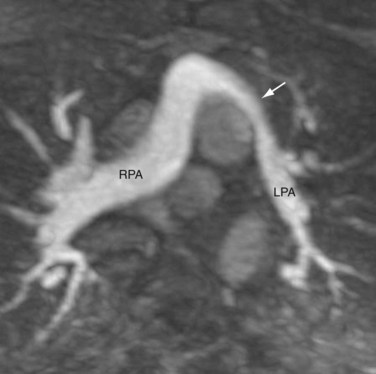
 FIGURE 44-15
FIGURE 44-15The atrial switch procedure, having a less favorable outcome than the arterial switch procedure, is now reserved for special cases in whom the latter is contraindicated. It may also be seen in patients whose surgery predated the arterial switch procedure. The atrial switch operations developed by Senning and Mustard differ only on the methods of constructing the atrial baffle. Senning used native atrial septal tissue for the baffle construction1 whereas Mustard used pericardium or Dacron material.2 The postoperative anatomy and physiology of the two operations are the same. In this procedure, the normal atrial septum is resected. An atrial baffle is sutured to the atrial wall to create two compartments of very complex geometry. The posterior compartment collects flow from the pulmonary veins and channels it to the right ventricle (Fig. 44-16). The anterior compartment collects flow from the two cavae and channels it the left ventricle (Fig. 44-17). In effect, the atrial baffle creates an AV discordance, thereby converting the CTGA to a CCTGA.
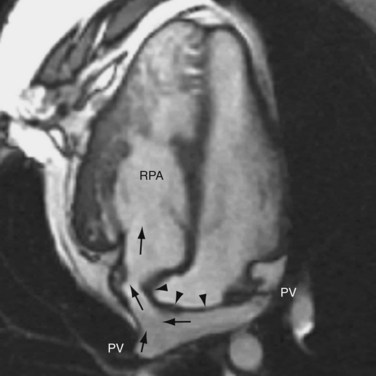
 FIGURE 44-16
FIGURE 44-16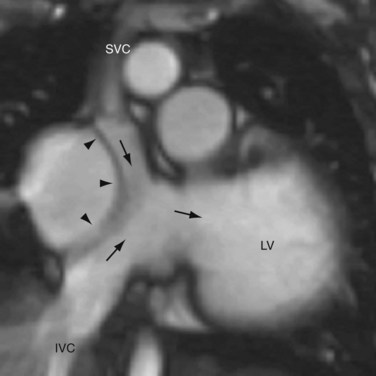
 FIGURE 44-17
FIGURE 44-17Because the atrial baffle does not grow with the patient, the atrial switch procedure is performed later than the arterial switch procedure, typically at 1 month to 1 year of age. The surgical mortality rate has been reported to be 1% to 10%, higher in patients with VSD.21 Late mortality of 7% to 15% sudden death is significantly higher than that of the arterial switch procedure. Survival rate is 90% at 10 years and 76% to 80% at 20 years.22 RV failure, as in CCTGA, is a late complication. Abnormal stretching of the atrial baffle may result in flow obstruction from channel narrowing or baffle leak from suture dehiscence. Although baffle leak is more common than flow obstruction, the former rarely requires reoperation. Flow obstruction typically occurs at the superior vena cava (SVC) channel (5% to 10%) and less commonly at the inferior vena cava (IVC) channel (1% to 2%). Pulmonary hypertension is associated with 7% of patients who had late repair of CTGA-VSD. Arrhythmia in postatrial switch patients is particularly troublesome. At 10 years, 50% of the patients develop bradyarrhythmia from sinus node dysfunction, requiring a pacemaker. Tachyarrhythmia may result from incisional atrial reentry tachycardia. Nearly 50% of patients experience one or more episodes of supraventricular tachycardia and 73% experience atrial flutter.
In CTGA patients with pulmonary stenosis, the Rastelli procedure is the indicated operation. In this procedure, the native pulmonary trunk is transected and the proximal trunk is surgically closed. An intracardiac baffle is placed in the RV to connect the VSD to the RVOT, effectively channeling the LV outflow to the transposed aorta (Fig. 44-18). An extracardiac valved pulmonary conduit is anastomosed to the RV after a ventriculostomy, and the conduit is connected to the distal pulmonary trunk, establishing the pulmonary flow (Fig. 44-19). After surgery, the LV becomes the systemic ventricle. A prerequisite for the Rastelli procedure is that the VSD must be large enough to conduct the left ventricular flow without obstruction. Surgical enlargement of the VSD risks damaging the conduction system. Surgical mortality has been reported at less than 5%.23 Survival rates are 82% at 5 years, 80% at 10 years, 68% at 15 years, and 52% at 20 years. Causes of late mortality include sudden death secondary to arrhythmia and LV failure. Reoperation rate for the Rastelli procedure is high mainly because the pulmonary conduit develops pulmonary stenosis or regurgitation, requiring replacement every 10 to 20 years.
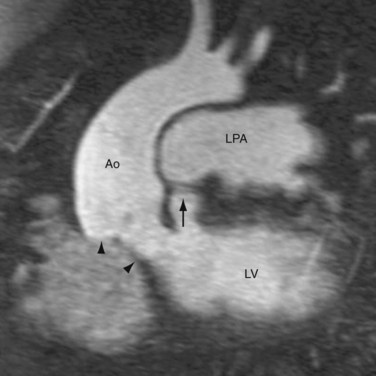
 FIGURE 44-18
FIGURE 44-18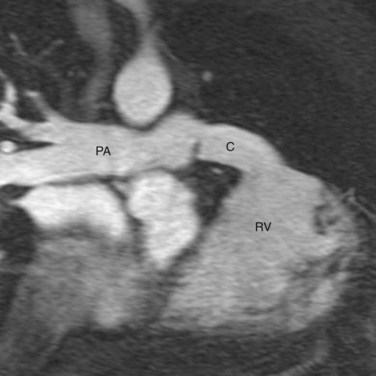
 FIGURE 44-19
FIGURE 44-19The Damus-Kaye-Stansel procedure is infrequently used in patients with TGA or Taussig-Bing anomalies. Indications are difficult coronary anatomy, subaortic stenosis, and absence of pulmonary stenosis.24 In patients who had atrial switching, pulmonary artery banding may improve the systemic RV function by increasing LV pressure and shifting the ventricular septum to the right. In patients whose anatomy is amendable, a reverse switch procedure may be performed to convert the atrial switching to arterial switching. However, the pulmonary LV must first be conditioned, or trained, to handle the systemic pressure load. This is accomplished by raising the pulmonary resistance with a pulmonary artery band (Fig. 44-20) and forcing the LV to pump at a higher pressure. The band is gradually tightened until the LV pumps at near systemic pressure.
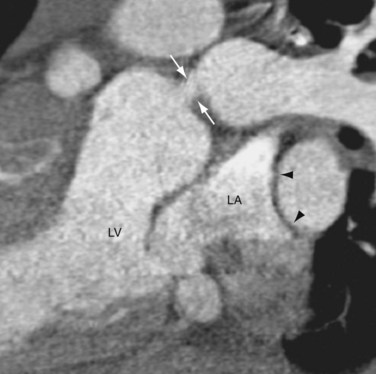
 FIGURE 44-20
FIGURE 44-20Surgery for Congenitally Corrected Transposition of the Great Arteries
In infants with CCTGA, palliative procedures are geared toward finding the right balance for the pulmonary flow. In patients with a large VSD, pulmonary artery banding is indicated to restrict the pulmonary flow and reduce the risk of developing pulmonary obstructive vascular disease. In patients with severe pulmonary stenosis and VSD, a Blalock-Taussig shunt is indicated to improve pulmonary flow and reduce cyanosis. Beyond palliation, operations are commonly performed to correct the anomalies other than CCTGA. These include VSD closure to remove shunting, resection to relieve pulmonary stenosis, and repair or annuloplasty of the tricuspid valve to reduce tricuspid regurgitation. The goals of these procedures are to reduce ventricular workloads and preserve ventricular functions. Compared with repairs of the same lesions in normal ventricles, the postoperative prognosis is worse for CCTGA. This may be related to conduction abnormality, worsening tricuspid regurgitation, and coronary anomalies inherent in CCTGA. Thirty-eight percent of postoperative patients develop complete AV block. Most patients undergo reoperation for atrioventricular valve regurgitations and pulmonary stenosis. The overall surgical mortality is 6%; the late survival rate is 55% to 85% at 10 years and 48% at 20 years.25 Causes of death include reoperation, sudden death from arrhythmia, and progressive systemic RV failure.
In light of the long-term risk of RV failure, Ilbawi and colleagues5 and others26 have advocated the double switch procedure, which combines atrial and the arterial switching, as the definitive treatment for CCTGA. The purpose of this operation is to return the LV as the systemic ventricle. However, just as in the reverse switch procedure, the LV must be trained first to handle the systemic pressure load by pulmonary artery banding. The time required for LV training depends on the age of the patient, from a few weeks for infants to 1 year for older children. There is evidence that the LV responds poorly to training in patients older than 16 years.27 The reported surgical mortality is 0% to 11%. Patients are also at risk for the complications associated with the atrial switch procedure. Although the immediate postoperative survival rate of the double switch procedure is good, it has yet to demonstrate long-term benefits over conventional operations that leave the RV as the systemic ventricle.28
IMAGING INDICATIONS AND REPORTING
Imaging for Complete Transposition of the Great Arteries
The manifestations of CTGA on chest radiographs are variable, from normal to shunt vascularity and increased heart size, depending on the size of the VSD. The classic cardiomediastinal silhouette of “egg on a string” is seen only in one third of cases (Fig. 44-21). Echocardiography is the definitive imaging modality. In the parasternal long-axis view, the LV is connected to the pulmonary trunk. In the short-axis view, the aorta is anterior and to the right of the pulmonary trunk. In the suprasternal view, the ascending aorta lies parallel to the pulmonary trunk. Echocardiography should assess for secondary lesions, such as ASD, VSD, and PDA, and the coronary anatomy, if possible. Doppler technique is used to detect any hemodynamically significant subvalvular and valvular stenoses of the outlet valves. Catheter angiography, CT, and MRI are generally not necessary for the initial diagnosis and management, except for the evaluation of the coronary anatomy when echocardiography is unclear. In rare cases, when the ventriculoarterial alignments and their relationship to the VSD are complex, three-dimensional imaging with cardiac-gated CT or MRI can be helpful for surgical planning.
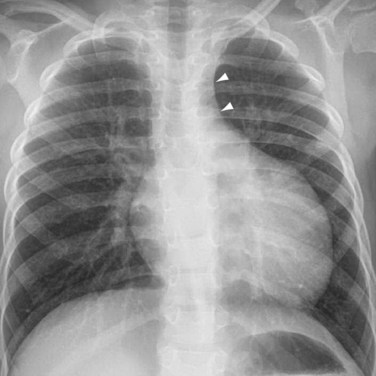
 FIGURE 44-21
FIGURE 44-21In the early days, after the development of the arterial switch procedure, catheter coronary angiography was routinely performed in postoperative patients to assess coronary patency. With experience and improved surgical outcome, coronary angiography is now reserved for patients presenting with signs or symptoms of cardiac ischemia. Because the obstructions likely occur in the proximal coronary arteries where they have been surgically manipulated, the stenoses can be evaluated noninvasively with MRI or CT coronary angiography (Fig. 44-22). For patients with detected coronary stenoses, a stress test such as a stress myocardial perfusion study or stress echocardiography should be performed to determine the clinical significance of the lesions. On occasion, aortic regurgitation or pulmonary stenosis develops after the arterial switch operation. These complications, along with their ventricular responses, are monitored with echocardiography, supplemented by MRI quantifications of ventricular volumes and valvular regurgitant fractions if necessary.
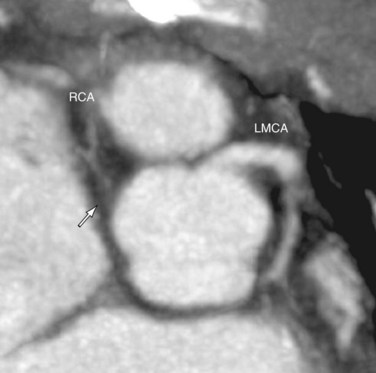
 FIGURE 44-22
FIGURE 44-22After pulmonary artery banding for a planned reverse switch procedure, Doppler imaging is used to estimate the pressure gradient at the pulmonary band and the left ventricular systolic pressure from a mitral regurgitant jet. The latter helps monitor whether the trained left ventricle is ready to take on systemic pressure load. This pressure load should be confirmed by catheter measurement before surgery. In some centers, cardiac MRI is used to monitor the increase in the left ventricular mass during the LV training (Fig. 44-23). This is calculated as the epicardial volume of the LV minus the endocardial volume of the LV, multiplied by myocardial tissue density, 1.05 g/mL.29 The goal of training is to attain an LV mass comparable to that of a normal systemic LV, estimated at 60 g/m2 body surface area or higher.30
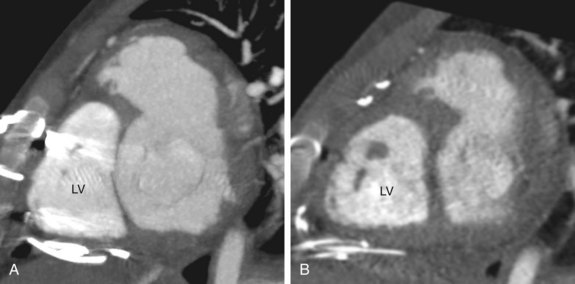
 FIGURE 44-23
FIGURE 44-23The imaging management of patients who have undergone the Rastelli procedure is similar to that of those who had complete surgical repair for tetralogy of Fallot. Complications center on the pulmonary artery conduit, stenosis causing excessive pressure load to the RV (Fig. 44-24) and regurgitation causing excessive volume load to the RV. Echocardiography and MRI are indicated to monitor RV volume, RV ejection fraction, and pulmonary regurgitation severity. MRI has the advantage of providing accurate quantitative measurements of these parameters for serial comparison. As in the case of repaired tetralogy of Fallot, the goal is to postpone conduit replacement until these parameters suggest potentially irreversible deterioration of the RV function. The critical values of these parameters are under ongoing investigation. In addition, the intracardiac baffle created by the Rastelli procedure may leak or it may cause obstruction between the VSD and transposed aortic valve. Rarely, clots can form in the blind-ended pulmonary trunk. Dislodgment of these clots into the baffled channel causes systemic thromboembolism. These complications are typically evaluated with echocardiography.
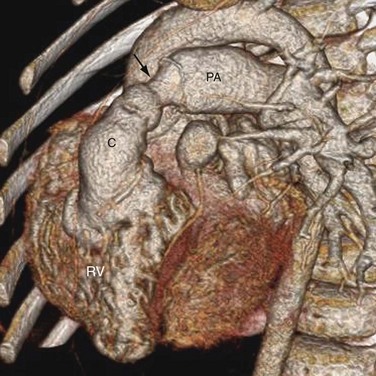
 FIGURE 44-24
FIGURE 44-24Imaging for Congenitally Corrected Transposition of the Great Arteries
Second, the ventricular morphology is evaluated. A right ventricle typically has course trabeculations (see Fig. 44-3), a prominent moderator band, and separation of the inlet-outlet valves (see Fig. 44-1). In contrast, a left ventricle has fine trabeculations except at the apex, absence of a moderator band, and fibrous continuity of the inlet-outlet valves without muscle in between (see Fig. 44-2). The diagnosis of CCTGA requires that the right atrium is aligned with the left ventricle and the left atrium aligned with the right ventricle, or atrioventricular discordance. In addition, it requires that the right ventricle be aligned with the aorta and the left ventricle is aligned with the pulmonary trunk, or ventriculoarterial discordance. Differentiation of the aorta and pulmonary trunk is usually straightforward by echocardiography and MRI.







Graham TP. Congenitally corrected transposition. In: Gatzoulis MA, Webb GD, Daubeney PEF, editors. Diagnosis and management of adult congenital heart disease. Edinburgh: Churchill Livingstone; 2003:379-387.
Hornung T. Transposition of the great arteries. In: Gatzoulis MA, Webb GD, Daubeney PEF, editors. Diagnosis and management of adult congenital heart disease. Edinburgh: Churchill Livingstone; 2003:349-362.
Warnes CA. Transposition of the great arteries. Circulation. 2006;114:2699-2709.
Yuh DD, Reitz BA. Cyanotic defects. In: Reitz BA, Yuh DD, editors. Congenital Cardiac Surgery. New York: McGraw-Hill; 2002:137-143.
1 Senning A. Surgical correction of transposition of the great vessels. Surgery. 1959;45:966-980.
2 Mustard WT. Successful two-stage correction of transposition of the great vessels. Surgery. 1964;55:469-472.
3 Jatene AD, Fontes VF, Paulista PP, et al. Anatomic correction of transposition of the great vessels. J Thorac Cardiovasc Surg. 1976;72:364-370.
4 Rastelli GC, McGoon DC, Wallace RB. Anatomic correction of transposition of the great arteries with ventricular septal defect and subpulmonary stenosis. J Thorac Cardiovasc Surg. 1969;58:545-552.
5 Ilbawi MN, DeLeon SY, Backer CL, et al. An alternative approach to the surgical management of physiologically corrected transposition with ventricular septal defect and pulmonary stenosis or atresia. J Thorac Cardiovasc Surg. 1990;100:410-415.
6 Van Praagh R, Weinberg PM, Smith SD. Malpositions of the heart. In: Adams FH, Emmanoulides GC, Riemenschneider TA, editors. Moss’ Heart Disease in Infants, Children and Adolescents. Baltimore: Lippincott Williams & Wilkins; 1989:530-580.
7 Van Praagh R, Papagiannis J, Grunenfelder J, et al. Pathologic anatomy of corrected transposition of the great arteries: medical and surgical implications. Am Heart J. 1998;135:772-785.
8 Pierpont ME, Basson CT, Benson DWJr, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015-3038.
9 Burn J, Brennan P, Little J, et al. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet. 1998;351:311-316.
10 Development of the cardiovascular and lymphatic systems. In: Standring S, editor. Gray’s anatomy: the anatomical basis of clinical practice. 39th ed. Edinburgh: Churchill Livingstone; 2005:1029-1055.
11 Wernovsky G, Sanders SP. Coronary artery anatomy and transposition of the great arteries. Coron Artery Dis. 1993;4:148-157.
12 Lundstrom U, Bull C, Wyse RK, Somerville J. The natural and “unnatural” history of congenitally corrected transposition. Am J Cardiol. 1990;65:1222-1229.
13 McKay R, Anderson RH, Smith A. The coronary arteries in hearts with discordant atrioventricular connections. J Thorac Cardiovasc Surg. 1996;111:988-997.
14 Liebman J, Cullum L, Belloc NB. Natural history of transpositon of the great arteries. Anatomy and birth and death characteristics. Circulation. 1969;40:237-262.
15 Graham TPJr, Bernard YD, Mellen BG, et al. Long-term outcome in congenitally corrected transposition of the great arteries: a multi-institutional study. J Am Coll Cardiol. 2000;36:255-261.
16 Rashkind WJ, Miller WW. Creation of an atrial septal defect without thoracotomy. A palliative approach to complete transposition of the great arteries. JAMA. 1966;196:991-992.
17 McQuillen PS, Hamrick SE, Perez MJ, et al. Balloon atrial septostomy is associated with preoperative stroke in neonates with transposition of the great arteries. Circulation. 2006;113:280-285.
18 Castaneda AR, Trusler GA, Paul MH, et al. The early results of treatment of simple transposition in the current era. J Thorac Cardiovasc Surg. 1988;95:14-28.
19 Losay J, Touchot A, Serraf A, et al. Late outcome after arterial switch operation for transposition of the great arteries. Circulation. 2001;104:I121-I126.
20 Bonhoeffer P, Bonnet D, Piechaud JF, et al. Coronary artery obstruction after the arterial switch operation for transposition of the great arteries in newborns. J Am Coll Cardiol. 1997;29:202-206.
21 Trusler GA, Williams WG, Duncan KF, et al. Results with the Mustard operation in simple transposition of the great arteries 1963-1985. Ann Surg. 1987;206:251-260.
22 Wilson NJ, Clarkson PM, Barratt-Boyes BG, et al. Long-term outcome after the mustard repair for simple transposition of the great arteries: 28-year follow-up. J Am Coll Cardiol. 1998;32:758-765.
23 Kreutzer C, De VJ, Oppido G, et al. Twenty-five-year experience with rastelli repair for transposition of the great arteries. J Thorac Cardiovasc Surg. 2000;120:211-223.
24 DeLeon SY, Ilbawi MN, Tubeszewski K, et al. The Damus-Stansel-Kaye procedure: anatomical determinants and modifications. Ann Thorac Surg. 1991;52:680-687.
25 Yeh TJr, Connelly MS, Coles JG, et al. Atrioventricular discordance: results of repair in 127 patients. J Thorac Cardiovasc Surg. 1999;117:1190-1203.
26 Devaney EJ, Charpie JR, Ohye RG, Bove EL. Combined arterial switch and Senning operation for congenitally corrected transposition of the great arteries: patient selection and intermediate results. J Thorac Cardiovasc Surg. 2003;125:500-507.
27 Winlaw DS, McGuirk SP, Balmer C, et al. Intention-to-treat analysis of pulmonary artery banding in conditions with a morphological right ventricle in the systemic circulation with a view to anatomic biventricular repair. Circulation. 2005;111:405-411.
28 Shin’oka T, Kurosawa H, Imai Y, et al. Outcomes of definitive surgical repair for congenitally corrected transposition of the great arteries or double outlet right ventricle with discordant atrioventricular connections: risk analyses in 189 patients. J Thorac Cardiovasc Surg. 2007;133:1318-1328.
29 Vinnakota KC, Bassingthwaighte JB. Myocardial density and composition: a basis for calculating intracellular metabolite concentrations. Am J Physiol Heart Circ Physiol. 2004;286:H1742-H1749.
30 Lorenz CH, Walker ES, Morgan VL, et al. Normal human right and left ventricular mass, systolic function, and gender differences by cine magnetic resonance imaging. J Cardiovasc Magn Reson. 1999;1:7-21.
