[level-membership-for-pathology-category]
Chapter 14 Respiratory tract
COMMON CLINICAL PROBLEMS FROM RESPIRATORY TRACT DISEASE
Pathological basis of respiratory signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Sputum |
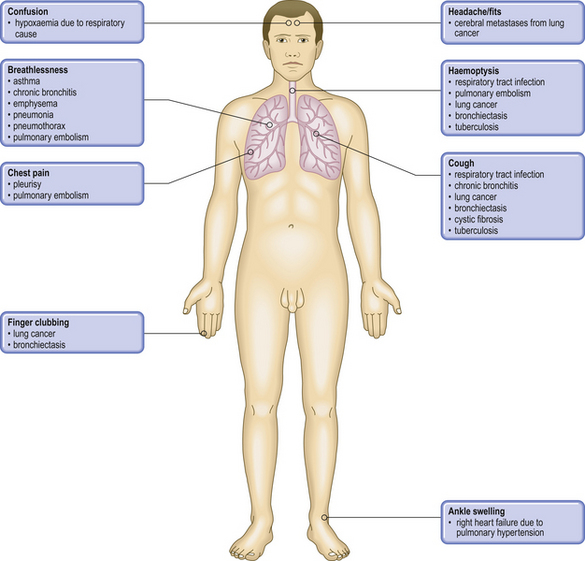
CoughPhysiological refl ex response to presence of mucus, exudate, tumour or foreign materialWheezing
DyspnoeaDecreased oxygen in the blood from impaired alveolar gas exchange, left heart failure or anaemiaCyanosisIncreased non-oxygenated haemoglobin, e.g. circulatory bypassing of lungs in congenital heart diseases or impaired alveolar gas exchangePleuritic painIrritation of the pleura due to pulmonary infl ammation, infarction or tumourPleural effusion
ClubbingOften accompanies carcinoma of lung and pulmonary fi brosis, as well as, less commonly, cirrhosis and chronic infl ammatory bowel diseaseWeight lossProtein catabolic state induced by chronic infl ammatory disease (e.g. tuberculosis) or tumoursAuscultation signs
Percussion signs
Globally respiratory diseases, particularly lung infections, together with gastrointestinal infection, account for most deaths in the developing world. Respiratory disease is also a common cause of death in the industrialised nations, accounting for about 14% of deaths in each sex. Out of a global total of 55.69 million deaths in 2000, 3.86 million were due to acute lower respiratory tract infections, 1.66 million to tuberculosis, 2.94 million to HIV/AIDS, 1.21 million to lung cancer and 3.54 million to a variety of other respiratory diseases, mainly chronic obstructive pulmonary disease (2.52 million). There is also considerable morbidity due to respiratory diseases: it is estimated that, in the UK, about 40% of absence from work is the result of such diseases, approximately 85% of which are transient infections of the upper respiratory tract (Table 14.1).
Table 14.1 Major aetiological factors in respiratory disease
| Aetiological factor | Disease |
|---|---|
| Genetic | |
| Air pollution | |
| Occupation | |
| Infection |
NORMAL STRUCTURE AND FUNCTION
The respiratory system extends from the nasal orifices to the periphery of the lung and the surrounding pleural cavity. From the nose to the distal bronchi, the mucosa is lined by mainly pseudostratified ciliated columnar epithelium with mucus-secreting goblet cells; this is respiratory mucosa (Fig. 14.1). A portion of the larynx is covered with stratified squamous epithelium.
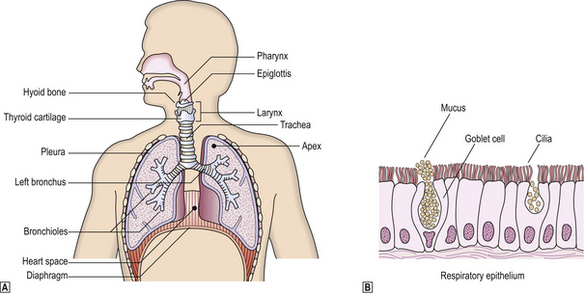
Fig. 14.1 The respiratory system.  Anatomy of the respiratory tract.
Anatomy of the respiratory tract.  Histology of respiratory epithelium. With the exception of the pharynx, epiglottis and vocal cords, the respiratory tract is lined by specialised epithelium compring ciliated columnar epithelia cells with admixed mucus-secreting goblet cells and scattered neuroendocrine cells.
Histology of respiratory epithelium. With the exception of the pharynx, epiglottis and vocal cords, the respiratory tract is lined by specialised epithelium compring ciliated columnar epithelia cells with admixed mucus-secreting goblet cells and scattered neuroendocrine cells.
Nasal passages and sinuses
These constitute the upper respiratory tract. The nasal passages and sinuses are in continuity and are lined with respiratory mucosa. The hairs in the nose trap large particles of foreign material, thereby filtering the air. The air is also warmed and humidified as it passes through the nasal cavity. The middle ear, also lined with respiratory epithelium, connects with the nasal cavity via the Eustachian tube.
Larynx
The larynx connects the trachea to the pharynx. Consisting of a complicated system of cartilages and muscles, it allows air into the trachea, with the epiglottis preventing the passage of food into the lungs, and also produces sound for speaking. Part of the larynx, including the vocal cords and epiglottis, is covered with non-keratinising squamous epithelium similar to that lining the oral cavity, pharynx and oesophagus.
Lungs
The lower respiratory tract consists of the trachea, bronchi, bronchioles, alveolar ducts and alveoli (Fig. 14.2). (Table 14.2).
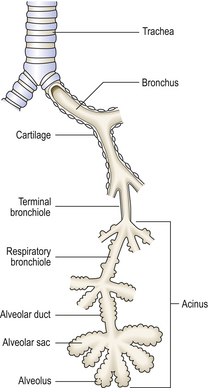
Table 14.2 Structure of the respiratory tree
| Part of respiratory tract | Structure |
|---|---|
| Trachea | Anterior C-shaped plates of cartilage with posterior smooth muscle. Mucous glands |
| Bronchi | Discontinuous foci of cartilage with smooth muscle. Mucous glands |
| Bronchioles | No cartilage or submucosal mucous glands. Clara cells secreting proteinaceous fluid. Ciliated epithelium |
| Alveolar duct | Flat epithelium. No glands. No cilia |
| Alveoli | Type I and II pneumocytes |
The lungs develop from an outpouching of the anterior wall of the primitive foregut at about the fifth week of development. From this tube, two lateral outgrowths appear which eventually form the right and left lungs. These outgrowths are surrounded by mesenchyme from which forms the connective tissue of the respiratory tree. Thus the lungs, like the gastrointestinal tract, develop from endoderm, and developmental abnormalities such as cysts can therefore be lined by either respiratory or gastrointestinal mucosa.
The lungs are divided into lobes: the right lung has three lobes (upper, middle, lower); the left lung has only two lobes (upper and lower). Each lung is formed of 10 anatomically defined bronchopulmonary segments. Each segment is supplied by a segmental branches of the pulmonary artery and bronchus (the bronchovascular bundle). The veins draining adjacent segments often anastomose before they reach the hilum and run principally in the fibrous septae of the lungs.
The respiratory tree is designed to transport clean, humidified air into distal airways and alveoli, where the waste product of metabolism (CO2) is exchanged for O2.
Bronchioles branch until they form terminal bronchioles less than 2 mm in diameter. The respiratory system distal to the terminal bronchiole is called the acinus or terminal respiratory unit, where gas exchange occurs. Small airways, defined as having an internal diameter of less than 2 mm, consist of terminal and respiratory bronchioles. Respiratory bronchioles are involved with gas exchange, having alveoli in their walls. A group of three to five respiratory acini is called a lobule.
The alveoli are lined by flattened type I pneumocytes with occasional type II pneumocytes; the latter are rounded cells with surface microvilli and are believed to be the stem cell population for the alveolus. Type II cells secrete surfactant, and replicate quickly after injury to alveolar walls. Beneath the alveolar cells lie a basement membrane which is shared by the alveolar capillary epithelial cells and some interstitial matrix, including elastin fibres. The structure of the alveolar–capillary membrane permits rapid and efficient diffusion of oxygen and carbon dioxide.
The lung is encased by the visceral pleura which is a thin layer of fibroconnective tissue and elastin with overlying mesothelial cells. The lungs sit within the chest cavity surrounded by the parietal pleura, by the diaphragm, ribs and intercostal muscles, vertebral column and sternum.
Blood supply and lymphatic drainage
The lungs are perfused by a dual arterial blood supply. The trunk of the pulmonary artery arises from the right ventricle, splits into main right and left pulmonary arteries and thence follows the airways, forming the bronchovascular bundles. The bronchial arteries arise from the descending thoracic aorta and supply oxygenated blood to lung parenchyma around the hilum. Pulmonary veins take all the blood from the lungs back to the left atrium.
Pulmonary veins course along the interlobular septa with lymphatics.These lymphatic channels and those in the pleura drain into the thoracic duct.
Control of respiration
Respiration is controlled by the respiratory centre in the medulla oblongata, and the carotid bodies situated at the carotid bifurcations. The medullary centre senses any change in CO2 concentration in the cerebrospinal fluid, and modifies respiration by nervous stimulation of respiratory muscles and the diaphragm. The partial pressure of O2 in the blood is monitored by the carotid bodies, which can then stimulate the respiratory centre through the glossopharyngeal nerves. Carotid bodies can become hyperplastic in response to chronic arterial hypoxaemia, such as occurs in:
Gas exchange
Air is drawn into the lungs by contraction of the diaphragm and intercostal muscles, creating a negative intrapleural pressure. On relaxation of these muscles, air is expelled as the lungs contract under the action of gravity and the elasticity in the lung connective tissue. The stiffness of the lungs, or compliance, is a measure of change in volume per unit change in pressure, and is therefore a measure of compressibility; for example, in pulmonary fibrosis the lungs cannot be easily compressed and therefore the compliance is decreased.
Clearly, gas exchange occurs only in alveoli that are both perfused and ventilated. Ventilation of non-perfused alveoli increases the ‘dead space’, that proportion of inspired air not involved with gas exchange. Perfusion of non-ventilated alveoli leads to physiological right-to-left shunting of non-oxygenated blood as it passes through the pulmonary circulation.
Acid–base balance
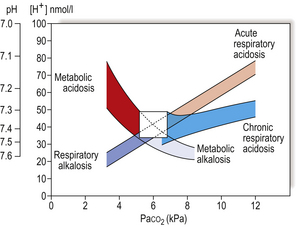
Normal acid–base balance in blood is dependent on both efficient alveolar ventilation and perfusion, with consequent successful gas exchange. This leads to the normal partial pressures of O2 and CO2 in arterial blood (Pao2 and Paco2), and a normal blood pH. Various metabolic disease states lead to disturbances in acid–base balance (Fig. 14.3). If the disease becomes chronic, compensatory mechanisms by both the lungs and kidneys operate in an attempt to restore blood pH.
PULMONARY FUNCTION TESTS
In normal quiet respiration only a relatively small proportion of the total lung capacity (TLC) is inhaled and exhaled; this is the tidal volume (TV). TLC is made up of the amount of air totally exhaled after maximum inspiration (the vital capacity or VC) and the residual volume (RV). TLC, RV, TV and VC are all easily measured in the laboratory using helium dilution techniques.
In addition to calculating volume parameters, some techniques also assess actual pulmonary function. Spiro-metry measures the amount of exhaled air per second. The maximum volume of air blown from the lungs within the first second after a previous maximum inspiration is called the forced expiratory volume (FEV1). This figure, highly reproducible in each individual, is a measure of small airway resistance. It is also dependent on the patient’s age, sex and size; for example, the small lungs of a child obviously cannot expel as much air as those of an adult. The ratio FEV1:VC compensates to a degree for the variability of lung size. It is possible to inhale more rapidly than exhale because, during inspiration, forces on the airways tend to open them further; during expiration, opposite forces tend to close the airways and thus restrict airflow. For a given lung volume, the expiratory flow rate reaches a peak (PEFR), which is again a measure of airways resistance.
An assessment of the ability of the lungs to exchange gas efficiently can be made by measuring the transfer factor for carbon monoxide (TCO). Air containing a known concentration of carbon monoxide is inhaled; the breath is held for 15 seconds and then exhaled. The amount of carbon monoxide absorbed is a measure of pulmonary gas exchange. TCO is dependent on the concentration of blood haemoglobin, which has a strong affinity for carbon monoxide. Diseases that diffusely affect the alveolar–capillary membrane (such as diffuse pulmonary fibrosis or emphysema where there is loss of alveolar surface area) will result in a low TCO.
Recently the level of nitric oxide (NO) in exhaled air has been added as a useful test; increased levels have been associated with asthma and other causes of bronchial irritation, while decreased levels have been found in cigarette smokers, patients with pulmonary hypertension and during treatment with corticosteroids.
Obstructive and restrictive defects
There are two major patterns of abnormal pulmonary function tests: obstructive defects (e.g. asthma) and restrictive defects (e.g. pulmonary fibrosis) (Table 14.3).
Table 14.3 Respiratory function tests and their diagnostic significance
| Test | Diagnostic significance |
|---|---|
| Peak expiratory flow rate (PEFR) | Reduced with obstructed airways or muscle weakness |
| Forced expiratory volume in 1 second (FEV1) | Reduced with obstructed airways, pulmonary fibrosis or oedema, or muscle weakness |
| Vital capacity (VC) |
Forced expiratory ratio (FEV1:VC)
Carbon monoxidetransfer (TCO)Reduced in pulmonary fibrosis, emphysema, oedema, embolism and anaemiaExhaled nitric oxide (NO)Increased in asthma, bronchiectasis and infections Decreased in pulmonary hypertension, cigarette smokers and after treatment with corticosteroids
In obstructive airways disease, RV and TLC are mildly increased due to hyperinflation of the lung while FEV1, FVC and the FEV1: VC ratio is decreased. Clearly, in conditions such as asthma, the results of pulmonary function tests will depend on the clinical state of the patient, whether in an acute attack of asthma or in remission. Restrictive diseases are those that restrict normal lung movement during respiration and are associated with reduced RV and TLC. The FEV1 and VC may be reduced but their ratio remains normal.
These tests are of most value in the follow-up of patients. They can also give an indication as to the possible benefits of treatment; for example, observing the improved FEV1:VC and PEFR after treatment with a bronchodilator would be a measure of the reversibility of the airways obstruction.
RESPIRATORY FAILURE
Respiratory failure can occur as a result of:
The effects of respiratory failure include impaired clearance of CO2 from the lungs, resulting in hypercapnia, and impaired absorption of O2 from the air, resulting in hypoxaemia. The patient is typically dyspnoeic, cyanosed and lapsing into coma. Hypercapnia (high blood CO2 concentration) is associated with a bounding pulse and warm, moist extremities.
DISEASES OF INFANCY AND CHILDHOOD
Respiratory diseases of infancy and childhood are predominantly infectious; such diseases, together with diarrhoea, are the primary cause of death in childhood in the developing world. Rarely disease may arise as a result of either developmental abnormalities or immaturity.
Developmental abnormalities
Developmental abnormalities include:
Tracheo-oesophageal fistula
Embryologically, the oesophagus and the trachea begin as a single tube; the trachea then buds off to form the pulmonary tree. In a tracheo-oesophageal fistula, the oesophagus ends in a blind pouch; the trachea then usually connects to the stomach via a fistula. On ingestion of food, the upper pouch quickly fills and overflows into the pulmonary tree, with choking and coughing, leading to aspiration pneumonia. Treatment is by surgery, usually after a period of feeding via gastrostomy.
Congenital diaphragmatic hernia with pulmonary hypoplasia
This presents as neonatal respiratory distress due to herniation of the stomach and loops of bowel into the thorax; usually the left diaphragm is defective. Surgical correction to restore normal thoracic and abdominal anatomy is essential at the earliest possible opportunity. However, even after this there is still a considerable mortality from the associated severe pulmonary hypoplasia, usually of the left lung.
Congenital cystic adenomatoid malformations
These are characterised by abnormalities in the development of small airways and the alveolar tissue of the lung. This results in the development of cysts within the lung which may be of varying size and can be localised to one lobe or be extensive and bilateral. The prognosis depends on the pattern of abnormality present and any other associated abnormalities. Localised areas are cured by surgical excision of the affected lobe.
Bronchogenic/foregut cysts
These occur in the lung or mediastinum and may be lined either by bronchial elements such as cartilage, smooth muscle and ciliated respiratory epithelium (bronchogenic cysts), or by squamous or even gastric or pancreatic type epithelium (foregut cysts). Usually, such cysts are asymptomatic, although complications may occur.
Pulmonary sequestration
A sequestered piece of lung is a mass of abnormal lung that does not communicate anatomically with the tracheo-bronchial tree; it is supplied by an anomalous artery, usually from the aorta. Sequestered segments of lung are found most often within the left lower lobe. Histology shows a multilobulated cystic mass with fibrosis and variable inflammation. An endogenous lipid pneumonia may result.
Congenital lobar emphysema
This condition is characterised by overdistension of a lobe due to intermittent bronchial obstruction. Symptoms arise due to pressure effects caused by the massively distended lobe. Usually, the left upper lobe is affected. The pathogenesis is thought to be abnormal bronchial cartilage allowing inspiration of air but not expiration. Extrabronchial compression by enlarged lymph nodes may produce a similar clinical picture. Treatment is surgical removal of the diseased lobe.
lmmaturity
Diseases due to immaturity include:
Hyaline membrane disease or idiopathic respiratory distress syndrome






Hyaline membrane disease (HMD) is almost always seen in premature infants of birth weight less than 2.5 kg. Infants are usually of less than 36 weeks’ gestation, and the incidence of HMD rises as the gestational age decreases. The risk of developing HMD may be decreased by giving mothers oral corticosteroids prior to delivery of the baby as this appears to stimulate surfactant production in the lungs.
Clinical features
After a few hours of relatively normal respiration, symptoms of tachypnoea and dyspnoea with expiratory grunting appear. Cyanosis quickly follows, with worsening respiratory distress. Hypoxaemia refractory to high concentration of inhaled oxygen is one hallmark of the disease, a finding also characteristic of adult respiratory distress syndrome (ARDS).
Pathogenesis
The pathogenesis is thought to be due to a deficiency of surfactant. This is secreted by type II pneumocytes, and normally lines distal airways; it reduces surface tension, thereby allowing airway opening during inspiration. Without normal quantities of surfactant, airways need greater effort to open, leading to respiratory distress.
Morphology
At autopsy the lungs are heavy, purple and solid, and sink in water. Histology shows collapsed alveoli with hyaline membranes lining alveolar ducts. Pulmonary lymphatics are dilated. As in ARDS, if the infant survives, resolution follows within the next few days, although pulmonary fibrosis may occur. Treatment is with oxygen and artificial ventilation.
Bronchopulmonary dysplasia
Bronchopulmonary dysplasia is the term used to describe the picture of lung organisation after HMD. Often, infants have been previously treated with high levels of oxygen, and it is not clear whether bronchopulmonary dysplasia is a separate disorder, solely related to oxygen toxicity, or merely a result of organisation after HMD. Certainly, the features are almost identical to those seen with organisation of ARDS; there is interstitial fibrosis, peribronchial fibrosis, and features of pulmonary hypertension. The airways may show extensive squamous metaplasia.



INFLAMMATORY DISORDERS
Rhinitis (the common cold) is caused by many different viruses, especially rhinoviruses, although respiratory syncytial virus (RSV), para-influenza viruses, coronaviruses, coxsackieviruses, echoviruses and bacteria, such as Haemophilus influenzae, may also be implicated. Rhinitis may also be caused by inhaled allergens as in ‘hay fever’ where the inflammatory reaction is mediated via type I and type III hypersensitivity reactions (Chs 9 and 10).
Nasal polyps may result from either chronic infective inflammation or chronic allergic inflammation. They consist of polypoid oedematous masses of mucosal tissue infiltrated with chronic inflammatory cells, especially plasma cells; eosinophils may be numerous if allergy is the cause.
Sinusitis is inflammation of the paranasal sinuses; it may be acute or chronic. If the drainage orifice is blocked by inflamed swollen mucosa, an abscess may follow. Cranial osteomyelitis, meningitis or cerebral abscess may then result from sinusitis by direct extension.
Wegener’s granulomatosis, a granulomatous form of vasculitis may involve the nose and upper respiratory tract and present with septal perforation or collapse of the nasal cartilages.
Otitis media is infection of the middle ear, often associated with generalised upper respiratory tract infection (URTI). The Eustachian tube may become swollen and blocked, leading to trapping of exudate in the middle ear. Eardrum perforation may ensue, followed by drainage of the effusion. More serious complications include mastoiditis, meningitis and brain abscess.
TUMOURS
Tumours of the nasal passages and sinuses are uncommon. They may be:
Haemangioma and squamous papilloma are benign lesions, the former often presenting with troublesome epistaxis (nosebleeds). Some squamous papillomas may be caused by human papillomavirus.
Juvenile angiofibromas are rare and occur exclusively in males, usually during adolescence. They are extremely vascular, and surgical removal can be difficult. These tumours contain androgen receptors, explaining the male preponderance.
Squamous cell carcinoma may be well differentiated, producing keratin, or very poorly differentiated. The latter may contain many lymphocytes and have been misnamed ‘lympho-epitheliomas’. Such tumours are most common in South-East Asia and account for 18% of all cancers in China; evidence suggests the Epstein–Barr virus is involved in the aetiology and pathogenesis of squamous cell carcinoma.
Adenocarcinoma of the nasal passages and sinuses occurs more frequently in people who have worked in woodwork and furniture industries. These tumours may present clinically up to 40 years after initial exposure.
Primary mucosal melanomas of the nose and sinuses are rare but have a very poor prognosis.
Primary extranodal lymphomas are almost always of non-Hodgkin’s type.
Plasmacytomas are tumours composed of plasma cells. They can occur as part of multiple myeloma or as isolated lesions without systemic disease.



INFLAMMATORY DISORDERS
Laryngitis may occur in association with viral or bacterial inflammation of trachea and bronchi; this is laryngotracheo-bronchitis. Diphtheria was once a common, and serious, bacterial cause of laryngitis, leading to the formation of a fibrinopurulent membrane that could cause airway obstruction. Now, partly as a result of immunisation in infancy with diphtheria toxoid, the disease is rare.
Epiglottitis is caused by capsulated forms of Haemophilus influenzae type B. The epiglottis becomes inflamed and greatly swollen, leading to airway obstruction (Fig. 14.4). Treatment is by intubation, although, rarely, tracheostomy may be necessary; antibiotics are also given to treat the infection.
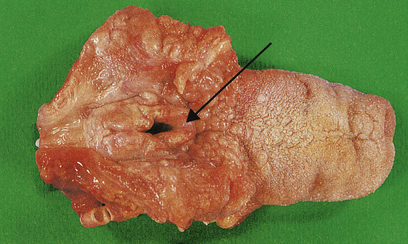
Fig. 14.4 Acute epiglottitis. Gross swelling of the epiglottis (arrowed) leading to respiratory obstruction in a child.
Allergic laryngitis occurs after inhalation of an allergen. There may be gross oedema leading to airway obstruction. Irritative laryngitis may be due to cigarette smoke or mechanical factors, e.g. endotracheal intubation.
Laryngeal polyps often develop in singers and are thus sometimes referred to as ‘singer’s nodes’. Even when only a few millimetres in diameter they can alter the character of the voice. They consist of oedematous myxoid connective tissue covered with squamous mucosa with amyloid-like material in the stroma.
TUMOURS
Papilloma may be caused by types of human papillomavirus. Papillomas consist of squamous epithelium covering fibrovascular cores of stroma. They may be multiple and recurrent, especially in children, but are usually single in adults. Such papillomas can extend into the trachea and bronchi.
Squamous cell carcinoma of the larynx typically affects males over 40 years of age and is associated with cigarette smoking. There may also be an increased risk in asbestos workers. As in squamous epithelium of the cervix, neoplasia is thought to be preceded by a phase of dysplasia. The dysplasia, especially if low grade, may be reversible on withdrawal of causative factors.
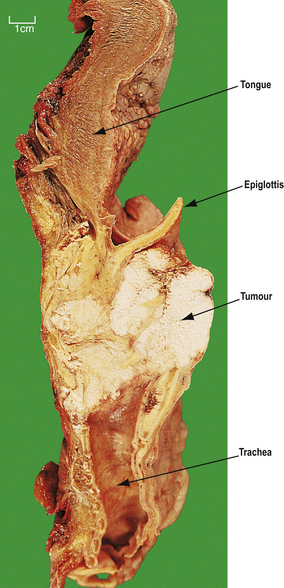
Most laryngeal carcinomas arise on the vocal cords (Fig. 14.5), although they may arise above, in the pyriform fossa, or below, as upper tracheal carcinomas. The lesions ulcerate, fungate and invade locally, later causing metastases in regional lymph nodes in the neck and beyond. Symptoms are hoarseness of voice and, later, pain, haemoptysis and dysphagia. Treatment is by chemo-radiotherapy and/or resection. These patients often have widespread mucosal abnormalities throughout the respiratory tract and are at high risk of developing further cancers, especially if they continueto smoke.
THE LUNGS
RESPIRATORY INFECTIONS
The lungs have an internal surface area of approximately 500 m2 which is exposed to the external environment and potentially subjected to inhaled microbes with every breath. It is therefore not surprising that respiratory infections are relatively common, with the World Health Organization projecting such infections to continue as one of the global leading causes of death and disability. Countering the threat of pathogens are the defence mechanisms, any abnormality in which will predispose to infection. Such abnormalities include:
Infections can be classified as primary, with no underlying predisposing condition in a healthy individual, or secondary, when local or systemic defences are weakened. The latter are by far the most common types of respiratory infection in developed countries, and are becoming yet more important with the spread of AIDS.
Bronchitis


In acute bronchitis, the trachea and larynx are involved as well as the lungs, and the disease is then known as acute laryngotracheobronchitis (or ‘croup’). The disease is most severe in children, with symptoms of cough, dyspnoea and tachypnoea. Viruses are usually the cause, especially respiratory syncytial virus (RSV), although Haemophilus influenzae and Streptococcus pneumoniae are frequent bacterial causes. Episodes of acute bronchitis are common in chronic obstructive airways disease, and cause a sudden deterioration in pulmonary function with cough and the production of purulent sputum. Acute bronchitis may be caused by direct chemical injury from air pollutants, such as smoke, sulphur dioxide and chlorine.
Chronic bronchitis is a clinical term defined as cough and sputum for 3 months in 2 consecutive years; it is discussed below under diffuse obstructive airways disease (p. 338).
Bronchiolitis



Primary bronchiolitis is an uncommon respiratory infection caused by viruses, especially RSV, in infants. Symptoms are of acute respiratory distress with dyspnoea and tachypnoea. Most cases resolve within a few days, although a minority may develop secondary pneumonia.
Follicular bronchiolitis with lymphoid aggregates and germinal centres, compressing the airway, can occur in rheumatoid disease.
Bronchiolitis obliterans is characterised by concentric fibrosis of the submucosa of small bronchioles resulting in obliteration of the lumen. It may occur in viral infections, especially RSV, after inhalation of toxic fumes, in lung transplant rejection, after aspiration, and with collagen vascular diseases.
Pneumonia




Pneumonia is usually due to infection affecting distal airways and alveoli, with the formation of an inflammatory exudate. It may be classified according to several criteria (Table 14.4, Fig. 14.6).
Table 14.4 Classifications of pneumonia
| Criterion | Type | Example/comment |
|---|---|---|
| Clinical circumstances | Primary | In an otherwise healthy person |
| Secondary | With local or systemic defects in defence | |
| Aetiological agent | Bacterial | Streptococcuspneumoniae,Staphylococcus aureus,Mycobacteriumtuberculosis, etc. |
| Viral | Influenza, measles, etc. | |
| Fungal | Cryptococcus, Candida,Aspergillus,etc. | |
| Other | Pneumocystis jiroveci, Mycoplasma, aspiration, lipid, eosinophilic | |
| Host reaction |
According to dominant component of exudateAnatomicalpattern
Most widely usedclassification before identifying aetiological agent
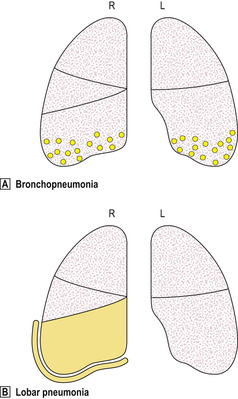
Fig. 14.6 Distribution of lesions in lobar and bronchopneumonia.  Bronchopneumonia is characterised by focal inflammation centred on the airways; it is often bilateral.
Bronchopneumonia is characterised by focal inflammation centred on the airways; it is often bilateral.  Lobar pneumonia is characterised by diffuse inflammation affecting the entire lobe. Pleural exudate is common.
Lobar pneumonia is characterised by diffuse inflammation affecting the entire lobe. Pleural exudate is common.
Bronchopneumonia




Bronchopneumonia has a characteristic patchy distribution, centred on inflamed bronchioles and bronchi with subsequent spread to surrounding alveoli (Fig. 14.6). It occurs most commonly in old age, in infancy and in patients with debilitating diseases, such as cancer, cardiac failure, chronic renal failure or cerebrovascular accidents. Bronchopneumonia may also occur in patients with acute bronchitis, chronic obstructive airways disease or cystic fibrosis. Failure to clear respiratory secretions, such as is common in the post-operative period, also predisposes to the development of bronchopneumonia.
Typical organisms include staphylococci, streptococci and Haemophilus influenzae. Patients often become septicaemic and toxic, with fever and reduced consciousness.
Affected areas of the lung tend to be basal and bilateral, and appear focally grey or grey–red at postmortem (Fig. 14.7). The inflamed lung parenchyma can be demonstrated by gently pressing on an affected area; normal lung recoils like a sponge, whereas pneumonic lung offers little resistance. Histology shows typical acute inflammation with exudation. With antibiotics and physiotherapy, the areas of inflammation most commonly resolve but may heal by organisation with scarring.

Lobar pneumonia





Pneumococcal pneumonia typically affects otherwise healthy adults between 20 and 50 years of age; however, lobar pneumonia caused by Klebsiella typically affects the elderly, diabetics or alcoholics. Symptoms include a cough, fever and production of sputum. The sputum appears purulent and may contain flecks of blood, so-called ‘rusty’ sputum. Fever can be very high (over 40°C), with rigors. Acute pleuritic chest pain on deep inspiration reflects involvement of the pleura. As the lung becomes consolidated (Fig. 14.8), the chest signs are dullness to percussion with bronchial breathing. The dullness recedes with resolution of the exudate.
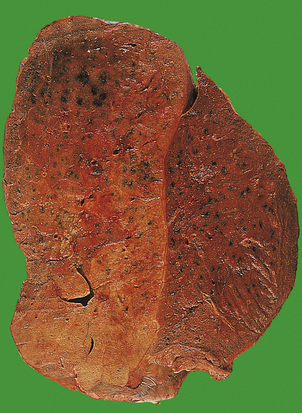
Fig. 14.8 Lobar pneumonia. An entire lobe, paler than the other, has become consolidated due to accumulation of acute inflammatory exudate within the alveoli. Note the abrupt demarcation at the interlobar fissure.
The pathology of lobar pneumonia is a classic example of acute inflammation, involving four stages:
Special pneumonias
Special pneumonias may be sub-classified into those occurring in non-immunosuppressed hosts and those occurring in immunosuppressed hosts.
In non-immunosuppressed hosts
Viral and mycoplasma pneumonia
The clinical course is varied depending on the extent and severity of the disease. In fatal cases, the lungs appear heavy, red and consolidated, as in adult respiratory distress syndrome (ARDS). Histology shows interstitial inflammation consisting of lymphocytes, macrophages and plasma cells. Hyaline membranes of fibrinous exudate are prominent. The alveoli may be relatively free of cellular exudate. Secondary bacterial infection is, however, common and may be severe, for example staphylococcal pneumonia complicating flu.
Mycoplasma pneumonia tends to cause a more low-grade pneumonia, with interstitial inflammation and less exudation.
Legionnaires’ disease
Since the first well-described outbreak in 1976, in a group of American Legion conventioneers, this disease has become increasingly recognised; 201 cases were reported in 1996 in the United Kingdom. It is caused by a bacillus, Legionella pneumophila, transmitted in water droplets from contaminated air humidifiers and water cisterns. Patients may be previously well, although a proportion have an underlying chronic illness, such as heart failure or carcinoma. Symptoms include cough, dyspnoea and chest pain, together with more systemic features, such as myalgia, headache, confusion, nausea, vomiting and diarrhoea. About 5–20% of cases are fatal depending on the age of the population affected. At autopsy the lungs are very heavy and consolidated.
In immunosuppressed hosts
Immunosuppression may be relative, as occurs in patients at the extremes of age, diabetics or those who are malnourished, or severe, as in those on high-dose steroid therapy, undergoing chemotherapy for malignancy, immunosuppression for transplantation or those with HIV/AIDS infection.
Most lung infections in these patients are with organisms similar to those seen in the general population. In addition, however, patients with severe immunosuppression are prone to infection with unusual organisms that are usually non-pathogenic in non-immunosuppressed individuals; these are known as ‘opportunistic’ infections. In any immunosuppressed patient, the onset of fever, shortness of breath and cough, together with pulmonary infiltrates, is an ominous event.
Common offending ‘opportunistic’ agents include:
Pneumocystis jiroveci
Alveoli are filled with a bubbly pink exudate. Round or crescent-shaped organisms are seen using a silver impregnation stain. There may also be diffuse alveolar damage.
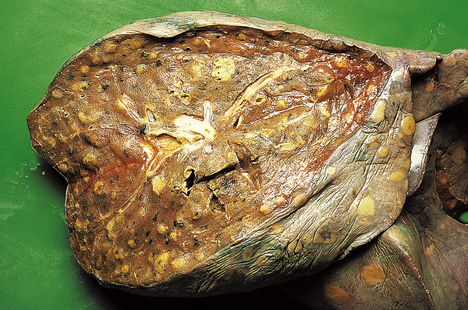
Fungi
Both Candida and Aspergillus species can cause widespread areas of necrosis (Fig. 14.9). Micro-abscesses contain the characteristic fungal filaments (hyphae).
Viruses
Viral infection may produce diffuse alveolar damage and areas of lung necrosis. Characteristic intranuclear inclusions are seen with infections by cytomegalovirus (CMV) and herpes viruses.
HIV lung disease
Pulmonary disease accounts for up to 70% of AIDS-defining illnesses and is the cause of death in at least a third of all AIDS patients.
The lung is frequently involved with infections, the commonest globally being tuberculosis, although combinations of common bacteria, Pneumocystis jiroveci, viruses and even fungi are commonly seen in AIDS patients in Western society.
Non-infective pneumonias
Cryptogenic organising pneumonia
Cryptogenic organising pneumonia is described in the Chronic interstitial diseases section (p. 341).
Aspiration pneumonia
Aspiration pneumonia occurs when fluid or food is aspirated into the lung, resulting in secondary inflammation and consolidation. Clinical situations where patients are at risk include sedation, operations, coma, stupor, laryngeal and oesophageal carcinoma, and severe debility. The parts of the lung affected vary according to the patient’s posture: lying on the back, the affected area is the apical segment of the lower lobe; lying on the right side, the posterior segment of the upper lobe is affected. Often, such areas of aspiration pneumonia contain anaerobic organisms, and a lung abscess containing foul material may ensue.
Lipid pneumonia
Lipid pneumonia may be endogenous, associated with airway obstruction causing distal collections of foamy macrophages and giant cells. This is often seen distal to bronchial carcinoma or an inhaled foreign body. Alternatively, lipid pneumonia may be exogenous, due to aspiration of material containing a high concentration of lipid. Such materials include liquid paraffin or oily nose drops. Vacuoles of lipid are ingested by foreign-body giant cells; there may be some interstitial fibrosis.
Eosinophilic pneumonia
Acute eosinophilic pneumonia is usually idiopathic and associated with a blood eosinophilia (Löffler’s syndrome); it is characterised by numerous eosinophils in the interstitium and alveoli. There is usually a swift response to steroid therapy.
Chronic eosinophilic pneumonia is less frequently associated with a peripheral eosinophilia. The lung shows extensive infiltration with eosinophils and the presence of organising exudates which may go on to give rise to fibrosis. The aetiology is often unclear but can be the result of allergic type reaction to drugs or other environmental agents.
Pulmonary tuberculosis




Pulmonary tuberculosis (TB) is the leading cause of death globally from a single infectious agent; it has been estimated that a third of the world’s population has been infected with the organism. Most cases of pulmonary TB are the result of infection with M. tuberculosis although other so-called ‘atypical mycobacteria’ may be encountered (e.g. M. avian intracellulare, M. kansasii, etc.). The number with active disease is approximately 22 million, and about 1.66 million people die annually from TB.
Disease, however, occurs in only about 10% of cases of infection, when the balance between host resistance and the pathogenicity of the bacteria tips in favour of the latter. TB is therefore the principal cause of HIV-related death in Africa and the Far East, with 33% of people living with HIV/AIDS being co-infected with TB. In 1990, 4.2% of TB cases were attributed to HIV infection; this rose to 11% by the year 2000. Before the advent of antituberculous treatment, therapy was aimed at improving host resistance using special diets and bed rest together with a change in socio-economic factors such as improved living conditions. Now therapy is aimed at killing the organism using combination antibacterial chemotherapy, and immunisation against infection using BCG (bacille Calmette–Guérin).
Clinicopathological features
Clinical and pathological features of pulmonary tuberculosis are extremely variable, and depend on the extent, stage and activity of the disease (Fig. 14.10). Symptoms may vary from insidious weight loss with night sweats and a mild chronic cough, to rampant bronchopneumonia with fever, dyspnoea and respiratory distress (‘galloping consumption’). Most early cases of primary tuberculosis are clinically silent.
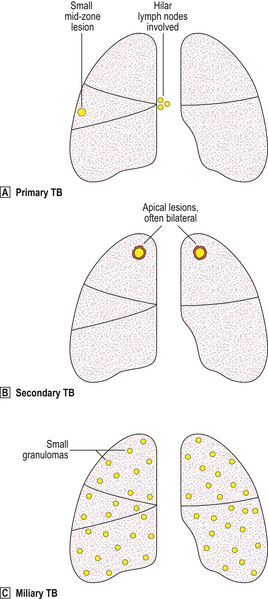
Fig. 14.10 Types of pulmonary tuberculosis.  Primary TB produces a small mid-zone lesion with involvement of hilar lymph nodes.
Primary TB produces a small mid-zone lesion with involvement of hilar lymph nodes.  In secondary TB, the lesions are usually apical and often bilateral.
In secondary TB, the lesions are usually apical and often bilateral.  In miliary TB, the lungs and many other organs contain numerous small granulomas.
In miliary TB, the lungs and many other organs contain numerous small granulomas.
Primary tuberculosis
The lungs are usually the initial site of contact between tubercle bacilli and humans. The focus of primary infection, which is usually asymptomatic, is called a Ghon focus. The pulmonary lesion is usually about 10 mm in diameter, and consists of a central zone of caseous necrosis surrounded by palisaded epithelioid histiocytes, the occasional Langhans’ giant cell and lymphocytes. Similar granulomas are seen in lymph nodes that drain the affected portion of the lung.
In almost all cases, a primary lesion will organise, leaving a fibrocalcific nodule in the lung, and there will be no clinical sequelae. However, tubercle bacilli may still be present within such scarred foci and may persist as viable organisms for years. In a few cases the infection may progress with systemic spread.
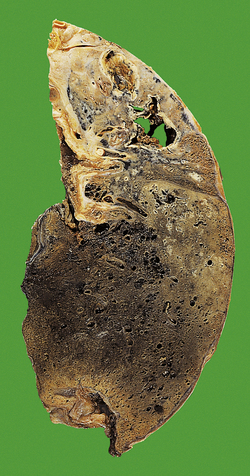
Secondary tuberculosis
As indicated above, most secondary TB represents re-activation of old primary infection. These lesions are nearly always located in the lung apices, sometimes bilaterally (Fig. 14.11). Histologically, typical granulomas are seen, most having central zones of caseous necrosis. Progression of the disease depends on the balance between host sensitivity and organism virulence. Most lesions are converted to fibrocalcific scars, a frequent finding in the lungs of elderly people at autopsy. However, as in primary TB, many complications can ensue.
Miliary tuberculosis
Miliary TB may be a consequence of either primary or secondary TB in which there is severe impairment of host resistance. The disease becomes widely disseminated, resulting in numerous small granulomas in many organs. Lesions are commonly found in the lungs, meninges, kidneys, bone marrow and liver, but no organ is exempt. The granulomas often contain numerous mycobacteria, and the Mantoux test is frequently negative (see below). This is an acute medical emergency necessitating prompt treatment with antituberculous chemotherapy if a fatal outcome is to be averted.
Host resistance to tuberculosis and the tuberculin (Mantoux or Heaf) test
A delicate balance exists between the properties of the tubercle bacillus and host (human) resistance. Tuberculosis is the classical infective example of the type IV delayed hypersensitivity reaction (Ch. 9). Killing is mediated by T-lymphocyte recruitment and activation of macrophages by cytokines, such as interferon-gamma. This process takes time; sensitivity to tubercle bacilli becomes detectable only about 2–4 weeks after inoculation. Antigenicity and virulence are probably related to the lipid properties of the bacillus cell wall; hence hypersensitivity can usually be induced by immunisation with BCG, a vaccine made from non-virulent tubercle bacilli.
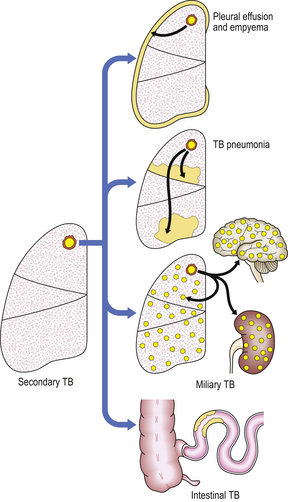
It follows that, in a primary infection, there is no specific hypersensitivity to tubercle bacilli and the inflammatory reaction is relatively mild with little caseous necrosis. In secondary tuberculosis, sensitised T-cells recognise the organism and recruit macrophages which form large granulomas; caseous tissue necrosis is extensive. The disease may disseminate when resistance becomes lowered (Fig. 14.12). Nevertheless, even when host resistance is strong, tubercle bacilli remain extremely difficult to eradicate, and may survive and replicate within the same macrophages recruited as their executioners.
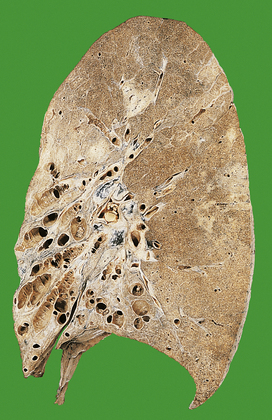
Bronchiectasis
Bronchiectasis is characterised by permanent dilatation of bronchi and bronchioles (Fig. 14.13).
 Results from pulmonary inflammation and scarring due to infection, bronchial obstruction or lung fibrosis (e.g. following radiotherapy)
Results from pulmonary inflammation and scarring due to infection, bronchial obstruction or lung fibrosis (e.g. following radiotherapy)



Aetiology
Bronchiectasis arises as a result of chronic inflammation that damages the alveolated lung around the airways and the airway walls. The resulting scarring causes airway distortion and dilatation with further inflammatory process in the wall of the damaged airway due to secondary infection. Bronchiectasis can arise from a wide range of lung insults:
Clinical features
Usually, the lower lobes are affected, leading to pooling of bronchial secretions with further infection. Symptoms are usually a chronic cough with expectoration of large quantities of foul-smelling sputum, sometimes flecked with blood. Patients may have finger-clubbing. Recurrent respiratory tract infections result from the inability of the patient to clear pooled secretions. Infective processes may remain localised to the bronchi, or spread.
Morphology
There is dilatation of bronchi and bronchioles, with inflammatory infiltration, especially polymorphs, during acute exacerbations. The inflammation and associated fibrosis extend into the adjacent lung tissue. The dilated bronchi and bronchioles can appear cylindrical, saccular or fusiform; these terms are purely descriptive of the variable morphology and are of no aetiological or prognostic significance.
Lung abscess
Lung abscesses may arise as a result of:
Lung abscesses are essentially identical to those found at other sites. They comprise a thick fibrous wall containing mixed inflammatory cells with acute inflammatory debris in the centre. Radiologically the appearances are of a cavitating mass and the differential diagnosis is that of tumour. These patients will, however, usually be clinically septic. Treatment is with antibiotics including cover for anaerobic organisms. The lesion will usually shrink with time to leave an area of fibrous scarring.
Aspergilloma
An aspergilloma is a fungal ball of Aspergillus organisms that grows in a saprophytic manner, usually in a pre-existing cavity. It most commonly complicates old tuberculous cavities but can also complicate cavitated infarcts, abscess cavities or areas of cystic bronchiectasis. The wall of the cavity is fibrotic and inflamed, and there is commonly ulceration of the lining mucosa; this can result in haemoptysis which may be severe. Aspergillomas may remain static for years or increase in size. Treatment with antifungal drugs may be helpful but some cases require surgical excision.
VASCULAR DISEASE OF THE LUNGS
Vascular disease of the lungs may be caused by:
Damage to vessel walls


Diseases of the lungs due to vessel wall damage are uncommon; most are thought to be immunologically mediated. Wegener’s granulomatosis is a necrotising vasculitis affecting the lungs, upper respiratory tract and kidneys (Ch. 21). The aetiology is unknown. Pulmonary involvement is characterised by large areas of necrosis associated with a granulomatous vasculitis affecting veins and arterioles. Churg–Strauss syndrome (allergic angiitis and granulomatosis) may lead to similar necrotising granulomas in the lungs. There is often a history of bronchial asthma. The kidneys and upper respiratory tract are not involved. Goodpasture’s syndrome is associated with the development of circulating antibodies which bind to antigens on the basement membranes of the alveoli and the glomerulus (Ch. 21). In the lung this is associated with the development of extensive lung haemorrhage.
Obstruction




Thrombo-embolism
Thrombo-embolism is the commonest pulmonary vascular lesion. Most emboli are thrombotic, originating in veins (Ch. 8); typical sites are the deep pelvic veins or the deep veins of the calf (Fig. 14.14).
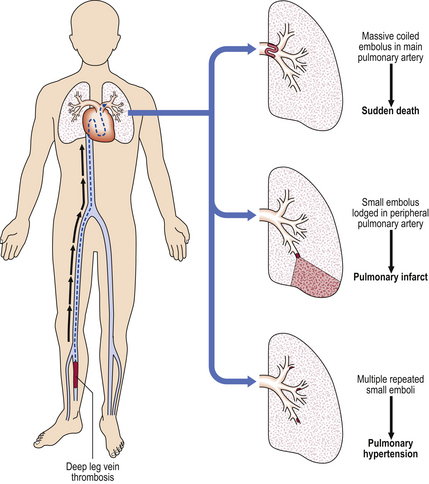
Fig. 14.14 Pathogenesis of pulmonary thrombo-embolism. The thrombus usually originates from the deep leg veins and, after detachment, becomes lodged in the pulmonary artery vasculature, causing sudden death (if massive), pulmonary infarction (if small), or pulmonary hypertension (if small and multiple).
Depending on the size, emboli may lodge in various sites in the pulmonary arterial tree. Symptoms will be related to the volume of lung tissue deprived of blood:
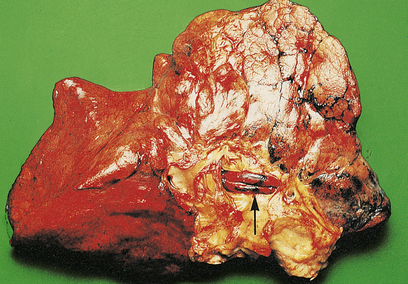
Fig. 14.15 Pulmonary embolism. A massive fatal embolus (arrowed) lodged in a major branch of the pulmonary artery.
Clearly, prevention of deep vein thrombosis is of major importance. Encouragement of improved venous flow in deep leg veins is effected by early ambulation of patients after operations, the use of tight elastic stockings, and leg exercises. Prophylactic anticoagulation is also used in some high-risk patients. Treatment of a major pulmonary embolus includes fibrinolytic agents and even surgical embolectomy. Such heroic measures must be tempered by the fact that patients with a pulmonary embolus have about a 30% chance of developing further emboli. Some patients experiencing repeated pulmonary embolism have an inherited thrombotic tendency (Ch. 23) or may be harbouring an occult malignancy.
Fat emboli
Fat emboli may occlude pulmonary arterioles, leading to breathlessness and sudden death. Such emboli result from fractures of bones containing fatty marrow, or from massive injury to subcutaneous fat. Globules of lipid enter the torn veins and thereby lead to embolism (Fig. 14.16). Marrow tissue may also be seen within pulmonary vessels following trauma but is also frequently seen in autopsy histology in cases of failed CPR.
Air emboli
Air emboli occur occasionally during childbirth or with abortion. Bubbles in the circulation can also occur when dissolved nitrogen comes out of solution, for example in divers during rapid decompression (Caisson disease or ‘the bends’). These micro-emboli can cause tiny infarcts in several organs, including muscle, bone, brain and lung.
Amniotic fluid emboli
Amniotic fluid emboli may occur during delivery or abortion. Flakes of keratin and vernix from fetal skin are seen in pulmonary arterioles.
Tumour emboli
Tumour emboli are, of course, very common; this is an important mechanism in the development of metastases (Ch. 11).
Variations in intravascular pressure
Several disorders are associated with changes in intravascular pressure:
Venous congestion and pulmonary oedema
Pulmonary oedema can result from:
An initial increase in venous hydrostatic pressure leads to pulmonary venous congestion. Common causes are:
Secondary pulmonary venous hypertension follows, with congestion of alveolar wall capillaries. Fluid is then forced out of the venous circulation into the alveoli to form pulmonary oedema.
The lungs are heavy, congested and contain bubbly fluid. In chronic congestion, recurrent alveolar haemorrhages lead to the accumulation of haemosiderin-laden macrophages (heart-failure cells) with some interstitial fibrosis, so-called ‘brown induration of the lung’.
Clinically, there is dyspnoea with a cough, producing bubbly fluid. Auscultation reveals fine crackles in the chest due to air bubbling through numerous fluid-soaked airways. There is respiratory impairment with hypoxaemia. The boggy lungs are prone to secondary infection.
Pulmonary hypertension and ‘cor pulmonale’
Pulmonary hypertension may be:
All the above mechanisms may lead to ‘cor pulmonale’ or heart failure caused primarily by respiratory disease, which is manifested by pulmonary hypertension and right ventricular hypertrophy.
Pre-capillary pulmonary hypertension may be:
Capillary pulmonary hypertension is due to disease in the pulmonary vascular bed. Severe chronic obstructive airways disease may also cause pulmonary hypertension and ‘cor pulmonale’.
Post-capillary pulmonary hypertension is due to high pressure in the pulmonary venous system causing secondary back pressure into the arterial tree. Examples include mitral stenosis, left ventricular failure from any cause, and the rare pulmonary veno-occlusive disease.
Any cause of chronic hypoxaemia may lead to pulmonary hypertension, including living at high altitude. The Pickwickian syndrome is characterised by chronic hypoxaemia and pulmonary hypertension caused by poor respiration associated with gross obesity.
Obviously, the gross and microscopic pathology in each instance is determined by the underlying cause. There are also relatively constant changes seen in the pulmonary arterial tree, including muscular hypertrophy, intimal proliferation, capillary dilatation and, in severe cases, necrotising arteritis.
Pulmonary veno-occlusive disease
Pulmonary veno-occlusive disease is rare; it leads to chronic venous congestion with interstitial fibrosis and many haemosiderin-laden macrophages in alveolar spaces. The left heart is normal, and disease is caused by internal thickening and occlusion of pulmonary veins in the septa. The aetiology is largely unknown, although some cases have been reported in association with drugs and connective tissue disorders. Symptoms are of progressive dyspnoea and ‘cor pulmonale’.
Pulmonary arterial hypertension with right-to-left shunt
In patients with a congenital atrial septal defect (Ch. 13), often asymptomatic, who subsequently develop pulmonary hypertension, the raised right intra-atrial blood pressure causes blood to flow through the defect into the left atrium (right-to-left shunt). Right-to-left shunts may also occur in patients with longstanding left-to-right shunts (i.e. the shunt reverses), for example in ventricular septal defects following the development of secondary pulmonary hypertension (Eisenmenger’s syndrome).
This has two important consequences:
OBSTRUCTIVE AIRWAYS DISEASE
Obstructive airways disease falls into two major groups:
Localised obstructive airways disease




Localised obstructive airways disease is caused by mechanical factors, for example a foreign body or tumour obstructing an airway. The area involved is limited and may be associated with little respiratory embarrassment, unless the patient has underlying lung disease.
When a bronchus or bronchiole becomes obstructed, the distal lung usually collapses. Numerous lipid-laden macrophages may fill the alveolar spaces distal to the obstruction, with possible secondary infection leading to bronchopneumonia (Fig. 14.17). Bronchiectasis may result if the obstruction is not relieved. Occasionally, the lung distal to an obstruction may become over-expanded, perhaps due to a valve effect caused by the obstruction.
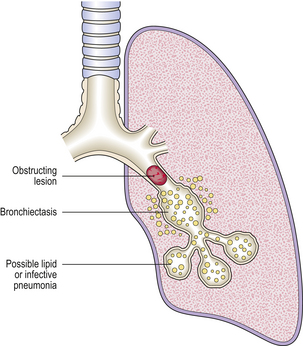
Fig. 14.17 Bronchial obstruction. The obstructing lesion causes a lipid or infective pneumonia in the distal lung and, if unrelieved, distal bronchiectasis.
Clinical symptoms are related to the underlying pathology and to secondary obstructive events. A localised expiratory wheeze may be heard over the affected lobe. Bronchoscopy usually identifies a proximal cause. Partial obstruction of the trachea (or larynx) may be associated with stridor (inspiratory ‘wheeze’) with severe respiratory distress and requires urgent intervention to prevent possible complete respiratory tract obstruction.
Diffuse obstructive airways disease




Diffuse obstructive airways disease is due to reversible or irreversible abnormalities in numerous small bronchi and/or bronchioles as these are the main ‘resistance vessels’ for air movement in the lungs. There is, therefore, significant respiratory impairment causing chronic airflow limitation and a characteristic obstructive pattern of pulmonary function tests:
Chronic obstructive pulmonary disease (COPD)
COPD is a clinical syndrome that pathologically is associated with two patterns of disease in the lung—chronic bronchitis and emphysema. Although pathologically these are two distinct processes, they almost always co-exist to some degree. Together they rank fifth in the global burden of disease: in the UK COPD affects approximately 6% of men and 4% of women over the age of 45, and in the USA it is the fourth leading cause of death, claiming ~120 000 lives annually. COPD is closely linked with cigarette smoking and is associated with progressive loss of lung function: increasing breathlessness, hypoxia and respiratory failure with cor pulmonale.
Clinical features
The clinical features of COPD are essentially cough that is productive of sputum, breathlessness and, in some patients, respiratory failure and cor pulmonale. The clinical features are the result of the presence in most patients of both chronic bronchitis and emphysema, although on occasion one pattern of disease may predominate. Chronic bronchitis is defined clinically as chronic cough and sputum for at least 3 months each year for 2 consecutive years. Emphysema can be demonstrated and assessed on CT scans and by measuring gas transfer (Tco). Breathlessness is contributed to by the small airways obstruction induced by chronic bronchitis and the loss of alveolar walls seen in emphysema. It is important to note that patients with COPD who develop respiratory failure may become hypocapnic (type 1 respiratory failure) while others hypoventilate and become hypercapnic (type 2 respiratory failure). This latter group are especially at risk of developing respiratory pulmonary hypertension and right heart failure (cor pulmonale). Patients with COPD have an increased risk of spontaneous pneumothorax due to rupture of bullae on the surface of the lung and, due to their smoking history, lung cancer.
Clinical exacerbations are associated with recurrent bronchial infections caused by bacteria such as Haemophilus influenzae and Streptococcus pneumoniae, or viruses such as respiratory syncytial virus and adenovirus. Treatment is with physiotherapy, controlled oxygen therapy and, sometimes, short-term use of antibiotics. During such exacerbations there may also be a reversible element to the airways obstruction due to local bronchial irritation causing bronchoconstriction; bronchodilators, such as salbutamol, are therefore also used in the treatment of an attack of chronic bronchitis. Although disease progression and prognosis are improved significantly in patients who stop smoking, it is important to recognise that in patients who develop respiratory failure the 3-year survival rate is less than 50%.
Chronic bronchitis




Aetiology
There is no doubt that chronic bronchitis is almost always entirely due to cigarette smoking. In the United Kingdom, before the Clean Air Act of 1956, urban air pollution was a significant factor. However, the incidence of chronic bronchitis over the last 15 years has remained steady in spite of ever-reducing air pollution; the only change has been a small reduction in male chronic bronchitis, undoubtedly resulting from less cigarette smoking in males.
Morphology
Histologically, chronic bronchitis is characterised by a rather non-specific chronic inflammatory infiltrate within the walls of bronchi of all sizes and bronchioles. This may be associated with the development of bronchial associated lymphoid tissue (BALT) with identifiable lymphoid aggregates and germinal centres. There is often marked hyperplasia of the submucosal glands in the larger airways and goblet cell metaplasia of the surface epithelium in smaller airways. The walls of these smaller airways show evidence of scarring. Hypersecretion of mucus may lead to mucous plugging of airways resulting in yet further airways obstruction. During exacerbations there may be evidence of more florid inflammation including neutrophils and even bronchopneumonia. Squamous metaplasia is also a common finding in these patients as a result of their smoking but this is not specifically associated with chronic bronchitis.
Emphysema


There are various patterns of emphysema (Fig. 14.18). Although each category has a precise anatomical definition, it must be emphasised that in advanced cases there is usually a mixed picture, and an accurate classification in an individual patient is therefore not possible. Suffice to say that all forms of pulmonary emphysema show loss of distal lung parenchyma with resultant airspace enlargement (Fig. 14.19). The pathogenesis is poorly understood but may result from an imbalance of tissue remodelling favouring removal of connective tissue as a result of smoking-induced inflammation. The fact that there are different patterns present may, however, suggest that different pathogenetic process occur.
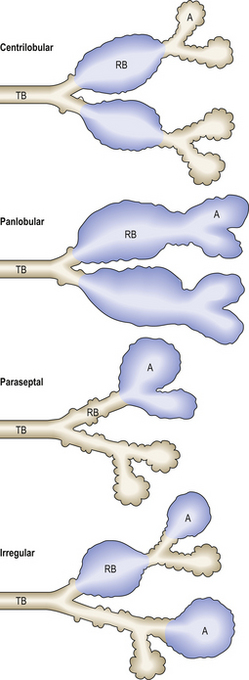
Fig. 14.18 Classification of emphysema. Emphysema is classified according to the pattern of distribution of lesions. These can, to some extent, be correlated with specific aetiological factors, e.g. centrilobular emphysema and cigarette smoke. A, alveolus; RB, respiratory bronchiole; TB, terminal bronchiole.
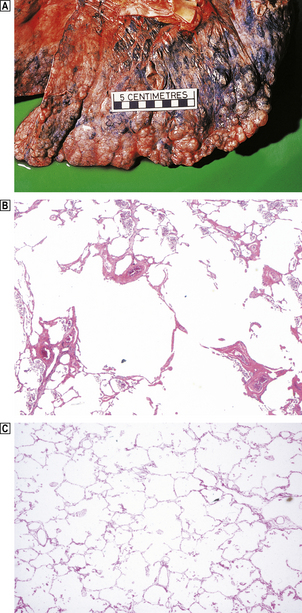
Fig. 14.19 Emphysema.  This is severe emphysematous change characterised by large bullae at the pleural surface.
This is severe emphysematous change characterised by large bullae at the pleural surface.  Histology shows the presence of enlarged alveolar spaces characteristic of emphysema in comparison with that in a normal lung
Histology shows the presence of enlarged alveolar spaces characteristic of emphysema in comparison with that in a normal lung  .
.
Centrilobular emphysema
Centrilobular (or centriacinar) emphysema involves airspaces in the centre of lobules. This lesion is commonest in men, and is closely associated with cigarette smoking, although centrilobular emphysema may also be seen in patients exposed to coal dust. In Britain coal mine dust is accepted to be a cause of centrilobular emphysema in the absence of coal-worker’s pneumoconiosis in miners who have worked underground for 20 years or more. In centrilobular emphysema the lesions are most common in the upper lobes. As noted above, a respiratory bronchiolitis with scarring is also frequently present, together with some large airways disease such as is seen in chronic bronchitis.
Panlobular emphysema
Panlobular (panacinar) emphysema involves all airspaces distal to the terminal bronchioles. Usually, lower lobes are affected, the bases being most severely involved, although the distribution can be patchy. Grossly, the lungs appear overdistended and voluminous. In cases where this is seen predominantly in the upper lobes, it is likely to represent severe centrilobular emphysema that has extended to involve the whole lobule. This pattern of emphysema is, however, seen in 70–80% of patients with homozygous alpha-1-antitrypsin deficiency. These patients often develop severe emphysema, usually before the age of about 50 years, especially if they smoke. These patients also have an increased risk of developing cirrhosis. Alpha-1 antitrypsin is an acute phase serum protein which inhibits the actions of collagenase, elastase and other proteases, including trypsin. One action of alpha-1 antitrypsin is to inhibit enzymes released from dying neutrophils and macrophages. Any stimulus, such as smoking, that leads to increased numbers of inflammatory cells in the lung will lead to alveolar wall destruction (emphysema) in patients with alpha-1 antitrypsin deficiency. The enzyme deficiency is inherited as an autosomal dominant trait, and the homozygous deficiency state is said to affect 1 in 3630 Caucasians; the defect is even rarer in black people.
Paraseptal emphysema
Paraseptal (distal acinar) emphysema involves airspaces at the periphery of the lobules, typically adjacent to pleura. There is often adjacent scarring and fibrosis. The dilated airspaces can become large and, if over 10 mm in diameter, are termed bullous. Upper lobes are more frequently involved.
Irregular emphysema
Irregular emphysema irregularly involves the respiratory acinus. This type is almost always associated with scarring and there is almost certainly an overlap with paraseptal emphysema. The pathogenesis is thought to be air trapping caused by fibrosis; this irregular pattern of emphysema is therefore commonly present around old healed tuberculous scars at lung apices.
Other pathological types
In addition to the four anatomical types of pulmonary emphysema discussed above, some other categories exist.
Bullous emphysema
This is not a separate category of emphysema but refers merely to the presence of balloon-like foci of emphysema over 10 mm in diameter. Cases of emphysema with bullae should, where possible, be classified into one of the four anatomical types discussed above. Bullae are prone to rupture, causing spontaneous pneumothorax. They are typically subpleural and apical.
Interstitial emphysema
This refers to inflation of the interstitium of the lung by air, and is most commonly due to traumatic rupture of an airway or spontaneous rupture of an emphysematous bulla. Interstitial emphysema may spread to the mediastinum or subcutis, giving the characteristic spongy crepitus on palpation.
Senile emphysema
This is also a misnomer, as there is no destruction of alveolar walls. Alveolar surface area decreases, and alveolar ductular size increases, progressively after the age of 30 years, leading to the overdistended, apparently voluminous lungs seen at autopsy in aged patients. This process is one of normal senile involution and is not a disease.
Asthma





Asthma is defined as hyper-reactivity of the bronchial tree with paroxysmal narrowing of the small airways (Fig. 14.20), which may reverse spontaneously or after treatment. Asthma is increasingly common in many countries, but it is a relatively rare cause of death.
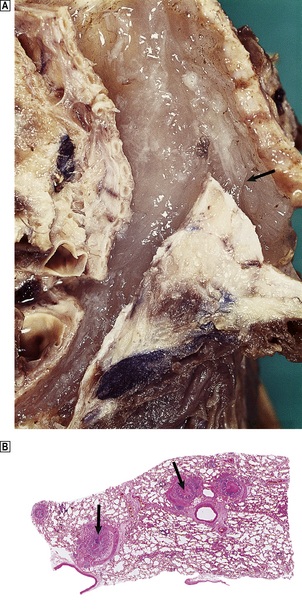
Fig. 14.20 Asthma.  Tracheal mucous plug (arrowed) in death from status asthmaticus.
Tracheal mucous plug (arrowed) in death from status asthmaticus.  Histological section of lung at autopsy showing occlusion of airways by oedema and mucous plugs (arrowed) accompanied by alveolar distension with entrapped gas.
Histological section of lung at autopsy showing occlusion of airways by oedema and mucous plugs (arrowed) accompanied by alveolar distension with entrapped gas.
There are five major clinical categories of asthma:
Each type has different predisposing factors, and is mediated in different ways. However, the resulting clinical symptoms and pathology are similar to those seen in atopic asthma. Any important differences are outlined below.
Atopic asthma
Atopic asthma is triggered by a variety of environmental agents, including dust, pollens, foods and animal danders, e.g. faecal pellets from housedust mites. There is often a family history of asthma, hay fever or atopic eczema. Patients with atopic asthma may also suffer from atopic disorders such as hay fever or eczema. Increased levels of endogenous nitric oxide (NO) produced by a variety of cells in the respiratory tract have been found in exhaled air from asthmatics when compared with normal controls; whether NO is merely a marker of airway irritation or has a more fundamental role in the pathogenesis of asthma is not known. Recently the gene labelled ADAM33 (a disintegrin and metalloproteinase) has been identified that may underlie the variation in bronchial hypersensitivity between individuals.
Bronchoconstriction is mediated by a type I hypersensitivity reaction (Ch. 9); bronchoconstriction leads to the clinical effects of wheezing, tachypnoea and dyspnoea (Fig. 14.21). Rarely, symptoms persist for days (status asthmaticus), leading to respiratory failure and even death. Release of histamine and slow-reacting substance of anaphylaxis (SRS-A) leads to bronchoconstriction, increased vascular permeability and mucus hypersecretion. Eosinophil chemotactic factor of anaphylaxis (ECF-A) attracts numerous eosinophils to the bronchial walls.
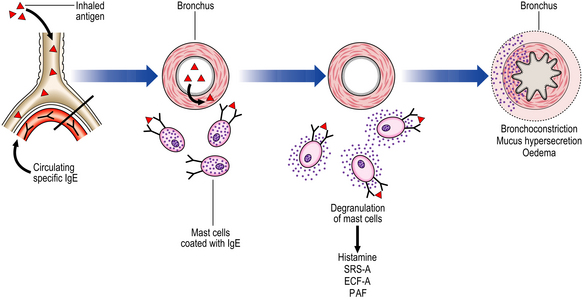
Fig. 14.21 Pathogenesis of allergic asthma. Inhalation of allergen (antigen) causes degranulation of mast cells bearing specific IgE molecules. Release of vasoactive substances from the mast cells causes bronchial constriction, oedema and mucus hypersecretion.
Platelet activating factor (PAF) leads to the aggregation of platelets with the release of further histamine and 5-hydroxytryptamine (5-HT) from their granules. The hypersensitivity reaction results in acute and chronic changes, with the former being identifiable during an acute episode and the latter being the result of airway remodelling following repeated attacks:
Non-atopic asthma
Non-atopic asthma is frequentely characterised by episodes of bronchospasm, often associated with respiratory tract infections. Testing for allergens by skin patching is negative. Bronchoconstriction may be due to local irritation in patients with unusually reactive airways.
Aspirin-induced asthma
Patients with this form of asthma may also have recurrent rhinitis with nasal polyps, and skin urticaria. The mechanism of induction of asthma by aspirin is unknown but may involve locally decreased prostaglandins or increased leukotrienes leading to airway hyper-reactivity.
Occupational asthma
Occupational asthma is induced by hypersensitivity to an agent inhaled at work. Inhaled agents may act as non-specific irritants precipitating bronchospasm in those with hyper-reactive airways, or they may act as agents capable of inducing asthma and airway hyper-reactivity. There are many different occupationally inhaled agents that can cause asthma. The diagnosis is often difficult and relies on demonstrating variation in lung function and symptoms during time at work and away from work. The mechanism of airway reaction is thought to be a combination of type I and type III hypersensitivity (Ch. 9).
In the United Kingdom, if asthma can be proved to be the result of an agent inhaled at work, the patient is entitled to statutory compensation.
Allergic bronchopulmonary aspergillosis
Allergic bronchopulmonary aspergillosis is a type of proximal bronchiectasis seen in patients with asthma and is due to inhalation of spores of the fungus Aspergillus fumigatus, inducing an immediate type I and delayed immune complex type III hypersensitivity reaction. Mucous plugs in bronchi contain the hyphae of aspergilli.
Pathogenesis of asthma
As noted above, some forms of asthma are mediated by type I and type III hypersensitivity reactions, yet in others the mechanisms are unknown. One hypothesis that would explain all types of asthma is that patients have bronchial beta-receptors relatively insensitive to catecholamines; thus, bronchi are partially constricted in the non-challenged resting state, and do not dilate well to beta-agonists, such as adrenaline (epinephrine). In addition to reacting to specific agents, these hyper-reactive airways also react to non-specific factors, such as cold, exercise and emotional stress. Why this should happen remains a mystery.
Obliterative bronchiolitis
This is a rare condition where the lumina of small bronchioles become progressively obliterated by fibrous tissue. It may follow episodes of acute airway injury following inhalation of gases (e.g. chlorine or ammonia), smoke or following severe viral bronchiolitis in children. It may also occur in patients with connective tissue disorders or inflammatory bowel disease. The most frequent setting in which this pattern of lung disease is observed is, however, in transplantation. Obliterative bronchiolitis is seen as a feature of chronic lung allograft rejection and is the commonest cause of ultimate graft failure. It also seen as part of the spectrum of graft-versus-host disease that may occur following bone marrow transplantation.
INTERSTITIAL DISEASES OF THE LUNG
The interstitial lung diseases (ILDs) or diffuse parenchymal lung diseases (DPLDs) are a heterogeneous group of conditions that present with a history of increasing breathlessness, hypoxia, restrictive lung function tests and bilateral shadowing in the lungs on chest X-ray and CT scans. These conditions primarily involve the alveoli of the lung. Diseases may be grouped into acute and chronic categories on the basis of clinical history and histological findings, with each disorder showing a basic pattern of either acute alveolar injury or chronic pulmonary fibrosis. In some instances they may display characteristic features, allowing the specific aetiology to be identified, but in many instances the trigger is unclear (idiopathic). It is also important to recognise that different aetiologies may result in a clinically and pathologically identical pattern in the lung.





Acute interstitial diseases
Acute interstitial diseases are characterised by a short history of dyspnoea, tachypnoea and respiratory distress. There is diffuse alveolar damage with alveolar exudation, formation of hyaline membranes and type II pneumocyte hyperplasia. Pathologically this is termed acute interstitial pneumonitis or diffuse alveolar damage syndrome, and clinically is seen most frequently in the context of adult respiratory distress syndrome (ARDS). This syndrome came to prominence during the Vietnam War when it was found that many soldiers were dying from respiratory failure during the first few days of recovery from severe wounds; most had suffered shock from blood loss, but this had been successfully treated at the battleground. Clinically, there was respiratory distress with tachypnoea, dyspnoea and hypoxaemia refractory to oxygen therapy.
Aetiology
Severe acute interstitial pneumonitis or ARDS can arise as a result of direct or indirect lung injury:
Pathogenesis
All the clinical situations listed above deliver a massive insult to alveolar–capillary walls, leading to diffuse alveolar damage. In many cases, the exact pathogenesis is unknown. Polymorphs are thought to be important in the pathogenesis, with release of enzymes, activation of complement and massive cytokine release. In the acute stages the lungs are heavy, oedematous and congested with areas of haemorrhage. Exudation of protein-rich fluid results in the formation of hyaline membranes lining alveolar ducts and alveoli, together with pulmonary oedema and extravasation of red cells. In most cases this exudate progresses to give rise to a fibro-proliferative phase and, if the patient survives long enough, extensive alveolar fibrosis. Resolution may occur via resorption of the oedema, and ingestion of red cells and hyaline membranes by alveolar macrophages; there is then regeneration of type II pneumocytes, which later differentiate to type I flattened pneumocytes.
Prognosis
About 40% of patients with severe acute lung injury (ARDS) die within the first few days despite intensive therapy. Most of the survivors progress to full recovery with resolution of the inflammation and restoration of the normal alveolar architecture; however, some survivors heal by organisation, leading to pulmonary fibrosis.
Radiation pneumonitis
The clinical effects of radiation toxicity to the lungs are highly dependent on the dose given (Ch. 6), the volume of lung irradiated and the length of treatment. If heavily exposed, a picture of diffuse alveolar damage may be seen as described above. If exposure is less severe and occurs over a longer period, progressive pulmonary fibrosis is seen with the typical restrictive defect of pulmonary function.
Diffuse intrapulmonary haemorrhage
Goodpasture’s syndrome is characterised by haemoptysis, haematuria, anaemia and pulmonary infiltrates. Most cases have circulating anti-glomerular basement membrane antibody in their blood. This antibody causes glomerulonephritis, and also acts on alveolar membranes leading to pulmonary haemorrhage.
Idiopathic pulmonary haemosiderosis is a rare condition presenting most often in children. Clinically, patients may present with recurrent episodes of intra-alveolar haemorrhage associated with haemoptysis, cough and dyspnoea; alternatively, they may present with insidious pulmonary fibrosis. The more acute form shows evidence of diffuse alveolar damage, with type II pneumocyte hyperplasia.
Chronic interstitial diseases
Chronic interstitial lung diseases give a clinical history lasting months or years with slowly increasing respiratory insufficiency, hypoxia and bilateral changes on chest X-ray. These conditions are essentially inflammatory/fibrotic in nature although other processes such as pulmonary oedema and lymphangitic carcinomatosis can occasionally mimic these ILDs. Examples include:
Idiopathic pulmonary fibrosis (IPF)
IPF is a progressive chronic pulmonary fibrosis of unknown aetiology, although approximately 20% of patients give a history of occupational exposure to metals and wood dusts. Most patients are aged over 60 years and present with increasing dyspnoea and a dry cough. Men are affected twice as often as women. The disease progresses to respiratory failure, with or without cor pulmonale, within about 5 years. Fatigue and considerable weight loss may occur, raising the clinical suspicion of malignancy. Examination often shows finger- and toe-clubbing; auscultation of the chest reveals dry crackles, reflecting the opening and closing of fibrotic airspaces. Signs of right ventricular strain or failure may be present. These patients also have an excess risk of carcinoma of the lung and ischaemic heart disease.
Pathogenesis
The pathogenesis of IPF is unknown. It is important to realise, however, that a very similar if not identical pattern of disease may be seen in association with connective tissue disorders and with asbestosis. Pulmonary function tests show the characteristic restrictive pattern.
Morphology
The majority of patients with IPF show a pattern of lung disease described as usual interstitial pneumonitis (UIP). This pattern of disease is characteristically patchy with the subpleural regions of the lower lobes predominantly affected. There is a variable interstitial inflammatory infiltrate in the affected areas of the lung with collapse of the lung architecture and the development of cystically dilated spaces within the fibrotic areas of the lung (honeycombing). A characteristic feature is the presence of areas of immature fibrous tissue or ‘fibroblastic foci’ in the less severely affected areas.
More recently it has been recognised that a group of patients with clinical IPF has a different pattern of lung fibrosis called non-specific interstitial pneumonitis (NSIP). This is characterised by a more even and diffuse pattern of alveolar wall inflammation and fibrosis with none of the ‘fibroblastic foci’ or honeycomb fibrosis typical of UIP. There is a suggestion that this pattern of disease may have a better prognosis than the more typical pattern of UIP.
Pneumoconioses
Exposure to inhaled materials may have several potential consequences depending on the nature of the dust and the degree of exposure:
These conditions are often all regarded as ‘occupational’ or ‘industrial’ lung disease although it is important to remember that exposure can occur outwith the workplace. Pneumoconiosis is, however, defined as lung fibrosis secondary to inhaled inorganic dust (e.g. coal dust, silica, metals, etc.). In many industrial settings patients may be exposed to a wide variety of different dusts and identifying which dust might be responsible for lung disease may be difficult.
The distribution of lung disease depends on the physical properties of each separate type of dust, which determine where the particles settle in the lung. Only very small particles reach distal alveoli, as larger particles are trapped in the nose or excreted by mucociliary clearance from the larger airways.
Dust particles are phagocytosed by alveolar macrophages, which then collect and drain into peribronchiolar lymphatics and thence to hilar lymph nodes. Not surprisingly, dusts causing disease in the lungs are also often present in the sinuses of hilar lymph nodes.
Coal-worker’s pneumoconiosis
In coal-worker’s pneumoconiosis (CWP), coal dust is phagocytosed by alveolar macrophages (dust cells), which then aggregate around bronchioles; the degree of black pigment in the lung (anthracosis) is related to the amount of inhaled carbon. Anthracosis is also commonly seen in smokers and those living in urban environments. The consequences of coal-dust inhalation are variable, ranging from trivial to fatal.
Simple CWP is associated with minimal scarring and no significant disability. The predominant pattern is of dust macules consisting of focal aggregates of dust-laden macrophages in and around the walls of respiratory bronchioles, pulmonary arterioles and pulmonary veins. Similar cells are seen in lymphatics and hilar lymph nodes. There may be some associated airspace enlargement consistent with emphysema. To qualify as pneumoconiosis there must, however, be evidence of fibrosis and this is characterised by the development of fibrous nodules. This is usually regarded as a progression from the macular stage. In simple CWP nodules less than 10mm in diameter are seen in a background of macular CWP.
Complicated CWP or progressive massive fibrosis (PMF) is characterised by large, irregular nodules with scarring (Fig. 14.22); they are greater than 10 mm in diameter and can be very large. These fibrotic black nodules may show central liquefaction and, when cut at autopsy, exude viscid jet-black liquid. They may contract, leading to adjacent irregular emphysema. Large nodules are usually mid-zonal or in upper lobes, and may be bilateral. The associated emphysema is always severe, often with the formation of bullae. Progression of the disease leads to further scarring and lung destruction with respiratory failure, or cor pulmonale.

Fig. 14.22 Coal-worker’s pneumoconiosis. This transilluminated thin slice of lung shows several large black fibrotic nodules. which measure more than 10 mm, consistent with complicated coal-worker’s pneumoconiosis (progressive massive fibrosis). In some cases these may cavitate as shown in this example.
It is recognised that in a group of miners working at the same pit for the same length of time, some will develop PMF and die, while others develop little respiratory impairment. The reasons why only some miners develop PMF are unknown and some investigators believe that the determining factor is merely the amount of coal dust inhaled. It is clear, however, that PMF is seen more commonly in some coal fields compared with others, which may suggest that exposure to other dusts such as silica in the rocks may play a role. Whatever the cause, the progression of CWP to PMF is an ominous event.
Caplan’s syndrome is characterised by the presence of large pigmented necrobiotic nodules in patients with CWP. This occurs in the presence of seropositive rheumatoid disease, although lung nodules may precede the development of systemic features. The nodules may regress.
Silicosis
Silicates are inorganic minerals abundant in stone and sand. Consequently, individuals working in a wide range of occupations involving the cutting, drilling, grinding or mining of stone or sand will be at risk from silicosis. Small particles of silica reach the distal lung where they are ingested by alveolar macrophages. However, in contrast to pure coal dust, silicates are toxic to macrophages, leading to their death with release of proteolytic enzymes and the undigested silica particles. The enzymes cause local inflammation, tissue destruction and subsequent fibrosis; the silica particles are ingested by other macrophages and the cycle repeats itself.
Nodules tend to form in the lungs after many years of exposure. With progressive fibrosis and increasing numbers of nodules, respiratory impairment increases. Pulmonary function tests show a restrictive defect like any other chronic interstitial lung disease. There is a recognised increased risk of reactivation of tuberculosis and, more controversially, a possible excess risk of the development of lung cancer.
The lungs show scattered nodules of hard, fibrous tissue with surrounding irregular emphysema. Similar changes may be apparent in hilar and mediastinal lymph nodes. Advanced cases show extensive diffuse pulmonary fibrosis, together with numerous large silicotic nodules.
Asbestosis
The name ‘asbestos’ is derived from a Greek word meaning ‘inconsumable’, and, indeed, asbestos has been used for its fire-resistant qualities for many centuries. Asbestos is used for insulation and the manufacture of brake linings and other friction materials. There are several types of asbestos: amphiboles are the fibres that cause pulmonary disease in humans, and of these crocidolite (blue asbestos) is probably the most dangerous.
Inhaled asbestos fibres collect in alveoli at the lung bases. Many become coated in acid mucopolysaccharide and encrusted with haemosiderin to form ‘asbestos bodies’ or ‘ferruginous bodies’, appearing as characteristic beaded structures. The majority of fibres are, however, detectable only by electron microscopy.
Asbestosis is characterised by the development of a pattern of lung fibrosis that is essentially similar to UIP described above with destructive subpleural honeycomb changes (Fig. 14.23). It is usually seen only in patients with relatively heavy asbestos exposure and is therefore most common in those who have worked extensively with insulation materials, for example in shipyards or power stations. Disease progression is much slower and the prognosis in most cases considerably better than for IPF. Differentiation of asbestosis from IPF may be important in terms of compensation but can be difficult. Quantification of the number of asbestos fibres may be helpful in this regard but clearly the finding of fibres by itself only proves previous exposure, not causation of the disease present.
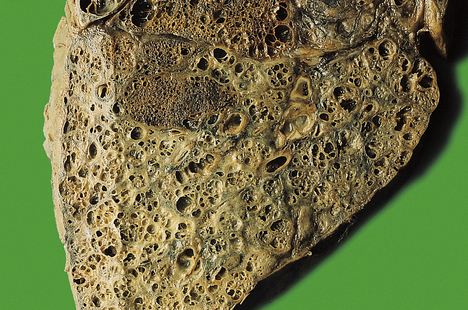
Extrinsic allergic alveolitis (hypersensitivity pneumonitis)
Interstitial lung disease caused by inhalation of organic dusts results from the individual being sensitised (hypersensitive) to the inhaled antigen (see also occupational asthma). Many antigens have been described as causing an allergic interstitial lung disease known as extrinsic allergic alveolitis or hypersensitivity pneumonitis. These include: cotton fibres, causing byssinosis; sugar cane fibres, causing bagassosis; and bird faeces, causing bird fancier’s lung. Whilst many of these may be associated with occupational exposure and thus can be regarded as an ‘occupational lung disease’, exposure to antigens in the domestic environment and even drugs can give an identical pattern of disease in the lung.
The best known and most typical example of an occupational extrinsic allergic alveolitis is farmer’s lung. In this disorder, a fungus present in poorly stored, mouldy hay is inhaled by whoever disturbs the hay. If the individual is already sensitised to the organism, a type III immune complex hypersensitivity reaction follows. One of the earliest features is a bronchiolitis. Later, chronic inflammatory cells are seen in the interstitium, together with non-caseating granulomas. The inflammatory process may resolve on withdrawal of the antigen but if there is chronic exposure then pulmonary fibrosis will develop.
Clinically, there is acute dyspnoea and cough a few hours after inhalation of the antigen. Corticosteroid treatment helps to ameliorate the inflammatory reaction and to prevent the onset of pulmonary fibrosis.
Sarcoidosis
Sarcoidosis is a multisystem inflammatory disorder which most commonly involves the mediastinal lymph nodes and lung. The aetiology is unknown but it is possible that the granulomas arise as a result of an aberrant immune response due to abnormal T-cell–macrophage interactions. The possibility of an infective aetiology has been considered for many years but no convincing demonstration of an infective agent has been found. Clinical symptoms are variable depending on the extent of the disease. Many patients may be asymptomatic while others may have systemic upset with tiredness and cough. Common extrapulmonary manifestations include erythema nodosum, iritis and arthralgia. In most cases the disease is either self-limiting or responds to steroid therapy. In a minority of cases progression to end-stage pulmonary fibrosis occurs. Involvement of other organs, including the heart and central nervous system, occurs more rarely and can cause severe morbidity and death.
Histologically, sarcoid in the lung is characterised by the presence of relatively discrete non-caseating granulomas (Fig. 14.24) which may coalesce to form larger nodules. Predominantly they are found in relation the bronchovascular bundles, septae and the pleura. The principal differential diagnosis is that of infective disorders such as tuberculosis and histoplasmosis.
Cryptogenic organising pneumonia (COP)
COP (or bronchiolitis obliterans organising pneumonia, BOOP) is a clinical syndrome characterised by mild systemic upset with possible cough, low-grade fever and breathlessness which radiologically shows evidence of focal lung consolidation that may ‘flit’ within the lungs over time. The pathogenesis is poorly understood. Pathologically, the lungs show nodular foci of organising exudates within alveolar spaces and terminal bronchioles, and variable, usually mild, interstitial inflammation. In most cases the condition responds well to steroid therapy and there is resolution with no significant fibrosis. Some patients may, however, relapse and more progressive cases with lung fibrosis have been described.
Langerhans’ cell histiocytosis (LCH)
Langerhans’ cell granulomatosis (previously known as histiocytosis X) is a disease principally of smokers, characterised by the proliferation of Langerhans’ cells. This is believed to occur as a response to cigarette smoke and increased numbers have been described in the lungs of smokers compared with non-smokers, even in the absence of LCH. The Langerhans’ cells are specialised histiocytes which are involved in antigen presentation. Inflammatory infiltrates are seen in the pulmonary interstitium with the Langerhans’ cells admixed with lymphocytes and eosinophils. These may form nodular masses with areas of cystic change. In most cases smoking cessation results in complete resolution of the nodules or healing, leaving small areas of fibrosis. In some cases, however, the disease may be progressive and result in end-stage pulmonary fibrosis.
Alveolar lipoproteinosis
Alveolar lipoproteinosis (or proteinosis) is a rare condition characterised by the accumulation of eosinophilic material within alveoli. In most instances the aetiology is unknown and the pathogenesis is uncertain but there appears to be an association with haematological disorders in some patients. Symptoms include dyspnoea and cough, when gelatinous material may be expectorated.
Connective tissue disorders and interstitial lung disease
Patients with connective tissue disorders such as rheumatoid arthritis and scleroderma may show a variety of pulmonary complications which fall into the ILD categories described above. The commonest pattern associated with these conditions is non-specific interstitial pneumonitis but patients may also develop cryptogenic organising pneumonia and usual interstitial pneumonitis. Drug treatments may also be associated with acute interstitial pneumonitis/ARDS (e.g. methotrexate), hypersensitivity reactions or opportunistic infection following immunosuppression. Other lung manifestations include rheumatoid nodules, Caplan’s syndrome, vasculitis and obliterative bronchiolitis.
LUNG TUMOURS
Lung tumours may be primary or secondary. Both are common.
Primary carcinoma of the lung





Over 90% of primary lung tumours are carcinomas. Lung cancer is the leading cause of death from cancer in the world, with a poor overall prognosis—typically around 5% 5-year survival. This is due to the average age of the patients, a high incidence of comorbid disease (e.g. ischaemic heart disease, COPD) and late presentation with advanced dissemination at the time of diagnosis. In the UK only about 10% of cases are operable at the time of diagnosis.
About one-third of all cancer deaths in males in the UK are due to lung cancer. The disease is also increasing in incidence among women; it now ranks as the commonest lethal cancer in females in the UK. Typically, patients are aged between 40 and 70 years; the disease rarely affects those less than 30 years of age.
Aetiology
Major risk factors for the development of lung cancer are:
Cigarette smoking
There is overwhelming evidence implicating cigarette smoking as the major risk factor for the development of lung cancer (Ch. 11). The rise in the incidence of lung cancer over the last century has closely paralleled the increase in cigarette smoking. Similar changes in the incidence of lung cancer have occurred in the developing world with an incidence of <5 per 100000 population 50 years ago, rising to 14 per 100000 by the end of the 20th century. Two-thirds of the world’s smokers reside in China and it is estimated that by the mid 21st century the annual death toll from lung cancer in China alone may run into millions.
Occupational hazards
There are several occupational hazards associated with an increased incidence of lung cancer. The most important are:
Fibrosis
Some peripheral lung cancers (usually adenocarcinomas) apparently arise in areas of fibrous scarring, e.g. wounds, old tuberculous foci or infarcts. The theory is that metaplastic and dysplastic changes occur in pneumocytes within the scar. Such ideas have recently been challenged, the so-called ‘scar cancers’ being considered carcinomas with a pronounced central desmoplastic (fibroblastic) reaction. Despite this argument, there is undoubtedly a significant increase of lung adenocarcinoma in patients with pulmonary fibrosis and honeycomb lung.
Clinical features
Weight loss, cough and haemoptysis are common presenting features. Weight loss is often severe and may be due to humoral factors from the tumour. Dyspnoea and chest pain are also common; the latter is often pleuritic and due to obstructive changes. Patients may present with, or ultimately develop, metastases; common sites include lymph nodes, bone, brain, liver and adrenals. Paraneoplastic effects are common and are due to ectopic hormones: ACTH and ADH from small cell lung carcinomas, PTH from squamous cell carcinomas. Finger-clubbing and hypertrophic pulmonary osteoarthropathy are common (Fig. 14.25).
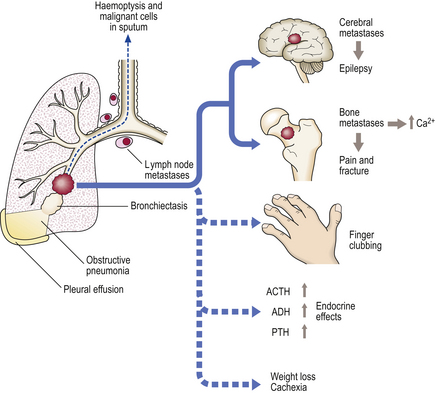
Lung carcinomas may arise centrally in the lung or towards the periphery. Central tumours may cause airway obstruction with collapse or distal consolidation of a lobe or the whole lung. These tumours may infiltrate medially into the mediastinum at a relatively early stage and patients can present as a result of this rather than the lesion in the lung (e.g. hoarseness from involvement of the left recurrent laryngeal nerve). More peripheral tumours are often asymptomatic but pleural involvement may lead to pleural effusion or the tumour may extend into the chest wall causing pain. Larger tumours, especially squamous carcinomas, often cavitate. Small cell carcinoma has usually disseminated by the time of diagnosis and the primary lesion in the lung may be small compared to the bulky nodal deposits in the mediastinum and liver.
Morphology
Most tumours arise from bronchi close to the hilum (Fig. 14.26); usually an upper lobe or main bronchus is involved. Ulceration is common, so the sputum may be bloodstained and contain malignant cells which can be detected cytologically (Fig. 14.27) or by biopsy at bronchoscopy (Fig. 14.28). Distally, the lung may be consolidated with foamy macrophages, the usual result of proximal obstruction, and patients may present with what appears to be a pneumonia.
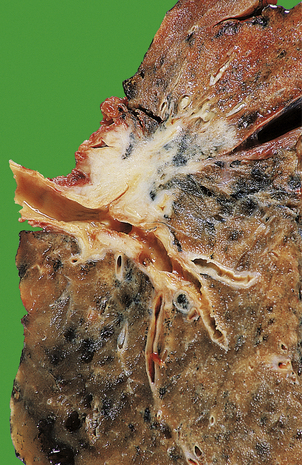
Fig. 14.26 Carcinoma of the lung. The tumour is arising at the hilum of the affected lobe and is invading the adjacent lung tissue.
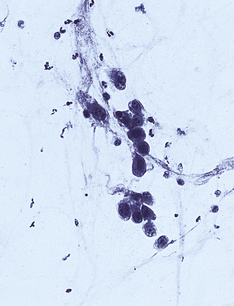
Fig. 14.27 Carcinoma of the lung. Tumour cells found in diagnostic bronchial washings, the appearances of which are consistent with non-small cell carcinoma of the lung.
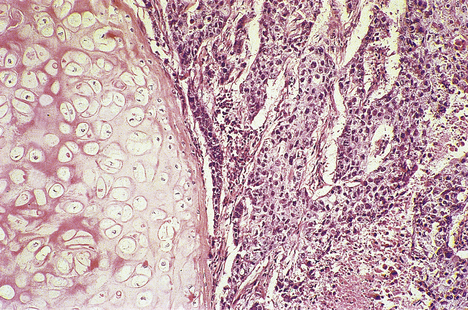
Fig. 14.28 Lung carcinoma. Histology of a poorly differentiated squamous cell carcinoma. Bronchial cartilage (left) is relatively resistant to neoplastic invasion.
Histological classification
There are four major types of lung cancer, classified according to their appearance on light microscopy; their approximate incidences are:
The lung cancers are discussed below according to this classification. For practical purposes the tumours are often divided into non-small cell carcinomas (SqCC, AC and LCUC) and small cell carcinomas as this distinction largely directs management. About 20% of lung carcinomas may show evidence of a mixed pattern of differentiation, e.g. SqCC and SCLC, or AC and SqCC.
Squamous cell carcinoma.
This is the type of lung cancer most closely associated with cigarette smoking although the relative incidence has been decreasing over the last few years. The tumours are usually central in location and are thought to arise from areas of squamous metaplasia through increasing grades of dysplasia. There is often haemorrhage and necrosis with cavitation. Tumours may be well, moderately or poorly differentiated. SqCC tends to metastasise locally to hilar lymph nodes; distant metastases are a later feature.
Small cell lung carcinomas.
These tumours were previously also known as ‘oat cell’ carcinoma because the small nuclei were thought to resemble oat grains; SCLC usually arise in a hilar bronchus. Unlike non-small cell carcinomas, they metastasise very early, producing widespread bulky secondary deposits. Sometimes, the primary tumour can be small and difficult to find. The histology is of a highly cellular tumour composed of small cells with hyperchromatic nuclei and indistinct nucleoli. The cells are very delicate and the chromatin may appear smudged. The cells commonly express neuroendocrine markers, suggesting that this is a form of very poorly differentiated neuroendocrine carcinoma.
Adenocarcinomas.
These may be central or peripheral and can show a wide range of morphological patterns. The relative incidence of adenocarcinoma has been increasing over the last two decades and this type of tumour has overtaken squamous carcinoma as the most common subtype. Adenocarcinomas are usually single lesions but they can arise in a multifocal pattern, sometimes bilaterally. In this situation the tumour usually has a broncho-alveolar configuration where the cells grow along the alveolar walls, giving rise to areas that appear consolidated (Fig. 14.29). There is now some evidence to suggest that most peripheral adenocarcinomas may arise from an area of broncho-alveolar carcinoma and that this may therefore represent an early form of these tumours.

Large cell undifferentiated carcinomas.
Usually central, these are highly aggressive and destructive lesions with necrosis and haemorrhage. Histologically, there is often gross nuclear pleomorphism with numerous bizarre mitoses. By definition no squamous or glandular differentiation is seen on light microscopy, although such evidence is often found ultrastructurally. Some of these tumours may have features suggesting neuroendocrine differentiation and the term large cell neuroendocrine carcinoma has been adopted to distinguish them from SCLC and carcinoid tumours.
Bronchial carcinoid tumours.
These are low-grade malignant tumours arising usually in the central airways. They are highly vascular and commonly present with haemoptysis or result in bronchial obstruction. These tumours generally have a good prognosis and, whilst metastatic spread to hilar lymph nodes can occur, this is rare and resection is usually curative. Some of these tumours may produce ACTH and are a rare cause of Cushing’s syndrome.
Staging and treatment
As with all tumours, the stage of the tumour at presentation is of great prognostic significance. Lung cancers are staged pathologically using the TNM system although clinically the Mountain classification is also widely used to group patients together for trials (Table 14.5). The main chance of survival in non-small cell carcinoma is complete surgical resection which is achievable only in tumours confined to the lung or with limited hilar node involvement. Spread to mediastinal nodes, direct invasion of mediastinal structures or metastatic spread to other sites is usually a contraindication. Chemotherapy and radiotherapy may be used radically or palliatively in patients who have either inoperable disease or who are unfit on medical grounds for resection. As previously indicated, SCLC has almost always metastasised at the time of diagnosis and is therefore not usually amenable to surgery. Combination chemotherapy/radiotherapy can induce remission in SCLC, but this is sustained in only in a few patients.
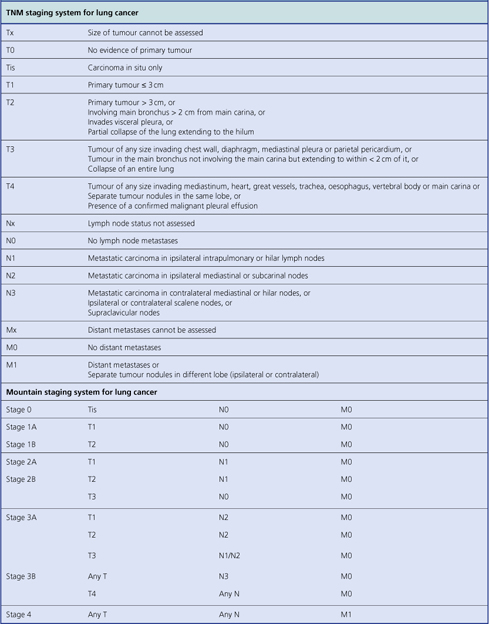
Other primary tumours
Primary tumours other than carcinomas are rare. They can be classified as:
Benign tumours
Adenomas may arise from bronchial mucous glands. They present as polypoid or sessile lesions in a bronchus. Symptoms are related to obstruction.
Benign mesenchymal tumours may arise anywhere that mesenchyme (connective tissue) occurs. Thus, neurofibromas, lipomas, etc. may be found in the lung. More common is the chondroma. The lesion is hard, white and well circumscribed, and is discovered as an isolated ‘coin’ lesion on chest X-ray. It is composed of nodules of cartilage with infoldings and clefts lined with bronchial or bronchiolar epithelium.
Malignant tumours
Malignant mesenchymal tumours (sarcomas) are extremely rare but the commonest primary type is angiosarcoma.
Primary pulmonary lymphomas are rare tumours presenting as pulmonary disease with or without hilar lymph node involvement, but without clinical evidence of disease elsewhere. HIV/AIDS patients have an increased incidence of pulmonary lymphoma. These tumours arise from bronchus- and bronchiole-associated lymphoid tissue.
Secondary lung tumours
Secondary lung tumours are more common than primary tumours, although a patient presenting first with a lung tumour is more likely to have a primary. Metastases may arise from blood or lymphatic spread. Usually, discrete nodules are seen scattered throughout both lungs; however, the lymphatics may be diffusely involved, leading to the appearance of lymphangitis carcinomatosa. Sarcomas, carcinomas and lymphomas can lead to pulmonary metastases. Carcinomas that commonly give rise to lung secondaries include those from the breast, kidney and gastrointestinal tract.
PLEURA
The pleura is composed of connective tissue lined with mesothelial cells forming two apposing surfaces; the visceral pleura covers the lungs and the parietal pleura covers the thoracic cage wall, diaphragm, heart and mediastinum.
EFFUSIONS AND PNEUMOTHORAX
Various fluids (effusions) and air (pneumothorax) can collect between the two layers of pleura (Table 14.6).
Table 14.6 Disorders due to collection of fluid and air in the pleural cavities
| Disorder | Collection | Causes |
|---|---|---|
| Haemothorax | Blood | Chest injury; ruptured aortic aneurysm |
| Hydrothorax | Low protein fluid (transudate) | Liver failure; cardiac failure; renal failure |
| High protein fluid (exudate) | Tumours; infection; inflammation | |
| Chylothorax | Lymph | Neoplastic obstruction of thoracic lymphatics |
| Pneumothorax | Air | Spontaneous, following rupture of alveolus or bulla in emphysema or tuberculosis |
| Traumatic, e.g. following penetrating injuries of the chest | ||
| Spontaneous idiopathic (in young healthy people without pulmonary disease); cause unknown | ||
| Pyothorax (empyema) | Pus | Infection |
Patients with pleural effusions or pneumothorax suffer shortness of breath and respiratory distress; these symptoms can be relieved by draining the fluid or air from the pleural cavity. Clinically, an effusion is dull to percussion; this is in contrast to a pneumothorax, which is hyper-resonant. To investigate the possibility that a pleural effusion might be due to primary or metastatic neoplasia, it is essential to perform a cytological examination of the cells within the fluid, with possible pleural biopsy.
INFLAMMATORY DISORDERS
Inflammation of the pleura (pleuritis or pleurisy) is common. It can be seen with:
The inflammation is nearly always accompanied by an effusion. Symptoms are usually of sharp, localised chest pain, worse on breathing. Treatment is of the underlying disorder causing the pleurisy. Depending on the degree of inflammation, pleurisy may resolve or organise to leave an area of fibrosis, sometimes with dystrophic calcification.
Pleural plaques are markers of asbestos exposure. They are asymptomatic patches of thickened fibrotic pleura on the diaphragm and posterior thoracic wall. Histologically, they consist of hyaline acellular connective tissue with a few inflammatory cells at the periphery. Diffuse pleural fibrosis and pleural effusions may both be related to asbestos exposure in the absence of lung cancer, mesothelioma or asbestosis.
TUMOURS
Benign tumours of the pleura are rare. The solitary fibrous tumour is composed of a tangled network of fibroblast-like cells in a collagenous stroma. It grows as a solitary lump in the pleura, sometimes becoming very large. Hypertrophic pulmonary osteoarthropathy is a frequent association.
Malignant tumours are most often secondary deposits from primary lung adenocarcinomas and breast carcinomas, although other sites may also spread to involve the pleura. These secondary deposits may grow and encase the lung, thus mimicking a mesothelioma (pseudo-mesothelioma).
Mesothelioma
Primary malignant mesothelioma is strongly associated with occupational exposure to asbestos, especially fibres such as crocidolite (‘blue’ asbestos) and amosite (‘brown’ asbestos).
Commonly confused conditions and entities relating to respiratory tract pathology
| Commonly confused | Distinction and explanation |
|---|---|
| Emphysema and empyema | Pulmonary emphysema is characterised by large thin-walled bullae formed from fusion of alveolar spaces, and is entirely unrelated to empyema, which is the name given to a pus-filled cavity (e.g. pleura) or hollow organ (e.g. gallbladder). |
| Bronchitis and bronchiectasis | Both conditions affect bronchi. Bronchitis is simply bronchial inflammation, acute or chronic. Bronchiectasis is permanent abnormal dilatation of bronchi. |
| Bronchopneumonia and lobar pneumonia | In bronchopneumonia the inflammation is centred on airways, may be due to a wide range of bacteria, and is common in patients debilitated through age or serious disease. Lobar pneumonia affects diffusely an entire lobe, is due to a smaller range of bacteria (e.g. Strep. pneumoniae, Klebsiella), and often appears without antecedent cause. |
| Primary and secondary tuberculosis | Both are due to Mycobacterium tuberculosis, but the differences are determined by the host reaction. Primary tuberculosis is a benign self-limiting condition typically affecting the mid-zones in children. Secondary tuberculosis occurs in adults in whom a hypersensitivity reaction to M. tuberculosis results in extensive tissue destruction and fibrosis. |
Other non-asbestos fibres, such as volcanic silicate erionite in Turkey, may also cause mesothelioma. The latent interval between exposure and the development of mesothelioma is often about 30 years. There has been a large increase in the incidence of mesothelioma in the Western world over the last decade, reflecting asbestos use and thus exposure in the previous decades. This is, however, expected to fall in the next decade or so following increasing restriction of its use in the 1970s and 1980s.
The tumour begins as nodules in the pleura which extend as a confluent sheet to surround the lung and extend into fissures (Fig. 14.30). The chest wall is often invaded, with infiltration of intercostal nerves, giving severe intractable pain. Lymphatics may be invaded, giving hilar node metastases. Distant metastases may be found in up to 30% of patients at autopsy.

Histology is varied; most commonly the appearance is of a mixed epithelial and spindle cell tumour although pure epithelial or spindle cell (sarcomatous) mesotheliomas can occur. Special histological techniques are often necessary to distinguish between mesothelioma and adenocarcinoma.
There is no proven effective treatment for malignant mesothelioma, and the median survival from diagnosis is around 11 months.
Britton M.. The epidemiology of mesothelioma. Seminars in Oncology. 2002;29:18-25.
Corrin B., Nicholson A.G.. Pathology of the lungs, 2nd edn. Edinburgh: Churchill Livingstone, 2006.
Hill A.T., Wallace W.A.H., Emmanuel X.. An atlas of investigation and therapy. Pulmonary infection. Oxford: Clinical Publishing, 2004.
Katzenstein A.A.. Katzenstein and Askin’s surgical pathology on non-neoplastic lung disease, 4th edn. Philadelphia: Saunders, 2006.
Khalil N., Churg A., Muller N.. O’Conner R 2007 Environmental, inhaled and ingested causes of pulmonary fibrosis. Toxicology and Pathology. 2007;35:86-96.
MacNee W.. Pathogenesis of chronic obstructive airways disease. Proceedings of the American Thoracic Society. 2005;2:258-266.
National Institute for Health and Clinical Excellence. Lung cancer diagnosis and treatment. London: NICE, 2005. http://www.nice.org.uk
Nicholson A.G.. Classification of the idiopathic interstitial pneumonias: making sense of the alphabet soup. Histopathology. 2002;41:381-391.
Scottish Intercollegiate Guidelines Network. SIGN 80. Management of patients with lung cancer. A national clinical guideline. SIGN Edinburgh, 2005 http://www.sign.ac.uk
Travis W.D., Colby T.V., Koss M.N., Rosada-de-Christenson M.L., King T.E.. Non-neoplastic disorders of the lower respiratory tract. Washington, DC: Fascicle 2. Armed Forces Institute of Pathology, 2002.
Travis W.D., Brambilla E., Muller-Hermelink H.K., Harris C.C.. WHO classification of tumours. Tumours of the lung, pleura, thymus and heart. Lyons: IARC Press, 2004.
Tweedle G.. Asbestos and its lethal legacy. Nature Reviews. Cancer. 2002;2:311-315.
World Health Organization. Burden of disease statistics. Geneva: WHO, 2008. http://www.who.int/healthinfo/bod/en/index.html
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 14 Respiratory tract
COMMON CLINICAL PROBLEMS FROM RESPIRATORY TRACT DISEASE
Pathological basis of respiratory signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Sputum |
CoughPhysiological refl ex response to presence of mucus, exudate, tumour or foreign materialWheezing
DyspnoeaDecreased oxygen in the blood from impaired alveolar gas exchange, left heart failure or anaemiaCyanosisIncreased non-oxygenated haemoglobin, e.g. circulatory bypassing of lungs in congenital heart diseases or impaired alveolar gas exchangePleuritic painIrritation of the pleura due to pulmonary infl ammation, infarction or tumourPleural effusion
ClubbingOften accompanies carcinoma of lung and pulmonary fi brosis, as well as, less commonly, cirrhosis and chronic infl ammatory bowel diseaseWeight lossProtein catabolic state induced by chronic infl ammatory disease (e.g. tuberculosis) or tumoursAuscultation signs
Percussion signs
Globally respiratory diseases, particularly lung infections, together with gastrointestinal infection, account for most deaths in the developing world. Respiratory disease is also a common cause of death in the industrialised nations, accounting for about 14% of deaths in each sex. Out of a global total of 55.69 million deaths in 2000, 3.86 million were due to acute lower respiratory tract infections, 1.66 million to tuberculosis, 2.94 million to HIV/AIDS, 1.21 million to lung cancer and 3.54 million to a variety of other respiratory diseases, mainly chronic obstructive pulmonary disease (2.52 million). There is also considerable morbidity due to respiratory diseases: it is estimated that, in the UK, about 40% of absence from work is the result of such diseases, approximately 85% of which are transient infections of the upper respiratory tract (Table 14.1).
Table 14.1 Major aetiological factors in respiratory disease
| Aetiological factor | Disease |
|---|---|
| Genetic | |
| Air pollution | |
| Occupation | |
| Infection |
NORMAL STRUCTURE AND FUNCTION
The respiratory system extends from the nasal orifices to the periphery of the lung and the surrounding pleural cavity. From the nose to the distal bronchi, the mucosa is lined by mainly pseudostratified ciliated columnar epithelium with mucus-secreting goblet cells; this is respiratory mucosa (Fig. 14.1). A portion of the larynx is covered with stratified squamous epithelium.
Fig. 14.1 The respiratory system. Anatomy of the respiratory tract. Histology of respiratory epithelium. With the exception of the pharynx, epiglottis and vocal cords, the respiratory tract is lined by specialised epithelium compring ciliated columnar epithelia cells with admixed mucus-secreting goblet cells and scattered neuroendocrine cells.
Nasal passages and sinuses
These constitute the upper respiratory tract. The nasal passages and sinuses are in continuity and are lined with respiratory mucosa. The hairs in the nose trap large particles of foreign material, thereby filtering the air. The air is also warmed and humidified as it passes through the nasal cavity. The middle ear, also lined with respiratory epithelium, connects with the nasal cavity via the Eustachian tube.
Larynx
The larynx connects the trachea to the pharynx. Consisting of a complicated system of cartilages and muscles, it allows air into the trachea, with the epiglottis preventing the passage of food into the lungs, and also produces sound for speaking. Part of the larynx, including the vocal cords and epiglottis, is covered with non-keratinising squamous epithelium similar to that lining the oral cavity, pharynx and oesophagus.
Lungs
The lower respiratory tract consists of the trachea, bronchi, bronchioles, alveolar ducts and alveoli (Fig. 14.2). (Table 14.2).
Table 14.2 Structure of the respiratory tree
| Part of respiratory tract | Structure |
|---|---|
| Trachea | Anterior C-shaped plates of cartilage with posterior smooth muscle. Mucous glands |
| Bronchi | Discontinuous foci of cartilage with smooth muscle. Mucous glands |
| Bronchioles | No cartilage or submucosal mucous glands. Clara cells secreting proteinaceous fluid. Ciliated epithelium |
| Alveolar duct | Flat epithelium. No glands. No cilia |
| Alveoli | Type I and II pneumocytes |
The lungs develop from an outpouching of the anterior wall of the primitive foregut at about the fifth week of development. From this tube, two lateral outgrowths appear which eventually form the right and left lungs. These outgrowths are surrounded by mesenchyme from which forms the connective tissue of the respiratory tree. Thus the lungs, like the gastrointestinal tract, develop from endoderm, and developmental abnormalities such as cysts can therefore be lined by either respiratory or gastrointestinal mucosa.
The lungs are divided into lobes: the right lung has three lobes (upper, middle, lower); the left lung has only two lobes (upper and lower). Each lung is formed of 10 anatomically defined bronchopulmonary segments. Each segment is supplied by a segmental branches of the pulmonary artery and bronchus (the bronchovascular bundle). The veins draining adjacent segments often anastomose before they reach the hilum and run principally in the fibrous septae of the lungs.
The respiratory tree is designed to transport clean, humidified air into distal airways and alveoli, where the waste product of metabolism (CO2) is exchanged for O2.
Bronchioles branch until they form terminal bronchioles less than 2 mm in diameter. The respiratory system distal to the terminal bronchiole is called the acinus or terminal respiratory unit, where gas exchange occurs. Small airways, defined as having an internal diameter of less than 2 mm, consist of terminal and respiratory bronchioles. Respiratory bronchioles are involved with gas exchange, having alveoli in their walls. A group of three to five respiratory acini is called a lobule.
The alveoli are lined by flattened type I pneumocytes with occasional type II pneumocytes; the latter are rounded cells with surface microvilli and are believed to be the stem cell population for the alveolus. Type II cells secrete surfactant, and replicate quickly after injury to alveolar walls. Beneath the alveolar cells lie a basement membrane which is shared by the alveolar capillary epithelial cells and some interstitial matrix, including elastin fibres. The structure of the alveolar–capillary membrane permits rapid and efficient diffusion of oxygen and carbon dioxide.
The lung is encased by the visceral pleura which is a thin layer of fibroconnective tissue and elastin with overlying mesothelial cells. The lungs sit within the chest cavity surrounded by the parietal pleura, by the diaphragm, ribs and intercostal muscles, vertebral column and sternum.
Blood supply and lymphatic drainage
The lungs are perfused by a dual arterial blood supply. The trunk of the pulmonary artery arises from the right ventricle, splits into main right and left pulmonary arteries and thence follows the airways, forming the bronchovascular bundles. The bronchial arteries arise from the descending thoracic aorta and supply oxygenated blood to lung parenchyma around the hilum. Pulmonary veins take all the blood from the lungs back to the left atrium.
Pulmonary veins course along the interlobular septa with lymphatics.These lymphatic channels and those in the pleura drain into the thoracic duct.
Control of respiration
Respiration is controlled by the respiratory centre in the medulla oblongata, and the carotid bodies situated at the carotid bifurcations. The medullary centre senses any change in CO2 concentration in the cerebrospinal fluid, and modifies respiration by nervous stimulation of respiratory muscles and the diaphragm. The partial pressure of O2 in the blood is monitored by the carotid bodies, which can then stimulate the respiratory centre through the glossopharyngeal nerves. Carotid bodies can become hyperplastic in response to chronic arterial hypoxaemia, such as occurs in:
Gas exchange
Air is drawn into the lungs by contraction of the diaphragm and intercostal muscles, creating a negative intrapleural pressure. On relaxation of these muscles, air is expelled as the lungs contract under the action of gravity and the elasticity in the lung connective tissue. The stiffness of the lungs, or compliance, is a measure of change in volume per unit change in pressure, and is therefore a measure of compressibility; for example, in pulmonary fibrosis the lungs cannot be easily compressed and therefore the compliance is decreased.
Clearly, gas exchange occurs only in alveoli that are both perfused and ventilated. Ventilation of non-perfused alveoli increases the ‘dead space’, that proportion of inspired air not involved with gas exchange. Perfusion of non-ventilated alveoli leads to physiological right-to-left shunting of non-oxygenated blood as it passes through the pulmonary circulation.
Acid–base balance
Normal acid–base balance in blood is dependent on both efficient alveolar ventilation and perfusion, with consequent successful gas exchange. This leads to the normal partial pressures of O2 and CO2 in arterial blood (Pao2 and Paco2), and a normal blood pH. Various metabolic disease states lead to disturbances in acid–base balance (Fig. 14.3). If the disease becomes chronic, compensatory mechanisms by both the lungs and kidneys operate in an attempt to restore blood pH.
PULMONARY FUNCTION TESTS
In normal quiet respiration only a relatively small proportion of the total lung capacity (TLC) is inhaled and exhaled; this is the tidal volume (TV). TLC is made up of the amount of air totally exhaled after maximum inspiration (the vital capacity or VC) and the residual volume (RV). TLC, RV, TV and VC are all easily measured in the laboratory using helium dilution techniques.
In addition to calculating volume parameters, some techniques also assess actual pulmonary function. Spiro-metry measures the amount of exhaled air per second. The maximum volume of air blown from the lungs within the first second after a previous maximum inspiration is called the forced expiratory volume (FEV1). This figure, highly reproducible in each individual, is a measure of small airway resistance. It is also dependent on the patient’s age, sex and size; for example, the small lungs of a child obviously cannot expel as much air as those of an adult. The ratio FEV1:VC compensates to a degree for the variability of lung size. It is possible to inhale more rapidly than exhale because, during inspiration, forces on the airways tend to open them further; during expiration, opposite forces tend to close the airways and thus restrict airflow. For a given lung volume, the expiratory flow rate reaches a peak (PEFR), which is again a measure of airways resistance.
An assessment of the ability of the lungs to exchange gas efficiently can be made by measuring the transfer factor for carbon monoxide (TCO). Air containing a known concentration of carbon monoxide is inhaled; the breath is held for 15 seconds and then exhaled. The amount of carbon monoxide absorbed is a measure of pulmonary gas exchange. TCO is dependent on the concentration of blood haemoglobin, which has a strong affinity for carbon monoxide. Diseases that diffusely affect the alveolar–capillary membrane (such as diffuse pulmonary fibrosis or emphysema where there is loss of alveolar surface area) will result in a low TCO.
Recently the level of nitric oxide (NO) in exhaled air has been added as a useful test; increased levels have been associated with asthma and other causes of bronchial irritation, while decreased levels have been found in cigarette smokers, patients with pulmonary hypertension and during treatment with corticosteroids.
Obstructive and restrictive defects
There are two major patterns of abnormal pulmonary function tests: obstructive defects (e.g. asthma) and restrictive defects (e.g. pulmonary fibrosis) (Table 14.3).
Table 14.3 Respiratory function tests and their diagnostic significance
| Test | Diagnostic significance |
|---|---|
| Peak expiratory flow rate (PEFR) | Reduced with obstructed airways or muscle weakness |
| Forced expiratory volume in 1 second (FEV1) | Reduced with obstructed airways, pulmonary fibrosis or oedema, or muscle weakness |
| Vital capacity (VC) |
Forced expiratory ratio (FEV1:VC)
Carbon monoxidetransfer (TCO)Reduced in pulmonary fibrosis, emphysema, oedema, embolism and anaemiaExhaled nitric oxide (NO)Increased in asthma, bronchiectasis and infections Decreased in pulmonary hypertension, cigarette smokers and after treatment with corticosteroids
In obstructive airways disease, RV and TLC are mildly increased due to hyperinflation of the lung while FEV1, FVC and the FEV1: VC ratio is decreased. Clearly, in conditions such as asthma, the results of pulmonary function tests will depend on the clinical state of the patient, whether in an acute attack of asthma or in remission. Restrictive diseases are those that restrict normal lung movement during respiration and are associated with reduced RV and TLC. The FEV1 and VC may be reduced but their ratio remains normal.
These tests are of most value in the follow-up of patients. They can also give an indication as to the possible benefits of treatment; for example, observing the improved FEV1:VC and PEFR after treatment with a bronchodilator would be a measure of the reversibility of the airways obstruction.
RESPIRATORY FAILURE
Respiratory failure can occur as a result of:
The effects of respiratory failure include impaired clearance of CO2 from the lungs, resulting in hypercapnia, and impaired absorption of O2 from the air, resulting in hypoxaemia. The patient is typically dyspnoeic, cyanosed and lapsing into coma. Hypercapnia (high blood CO2 concentration) is associated with a bounding pulse and warm, moist extremities.
DISEASES OF INFANCY AND CHILDHOOD
Respiratory diseases of infancy and childhood are predominantly infectious; such diseases, together with diarrhoea, are the primary cause of death in childhood in the developing world. Rarely disease may arise as a result of either developmental abnormalities or immaturity.
Developmental abnormalities
Developmental abnormalities include:
Tracheo-oesophageal fistula
Embryologically, the oesophagus and the trachea begin as a single tube; the trachea then buds off to form the pulmonary tree. In a tracheo-oesophageal fistula, the oesophagus ends in a blind pouch; the trachea then usually connects to the stomach via a fistula. On ingestion of food, the upper pouch quickly fills and overflows into the pulmonary tree, with choking and coughing, leading to aspiration pneumonia. Treatment is by surgery, usually after a period of feeding via gastrostomy.
Congenital diaphragmatic hernia with pulmonary hypoplasia
This presents as neonatal respiratory distress due to herniation of the stomach and loops of bowel into the thorax; usually the left diaphragm is defective. Surgical correction to restore normal thoracic and abdominal anatomy is essential at the earliest possible opportunity. However, even after this there is still a considerable mortality from the associated severe pulmonary hypoplasia, usually of the left lung.
Congenital cystic adenomatoid malformations
These are characterised by abnormalities in the development of small airways and the alveolar tissue of the lung. This results in the development of cysts within the lung which may be of varying size and can be localised to one lobe or be extensive and bilateral. The prognosis depends on the pattern of abnormality present and any other associated abnormalities. Localised areas are cured by surgical excision of the affected lobe.
Bronchogenic/foregut cysts
These occur in the lung or mediastinum and may be lined either by bronchial elements such as cartilage, smooth muscle and ciliated respiratory epithelium (bronchogenic cysts), or by squamous or even gastric or pancreatic type epithelium (foregut cysts). Usually, such cysts are asymptomatic, although complications may occur.
Pulmonary sequestration
A sequestered piece of lung is a mass of abnormal lung that does not communicate anatomically with the tracheo-bronchial tree; it is supplied by an anomalous artery, usually from the aorta. Sequestered segments of lung are found most often within the left lower lobe. Histology shows a multilobulated cystic mass with fibrosis and variable inflammation. An endogenous lipid pneumonia may result.
Congenital lobar emphysema
This condition is characterised by overdistension of a lobe due to intermittent bronchial obstruction. Symptoms arise due to pressure effects caused by the massively distended lobe. Usually, the left upper lobe is affected. The pathogenesis is thought to be abnormal bronchial cartilage allowing inspiration of air but not expiration. Extrabronchial compression by enlarged lymph nodes may produce a similar clinical picture. Treatment is surgical removal of the diseased lobe.
lmmaturity
Diseases due to immaturity include:
Hyaline membrane disease or idiopathic respiratory distress syndrome
Hyaline membrane disease (HMD) is almost always seen in premature infants of birth weight less than 2.5 kg. Infants are usually of less than 36 weeks’ gestation, and the incidence of HMD rises as the gestational age decreases. The risk of developing HMD may be decreased by giving mothers oral corticosteroids prior to delivery of the baby as this appears to stimulate surfactant production in the lungs.
Clinical features
After a few hours of relatively normal respiration, symptoms of tachypnoea and dyspnoea with expiratory grunting appear. Cyanosis quickly follows, with worsening respiratory distress. Hypoxaemia refractory to high concentration of inhaled oxygen is one hallmark of the disease, a finding also characteristic of adult respiratory distress syndrome (ARDS).
Pathogenesis
The pathogenesis is thought to be due to a deficiency of surfactant. This is secreted by type II pneumocytes, and normally lines distal airways; it reduces surface tension, thereby allowing airway opening during inspiration. Without normal quantities of surfactant, airways need greater effort to open, leading to respiratory distress.
Morphology
At autopsy the lungs are heavy, purple and solid, and sink in water. Histology shows collapsed alveoli with hyaline membranes lining alveolar ducts. Pulmonary lymphatics are dilated. As in ARDS, if the infant survives, resolution follows within the next few days, although pulmonary fibrosis may occur. Treatment is with oxygen and artificial ventilation.
Bronchopulmonary dysplasia
Bronchopulmonary dysplasia is the term used to describe the picture of lung organisation after HMD. Often, infants have been previously treated with high levels of oxygen, and it is not clear whether bronchopulmonary dysplasia is a separate disorder, solely related to oxygen toxicity, or merely a result of organisation after HMD. Certainly, the features are almost identical to those seen with organisation of ARDS; there is interstitial fibrosis, peribronchial fibrosis, and features of pulmonary hypertension. The airways may show extensive squamous metaplasia.
INFLAMMATORY DISORDERS
Rhinitis (the common cold) is caused by many different viruses, especially rhinoviruses, although respiratory syncytial virus (RSV), para-influenza viruses, coronaviruses, coxsackieviruses, echoviruses and bacteria, such as Haemophilus influenzae, may also be implicated. Rhinitis may also be caused by inhaled allergens as in ‘hay fever’ where the inflammatory reaction is mediated via type I and type III hypersensitivity reactions (Chs 9 and 10).
Nasal polyps may result from either chronic infective inflammation or chronic allergic inflammation. They consist of polypoid oedematous masses of mucosal tissue infiltrated with chronic inflammatory cells, especially plasma cells; eosinophils may be numerous if allergy is the cause.
Sinusitis is inflammation of the paranasal sinuses; it may be acute or chronic. If the drainage orifice is blocked by inflamed swollen mucosa, an abscess may follow. Cranial osteomyelitis, meningitis or cerebral abscess may then result from sinusitis by direct extension.
Wegener’s granulomatosis, a granulomatous form of vasculitis may involve the nose and upper respiratory tract and present with septal perforation or collapse of the nasal cartilages.
Otitis media is infection of the middle ear, often associated with generalised upper respiratory tract infection (URTI). The Eustachian tube may become swollen and blocked, leading to trapping of exudate in the middle ear. Eardrum perforation may ensue, followed by drainage of the effusion. More serious complications include mastoiditis, meningitis and brain abscess.
TUMOURS
Tumours of the nasal passages and sinuses are uncommon. They may be:
Haemangioma and squamous papilloma are benign lesions, the former often presenting with troublesome epistaxis (nosebleeds). Some squamous papillomas may be caused by human papillomavirus.
Juvenile angiofibromas are rare and occur exclusively in males, usually during adolescence. They are extremely vascular, and surgical removal can be difficult. These tumours contain androgen receptors, explaining the male preponderance.
Squamous cell carcinoma may be well differentiated, producing keratin, or very poorly differentiated. The latter may contain many lymphocytes and have been misnamed ‘lympho-epitheliomas’. Such tumours are most common in South-East Asia and account for 18% of all cancers in China; evidence suggests the Epstein–Barr virus is involved in the aetiology and pathogenesis of squamous cell carcinoma.
Adenocarcinoma of the nasal passages and sinuses occurs more frequently in people who have worked in woodwork and furniture industries. These tumours may present clinically up to 40 years after initial exposure.
Primary mucosal melanomas of the nose and sinuses are rare but have a very poor prognosis.
Primary extranodal lymphomas are almost always of non-Hodgkin’s type.
Plasmacytomas are tumours composed of plasma cells. They can occur as part of multiple myeloma or as isolated lesions without systemic disease.
INFLAMMATORY DISORDERS
Laryngitis may occur in association with viral or bacterial inflammation of trachea and bronchi; this is laryngotracheo-bronchitis. Diphtheria was once a common, and serious, bacterial cause of laryngitis, leading to the formation of a fibrinopurulent membrane that could cause airway obstruction. Now, partly as a result of immunisation in infancy with diphtheria toxoid, the disease is rare.
Epiglottitis is caused by capsulated forms of Haemophilus influenzae type B. The epiglottis becomes inflamed and greatly swollen, leading to airway obstruction (Fig. 14.4). Treatment is by intubation, although, rarely, tracheostomy may be necessary; antibiotics are also given to treat the infection.
Fig. 14.4 Acute epiglottitis. Gross swelling of the epiglottis (arrowed) leading to respiratory obstruction in a child.
Allergic laryngitis occurs after inhalation of an allergen. There may be gross oedema leading to airway obstruction. Irritative laryngitis may be due to cigarette smoke or mechanical factors, e.g. endotracheal intubation.
Laryngeal polyps often develop in singers and are thus sometimes referred to as ‘singer’s nodes’. Even when only a few millimetres in diameter they can alter the character of the voice. They consist of oedematous myxoid connective tissue covered with squamous mucosa with amyloid-like material in the stroma.
TUMOURS
Papilloma may be caused by types of human papillomavirus. Papillomas consist of squamous epithelium covering fibrovascular cores of stroma. They may be multiple and recurrent, especially in children, but are usually single in adults. Such papillomas can extend into the trachea and bronchi.
Squamous cell carcinoma of the larynx typically affects males over 40 years of age and is associated with cigarette smoking. There may also be an increased risk in asbestos workers. As in squamous epithelium of the cervix, neoplasia is thought to be preceded by a phase of dysplasia. The dysplasia, especially if low grade, may be reversible on withdrawal of causative factors.
Most laryngeal carcinomas arise on the vocal cords (Fig. 14.5), although they may arise above, in the pyriform fossa, or below, as upper tracheal carcinomas. The lesions ulcerate, fungate and invade locally, later causing metastases in regional lymph nodes in the neck and beyond. Symptoms are hoarseness of voice and, later, pain, haemoptysis and dysphagia. Treatment is by chemo-radiotherapy and/or resection. These patients often have widespread mucosal abnormalities throughout the respiratory tract and are at high risk of developing further cancers, especially if they continueto smoke.
THE LUNGS
RESPIRATORY INFECTIONS
The lungs have an internal surface area of approximately 500 m2 which is exposed to the external environment and potentially subjected to inhaled microbes with every breath. It is therefore not surprising that respiratory infections are relatively common, with the World Health Organization projecting such infections to continue as one of the global leading causes of death and disability. Countering the threat of pathogens are the defence mechanisms, any abnormality in which will predispose to infection. Such abnormalities include:
Infections can be classified as primary, with no underlying predisposing condition in a healthy individual, or secondary, when local or systemic defences are weakened. The latter are by far the most common types of respiratory infection in developed countries, and are becoming yet more important with the spread of AIDS.
Bronchitis
In acute bronchitis, the trachea and larynx are involved as well as the lungs, and the disease is then known as acute laryngotracheobronchitis (or ‘croup’). The disease is most severe in children, with symptoms of cough, dyspnoea and tachypnoea. Viruses are usually the cause, especially respiratory syncytial virus (RSV), although Haemophilus influenzae and Streptococcus pneumoniae are frequent bacterial causes. Episodes of acute bronchitis are common in chronic obstructive airways disease, and cause a sudden deterioration in pulmonary function with cough and the production of purulent sputum. Acute bronchitis may be caused by direct chemical injury from air pollutants, such as smoke, sulphur dioxide and chlorine.
Chronic bronchitis is a clinical term defined as cough and sputum for 3 months in 2 consecutive years; it is discussed below under diffuse obstructive airways disease (p. 338).
Bronchiolitis
Primary bronchiolitis is an uncommon respiratory infection caused by viruses, especially RSV, in infants. Symptoms are of acute respiratory distress with dyspnoea and tachypnoea. Most cases resolve within a few days, although a minority may develop secondary pneumonia.
Follicular bronchiolitis with lymphoid aggregates and germinal centres, compressing the airway, can occur in rheumatoid disease.
Bronchiolitis obliterans is characterised by concentric fibrosis of the submucosa of small bronchioles resulting in obliteration of the lumen. It may occur in viral infections, especially RSV, after inhalation of toxic fumes, in lung transplant rejection, after aspiration, and with collagen vascular diseases.
Pneumonia
Pneumonia is usually due to infection affecting distal airways and alveoli, with the formation of an inflammatory exudate. It may be classified according to several criteria (Table 14.4, Fig. 14.6).
Table 14.4 Classifications of pneumonia
| Criterion | Type | Example/comment |
|---|---|---|
| Clinical circumstances | Primary | In an otherwise healthy person |
| Secondary | With local or systemic defects in defence | |
| Aetiological agent | Bacterial | Streptococcuspneumoniae,Staphylococcus aureus,Mycobacteriumtuberculosis, etc. |
| Viral | Influenza, measles, etc. | |
| Fungal | Cryptococcus, Candida,Aspergillus,etc. | |
| Other | Pneumocystis jiroveci, Mycoplasma, aspiration, lipid, eosinophilic | |
| Host reaction |
According to dominant component of exudateAnatomicalpattern
Most widely usedclassification before identifying aetiological agent
Fig. 14.6 Distribution of lesions in lobar and bronchopneumonia. Bronchopneumonia is characterised by focal inflammation centred on the airways; it is often bilateral. Lobar pneumonia is characterised by diffuse inflammation affecting the entire lobe. Pleural exudate is common.
Bronchopneumonia
Bronchopneumonia has a characteristic patchy distribution, centred on inflamed bronchioles and bronchi with subsequent spread to surrounding alveoli (Fig. 14.6). It occurs most commonly in old age, in infancy and in patients with debilitating diseases, such as cancer, cardiac failure, chronic renal failure or cerebrovascular accidents. Bronchopneumonia may also occur in patients with acute bronchitis, chronic obstructive airways disease or cystic fibrosis. Failure to clear respiratory secretions, such as is common in the post-operative period, also predisposes to the development of bronchopneumonia.
Typical organisms include staphylococci, streptococci and Haemophilus influenzae. Patients often become septicaemic and toxic, with fever and reduced consciousness.
Affected areas of the lung tend to be basal and bilateral, and appear focally grey or grey–red at postmortem (Fig. 14.7). The inflamed lung parenchyma can be demonstrated by gently pressing on an affected area; normal lung recoils like a sponge, whereas pneumonic lung offers little resistance. Histology shows typical acute inflammation with exudation. With antibiotics and physiotherapy, the areas of inflammation most commonly resolve but may heal by organisation with scarring.
Lobar pneumonia
Pneumococcal pneumonia typically affects otherwise healthy adults between 20 and 50 years of age; however, lobar pneumonia caused by Klebsiella typically affects the elderly, diabetics or alcoholics. Symptoms include a cough, fever and production of sputum. The sputum appears purulent and may contain flecks of blood, so-called ‘rusty’ sputum. Fever can be very high (over 40°C), with rigors. Acute pleuritic chest pain on deep inspiration reflects involvement of the pleura. As the lung becomes consolidated (Fig. 14.8), the chest signs are dullness to percussion with bronchial breathing. The dullness recedes with resolution of the exudate.
Fig. 14.8 Lobar pneumonia. An entire lobe, paler than the other, has become consolidated due to accumulation of acute inflammatory exudate within the alveoli. Note the abrupt demarcation at the interlobar fissure.
The pathology of lobar pneumonia is a classic example of acute inflammation, involving four stages:
[/not-level-membership-for-pathology-category]
