CHAPTER 4 Transduction, Transmission and Perception of Pain
Painful spinal disorders are common problems in society, affecting an estimated 50 million Americans. The societal costs (including litigation, work lost, treatment, and disability) for such disorders of the spine are staggering. For example, the cost of low back pain alone has been estimated at US$40–50 billion annually.1,2 Chronic neck pain has a similarly high cost of nearly US$30 billion in health-related expenses.3 Until a better understanding of the pathomechanisms of pain and the injuries which produce them are defined, the effective prevention and treatment of these disorders and their symptoms will remain elusive. Further, distinguishing those physiologic mechanisms which lead to persistent pain from those which differentially produce only transient symptoms is also important in understanding and managing these syndromes. It is the intent of this chapter to highlight traditional and emerging theories of pain detection and transmission in the context of spine-related syndromes. A brief discussion of the neurophysiology of pain highlights concepts of local responses, pain transduction, signal transmission, and processing and is integrated with more recent hypotheses of the central nervous system’s (CNS) neuroimmunologic involvement in persistent pain.
It is important to define, at the outset, relevant distinctions in terminology. ‘Pain’ is a complex perception that is influenced by prior experience and by the context within which the noxious stimulus occurs. Likewise, ‘nociception’ is the physiologic response to tissue damage or prior tissue damage. Similarly, for discussion in this chapter, ‘hyperalgesia’ is defined as enhanced pain to a noxious stimulus.4 Strictly speaking, this is a leftward shift of the stimulus–response function relating pain to intensity. The corresponding pain threshold is lowered and there is enhanced response to a given stimulus. Hyperalgesia is mediated by nociceptor sensitization, where ‘sensitization’ describes a corresponding shift in the neural response curve for stimulation. Sensitization is characterized by a decrease in threshold, an increased response to suprathreshold stimulus, and spontaneous neural activity.
RELEVANT NEURAL ANATOMY
Before presenting and discussing pain mechanisms, it is first necessary to describe the relevant anatomical structures, biological connections, and relationships of neural sensory and processing components. These are reviewed only briefly here to provide appropriate context; a more detailed presentation can be found in texts specifically focused in neural science and pain.4,5
The primary afferents, which relay pain signals from injured or stimulated tissues, terminate in the dorsal horn of the spinal cord. At each level in the spinal cord, the dorsal nerve roots carry sensory information from the periphery into the spinal cord. Dorsal roots contain sensory neurons, whose cell bodies make up the enlarged dorsal root ganglion (DRG) (Fig. 4.1). In contrast, the ventral root contains the axons of neurons whose cell bodies are within the ventral horn of the spinal cord and transmits efferent signals. At each spinal level, the dorsal and ventral nerve roots come together, outside of the spinal column and distal to the DRG, and combine to form the nerve which communicates with the peripheral nervous system. The spinal nerves further branch into smaller nerves in the periphery and innervate bones, ligaments, joints, discs, muscles, organs, and many other tissue types.
Structurally, three protective layers surround the spinal cord, which are themselves extensions of the cranial meninges: the dura mater (outermost), the arachnoid mater, and the pia mater (innermost layer closest to the spinal cord). Within the spinal column, the lumbar dorsal and ventral nerve roots extend below the spinal cord and this neural tissue, collectively called cauda equina, fills the sacral spinal column. The spinal cord is anatomically composed of two regions (Fig. 4.2). These are distinguished by their appearance, function, and cell populations. The gray matter, which has a darker appearance, contains the cell bodies of spinal neurons and makes up the central region of the spinal cord. It is surrounded by the white matter which contains the axons of the spinal neurons. The columnar tracts of the spinal cord are regionally specialized according to information they carry (see Fig. 4.2). The lateral column contains motor neurons; the dorsal column carries information related to mechanoreception; and the ventrolateral column houses neurons which communicate information regarding pain, temperature, and motor signals. In general, the sensory system ascending pathway comprises the dorsal portion of the spinal cord, while the descending pathway of the motor control system comprises the ventral aspect of the cord.
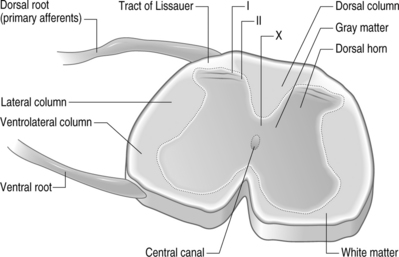
Afferents of the dorsal nerve root enter the spinal cord dorsolaterally and branch in the white matter, with collaterals which terminate in the gray matter. Nerve fibers mediating pain pass through the tract of Lissauer and have branches which terminate in the most superficial regions of the dorsal horn, laminae I and II. Neurons in these laminae synapse on secondary neurons in laminae IV–VI of the dorsal horn and these secondary neurons cross the midline before ascending to the brain contralaterally in the anterolateral region of the cord. Lamina X, which is located in the gray matter region closest to the central canal, also receives sensory inputs related to pain. The neurons of the substantia gelatinosa receive information from Aδ and C fibers; Aβ afferents terminate in the deeper laminae. After injury, it is believed that Aβ afferents sprout from the deeper lamina into the dorsal horn where they make synaptic contacts with neurons.6,7
Each regional level of the spinal cord receives sensory information from specific regions of the body, known as dermatomes. Typically, nerves from approximately two spinal levels innervate any given region of the skin’s surface. These surfaces have been divided into discrete regions, providing a dermatomal map relating each region of the skin to a corresponding spinal level.8 Clinically, dermatomal maps are used to identify the origin of painful symptoms. However, nerve endings which innervate internal organs can also produce cutaneous sensation. This ‘referred pain’ sensation is experienced at sites other than its source and is due to the fact that nearly all spinal neurons that innervate internal organs also are associated with cutaneous sensation.
TRANSDUCTION
Nociceptive afferents are specific for sensing different noxious stimuli: thermal, mechanical, and chemical stimuli. Some nociceptors are polymodal and sense all types of stimuli. Broadly, sensory nerve fibers range in diameter from <0.05 μm to 20 μm. These fibers are either unmyelinated or myelated with thick or thin myelin sheathes enclosing them. Their conduction velocities can range from 0.5 m/s to 120 m/s, depending on their axon diameter and/or the presence of myelination. A fibers are myelinated and can evoke sharp and pricking pain sensation; they can also convey sensations of aching pain. The largest, myelinated sensory axons, Aβ, are generally classified as mechanoreceptive. Aβ fibers are primarily proprioceptive (sensing mechanical movement in joints and muscles). Their diameters are approximately 10 μm; these fibers have slower conduction velocities than Aδ fibers. The smallest myelinated fibers are Aγ fibers, which primarily mediate pin-prick, itching, and other mechanical sensations of pain. Unmyelinated C fibers mediate thermal sensation, in addition to mechanical pain. Stimulation of C fibers can also induce a burning sensation. C and some A fibers are the primary high-threshold noceiceptors. The A fibers exhibit greater response frequencies than C fibers and have more communications to the spinal cord.
Pain detection and signaling begin at the injury site where Aδ and C fibers are activated by thermal, chemical, mechanical, and/or electrical stimuli. These nociceptors can become sensitized, lowering their thresholds for firing and increasing their firing rates when stimulated.9 Further, damaged cells at the site of injury release ATP and free protons. Briefly, a complex series of reactions also occur, including altered blood flow, release of neuropeptides from the afferent fiber itself, and a generalized host of biochemical and cellular responses typical of an immune response. For painful tissue injuries, inflammation is induced in an effort to promote healing and recovery. In this process, inflammatory mediators such as prostaglandin E, serotonin, bradykinin, and histamine, among others, can alter fiber responses. These and other mediators directly activate nociceptors or can sensitize those nociceptors which are already responding to stimuli. This further induces increased neuronal activity. Specifically, bradykinin, serotonin, excitatory amino acids, and hydrogen ions are all responsible for directly activating afferents.9,10 Similarly, prostaglandins, serotonin, noradrenaline, adenosine, nitric oxide, and nerve growth factors can sensitize nociceptors.
In addition to altered electrical activity, and the local synthesis and release of inflammatory mediators that induce inflammation and edema as part of the healing process, these same immune processes that provide healing also sensitize nociceptors and recruit new nociceptors that enhance pain.11,12 In particular, cytokines are released in the periphery in association with tissue injury and inflammation. These small proteins, in turn, contribute to the local inflammatory response, while further modulating electrophysiologic responses of nerve fibers and altering nociception. Additional details regarding specific biochemical mediators of pain and their mechanisms of action throughout the pain signaling pathway (i.e. injury site and spinal cord) are provided in a later section specifically addressing biochemical mediators of pain.
TRANSMISSION
In general, injury to a variety of tissues, including muscle, disc, ligament, and neural tissue, can induce many cascades and physiological signals which lead to pain transmission (Fig. 4.3). During this process, neuroplasticity and subsequent CNS sensitization responses produce altered functions of chemical, electrophysiological, and pharmacological systems throughout the periphery and central systems.4,13–17 This cascade of biochemical and molecular reactions causes changes in gene transcription, post-translational modifications, and culminates with the transmission of pain.18 The interplay of these system responses involves complicated cross-system effects between injury and changes in both the peripheral and central nervous systems. The integration of multiple physiologic systems, as occurs with pain transmission (see Fig. 4.3), contributes to the overall challenges in preventing such syndromes since a given insult may initiate a host of different responses which can be established and maintained remote from the actual site of injury. Regardless, there is a generalized series of responses which occurs following a painful injury in the periphery.
A chemical insult in the periphery causes direct activation of sensory neurons; voltage-gated sodium channels are integral for initiating and propagating pain signals in these sensory neurons. Two separate types of such channels have been designated as SNS and SNS-2, respectively. The SNS subtype is present on both C and some A fibers; SNS-2 channels are exclusively present on unmyelinated neurons. The vanilloid receptor (VR1) is spatially co-localized with these channels.
Calcium concentrations play a critical role in mediating pain in the central nervous system by controlling and regulating levels of nitric oxide synthase (NOS), prostaglandins, and changes in their gene transcription.19 An increase in calcium causes enhanced release of neurotransmitters, as well as an increase in activity of neuronal nitric oxide synthase. Because changes in intracellular calcium occur through the actions of several different channel types, it is possible to isolate the effects of calcium based on which channel opened. For example, in striatal neurons, the population of calcium that activates nNOS is derived from opening of a voltage-gated calcium channel. In addition, increases in intracellular calcium via NMDA receptor influx causes increases in arachidonic acid which can enable COX and prostaglandin production. This process sensitizes neurons of the dorsal horn by a protein kinase C pathway; this second messenger can induce neurons to release substance P and glutamate and provide nociceptive transmission.
Primary afferents communicate with spinal neurons via synaptic transmission. A variety of neurotransmitters (i.e. glutamate, NMDA, substance P) modulate postsynaptic responses, with further transmission to postsynaptic spinal neurons and supraspinal sites via the ascending pathways.9 Peripheral signals of injury pain generate increased neuronal excitability in the spinal cord.17 Associated with this sensitization are a decreased activation threshold, increased response magnitude, and increased recruitment of receptive fields.4 The continuous input from nociceptive afferents can drive spinal circuits and lead to central sensitization, maintaining a chronic pain state.15 These neuroplastic changes are accompanied by other electrophysiological manifestations that cause neurons to fire with increased frequency and even to fire spontaneously.16 Spinal processing is further directly altered by descending inhibitory and facilitory pathways that can provide additional modulation of spinal interneurons.20
Ultimately, persistent pain results from the sensitization of the central nervous system. While the exact mechanism by which the spinal cord reaches a ‘hyperexcitable’ state of sensitization is not fully known, many hypotheses have emerged. Here, only highlights of these theories are provided as an overview. More extensive and detailed discussions can be found elsewhere in the literature.17,21–23 Central hyperexcitability is characterized by a ‘wind-up’ response of repetitive C fiber stimulation, expanding receptive field areas, and spinal neurons taking on properties of wide dynamic-range neurons.24 Low threshold Aβ afferents, which normally do not transmit pain, begin to signal spontaneous and movement-induced pain.22 Aβ fibers stimulate postsynaptic neurons to transmit pain, where these Aβ fibers previously had no effect. This plasticity of neuronal function in the spinal cord contributes to central sensitization.
BIOCHEMICAL MEDIATORS OF PAIN
Many chemical mediators are involved in the transduction and transmission of pain, including local sensory terminals, synaptic transmitters, afferent sensitizers in the periphery, neuronal activators and modulators, and mediators of spinal cord signaling. Many of these mediators are endogenous; some are inducible. In addition, many of these chemicals are directly involved in pain, while some are primarily inflammatory with secondary actions of nociception. Their properties and interactions are eloquently detailed in the previous chapter.4 A brief synopsis of the primary mediators affecting nociception is provided to create context for understanding the complexities of pain signaling.
Bradykinin is a hormone intimately involved in inflammation and vascular responses that also contributes to pain signaling. This protein works through a series of transmembrane G protein-coupled receptors: bradykinin B1 is implicated in chronic pain and bradykinin B2 is involved in acute inflammation and pain.25 Both receptors are expressed on primary sensory neurons where they can directly participate in the primary effects of nociception. Bradykinin sensitizes nociceptors, which produces hyperalgesia. While the B2 receptor is constitutively expressed, the B1 receptor is induced in tissue that is damaged by either infection, disease, or injury. For example, as early as 48 hours after a peripheral nerve injury, B2 receptor mRNA increases, while B1 expression does not reach similar levels until 14 days later.25 B2 receptor kinetics are comparatively fast relative to B1, including the processes of ligand binding, release, and internalization. Through the B2 receptor, bradykinin causes the release of substance P and calcitonin gene-related peptide from primary sensory neurons. This release is further increased in the presence of prostaglandins, which are upregulated at the site of insult.
Substance P is a pronociceptive neurotransmitter, which is released in the periphery following afferent activation. It mediates inflammation by causing vasodilation and the release of prostaglandin E2 and cytokines. These actions impact local inflammation, directly modulating nociception. Substance P can cause a release of calcium from intracellular stores and lead to NO production, neuronal excitability, and long-term sensitization.26,19 Substance P has been shown to have a role in pain in the central nervous system. Administration of antagonists to the substance P receptor, neurokinin-1 (NK-1), can induce antinociception in the central nervous system following chronic painful mechanical nerve constriction.28
Calcitonin gene-related peptide (CGRP), a neuropeptide often co-localized with substance P in the spinal cord, contributes to sensiti zation. It regulates nociceptive responses by promoting the release of substance P and glutamate from primary afferents, while impeding the metabolism of substance P.28,29 CGRP alone causes a slow membrane depolarization in sensory neurons and an influx of calcium through voltage-gated calcium channels. These actions enhance sensitization and promote NK-1 and NMDA activation.19 Antibodies to substance P and CGRP can attenuate pain symptoms in inflammatory models of carageenan-induced hyperalgesia and painful nerve injury;26,30 these studies strongly implicate both such neuropeptides in pain transmission.
Excitatory amino acids, such as glutamate, have potent roles in pain, both at the site of injury and in the central nervous system. Indeed, both non-neuronal and neuronal cells in the periphery produce glutamate. Both of these sources act on primary afferents by activating them when bound to any of the NMDA, kainite, AMPA, or metabotropic glutamate receptors.4 Certainly, this positive feedback mechanism of nociceptor excitation leads to peripheral sensitization and to altered afferent signaling into the spinal cord. Glutamate receptors are expressed in dorsal root ganglion cells,30 suggesting its direct involvement in afferent signaling to the cord. As the NMDA glutamate receptor has a key role in regulating synaptic efficacy, there is also a direct role for this receptor in the spinal plasticity changes associated with central sensitization and persistent pain.
In a normal resting state, NMDA receptors are blocked by the presence of a magnesium ion, and glutamate is free to bind only to low-affinity AMPA receptors. For a neuronal depolarization, the magnesium ion blocking the NMDA receptor becomes liberated, which allows glutamate to bind in its place. This binding activates a complicated cascade, allowing an influx of a host of ions, including calcium. These events, in turn, activate a kinase cascade. Each NMDA receptor is composed of two subunits: the NR1 subunit and the NR2 subunit. The NR2B subunit has been specifically implicated in pain transmission via research findings from a variety of knockout studies,31 and its ability to be more readily phosphorylated than the other NR subunits. NMDA receptors are found both post- and presynaptically. Presynaptic NMDA receptor activation causes the release of substance P from small-diameter primary afferent fibers. The opening of the NMDA receptor also increases the synaptic strength of neurons in the dorsal horn by activating the kinase cascade. These events, which can directly potentiate neuronal fiber sensitization, produce the ‘wind-up’ responses described for dorsal horn neurons.17,24,32 However, despite the known involvement of the NMDA receptor in pain signaling, little clinical success has been demonstrated for NMDA antagonists in ameliorating pain, due to profound side effects such as memory or coordination deficits.
Cytokines can directly and indirectly regulate biochemical cascades leading to the transmission and modulation of pain. Broadly, cytokines include both proinflammatory and antiinflammatory proteins, both of which are upregulated in painful injuries.33–37 In particular, in models of neural injury either distal (peripheral injury) or proximal (nerve root injury) to the DRG, spinal IL-1β, IL-6, IL-10, and TNF-mRNA have all been observed to be significantly elevated for persistent pain.38 Not all of these factors are proinflammatory. For example, IL-10 has been shown to suppress NO production in cultured astrocytes and also suppress proliferation in macrophages,39 which in turn has antiinflammatory effects. The presence of cytokines can further stimulate their own production, as demonstrated by IL-1 stimulating its own production.36 Cytokines mediate cellular processes through the production or suppression of nitric oxide. NO has an immunoregulatory role in the central nervous system. Its production leads directly to hyperalgesia, as previously mentioned. Cytokines regulate NO by interfering with the production of NOS. Astrocytes produce each of the two main forms of NOS, inducible (iNOS) and constitutive (cNOS), whereas microglia are only responsible for inducible NOS.39 TNF-α and IL-1β control the stimulation of iNOS in both astrocytes and glia, therefore inducing the production of NO from those cell types. Alternatively, TGF-β suppresses NO production in both astrocytes and microglia whereas IL-10 only affects NO production in astrocytes.
CNS NEUROIMMUNE RESPONSES IN PAIN
While central sensitization contributes to nociceptive mechanisms of persistent pain in the CNS, recent research has demonstrated the potent role of spinal neuroimmune responses in the generation of chronic pain.40 CNS immune changes have been demonstrated to occur in the setting of persistent pain.33,36–38,41,42 Among the disorders in which this phenomena has been observed are radiculopathy, neuropathy, diabetes, and HIV. These immunological alterations provide evidence that implicates a role for centrally produced proinflammatory cytokines, glial activation, and leukocyte trafficking in rodent pain models of lumbar radiculopathy.38,43–46 From this body of work, a cascade of events in the CNS has been proposed following injury.14,40 Glial cells and neurons become activated and can produce and release cytokines, which in turn can lead to increased activation of these cell types. As well, they lead to increased release of pain mediators.33,34,36 Glial and/or neuronal proinflammatory cytokines can sensitize peripheral nociceptive fields47 and cells in the dorsal root ganglia.48
Events that induce behavioral hypersensitivity also activate central and peripheral immune cells that mediate chronic pain.25,34,36,49,50 Cytokines and growth factors have been strongly implicated in the generation of pathological pain states throughout the nervous system. Specifically, proinflammatory cytokines, such as IL-1, IL-6, and TNF, are upregulated both locally and in the spinal cord.32,33,35 Immune activation with cytokine production may indirectly induce the expression of many pain mediators such as glutamate, nitric oxide, and prostaglandins in the CNS. In conjunction with this neuroimmune activation, neuroinflammatory actions, in which immune cells migrate from the periphery into the CNS in association with pain, occur.14,40,45 Such infiltration may lead to further nociceptive changes in the CNS and potentially to central sensitization. Infiltrating immune cells alter and compound neuronal activation, and promote algesic mediator release, further perpetuating the maintained excitability and sensitization in the CNS, leading to pain and behavioral sensitivity in the case of in vivo models. The spinal immune response of nociception has many facets, forming a complicated cascade of events leading to pain (see Fig. 4.3). It is important to recognize, though, that while quite potent, these immune responses are only one aspect of nociception and the effects of these cellular and chemical sequelae can themselves directly modulate many other potent pathways of pain signaling in the CNS.
INJURY AND BIOMECHANICAL FACTORS MODULATING NOCICEPTION AND PAIN PERCEPTION
Electrophysiologic and neuroimmune responses of the CNS likely work together to affect pain for spinal syndromes, with local biomechanics at the site of an injury modulating both such responses. Low back pain is an ideal syndrome to use as an illustrative example for discussing these mechanisms and cellular response cascades of pain. In this discussion, injury conditions are presented as examples of how mechanical loading modulates nociception in low back pain, with particular emphasis on nerve root injury (i.e. radiculopathy). In addition, throughout the following text comments will be made where applicable to other spinal regions such as the cervical spine.
In vivo studies of animal models of pain report altered electrophysiologic and cellular function for graded cauda equina compression. Applied compression increases endoneurial pressure locally in the rat sciatic nerve and DRG in proportion to the degree of mechanical loading.51,52 It has been shown that edema patterns and intensity are modulated by the nature of the mechanical insult.52–55 Loading to the nerve root can produce changes in electrical impulse propagation and conduction velocity56–58 and repetitive neuronal firing in the dorsal horn of the spinal cord,57,59 which are physiologic correlates leading to spinal sensitization and, consequently, persistent pain. This collective body of research suggests an electrophysiological and neuronal mechanism of spinal cord plasticity and central sensitization for mechanical injuries. It is only inferential for understanding the production and maintenance of pain symptoms.
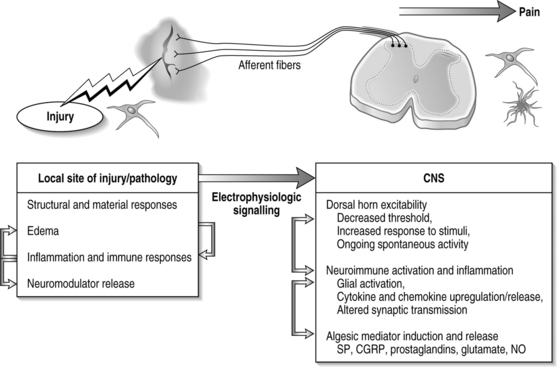
Imaging techniques have been used in a rodent model of painful lumbar radiculopathy to quantify nerve root tissue deformation for an applied root ligation. In that study, Winkelstein et al. examined the injury parameter of tissue deformation in the context of pain (behavioral hypersensitivity).60,61 Local injury mechanics were found to modulate pain behaviors. There was a significant positive correlation in that pain model between behavioral sensitivity and the amount of tissue compression.60 This observation led to a mechanical compressive deformation threshold for pain behaviors and hypersensitivity that were defined based on the amount of nerve root compression.27 More recent work examining cervical nerve root compression magnitude and behavioral sensitivity in the forepaw suggests that a force-based threshold may be more sensitive than deformation for predicting pain symptoms. It has been suggested that a load below 10 g may be sufficient to elicit persistent pain for cervical dorsal nerve root compression.49 Interestingly, while work in the cervical spine suggests that tissue loading may more directly influence pain symptoms, neural tissue is a very soft material and, as such, can undergo extreme tissue deformations before establishing any perceivable load. Continued research is needed to fully define those exact mechanisms through which direct mechanical loading/deformation of the cervical root can be transduced to produce persistent pain. Nonetheless, recent findings of evidence of neuronal degeneration local to the insult (Fig. 4.4) suggest that local changes in the region of injury are robust and occur as early as 1 day after direct insult. Together, mechanical parameters defining painful injuries provide added utility for clinicians in diagnosing painful injuries, directly linking the injury event to the likelihood of pain symptoms. Moreover, in the future, it will hopefully provide insight into predicting clinical outcomes for this class of injuries.
While defining the relationship between spinal tissue insult and pain is necessary for understanding the clinical context of painful pathologies, understanding the specific and relevant nociceptive responses is crucial for characterizing the central mechanisms of persistent spine pain. Using RNase Protection Assays to detect spinal mRNA of a panel of cytokines (TNF-α, IL-1αβ, IL-6, IL-10) in lumbar radiculopathy, a statistically significant correlation was found between mRNA levels at postoperative day 7 and the degree of tissue deformation for lumbar root compression.60 This observation suggests a modulatory effect of injury magnitude on one aspect of spinal nociception. IL-1β has been reported to depend on nerve root compression intensity.44 This observation suggests preservation of these changes at both the message and protein level spinal cytokines involved in chronic low back pain. In comparative models of cervical nerve root injury created by either compression or transection, spinal IL-6 protein at day 1 following radicular injury was elevated, implying a potential relationship to hypersensitivity on the day of assay.35 While continued research into these and other cytokine responses is needed for understanding cervical nerve root injuries and pain mechanisms, findings suggest similar cytokine responses may be evident throughout the spine (i.e. lumbar versus cervical injuries).
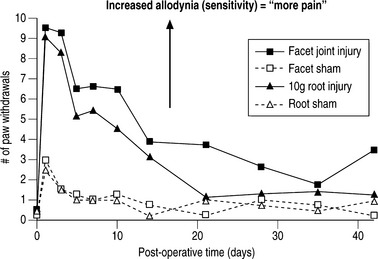
Spinal microglial activation has been demonstrated to be more intense for greater nerve root deformation at injury in lumbar injuries,44,62 which is consistent with the graded behavioral responses and spinal cytokine expression according to injury severity.44,60,61 Interestingly, in these same studies, astrocytic activation did not follow injury magnitude, suggesting that biomechanics at injury in lumbar radiculopathy models may differentially modulate some neuroimmune responses and not others.62 Recent work from the authors’ laboratory has examined these same spinal glial responses in two different cervical spine injuries.49,50 In these studies of nerve root compression and facet joint tension different astrocytic responses were observed. In the nerve root compression model spinal astrocytic expression was elevated compared to sham and followed the behavioral hypersensitivity patterns.49 Astrocytic activation did not show a dependence on mechanical force magnitude. In our facet-mediated painful injury model spinal astrocytic activation did demonstrate a significant correlation with injury magnitude and behavioral sensitivity.50 This finding may suggest that different spinal immune cascades exist for mechanical injuries to different tissue types. In a study directly comparing behavioral hypersensitivity produced for these nerve root compressions and facet-mediated injuries, no differences were observed (Fig. 4.5). Both insults produced symptoms immediately that were sustained for almost 5 weeks depending on the insult. Moreover, within the first 2 weeks following injury, there was no difference in symptom intensity between the two scenarios. However, for this pilot study, the joint-mediated pain appeared to be sustained for a longer duration than the neural tissue injury. Together with the CNS glial findings for these two models of neck pain, these behavioral outcomes suggest that pain symptoms (perception) may be mediated differently depending on the source of the insult or signaling. This comparative work from the authors’ lab suggests that for injuries in the cervical spine direct damage to neural tissue may be perceived the same as for a nondestructive ligament loading scenario.63 These observations and suppositions highlight the need for continued integrative research to identify common and different physiologic mechanisms for injuries within the musculoskeletal system.
SUMMARY
In the typical response of an acutely painful episode for spinal conditions, the balance of injury, repair, and healing is achieved and the cascade of electrophysiologic and chemical events resolves following inflammation and injury. However, for persistent pain, the local, spinal, and even supraspinal, responses are undoubtedly altered from that described above (Fig. 4.6). Based on the discussion presented here regarding persistent pain, a comprehensive picture is emerging for spinal injuries and CNS responses of nociception: spinal cytokine upregulation, microglial and astrocytic activation, altered neuronal–glial interactions, cellular adhesion molecule upregulation, and immune cell infiltration into the spinal cord.14,36,40,46,64 These aspects of neuroimmune activation induce the expression and release of pain mediators (substance P, glutamate, nitric oxide) and also lead to neuronal hypersensitivity. Many of these may be locally and centrally mediated (see Fig. 4.6).
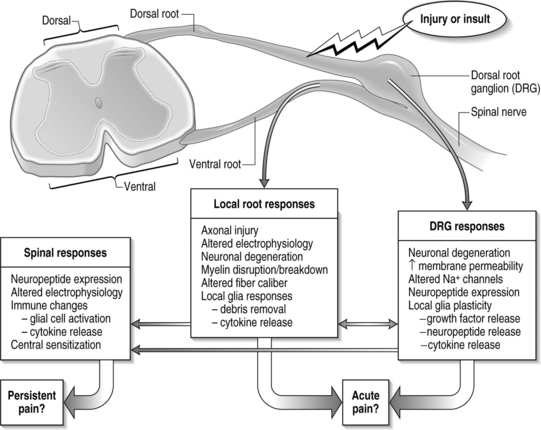
In this context, it is important to consider novel methods for preventing and treating painful injuries. Clinical emphasis has largely been focused on local interventions at the injury site. However, the previous discussion points to the spinal cord physiology as having equal, if not stronger, contribution for maintenance of pain (see Fig. 4.6). Continued understanding of spinal mechanisms of central sensitization can hopefully provide valuable contributions to this understanding. It was the intent of this chapter to highlight existing theories of persistent pain and illuminate interesting new work within the study of pain, highlighting the complications and intricacies of its nature. The extremely complicated mechanisms of pain detection and signaling and their extensive integration with each other point to an equally complicated clinical presentation.
1 Frymoyer J, Cats-Baril W. An overview of the incidences and costs of low back pain. Orthoped Clin N Am. 1991;22:263-271.
2 Frymoyer J, Durett C. The economics of spinal disorders. In: Frymoyer JW, editor. The adult spine: principles and practice. Philadelphia: Lippincott-Raven, 1997.
3 Kivioja J, Rinaldi L, Ozenci V, et al. Chemokines and their receptors in whiplash injury: elevated RANTES and CCR-5. J Clin Immunol. 2001;21:272-277.
4 Wall P, Melzack R. Textbook of pain, 3rd Edn. London: Churchill Livingstone, 1994.
5 Kandel ER, Schwartz JH, Jessell TM. Principles of neural science, 3rd Edn. New York: Elsevier, 1991.
6 Furue H, Katafuchi T, Yoshimura M. Sensory processing and functional reorganization of sensory transmission under pathological conditions in the spinal dorsal horn. Neurosci Res. 2004;48:361-368.
7 Kohno T, Moore K, Baba H, et al. Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J Physiol. 2003;548:131-138.
8 Slipman C, Plastaras C, Palmitier R, et al. Symptom provocation of fluoroscopically guided cervical nerve root stimulation. Are dynatomal maps identical to dermatomal maps? Spine. 1998;23:2235-2242.
9 Cavanaugh JM. Neurophysiology and neuroanatomy of neck pain. In: Yoganandan N, Pintar FA, editors. Frontiers in whiplash trauma: clinical and biomechanical. Amsterdam: IOS Press; 2000:79-96.
10 Kawakami M, Weinstein JN. Associated neurogenic and nonneurogenic pain mediators that probably are activated and responsible for nociceptive input. In: Weinstein J, Gordon S, editors. Low back pain: a scientific and clinical overview. Rosemont, IL: AAOS; 1986:265-273.
11 Dray A, Perkins M. Bradykinin and inflammatory pain. Trends Neurosci. 1993;16(3):99-104.
12 Dubner R, Hargreaves KM. The neurobiology of pain and its modulation. Clin J Pain. 1989;S2:S1-S6.
13 Black J, Langworthy K, Hinson A, et al. NGF has opposing effects on Na+ channel III and SNS gene expression in spinal sensory neurons. Neuroreport. 1997;8:2331-2335.
14 DeLeo J, Winkelstein B. Physiology of chronic spinal pain syndromes: From animal models to biomechanics. Spine. 2002;27(22):2526-2537.
15 Devor M. Neuropathic pain and injured nerve: peripheral mechanisms. Br Med Bull. 1991;47:619-630.
16 Waxman S, Dib-Hajj S, Cummins T, et al. Sodium channels and pain. Proc Nat Acad Sci USA. 1999;96:7635-7639.
17 Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306:686-688.
18 Costigan M, Woolf C. Pain: molecular mechanisms. J Pain. 2000;1:35-44.
19 Millan M. The induction of pain: An integrative review. Progress Neurobiol. 1999;57:1-164.
20 Vanderah TW, Ossipov MH, Lai J, et al. Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain. 2001;92:5-9.
21 Coderre TJ, Katz J, Vaccarino A, et al. Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain. 1993;52:259-285.
22 Devor M. Pain arising from the nerve root and the dorsal root ganglion. In: Weinstein J, Gordon S, editors. Low back pain: a scientific and clinical overview. Rosemont, IL: AAOS; 1986:187-208.
23 Dubner R, Basbaum AI. Spinal dorsal horn plasticity following tissue or nerve injury. In: Wall PD, Melzak R, editors. Textbook of pain. Edinburgh: Churchill-Livingstone; 1994:225-241.
24 Cook AJ, Woolf CJ, Wall PD, et al. Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature. 1987;325:151-153.
25 Couture R, Harrisson M, Vianna RM, et al. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001;429:161-176.
26 Ma W, Eisenach J. Intraplantar injection of a cyclooxygenase inhibitor ketorolac reduces immunoreactivities of substance P, calcitonin gene-related peptide, and dynorphin in the dorsal horn of rats with nerve injury or inflammation. Neuroscience. 2003;121:681-690.
27 Winkelstein BA, DeLeo JA. Mechanical thresholds for initiation and persistence of pain following nerve root injury: Mechanical and chemical contributions at injury. J Biomech Engineer. 2004;126:258-263.
28 Meert T, Vissers K, Geenan F, et al. Functional role of exogenous administration of substance P in chronic constriction injury model of neuropathic pain in gerbils. Pharmacol Biochem Behav. 2003;76(1):17-25.
29 Allen B, Li J, Menning P, et al. Primary afferent fibers that contribute to increased substance P receptor internalization in the spinal cord after injury. J Neurophysiol. 1999;81(3):1379-1390.
30 Satoh M, Kuraishi Y, Kawamura M. Effects of intrathecal antibodies to substance P, calcitonin gene-related peptide and galanin on repeated cold stress-induced hyperalgesia: comparison with carrageenan-induced hyperalgesia. Pain. 1992;49(2):273-278.
31 Bursztajn S, Rutkowski M, Deleo J. The role of the N-methyl-D-aspartate receptor NR1 subunit in peripheral nerve injury-induced mechanical allodynia, glial activation and chemokine expression in the mouse. Neuroscience. 2004;125:269-275.
32 Woolf C, Safieh-Garabedian B, Ma Q, et al. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:277-331.
33 DeLeo J, Colburn R. The role of cytokines in nociception and chronic pain. In: Weinstein J, Gordon S, editors. Low back pain: a scientific and clinical overview. Rosemont, IL: AAOS; 1986:163-185.
34 DeLeo J, Colburn R, Nichols M, et al. Interleukin (IL)-6 mediated hyperalgesia/allodynia and increased spinal IL-6 in two distinct mononeuropathy models in the rat. J Interferon Cytokine Res. 1996;16:695-700.
35 Hubbard R, Rothman S, Winkelstein B. Mechanisms of persistent neck pain following nerve root compression injury: understanding behavioral hypersensitivity in the context of spinal cytokine responses and tissue biomechanics. North American Spine Society 19th Annual Meeting, #P49, Chicago, IL, October, 2004.
36 Watkins L, Maier S, Goehler L. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses, and pathological pain states. Pain. 1995;63:289-302.
37 Watkins L, Wiertelak E, Goehler L, et al. Characterization of cytokine-induced hyperalgesia. Brain Res. 1994;654:15-26.
38 Winkelstein B, Rutkowski M, Sweitzer S, et al. Nerve injury proximal or distal to the DRG induces florid spinal neuroimmune activation related to enhanced behavioral sensitivity. J Comparative Neurol. 2001;439:127-139.
39 Xiao BG, Link H. Immune regulation within the central nervous system. J Neurol Sci. 1998;157:1-12.
40 DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;91:1-6.
41 Rutkowski M, Pahl J, Sweitzer S, et al. Limited role of macrophages in generation of nerve injury-induced mechanical allodynia. Physiol Behav. 2000;71:225-235.
42 Sweitzer S, Martin D, DeLeo J. IL-1ra and sTNFr reduces mechanical allodynia and spinal cytokine expression in a model of neuropathic pain. Neuroscience. 2001;103:529-539.
43 Colburn R, Rickman A, DeLeo J. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exper Neurol. 1999;157:289-304.
44 Hashizume H, DeLeo J, Colburn R, et al. Spinal glial activation and cytokine expression following lumbar root injury in the rat. Spine. 2000;25:1206-1217.
45 Rutkowski MD, Winkelstein BA, Hickey WF, et al. Lumbar nerve root injury induces CNS neuroimmune activation and neuroinflammation in the rat: Relationship to painful radiculopathy. Spine. 2002;27(15):1604-1613.
46 Sweitzer S, Arruda J, DeLeo J. The cytokine challenge: methods for the detection of central cytokines in rodent models of persistent pain. In: Kruger L, editor. Methods in pain research. Boca Raton: CRC Press; 2001:109-132.
47 Junger H, Sorkin LS. Nociceptive and inflammatory effects of subcutaneous TNF alpha. Pain. 2000;85(1–2):145-151.
48 Ozaktay AC, Cavanaugh JM, Asik I, et al. Dorsal root sensitivity to interleukin-1 beta, interleukin-6 and tumor necrosis factor in rats. Eur Spine J. 2002;11(5):467-475.
49 Hubbard RD, Winkelstein BA. Transient cervical nerve root compression in the rat induces bilateral forepaw allodynia and spinal glial activation: mechanical factors in painful neck injuries. Spine. 2005;30(17):1924-1932.
50 Lee K, Davis M, Mejilla R, et al. In vivo cervical facet capsule distraction: mechanical implications for whiplash and neck pain. Proceedings of 48th Stapp Car Crash Conference, Paper #2004-22-0016, 48:373-393, 2004.
51 Lundborg G, Myers R, Powell H. Nerve compression injury and increased endoneurial fluid pressure: a ‘miniature compartment syndrome.’. J Neurol Neurosurg Psychiatry. 1983;46:1119-1124.
52 Rydevik B, Myers R, Powell H. Pressure increase in the dorsal root ganglion following mechanical compression. Closed compartment syndrome in nerve roots. Spine. 1989;14:574-576.
53 Olmarker K, Holm S, Rydevik B. Importance of compression onset rate for the degree of impairment of impulse propagation in experimental compression injury of the porcine cauda equina. Spine. 1990;15:416-419.
54 Olmarker K, Rydevik B, Holm S. Edema formation in spinal nerve roots induced by experimental, graded compression. An experimental study on the pig cauda equina with special reference to differences in effects between rapid and slow onset of compression. Spine. 1989;14:569-573.
55 Pedowitz R, Garfin S, Massie J, et al. Effects of magnitude and duration of compression on spinal nerve root conduction. Spine. 1992;17:194-199.
56 Cornefjord M, Sato K, Olmarker K, et al. A model for chronic nerve root compression studies. Presentation of a porcine model for controlled slow-onset compression with analyses of anatomic aspects, compression onset rate, and morphologic and neurophysiologic effects. Spine. 1997;22:946-957.
57 Hanai F, Matsui N, Hongo N. Changes in responses of wide dynamic range neurons in the spinal dorsal horn after dorsal root or dorsal root ganglion compression. Spine. 1996;21:1408-1415.
58 Skouen J, Brisby H, Otami K, et al. Protein markers in cerebrospinal fluid experimental nerve root injury. A study of slow-onset chronic compression effects or the biochemical effects of nucleus pulposus on sacral nerve roots. Spine. 1999;24:2195-2200.
59 Yoshizawa H, Kobayashi S, Kubota K. Effects of compression on intraradicular blood flow in dogs. Spine. 1989;14:1220-1225.
60 Winkelstein B, Rutkowski M, Weinstein J, et al. Quantification of neural tissue injury in a rat radiculopathy model: comparison of local deformation, behavioral outcomes, and spinal cytokine mRNA for two surgeons. J Neurosci Methods. 2001;111:49-57.
61 Winkelstein B, Weinstein J, DeLeo J. Local biomechanical factors in lumbar radiculopathy: An in vivo model approach. Spine. 2001;27:27-33.
62 Winkelstein BA, DeLeo JA. Nerve root tissue injury severity differentially modulates spinal glial activation in a rat lumbar radiculopathy model: Considerations for persistent pain. Brain Res. 2002;956(2):294-301.
63 Lee K, Thinnes J, Gokhin D, et al. A novel rodent neck pain model of facet-mediated behavioral hypersensitivity: implications for persistent pain and whiplash injury. J Neurosci Methods. 2004;137:151-159.
64 Sweitzer S, White KA, Dutta C, et al. The differential role of spinal MHC class II and cellular adhesion molecules in peripheral inflammatory versus neuropathic pain in rodents. J Neuroimmunol. 2002;125(1-2):82-93.