Chapter 24 Transfusion Medicine and Coagulation Disorders
OVERVIEW OF HEMOSTASIS
Proper hemostasis requires the participation of innumerable biological elements (Box 24-1). They can be divided into four topics to facilitate understanding: coagulation factors, platelet function, the endothelium, and fibrinolysis. The reader must realize this is for simplicity of learning and that in biology the activation creates many reactions and control mechanisms, all interacting simultaneously. The interaction of the platelets, endothelial cells, and proteins to either activate or deactivate coagulation is a highly buffered and controlled process. It is perhaps easiest to think of coagulation as a wave of biological activity occurring at the site of tissue injury (Fig. 24-1). Although there are subcomponents to coagulation itself the injury/control leading to hemostasis is a four-part event: initiation, acceleration, control, and lysis (recanalization/fibrinolysis). The initiation phase begins with tissue damage, which is really begun with endothelial cell destruction or dysfunction. This initiationphase leads to binding of platelets, as well as protein activations; both happen nearly simultaneously and each has feedbacks into the other. Platelets adhere and create an activation or acceleration phase that gathers many cells to the site of injury and creates a large number of biochemical protein cascade events. As the activation phase ramps up into an explosive set of reactions, counter-reactions are spun off, leading to control proteins damping the reactions. The surrounding normal endothelium exerts control over the reactions. Eventually the control reactions overpower the acceleration reactions and lysis comes into play.
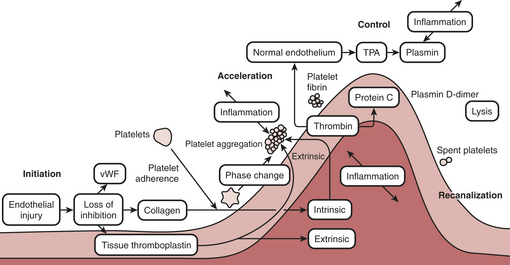
Protein Coagulation Activations
Coagulation Pathways
With few exceptions, the coagulation factors are glycoproteins synthesized in the liver, which circulate as inactive molecules termed zymogens. Factor activation proceeds sequentially, with each factor serving as substrate in an enzymatic reaction catalyzed by the previous factor in the sequence. Hence, this has classically been called a “cascade” or “waterfall” sequence. Cleavage of a polypeptide fragment changes an inactive zymogen to an active enzyme. The active form is termed a serine protease because the active site for its protein-splitting activity is a serine amino acid residue. Many reactions require the presence of calcium ion (Ca2+) and a phospholipid surface (platelet phosphatidylserine). The phospholipids occur most often either on the surface of an activated platelet or endothelial cell and occasionally on the surface of white cells. So anchored, their proximity to one another permits reaction rates profoundly accelerated (up to 300,000-fold) from those measured when the enzymes remain in solution. The factors form four interrelated arbitrary groups (Fig. 24-2): the contact activation and the intrinsic, extrinsic, and common pathways.
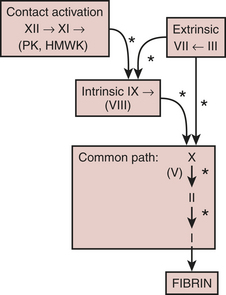
extrinsic system
Activation of factor X can proceed independently of factor XII by substances classically thought to be extrinsic to the vasculature. Any number of endothelial cell insults can lead to the production of tissue factor by the endothelial cell. At rest, the endothelial cell is very antithrombotic. However, with ischemia, reperfusion, sepsis, or cytokines, the endothelial cell will stimulate its production of intracellular NFκb and send messages for the production of messenger RNA for tissue factor production. This can happen quickly, and the resting endothelial cell can turn out large amounts of tissue factor. It is widely held today that the activation of tissue factor is what drives many of the abnormalities of coagulation after cardiac surgery, rather than contact activation.1 Thromboplastin, also known as tissue factor, released from tissues into the vasculature, acts as a cofactor for initial activation of factor X by factor VII. Factors VII and X then activate one another with the help of platelet phospholipid and Ca2+, thus rapidly generating factor Xa. Factor VIIa also activates factor IX, thus linking the extrinsic and intrinsic paths.
Modulators of the Coagulation Pathway
Regulation of the extrinsic limb of the coagulation pathway occurs via tissue factor pathway inhibitor (TFPI), a glycosylated protein that associates with lipoproteins in plasma.2 TFPI is not a serpin. It impairs the catalytic properties of the factor VIIa/tissue factor complex on factor X activation. Both vascular endothelium and platelets appear to produce TFPI. Heparin releases TFPI from endothelium, increasing TFPI plasma concentrations by as much as sixfold.2
Platelet Function
Most clinicians think first of the coagulation proteins when considering hemostasis. Although no one element of the many that participate in hemostasis assumes dominance, platelets may be the most complex. Without platelets, there is no coagulation and no hemostasis. Without the proteins, there is hemostasis, but it lasts only 10 to 15 minutes as the platelet plug is inherently unstable and breaks apart under the shear stress of the vasculature. Platelets provide phospholipid for coagulation factor reactions; contain their own microskeletal system and coagulation factors; secrete active substances affecting themselves, other platelets, the endothelium, and other coagulation factors; and alter shape to expose membrane glycoproteins essential to hemostasis. Platelets have perhaps as many as 30 to 50 different types of cell receptors. The initial response to vascular injury is formation of a platelet plug. Good hemostatic response depends on proper functioning of platelet adhesion, activation, and aggregation (Fig. 24-3).
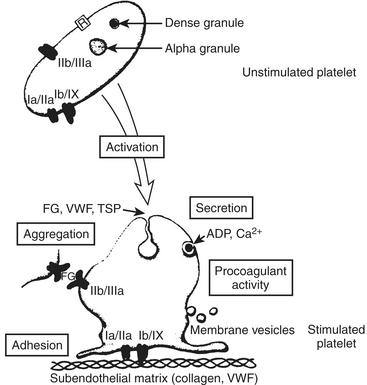
Platelet Adhesion
Platelet adhesion begins rapidly—within 1 minute of endothelial injury—and completely covers exposed subendothelium within 20 minutes. It begins with decreased platelet velocity when GPIb/IX and vWF mediate adhesion, followed by platelet activation, GPIIb/IIIa conformational change, and then vWF binding and platelet arrest on the endothelium at these vWF ligand sites.3
Drug-Induced Platelet Abnormalities
Many other agents inhibit platelet function.4 β-Lactam antibiotics coat the platelet membrane, whereas the cephalosporins are rather profound but short-term platelet inhibitors. Many cardiac surgeons may not realize that their standard drug regimen for antibiotics may be far more of a bleeding risk than aspirin. Hundreds of drugs can inhibit platelet function. Calcium channel blockers, nitrates, and β-blockers are ones commonly utilized in cardiac surgery. Nitrates are effective antiplatelet agents and that may be part of why they are of such benefit in angina, not just for their vaso-relaxing effect on large blood vessels. Nonsteroidal anti-inflammatory drugs (NSAIDs) reversibly inhibit both endothelial cell and platelet cyclooxygenase.
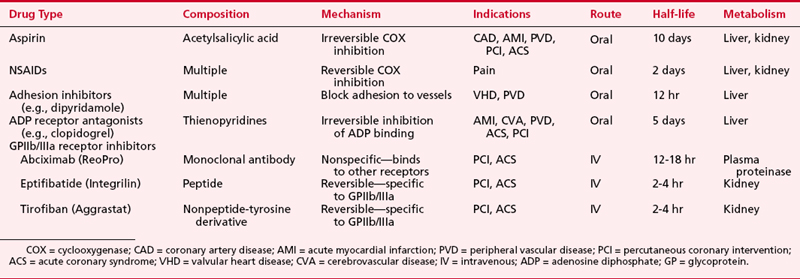
In addition to the partial inhibitory effects of aspirin and the other drugs just mentioned, new therapies have been developed that inhibit platelet function in a more specific manner. These drugs include platelet adhesion inhibitor agents, platelet-ADP-receptor antagonists, and GPIIb/IIIa receptor inhibitors (Table 24-1).
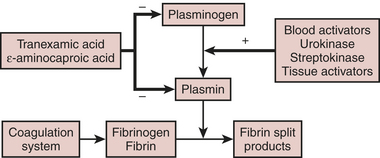
Fibrinolysis
Clinical Applications
Figure 24-4 illustrates the fibrinolytic pathway, with activators and inhibitors. Streptokinase, ASPAC, and t-PA find application in the lysis of thrombi associated with myocardial infarction. These intravenous agents “dissolve” clots that form on atheromatous plaque. Clinically significant bleeding may result from administration of any of these exogenous activators or streptokinase.
HEPARIN
Pharmacology
Chemical Structure
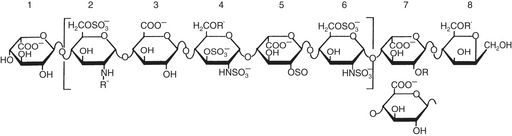
The N-sulfated-D-glucosamine and L-iduronic acid residues of heparin alternate in copolymer fashion to form chains of varying length (Fig. 24-5). As a linear anionic polyelectrolyte, with the negative charges being supplied by sulfate groups, heparin demonstrates a wide spectrum of activity with enzymes, hormones, biogenic amines, and plasma proteins. A pentasaccharide segment binds to antithrombin. Heparin is a heterogeneous compound: the carbohydrates vary in both length and side chain composition, yielding a range of molecular weights from 5 000 to 30,000, with most chains between 12,000 and 19,000. Today, the standard heparin is called unfractionated heparin (UFH).
Pharmacokinetics and Pharmacodynamics
Actions and Interactions
Antithrombin inhibits serine proteases even without heparin. The extent to which heparin accelerates antithrombin inhibition depends on the substrate enzyme: UFH accelerates the formation of the thrombin-antithrombin complex by 2000-fold but accelerates formation of the factor Xa-antithrombin complex by only 1200-fold. In contrast, LMWH fragments preferentially inhibit factor Xa. Enzyme inhibition proceeds by formation of a ternary complex consisting of heparin, antithrombin, and the proteinase to be inhibited (e.g., thrombin, factor Xa). For UFH, inhibition of thrombin occurs only on simultaneous binding to both antithrombin and thrombin. This condition requires a heparin fragment of at least 18 residues. A pentasaccharide sequence binds to antithrombin. LMWHs, consisting of chains 8 to 16 units long, preferentially inhibit factor Xa. In this case, the heparin fragment activates antithrombin, which then sequentially inactivates factor Xa; heparin and factor Xa do not directly interact (Fig. 24-6).

Heparin Resistance
Patients receiving UFH infusions exhibit a much diminished ACT response to full anticoagulating doses of UFH for CPB (200 to 400 units/kg). With widespread use of heparin infusions to treat myocardial ischemia and infarction, heparin resistance or, more appropriately, “altered heparin responsiveness” has become more problematic during cardiac surgery (Box 24-2).5
Treatment
Antithrombin concentrate specifically addresses antithrombin deficiency. It is a solvent detergent-treated and heat-inactivated product providing greater protection against infectious disease transmission than that provided by FFP. The literature supports its success in treating heparin resistance during cardiac surgery. A multicenter study on the efficacy of using a recombinant human antithrombin (rhAT) in heparin-resistant patients undergoing CPB was published.6 The patients received 75 units/kg of rhAT, which was effective in restoring heparin responsiveness in most patients. However, some patients still required FFP and the patients bled more than did a control group postoperatively.
Heparin Rebound
Heparin Effects other Than Anticoagulation
Unfractionated heparin was never biologically intended to circulate freely in plasma.7 As such it has a number of underappreciated and untoward effects. All too often the effects of CPB have been asserted as causing a coagulopathy; however, the effect of heparin contributing to this has not been widely studied. That is because there has not been an alternative anticoagulant to compare with heparin until now. In the future, there may be better anticoagulants to use during cardiac surgery.
Heparin-Induced Thrombocytopenia
Mechanism
These patients demonstrate a heparin-dependent antibody, usually IgG, although others are described, which aggregates platelets in the presence of heparin.8 During heparin therapy, measured antibody titers remain low, owing to antibody binding to platelets. Titers rise after heparin therapy ceases; but paradoxically, antibody may be undetectable a few months later. Two other features are unexpected: first, the antibody does not aggregate platelets in the presence of excess heparin; and, second, not all reexposed patients develop thrombocytopenia.
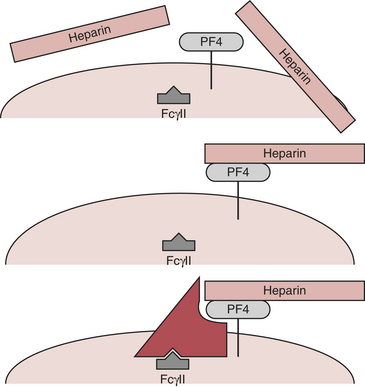
The platelet surface contains complexes of heparin and PF4. Affected patients have antibody to this complex. Antibody binding activates platelets via their FcγII receptors and activates endothelium (Fig. 24-7). The activation of the platelet surface triggers a secondary thrombin release. Platelets can attach to each other creating what is known as a white clot syndrome. But if secondary thrombin generation is created through antibody activation of the platelets then a fibrin clot can be the result. In the absence of heparin, the heparin-PF4 antigen cannot form.
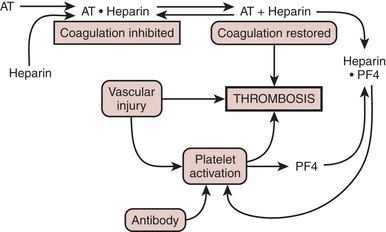
In the absence of an endothelial defect, the only responses to the antibody-antigen interaction are platelet consumption and thrombocytopenia. Atheroma rupture, endovascular interventions such as balloon angioplasty, vascular surgery, and other procedures that disrupt endothelium can provide a nidus for platelet adhesion and subsequent activation. PF4, released with platelet activation, binds to heparin locally, thus not only removing the inhibition of coagulation but also generating additional antigenic material (Fig. 24-8). Clumps of aggregated platelets thrombose vessels, resulting in organ and limb infarction. Amputation and/or death often occurs with established HIT with thrombosis (HITT). The presence of heparin-PF4 antibodies has recently been associated with other adverse effects. It appears that if a patient undergoes cardiac surgery with positive antibodies, the risk of mortality and/or myocardial infarction may at least double.
Treatment and Prevention
LMWH heparin, as an alternative to UFH, has been used for urgent surgery. While LMWHs can also induce thrombocytopenia, by displaying different antigenic determinants, they may prove acceptable alternatives for patients who develop HIT from UFH. Table 24-2 summarizes the therapeutic options available for urgent cardiac surgery in patients with HIT.
Table 24-2 Therapeutic Options for Anticoagulation for Bypass in Patients with Heparin-Induced Thrombocytopenia*
* No agent is indicated for anticoagulation in CPB at this time.
New Modes of Anticoagulation
The hemostatic goal during CPB is complete inhibition of the coagulation system. Unfortunately, even large doses of heparin do not provide this, as evidenced by formation of fibrinopeptides during surgery. Despite being far from the ideal anticoagulant, heparin still performs better than its alternatives. Current substitutes for heparin include ancrod, a proteinase obtained from snake venom that destroys fibrinogen; heparin fragments, which provide less thrombin inhibition than the parent, unfractionated molecule; direct factor Xa inhibitors; and direct thrombin inhibitors (Box 24-4).
Direct Thrombin Inhibitors
New direct thrombin inhibitors are now available (Fig. 24-9).9 These include argatroban and bivalirudin. Argatroban is a derivative of arginine and is a relatively small molecule. It binds at the active cleavage site of thrombin and stops thrombin’s action on serine proteases. It is completely hepatically cleared and has a half-life of 45 to 55 minutes with prolongation when liver function is depressed or liver blood flow is decreased. There is no reversal agent for argatroban. It has been approved by the U.S. Food and Drug Administration (FDA) for anticoagulation in the face of HITT, but there has not been, to date, a large-scale prospective randomized trial for cardiac surgery or any type of comparison to heparin/protamine. Some case reports do exist of successful usage of argatroban in HITT patients with acceptable amounts of postoperative bleeding. It has been more commonly utilized in the ICU for patients with hypercoagulable syndromes and HITT.
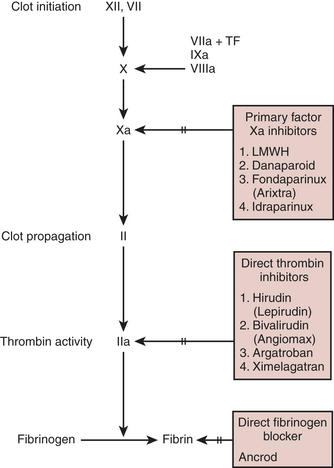
Several clinical trials of bivalirudin for cardiology procedures or cardiac surgery have been completed and published. Other ongoing and pivotal trials aiming for FDA approval are under way comparing bivalirudin to heparin/protamine for both on- and off-pump CABG cases as well as for patients with HITT. Bivalirudin has been FDA approved as a primary anticoagulant for angioplasty. In trials comparing bivalirudin to either heparin/protamine alone or heparin plus the use of a IIb/IIIa inhibitor, bivalirudin was found to have at least equal or better safety and less bleeding than either of the other therapies. When compared with heparin/protamine alone in PCI, bivalirudin was found to be superior, not just in bleeding, but also in terms of morbidity and mortality (as a combined endpoint). In a trial of 100 off-pump CABG patients randomized to receive either bivalirudin or heparin/protamine, bleeding and outcome were equal between the groups.10 These patients underwent recatheterization at 3 months, and it was found that the bivalirudin patients had overall better flow down their grafts than did the patients who had received heparin/protamine. A phase I/II safety trial of bivalirudin in 30 on-pump CABG patients has also shown good safety, but no comparison was carried out to look at advantages against heparin/protamine. At this time, considerable research is ongoing and whether bivalirudin will prove superior to heparin/protamine for routine CABG is yet to be seen.11 When used, the doses for CPB have been a 0.75-mg/kg bolus followed by an infusion at 1.75 mg/kg/hr titrated to the ACT (target, 2.5 times baseline). The CPB system is also primed with 50 mg, and no stasis can be allowed in the CPB circuit due to metabolism of bivalirudin during CPB. The infusion is stopped 15 to 30 minutes before CPB is discontinued, and patients bleed for up to 4 to 6 hours. In off-pump coronary artery bypass grafting similar doses to ACT targets of 350 to 450 seconds have been used.
PROTAMINE
Adverse Reactions
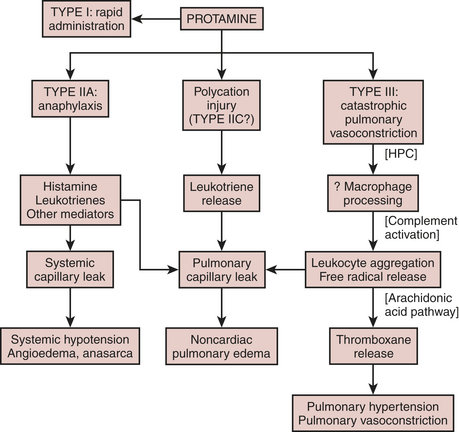
Rapid Administration
peripheral cardiovascular changes
Systemic hypotension from protamine occurs with rapid injection.12 Decreased systemic vascular resistance (SVR) accompanies the systemic hypotension, whereas venous return and cardiac filling pressures decrease.
Anaphylactoid Reaction
allergy, anaphylaxis, and adverse responses
Not all adverse responses to protamine are allergic reactions. Hypersensitivity allergic reactions involve release of vasoactive mediators resulting from antigen-antibody interaction. The broader term anaphylactoid reaction includes not only severe immediate hypersensitivity allergy, termed anaphylaxis, but also other life-threatening idiosyncratic responses of nonimmunologic origin.13
Pulmonary Vasoconstriction
Several years after pulmonary artery catheters achieved common usage and case reports sensitized clinicians to adverse responses to protamine, Lowenstein and associates reported a series of cases in which protamine caused systemic hypotension, decreased left atrial pressure, elevated rather than decreased pulmonary artery pressure, and right ventricular distention and failure.14 Unlike in anaphylaxis, plasma histamine levels do not change during this idiosyncratic, catastrophic pulmonary vasoconstriction, thus justifying a separate classification for this unusual response. The duration of pulmonary hypertension may vary substantially from brief episodes to those requiring reinstitution of CPB.
Animal models of type III protamine responses demonstrate that heparin must precede the protamine, that heparin/protamine complexes activate the complement pathway, and that blockade of complement activation attenuates pulmonary damage. Furthermore, leukocytes respond to complement activation by forming free radicals, which stimulate the arachidonate pathway. Blockade of this pathway mitigates the pulmonary response, whereas antihistamines do not. Figure 24-10 summarizes the speculative mechanisms of various adverse responses to protamine.
Alternatives to Protamine
This section discusses techniques for neutralizing heparin other than administration of protamine.
Heparinase
Systemic administration of the enzyme heparinase I, produced by Flavobacterium, results in a return of the ACT to normal in an ex vivo model, animal models of CPB, and human volunteers. Initial investigation in patients undergoing elective coronary artery bypass grafting operations confirms the utility of heparinase in neutralizing heparin-induced anticoagulation.15
BLEEDING PATIENT
The TEG has been extensively tested both alone and in conjunction with a number of other tests including prothrombin time (PT), platelet count, and fibrinogen. The TEG has been shown to have the best predictive accuracy for postoperative bleeding.16 Using an algorithm based on the TEG and other tests, blood product utilization was cut considerably. Chest tube bleeding was not different, but the TEG did predict which patients might bleed abnormally. Work with TEG monitoring has shown that it can detect both hypocoagulable states as well as hypercoagulable states. New additives to the testing make it sensitive to the ADP receptor platelet antagonists as well as the GPIIb/IIIa inhibitors.
Prevention of Bleeding
Desmopressin
This analog of vasopressin provides more potent and longer lasting antidiuretic activity than vasopressin, with little vasoconstriction (Box 24-5). Like the parent compound and like epinephrine and insulin, desmopressin releases coagulation system mediators from vascular endothelium. Factor VIII coagulant activity increases 2- to 20-fold and is maximal 30 to 90 minutes after injection. Factor XII levels also increase. In response to desmopressin, endothelium releases the larger multimers of vWF, as well as t-PA and prostacyclin. Nevertheless, the overall effect of desmopressin is procoagulant, perhaps because of the impact of factor VIII and vWF.
synthetic antifibrinolytics
Several investigations, using prophylactic antifibrinolytics, document a savings in blood loss, as well as in blood transfused in a general population of cardiac surgery patients. By commencing administration of tranexamic acid before CPB, chest tube drainage in the first 12 hours after operation decreased by 30% and the likelihood of receiving banked blood within 5 days of operation decreased from 41% to 22%. Prophylactic antifibrinolytics may spare platelet function by inhibiting the deleterious effects of plasmin. Administration of very large doses of antifibrinolytics appears to offer no greater savings. Cardiac surgery patients undergoing repeat operation may benefit particularly from prophylactic antifibrinolytic administration.17
aprotinin
Bovine lung provides the source of this 58-residue polypeptide serine protease inhibitor. Aprotinin inhibits a host of proteases, including trypsin, plasmin, kallikrein, and factor XIIa activation of complement (Fig. 24-11). The adult intravenous dose for surgical hemostasis is 2 million kallikrein inhibitor units (KIU) for both patient and bypass pump, followed by 600,000 KIU/hr. The elimination half-life of aprotinin, 7 hours, is considerably longer than that of the synthetic antifibrinolytics; after 6 days, aprotinin continues to be excreted in the urine.
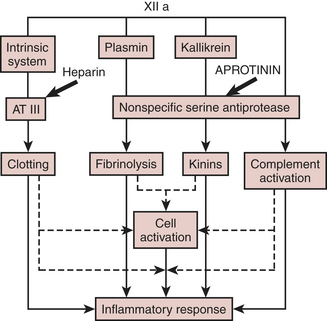
Figure 24-11 Actions of aprotinin on the contact coagulation system, fibrinolytic pathway, and complement activation.
(From Royston D: The serine antiprotease aprotinin [Trasylol]: A novel approach to reducing postoperative bleeding. Blood Coag Fibrin 1:55, 1990.)
High-dose aprotinin alone prolongs the celite ACT. Most investigators simply avoid the celite ACT and use kaolin ACT. The kaolin ACT adsorbs about 98% of aprotinin and any intrinsic antithrombin effect that aprotinin has is therefore mitigated. It is recommended to use the kaolin ACT and keep the length of ACT time the same as if aprotinin was not being used. An animal protein, aprotinin can cause anaphylaxis, although this is uncommon (<1 in 1000). Aprotinin costs significantly more than equivalent doses of synthetic antifibrinolytic drugs. The discussion and argument about cost effectiveness of aprotinin versus other antifibrinolytics has continued for more than 10 years. The major concerns today with aprotinin are its adverse effects on renal function and overall cardiovascular outcome; These concerns led to its removal from the market in 2007.18
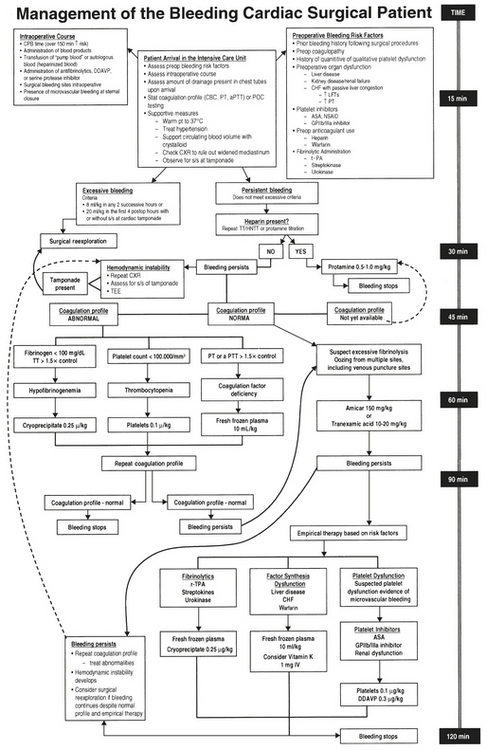
Determine the Cause
Table 24-3 lists a treatment plan for excessive bleeding after cardiac surgery. Interventions appear not in order of likelihood but rather by priority of consideration. Thus, surgical causes should be ruled out before seizing on the diagnosis of a consumptive coagulopathy. The priority will also vary among institutions, depending on the availability and cost of resources. This table provides a simple algorithm for treating postoperative bleeding. More complete schemes present a daunting level of complexity that deter implementation (Fig. 24-12).
Table 24-3 A Treatment Plan for Excessive Bleeding after Cardiac Surgery
| Action | Amount | Indication |
|---|---|---|
| Rule out surgical cause | — | No oozing at puncture sites; chest radiograph |
| More protamine | 0.5 to 1 mg/kg | ACT > 150 s or aPTT > 1.5 times control |
| Warm the patient | — | “Core” temperature < 35°C |
| Apply PEEP* | 5 to 10 cm H2O | — |
| Desmopressin | 0.3 μg/kg IV | Prolonged bleeding time |
| Aminocaproic acid | 50 mg/kg, then 25 mg/kg/hr | Elevated D‑dimer or teardrop-shaped TEG tracing |
| Tranexamic acid | 10 mg/kg, then 1 mg/kg/hr | Elevated D-dimer or teardrop-shaped TEG tracing |
| Platelet transfusion | 1 U/10 kg | Platelet count < 100,000/mm3 |
| Fresh frozen plasma | 15 mL/kg | PT or aPTT > 1.5 times control |
| Cryoprecipitate | 1 U/4 kg | Fibrinogen < 1 g/L or 100 mg/dL |
ACT = activated coagulation time; PT = prothrombin time; aPTT = activated partial thromboplastin time; TEG = thromboelastographic.
* Positive end-expiratory pressure (PEEP) is contraindicated in hypovolemia.
Blood Products
Banked blood should be infused to maintain a hemoglobin concentration that allows appropriate oxygen delivery to tissues. Because patients undergoing cardiac surgery also experience the hemodiluting and antihemostatic effects of CPB, the prudent clinician will commence platelet and factor replenishment earlier in the course of hemorrhage while awaiting laboratory confirmation. Each unit of platelet concentrate supplies about 1011 platelets, which increases the platelet count by about 20,000/mm3 in the adult. Enough plasma accompanies platelet concentrates to supply the equivalent of 1.0 to 1.5 units of plasma for each 6 units of platelets.
Drugs
Recombinant factor VIIa (rFVIIa NovoSeven) has been approved for use in hemophiliacs who are resistant to factor VIII concentrates. When rFVIIa is administered, it binds to tissue factor and activates factor X leading to thrombin and fibrin formation. These then activate platelets. Thrombin generation and clotting take place on the surface of platelets and at sites of injury.19,20 Numerous reports have been published of the off-label use of this “rescue agent” to stop bleeding in surgical patients, including cardiac surgical patients after CPB.
SUMMARY
1. Edmunds L.H.Jr. Blood-surface interactions during cardiopulmonary bypass. J Card Surg. 1993;8:404.
2. Fischer R., Kuppe H., Koster A. Impact of heparin management on release of tissue factor pathway inhibitor during cardiopulmonary bypass. Anesthesiology. 2004;100:1040.
3. Savage B., Saldivar E., Ruggeri Z.M. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 1996;84:289.
4. Lange R.A., Hillis L.D. Antiplatelet therapy for ischemic heart disease. N Engl J Med. 2004;350:277.
5. Koster A., Fischer T., Grunendel M., et al. Management of heparin: Resistance and cardiopulmonary bypass: The effect of 5 different anticoagulation strategies on hemostatic activation. J Cardiothorac Vasc Anesth. 2004;18:131.
6. Avidan M., Levy J., Scholz J., et al. A phase III, double blind, placebo-controlled, multicenter study on the efficacy of recombinant human antithrombin in heparin-resistant patients scheduled to undergo cardiac surgery necessitating cardiopulmonary bypass. Anesthesiology. 2005;102:276.
7. Day J.R.S., Landis R.C., Taylor K.M. Heparin is more than just an anticoagulant. J Cardiothorac Vasc Anesth. 2004;18:93.
8. Godal H.C. Heparin-induced thrombocytopenia. In: Lane D.A., Lindahl U., editors. Heparin. Boca Raton, FL: CRC Press; 1989:533.
9. Dinisio M., Middeldorp S., Buller H. Direct thrombin inhibitors. N Engl J Med. 2005;353:1028.
10. Merry A.F., Raudkivi P., White H.D., et al. Anticoagulation with bivalirudin (a direct thrombin inhibitor) vs heparin: A randomized trial in OPCAB graft surgery. Ann Thorac Surg. 2004;77:925.
11. Koster A., Spiess B.D., Chew D.P., et al. Effectiveness of bivalirudin as a replacement for heparin during cardiopulmonary bypass in patients undergoing coronary artery bypass grafting. Am J Cardiology. 2004;93:356.
12. Welsby I., Newman M., Phillips-Bute B., et al. Hemodynamic changes after protamine administration. Anesthesiology. 2005;102:308.
13. Horrow J.C. Protamine allergy. J Cardiothorac Vasc Anesth. 1988;2:225.
14. Kirklin J.K. Prospects for understanding and eliminating deleterious effects of cardiopulmonary bypass. Ann Thorac Surg. 1991;51:529.
15. Michelsen L.G., Kikura M., Levy J.H., et al. Heparinase I (Neutralase) reversal of systemic anticoagulation. Anesthesiology. 1996;85:339.
16. Cammerer U., Dietrich W., Rampf T., et al. The predictive value of modified computerized thromboelastography and platelet function analysis for postoperative blood loss in routine cardiac surgery. Anesth Analg. 2003;96:51.
17. Shore-Lesserson L., Reich D.L., Vela-Cantos F., et al. Tranexamic acid reduces transfusions and mediastinal drainage in repeat cardiac surgery. Anesth Analg. 1996;83:18.
18. Smith P.K., Shah A.S. The role of aprotinin in a blood-conservation program. J Cardiothoracic Vasc Anesth. 2004;18(suppl):S24.
19. Roberts H., Monroe D., Hoffman M. Safety profile of recombinant factor VIIa. Semin Hematol. 2004;41(suppl 1):101.
20. Tanaka K., Waly A., Cooper W., Levy J. Treatment of excessive bleeding in Jehovah’s Witness patients after cardiac surgery with recombinant factor VIIa. Anesthesiology. 2003;98:1513.