http://evolve.elsevier.com/McCuistion/pharmacology/
Organ Transplantation
Organ transplantation is a life-saving procedure. In cadaveric transplantation, a healthy organ donated at the time of a person’s death is transplanted into the body of a patient with end-stage organ failure. In living-donor transplantation, a kidney or a portion of liver donated by a living person is transplanted into the body of a patient with end-stage kidney or liver disease. Organ transplant is an acceptable treatment option when organs fail (e.g., kidney, heart, liver, and lung). More than 122,000 people are currently waiting for an organ transplant. Every 10 minutes, another person is added to the wait list, and over 8000 people die each year while waiting for a donor organ.
Principles of Immunosuppression
The immune system remains the biggest barrier to transplantation as a routine medical treatment because it has effective mechanisms to fight off foreign organisms. These same mechanisms are involved in the rejection of transplanted organs, which are recognized as foreign by the recipient’s immune system. The underlying premise of immunosuppression is to use multiple drugs that alter different aspects of the immune system (Fig. 30.1), thereby reducing the chances of transplant rejection and enabling the use of lower doses of individual drugs, which reduces the likelihood of drug toxicity. Transplantation has revolutionized care for patients with end-stage organ failure, yet significant problems remain with treatments designed to promote transplant survival and prevent rejection. Immunosuppressant drugs are not always effective; in addition, they are expensive and must be taken daily, and they are associated with toxic effects.
Immunosuppressant Drugs
Induction Therapy
Induction therapy provides intense immunosuppression with drugs designed to diminish antigen presentation and T-cell response, thus reducing the risk for acute rejection during the initial transplant period.
Basiliximab is a monoclonal antibody that inhibits interleukin 2 (IL-2)–mediated activation of lymphocytes, a critical component of the cellular immune response involved in transplant rejection. By inhibiting activation of lymphocytes, it prevents the body from mounting an immune response against the transplanted organ. Basiliximab has been approved for induction therapy in kidney transplants. Complete pharmacokinetic data are not available, but drug half-life is known to be 7.2 days in adults and 9.5 days in children.
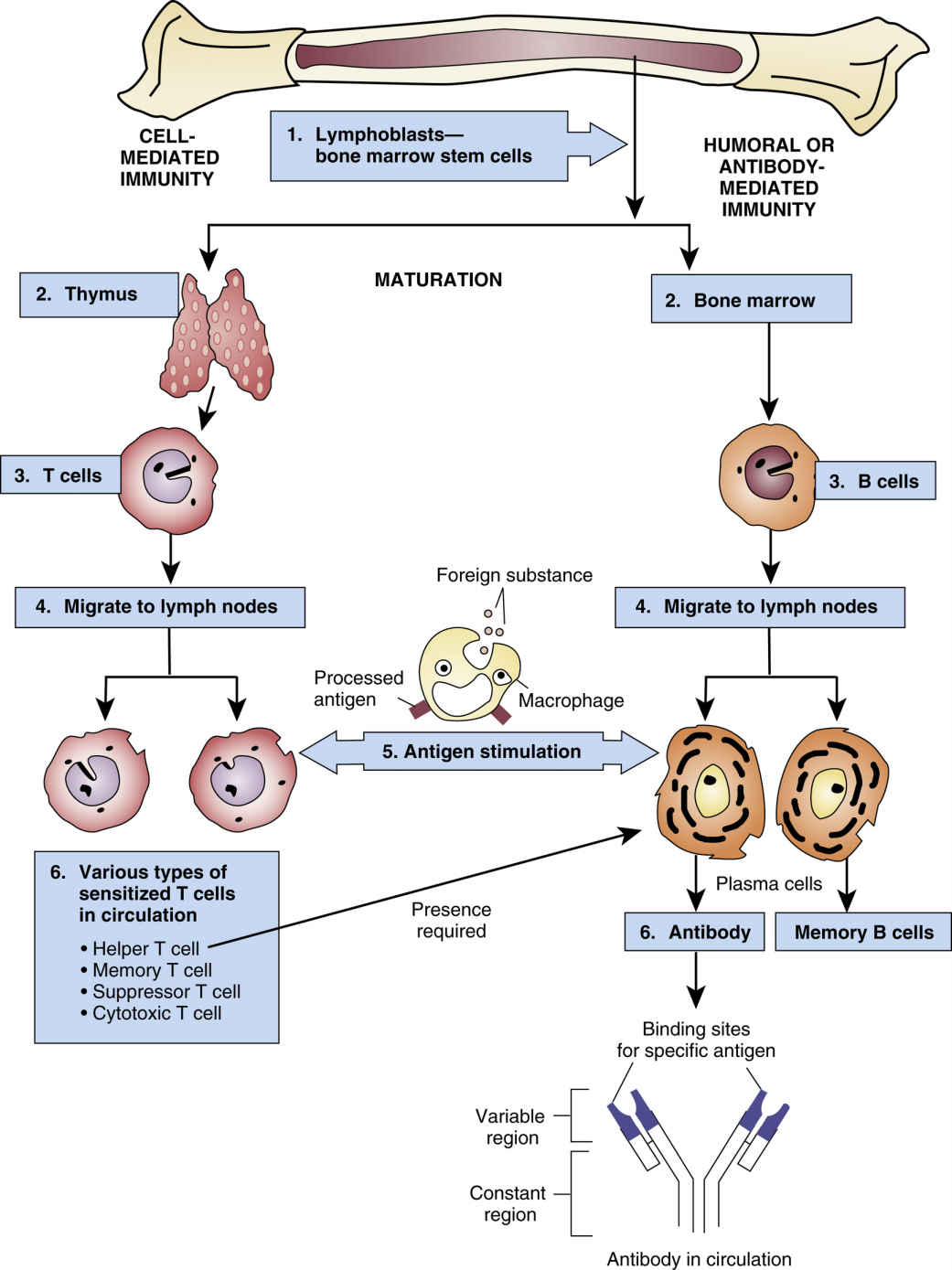
FIG. 30.1 The Immune Response.
From Gould, B., & Dyer, R. (2011). Pathophysiology for the health professions (4th ed.). St Louis: Saunders.
Basiliximab is administered intravenously (IV), 20 mg within 2 hours before transplant surgery, followed by a second 20-mg dose 4 days after transplantation. The second dose should be withheld if complications occur (including severe hypersensitivity reactions or loss of the transplanted organ). Children under 35 kg should receive 10 mg IV within 2 hours before transplant surgery, followed by a second 10-mg dose 4 days after transplantation; the second dose should be withheld if complications occur (including severe hypersensitivity reactions or loss of the transplanted organ); children over 35 kg should receive the adult dose.
Side effects of basiliximab include abdominal and back pain, coughing, dizziness, fever or chills, fatigue, weakness, dysuria, dyspnea, sore throat, edema, tremor, nausea and vomiting, and anemia. Serious reactions include sepsis, opportunistic infections, malignancy, lymphoproliferative disorders, thrombocytopenia, leukopenia, diabetes mellitus, anaphylaxis, capillary leak syndrome, and cytokine release syndrome (Box 30.1).
Transplant recipients who receive basiliximab should not receive live vaccines because they may produce an inadequate immune response and are at risk for disseminated infection resulting from the live virus. Caution is advised when basiliximab is administered with other drugs that lower the immune response because of the increased risk of serious infection.
Basiliximab was designated pregnancy category B; no adequate and well-controlled studies have been done on the drug’s use in pregnant women, therefore women of childbearing potential should use effective contraceptive measures before beginning treatment, during treatment, and for 4 months after completion of therapy. It is not known whether basiliximab is excreted in breast milk. Because of the potential for adverse drug reactions, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Maintenance Therapy
Calcineurin Inhibitors
The calcineurin inhibitors (CNIs) suppress the immune system by binding to cytoplasmic proteins that inhibit calcineurin phosphatase, resulting in a reduction in cytokine synthesis and inhibition of T-lymphocyte proliferation. There are two CNIs, cyclosporine and tacrolimus; the prototype CNI is cyclosporine. The drug became available in 1983 and was modified to improve bioavailability in 1994.  Cyclosporine oral solution USP modified and cyclosporine oral solution USP are not bioequivalent and cannot be used interchangeably.
Cyclosporine oral solution USP modified and cyclosporine oral solution USP are not bioequivalent and cannot be used interchangeably.
Cyclosporine oral solution USP modified and cyclosporine oral solution USP are not bioequivalent and cannot be used interchangeably.After oral administration, absorption of cyclosporine is incomplete and varies widely from patient to patient. Drug distribution is concentration dependent; cyclosporine is 90% protein bound. The drug is extensively metabolized by the cytochrome P450 (CYP450) 3A enzyme system in the liver. Elimination is primarily biliary with only 6% excreted in urine. The average drug half-life is 8.4 hours.
Initial dosing of cyclosporine is 7 to 9 mg/kg orally per day in two divided doses; the first dose is given 4 to 12 hours before the transplant or postoperatively in both adult and pediatric patients. Adjustments to drug dosing are based on therapeutic drug monitoring (TDM), with the desired level between 100 and 500 ng/mL, depending on the organ transplanted and the length of time posttransplant.
The daily dose of cyclosporine oral solution USP modified should always be given in two divided doses and on a consistent schedule with regard to time of day and relation to meals. Grapefruit and grapefruit juice affect drug metabolism, increasing blood concentration of cyclosporine; for this reason, grapefruit should be avoided. To make cyclosporine oral solution USP modified more palatable, it should be diluted with room-temperature orange or apple juice; patients should avoid switching diluents frequently. When mixed with juice, the cyclosporine solution may appear cloudy. Cyclosporine is also available as immediate-release capsules (25 mg and 100 mg) and as an IV solution (250 mg/5 mL).
Common side effects of cyclosporine include elevated blood urea nitrogen (BUN) and creatinine, hypertension, hirsutism, infection, tremor, gingival hyperplasia, headache, hypertriglyceridemia, nausea and vomiting, diarrhea, hyperuricemia, hyperglycemia, arthralgia, edema, acne, hypomagnesemia, and hyperkalemia. Serious reactions include opportunistic infections, nephropathy and nephrotoxicity, diabetes mellitus, leukopenia, thrombocytopenia, hemolytic anemia, malignancy, and seizures.
Transplant recipients taking cyclosporine should not receive live vaccines because they may have inadequate immune response and are at risk for disseminated infection resulting from the live virus.
Multiple antibiotics, melphalan (an antineoplastic drug), antifungals, antiinflammatory drugs, cimetidine and ranitidine (histamine 2–receptor blockers), tacrolimus (an immunosuppressant), fibric acid derivatives, and methotrexate may potentiate kidney dysfunction when administered with cyclosporine. Calcium channel blockers, azole antifungals, macrolide antibiotics, and glucocorticoids can increase cyclosporine concentrations, as can allopurinol, amiodarone, bromocriptine, colchicine, danazol, imatinib, metoclopramide, nefazodone, and oral contraceptives. Other drugs—nafcillin, rifampin, anticonvulsants, bosentan, octreotide, orlistat, sulfinpyrazone, terbinafine, ticlopidine—and St. John’s wort can decrease cyclosporine concentrations.
Concomitant administration of cyclosporine with 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors (e.g., lovastatin, simvastatin, pravastatin, fluvastatin, and atorvastatin) may increase the risk for rhabdomyolysis. The dosage of HMG-CoA reductase inhibitors should be reduced.
In addition to TDM (drug levels should be drawn just prior to the dose), patients taking cyclosporine should have frequent monitoring of BUN, creatinine, potassium, and magnesium levels in addition to liver function tests (LFTs) and lipid profiles.
Cyclosporine was designated pregnancy category C; it does not appear to be a major human teratogen; however, it may be associated with increased rates of prematurity. Cyclosporine is present in breast milk. Because of the potential for serious adverse drug reactions in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Tacrolimus
Approved by the FDA in 1994, tacrolimus is the second calcineurin inhibitor approved for prophylaxis of rejection in heart, liver and kidney transplants. It carries a boxed warning for malignancies and serious infections. Patients taking this drug are at increased risk for developing lymphoma and other malignancies. Additionally, patients taking tacrolimus are at risk for developing bacterial, viral, fungal, and protozoal infections.
Dosing for heart transplant recipients begins no sooner than 6 hours posttransplant with a continuous infusion at 0.01 mg/kg/day IV. When oral dosing begins, it should be at a dose of 0.075 mg/kg/day orally divided every 12 hours. For patients with a liver transplant, tacrolimus is administered with a continuous infusion at 0.03 to 0.05 mg/kg/day IV. When oral dosing begins, it should be dosed 0.1 to 0.15mg/kg/day orally divided every two hours. Kidney transplant recipients should begin tacrolimus within 24 hours of receiving the transplanted organ. Dosing ranges from 0.1 to 0.2 mg/kg/day orally divided every 12 hours, and may vary based on drugs used for induction.
For all transplant recipients, dosing is adjusted based on serum drug levels with trough levels ranging between 5 and 20 ng/mL, depending on organ transplanted and length of time since transplant.
The drug is highly nephrotoxic and should be administered at the lowest recommended dose for those with renal impairment. For those with postoperative oliguria, use of tacrolimus may be delayed until kidney function is adequate. All patients on tacrolimus should have their kidney function monitored periodically during therapy.
Tacrolimus may prolong the QT/QTc interval and may cause Torsade de Pointes. Avoid Tacrolimus in patients with congenital long QT syndrome. In patients with congestive heart failure, bradyarrhythmias, those taking certain antiarrhythmic medications or other medicinal products that lead to QT prolongation, and those with electrolyte disturbances such as hypokalemia, hypocalcemia, or hypomagnesemia, consider obtaining electrocardiograms and monitoring electrolytes (magnesium, potassium, calcium) periodically during treatment.
Tacrolimus is slowly absorbed in the GI tract. Bioavailability averages 25% and peak serum concentration is reached in 3 hours. Food, especially food high in fat, slows absorption and reduces bioavailability. It is metabolized extensively in the liver and is over 90% excreted in the feces, with the remaining excreted in the urine. Genetic variations in activity of the CYP3A5 protein can affect serum concentrations of tacrolimus. Its half-life is variable depending on organ transplanted (3.5 to 40.6 hours).
Tacrolimus has multiple common side effects, including tremor, diarrhea, headache, hypertension, nephrotoxicity, infection, insomnia, electrolyte, metabolic and lipid abnormalities, constipation, edema, fever, anemia, hyperglycemia, hepatotoxicity, anorexia, dyspepsia, dyspnea, pruritus, dizziness, cough, leukopenia, and photosensitivity. Serious reactions include malignancy, posttransplant lymphoproliferative disorder, severe infections, Stevens-Johnson syndrome, toxic epidermal necrolysis, anaphylaxis, neurotoxicity, seizures, myocardial hypertrophy, QT prolongation, torsades de pointes, pericardial effusion, diabetes, myelosuppression, DIC, thrombocytopenic purpura, and hemolytic anemia.
Persons receiving tracrolimus have absolute contraindication for live vaccines, mifepristone, pimozide, quinidine, saquinavir, streptozocin, talimogene laherparepvec (a genetically modified oncolytic viral therapy used in patients with recurrent melanoma), and ziprasidone. Multiple other drugs should be used with caution. Protease inhibitors may increase serum drug levels, as can antifungal agents, calcium channel blockers, gastric acid suppressors/antacids, and antibacterials. Anticonvulsants can decrease serum drug levels, as can St. John’s wort. Patients receiving tacrolimus should avoid grapefruit juice, as it too, may increase serum drug levels.
Costimulation Blockers
Belatacept is a first-in-class selective T-cell costimulation blocking agent indicated for use in combination with basiliximab induction, mycophenolate mofetil, and corticosteroids to prevent kidney transplant rejection. It inhibits T-cell CD28 activation and proliferation by binding costimulatory ligands (CD80, CD86) of antigen-presenting cells, thereby inhibiting T-lymphocyte proliferation and the production of the cytokines IL-2 and IL-4, interferon-alfa, and tumor necrosis factor alpha (TNF-α), which are involved in systemic inflammation. Metabolism and excretion of belatacept are unknown, but half-life is 8.2 to 9.8 days.
The prescribed dose must be divisible by 12.5 to accurately prepare the dose from the reconstituted solution. Initial dosing is 10 mg/kg IV prior to surgery on the day of transplant; the dose is repeated on day 5 and at the end of weeks 2, 4, 8, and 12 after transplantation. Maintenance dosing is 5 mg/kg IV at the end of week 16 after transplantation and then every 4 weeks thereafter. Belatacept is not approved for pediatric use.
 This drug carries a boxed warning for increased risk of developing posttransplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS). Additionally, recipients without immunity to EBV are at a particularly increased risk, therefore belatacept is for use in EBV-seropositive patients only.
This drug carries a boxed warning for increased risk of developing posttransplant lymphoproliferative disorder (PTLD), predominantly involving the central nervous system (CNS). Additionally, recipients without immunity to EBV are at a particularly increased risk, therefore belatacept is for use in EBV-seropositive patients only.Common side effects of belatacept include infection, anemia, diarrhea, peripheral edema, hypertension, constipation, fever, cough, nausea and vomiting, altered potassium levels, headache, leukopenia, abdominal pain, dyslipidemia, hypophosphatemia, arthralgia, hyperglycemia, proteinuria, increased creatinine, insomnia, hypocalcemia, back pain, dysuria, and anxiety. Serious reactions include PTLD, malignancy, serious infections, progressive multifocal leukoencephalopathy (PML), neutropenia, acute renal failure, nephropathy, and diabetes mellitus.
[level-membership-for-nursing-midwifery-medical-assistant-category]
Transplant recipients receiving belatacept should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when belatacept is administered with other drugs that lower the immune response because of the increased risk of serious infection.
Belatacept was designated pregnancy category C, and adverse events have been observed in animal studies. According to the manufacturer, belatacept should not be used in pregnancy unless the potential benefit to the mother outweighs the potential risk to the fetus.  A pregnancy registry has been established to monitor outcomes of women exposed to belatacept during pregnancy (1-877-955-6877). Safety during breastfeeding is unknown, therefore a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
A pregnancy registry has been established to monitor outcomes of women exposed to belatacept during pregnancy (1-877-955-6877). Safety during breastfeeding is unknown, therefore a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
A pregnancy registry has been established to monitor outcomes of women exposed to belatacept during pregnancy (1-877-955-6877). Safety during breastfeeding is unknown, therefore a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.Mammalian Target of Rapamycin Inhibitors
There are two mammalian target of rapamycin (mTOR) inhibitors approved for the prevention of organ rejection in kidney transplant recipients aged 13 years and older: sirolimus and everolimus. Sirolimus is the prototype drug: it inhibits T-lymphocyte activation and proliferation and inhibits antibody production by binding to the FK binding protein, thereby inhibiting IL-2–mediated signal transduction. This results in cell-cycle arrest in the G1-S phase (growth phase and DNA replication), blocking T- and B-cell activation by cytokines. Time to peak concentration is 1 to 3 hours for sirolimus solution and 1 to 6 hours for tablets. Systemic bioavailability is 14% for solution and 47% for tablets. Sirolimus is extensively protein bound (92%) and is extensively metabolized in the intestine and liver. Excretion is 91% in the feces and 2.2% in the urine, and drug half-life is 2.5 days.
 There are two boxed warnings for sirolimus: use of sirolimus increases susceptibility to infection and the possible development of lymphoma and other malignancies, and the safety and efficacy has not been established in liver or lung transplant patients, therefore use is not advised in liver or lung transplant patients due to increased morbidity and mortality.
There are two boxed warnings for sirolimus: use of sirolimus increases susceptibility to infection and the possible development of lymphoma and other malignancies, and the safety and efficacy has not been established in liver or lung transplant patients, therefore use is not advised in liver or lung transplant patients due to increased morbidity and mortality.For patients who weigh less than 40 kg, the loading dose should be 3 mg/m2 once orally as soon as possible after transplantation; maintenance dosing should be adjusted based on body surface area (BSA) to 1 mg/m2 per day orally. In patients who weigh more than 40 kg, the loading dose is 6 mg once orally as soon as possible after transplantation, and maintenance dosing is 2 mg/day orally.
It is recommended that the maintenance dose of sirolimus tablets be reduced by approximately one third in patients with mild or moderate hepatic impairment and by half in patients with severe hepatic impairment. Dosing should not exceed 40 mg/day.
TDM is recommended for all patients. Target whole-blood trough levels should range between 16 and 24 ng/mL for the first year following transplantation; thereafter, trough levels should range between 12 and 20 ng/mL. Sirolimus tablets are to be administered orally once daily, consistently with or without food. Tablets should not be crushed, chewed, or split. It is recommended that sirolimus tablets be taken 4 hours after administration of cyclosporine.
The most common side effects of sirolimus are peripheral edema, hypertriglyceridemia, hypertension, hypercholesterolemia, increased creatinine, constipation, abdominal pain, diarrhea, headache, fever, urinary tract infection, anemia, nausea, arthralgia, pain, and thrombocytopenia. Serious reactions include malignancy, lymphoma, severe infection, PML, nephropathy, hemolytic uremic syndrome, thrombotic microangiopathy, venous thromboembolism, myelosuppression, exfoliative dermatitis, pericardial effusion, ascites, interstitial lung disease, hepatotoxicity, and osteonecrosis.
Concurrent administration of sirolimus with ketoconazole, mifepristone, or voriconazole is contraindicated due to increased serum drug levels secondary to reduced metabolism of sirolimus.
Transplant recipients taking sirolimus should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when sirolimus is administered with other drugs that lower the immune response because of the increased risk of serious infection.
Sirolimus was designated pregnancy category C; no adequate and well-controlled studies have been done in pregnant women, therefore effective contraception must be initiated before sirolimus therapy, and it should continue during sirolimus therapy and for 12 weeks after therapy has been stopped. Sirolimus should be used during pregnancy only if the potential benefit outweighs the potential risk. Whether sirolimus is excreted in breast milk is unknown, therefore a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Purine Antimetabolites
Azathioprine, a purine antimetabolite, is converted into 6-mercaptopurine in the body, where it blocks purine metabolism and DNA synthesis, thereby suppressing T- and B-lymphocyte proliferation. It is indicated for the prevention of kidney transplant rejection. Azathioprine is well absorbed following oral administration. Both azathioprine and the metabolite 6-mercaptopurine are moderately bound to serum proteins (30%) and undergo extensive metabolism in the liver; both are primarily excreted in bile, and drug half-life is 5 hours.
 Azathioprine has a boxed warning that chronic immunosuppression with azathioprine increases risk of malignancy in humans.
Azathioprine has a boxed warning that chronic immunosuppression with azathioprine increases risk of malignancy in humans.Dosing ranges from 1 to 3 mg/kg per day orally (an IV form is not currently available in the United States). In the setting of kidney dysfunction, the dose should be reduced by 25% for a creatinine clearance (CrCl) between 10 and 50 mL/min; if CrCl is less than 10 mL/min, the dose should be reduced by 50%.
Common side effects include leukopenia, thrombocytopenia, anemia, infection, nausea and vomiting, anorexia, diarrhea, elevated LFTs, malaise, myalgia, fever, and rash. Serious side effects include myelosuppression, PML, pancreatitis, hepatotoxicity, lymphomas, and other malignancies.
Transplant recipients taking azathioprine should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when azathioprine is administered with other drugs that lower the immune response because of an increased risk of serious infection. Combining azathioprine with antihypertensive drugs increases the risk of leukopenia.
Patients who receive azathioprine should have a complete blood count (CBC), including a platelet count, taken weekly during the first month, twice monthly for the second and third months of treatment, then monthly. Creatinine and LFTs should be monitored.
Azathioprine was designated pregnancy category D because it can cause fetal harm when given to pregnant women and should be avoided; however, maternal benefit may outweigh fetal risk in serious or life-threatening situations. Azathioprine is found in breast milk and is possibly unsafe during breastfeeding, therefore a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Inosine Monophosphate Dehydrogenase Inhibitors
Mycophenolate mofetil blocks synthesis of purine nucleotides by inhibition of the enzyme inosine monophosphate dehydrogenase (IMPDH), thereby preventing the proliferation of T cells and lymphocytes and preventing the formation of antibodies from B cells; it also may inhibit recruitment of leukocytes to inflammatory sites. Mycophenolate mofetil is rapidly absorbed following oral administration with 94% bioavailability, and it is 98% protein bound. It is a prodrug that is metabolized in the liver to mycophenolic acid (MPA); it is excreted in the urine (93%) and in feces (6%). Drug half-life for the oral formulation is 17.9 hours; the half-life for the IV formulation is 16.6 hours.
Recommended dosing for kidney transplant recipients is 1 g orally twice daily; for heart and liver transplant recipients, the dosage is 1.5 g orally twice daily. Kidney function should be monitored; for a CrCl less than 25 mL/min, the maximum dosage is 1 g twice daily. In pediatric transplant recipients, the recommended dose of mycophenolate mofetil oral suspension is 600 mg/m2 twice daily, up to a maximum of 1 g twice daily.
The dose of mycophenolate mofetil should be reduced or interrupted for an absolute neutrophil count (ANC) of less than 1300. To calculate the ANC, the following formula is used (WBC is white blood cell count):
WBC×totalneutrophils(segmentedneutrophils% +segmentedbands%) ×10=ANC
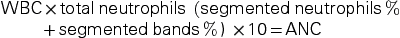
A normal ANC is over 1500. An ANC of 500 to 1500 is considered neutropenic, and an ANC less than 500 indicates severe neutropenia and significantly increases a person’s risk for infection (Box 30.2).
Common side effects of mycophenolate mofetil include hypertension, infection, diarrhea, edema, anemia, abdominal pain, constipation, headache, nausea and vomiting, dyspnea and cough, hypercholesterolemia, tremor, hypokalemia, acne, and insomnia. Serious reactions include thrombocytopenia, leukopenia, neutropenia, severe infection, viral reactivation, nephropathy, PML, lymphoma, lymphoproliferative disorders, malignancy, gastrointestinal (GI) bleeding, acute renal failure, and interstitial lung disease.
Transplant recipients taking mycophenolate mofetil should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when mycophenolate mofetil is administered with other drugs that lower the immune response because of the increased risk of serious infection. Combining mycophenolate mofetil with nonsteroidal antiinflammatory drugs (NSAIDs) increases the risk of GI bleeding.
Patients taking mycophenolate mofetil should have a baseline creatinine level drawn and should have CBCs done weekly during the first month, twice monthly for the second and third months of treatment, then monthly throughout the first year.
Mycophenolate mofetil was designated pregnancy category D; it is associated with an increased risk of first-trimester pregnancy loss and an increased risk of congenital malformations.  Females of reproductive potential must be made aware of the increased risk of first-trimester pregnancy loss and congenital malformations and must be counseled regarding pregnancy prevention and planning. It is unknown whether mycophenolate mofetil is found in breast milk, therefore a decision should be made whether to discontinue breastfeeding or to discontinue the drug, taking into account the importance of the drug to the mother.
Females of reproductive potential must be made aware of the increased risk of first-trimester pregnancy loss and congenital malformations and must be counseled regarding pregnancy prevention and planning. It is unknown whether mycophenolate mofetil is found in breast milk, therefore a decision should be made whether to discontinue breastfeeding or to discontinue the drug, taking into account the importance of the drug to the mother.
Females of reproductive potential must be made aware of the increased risk of first-trimester pregnancy loss and congenital malformations and must be counseled regarding pregnancy prevention and planning. It is unknown whether mycophenolate mofetil is found in breast milk, therefore a decision should be made whether to discontinue breastfeeding or to discontinue the drug, taking into account the importance of the drug to the mother.Corticosteroids
Prednisone, a corticosteroid, is a glucocorticoid receptor agonist. It decreases inflammation by suppression of migration of polymorphonuclear leukocytes and reversal of increased capillary permeability; it suppresses the immune system by reducing the activity and volume of the lymphatic system; prednisone suppresses adrenal function at high doses and is readily absorbed from the GI tract (up to 90%). Plasma protein binding is less than 50% but is concentration dependent. Prednisone is metabolized by the liver to its active metabolite, prednisolone. It is excreted in the urine as sulfate and glucuronide conjugates. Prednisone has a plasma half-life of 2 to 4 hours and should be administered after meals or with food or milk to decrease GI upset.
In addition to their use in maintenance therapy, corticosteroids are used in high doses for the treatment of acute transplant rejection. The drug, methylprednisolone sodium succinate, is administered IV in doses that range from 250 mg to 500 mg daily for 3 to 5 days.
Common side effects of corticosteroid use include sodium retention, edema, hypokalemia, hypertension, diaphoresis, muscle atrophy, nausea and vomiting, dyspepsia, petechiae and ecchymosis, facial erythema, acne, rash, headache, dizziness and vertigo, insomnia, emotional lability, depression, anxiety, glucose intolerance, menstrual irregularities, hirsutism, appetite changes, and weight gain. Serious reactions include anaphylaxis, adrenal insufficiency, steroid psychosis, infection, diabetes mellitus, seizures, heart failure, peptic ulcer disease and GI bleeding, osteonecrosis, and tendon rupture. Long-term use may lead to impaired wound healing, skin atrophy, Cushing syndrome, glaucoma and cataracts, Kaposi sarcoma, and growth suppression in children. Cessation of corticosteroids may lead to withdrawal symptoms with high doses or long-term use.
Transplant recipients taking corticosteroids should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when corticosteroids are administered with other drugs that lower the immune response because of the increased risk of serious infection. Combining corticosteroids with certain antibiotics increases the risk of QT prolongation and arrhythmias; combining corticosteroids with diuretics increases the risk of hypokalemia.
Transplant recipients taking corticosteroids should have periodic monitoring of their electrolytes, blood pressure, weight, and glucose levels. Pediatric patients should have their height monitored. Chest x-rays and ophthalmic examinations are indicated with long-term use.
Caution is advised during pregnancy, especially in the first trimester or with long-term use, because of the possible risk of low birthweight and premature birth. Corticosteroids are probably safe during breastfeeding.
Drugs for Transplant Rejection
Transplant rejection occurs when the immune system of the transplant recipient attacks the transplanted organ. This happens because the immune system recognizes foreign tissues and attempts to destroy them, just as it attempts to destroy infecting organisms, such as bacteria and viruses. Treatment of rejection with an anti–T-cell antibody is used when corticosteroids have failed to reverse rejection or for treatment of a recurrent rejection.
Antithymocyte globulin (rabbit), or ATG rabbit, is a polyclonal (depleting) antibody that blocks T-cell membrane proteins CD2, CD3, and CD45; this causes altered T-cell function and lysis and prolonged T-cell depletion, which begins within 24 hours. Complete pharmacokinetic data are not available, but drug half-life is 2 to 3 days.
Dosing is 1.5 mg/kg IV each day for 7 to 14 days in adults. This drug is not approved for use in children. Dosage should be decreased by 50% if the white blood cell (WBC) count decreases to 2000 to 3000 or the platelet count is 50,000 to 75,000. Treatments should be discontinued if the WBC count falls to less than 2000 or the platelet count falls to less than 50,000.
Common side effects of ATG rabbit include high fever, chills, nausea and vomiting, headache, diarrhea, malaise, shortness of breath, leukopenia, thrombocytopenia, peripheral edema, and increased risk for infection. Serious side effects include anaphylaxis, severe infusion reaction, cytokine release syndrome, serum sickness, sepsis, cytomegalovirus (CMV), malignancy, and lymphoproliferative disorders. Premedication with corticosteroids and antihistamines decreases the incidence and severity of adverse reactions.  Close supervision of the patient is required during and after IV infusion, to include frequent vital signs and assessment of the site for signs of extravasation.
Close supervision of the patient is required during and after IV infusion, to include frequent vital signs and assessment of the site for signs of extravasation.
Close supervision of the patient is required during and after IV infusion, to include frequent vital signs and assessment of the site for signs of extravasation.Transplant recipients receiving ATG rabbit should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when ATG rabbit is administered with other drugs that lower the immune response because of the increased risk of serious infection.
Patients should have WBC and platelets monitored frequently during treatment. ATG rabbit was designated pregnancy category C, and there are no adequate and well-controlled studies in pregnant women. Inadequate information is available to assess the risk of ATG rabbit when breastfeeding, therefore a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Muromonab-CD3 is a monoclonal antibody that binds specifically to the CD3 complex on the surface of T lymphocytes; the CD3 complex is involved in antigen recognition and cell stimulation. Immediately after administration, CD3-positive T lymphocytes are abruptly removed from circulation. Complete pharmacokinetic data are not available, but drug half-life is 18 hours.
Dosing is 5 mg IV push daily for 10 to 14 days in adults. In pediatric patients under 30 kg, dosage is 2.5 mg IV push daily for 10 to 14 days; pediatric patients over 30 kg are dosed the same as adults.
 Muromonab-CD3 carries a boxed warning for the risk of anaphylactic reactions occurring with any dose and life-threatening or lethal systemic, cardiovascular, and CNS reactions.
Muromonab-CD3 carries a boxed warning for the risk of anaphylactic reactions occurring with any dose and life-threatening or lethal systemic, cardiovascular, and CNS reactions.Common side effects of muromonab-CD3 include fever, chills, nausea and vomiting, diarrhea, headache, tachycardia, hypotension, dyspnea, tremor, rash, edema, fatigue, diaphoresis, dyspepsia, arthralgia, pruritus, leukopenia, and increased risk of infection. Serious reactions include anaphylaxis, Stevens-Johnson syndrome, cytokine release syndrome, cardiorespiratory arrest, seizures, encephalopathy, aseptic meningitis, opportunistic infection, malignancy, lymphoproliferative disorders, thrombosis, thrombocytopenia, anemia, neutropenia, and leukopenia.
Transplant recipients receiving muromonab-CD3 should not receive live vaccines because their immune response may be inadequate, and they are at risk for disseminated infection resulting from the live virus. Caution is advised when muromonab-CD3 is administered with other drugs that lower the immune response because of the increased risk of serious infection.
Transplant recipients receiving muromonab-CD3 should have BUN, creatinine, LFTs, and a CBC with differential drawn at baseline.
Muromonab-CD3 was designated pregnancy category C, and there are no adequate and well-controlled studies in pregnant women; however, potential benefits may warrant use of the drug in pregnant women despite potential risks. The drug is unsafe during breastfeeding.
Drugs for Infection
Bacterial
Pneumocystis jiroveci pneumonia (PJP) is a life-threatening illness in immunocompromised patients. Routine prophylaxis with trimethoprim-sulfamethoxazole (TMP-SMZ) has significantly reduced the morbidity and mortality of PJP following transplantation. TMP-SMZ is dosed once every morning or one tablet three times a week (Monday, Wednesday, and Friday). TMP-SMZ can make the skin more sensitive to sunlight, therefore patients should be instructed to use a lotion with a minimum sun protection factor (SPF) of 25 when in the sun. See Chapter 26 for further information regarding antibacterial drugs.
Fungal
Transplant recipients use nystatin to prevent or treat thrush in the mouth and esophagus. This is usually given when the patient is on a high-dose immunosuppression regimen and is stopped when the steroid dose is reduced below 20 mg per day. Nurses must instruct the transplant recipient in proper administration of nystatin liquid:
• Shake the preparation well before measuring the dose.
• Swish the dose around in the mouth for at least 2 minutes before swallowing.
• Allow the nystatin to coat the mouth for as long as possible.
• Do not eat or drink anything for 30 minutes after taking the medication.
See Chapter 27 for further information regarding antifungal drugs.
Viral
One common virus that patients develop after transplantation is CMV. It may present as a viral syndrome or as invasive disease, and it plays a role in organ rejection. Transplant recipients receive antiviral prophylaxis with oral ganciclovir or valganciclovir for 3 to 6 months following surgery. If untreated, CMV can cause serious complications, particularly in the liver, intestine, kidneys, heart, lungs and eyes. See Chapter 27 for further information regarding antiviral drugs.
Promoting Adherence
On discharge from the hospital, transplant recipients begin a lifelong journey of close medical supervision that includes frequent visits to their health care provider, monitoring of blood work, and maintenance of a complex drug regimen. The 1-year survival rates are over 80% for liver transplants and over 90% for kidney transplants; most recipients experience an improved quality of life. However, long-term adherence is a problem, with reports of nonadherence ranging from a low of 2% to as high as 68%. Nonadherence to the posttransplant regimen (e.g., drug regimen, exercise and health promotion) is one of the top three reasons for transplant failure. Factors that affect adherence include episodes of rejection, comorbid illness and disease, side effects of drugs, and health care costs. Nurses play a key role in promoting adherence by incorporating education, motivational strategies, and coping skills into an individually tailored posttransplant plan of care.
 Nursing Process: Patient-Centered Collaborative Care
Nursing Process: Patient-Centered Collaborative CareOrgan Transplants: Immunosuppression
Assessment
• Assess for clinical signs of rejection including malaise, fever, edema, pain over the transplant site, and increased weight.
• Assess for presence of risk factors for infection, including drugs, travel, and exposure to individuals with active infections.
• Assess for signs and symptoms of infection, including redness, swelling, pain, and elevated temperature.
• Assess immunization status.
• Assess nutritional status, including weight and history of weight loss.
Nursing Diagnosis
• Protection, Ineffective related to the possibility of transplant rejection
• Infection, Risk for
Planning
• Infection is recognized early to allow prompt treatment.
• The transplant recipient and family members will verbalize understanding of early signs and symptoms of rejection.
• The transplant recipient will remain free of infection.
Nursing Interventions
• Instruct the transplant recipient on the benefits of a balanced and healthy diet accompanied by exercise.
• Patients should be instructed to avoid anyone with an active infection and to be careful not to injure themselves, which may increase the chance of acquiring a wound infection. They should stay away from anyone who has a cold, mumps, measles, chickenpox, or other communicable diseases.
• Promote health education with patients and families in order to recognize and minimize the risk of complications and rejection and to facilitate optimum quality of life.
• Teach the transplant recipient the signs and symptoms of rejection and infection, and instruct them on when to call their health care provider.
• Teach the transplant recipient to self-monitor vital signs, daily weights, and blood glucose (if appropriate); ensure the recipient knows when to call the health care provider.
• Transplant recipients and their families should be taught to wash hands properly, especially after toileting, before meals, and before administering drugs.
Evaluation
• Evaluate effectiveness of the plan, including adherence to the immunosuppressive drug regimen and freedom from infection and transplant rejection.
Critical Thinking Case Study
EM is a 24-year-old female kidney transplant recipient 6 months out from surgery. She is experiencing an episode of acute rejection. In addition to cyclosporine oral solution, mycophenolate mofetil, and prednisone, she is now receiving antithymocyte globulin (rabbit), having failed high-dose steroid treatment for the rejection. EM has had a CBC with differential drawn and her WBC counts are 2.0 × 103/μL, segmented neutrophils are 14.8%, and the segmented bands are 5%.
1. What would be your primary nursing diagnosis? Why?
2. Why is EM prescribed multiple immunosuppressant drugs?
3. When questioning EM about her drug regimen, she states she mixes her cyclosporine in orange juice. She has also been drinking a small glass of grapefruit juice at night because she’s heard it helps to lower lipid levels, and she knows cyclosporine can cause elevated lipids. How would you respond to EM?
4. Calculate the ANC. What precautions are necessary if the ANC is less than 500?
[/level-membership-for-nursing-midwifery-medical-assistant-category][not-level-membership-for-nursing-midwifery-medical-assistant-category]
Buy Membership for Nursing & Midwifery & Medical Assistant Category to continue reading. Learn more here
[/not-level-membership-for-nursing-midwifery-medical-assistant-category]
