[level-membership-for-hematology-oncology-and-palliative-medicine-category]
15 Total Body Irradiation
History
Only a decade after Roentgen described the “x-ray,” German biophysical engineer Friedrich J. Dessauer first described a “new technique of radiotherapy” that involved homogenous irradiation of the entire body. In his initial report describing the technique in 1905, he proposed irradiating a supine patient using three simultaneously active low-voltage Roentgen ray sources (Fig. 15-1).1,2 In 1907, Aladár Elfer, a medical professor in Hungary, reported his experience using a TBI technique that spared the head in three patients with leukemia.3 Although there is a paucity of data regarding the early use of the technique, some have speculated that untoward hematologic toxicity probably limited its application.4
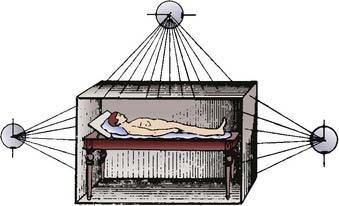
FIGURE 15-1 • Diagram demonstrating the total body irradiation technique proposed by Dessauer in 1905.
Early success using TBI to treat hematopoietic and lymphoid malignancies in Europe (there named the Teschendorf method) prompted development of the technique in the United States.5–7 Arthur C. Heublein, in collaboration with Gioacchino Failla, is credited with the development of the first TBI unit in North America, located at Memorial Hospital in New York City. In the United States, the technique became known as Heublein therapy.8 A specially constructed treatment ward was designed to treat four patients at an extended distance (5-7 meters) simultaneously at an exposure rate of 0.7 R per hour for approximately 20 hours per day, typically over 1 to 2 weeks, using a 185 kV x-ray tube at 3 mA, with a 2-mm copper filter. The goal was to deliver 25% of erythema dose (750 R). Remarkably, in Heublein’s initial report, no hematopoietic toxicity was noted with this treatment schedule. Of 12 patients with advanced lymphomas and leukemias, 7 (58%) demonstrated some form of improvement after treatment; 2 of 8 patients (25%) with metastatic breast, melanoma, and kidney cancers were also noted to demonstrate some form of improvement.9,10 A later report of the experience with 270 cancer patients from Memorial Hospital treated with TBI between 1931 and 1940 confirmed that the technique was more successful in patients with hematopoietic and lymphoid malignancies, compared with patients with carcinomas or sarcomas, for whom it was ineffective. The authors emphasized that the technique was safe if doses were prescribed cautiously. They did not recommend exposures greater than 300 R, and noted hematopoietic and gastrointestinal toxicity with exposures as low as 50 to 100 R.8
In the early 1940s, World War II prompted the Manhattan Project, an initiative to develop nuclear weapons. Part of this endeavor sponsored research into the human biologic response to ionizing radiation, including TBI. The military’s interest in TBI was primarily to help understand human tolerance for radiation exposure during occupational duties and warfare, and to develop radiation biodosimetric assays. Several research studies coordinated through the Manhattan Project were initiated in patients with advanced malignancies,11–13 as well as in patients with benign diseases.13 For example, studies of dose escalation, radiation biologic dosimetry, and cognitive and psychomotor function were carried out at M.D. Anderson Hospital for Cancer Research.14 A detailed report of 30 patients treated at the maximum exposure level (200 R) in the initial study concluded that side effects primarily consisted of nausea, vomiting, and myelosuppression, and that intervention was necessary in 10% of patients treated with this dose of TBI.15 At Baylor University College of Medicine, studies using 25 to 250 R of TBI with 250 kV to 2 MV energy machines were performed to find a biologic dosimeter, as well as to study acute effects of radiotherapy.16 The Sloan-Kettering Institute for Cancer Research also participated in similar studies of at least 20 patients, although the results were never published. The military conducted similar studies at the Naval Hospital in Bethesda, Maryland, and reported palliation of patients with radiosensitive diseases treated with fractionated TBI.17
The most recent Department of Defense–sponsored research study of TBI was at the University of Cincinnati and focused on identifying biochemical markers in the urine that predicted response to TBI. Later, studies of the neuropsychiatric effects of TBI were initiated. Ultimately, only results regarding the palliation of advanced malignancies were reported.18 Patients with advanced metastatic radioresistant malignancies for whom chemotherapy was unavailable were often treated with TBI in the absence of any clear anticipated benefit. Patients treated with TBI in this manner were included in research studies, often without consenting to participate. The ethics of this practice were called into question by a report written in 1995 by the United States Department of Energy’s Advisory Committee on Human Radiation Experiments,19 which could be a contributor to the public’s uneasiness with radiation in general.20
TBI was used not only in malignant diseases; it was considered the critical immunosuppressant in the first successful solid organ transplant. In 1959, a kidney was successfully transplanted between dizygotic twins after TBI to 450 R to the recipient.21 Around the same time in France, successful kidney transplants after TBI were being reported.22,23 Of the first seven patients who underwent kidney transplant following TBI or pharmacologic immunosuppression worldwide between 1959 and 1962, the two who did not experience kidney failure were treated with TBI alone (without chemical immunosuppression) prior to transplant, and each survived in excess of 20 years after transplant.21–23 However, successful preclinical studies with pharmacologic therapy prompted the use of chemical immunosuppressants (corticosteroids, 6-mercaptopurine, and azathioprine) for solid organ transplantation after 1963.24
With an increased understanding of the human response to TBI, and a rapidly growing body of preclinical in vivo studies of TBI, therapeutic protocols were developed to maximize benefit in patients with malignant diseases. In 1957, Nobel laureate E. Donnall Thomas first reported the use of bone marrow infusion in humans following whole-body irradiation or chemotherapy,25 and less than one year later he published his experience using TBI with exposures up to 600 R, followed by bone marrow transplantation.26 In the series of the first five patients with leukemia treated with TBI, who then received intravenous infusion of normal donor marrow, Thomas et al. noted the difficulty of acute myelosuppression and resultant hemorrhage and infection during the period leading up to engraftment. The report also comments that low dose rates (delivery over 2-3 days) appeared preferable to higher dose rates, for metabolic and immunologic reasons. In addition, patients receiving 200 to 300 R fared better than those receiving 400 to 600 R. The problem of delivering an adequately homogenous dose was raised, and suggestions to use higher-energy photons were proposed.26 Thomas et al. later reported on syngeneic bone marrow transplantation in two children after 850 to 1140 R was delivered in a single fraction over 22 to 25 hours, using 60Co sources. The authors concluded that 1000 R of TBI did not produce “troublesome” acute radiation sickness, and produced remission of leukemia, but not cure.27 The first report of successful cure of a patient with leukemia with allogeneic transplantation after TBI was reported in 1969. The technique involved opposed 60Co sources, which operated at 5.8 R per minute, to a total exposure of 1620 R, calculated to be 954 rad at midline. With appropriate supportive care, no major acute radiation sickness was noted, but the patient died of overwhelming cytomegalovirus (CMV) infection, without evidence of leukemia.28
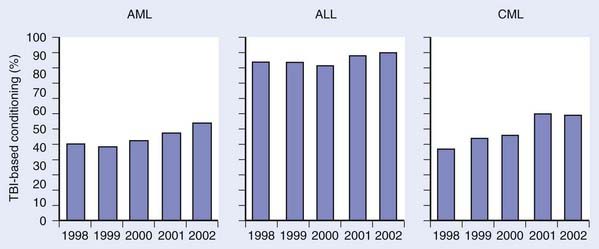
Over the next several years, techniques of combining chemotherapy and TBI were developed and refined, with promising results.29,30 Success in the treatment of advanced leukemias and severe aplastic anemia (AA) was achieved. Departure from the use of TBI alone was primarily fostered by the development of more effective cytotoxic chemotherapeutics and immunologic therapies, which, when combined with TBI, yielded fewer leukemic recurrences.29 Although the use of TBI without HSCT has largely been abandoned, primarily for fear of inducing secondary malignant neoplasms (SMNs) and limiting later therapeutic options, some question the validity of this fear and still contend that very low-dose TBI (1.5-2 Gy in 10-20 fractions over several weeks) is a viable option for upfront therapy in advanced indolent lymphomas.31 Fig. 15-2 demonstrates how the use of TBI during HSCT has remained constant or increased during preceding years.32
Background on Hematopoietic Stem Cell Transplant
HSCT has evolved into a highly complex clinical discipline, firmly rooted in immune system and cancer biology, the details of which are beyond the scope of this chapter. When HSCT was initially performed (see “History”), bone marrow was extracted from the donor, filtered, and infused intravenously to the recipient. Later, peripheral blood stem cells, instead of bone marrow, were collected from the donor, and found to be an alternative to bone marrow grafts. For this reason, “bone marrow transplants” are now more appropriately termed hematopoietic stem cell transplants, because the critical component of the graft is the hematopoietic stem cell (HSC), independent of the source. Oftentimes, peripheral blood stem cells are “mobilized” from the donor using hematopoietic growth stimulating factors, and removed from the donor by apheresis. In addition, recent success using HSCs derived from human umbilical cord blood has been described. Depending on the source of HSCs, various postcollection processing measures (cell selection and depletion) may be undertaken to optimize the outcome of the transplant.
When transplant occurs between different individuals, the hematopoietic graft is said to be allogeneic. This is in contradistinction to reinfusion of native HSCs back into the donor in an “autologous” transplant, more appropriately termed an autoplant, because nothing is being transferred between different individuals.33 A rare but alternative situation is of a patient with a genetically identical twin in the case of a “syngeneic” transplant. Of these three methods of HSCT, autologous and syngeneic transplant are generally associated with less risk because issues related to immunocompatibility are minimized. Allogeneic transplants requiring the “matching” of donor and recipient are typically carried out through identification of human leukocyte antigen compatibility. The donor may be related to the recipient, or identified through registries of volunteers, such as the National Marrow Donor Program.
Prior to undergoing HSCT, most patients require intensive antineoplastic or immunosuppressive therapy in preparation for HSCT, often referred to as conditioning. Conditioning can involve cytotoxic chemotherapy, immunosuppressants, antibody therapy, or radiation therapy. The nature of the conditioning regimen can be referred to as high or reduced intensity, myeloablative, submyeloablative, or nonmyeloablative, to carry out conventional or mini transplants.33 Although agreed-upon formal definitions of these regimens do not exist,34 the goal of high-intensity, myeloablative, or conventional transplant is to completely eliminate the recipient’s native HSC compartment, which necessitates HSCT (autologous or allogeneic) for survival. High-intensity, myeloablative, or conventional transplants may or may not involve TBI, to high doses (>5 Gy in a single fraction, >8 Gy in multiple fractions). Reduced-intensity, nonmyeloablative, or mini-transplant conditioning regimens are often used for older patients or for those with medical problems for whom a high-intensity, myeloablative, or conventional transplant would cause excessive morbidity or mortality, and may or may not involve TBI to lower doses. During and immediately after conditioning, the transplant recipient is at significant risk of infections and other hematologic complications. For this reason, the supportive care of HSCT recipients is very complex, and should only be undertaken in specialized facilities. Nonetheless, some groups have developed reduced-intensity and myeloablative HSCT regimens, including TBI, which have been safely undertaken on an outpatient basis.35,36
According to data summarized by the Center for International Blood and Marrow Transplant Research (CIBMTR) in 2005, the diseases most commonly treated with HSCT are (in decreasing order of frequency): multiple myeloma (MM); non-Hodgkin lymphoma (NHL); acute myelogenous leukemia (AML); Hodgkin disease (HD); acute lymphoid leukemia (ALL); myelodysplastic and myeloproliferative disease (MDS); chronic myelogenous leukemia (CML); AA, and various other leukemias, cancers, and nonmalignant diseases.37 The 2009 National Comprehensive Cancer Network (NCCN) guidelines for therapy indicate that allogeneic or autologous HSCT may be a treatment option for AML, MM, MDS, CML, HD, and NHL, depending on the clinical situation. Detailed discussion of these diseases and their management is beyond the scope of this chapter, and the reader is referred to appropriate chapters in this text and others for further review.38–41 The role of TBI in nonmalignant diseases is discussed further below.
Radiobiology
Preclinical studies have helped define some of the fundamental radiobiologic properties of the normal lymphocytes. The D0 of normal lymphocytes has been reported to be 0.5 to 1.4 Gy,42–45 depending on the in vitro or in vivo model used to calculate this parameter. This D0 suggests that normal lymphocytes cells are very sensitive to ionizing radiation. A very small shoulder on the radiation cell survival curve has been noted,46,47 suggesting little repair between fractions of radiation. Clinical data have revealed similar findings in patients undergoing hyperfractionated TBI (HTBI), with lymphocyte survival demonstrating an effective D0 of 3.8 Gy according to one study.48 Other radiobiologic phenomena have been ill-defined in other normal hematopoietic cells. Radiobiologically relevant levels of hypoxia are unlikely in the hematopoietic compartment. Repopulation is not likely to influence hematopoietic cell survival, given the short duration of most TBI regimens (1-5 days), although, given the variable lifespan of leukocytes (days to years), it may be of some relevance. Redistribution appears to be of significance, given the time scale for TBI; however, this has been difficult to assess.49
The radiobiology of malignant hematopoietic cells has been described. The D0 of leukemic cells generally ranges between 0.8 and 1.5 Gy, however, compared with normal hematopoietic cells, a wider range of radiosensitivities have been described.50–61 Many have cited the technical nuances and variations in assay technique for this great range.49,62 Similar to normal hematopoietic cells, their malignant counterparts are thought to demonstrate little sublethal damage repair,63–67 although split-dose and low-dose-rate experiments have demonstrated the capacity for leukemic cells to repair radiation-induced damage.55,58,68,69 Generally, leukemic cells are believed to have a cell survival curve with minimal or no shoulder, although this varies across cell types and cell lines.47,51,54 For example, Cosset and colleagues summarized preclinical and clinical findings, concluding that AML demonstrates little repair, whereas CML does demonstrate repair, and ALL, myeloma, and lymphomas have not been well studied but appear to demonstrate a wide range of repair capacity.62,70 Similar to normal hematopoietic cells, reoxygenation is unlikely to be radiobiologically relevant to malignant hematopoietic cells during TBI. Redistribution and repopulation, however, may be relevant, but have not been systematically studied.
In vivo preclinical research laid the foundation for the first successful HSCT in humans. Studies in rats,71 dogs,72 and nonhuman primates73 demonstrated that reconstitution of the hematopoietic system was possible after TBI with supralethal doses of radiation. Later work in animals revealed that delivering TBI in several fractions required a higher total dose relative to the biologically isoeffective dose given in a single fraction.74–76 Another model demonstrated no significant difference in the effect of a low-dose-rate (0.04 Gy/min) single fraction of TBI compared with a hyperfractionated course of TBI given thrice daily to the same total dose.77
Although the hematopoietic system is the target of TBI, normal tissues effectively limit the dose that can be safely delivered. The sparing of normal tissues with fractionated TBI was proposed by Peters and colleagues,63,64 and subsequently supported by preclinical data in mice78,79 and dogs80 showing less lung injury with fractionated TBI regimens.
Immediate Toxicity and Management
Although a good deal of what has been learned about the acute in vivo biologic effects of TBI is derived from laboratory-based animal studies, whole-body irradiation also has been studied in people exposed during accidental or wartime nuclear events.81,82 These large-scale studies are valuable because they deal with apparently normal subjects; however, the retrospective nature limits the quality of the data. The reader is referred to several excellent texts for a review of the acute and fatal radiation syndromes (gastrointestinal, hematopoietic, and cerebrovascular) that can be caused by TBI in an uncontrolled setting.83–85
Acute side effects of therapeutic TBI can be difficult to distinguish from other HSCT-related morbidity. However, Chaillet and colleagues conducted a very informative prospective clinical study of the symptoms and signs that occur in patients after TBI, prior to the initiation of any other HSCT related therapy.86 Thirty-one patients between 4.5 and 55 years of age were treated using parallel-opposed anteroposterior (AP) 18 MV photons from a linear accelerator (linac). Shielding was used to limit the lung dose to 8 Gy. A total dose of 10 Gy was given as a “single dose” as six discrete fractions of 1.6 Gy, each given over 15 minutes, with a 30-minute break between fractions, for a mean dose rate of 0.04 Gy per minute and instantaneous dose rate of 0.11 to 0.12 Gy per minute. Symptoms and signs were assessed regularly during the 4 hour TBI, and for 20 hours after the completion of TBI. Antiemetics, but no chemotherapy or steroids, were given prior to the start of TBI. Table 15-1 displays the symptoms and signs experienced by patients during the 4 hours of TBI, and within 24 hours of starting TBI. Fever was a very common finding, and a maximum of 40.8° C was noted in one patient. Tachycardia frequently paralleled febrile episodes; a maximum rate of 130 beats per minute was noted. Drowsiness was noted only in patients who received sedating antiemetics. Parotid gland pain was common, and bilateral parotid swelling was noted in 29% of patients. Marked lacrimation was noted in 6% of patients, whereas 16% of patients experienced ocular dryness. Two patients experienced mild conjunctival edema. Hypertension was noted only during TBI. The results of a similar study conducted by Buchali and colleagues of patients who were treated with a fractionated course of TBI delivered mostly to a total dose of 12 Gy using 2 Gy per fraction, twice daily, 8 hours apart, with lung doses limited to 10 Gy, is also summarized in Table 15-1.87
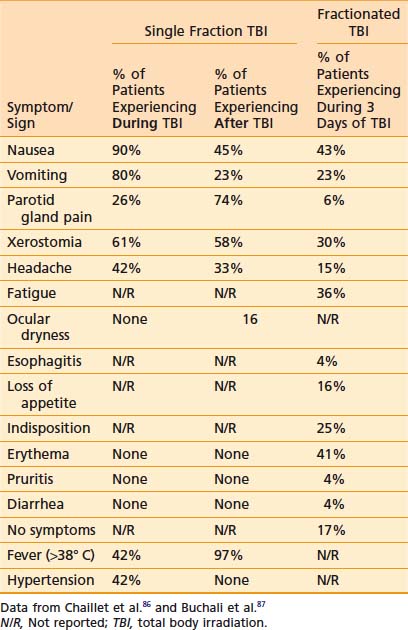
A prospective clinical study showed that fractionation of TBI can reduce acute nausea, vomiting, mucositis, diarrhea, and parotitis, although these differences were not statistically significant. Late cutaneous eruptions were more common in patients undergoing fractionated TBI, although this was not statistically significant. This same study, which randomized patients to high- or low-dose-rate TBI, revealed no differences in the acute toxicities mentioned when comparing dose rate.88 Another randomized controlled trial (RCT) reported that fractionating TBI revealed “no apparent difference in acute toxicity” compared with single-fraction TBI, with both regimens being “well-tolerated.”89
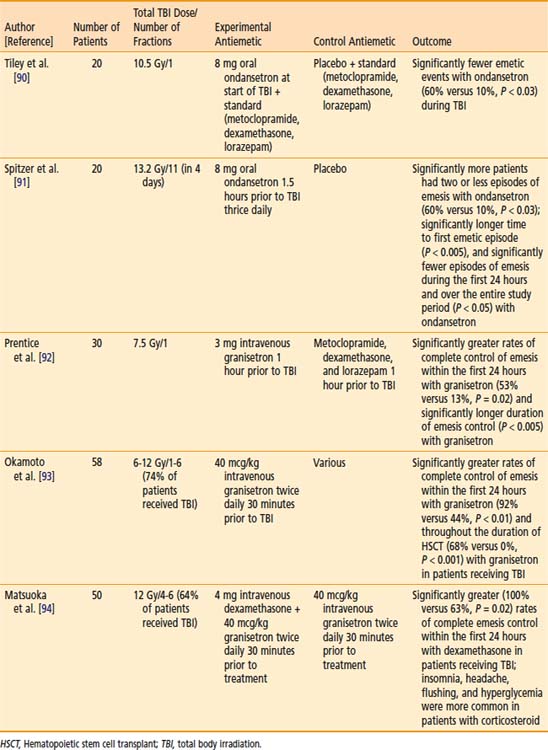
Older studies cite nausea and vomiting as frequent side effects of TBI. These symptoms have been substantially minimized with the advent of more effective antiemetics, such as the 5-hydroxytryptamine (serotonin) receptor-3 (5HT-3) antagonists. Several small, high-quality controlled clinical studies support the prophylactic use of 5HT-3 antagonists to reduce nausea and vomiting during TBI, and are summarized in Table 15-2.90–94 The use of corticosteroids in conjunction with 5-HT3 antagonists is supported by a trial listed in Table 15-2; however, given the toxicity associated with this approach, consensus regarding routine administration in conjunction with TBI is lacking.95–97 Of note, less nausea and vomiting has been noted in myeloablative conditioning regimens involving TBI, compared with those which use chemotherapy alone, even with modern antiemetics.98
Table 15-2 Randomized Controlled Trials of Prophylactic Antiemetics in Patients Undergoing Total Body Irradiation
Oral mucositis is a side effect of TBI in up to 75% of patients undergoing myeloablative TBI, causing mouth pain, odynophagia, necessitating intensive supportive care such as total parenteral nutrition and opioid analgesics.99 In one study, intensive dental hygiene conferred a reduction in the rate of moderate and severe mucositis, although the authors felt it to be clinically insignificant.100 Topical oral agents, such as chlorhexidine digluconate and neutral calcium phosphate in conjunction with topical fluoride treatments, can decrease the duration and severity of oral mucositis as well as pain and the need for opioid analgesics.101–103 Similarly, prophylactic oral sucralfate and clarithromycin have reduced moderate and severe oral mucositis rates.104,105 When given prophylactically, one study showed that amifostine limited the duration of mucositis, with an associated decrease in the rate of moderate and severe infections, with no effect on HSCT outcome.106 One study noted that short-term intravenous recombinant granulocyte-macrophage–colony stimulating factor decreased rates of moderate to severe mucositis,107 whereas another found no effect when this agent was delivered topically.108 Recently, Spielberger et al. reported the results of a trial of the recombinant human keratinocyte growth factor, palifermin, given before and after conditioning with 12 Gy of fractionated TBI. Palifermin reduced the rate and duration of moderate and severe mucositis by 35% and 3 days, respectively, and decreased mouth and throat pain, as reflected in reduced morphine usage and decreased the need for total parenteral nutrition (by 24%).109 This study dealt only with patients undergoing autologous HSCT; however, in the setting of TBI for allogeneic HSCT, palifermin may also confer a protective effect on the mucosa, although this has not been studied in an RCT.110
Skin erythema may also be noted toward the end of a course of TBI; desquamation is rare. Hyperpigmentation may be noted in the long term. Alopecia typically occurs 7 to 14 days after TBI is complete, and hair typically returns 3 to 6 months after treatment.111 Changes in the color or texture of regrown hair have been noted by some. Of note, myeloablative conditioning regimens using chemotherapy alone have noted a significantly higher incidence of permanent alopecia.112
Later Toxicity and Management
As previously mentioned (see “Radiobiology”), the hematopoietic system is particularly sensitive to TBI, and lymphopenia is often seen with doses of 0.5 Gy, and can be seen with doses of 0.3 Gy. Lymphopenia is typically followed by neutropenia, thrombocytopenia, and finally anemia. Soon after a TBI dose of 4 to 6 Gy, lymphocytosis can be seen, but typically is followed by neutropenia within 1 week. Neutrophils fall to their minimum 3 to 4 weeks after TBI (Fig. 15-3).113 Regeneration of the HSC compartment depends on the total dose used, because higher doses cause more rapid myelosuppression, of greater duration. Administration of hematopoietic growth factors after TBI have the theoretical potential to alter hematopoietic system reconstitution, although reports in the setting of allogeneic HSCT have demonstrated an increased risk of GVHD and compromised survival,114 and therefore routine use is controversial.115,116 Of note, hematopoietic growth factors have only been used in the post-TBI period, given the concerns raised by a trial in lung cancer, in which growth factors increased pulmonary toxicity and thrombocytopenia when given concurrent with chemoradiation therapy.117
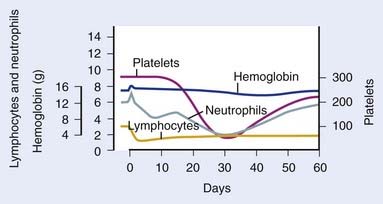
FIGURE 15-3 • Representative hematologic response to total body irradiation, given as a single fraction of 200 cGy on day 0.
(From Andrews GA: Radiation accidents and their management, Radiat Res Suppl 7:390–397, 1967.)
As noted previously, salivary glands frequently are affected by TBI. Although acute parotitis is typically self-limited and can be managed with anti-inflammatory medicines, long-term salivary gland dysfunction can result in xerostomia, which may lead to dental caries. In a study of children who underwent allogeneic HSCT, the risk of developing impaired salivary function was 22% in those who receive TBI as part of conditioning versus 1% in those who did not.118 Studies have shown that salivary flow can improve up to 1 year after the completion of TBI.119 Fractionating TBI was shown to reduce salivary dysfunction by 54% in one study.120 Myeloablative conditioning regimens with and without TBI have been associated with tooth development abnormalities in children.120–122 In one series, myeloablative conditioning regimens using chemotherapy alone were associated with significantly higher rates of tooth developmental abnormalities than those involving TBI, although rates of salivary gland dysfunction were highest amongst the patients treated with single-fraction TBI.123 Because of the increased risk of oral pathologic conditions associated with TBI, careful pretransplant evaluation by a dental specialist is recommended to minimize the risk of serious morbidity.124 Pilocarpine has been noted to help relieve symptoms of xerostomia in patients treated with TBI.125
The major dose-limiting toxicity of TBI is pneumonopathy, which can manifest early as pneumonitis and later as pulmonary fibrosis. In the setting of HSCT, radiation pneumonopathy can be difficult to distinguish from other causes of pathologic conditions of the lung; moreover, lung damage is likely multifactorial, with risk of acute lung complications estimated to be 30% to 60%, depending on factors like infection, conditioning regimen, GVHD, age, and diagnosis (see Peters and Afessa126 for review). Likewise, late pneumopathy occurs in 10% to 26% of patients, and is associated with underlying lung dysfunction, type of conditioning regimen, acute and chronic GVHD and prophylaxis, donor and recipient age and immunocompatibility, stage of disease, and genetic predisposition (see Patriarca et al.127 for review). TBI has been shown to be a risk factor for the idiopathic pneumonia syndrome128 as well as diffuse alveolar hemorrhage.129 Although rates of pneumonopathy in patients receiving TBI vary widely (10%-84%),130 some series have reported pneumonitis in up to 20% of HSCT patients who never received TBI.131 In the modern era, with appropriate TBI techniques, the risk of pneumonopathy in patients treated with TBI may not be increased at all.132 Nevertheless, the significance of the problem is clear, given that mortality related to interstitial pneumonitis in patients treated with TBI can be 60% to 80%.131,133,134
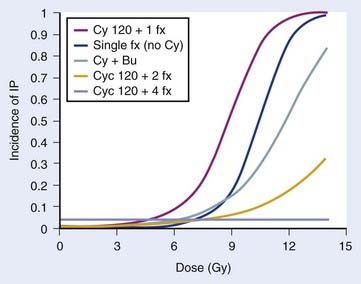
Several TBI-specific factors (total dose, fractionation, dose rate, and the use of lung shielding) have been shown to have significant bearing on the development of pulmonary complications. The total dose used during TBI has frequently been cited as a major factor influencing lung complications.130,135,136 In two prospective RCTs using 12 Gy versus 15.75 Gy, higher rates of mortality were noted within the first 6 months in patients treated with 15.75 Gy, although pulmonary complications were not specifically cited as the excess cause of deaths.137–140 In a retrospective dosimetric study, mean lung dose larger than 9.4 Gy was found to be an independent predictor for lethal pulmonary complications in patients receiving TBI to a total dose of 10 Gy in three daily fractions, at 0.055 Gy per minute using parallel opposed lateral fields.141 Two RCTs have demonstrated that fractionated TBI can reduce pneumonitis, compared with single-fraction TBI, although only one study was statistically significant.89,142,143 A retrospective study found no difference in pneumonitis rates when comparing a single fraction of 6 Gy and three daily fractions of 3.33 Gy, suggesting that total doses smaller than 10 Gy may not require fractionation to prevent toxicity, although no randomized data support this.144 The necessity of hyperfractionation to prevent lung toxicity is unclear: a comparison of two prospective single-arm trials at the same institution revealed that conventional fractionation given with AP fields and lung blocks to a total dose of 12 Gy in daily 3-Gy fractions may not be any different than HTBI given twice daily with 1.7 Gy per fraction to a total dose of 10.2 Gy for 3 days, using parallel opposed lateral fields and no blocks.145 Sampath et al. recently reviewed 26 studies involving 1096 patients to create a dose-response model to predict risk of pneumonitis from TBI, taking other factors into consideration. Although unable to estimate the risk of pneumonitis for hyperfractionated regimens, they were able to determine the effect of fractionation and cyclophosphamide (CY) and busulfan (Bu) on the risk of developing pneumonitis, in a dose-response model, as seen in Fig. 15-4.146 Pneumonitis rates were not significantly different in a trial that randomized patients to high- or low-dose-rate TBI.88 However, there is an abundance of retrospective clinical data suggesting that lowering the dose rate (<0.025-0.09 Gy/min) does decrease the likelihood of pulmonary complications,130,136,140,147,148 especially if TBI is delivered as a single fraction.149 If TBI is fractionated, some report that low dose rate (<0.069 Gy/min) is not necessary,134 although others found a beneficial effect.150,151 Studies of patients receiving fractionated TBI with lung shielding have demonstrated a reduction in pneumonopathy152,153; however, one RCT found no difference in pneumonopathy if the shielding allowed a lung dose of either 6 or 8 Gy in a single fraction.154
Pulmonary function tests (PFTs; spirometry and diffusion capacity) are often helpful in the assessment of patients with pulmonary symptoms or radiographic abnormalities. Studies of PFTs in patients treated with HSCT have demonstrated a deleterious effect on spirometry and diffusion capacity, which often resolves in the absence of other complicating factors,155–157 and is related to TBI dose.158 A retrospective study found that lung shielding had a small but significantly beneficial effect on PFTs one year after HSCT, especially in patients with abnormal function prior to HSCT.159 In one study, Bu, but not TBI, was not associated with a negative effect on PFTs.160 There is no evidence that PFTs improve pulmonary outcome after HSCT in adulthood, and for this reason, they are not recommended.161 However, some groups recommend baseline PFTs as part of long-term follow-up care for children treated with TBI.162 Counseling patients regarding smoking cessation is of critical importance in all patients, especially those who are at increased risk of developing lung injury. In the event of acute pneumonitis, high-dose steroids (30-60 mg of prednisone/day) typically alleviate symptoms within 24 to 48 hours.
Cardiovascular toxicity as a result of HSCT has been relatively rare, given the stringent selection criteria for patients treated with this aggressive therapeutic modality. Nevertheless, among patients who survive HSCT, cardiac events were responsible for 2.4% and 3.0% of deaths in patients that underwent autologous and allogeneic HSCT, respectively, which represents a greater than expected occurrence in some patients.163,164 Most of the recent literature has identified no association between TBI and development of cardiovascular disease.165,166 Several detailed prospective analyses using plasma cardiac troponin and brain natriuretic peptide levels, electrocardiography and echocardiography revealed no evidence of cardiac dysfunction in previously healthy individuals treated with TBI.167,168 However, a prospective study of children who underwent allogeneic HSCT found a 12% and 26% cumulative incidence of abnormalities in ejection fraction (<30%) on echocardiography prior to and five years after HSCT, respectively. This study revealed that TBI was associated with abnormalities in cardiac function on univariate analysis, but not multivariate analysis; the five-year cumulative incidence of cardiac abnormality was 26% in children treated with TBI with prior anthracycline therapy and 2% in children treated with TBI without prior anthracycline therapy.169 For these reasons, it appears prudent to screen patients who have undergone HSCT for cardiovascular disease or risk factors, whether or not this involved TBI, to minimize late morbidity and mortality.
Hepatotoxicity from TBI manifests primarily as veno-occlusive disease (VOD), also known as sinusoidal obstructive syndrome, of the liver. This clinicopathologic phenomenon was first described in 1977 by Shulman et al.,170 who noted the onset of weight gain from ascites, painful hepatomegaly, and jaundice from centrilobular liver acinus necrosis 1 to 4 weeks after HSCT. Overall, up to 70% of patients that undergo HSCT can be affected by VOD. TBI, along with many other risk factors, has been implicated in the development of VOD (see Wadleigh et al.171 for review). Notably, a RCT and meta-analysis found that patients treated with Bu instead of TBI were significantly more likely to develop VOD.172,173 Two RCTs have concluded that fractionated TBI reduces the incidence of VOD compared with single-dose TBI.142,143 Another RCT found no difference in the rate of VOD when the dose rate was either 0.06 Gy per minute or 0.15 Gy per minute,88 although a retrospective study of single-dose TBI found that dose rates of 0.07 Gy per minute were associated with less VOD than dose rates of 0.18 to 1.2 Gy per minute.174 TBI dose greater than 13.2 Gy has been reported to be associated with higher rates of VOD on univariate analysis,175 although dose greater than 12 Gy was not associated with VOD in another retrospective study.176 Lawton et al. reported a non–statistically significant 10% decrease in the rate of fatal VOD in patients treated with TBI as part of HSCT, when a 10% attenuation liver block was employed.177 Ursodeoxycholic acid was effective in preventing VOD in a RCT of patients undergoing TBI followed by HSCT,178 although another RCT did not support this finding.179 Reduced-intensity conditioning regimens may also prevent VOD. Treatment of VOD can include the fibrinolytic antithrombotic agent, defibrotide; decompressing the sinusoids by a transjugular intrahepatic portosystemic shunt and liver transplantation are more invasive options for management of severe disease.180
Cataracts are one of the most common complications of TBI. Patients may present with painless vision loss, and may be noted to have opacification of the lens on examination. In one series of patients treated with TBI in one or two fractions, severe visual impairment was noted in approximately half of patients.181 This problem has been noted to arise in a large proportion of patients treated with TBI, depending on total dose, fractionation, and dose-rate used. When considering risk factors associated with HSCT, steroid use,182–184 prior cranial irradiation,185,186 and the development of GVHD184 have been shown to predispose to cataractogenesis, whereas heparin use appears to be protective.187 Delivering TBI in a single fraction is the single biggest risk factor for developing a cataract after HSCT.187–192 High dose rate (>0.035-0.048 Gy/min) TBI also appears to increase the risk of cataract formation.185,187,190,193 A prospective study that randomized patients to high- or low-dose-rate TBI found that the incidence of cataract 5 years after treatment was 12% and 34% in the low- and high-dose-rate arms, with 13% and 39% of the cataracts occurring in patients who received fractionated or single-dose TBI, respectively.194 Kal and van Kempen-Harteveld recently reviewed the subject of TBI-induced cataracts and concluded that a biologically equivalent dose of 40 Gy yields a 10% chance of developing cataracts, using linear-quadratic modeling that included corrections for dose rate, with an α/β of 0.65 for late effects on the lens. On the basis of this, they suggest considering lens shielding for single-fraction TBI regimens,195 although this is controversial in the setting of malignant disease.196 Given the frequency of cataracts occurring after TBI, patients should be monitored for the development of this complication.197 Management of cataracts that impair vision or degrade quality of life may involve phacoemulsification or extraction; recent data suggest these procedures are safe, with an adverse event rate of 0.1% for experienced surgeons,198 and effective, with a 90% chance of 20/40 vision postoperatively.199
Kidney dysfunction occurs in approximately 17% of survivors of HSCT,200 and can manifest in a number of ways, with the most- to least-frequent syndromes being idiopathic chronic kidney disease, nephrotic syndrome, thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome), and acute renal failure (see Hingorani201 for review). The syndrome best associated with TBI is thrombotic microangiopathy, which can manifest as nephritis, hypertension, proteinuria, or anemia 6 to 12 months after HSCT. HSCT-related risk factors for nephropathy can include GVHD, infections with CMV or BK virus, nephrotoxic medications like cytotoxic chemotherapy (cytarabine, CY, ifosfamide, cisplatin, retinoic acid, carmustine, actinomycin D, melphalan), antibiotics (acyclovir, ganciclovir, foscarnet, vancomycin, amphotericin, aminoglycosides), and immunosuppressants (cyclosporine, tacrolimus, methotrexate). It is worth noting that the larger and more contemporary studies of chronic kidney disease after HSCT, have not demonstrated an association with TBI.202–206 Total dose has been implicated as the most important in predicting renal morbidity from TBI; a retrospective study found that GVHD and high dose (13.5 Gy) was associated with elevated serum creatinine.207 A prospective evaluation of renal function using radioisotopes found that early nephropathy was associated with age younger than 40 years, use of kidney blocks (possibly related to nephrotoxic contrast media given during simulation), and nephrotoxic drug use, while late nephropathy was associated with nephrotoxic drug used, but not TBI dose.208 The benefit of kidney shielding has been assessed in two retrospective studies, which both demonstrated significant improvement in long-term kidney function, with no evidence of dysfunction when the hyperfractionated dose was limited to 9.8 to 10 Gy.209,210 Two recent dose-effect modeling studies have demonstrated that nephropathy is unlikely after a biologically equivalent total dose of 16 Gy (calculated using a linear quadratic model, with corrections for dose rate and an α/β of 2.5 Gy) and that fractionating TBI and delivering a low dose rate (<0.10 Gy/min) prevent kidney dysfunction.195,211 Monitoring blood chemistry and counts, as well as blood pressure and urine studies are advised given the prevalence of kidney disease after HSCT. Although there are no proven therapies to treat HSCT-related nephropathy, medical therapies (antihypertensives, corticosteroids), plasma exchange, hemodialysis and renal transplant, and are possible options for management (see Hingorani201 for review). The angiotensin-converting enzyme inhibitor captopril is known to prevent chronic nephropathy from diabetes212 and was been hypothesized to reduce the risk of radiation nephropathy from TBI in preclinical studies.213,214 A small RCT of captopril to mitigate chronic renal failure in patients undergoing 12 Gy of fractionated TBI (with kidney dose of 9.8 Gy) prior to HSCT revealed less nephropathy; however, this was not statistically significant.215
Primary hypothyroidism is the most common endocrinopathy following TBI with an incidence of approximately 25% after fractionated therapy.216 The incidence is significantly higher in series of single-dose TBI and is known to increase with longer follow-up. It has been suggested that adults have a lower risk of hypothyroidism than children and that some patients develop only transient hypothyroidism. This condition is easily managed with thyroid hormone replacement. Following TBI, central hypothyroidism caused by thyrotropin (thyroid-stimulating hormone) deficiency is rare, as is deficiency of corticotropin (adrenocorticotrophic hormone), and hypogonadotropic hypogonadism.
Impaired growth and loss of adult height has been described as a consequence of TBI during childhood. This effect is multifactorial because radiation can cause poor nutritional conditions, skeletal dysplasia, and also temporary or permanent growth hormone (GH) deficiency. The effects of TBI on growth are inversely related to patient age at the time of treatment. Both the total dose and dose per fraction of TBI have shown to be related to the risk of growth impairment. These factors are also important when considering GH replacement for children proven to have deficiency. Sanders et al. observed that GH replacement was not effective after single-dose TBI or when TBI was delivered after age 10 years.217
Typical TBI regimens allow preservation of testicular Leydig cell function and therefore normal testosterone levels. However, almost all males will develop azoospermia as a result of germ cell irradiation during TBI. Although male patients are universally counseled to expect infertility, there are rare reports of spermatogenic activity in men many years after exposure to TBI during childhood. Age plays an even more important role in female fertility following TBI. Although approximately half of prepubescent girls who receive TBI will undergo spontaneous puberty, many will experience premature menopause in their twenties or thirties.218 Ovarian failure is expected in virtually all females receiving TBI after age 10 years. For the small percentage of women who become pregnant after TBI, some investigators have reported an increased risk for spontaneous abortion, whereas others do not.
Other endocrine and metabolic complications of HSCT include insulin resistance, lipid disorders and hypertension. Both type 1 and type 2 diabetes mellitus are associated with TBI.219The possible mechanisms involved are active areas of investigation.
Declines in neuropsychiatric function following TBI have been observed, but are subtle because of relatively low doses and modern fractionation. A recent prospective study at St. Jude tracked 158 children who had undergone HSCT, of whom 80 had received full-dose fractionated TBI. Patients were given a battery of neurocognitive examinations at 1, 3, and 5 years after transplant. Although TBI was associated with a 3-point decline in IQ compared with non-TBI patients, this difference was small compared with the 20 point difference observed between children from high and low socioeconomic groups.220
Beyond organ-specific toxicities, the risk of developing SMNs is increased in patients that have undergone HSCT. Typically, three groups of SMNs after HSCT are described, and follow a distinct pattern of development (Fig. 15-5):221 MDS and AML, post-transplant lymphoproliferative disorder (PTLD), and solid tumors.222 There are multiple risk factors that may predispose to development of a SMN after HSCT, including but not limited to genetic aberrations, pre-HSCT therapy, conditioning regimen, graft source and processing, post-transplant immunosuppression, and GVHD (for review see DeVita et al.38); only the association of TBI with SMNs is discussed here.
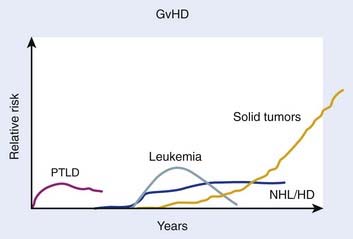
Secondary MDS or AML after HSCT has been reported to occur at rates between 1.1% at 20 months to 24.3% at 43 months. The World Health Organization classifies secondary MDS and AML as alkylating agent– or radiation-related, typically occurring 4 to 7 years after treatment, or topoisomerase-II inhibitor–related, typically occurring 6 months to 5 years after treatment.223 Several studies have attempted to quantify the risk of developing secondary MDS or AML after undergoing TBI, with conflicting results. Studies from research groups from Minnesota,224 Newcastle,225 the City of Hope,226 and France227 have demonstrated no increase in rates of MDS or AML after myeloablative HSCT using TBI. Other studies from Nebraska,228 the European Group for Bone Marrow Transplantation (EBMT),229 Paris,230 Barcelona,231 and France232 have found weak associations of borderline or no statistical significance. One study found an association of secondary MDS or AML with TBI on multivariate analysis when TBI was used with etoposide and CY.233 One of the more recent and detailed studies that investigated this subject found that, among 4000 patients treated with autologous HSCT for lymphoma, 57 developed MDS or AML, with a cumulative incidence rate of 3.7% 7 years after HSCT. A case-control analysis revealed a weak association of TBI and the subsequent development of MDS or AML, which was not statistically significant. In a small, subgroup analysis, a TBI dose of 13.2 Gy was associated with MDS or AML, but the authors cautioned that this group of patients may represent a population that shared other, non-TBI related factors that could have contributed to leukemogenesis. Older age also appeared to increase the risk.234 The dose threshold effect was not supported by a more recent study that found no differences in the rate of MDS or AML after TBI whether 12 or 14 Gy of TBI was given.235
The association of TBI with the subsequent development of a PTLD has been described in several studies. Because it is thought to be a consequence of immune system dysfunction, PTLD occurs primarily in patients that have undergone allogeneic, not autologous, HSCT. The cumulative incidence of PTLD at 12 years varies by the number of risk factors harbored by the patient, and ranged from 0.2 to 8.1%.236 Small, early studies from Seattle222 and the CIBMTR237 suggested an association between TBI use during HSCT and subsequent development of a PTLD. However, subsequent research from Minnesota,224 Vancouver,238 and Nebraska239 suggested no association of TBI and PTLD. A follow-up study from the CIBMTR, the largest and most comprehensive study available, involving 26,000 patients who underwent allogeneic HSCT over a 30-year period clearly demonstrated that no association between PTLD and TBI exists, in contrast to their previous findings.236
The risk of solid tumors after HSCT involving TBI has been studied extensively.240–250 The largest, and most recently updated analysis from the CIMBTR suggests that among patients who undergo allogeneic HSCT, the cumulative incidence of developing a solid malignancy (including non-skin carcinoma in situ carcinomas) is 1%, 2.2%, and 3.3% at 10, 15, and 20 years after transplant, based on the competing risk analysis method.251 The number of secondary solid tumors was double what would have been expected in this population; significantly higher rates of oral cavity and pharyngeal, liver, central nervous system (CNS), thyroid, bone, soft tissue cancers, and melanomas were observed. TBI was found to be a risk factor for developing a secondary solid tumor, with risk increased in those who survived more than 5 years from the HSCT, and those less than 30 years old at the time of HSCT. Although an earlier study found that TBI doses less than 10 Gy were associated with fewer secondary solid tumors,240 this dose association was not maintained in the subsequent analysis.251 There was no difference in secondary solid tumors in patients treated with fractionated or single-dose TBI. There was no increased risk of squamous cell carcinomas in patients treated with TBI. There was no increased risk of secondary solid tumors in patients treated with TBI after age 30. Chronic GVHD and male sex were also associated with the development of secondary solid tumors and squamous cell carcinoma, respectively.251
Given the increased risk of second malignant neoplasms after TBI, careful surveillance of long-term survivors is a desirable (but not yet proven effective) strategy to minimize secondary morbidity and mortality. Guidelines published by the American Cancer Society,252 the NCCN (www.nccn.org), and the Children’s Oncology Group162 provide important references when assessing patients at higher risk of developing a secondary solid tumor.
Physical Principles
A report in 1980 highlighted the fact that different institutions carry out TBI in a variety of ways.253 In fact, the physics of TBI has not changed significantly since 1986 when the American Association of Physicists in Medicine published “The Physical Aspects of Total and Half Body Photon Irradiation.”254 The data, formulas, and recommendations in this comprehensive report are still relevant today.
Geometry
The most common TBI technique, developed at Memorial Sloan-Kettering Cancer Center (MSKCC), uses opposing AP-posteroanterior (PA) fields with the patient standing several meters from the source and the beam pointed horizontally.255 AP-PA fields delivered using higher linac energies are recommended to optimize dose uniformity.254 A beam spoiler placed upstream from the patient’s surface showers the skin with electrons to reduce skin sparing. A TBI stand developed at MSKCC and commercially available provides support and comfort for the patient and reproducibility of setup; an acrylic scatter spoiler positioned upstream is available as an option. The stand also provides facility for lung blocks, film verification, and in vivo dosimetry (Fig. 15-6).
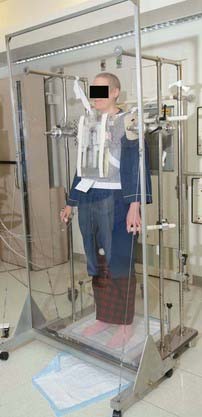
FIGURE 15-6 • Patient in position for total body irradiation using anteroposterior beam arrangement, with lung blocks in place.
Although the upright patient position is preferred to achieve full body coverage, this is not practical for all patients. Young children may not be able to stand and may require anesthesia for an accurate and reproducible treatment. For this group of patients, the standard geometry is lying on the floor with the gantry pointing downward. Other features of TBI (i.e., spoiler and blocks) are maintained. For taller children and adults who cannot tolerate the standing position, treatment on the floor is an option with abutting fields. Similarly, in some clinics where a large treatment room is unavailable, combinations of gantry or patient motion, possibly involving matching fields, are used.254,256
Alternatively, patients can be irradiated using right and left lateral fields.257 This technique has the advantage that the treatment distance can be reduced with the patient treated in a sitting position or partly reclining position on a stretcher. As the body part thickness variation is much greater for lateral fields than for the AP-PA fields, compensators attached to the head of the machine are used to improve the uniformity, especially for lower energies (≤6 MV). Arm positions are important to achieve the proper lung dose through self-shielding.
Planning
Electron boosts compensate the anterior and posterior chest walls for the blocked photons. Electron energy and surface bolus is based on CT planning of the chest wall-lung region such that the interface is 80% to 95% of Dmax, and the mid-lung dose is limited to 10 Gy for the photon-electron composite. A typical electron boost dose distribution is shown in Fig. 15-7.
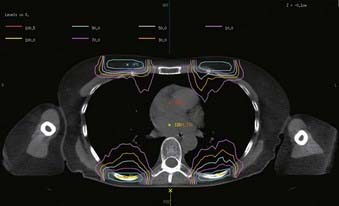
Treatment
A variety of TBI protocols are followed at MSKCC. The total dose is prescribed to midplane pelvis.
Commissioning and Quality Assurance
For commissioning, output (cGy/MU at Dmax) and tissue maximum ratio (TMR) are measured in a large water tank at 440 cm (220 cm for pediatric patients) for a 40 × 40 cm2 collimator size with the spoiler in place. Field flatness is verified over the area that will encompass the patient. Routine quality assurance checks of the output in both the adult and pediatric TBI geometries are performed monthly in a 25 cm3 polystyrene phantom. For adult TBI patients, a phantom factor of 1.009 corrects the output to that in a larger phantom based on published data.254
MU for the photon fields are then calculated as follows:
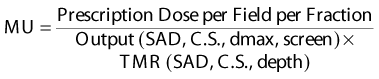
Role of Total Body Irradiation in Contemporary Hematopoietic Stem Cell Transplant
Necessity of Total Body Irradiation in Hematopoietic Stem Cell Conditioning
Although the earliest studies of HSCT for malignant hematopoietic diseases involved both chemotherapy and radiotherapy (see “History”), in the 1970s several non-TBI (chemotherapy only) regimens were developed, largely for logistical reasons—adequate facilities to perform TBI were not widely available.258 Soon thereafter, the availability of conditioning regimens for HSCT without TBI (primarily involving Bu and CY; see Santos259) gave rise to several important clinical studies that investigated the necessity of TBI in HSCT in various hematopoietic diseases.
Acute Myelogenous Leukemia
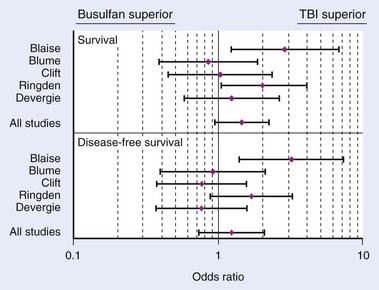
There have been two RCTs comparing TBI-based conditioning regimens to non-TBI–based conditioning regimens for AML. In 1987, the Groupe d’Etude des Greffes de Moelle Osseuse initiated a prospective, multicenter, RCT to evaluate whether TBI could be replaced by Bu in an effort to limit toxicity in allogeneic HSCT.260 A similar single-institution study was initiated at the University of Minnesota for patients receiving autologous HSCT, but accrual was poor and the study was terminated early.261 As noted in Table 15-3, superior overall survival, disease-free survival, survival, and fewer relapses were noted with the TBI conditioning regimen in both trials, with only the French trial demonstrating a statistically significant difference.260,262 An updated, combined analysis of three trials of allogeneic HSCT for AML that randomized patients to conditioning with or without TBI found marginally significant improvements in overall survival and disease-free survival. Patients treated with or without TBI had an estimated 10-year survival of 63% or 51% (P = 0.068), and 10-year disease free survival of 57% or 47% (P = 0.051). On multivariate analysis, TBI was associated with significantly less hair loss and no difference in the rates of GVHD, cataracts, avascular necrosis, or pulmonary complications.263 A retrospective analysis by the CIBMTR revealed that TBI-based conditioning yields fewer relapses (especially in extramedullary and CNS sites) and less hepatic VOD than Bu-based conditioning.264
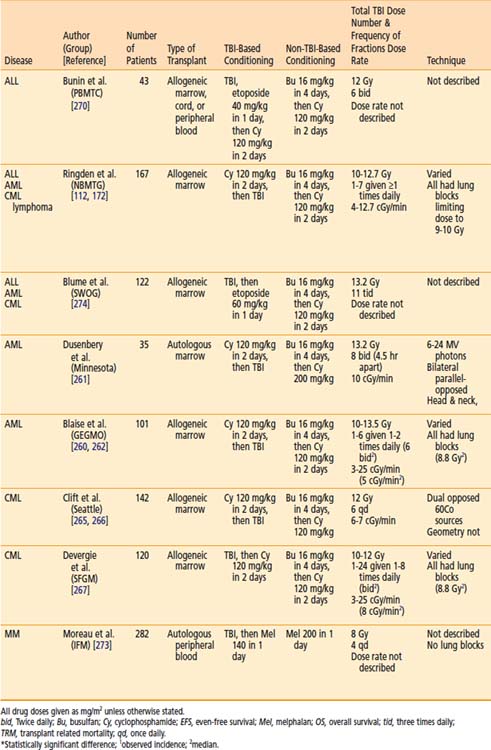
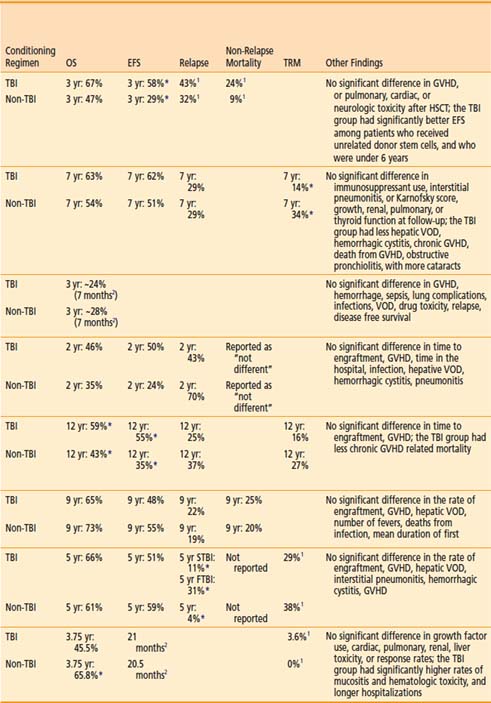
Chronic Myelogenous Leukemia
Two RCTs have compared the efficacy and toxicity of allogeneic HSCT in patients with CML, preceded by conditioning with either TBI and CY or Bu and CY (see Table 15-3).265–267 Both studies showed no significant difference in overall survival, event-free survival, or transplant-related mortality. The French study found more relapses in patients treated with TBI (especially fractionated TBI), but this was not seen in the study from Seattle, which used fractionated TBI exclusively. The Seattle trial did show significantly longer fevers, more blood cultures revealing bacteria or fungi, more hospitalizations in the 100 days after transplant, and higher incidence of grade 2 through 4 GVHD in patients treated with TBI. An updated, combined analysis of three studies that randomized patients with CML to different conditioning regimens prior to allogeneic HSCT confirmed no significant difference in survival (65% and 63%) and disease-free survival (52% and 42%) for patients treated with Bu-CY and CY-TBI, respectively. No differences in the 5-year cumulative incidence of chronic GVHD (37% and 39%). However, cataracts were more common among patients treated with TBI.263 Retrospective data also suggest that Bu-CY conditioning regimens prior to allogeneic HSCT for CML may be favored because of lower relapse rates,268 although higher rates of GVHD, hepatotoxicity, and hemorrhagic cystis have been reported with chemotherapy alone.269
Acute Lymphoid Leukemia
The Pediatric Blood and Marrow Transplant Consortium conducted the only RCT comparing the effect of preallogeneic HSCT conditioning using Bu or TBI, in children with ALL. A CNS boost of 6 Gy was given to patients with history of prior CNS disease, but no prior CNS irradiation if randomized to TBI; if randomized to Bu, patients with history of CNS disease received 18 Gy of CNS radiation therapy prior to Bu. The study showed no difference in overall survival; however, longer event-free survival was noted for patients treated with TBI. Patients 6 years old or younger, as well as those in first complete remission, appeared to experience the most benefit from the TBI regimen.270 A retrospective study using the CIBMTR data by Davies and colleagues of 627 children with ALL that underwent HSCT after conditioning with CY and TBI, or Bu and CY found that the risk of relapse was not significantly different in the two cohorts. However, the risk of treatment-related mortality was significantly greater in the Bu-CY group. The use of CY-TBI was associated with superior overall survival and leukemia-free survival. Based on this data, the authors concluded that less treatment-related mortality in the CY-TBI group led to greater overall survival and leukemia free survival.271 A multivariate analysis of adults with ALL who underwent HSCT preceded by conditioning with fractionated TBI to 12 Gy (lung dose of 9 Gy) or Bu found that shorter event-free survival and disease relapse were more likely among patients that did not receive TBI on multivariate analysis. There were no differences in the rates of transplant related mortality, hepatic VOD, or GVHD.272
Multiple Myeloma
The Intergroup Francophone du Myelome conducted a trial (9502) that randomized 399 patients with newly diagnosed MM to high-dose therapy in preparation for autologous HSCT after three cycles of vincristine-doxorubicin (Adriamycin)-dexamethasone chemotherapy. High-dose therapy consisting of melphalan 200 mg/m2 (HDM200) or melphalan 140 mg/m2 and 8 Gy of TBI delivered in four daily fractions without lung shielding (HDM140). Overall survival was longer in the HDM200 group (P = 0.05); however event-free survival was not different in the two groups. The authors speculated that the improved overall survival was attributed to the better available salvage regimens available after initial therapy with HDM200 on the protocol, and suggest that the new standard conditioning regimen for autologous HSCT for MM should be without TBI.273
Mixed Diseases
Two prospective trials of conditioning have been carried out in groups of patients with leukemias and lymphomas. As these trials included a heterogenous group of diseases, results should be interpreted with caution. Nevertheless, in 1988 the Nordic Bone Marrow Transplantation Group initiated a multicenter, RCT of 167 children (n = 27) and adults (n = 140) with AML (n = 68), ALL (n = 38), CML (n = 57), or lymphoma (n = 4) prior to allogeneic HSCT.172 With 7 years of follow-up, the authors found a survival benefit in the TBI group among patients with “advanced” disease, with less toxicity compared with Bu regimens; no differences were identified in the subset analysis of the different diseases that were treated.112 The Southwest Oncology Group’s RCT 8612 also investigated the role of TBI prior to allogeneic HSCT for ALL (n = 48), AML (n = 40), and CML (n = 34) not in the first remission. This regimen substituted etoposide for CY in the TBI group only, making a meaningful comparison difficult. Although overall survival and disease-free survival appeared superior for “good-risk” patients in the TBI group, these differences were not statistically significant.274
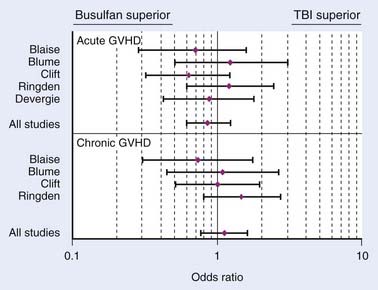
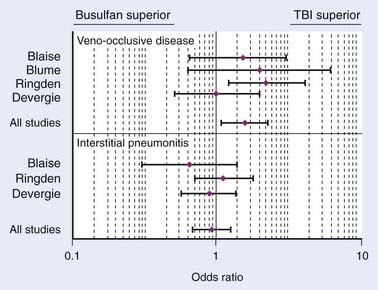
In conclusion, a meta-analysis by Hartman et al.173 summarizes how TBI influences the outcome of conditioning regimens prior to allogeneic HSCT.173 As demonstrated in Fig. 15-8, overall survival and disease-free survival are slightly better with TBI-based regimens, although this was not statistically significant. As noted in Fig. 15-9 and Fig. 15-10, GVHD was roughly equivalent in the studies, whereas hepatic VOD was clearly more common with Bu-based regimens. Interstitial pneumonitis appeared to also be very similar in both groups. In the setting of autologous HSCT, a non-TBI-based conditioning regimen is preferred for MM; however, no other trials have randomized patients to a TBI or non-TBI conditioning regimen prior to autologous HSCT, and therefore institutional preference primarily dictates use of the technique in these situations.
Total Body Irradiation Dose
The appropriate radiation dose to use during conditioning with TBI has been investigated by several groups. In 1985, researchers in Seattle conducted a RCT of two TBI regimens as part of HSCT for AML. Seventy-one patients were randomized to receive 12 Gy in six daily fractions (2 Gy/fraction/day), or to 15.75 Gy in seven daily fractions (2.25 Gy/fraction/day). TBI was delivered using opposed 60Co sources, at 0.06 to 0.07 Gy per minute. The use of shielding was not described. Higher-dose (15.75 Gy) TBI was associated with higher rates of mortality in patients that did not relapse, and higher rates of grade 2 through 4 acute GVHD (P = 0.02). Patients receiving higher-dose TBI were less likely to receive adequate immunosuppression. Nonetheless, patients treated with lower-dose TBI (12 Gy) appeared to be more likely to relapse, although this finding was marginally significant (P < 0.06).137 A report of the long-term results (minimum follow up of 7.5 years) of the trial revealed no difference in overall survival between the two TBI groups. Again, relapse was noted to be more common in patients with lower-dose TBI, with marginal statistical significance (P = 0.06). Nonrelapse mortality was again noted to be significantly higher in the higher-dose TBI group, although the authors note that this was limited to the first 6 months after transplant.139
In a similar trial, also from Seattle, 116 patients with CML were randomized to receive either 12 or 15.75 Gy, as described previously. Higher-dose (15.75 Gy) TBI was associated with a superior relapse-free survival. Higher-dose TBI was also associated with more grade 2 through 4 acute GVHD, although this was not statistically significant (P = 0.15). Initially, survival appeared superior in patients receiving lower-dose (12 Gy) TBI; however, further follow-up revealed no significant difference in survival. Similar to the experience with AML, the authors conclude that higher-dose TBI was associated with greater relapse-free survival at the cost of more acute GVHD, which likely led to transplant-related mortality that offset the survival benefit.138
Retrospective data do suggest a benefit to higher-dose (>12 Gy) TBI in certain clinical scenarios. Although the highest doses of TBI reported in the literature are 20 Gy prior to autologous HSCT,275 at least one study has reported lower relapse rates and less transplant-related mortality in a subset of patients with ALL who received a TBI dose larger than 13 Gy.276 Although modeling studies suggest that higher biologically effective doses are associated with lower relapse rates and longer overall survival,277 excess toxicity with higher doses in clinical dose-escalation studies does warrant caution.278–281 For these reasons, fractionated TBI doses used prior to myeloablative HSCT often range between 12 and 15 Gy.
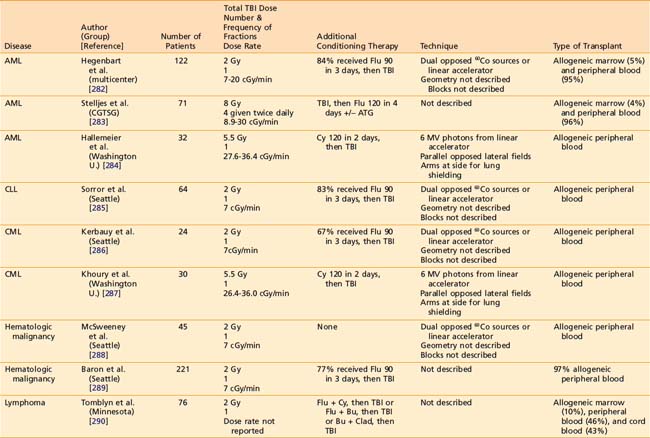
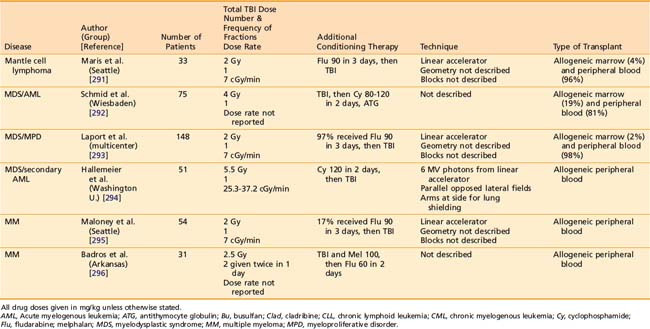
Reduced-intensity conditioning regimens have been used more often over the preceding decade for patients that might not be able to tolerate a myeloablative HSCT because of advanced age or comorbidities, but could benefit from the GVT effects of allogeneic HSCT. The role of TBI as part of reduced intensity regimens is ill-defined, as prospective RCTs of reduced intensity conditioning regimens involving TBI have not been completed. Likewise, the optimal dose of TBI as part of reduced intensity autologous HSCT has not been defined in an RCT. Several notable studies of allogeneic HSCT have employed TBI doses of 2 to 8 Gy, often in conjunction with chemotherapy.282–296 Table 15-4 lists some representative allogeneic HSCT regimens using low-dose TBI.
Fractionation and Total Body Irradiation Dose Rate
Laboratory-based radiobiological data (see “Radiobiology”) and clinical observations prompted trials investigating fractionation and dose-rate delivery in TBI. Thomas and colleagues were the first to conduct an RCT of single-dose or fractionated TBI in patients with AML. In this study, 53 patients with AML were randomized to receive TBI in a single 10 Gy dose, or 12 Gy in six daily fractions after receiving CY. TBI was delivered using opposed 60Co sources, at a rate of 80 R per minute in air at midpoint (≈0.06 Gy/min). Disease-free survival was significantly longer in the patients who were treated with fractionated TBI. There was no apparent difference in acute toxicity in the two treatment groups.89 A follow-up report also described the outcome of patients treated with the same transplant regimen prior to the RCT, but using a single fraction of 9.2 Gy using opposed 60Co sources at 0.04 Gy per minute. Among the patients randomized, those who received fractionated TBI survived significantly longer than those receiving single-fraction TBI. Patients receiving single-fraction TBI experienced twice as many leukemic recurrences, compared with those receiving fractionated TBI, although this was not statistically significant (P = 0.09). VOD of the liver was significantly more common in patients receiving single-fraction TBI (P = 0.02).142
Ozsahin et al.88 conducted a study investigating the effect of dose rate in patients undergoing single-dose or fractionated TBI as part of autologous or allogeneic HSCT for a variety of hematologic and lymphoid malignancies. In this trial, 157 patients receiving single-dose or multiple fractions of TBI were randomized to low dose rate (0.06 Gy/min or 0.03 Gy/min, respectively) or high dose rate (0.15 Gy/min or 0.06 Gy/min, respectively). Radiotherapy was delivered with 60Co or 6-MV photons from a linac. Single-dose TBI was delivered to a total dose of 10 Gy while limiting the lung dose to 8 Gy. Fractionated TBI was delivered to a total dose of 12 Gy in six fractions over 3 days, while limiting the lung dose to 9 Gy. A variety of chemotherapies were used. The authors found no significant differences in overall or relapse-free survival, pneumonitis, VOD, or GVHD. Cataracts were significantly more common among patients receiving single-fraction, high-dose-rate TBI.88 In a follow-up report of this study, with a median follow-up of 50 months, it was demonstrated that TBI in a single fraction or at a high-dose-rate was associated with the development of cataracts.194
Girinsky and colleagues conducted an RCT of 160 patients older than 15 years, undergoing autologous or allogeneic HSCT for a variety of hematologic and lymphoid malignancies. Patients were treated with a single fraction of TBI (STBI) with 10 Gy during 4 hours (average dose rate of 0.045 Gy/min; instantaneous dose rate of 0.125 Gy/min), or HTBI with 14.85 Gy in 11 fractions during 5 days at a dose rate of 0.25 Gy per minute. A linac was used to deliver 18-MV photons with lung shielding; patients receiving STBI received a lung dose of 8 Gy, and HTBI patients received a lung dose of 9 Gy. After TBI, CY or melphalan were given, followed by HSCT. With a median follow-up of 8 years in 147 assessable patients, overall survival and cause-specific survival were higher in the HTBI group; this was not statistically significant. However, a multivariate analysis that controlled for prognostic variables revealed greater cause-specific survival but not overall survival was longer in patients treated with HTBI (P = 0.04). There was no significant difference in the rates of lethal pneumonitis, hepatic VOD, GVHD, graft failure, infection, or organ failure. However, the rate of hepatic VOD was significantly higher in the STBI group.143
The necessity of hyperfractionation versus conventional fractionation has never been tested in a prospective trial. Investigators from M.D. Anderson Cancer Center compared the outcome of patients on different prospective HSCT protocols, and found that fewer recurrences occurred in the conventionally fractionated group. These results should be interpreted with caution, because this study was not randomized, and other factors not related to TBI may have influenced the results.145 Investigators in Seattle have conducted phase I and II studies of twice- and thrice-daily hyperfractionation protocols that did not suggest a significant benefit with hyperfractionation versus conventional fractionation when compared with historical results.280,297
Organ Shielding During Total Body Irradiation
The necessity of shielding to prevent normal tissue toxicity has been investigated in several studies; because pneumonopathy has been considered the dose-limiting toxicity of TBI, the studies have primarily been concerned with lung shielding. Labar et al.152 conducted a trial in 64 patients with AML, ALL, and CML undergoing TBI and CY conditioning. Patients were treated with 12 Gy of TBI in three fractions given once daily and were randomized to TBI with or without lung shielding, after stratifying by age group (<20, 21-30, 31-40, >40 years), sex, underlying disease, GVHD prophylaxis, and donor-recipient sex combination. TBI was delivered using a 60Co source, and blocks were used to limit the estimated lung dose to 9 Gy in the group of patients treated with lung shields. There was no difference in leukemia-free survival or rate of relapse. Although interstitial pneumonitis was more common in the group of patients treated without lung blocks (12%) compared with those treated with lung blocks (4%), this difference was not statistically significant.152
Girinsky et al.154 conducted a similar study in a group of 85 patients with various lymphoid or hematopoietic malignancies. Each patient received TBI with a dose of 10 Gy given in a single fraction during 4 hours (average dose rate 0.04 Gy/min, instantaneous dose rate 0.125 Gy/min). Patients were randomly assigned to receive a 6 or 8 Gy lung dose. The authors observed no differences in overall survival, or lung complications. However, chronic GVHD and infections were significantly less common in the low lung dose group. Interestingly, there were significantly more leukemia relapses in the low lung dose group. The authors speculated whether the additional lung shielding may have fostered survival of leukemic cells, which led to higher rates of relapse.154
Retrospective data support the benefit of shielding. Weshler et al.153 reported a series of 44 consecutive patients who received 12 Gy of TBI; the first 23 patients were treated with opposed lateral fields and had no lung shielding in place, whereas the subsequent 21 patients had half-value layer transmission block after 6 Gy. Pneumonitis occurred in 26% of patients without lung shielding, whereas no pneumonitis was noted in those who had lung shielding.153 Investigators from Wisconsin have demonstrated sparing of renal and hepatic function with shielding.177,209
Total Body Irradiation in Nonmalignant Diseases
Because radiotherapy is most commonly employed in the treatment of malignant diseases dealt with in other sections of this book, special attention is paid in this chapter to the role of TBI as part of HSCT for nonmalignant diseases. The basis of this role lies in preclinical studies, which have demonstrated that among the highest tolerable doses of TBI, CY or Bu, TBI was the most effective single conditioning agent in a rat model of autoimmune arthritis treated with pseudo-autologous HSCT.298 Although no RCT data exist to support the use of TBI in the conditioning regimen of patients undergoing autologous HSCT for nonmalignant, autoimmune conditions, several pilot studies have prompted the execution of phase III studies incorporating TBI.
Saccardi et al. summarized the results of phase I and II trials carried out by the EBMT using HSCT for multiple sclerosis (MS), which revealed stabilization or improvement of neurologic symptoms in 63% of patients who generally had severe or progressive disease.299 Collectively, TBI was part of the conditioning regimen in 16 of the 178 patients. Statistical analysis revealed that Bu, and not TBI, was associated with transplant-related mortality. Interestingly, Nash et al., studied 26 patients in a pilot study using high-dose myeloablative conditioning prior to autologous HSCT, including 8 Gy of TBI, and found that 3-year progression free survival was 73%, with 91% overall survival, with minimal toxicity.300 Concerns about the use of TBI in HSCT for MS were brought up by Samijin et al.301 and Burt et al.302 who speculated that radiotherapy may have contributed to axonal injury or caused dysfunction of neural precursor cells, although direct evidence of this is lacking. For these reasons, the ongoing RCT to compare autologous HSCT with mitoxantrone drug therapy for severe MS will not involve TBI (http://www.astims.org/). Similar concerns regarding the toxicity of radiotherapy in patients with inflammatory bowel disease has prompted investigators to omit TBI from ongoing RCTs of autologous HSCT in Crohn disease.303
Similar to MS and Crohn disease, autologous HSCT has been explored recently for patients with systemic sclerosis (SSc, scleroderma) in several pilot studies. Binks et al.304 reported the results of a phase I and II trial of autologous HSCT for poor prognosis SSc using a variety of conditioning regimens, some of which included TBI. In their initial report, two of nine patients who underwent 8 Gy of TBI died of interstitial pneumonitis 1 and 3 months after TBI.304 This prompted investigators to minimize TBI dose during further investigations of HSCT in this disease.305 After two of the first eight patients in a study by Nash et al. died of pneumonitis, these investigators shielded the lungs with five half-value layers after the first 2-Gy fraction.306 With shielding in place, no further TBI-related pulmonary mortality has occurred; however, two patients developed MDS and a former smoker was diagnosed with stage I lung cancer.307 Autologous HSCT for SSc is currently being studied in two RCTs; in the United States, the Scleroderma Cyclophosphamide or Transplantation (NCT00114530) trial will incorporate 8 Gy of TBI, whereas in the Autologous Stem Cell Transplantation International Scleroderma (http://www.astistrial.com) trial in Europe, TBI will not be used.
HSCT has been used successfully for management of a few of the varied genetically acquired metabolic diseases in children. Dramatic improvements after HSCT have been published for certain lysosomal storage diseases including Hurler syndrome and early cerebral adrenoleukodystrophy.308 Although typical TBI-containing regimens were used early in the experience, the trend is now for HSCT using umbilical cord blood without TBI in these very young children.309 Similarly, reduced-intensity allogeneic transplants without TBI have shown promise for children with disorders such as sickle cell disease, thalassemia, and other hemoglobinopathies as well as immunodeficiencies (SCID), and bone marrow failure syndromes.310 HSCT protocols for Fanconi anemia show great promise and continue to use TBI. Because the underlying disorder in this disease is characterized by an inability to repair sublethal DNA damage, patients are exquisitely sensitive to radiotherapy and do not derive benefits of fractionation. A prospective dose-escalation study revealed that no benefit was derived from 6 Gy; therefore, a low single dose of 4.5 Gy is the standard.311
1 Dessauer FJ. Beitrage zur Bestrahlung tiefliegender Prozesse. Medizinischen Klinik. 21, 1905.
2 Dessauer FJ. Eine neue Anordnung zur Rontgenbestrahlung. Archiv fur Physikalische Medizin und medizinische Technik. 1907;2:218-223.
3 Elfer A. Orvosi Hetilap. 1907;51:587.
4 Del Regato JA. Total-body irradiation in treatment of chronic lymphogenous leukemia—Janeway Lecture, 1973. Am J Roentgenol. 1974;120(3):504-520.
5 Teschendorf W. Uber Bestrahlungen des ganzen menshlichen Korpers bei Blutkrankenheiten. Strahlentherapie. 1925;26:720-728.
6 Sluys F. La roentgenisation totale dans la lymphogranulomatose maligne. J Belge de Radiol. 1930;19:297-317.
7 Sluys F. La roentgentherapie totale dans la lymphogranulomatose maligne. Scalpel. 1930;2(27):733-740.
8 Medinger FG, Craver LF. Total body irradiation with review of cases. Am J Roentgenol Radium Ther. 1942;48(5):651-671.
9 Heublein AC. A preliminary report on continuous irradiation of the entire body. Radiology. 1932;18(6):1051-1062.
10 Craver LF, MacComb WS. Heublein’s method of continuous irradiation of entire body for generalized neoplasms. Am J Roentgenol Radium Ther. 1934;32:654-674.
11 Craver LF. Tolerance to whole-body irradiation of patients with advanced cancer. In: Stone RS, editor. Industrial medicine on the plutonium project: survey and collected papers. New York: McGraw-Hill; 1951:485-498.
12 Nickson JJ. Blood changes in human beings following total-body irradiation. In: Stone RS, editor. Industrial medicine on the plutonium project: survey and collected papers. New York: McGraw-Hill; 1951:308-337.
13 Low-Beer BVA, Stone RS. Hematological studies on patients treated by total-body exposure to x-rays. In: Stone RS, editor. Industrial medicine on the plutonium project: survey and collected papers. New York: McGraw-Hill; 1951:338-418.
14 Miller LS, Fletcher GH, Gerstner HB. Systemic and clinical effects induced in 263 cancer patients by whole body x-irradiation with nominal air doses of 15 to 200 R. TX: Randolf AFB; 1957.
15 Miller LS, Fletcher GH, Gerstner HB. Radiobiologic observations on cancer patients treated with whole-body x-irradiation. Radiat Res. 1958;8(2):150-165.
16 Collins VP, Loeffler RK. The therapeutic use of single doses of total body radiation. Am J Roentgenol Radium Ther Nucl Med. 1956;75(3):542-547.
17 King ER. Use of total-body radiation in treatment of far-advanced malignancies. JAMA. 1961;177(9):610.
18 Saenger EL, et al. Whole-body and partial body radiotherapy of advanced cancer. Am J Roentgenol. 1973;117(3):670-685.
19 Advisory Committee on Human Radiation Experiments. Final Report, US Department of Energy. Washington, DC: U.S. Government Printing Office; 1995.
20 Thomas J. Rebuilding trust in established institutions. Health Phys. 2001;80(4):379-383.
21 Merrill JP, et al. Successful homotransplantation of the kidney between nonidentical twins. N Engl J Med. 1960;262(25):1251-1260.
22 Hamburger J, et al. Transplantation dun rein entre jumeaux non monozygotes—apres irradiation du receveur bon fonctionnement au quatrieme mois. Presse Medicale. 1959;67(48):1771-1775.
23 Hamburger J, et al. Renal homotransplantation in man after radiation of recipient—experience with 6 patients since 1959. Am J Med. 1962;32(6):854.
24 Murray JE, et al. Prolonged survival of human-kidney homografts by immunosuppressive drug therapy. N Engl J Med. 1963;268(24):1315.
25 Thomas ED, et al. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. 1957;257(11):491-496.
26 Thomas ED, Lochte HL, Ferrebee JW. Irradiation of the entire body and marrow transplantation—some observations and comments. Blood. 1959;14(1):1-23.
27 Thomas ED, et al. Supralethal whole body irradiation and isologous marrow transplantation in man. J Clin Invest. 1959;38(10):1709-1716.
28 Buckner CD, et al. Allogeneic marrow engraftment following whole body irradiation in a patient with leukemia. Blood. 1970;35(6):741.
29 Thomas ED, et al. Bone-marrow transplantation 1. N Engl J Med. 1975;292(16):832-843.
30 Thomas ED, et al. Bone-marrow transplantation 2. N Engl J Med. 1975;292(17):895-902.
31 Haas RL. Low dose radiotherapy in indolent lymphomas, enough is enough. Hematol Oncol. 2009;27(2):71-81.
32 Heinzelmann F, et al. Total-body irradiation—role and indications—results from the German Registry for Stem Cell Transplantation (DRST). Strahlentherapie Und Onkologie. 2006;182(4):222-230.
33 Deeg HJ. Transplantation conditioning regimens: can we say it better? Biology of Blood and Marrow Transplantation. 2009;15(6):653-655.
34 Giralt S, et al. Reduced-intensity conditioning regimen workshop: defining the dose spectrum. Report of a Workshop Convened by the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2009;15(3):367-369.
35 Bredeson C, et al. Outpatient total body irradiation as a component of a comprehensive outpatient transplant program. Bone Marrow Transplant. 2002;29(8):667-671.
36 Stiff P, et al. Autologous hematopoietic stem cell transplants that utilize total body irradiation can safely be carried out entirely on an outpatient basis. Bone Marrow Transplant. 2006;38(11):757-764.
37 Pasquini MCW, Wang Z, Schneider L. Current use and outcome of hematopoietic stem cell transplantation: Part I—CIBMTR Summary Slides, 2007. CIBMTR newsletter [serial online] School of Aviation Medicine, Air University, United States Air Force. 2007;13:5-9.
38 DeVita VT, Lawrence TS, Rosenberg SA. DeVita, Hellman, and Rosenberg’s Cancer: Principles & practice of Oncology, ed 8. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins; 2008.
39 Oringanje C, Nemecek E, Oniyangi O. Hematopoietic stem cell transplantation for children with sickle cell disease. Cochrane Database Syst Rev. 2009;1:1-11.
40 Michlitsch JG, Walters MC. Recent advances in bone marrow transplantation in hemoglobinopathies. Curr Mol Med. 2008;8(7):675-689.
41 Prasad V, Kurtzberg J. Emerging trends in transplantation of inherited metabolic diseases. Bone Marrow Transplant. 2008;41(2):99-108.
42 Testa NG, Hendry JH, Molineux G. Long-term bone-marrow damage in experimental systems and in patients after radiation or chemotherapy. Anticancer Res. 1985;5(1):101-110.
43 Uckun FM, Song CW. Radiobiological features of human pluripotent bone-marrow progenitor cells (Cfu-Gemm). Int J Radiat Oncol Biol Phys. 1989;17(5):1021-1025.
44 Vriesendorp HM, van Bekkum DW. Role of total body irradiation in conditioning for bone marrow transplantation. Vriesendorp HMavB, DW HJ, editors. Thierfelder S, Rodt H, Koldb. Haematology and blood transfusion. Springer Verlag. Berlin. 349.
45 Vriesendorp HM, van Bekkum DW. Susceptibility to total body irradiation. In: Broerse TJ, MacVittie TJ, editors. Response of different species to total body irradiation. Boston: Nijhoff; 1984:43.
46 Kwan DK, Norman A. Radiosensitivity of human lymphocytes and thymocytes. Radiat Res. 1977;69(1):143-151.
47 Szczylik C, Wiktorjedrzejczak W. The effects of x-irradiation in vitro on sub-populations of human-lymphocytes. Int J Radiat Biol. 1981;39(3):253-263.
48 Shank B, Andreeff M, Li D. Cell-survival kinetics in peripheral-blood and bone-marrow during total-body irradiation for marrow transplantation. Int J Radiat Oncol Biol Phys. 1983;9(11):1613-1623.
49 Wheldon TE. The radiobiological basis of total body irradiation. Br J Radiol. 1997;70(840):1204-1207.
50 Uckun FM, Aeppli D, Song CW. Radiation-resistance of primary clonogenic blasts from children with acute lymphoblastic-leukemia. Int J Radiat Oncol Biol Phys. 1993;27(4):899-906.
51 Steel GG, Wheldon TE. The radiation biology of paediatric tumors. In: Pinkerton PN, editor. Paediatric Oncology. London: John Wiley, 1991.
52 O’Donoghue JA. The implications of in-vitro radiation-survival curves for the optimal scheduling of total-body irradiation with bone marrow rescue in the treatment of leukaemia. Br J Radiol. 1987;60(711):279-283.
53 Song CW. Radiobiological basis of total body irradiation with different dose rate and fractionation: Repair capacity of hemopoietic cells. Int J Radiat Oncol Biol Phys. 1981;7(12):1695-1701.
54 Weichselbaum RR, et al. In vitro radiosensitivity of human-leukemia cell-lines. Radiology. 1981;139(2):485-487.
55 Rhee JG, et al. Effect of fractionation and rate of radiation-dose on human-leukemic cells, Hl-60. Radiat Res. 1985;101(3):519-527.
56 FitzGerald TJ. Effect of x-irradiation dose rate on the clonagenic survival of human and experimental animal hematopoietic tumor cell lines: evidence for heterogeneity. Int J Radiat Oncol Biol Phys. 1986;12(1):69-73.
57 FitzGerald TJ. Radiosensitivity of human bone marrow granulocyte-macrophage progenitor cells and stromal colony-forming cells: effect of dose rate. Radiat Res. 1986;107(2):205-215.
58 Lehnert S, et al. Radiation response of hematopoietic-cell lines of human-origin. Int J Radiat Biol. 1986;49(3):423-431.
59 Laver J. Effects of low dose rate irradiation on human marrow hematopoietic and microenvironmental cells: sparing effect upon survival of stromal and leukemic cells. Bone Marrow Transplant. 1987;2(3):271-278.
60 FitzGerald TJ. Radiosensitivity of permanent human bone marrow stromal cell lines: effect of dose rate. Int J Radiat Oncol Biol Phys. 1988;15(5):1153-1159.
61 FitzGerald TJ. Radiosensitivity of cloned permanent murine bone marrow stromal cell lines: nonuniform effect of low dose rate. Exp Hematol. 1988;16(10):820-826.
62 Cosset JM. Radiobiological and clinical bases for total body irradiation in the leukemias and lymphomas. Semin Radiat Oncol. 1995;5(4):301-315.
63 Peters L. Discussion: the radiobiological bases of TBI. Int J Radiat Oncol Biol Phys. 1980;6(6):785-787.
64 Peters LJ. Radiobiological considerations in the use of total-body irradiation for bone-marrow transplantation. Radiology. 1979;131(1):243-247.
65 Dutreix J. Time factors in total body irradiation. Pathologie Biologie. 1979;27(6):365-369.
66 Dutreix J. Biological problems of total body irradiation. Journal Européen de Radiothérapie. 1982;3(4):165-173.
67 Kimler BF, et al. Radiation response of human normal and leukemic hemopoietic cells assayed by in vitro colony formation. Int J Radiat Oncol Biol Phys. 1985;11(4):809-816.
68 Shank B. Hyperfractionation versus single-dose irradiation in human acute lymphocytic-leukemia cells—application to TBI for marrow transplantation. Radiother Oncol. 1993;27(1):30-35.
69 Uckun FM, et al. Intrinsic radiation-resistance of primary clonogenic blasts from children with newly diagnosed B-cell precursor acute lymphoblastic-leukemia. J Clin Invest. 1993;91(3):1044-1051.
70 Cosset JM, et al. Single-dose versus fractionated total-body irradiation before bone-marrow transplantation—radiobiological and clinical considerations. Int J Radiat Oncol Biol Phys. 1994;30(2):477-492.
71 Odell TT, et al. The homotransplantation of functional erythropoietic elements in the rat following total-body irradiation. Ann N Y Acad Sci. 1957;64(5):811-825.
72 Alpen EL, Baum SJ. Acute radiation protection of dogs by bone marrow autotransfusion. Radiat Res. 1957;7(3):298-299.
73 Crouch BG, Overman RR. Whole-body radiation protection in primates. Fed Proc. 1957;16(1):27.
74 Storb R, et al. Comparison of fractionated to single-dose total-body irradiation in conditioning canine littermates for Dla-identical marrow grafts. Blood. 1989;74(3):1139-1143.
75 Storb R, et al. Fractionated versus single-dose total-body irradiation at low and high-dose rates to condition canine littermates for Dla-identical marrow grafts. Blood. 1994;83(11):3384-3389.
76 Salomon O, et al. Induction of donor-type chimerism in murine recipients of bone-marrow allografts by different radiation regimens currently used in treatment of leukemia patients. Blood. 1990;76(9):1872-1878.
77 Girinski T, et al. Similar effects on murine hematopoietic compartment of low-dose rate single dose and high-dose rate fractionated total-body irradiation—Preliminary results after a unique dose of 750 cGy. Br J Cancer. 1990;61(6):797-800.
78 Vegesna V, et al. Multifraction radiation response of mouse lung. Int J Radiat Biol. 1985;47(4):413-422.
79 Penney DP, et al. Morphological correlates of fractionated radiation of the mouse lung—Early and late effects. Int J Radiat Oncol Biol Phys. 1994;29(4):789-804.
80 Mcchesney SL, Gillette EL, Powers BE. Response of the canine lung to fractionated irradiation—Pathologic changes and isoeffect curves. Int J Radiat Oncol Biol Phys. 1989;16(1):125-132.
81 Andrews GA. Criticality accidents in Vinca, Yugoslavia, and Oak Ridge, Tennessee—Comparison of radiation injuries and results of therapy. JAMA. 1962;179(3):191-193.
82 Cosset JM. ESTRO Breur Gold Medal Award Lecture 2001: irradiation accidents—lessons for oncology? Radiother Oncol. 2002;63(1):1-10.
83 International Atomic Energy Agency. Safety reports series. In: World Health Organization, editor. Diagnosis and Treatment of Radiation Injuries. Vienna: International Atomic Energy Agency; 1998:49.
84 Waselenko JK, et al. Medical management of the acute radiation syndrome: recommendations of the Strategic National Stockpile Radiation Working Group. Ann Intern Med. 2004;140(12):1037-1051.
85 Hall EJ, Giaccia AJ. Radiobiology for the radiologist, 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2006.
86 Chaillet MP, et al. Prospective study of the clinical symptoms of therapeutic whole-body irradiation. Health Phys. 1993;64(4):370-374.
87 Buchali A, et al. Immediate toxicity during fractionated total body irradiation as conditioning for bone marrow transplantation. Radiother Oncol. 2000;54(2):157-162.
88 Ozsahin M, et al. Total-body irradiation before bone-marrow transplantation—results of 2 randomized instantaneous dose-rates in 157 patients. Cancer. 1992;69(11):2853-2865.
89 Thomas ED, et al. Marrow transplantation for acute nonlymphoblastic leukemia in 1st remission using fractionated or single-dose irradiation. Int J Radiat Oncol Biol Phys. 1982;8(5):817-821.
90 Tiley C, et al. Results of a double-blind placebo-controlled study of ondansetron as an antiemetic during total-body irradiation in patients undergoing bone-marrow transplantation. Leuk Lymphoma. 1992;7(4):317-321.
91 Spitzer TR, et al. Randomized double-blind, placebo-controlled evaluation of oral ondansetron in the prevention of nausea and vomiting associated with fractionated total-body irradiation. J Clin Oncol. 1994;12(11):2432-2438.
92 Prentice HG, et al. Granisetron in the prevention of irradiation-induced emesis. Bone Marrow Transplant. 1995;15(3):445-448.
93 Okamoto S, et al. Granisetron in the prevention of vomiting induced by conditioning for stem cell transplantation: a prospective randomized study. Bone Marrow Transplant. 1996;17(5):679-683.
94 Matsuoka S, et al. Granisetron plus dexamethasone versus granisetron alone in the prevention of vomiting induced by conditioning for stem cell transplantation: a prospective randomized study. Int J Hematol. 2003;77(1):86-90.
95 Maranzano E, et al. Evidence-based recommendations for the use of antiemetics in radiotherapy. Radiother Oncol. 2005;76(3):227-233.
96 National Comprehensive Cancer Network. NCCN Clinical practice guidelines in oncology. antiemesis, V.3.2009. [cited 2009 August 18]; Available from http://www.nccn.org/professionals/physician_gls/PDF/antiemesis.pdf, 2009.
97 Kris MG, et al. American Society of Clinical Oncology guideline for antiemetics in oncology: Update 2006. J Clin Oncol. 2006;24(18):2932-2947.
98 Orchard PJ, et al. A prospective randomized trial of the anti-emetic efficacy of ondansetron and granisetron during bone marrow transplantation. Biol Blood Marrow Transplant. 1999;5(6):386-393.
99 Woo SB, et al. A longitudinal study of oral ulcerative mucositis in bone-marrow transplant recipients. Cancer. 1993;72(5):1612-1617.
100 Borowski B, et al. Prevention of oral mucositis in patients treated with high-dose chemotherapy and bone-marrow transplantation—a randomized controlled trial comparing 2 protocols of dental care. Eur J Cancer B Oral Oncol Part B. 1994;30B(2):93-97.
101 Ferretti GA, et al. Chlorhexidine for prophylaxis against oral infections and associated complications in patients receiving bone-marrow transplants. J Am Dent Assoc. 1987;114(4):461-467.
102 Ferretti GA, et al. Control of oral mucositis and candidiasis in marrow transplantation—a prospective, double-blind trial of chlorhexidine digluconate oral rinse. Bone Marrow Transplant. 1988;3(5):483-493.
103 Papas AS, et al. A prospective, randomized trial for the prevention of mucositis in patients undergoing hematopoietic stem cell transplantation. Bone Marrow Transplant. 2003;31(8):705-712.
104 Castagna L, et al. Prevention of mucositis in bone marrow transplantation: a double blind randomised controlled trial of sucralfate. Ann Oncol. 2001;12(7):953-955.
105 Yuen KY, et al. Effects of clarithromycin on oral mucositis in bone marrow transplant recipients. Haematologica. 2001;86(5):554-555.
106 Hwang WYK, et al. A randomized trial of amifostine as a cytoprotectant for patients receiving myeloablative therapy for allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2004;34(1):51-56.
107 Ifrah N, et al. Intensive short term therapy with granulocyte-macrophage-colony stimulating factor support, similar to therapy for acute myeloblastic leukemia, does not improve overall results for adults with acute lymphoblastic leukemia. Cancer. 1999;86(8):1496-1505. Wiley-Liss
108 Van der Lelie H, et al. Effect of locally applied GM-CSF on oral mucositis after stem cell transplantation: a prospective placebo-controlled double-blind study. Ann Hematol. 2001;80(3):150-154.
109 Spielberger R, et al. Palifermin for oral mucositis after intensive therapy for hematologic cancers. N Engl J Med. 2004;351(25):2590-2598.
110 Hensley ML, et al. American Society of Clinical Oncology 2008 clinical practice guideline update: use of chemotherapy and radiation therapy protectants. J Clin Oncol. 2009;27(1):127-145.
111 Thomas ED, et al. Marrow transplantation for patients with acute lymphoblastic leukemia in remission. Blood. 1979;54(2):468-476.
112 Ringden O, et al. Increased risk of chronic graft-versus-host disease, obstructive bronchiolitis, and alopecia with busulfan versus total body irradiation: long-term results of a randomized trial in allogeneic marrow recipients with leukemia. Blood. 1999;93(7):2196-2201.
113 Andrews GA. Radiation accidents and their management. Radiat Res. 1967;S:390.
114 Trivedi M, et al. Optimal use of G-CSF administration after hematopoietic SCT. Bone Marrow Transplant. 2009;43(12):895-908.
115 Smith TJ, et al. 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol. 2006;24(19):3187-3205.
116 Crawford J, et al. Hematopoietic growth factors: ESMO recommendations for the applications. Ann Oncol. 2009;20:162-165.
117 Bunn PA, et al. Chemoradiotherapy with or without granulocyte-macrophage colony-stimulating factor in the treatment of limited-stage small-cell lung-cancer—a prospective phase III randomized study of the Southwest Oncology Group. J Clin Oncol. 1995;13(7):1632-1641.
118 Dahllof G, et al. Risk factors for salivary dysfunction in children 1 year after bone marrow transplantation. Oral Oncol. 1997;33(5):327-331.
119 Bagesund M, et al. Longitudinal scintigraphic study of parotid and submandibular gland function after total body irradiation in children and adolescents. Int J Paediatr Dent. 2007;17(1):34-40.
120 Dahllof G. Oral and dental late effects after pediatric stem cell transplantation. Biol Blood Marrow Transplant. 2007;14(1):81-83.
121 Holtta P, et al. Agenesis and microdontia of permanent teeth as late adverse effects after stem cell transplantation in young children. Cancer. 2005;103(1):181-190.
122 Holtta P, et al. Disturbed root development of permanent teeth after pediatric stem cell transplantation—dental root development after SCT. Cancer. 2005;103(7):1484-1493.
123 Nasman M, Forsberg CM, Dahllof G. Long-term dental development in children after treatment for malignant disease. Eur J Orthod. 1997;19(2):151-159.
124 Jones LR, Toth BB, Keene HJ. Effects of total-body irradiation on salivary-gland function and caries-associated oral microflora in bone-marrow transplant patients. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1992;73(6):670-676.
125 Singhal S, et al. Pilocarpine hydrochloride for symptomatic relief of xerostomia due to chronic graft-versus-host disease or total-body irradiation after bone-marrow transplantation for hematologic malignancies. Leukema & Lymphoma. 1997;24(5-6):539-543. Harwood Acad Publ Gmbh
126 Peters SG, Afessa B. Acute lung injury after hematopoietic stem cell transplantation. Clin Chest Med. 2005;26(4):561.
127 Patriarca F, et al. Clinical presentation, outcome and risk factors of late-onset non-infectious pulmonary complications after allogeneic stem cell transplantation. Curr Stem Cell Res Ther. 2009;4:161-167.
128 Fukuda T, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood. 2003;102(8):2777-2785.
129 Robbins RA, et al. Diffuse alveolar hemorrhage in autologous bone-marrow transplant recipients. Am J Med. 1989;87(5):511-518.
130 Weiner RS, et al. Interstitial pneumonitis after bone-marrow transplantation: assessment of risk factors. Ann Intern Med. 1986;104(2):168-175.
131 Pino y Torres JL, et al. Risk factors in interstitial pneumonitis following allogenic bone marrow transplantation. Int J Radiat Oncol Biol Phys. 1982;8(8):1301-1307.
132 Savani BN, et al. Chronic GVHD and pretransplantation abnormalities in pulmonary function are the main determinants predicting worsening pulmonary function in long-term survivors after stem cell transplantation. Biol Blood Marrow Transplant. 2006;12(12):1261-1269.
133 Shank B, et al. Hyperfractionated total-body irradiation for bone-marrow transplantation 1. Early results in leukemia patients. Int J Radiat Oncol Biol Phys. 1981;7(8):1109-1115.
134 Gogna NK, et al. Lung dose rate and interstitial pneumonitis in total body irradiation for bone marrow transplantation. Australas Radiol. 1992;36(4):317-320.
135 Keane TJ, Vandyk J, Rider WD. Idiopathic interstitial pneumonia following bone-marrow transplantation—the relationship with total-body irradiation. Int J Radiat Oncol Biol Phys. 1981;7(10):1365-1370.
136 Bortin M, et al. Factors associated with interstitial pneumonitis after bone-marrow transplantation for acute leukaemia. Lancet. 1982;319(8269):437-439.
137 Clift RA, et al. Allogeneic marrow transplantation in patients with acute myeloid-leukemia in 1st remission—a randomized trial of 2 irradiation regimens. Blood. 1990;76(9):1867-1871.
138 Clift RA, et al. Allogeneic marrow transplantation in patients with chronic myeloid-leukemia in the chronic phase—a randomized trial of 2 irradiation regimens. Blood. 1991;77(8):1660-1665.
139 Clift RA, et al. Long-term follow-up of a randomized trial of two irradiation regimens for patients receiving allogeneic marrow transplants during first remission of acute myeloid leukemia. Blood. 1998;92(4):1455-1456.
140 Vandyk J, et al. Radiation pneumonitis following large single dose irradiation—a re-evaluation based on absolute dose to lung. Int J Radiat Oncol Biol Phys. 1981;7(4):461-467.
141 Della Volpe A, et al. Lethal pulmonary complications significantly correlate with individually assessed mean lung dose in patients with hematologic malignancies treated with total body irradiation. Int J Radiat Oncol Biol Phys. 2002;52(2):483-488.
142 Deeg HJ, et al. Marrow transplantation for acute nonlymphoblastic leukemia in 1st remission—toxicity and long-term follow-up of patients conditioned with single dose or fractionated total-body irradiation. Bone Marrow Transplant. 1986;1(2):151-157.
143 Girinsky T, et al. Prospective randomized comparison of single-dose versus hyperfractionated total-body irradiation in patients with hematologic malignancies. J Clin Oncol. 2000;18(5):981-986.
144 Morgan TL, et al. A comparison of single-dose and fractionated total-body irradiation on the development of pneumonitis following bone marrow transplantation. Int J Radiat Oncol Biol Phys. 1996;36(1):61-66.
145 Gopal R, et al. Comparison of two total body irradiation fractionation regimens with respect to acute and late pulmonary toxicity cancer. Cancer. 2001;92(7):1949-1958. John Wiley & Sons
146 Sampath S, Schultheiss TE, Wong J. Dose response and factors related to interstitial pneumonitis after bone marrow transplant. Int J Radiat Oncol Biol Phys. 2005;63(3):876-884.
147 Beyzadeoglu M, et al. Effect of dose-rate and lung dose in total body irradiation on interstitial pneumonitis after bone marrow transplantation. Tohoku J Exp Med. 2004;202(4):255-263.
148 Belkacemi Y, et al. Total-body irradiation before bone marrow transplantation for acute leukemia in first or second complete remission—results and prognostic factors in 326 consecutive patients. Strahlentherapie Und Onkologie. 1998;174(2):92-104.
149 Ozsahin M, et al. Interstitial pneumonitis following autologous bone-marrow transplantation conditioned with cyclophosphamide and total-body irradiation. Int J Radiat Oncol Biol Phys. 1996;34(1):71-77.
150 Carruthers SA, Wallington M. Total body irradiation and pneumonitis risk: a review of outcomes. Br J Cancer. 2004;90(11):2080-2084.
151 Corvo R, et al. Total body irradiation correlates with chronic graft versus host disease and affects prognosis of patients with acute lymphoblastic leukemia receiving an HLA identical allogeneic bone marrow transplant. Int J Radiat Oncol Biol Phys. 1999;43(3):497-503.
152 Labar B, et al. Total-body irradiation with or without lung shielding for allogeneic bone-marrow transplantation. Bone Marrow Transplant. 1992;9(5):343-347.
153 Weshler Z, et al. Interstitial pneumonitis after total-body irradiation—effect of partial lung shielding. Br J Haematol. 1990;74(1):61-64.
154 Girinsky T, et al. Consequences of 2 different doses to the lungs during a single-dose of total-body irradiation—results of a randomized study on 85 patients. Int J Radiat Oncol Biol Phys. 1994;30(4):821-824.
155 Gore EM, et al. Pulmonary function changes in long-term survivors of bone marrow transplantation. Int J Radiat Oncol Biol Phys. 1996;36(1):67-75.
156 Tait RC, et al. Subclinical pulmonary-function defects following autologous and allogeneic bone-marrow transplantation—relationship to total-body irradiation and graft-versus-host disease. Int J Radiat Oncol Biol Phys. 1991;20(6):1219-1227.
157 Gandola L, et al. Prospective evaluation of pulmonary function in cancer patients treated with total-body irradiation, high-dose melphalan, and autologous hematopoietic stem-cell transplantation. Int J Radiat Oncol Biol Phys. 1990;19(3):743-749.
158 Chien JW, et al. Comparison of lung function after myeloablative and 2 Gy of total body irradiation-based regimens for hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11(4):288-296.
159 Soule BP, et al. Pulmonary function following total body irradiation (with or without lung shielding) and allogeneic peripheral blood stem cell transplant. Bone Marrow Transplant. 2007;40(6):573-578.
160 Beinert T, et al. Late pulmonary impairment following allogeneic bone marrow transplantation. Eur J Med Res. 1996;1(7):343-348.
161 Carver JR, et al. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol. 2007;25(25):3991-4008.
162 Group CSO. Long-term follow-up guidelines for survivors of childhood, adolescent and young adult cancers, Version 3.0. [cited 2009 August 27]; Version 3.0. Available from www.survivorshipguidelines.org, October 2008.
163 Bhatia S, et al. Late mortality in survivors of autologous hematopoietic-cell transplantation: report from the Bone Marrow Transplant Survivor Study. Blood. 2005;105(11):4215-4222.
164 Bhatia S, et al. Late mortality after allogeneic hematopoietic cell transplantation and functional status of long-term survivors: report from the Bone Marrow Transplant Survivor Study. Blood. 2007;110(10):3784-3792.
165 Tichelli A, et al. Late cardiovascular events after allogeneic hematopoietic stem cell transplantation: a retrospective multicenter study of the Late Effects Working Party of the European Group for Blood and Marrow Transplantation. Haematologica. 2008;93(8):1203-1210.
166 Armenian SH, et al. Late congestive heart failure after hematopoietic cell transplantation. J Clin Oncol. 2008;26(34):5537-5543.
167 Auner HW, et al. Monitoring of cardiac function by serum cardiac troponin T levels, ventricular repolarisation indices, and echocardiography after conditioning with fractionated total body irradiation and high-dose cyclophosphamide. Eur J Haematol. 2002;69(1):1-6.
168 Snowden JA, et al. Assessment of cardiotoxicity during haemopoietic stem cell transplantation with plasma brain natriuretic peptide. Bone Marrow Transplant. 2000;26(3):309-313.
169 Uderzo C, et al. Impact of cumulative anthracycline dose, preparative regimen and chronic graft-versus-host disease on pulmonary and cardiac function in children 5 years after allogeneic hematopoietic stem cell transplantation: a prospective evaluation on behalf of the EBMT Pediatric Diseases and Late Effects Working Parties. Bone Marrow Transplant. 2007;39(11):667-675.
170 Shulman HM, et al. An analysis of hepatic venooclusive disease and centrilobular hepatic degeneration following bone-marrow transplantation. Gastroenterology. 1980;79(6):1178-1191.
171 Wadleigh M, et al. Hepatic veno-occlusive disease: pathogenesis, diagnosis and treatment. Curr Opin Hematol. 2003;10(6):451-462.
172 Ringden O, et al. A randomized trial comparing busulfan with total-body irradiation as conditioning in allogeneic marrow transplant recipients with leukemia—a report from the Nordic-Bone-Marrow-Transplantation-Group. Blood. 1994;83(9):2723-2730.
173 Hartman AR, Williams S, Dillon JJ. Survival, disease-free survival and adverse effects of conditioning for allogeneic bone marrow transplantation with busulfan/cyclophosphamide vs total body irradiation: a meta-analysis. Bone Marrow Transplant. 1998;22(5):439-443.
174 Shulman HM, Hinterberger W. Hepatic venoocclusive disease—liver toxicity syndrome after bone-marrow transplantation. Bone Marrow Transplant. 1992;10(3):197-214.
175 McDonald GB, et al. Veno-occlusive disease of the liver and multiorgan failure after bone marrow transplantation: a cohort study of 355 patients. Ann Intern Med. 1993;118(4):255-267.
176 Rozman C, et al. Risk factors for hepatic veno-occlusive disease following HLA-identical sibling bone marrow transplants for leukemia. Bone Marrow Transplant. 1996;17(1):75-80.
177 Lawton CA, et al. Technical modifications in hyperfractionated total-body irradiation for lymphocyte-t deplete bone-marrow transplant. Int J Radiat Oncol Biol Phys. 1989;17(2):319-322.
178 Ohashi K, et al. The Japanese multicenter open randomized trial of ursodeoxycholic acid prophylaxis for hepatic veno-occlusive disease after stem cell transplantation. Am J Hematol. 2000;64(1):32-38.
179 Park SH, et al. A randomized trial of heparin plus ursodiol vs heparin alone to prevent hepatic veno-occlusive disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;29(2):137-143.
180 Senzolo M, et al. Veno occlusive disease: Update on clinical management. World J Gastroenterol. 2007;13(29):3918-3924.
181 Van Kempen-Harteveld ML, et al. Cataract after total body irradiation and bone marrow transplantation: Degree of visual impairment. Int J Radiat Oncol Biol Phys. 2002;52(5):1375-1380.
182 Van Kempen-Harteveld ML, et al. Cataract-free interval and severity of cataract after total body irradiation and bone marrow transplantation: influence of treatment parameters. Int J Radiat Oncol Biol Phys. 2000;48(3):807-815.
183 Dunn JP, et al. Bone-marrow transplantation and cataract development. Arch Ophthal. 1993;111(10):1367-1373.
184 Deeg HJ, et al. Cataracts after total-body irradiation and marrow transplantation—a sparing effect of dose fractionation. Int J Radiat Oncol Biol. 1984;10(7):957-964.
185 Fife K, et al. Risk factors for requiring cataract surgery following total-body irradiation. Radiother Oncol. 1994;33(2):93-98.
186 Zierhut D, et al. Cataract incidence after total-body irradiation. Int J Radiat Oncol Biol Phys. 2000;46(1):131-135.
187 Belkacemi Y, et al. Cataractogenesis after total body irradiation. Int J Radiat Oncol Biol Phys. 1996;35(1):53-60.
188 Aristei C, et al. Cataracts in patients receiving stem cell transplantation after conditioning with total body irradiation. Bone Marrow Transplant. 2002;29(6):503-507.
189 Schneider RA, et al. Long-term outcome after static intensity-modulated total body radiotherapy using compensators stratified by pediatric and adult cohorts. Int J Radiat Oncol Biol Phys. 2008;70(1):194-202.
190 Belkacemi Y, et al. Cataracts after total body irradiation and bone marrow transplantation in patients with acute leukemia in complete remission: A study of the European Group for Blood and Marrow Transplantation. Int J Radiat Oncol Biol Phys. 1998;41(3):659-668.
191 Ferry C, et al. Long-term outcomes after allogeneic stem cell transplantation for children with hematological malignancies. Bone Marrow Transplant. 2007;40(3):219-224.
192 Fahnehjelm KT, et al. Visual outcome and cataract development after allogeneic stem-cell transplantation in children. Acta Ophthalmol Scand. 2007;85(7):724-733.
193 Beyzadeoglu M, et al. Evaluation of fractionated total body irradiation and dose rate on cataractogenesis in bone marrow transplantation. Haematologia. 2002;32(1):25-30.
194 Ozsahin M, et al. Total-body irradiation and cataract incidence—a randomized comparison of 2 instantaneous dose-rates. Int J Radiat Oncol Biol Phys. 1994;28(2):343-347.
195 Kal HB, Van Kempen-Harteveld ML. Induction of severe cataract and late renal dysfunction following total body irradiation: dose-effect relationships. Anticancer Res. 2009;29(8):3305-3309.
196 Van Kempen-Harteveld ML, et al. Eye shielding during total body irradiation for bone marrow transplantation in children transplanted for a hematological disorder: risks and benefits. Bone Marrow Transplant. 2003;31(12):1151-1156.
197 Gurney JG, et al. Visual, auditory, sensory, and motor impairments in long-term survivors of hematopoietic stem cell transplantation performed in childhood—results from the Bone Marrow Transplant Survivor study. Cancer. 2006;106(6):1402-1408.
198 Bell CM, et al. Surgeon volumes and selected patient outcomes in cataract surgery—a population-based analysis. Ophthalmology. 2007;114(3):405-410.
199 Powe NR, et al. Synthesis of the literature on visual-acuity and complications following cataract-extraction with intraocular-lens implantation. Arch Ophthalmol. 1994;112(2):239-252.
200 Ellis MJ, et al. Chronic kidney disease after hematopoietic cell transplantation: a systematic review. Am J Transplant. 2008;8(11):2378-2390.
201 Hingorani S. Chronic kidney disease in long-term survivors of hematopoietic cell transplantation: epidemiology, pathogenesis, and treatment. J Am Soc Nephrol. 2006;17(7):1995-2005.
202 Choi M, et al. Incidence and predictors of delayed chronic kidney disease in long-term survivors of hematopoietic cell transplantation. Cancer. 2008;113(7):1580-1587.
203 Weiss AS, et al. Chronic kidney disease following non-myeloablative hematopoietic cell transplantation. Am J Transplant. 2006;6(1):89-94.
204 Hingorani SR, et al. Risk factors for chronic kidney disease (CKD) after hematopoietic cell transplantation (HCT). Biology of Blood and Marrow Transplant. 2005;11(2):72-73. Carden Jennings
205 Kist-van Holthe JE, et al. Prospective study of renal insufficiency after bone marrow transplantation. Pediatr Nephrol. 2002;17(12):1032-1037.
206 Hingorani S, et al. Chronic kidney disease in long-term survivors of hematopoietic cell transplant. Bone Marrow Transplant. 2007;39(4):223-229.
207 Miralbell R, et al. Renal toxicity after allogeneic bone marrow transplantation: the combined effects of total-body irradiation and graft-versus-host disease. J Clin Oncol. 1996;14(2):579-585.
208 Miralbell R, et al. Renal insufficiency in patients with hematologic malignancies undergoing total body irradiation and bone marrow transplantation: a prospective assessment. Int J Radiat Oncol Biol Phys. 2004;58(3):809-816.
209 Lawton CA, et al. Long-term results of selective renal shielding in patients undergoing total body irradiation in preparation for bone marrow transplantation. Bone Marrow Transplant. 1997;20(12):1069-1074.
210 Igaki H, et al. Renal dysfunction after total-body irradiation—significance of selective renal shielding blocks. Strahlenther Onkol. 2005;181(11):704-708.
211 Cheng JC, Schultheiss TE, Wong JYC. Impact of drug therapy, radiation dose, and dose rate on renal toxicity following bone marrow transplantation. Int J Radiat Oncol Biol Phys. 2008;71(5):1436-1443.
212 Lewis EJ, et al. The effect of angiotensin-converting enzyme-inhibition on diabetic nephropathy. New Engl J Med. 1993;329(20):1456-1462.
213 Moulder JE, Fish BL, Cohen EP. Noncontinuous use of angiotensin converting enzyme inhibitors in the treatment of experimental bone marrow transplant nephropathy. Bone Marrow Transplant. 1997;19(7):729-735.
214 Cohen EP, Fish BL, Moulder JE. Successful brief captopril treatment in experimental radiation nephropathy. J Lab Clin Med. 1997;129(5):536-547.
215 Cohen EP, et al. Captopril to mitigate chronic renal failure after hematopoietic stem cell transplantation: a randomized controlled trial. Int J Radiat Oncol Biol Phys. 2008;70(5):1546-1551.
216 Chemaitilly W, et al. Final height in pediatric patients after hyperfractionated total body irradiation and stem cell transplantation. Bone Marrow Transplant. 2007;40(1):29-35.
217 Sanders JE, et al. Final adult height patients who received hematopoietic cell transplant in childhood. Blood. 2005;105(3):1348-1354.
218 Mertens AC, et al. Patterns of gonadal dysfunction following bone marrow transplantation. Bone Marrow Transplant. 1998;22(4):345-350.
219 Baker KS, et al. Diabetes, hypertension, and cardiovascular events in survivors of hematopoietic cell transplantation: a report from the bone marrow transplantation survivor study. Blood. 2007;109(4):1765-1772.
220 Phipps S, et al. Cognitive and academic consequences of stem-cell transplantation in children. J Clin Oncol. 2008;26(12):2027-2033.
221 Ades L, Guardiola P, Socie G. Second malignancies after allogeneic hematopoietic stem cell transplantation: new insight and current problems. Blood Rev. 2002;16(2):135-146.
222 Witherspoon RP, et al. Secondary cancers after bone-marrow transplantation for leukemia or aplastic-anemia. New Engl J Med. 1989;321(12):784-789.
223 Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292-2302.
224 Bhatia S, et al. Malignant neoplasms following bone marrow transplantation. Blood. 1996;87(9):3633-3639.
225 Taylor PRA, et al. Low incidence of myelodysplastic syndrome following transplantation using autologous non-cryopreserved bone marrow. Leukemia. 1997;11(10):1650-1653.
226 Krishnan A, et al. Predictors of therapy-related leukemia and myelodysplasia following autologous transplantation for lymphoma: an assessment of risk factors. Blood. 2000;95(5):1588-1593.
227 Sebban C, et al. Standard chemotherapy with interferon compared with CHOP followed by high-dose therapy with autologous stem cell transplantation in untreated patients with advanced follicular lymphoma: the GELF-94 randomized study from the Groupe d’Etude des Lymphomes de l’Adulte (GELA). Blood. 2006;108(8):2540-2544.
228 Darrington DL, et al. Incidence and characterization of secondary myelodysplastic syndrome and acute myelogenous leukemia following high-dose chemoradiotherapy and autologous stem-cell transplantation for lymphoid malignancies. J Clin Oncol. 1994;12(12):2527-2534.
229 Milligan DW, et al. Secondary leukaemia and myelodysplasia after autografting for lymphoma: results from the EBMT. Br J Haematol. 1999;106(4):1020-1026.
230 Park S, et al. Myelodysplasias and leukemias after autologous stem cell transplantation for lymphoid malignancies. Bone Marrow Transplant. 2000;26(3):321-326.
231 Sureda A, et al. Autologous stem-cell transplantation for Hodgkin’s disease: results and prognostic factors in 494 patients from the Grupo Espanol de Linfomas/Transplante Autolog de Medula Osea Spanish Cooperative Group. J Clin Oncol. 2001;19(5):1395-1404.
232 Beauchamp-Nicoud A, et al. Therapy-related myelodysplasia and/or acute myeloid leukaemia after autologous haematopoietic progenitor cell transplantation in a prospective single centre cohort of 221 patients. Br J Haematol. 2003;122(1):109-117.
233 Hosing C, et al. Risk of therapy-related myelodysplastic syndrome/acute leukemia following high-dose therapy and autologous bone marrow transplantation for non-Hodgkin’s lymphoma. Ann Oncol. 2002;13(3):450-459.
234 Metayer C, et al. Myelodysplastic syndrome and acute myeloid leukemia after autotransplantation for lymphoma: a multicenter case-control study. Blood. 2003;101(5):2015-2023.
235 Brown JR, et al. Increasing incidence of late second malignancies after conditioning with cyclophosphamide and total-body irradiation and autologous bone marrow transplantation for non-Hodgkin’s lymphoma. J Clin Onc. 2005;123(10):2208-2214.
236 Landgren O, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. 2009;113(20):4992-5001.
237 Curtis RE, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi-institutional study. Blood. 1999;94(7):2208-2216.
238 Micallef INM, et al. Lymphoproliferative disorders following allogeneic bone marrow transplantation: the Vancouver experience. Bone Marrow Transplant. 1998;22(10):981-987.
239 Gross TG, et al. B cell lymphoproliferative disorders following hematopoietic stem cell transplantation: Risk factors, treatment and outcome. Bone Marrow Transplant. 1999;23(3):251-258.
240 Curtis RE, et al. Solid cancers after bone marrow transplantation. New Engl J Med. 1997;336(13):897-904.
241 Kolb HJ, et al. Malignant neoplasms in long-term survivors of bone marrow transplantation. Ann Intern Med. 1999;131(10):738-744.
242 Socie G, et al. New malignant diseases after allogeneic marrow transplantation for childhood acute leukemia. J Clin Oncol. 2000;18(2):348-357.
243 Bhatia S, et al. Solid cancers after bone marrow transplantation. J Clin Oncol. 2001;19(2):464-471.
244 Baker KS, et al. New malignancies after blood or marrow stem-cell transplantation in children and adults: incidence and risk factors. J Clin Oncol. 2003;21(7):1352-1358.
245 Friedman DL, et al. Second malignant neoplasms following hematopoietic stem cell transplantation. Int J Hematol. 2004;79(3):229-234.
246 Shimada K, et al. Solid tumors after hematopoietic stem cell transplantation in Japan: incidence, risk factors and prognosis. Bone Marrow Transplant. 2005;36(2):115-121.
247 Leisenring W, et al. Nonmelanoma skin and mucosal cancers after hematopoietic cell transplantation. J Clin Oncol. 2006;24(7):1119-1126.
248 Gallagher G, Forrest DL. Second solid cancers after allogeneic hematopoietic stem cell transplantation. Cancer. 2007;109(1):84-92.
249 Cohen A, et al. Risk for secondary thyroid carcinoma after hematopoietic stem-cell transplantation: an EBMT late effects working party study. J Clin Oncol. 2007;25(17):2449-2454.
250 Friedman DL, et al. Increased risk of breast cancer among survivors of allogeneic hematopoietic cell transplantation: a report from the FHCRC and the EBMT-Late Effect Working Party. Blood. 2008;111(2):939-944.
251 Rizzo JD, et al. Solid cancers after allogeneic hematopoietic cell transplantation. Blood. 2009;113(5):1175-1183.
252 Smith RA, Cokkinides V, Brawley OW. Cancer screening in the United States, 2009: a review of current American Cancer Society guidelines and issues in cancer screening. CA Cancer J Clin. 2009;59(1):27-41.
253 Kim TH, Khan FM, Galvin JM. A report of the work party—comparison of total-body irradiation techniques for bone-marrow transplantation. Int J Radiat Oncol Biol Phys. 1980;6(6):779-784.
254 Americal Association on Physicists in Medicine, Committee on Radiation Therapy, Task Group 29. The physical aspects of total and half body photon irradiation. New York: American Institute of Physics; 1986. p 55
255 Shank B, et al. Hyperfractionated total-body irradiation for bone-marrow transplantation—results in 70 leukemia patients with allogeneic transplants. Int J Radiat Oncol Biol Phys. 1983;9(11):1607-1611.
256 Chui CS, et al. Total body irradiation with an arc and a gravity-oriented compensator. Int J Radiat Oncol Biol Phys. 1997;39(5):1191-1195.
257 Khan FM, et al. Basic data for dosage calculation and compensation. Int J Radiat Oncol Biol Phys. 1980;6(6):745-751.
258 Copelan EA, Deeg HJ. Conditioning for allogeneic marrow transplantation in patients with lymphohematopoietic malignancies without the use of total-body irradiation. Blood. 1992;80(7):1648-1658.
259 Santos GW. Busulfan (Bu) and cyclophosphamide (Cy) for marrow transplantation. Bone Marrow Transplant. 1989;4:236-239.
260 Blaise D, et al. Allogeneic bone-marrow transplantation for acute myeloid-leukemia in first remission—a randomized trial of a busulfan-cytoxan versus cytoxan-total body irradiation as preparative regimen—a report from the Groupe-Detudes-De-La-Greffe-De-Moelle-Osseuse. Blood. 1992;79(10):2578-2582.
261 Dusenbery KE, et al. Randomized comparison of cyclophosphamide total-body irradiation versus busulfan cyclophosphamide conditioning in autologous bone-marrow transplantation for acute myeloid-leukemia. Int J Radiat Oncol Biol Phys. 1995;31(1):119-128.
262 Blaise D, et al. Long-term follow-up of a randomized trial comparing the combination of cyclophosphamide with total body irradiation or busulfan as conditioning regimen for patients receiving HLA-identical marrow grafts for acute myeloblastic leukemia in first complete remission. Blood. 2001;97(11):3669-3671.
263 Socie G, et al. Busulfan plus cyclophosphamide compared with total-body irradiation plus cyclophosphamide before marrow transplantation for myeloid leukemia: long-term follow-up of 4 randomized studies. Blood. 2001;98(13):3569-3574.
264 Litzow MR, et al. Comparison of outcome following allogeneic bone marrow transplantation with cyclophosphamide-total body irradiation versus busulphan-cyclophosphamide conditioning regimens for acute myelogenous leukaemia in first remission. Br J Haematol. 2002;119(4):1115-1124.
265 Clift RA, et al. Marrow transplantation for chronic myeloid-leukemia—a randomized study comparing cyclophosphamide and total-body irradiation with busulfan and cyclophosphamide. Blood. 1994;84(6):2036-2043.
266 Clift RA, et al. Long-term follow-up of a randomized study comparing cyclophosphamide and total body irradiation with busulfan and cyclophosphamide for patients receiving allogeneic marrow transplants during chronic phase of chronic myeloid leukemia. Blood. 1999;94(11):3960-3962.
267 Devergie A, et al. Allogeneic bone-marrow transplantation for chronic myeloid-leukemia in first chronic phase—a randomized trial of busulfan-cytoxan versus cytoxan-total-body irradiation as preparative regimen—a report from the French Society of Bone Marrow Graft (SFGM). Blood. 1995;85(8):2263-2268.
268 Kim I, et al. Allogeneic bone marrow transplantation for chronic myeloid leukemia: a retrospective study of busulfan-cytoxan versus total body irradiation—cytoxan as preparative regimen in Koreans. Clin Transplant. 2001;15(3):167-172.
269 Kroger N, et al. Comparison of total body irradiation vs busulfan in combination with cyclophosphamide as conditioning for unrelated stem cell transplantation in CML patients. Bone Marrow Transplant. 2001;27(4):349-354.
270 Bunin N, et al. Randomized trial of busulfan vs total body irradiation containing conditioning regimens for children with acute lymphoblastic leukemia: a Pediatric Blood and Marrow Transplant Consortium study. Bone Marrow Transplant. 2003;32(6):543-548.
271 Davies S. Comparison of preparative regimens in transplants for children with acute lymphoblastic leukemia. J Clin Oncol. 2000;18(2):340-347.
272 Granados E, et al. Hematopoietic cell transplantation in acute lymphoblastic leukemia: better long-term event-free survival with conditioning regimens containing total body irradiation. Haematologica. 2000;85(10):1060-1067.
273 Moreau P, et al. Comparison of 200 mg/m(2) melphalan and 8 Gy total body irradiation plus 140 mg/m(2) melphalan as conditioning regimens for peripheral blood stem cell transplantation in patients with newly diagnosed multiple myeloma: final analysis of the Intergroupe Francophone du Myelome 9502 randomized trial. Blood. 2002;99(3):731-735.
274 Blume KG, et al. A prospective randomized comparison of total-body irradiation-etoposide versus busulfan-cyclophosphamide as preparatory regimens for bone-marrow transplantation in patients with leukemia who were not in 1st remission—a Southwest Oncology Group Study. Blood. 1993;81(8):2187-2193.
275 McAfee SL, et al. Dose-escalated total body irradiation and autologous stem cell transplantation for refractory hematologic malignancy. Int J Radiat Oncol Biol Phys. 2002;53(1):151-156.
276 Marks DI, et al. A comparison of cyclophosphamide and total body irradiation with etoposide and total body irradiation as patients conditioning regimens for undergoing sibling allografting for acute lymphoblastic leukemia in first or second complete remission. Biol Blood Marrow Transplant. 2006;12(4):438-453.
277 Kal HB, et al. Biologically effective dose in total-body irradiation and hematopoietic stem cell transplantation. Strahlenther Onkol. 2006;182(11):672-679.
278 Alyea E, et al. Effect of total body irradiation dose escalation on outcome following T-cell-depleted allogeneic bone marrow transplantation. Biol Blood Marrow Transplant. 2002;8(3):139-144.
279 Bieri S, et al. Total body irradiation before allogeneic bone marrow transplantation: is more dose better? Int J Radiat Oncol Biol Phys. 2001;49:1071-1077.
280 Demirer T, et al. Allogeneic marrow transplantation following cyclophosphamide and escalating doses of hyperfractionated total-body irradiation in patients with advanced lymphoid malignancies—a phase I/II trial. Int J Radiat Oncol Biol Phys. 1995;32(4):1103-1109.
281 Sobecks RM, et al. A dose escalation study of total body irradiation followed by high-dose etoposide and allogeneic blood stem cell transplantation for the treatment of advanced hematologic malignancies. Bone Marrow Transplant. 2000;25(8):807-813.
282 Hegenbart U, et al. Treatment for acute myelogenous leukemia by low-dose, total-body, irradiation-based conditioning and hematopoietic cell transplantation from related and unrelated donors. J Clin Oncol. 2006;24(3):444-453.
283 Stelljes M, et al. Conditioning with 8-Gy total body irradiation and fludarabine for allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia. Blood. 2005;106(9):3314-3321.
284 Hallemeier C, et al. Outcomes of adults with acute myelogenous leukemia in remission given 550 cGy of single-exposure and total body irradiation, cyclophosphamide, unrelated donor bone marrow transplants. Biol Blood Marrow Transplant. 2004;10(5):310-319.
285 Sorror ML, et al. Hematopoietic cell transplantation after nonmyeloablative conditioning for advanced chronic lymphocytic leukemia. J Clin Oncol. 2005;23(16):3819-3829.
286 Kerbauy FR, et al. Hematopoietic cell transplantation from HLA-identical sibling donors after low-dose radiation-based conditioning for treatment of CML. Leukemia. 2005;19(6):990-997.
287 Khoury H, et al. Low incidence of transplantation-related acute complications in patients with chronic myeloid leukemia undergoing allogeneic stem cell transplantation with a low-dose (550 cGy) total body irradiation conditioning regimen. Biol Blood Marrow Transplant. 2001;7(6):352-358.
288 McSweeney PA, et al. Hematopoietic cell transplantation in older patients with hematologic malignancies: Replacing high-dose cytotoxic therapy with graft-versus-tumor effects. Blood. 2001;97(11):3390-3400.
289 Baron F, et al. Graft-versus-tumor effects after allogeneic hematopoietic cell transplantation with nonmyeloablative conditioning. J Clin Oncol. 2005;23(9):1993-2003.
290 Tomblyn M, et al. Similar and promising outcomes in lymphoma patients treated with myeloablative or nonmyeloablative conditioning and allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2008;14(5):538-545.
291 Maris MB, et al. Allogeneic hematopoietic cell transplantation after fludarabine and 2 Gy total body irradiation for relapsed and refractory mantle cell lymphoma. Blood. 2004;104(12):3535-3542.
292 Schmid C, et al. Sequential regimen of chemotherapy, reduced-intensity conditioning for allogeneic stem-cell transplantation, and prophylactic donor lymphocyte transfusion in high-risk acute myeloid leukemia and myelodysplastic syndrome. J Clin Oncol. 2005;23(24):5675-5687.
293 Laport GG, et al. Reduced-intensity conditioning follow by allogeneic hematopoietic cell transplantation for adult patients with myelodysplastic syndrome and myeloproliferative disorders. Biol Blood Marrow Transplant. 2008;14(2):246-255.
294 Hallemeier CL, et al. Long-term remissions in patients with myelodysplastic syndrome and secondary acute myelogenous leukemia undergoing allogeneic transplantation following a reduced intensity conditioning regimen of 550 cGy total body irradiation and cyclophosphamide. Biol Blood Marrow Transplant. 2006;12(7):749-757.
295 Maloney DG, et al. Allografting with nonmyeloablative conditioning following cytoreductive autografts for the treatment of patients with multiple myeloma. Blood. 2003;102(9):3447-3454.
296 Badros A, et al. Improved outcome of allogeneic transplantation in high-risk multiple myeloma patients after nonmyeloablative conditioning. J Clin Oncol. 2002;20(5):1295-1303.
297 Petersen FB, et al. Marrow transplantation following escalating doses of fractionated total-body irradiation and cyclophosphamide—a phase I trial. Int J Radiat Oncol Biol Phys. 1992;23(5):1027-1032.
298 Van Bekkum DW. Conditioning regimens for the treatment of experimental arthritis with autologous bone marrow transplantation. Bone Marrow Transplant. 2000;25(4):357-364.
299 Saccardi R, et al. Autologous stem cell transplantation for progressive multiple sclerosis: update of the European Group for Blood and Marrow Transplantation autoimmune diseases working party database. Mult Scler. 2006;12(6):814-823.
300 Nash RA, et al. High-dose immunosuppressive therapy and autologous peripheral blood stem cell transplantation for severe multiple sclerosis. Blood. 2003;102(7):2364-2372.
301 Samijn JPA, et al. Intense T cell depletion followed by autologous bone marrow transplantation for severe multiple sclerosis. J Neurol Neurosurg Psychiatry. 2006;77(1):46-50.
302 Burt RK, et al. Hematopoietic stem cell transplantation for progressive multiple sclerosis: failure of a total body irradiation-based conditioning regimen to prevent disease progression in patients with high disability scores. Blood. 2003;102(7):2373-2378.
303 Fortun PJ, Hawkey CJ. The role of stem cell transplantation in inflammatory bowel disease. Autoimmunity. 2008;41(8):654-659.
304 Binks M, et al. Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: procedure related mortality and impact on skin disease. Ann Rheum Dis. 2001;60(6):577-584.
305 Farge D, et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis. 2004;63(8):974-981.
306 Nash RA, et al. High-dose immunosuppressive therapy and autologous hematopoietic cell transplantation for severe systemic sclerosis: Long-term follow-up of the US multicenter pilot study. Blood. 2007;110(4):1388-1396.
307 McSweeney PA, et al. High-dose immunosuppressive therapy for severe systemic sclerosis: Initial outcomes. Blood. 2002;100(5):1602-1610.
308 Orchard PJ, et al. Hematopoietic cell therapy for metabolic disease. J Pediatr. 2007;151(4):340-346.
309 Staba SL, et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N Engl J Med. 2004;350(19):1960-1969.
310 Jacobsohn D. Allogeneic transplantation for non-malignant disease in children. Haematologica. 2006;91(6):726A.
311 Macmillan ML, et al. Haematopoietic cell transplantation in patients with Fanconi anaemia using alternate donors: results of a total body irradiation dose escalation trial. Br J Haematol. 2000;109(1):121-129.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
15 Total Body Irradiation
History
Only a decade after Roentgen described the “x-ray,” German biophysical engineer Friedrich J. Dessauer first described a “new technique of radiotherapy” that involved homogenous irradiation of the entire body. In his initial report describing the technique in 1905, he proposed irradiating a supine patient using three simultaneously active low-voltage Roentgen ray sources (Fig. 15-1).1,2 In 1907, Aladár Elfer, a medical professor in Hungary, reported his experience using a TBI technique that spared the head in three patients with leukemia.3 Although there is a paucity of data regarding the early use of the technique, some have speculated that untoward hematologic toxicity probably limited its application.4
FIGURE 15-1 • Diagram demonstrating the total body irradiation technique proposed by Dessauer in 1905.
Early success using TBI to treat hematopoietic and lymphoid malignancies in Europe (there named the Teschendorf method) prompted development of the technique in the United States.5–7 Arthur C. Heublein, in collaboration with Gioacchino Failla, is credited with the development of the first TBI unit in North America, located at Memorial Hospital in New York City. In the United States, the technique became known as Heublein therapy.8 A specially constructed treatment ward was designed to treat four patients at an extended distance (5-7 meters) simultaneously at an exposure rate of 0.7 R per hour for approximately 20 hours per day, typically over 1 to 2 weeks, using a 185 kV x-ray tube at 3 mA, with a 2-mm copper filter. The goal was to deliver 25% of erythema dose (750 R). Remarkably, in Heublein’s initial report, no hematopoietic toxicity was noted with this treatment schedule. Of 12 patients with advanced lymphomas and leukemias, 7 (58%) demonstrated some form of improvement after treatment; 2 of 8 patients (25%) with metastatic breast, melanoma, and kidney cancers were also noted to demonstrate some form of improvement.9,10 A later report of the experience with 270 cancer patients from Memorial Hospital treated with TBI between 1931 and 1940 confirmed that the technique was more successful in patients with hematopoietic and lymphoid malignancies, compared with patients with carcinomas or sarcomas, for whom it was ineffective. The authors emphasized that the technique was safe if doses were prescribed cautiously. They did not recommend exposures greater than 300 R, and noted hematopoietic and gastrointestinal toxicity with exposures as low as 50 to 100 R.8
In the early 1940s, World War II prompted the Manhattan Project, an initiative to develop nuclear weapons. Part of this endeavor sponsored research into the human biologic response to ionizing radiation, including TBI. The military’s interest in TBI was primarily to help understand human tolerance for radiation exposure during occupational duties and warfare, and to develop radiation biodosimetric assays. Several research studies coordinated through the Manhattan Project were initiated in patients with advanced malignancies,11–13 as well as in patients with benign diseases.13 For example, studies of dose escalation, radiation biologic dosimetry, and cognitive and psychomotor function were carried out at M.D. Anderson Hospital for Cancer Research.14 A detailed report of 30 patients treated at the maximum exposure level (200 R) in the initial study concluded that side effects primarily consisted of nausea, vomiting, and myelosuppression, and that intervention was necessary in 10% of patients treated with this dose of TBI.15 At Baylor University College of Medicine, studies using 25 to 250 R of TBI with 250 kV to 2 MV energy machines were performed to find a biologic dosimeter, as well as to study acute effects of radiotherapy.16 The Sloan-Kettering Institute for Cancer Research also participated in similar studies of at least 20 patients, although the results were never published. The military conducted similar studies at the Naval Hospital in Bethesda, Maryland, and reported palliation of patients with radiosensitive diseases treated with fractionated TBI.17
The most recent Department of Defense–sponsored research study of TBI was at the University of Cincinnati and focused on identifying biochemical markers in the urine that predicted response to TBI. Later, studies of the neuropsychiatric effects of TBI were initiated. Ultimately, only results regarding the palliation of advanced malignancies were reported.18 Patients with advanced metastatic radioresistant malignancies for whom chemotherapy was unavailable were often treated with TBI in the absence of any clear anticipated benefit. Patients treated with TBI in this manner were included in research studies, often without consenting to participate. The ethics of this practice were called into question by a report written in 1995 by the United States Department of Energy’s Advisory Committee on Human Radiation Experiments,19 which could be a contributor to the public’s uneasiness with radiation in general.20
TBI was used not only in malignant diseases; it was considered the critical immunosuppressant in the first successful solid organ transplant. In 1959, a kidney was successfully transplanted between dizygotic twins after TBI to 450 R to the recipient.21 Around the same time in France, successful kidney transplants after TBI were being reported.22,23 Of the first seven patients who underwent kidney transplant following TBI or pharmacologic immunosuppression worldwide between 1959 and 1962, the two who did not experience kidney failure were treated with TBI alone (without chemical immunosuppression) prior to transplant, and each survived in excess of 20 years after transplant.21–23 However, successful preclinical studies with pharmacologic therapy prompted the use of chemical immunosuppressants (corticosteroids, 6-mercaptopurine, and azathioprine) for solid organ transplantation after 1963.24
With an increased understanding of the human response to TBI, and a rapidly growing body of preclinical in vivo studies of TBI, therapeutic protocols were developed to maximize benefit in patients with malignant diseases. In 1957, Nobel laureate E. Donnall Thomas first reported the use of bone marrow infusion in humans following whole-body irradiation or chemotherapy,25 and less than one year later he published his experience using TBI with exposures up to 600 R, followed by bone marrow transplantation.26 In the series of the first five patients with leukemia treated with TBI, who then received intravenous infusion of normal donor marrow, Thomas et al. noted the difficulty of acute myelosuppression and resultant hemorrhage and infection during the period leading up to engraftment. The report also comments that low dose rates (delivery over 2-3 days) appeared preferable to higher dose rates, for metabolic and immunologic reasons. In addition, patients receiving 200 to 300 R fared better than those receiving 400 to 600 R. The problem of delivering an adequately homogenous dose was raised, and suggestions to use higher-energy photons were proposed.26 Thomas et al. later reported on syngeneic bone marrow transplantation in two children after 850 to 1140 R was delivered in a single fraction over 22 to 25 hours, using 60Co sources. The authors concluded that 1000 R of TBI did not produce “troublesome” acute radiation sickness, and produced remission of leukemia, but not cure.27 The first report of successful cure of a patient with leukemia with allogeneic transplantation after TBI was reported in 1969. The technique involved opposed 60Co sources, which operated at 5.8 R per minute, to a total exposure of 1620 R, calculated to be 954 rad at midline. With appropriate supportive care, no major acute radiation sickness was noted, but the patient died of overwhelming cytomegalovirus (CMV) infection, without evidence of leukemia.28
Over the next several years, techniques of combining chemotherapy and TBI were developed and refined, with promising results.29,30 Success in the treatment of advanced leukemias and severe aplastic anemia (AA) was achieved. Departure from the use of TBI alone was primarily fostered by the development of more effective cytotoxic chemotherapeutics and immunologic therapies, which, when combined with TBI, yielded fewer leukemic recurrences.29 Although the use of TBI without HSCT has largely been abandoned, primarily for fear of inducing secondary malignant neoplasms (SMNs) and limiting later therapeutic options, some question the validity of this fear and still contend that very low-dose TBI (1.5-2 Gy in 10-20 fractions over several weeks) is a viable option for upfront therapy in advanced indolent lymphomas.31 Fig. 15-2 demonstrates how the use of TBI during HSCT has remained constant or increased during preceding years.32
Background on Hematopoietic Stem Cell Transplant
HSCT has evolved into a highly complex clinical discipline, firmly rooted in immune system and cancer biology, the details of which are beyond the scope of this chapter. When HSCT was initially performed (see “History”), bone marrow was extracted from the donor, filtered, and infused intravenously to the recipient. Later, peripheral blood stem cells, instead of bone marrow, were collected from the donor, and found to be an alternative to bone marrow grafts. For this reason, “bone marrow transplants” are now more appropriately termed hematopoietic stem cell transplants, because the critical component of the graft is the hematopoietic stem cell (HSC), independent of the source. Oftentimes, peripheral blood stem cells are “mobilized” from the donor using hematopoietic growth stimulating factors, and removed from the donor by apheresis. In addition, recent success using HSCs derived from human umbilical cord blood has been described. Depending on the source of HSCs, various postcollection processing measures (cell selection and depletion) may be undertaken to optimize the outcome of the transplant.
When transplant occurs between different individuals, the hematopoietic graft is said to be allogeneic. This is in contradistinction to reinfusion of native HSCs back into the donor in an “autologous” transplant, more appropriately termed an autoplant, because nothing is being transferred between different individuals.33 A rare but alternative situation is of a patient with a genetically identical twin in the case of a “syngeneic” transplant. Of these three methods of HSCT, autologous and syngeneic transplant are generally associated with less risk because issues related to immunocompatibility are minimized. Allogeneic transplants requiring the “matching” of donor and recipient are typically carried out through identification of human leukocyte antigen compatibility. The donor may be related to the recipient, or identified through registries of volunteers, such as the National Marrow Donor Program.
Prior to undergoing HSCT, most patients require intensive antineoplastic or immunosuppressive therapy in preparation for HSCT, often referred to as conditioning. Conditioning can involve cytotoxic chemotherapy, immunosuppressants, antibody therapy, or radiation therapy. The nature of the conditioning regimen can be referred to as high or reduced intensity, myeloablative, submyeloablative, or nonmyeloablative, to carry out conventional or mini transplants.33 Although agreed-upon formal definitions of these regimens do not exist,34 the goal of high-intensity, myeloablative, or conventional transplant is to completely eliminate the recipient’s native HSC compartment, which necessitates HSCT (autologous or allogeneic) for survival. High-intensity, myeloablative, or conventional transplants may or may not involve TBI, to high doses (>5 Gy in a single fraction, >8 Gy in multiple fractions). Reduced-intensity, nonmyeloablative, or mini-transplant conditioning regimens are often used for older patients or for those with medical problems for whom a high-intensity, myeloablative, or conventional transplant would cause excessive morbidity or mortality, and may or may not involve TBI to lower doses. During and immediately after conditioning, the transplant recipient is at significant risk of infections and other hematologic complications. For this reason, the supportive care of HSCT recipients is very complex, and should only be undertaken in specialized facilities. Nonetheless, some groups have developed reduced-intensity and myeloablative HSCT regimens, including TBI, which have been safely undertaken on an outpatient basis.35,36
According to data summarized by the Center for International Blood and Marrow Transplant Research (CIBMTR) in 2005, the diseases most commonly treated with HSCT are (in decreasing order of frequency): multiple myeloma (MM); non-Hodgkin lymphoma (NHL); acute myelogenous leukemia (AML); Hodgkin disease (HD); acute lymphoid leukemia (ALL); myelodysplastic and myeloproliferative disease (MDS); chronic myelogenous leukemia (CML); AA, and various other leukemias, cancers, and nonmalignant diseases.37 The 2009 National Comprehensive Cancer Network (NCCN) guidelines for therapy indicate that allogeneic or autologous HSCT may be a treatment option for AML, MM, MDS, CML, HD, and NHL, depending on the clinical situation. Detailed discussion of these diseases and their management is beyond the scope of this chapter, and the reader is referred to appropriate chapters in this text and others for further review.38–41 The role of TBI in nonmalignant diseases is discussed further below.
Radiobiology
Preclinical studies have helped define some of the fundamental radiobiologic properties of the normal lymphocytes. The D0 of normal lymphocytes has been reported to be 0.5 to 1.4 Gy,42–45 depending on the in vitro or in vivo model used to calculate this parameter. This D0 suggests that normal lymphocytes cells are very sensitive to ionizing radiation. A very small shoulder on the radiation cell survival curve has been noted,46,47 suggesting little repair between fractions of radiation. Clinical data have revealed similar findings in patients undergoing hyperfractionated TBI (HTBI), with lymphocyte survival demonstrating an effective D0 of 3.8 Gy according to one study.48 Other radiobiologic phenomena have been ill-defined in other normal hematopoietic cells. Radiobiologically relevant levels of hypoxia are unlikely in the hematopoietic compartment. Repopulation is not likely to influence hematopoietic cell survival, given the short duration of most TBI regimens (1-5 days), although, given the variable lifespan of leukocytes (days to years), it may be of some relevance. Redistribution appears to be of significance, given the time scale for TBI; however, this has been difficult to assess.49
The radiobiology of malignant hematopoietic cells has been described. The D0 of leukemic cells generally ranges between 0.8 and 1.5 Gy, however, compared with normal hematopoietic cells, a wider range of radiosensitivities have been described.50–61 Many have cited the technical nuances and variations in assay technique for this great range.49,62 Similar to normal hematopoietic cells, their malignant counterparts are thought to demonstrate little sublethal damage repair,63–67 although split-dose and low-dose-rate experiments have demonstrated the capacity for leukemic cells to repair radiation-induced damage.55,58,68,69 Generally, leukemic cells are believed to have a cell survival curve with minimal or no shoulder, although this varies across cell types and cell lines.47,51,54 For example, Cosset and colleagues summarized preclinical and clinical findings, concluding that AML demonstrates little repair, whereas CML does demonstrate repair, and ALL, myeloma, and lymphomas have not been well studied but appear to demonstrate a wide range of repair capacity.62,70 Similar to normal hematopoietic cells, reoxygenation is unlikely to be radiobiologically relevant to malignant hematopoietic cells during TBI. Redistribution and repopulation, however, may be relevant, but have not been systematically studied.
In vivo preclinical research laid the foundation for the first successful HSCT in humans. Studies in rats,71 dogs,72 and nonhuman primates73 demonstrated that reconstitution of the hematopoietic system was possible after TBI with supralethal doses of radiation. Later work in animals revealed that delivering TBI in several fractions required a higher total dose relative to the biologically isoeffective dose given in a single fraction.74–76 Another model demonstrated no significant difference in the effect of a low-dose-rate (0.04 Gy/min) single fraction of TBI compared with a hyperfractionated course of TBI given thrice daily to the same total dose.77
Although the hematopoietic system is the target of TBI, normal tissues effectively limit the dose that can be safely delivered. The sparing of normal tissues with fractionated TBI was proposed by Peters and colleagues,63,64 and subsequently supported by preclinical data in mice78,79 and dogs80 showing less lung injury with fractionated TBI regimens.
Immediate Toxicity and Management
Although a good deal of what has been learned about the acute in vivo biologic effects of TBI is derived from laboratory-based animal studies, whole-body irradiation also has been studied in people exposed during accidental or wartime nuclear events.81,82 These large-scale studies are valuable because they deal with apparently normal subjects; however, the retrospective nature limits the quality of the data. The reader is referred to several excellent texts for a review of the acute and fatal radiation syndromes (gastrointestinal, hematopoietic, and cerebrovascular) that can be caused by TBI in an uncontrolled setting.83–85
Acute side effects of therapeutic TBI can be difficult to distinguish from other HSCT-related morbidity. However, Chaillet and colleagues conducted a very informative prospective clinical study of the symptoms and signs that occur in patients after TBI, prior to the initiation of any other HSCT related therapy.86 Thirty-one patients between 4.5 and 55 years of age were treated using parallel-opposed anteroposterior (AP) 18 MV photons from a linear accelerator (linac). Shielding was used to limit the lung dose to 8 Gy. A total dose of 10 Gy was given as a “single dose” as six discrete fractions of 1.6 Gy, each given over 15 minutes, with a 30-minute break between fractions, for a mean dose rate of 0.04 Gy per minute and instantaneous dose rate of 0.11 to 0.12 Gy per minute. Symptoms and signs were assessed regularly during the 4 hour TBI, and for 20 hours after the completion of TBI. Antiemetics, but no chemotherapy or steroids, were given prior to the start of TBI. Table 15-1 displays the symptoms and signs experienced by patients during the 4 hours of TBI, and within 24 hours of starting TBI. Fever was a very common finding, and a maximum of 40.8° C was noted in one patient. Tachycardia frequently paralleled febrile episodes; a maximum rate of 130 beats per minute was noted. Drowsiness was noted only in patients who received sedating antiemetics. Parotid gland pain was common, and bilateral parotid swelling was noted in 29% of patients. Marked lacrimation was noted in 6% of patients, whereas 16% of patients experienced ocular dryness. Two patients experienced mild conjunctival edema. Hypertension was noted only during TBI. The results of a similar study conducted by Buchali and colleagues of patients who were treated with a fractionated course of TBI delivered mostly to a total dose of 12 Gy using 2 Gy per fraction, twice daily, 8 hours apart, with lung doses limited to 10 Gy, is also summarized in Table 15-1.87
A prospective clinical study showed that fractionation of TBI can reduce acute nausea, vomiting, mucositis, diarrhea, and parotitis, although these differences were not statistically significant. Late cutaneous eruptions were more common in patients undergoing fractionated TBI, although this was not statistically significant. This same study, which randomized patients to high- or low-dose-rate TBI, revealed no differences in the acute toxicities mentioned when comparing dose rate.88 Another randomized controlled trial (RCT) reported that fractionating TBI revealed “no apparent difference in acute toxicity” compared with single-fraction TBI, with both regimens being “well-tolerated.”89
Older studies cite nausea and vomiting as frequent side effects of TBI. These symptoms have been substantially minimized with the advent of more effective antiemetics, such as the 5-hydroxytryptamine (serotonin) receptor-3 (5HT-3) antagonists. Several small, high-quality controlled clinical studies support the prophylactic use of 5HT-3 antagonists to reduce nausea and vomiting during TBI, and are summarized in Table 15-2.90–94 The use of corticosteroids in conjunction with 5-HT3 antagonists is supported by a trial listed in Table 15-2; however, given the toxicity associated with this approach, consensus regarding routine administration in conjunction with TBI is lacking.95–97 Of note, less nausea and vomiting has been noted in myeloablative conditioning regimens involving TBI, compared with those which use chemotherapy alone, even with modern antiemetics.98
Table 15-2 Randomized Controlled Trials of Prophylactic Antiemetics in Patients Undergoing Total Body Irradiation
Oral mucositis is a side effect of TBI in up to 75% of patients undergoing myeloablative TBI, causing mouth pain, odynophagia, necessitating intensive supportive care such as total parenteral nutrition and opioid analgesics.99 In one study, intensive dental hygiene conferred a reduction in the rate of moderate and severe mucositis, although the authors felt it to be clinically insignificant.100 Topical oral agents, such as chlorhexidine digluconate and neutral calcium phosphate in conjunction with topical fluoride treatments, can decrease the duration and severity of oral mucositis as well as pain and the need for opioid analgesics.101–103 Similarly, prophylactic oral sucralfate and clarithromycin have reduced moderate and severe oral mucositis rates.104,105 When given prophylactically, one study showed that amifostine limited the duration of mucositis, with an associated decrease in the rate of moderate and severe infections, with no effect on HSCT outcome.106 One study noted that short-term intravenous recombinant granulocyte-macrophage–colony stimulating factor decreased rates of moderate to severe mucositis,107 whereas another found no effect when this agent was delivered topically.108 Recently, Spielberger et al. reported the results of a trial of the recombinant human keratinocyte growth factor, palifermin, given before and after conditioning with 12 Gy of fractionated TBI. Palifermin reduced the rate and duration of moderate and severe mucositis by 35% and 3 days, respectively, and decreased mouth and throat pain, as reflected in reduced morphine usage and decreased the need for total parenteral nutrition (by 24%).109 This study dealt only with patients undergoing autologous HSCT; however, in the setting of TBI for allogeneic HSCT, palifermin may also confer a protective effect on the mucosa, although this has not been studied in an RCT.110
Skin erythema may also be noted toward the end of a course of TBI; desquamation is rare. Hyperpigmentation may be noted in the long term. Alopecia typically occurs 7 to 14 days after TBI is complete, and hair typically returns 3 to 6 months after treatment.111 Changes in the color or texture of regrown hair have been noted by some. Of note, myeloablative conditioning regimens using chemotherapy alone have noted a significantly higher incidence of permanent alopecia.112
Later Toxicity and Management
As previously mentioned (see “Radiobiology”), the hematopoietic system is particularly sensitive to TBI, and lymphopenia is often seen with doses of 0.5 Gy, and can be seen with doses of 0.3 Gy. Lymphopenia is typically followed by neutropenia, thrombocytopenia, and finally anemia. Soon after a TBI dose of 4 to 6 Gy, lymphocytosis can be seen, but typically is followed by neutropenia within 1 week. Neutrophils fall to their minimum 3 to 4 weeks after TBI (Fig. 15-3).113 Regeneration of the HSC compartment depends on the total dose used, because higher doses cause more rapid myelosuppression, of greater duration. Administration of hematopoietic growth factors after TBI have the theoretical potential to alter hematopoietic system reconstitution, although reports in the setting of allogeneic HSCT have demonstrated an increased risk of GVHD and compromised survival,114 and therefore routine use is controversial.115,116 Of note, hematopoietic growth factors have only been used in the post-TBI period, given the concerns raised by a trial in lung cancer, in which growth factors increased pulmonary toxicity and thrombocytopenia when given concurrent with chemoradiation therapy.117
FIGURE 15-3 • Representative hematologic response to total body irradiation, given as a single fraction of 200 cGy on day 0.
(From Andrews GA: Radiation accidents and their management, Radiat Res Suppl 7:390–397, 1967.)
As noted previously, salivary glands frequently are affected by TBI. Although acute parotitis is typically self-limited and can be managed with anti-inflammatory medicines, long-term salivary gland dysfunction can result in xerostomia, which may lead to dental caries. In a study of children who underwent allogeneic HSCT, the risk of developing impaired salivary function was 22% in those who receive TBI as part of conditioning versus 1% in those who did not.118 Studies have shown that salivary flow can improve up to 1 year after the completion of TBI.119 Fractionating TBI was shown to reduce salivary dysfunction by 54% in one study.120 Myeloablative conditioning regimens with and without TBI have been associated with tooth development abnormalities in children.120–122 In one series, myeloablative conditioning regimens using chemotherapy alone were associated with significantly higher rates of tooth developmental abnormalities than those involving TBI, although rates of salivary gland dysfunction were highest amongst the patients treated with single-fraction TBI.123 Because of the increased risk of oral pathologic conditions associated with TBI, careful pretransplant evaluation by a dental specialist is recommended to minimize the risk of serious morbidity.124 Pilocarpine has been noted to help relieve symptoms of xerostomia in patients treated with TBI.125
The major dose-limiting toxicity of TBI is pneumonopathy, which can manifest early as pneumonitis and later as pulmonary fibrosis. In the setting of HSCT, radiation pneumonopathy can be difficult to distinguish from other causes of pathologic conditions of the lung; moreover, lung damage is likely multifactorial, with risk of acute lung complications estimated to be 30% to 60%, depending on factors like infection, conditioning regimen, GVHD, age, and diagnosis (see Peters and Afessa126 for review). Likewise, late pneumopathy occurs in 10% to 26% of patients, and is associated with underlying lung dysfunction, type of conditioning regimen, acute and chronic GVHD and prophylaxis, donor and recipient age and immunocompatibility, stage of disease, and genetic predisposition (see Patriarca et al.127 for review). TBI has been shown to be a risk factor for the idiopathic pneumonia syndrome128 as well as diffuse alveolar hemorrhage.129 Although rates of pneumonopathy in patients receiving TBI vary widely (10%-84%),130 some series have reported pneumonitis in up to 20% of HSCT patients who never received TBI.131 In the modern era, with appropriate TBI techniques, the risk of pneumonopathy in patients treated with TBI may not be increased at all.132 Nevertheless, the significance of the problem is clear, given that mortality related to interstitial pneumonitis in patients treated with TBI can be 60% to 80%.131,133,134
Several TBI-specific factors (total dose, fractionation, dose rate, and the use of lung shielding) have been shown to have significant bearing on the development of pulmonary complications. The total dose used during TBI has frequently been cited as a major factor influencing lung complications.130,135,136 In two prospective RCTs using 12 Gy versus 15.75 Gy, higher rates of mortality were noted within the first 6 months in patients treated with 15.75 Gy, although pulmonary complications were not specifically cited as the excess cause of deaths.137–140 In a retrospective dosimetric study, mean lung dose larger than 9.4 Gy was found to be an independent predictor for lethal pulmonary complications in patients receiving TBI to a total dose of 10 Gy in three daily fractions, at 0.055 Gy per minute using parallel opposed lateral fields.141 Two RCTs have demonstrated that fractionated TBI can reduce pneumonitis, compared with single-fraction TBI, although only one study was statistically significant.89,142,143 A retrospective study found no difference in pneumonitis rates when comparing a single fraction of 6 Gy and three daily fractions of 3.33 Gy, suggesting that total doses smaller than 10 Gy may not require fractionation to prevent toxicity, although no randomized data support this.144 The necessity of hyperfractionation to prevent lung toxicity is unclear: a comparison of two prospective single-arm trials at the same institution revealed that conventional fractionation given with AP fields and lung blocks to a total dose of 12 Gy in daily 3-Gy fractions may not be any different than HTBI given twice daily with 1.7 Gy per fraction to a total dose of 10.2 Gy for 3 days, using parallel opposed lateral fields and no blocks.145 Sampath et al. recently reviewed 26 studies involving 1096 patients to create a dose-response model to predict risk of pneumonitis from TBI, taking other factors into consideration. Although unable to estimate the risk of pneumonitis for hyperfractionated regimens, they were able to determine the effect of fractionation and cyclophosphamide (CY) and busulfan (Bu) on the risk of developing pneumonitis, in a dose-response model, as seen in Fig. 15-4.146 Pneumonitis rates were not significantly different in a trial that randomized patients to high- or low-dose-rate TBI.88 However, there is an abundance of retrospective clinical data suggesting that lowering the dose rate (<0.025-0.09 Gy/min) does decrease the likelihood of pulmonary complications,130,136,140,147,148 especially if TBI is delivered as a single fraction.149 If TBI is fractionated, some report that low dose rate (<0.069 Gy/min) is not necessary,134 although others found a beneficial effect.150,151 Studies of patients receiving fractionated TBI with lung shielding have demonstrated a reduction in pneumonopathy152,153; however, one RCT found no difference in pneumonopathy if the shielding allowed a lung dose of either 6 or 8 Gy in a single fraction.154
Pulmonary function tests (PFTs; spirometry and diffusion capacity) are often helpful in the assessment of patients with pulmonary symptoms or radiographic abnormalities. Studies of PFTs in patients treated with HSCT have demonstrated a deleterious effect on spirometry and diffusion capacity, which often resolves in the absence of other complicating factors,155–157 and is related to TBI dose.158 A retrospective study found that lung shielding had a small but significantly beneficial effect on PFTs one year after HSCT, especially in patients with abnormal function prior to HSCT.159 In one study, Bu, but not TBI, was not associated with a negative effect on PFTs.160 There is no evidence that PFTs improve pulmonary outcome after HSCT in adulthood, and for this reason, they are not recommended.161 However, some groups recommend baseline PFTs as part of long-term follow-up care for children treated with TBI.162 Counseling patients regarding smoking cessation is of critical importance in all patients, especially those who are at increased risk of developing lung injury. In the event of acute pneumonitis, high-dose steroids (30-60 mg of prednisone/day) typically alleviate symptoms within 24 to 48 hours.
Cardiovascular toxicity as a result of HSCT has been relatively rare, given the stringent selection criteria for patients treated with this aggressive therapeutic modality. Nevertheless, among patients who survive HSCT, cardiac events were responsible for 2.4% and 3.0% of deaths in patients that underwent autologous and allogeneic HSCT, respectively, which represents a greater than expected occurrence in some patients.163,164 Most of the recent literature has identified no association between TBI and development of cardiovascular disease.165,166 Several detailed prospective analyses using plasma cardiac troponin and brain natriuretic peptide levels, electrocardiography and echocardiography revealed no evidence of cardiac dysfunction in previously healthy individuals treated with TBI.167,168 However, a prospective study of children who underwent allogeneic HSCT found a 12% and 26% cumulative incidence of abnormalities in ejection fraction (<30%) on echocardiography prior to and five years after HSCT, respectively. This study revealed that TBI was associated with abnormalities in cardiac function on univariate analysis, but not multivariate analysis; the five-year cumulative incidence of cardiac abnormality was 26% in children treated with TBI with prior anthracycline therapy and 2% in children treated with TBI without prior anthracycline therapy.169 For these reasons, it appears prudent to screen patients who have undergone HSCT for cardiovascular disease or risk factors, whether or not this involved TBI, to minimize late morbidity and mortality.
Hepatotoxicity from TBI manifests primarily as veno-occlusive disease (VOD), also known as sinusoidal obstructive syndrome, of the liver. This clinicopathologic phenomenon was first described in 1977 by Shulman et al.,170 who noted the onset of weight gain from ascites, painful hepatomegaly, and jaundice from centrilobular liver acinus necrosis 1 to 4 weeks after HSCT. Overall, up to 70% of patients that undergo HSCT can be affected by VOD. TBI, along with many other risk factors, has been implicated in the development of VOD (see Wadleigh et al.171 for review). Notably, a RCT and meta-analysis found that patients treated with Bu instead of TBI were significantly more likely to develop VOD.172,173 Two RCTs have concluded that fractionated TBI reduces the incidence of VOD compared with single-dose TBI.142,143 Another RCT found no difference in the rate of VOD when the dose rate was either 0.06 Gy per minute or 0.15 Gy per minute,88 although a retrospective study of single-dose TBI found that dose rates of 0.07 Gy per minute were associated with less VOD than dose rates of 0.18 to 1.2 Gy per minute.174 TBI dose greater than 13.2 Gy has been reported to be associated with higher rates of VOD on univariate analysis,175 although dose greater than 12 Gy was not associated with VOD in another retrospective study.176 Lawton et al. reported a non–statistically significant 10% decrease in the rate of fatal VOD in patients treated with TBI as part of HSCT, when a 10% attenuation liver block was employed.177 Ursodeoxycholic acid was effective in preventing VOD in a RCT of patients undergoing TBI followed by HSCT,178 although another RCT did not support this finding.179 Reduced-intensity conditioning regimens may also prevent VOD. Treatment of VOD can include the fibrinolytic antithrombotic agent, defibrotide; decompressing the sinusoids by a transjugular intrahepatic portosystemic shunt and liver transplantation are more invasive options for management of severe disease.180
Cataracts are one of the most common complications of TBI. Patients may present with painless vision loss, and may be noted to have opacification of the lens on examination. In one series of patients treated with TBI in one or two fractions, severe visual impairment was noted in approximately half of patients.181 This problem has been noted to arise in a large proportion of patients treated with TBI, depending on total dose, fractionation, and dose-rate used. When considering risk factors associated with HSCT, steroid use,182–184 prior cranial irradiation,185,186 and the development of GVHD184 have been shown to predispose to cataractogenesis, whereas heparin use appears to be protective.187 Delivering TBI in a single fraction is the single biggest risk factor for developing a cataract after HSCT.187–192 High dose rate (>0.035-0.048 Gy/min) TBI also appears to increase the risk of cataract formation.185,187,190,193 A prospective study that randomized patients to high- or low-dose-rate TBI found that the incidence of cataract 5 years after treatment was 12% and 34% in the low- and high-dose-rate arms, with 13% and 39% of the cataracts occurring in patients who received fractionated or single-dose TBI, respectively.194 Kal and van Kempen-Harteveld recently reviewed the subject of TBI-induced cataracts and concluded that a biologically equivalent dose of 40 Gy yields a 10% chance of developing cataracts, using linear-quadratic modeling that included corrections for dose rate, with an α/β of 0.65 for late effects on the lens. On the basis of this, they suggest considering lens shielding for single-fraction TBI regimens,195 although this is controversial in the setting of malignant disease.196 Given the frequency of cataracts occurring after TBI, patients should be monitored for the development of this complication.197 Management of cataracts that impair vision or degrade quality of life may involve phacoemulsification or extraction; recent data suggest these procedures are safe, with an adverse event rate of 0.1% for experienced surgeons,198 and effective, with a 90% chance of 20/40 vision postoperatively.199
Kidney dysfunction occurs in approximately 17% of survivors of HSCT,200 and can manifest in a number of ways, with the most- to least-frequent syndromes being idiopathic chronic kidney disease, nephrotic syndrome, thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome), and acute renal failure (see Hingorani201 for review). The syndrome best associated with TBI is thrombotic microangiopathy, which can manifest as nephritis, hypertension, proteinuria, or anemia 6 to 12 months after HSCT. HSCT-related risk factors for nephropathy can include GVHD, infections with CMV or BK virus, nephrotoxic medications like cytotoxic chemotherapy (cytarabine, CY, ifosfamide, cisplatin, retinoic acid, carmustine, actinomycin D, melphalan), antibiotics (acyclovir, ganciclovir, foscarnet, vancomycin, amphotericin, aminoglycosides), and immunosuppressants (cyclosporine, tacrolimus, methotrexate). It is worth noting that the larger and more contemporary studies of chronic kidney disease after HSCT, have not demonstrated an association with TBI.202–206 Total dose has been implicated as the most important in predicting renal morbidity from TBI; a retrospective study found that GVHD and high dose (13.5 Gy) was associated with elevated serum creatinine.207 A prospective evaluation of renal function using radioisotopes found that early nephropathy was associated with age younger than 40 years, use of kidney blocks (possibly related to nephrotoxic contrast media given during simulation), and nephrotoxic drug use, while late nephropathy was associated with nephrotoxic drug used, but not TBI dose.208 The benefit of kidney shielding has been assessed in two retrospective studies, which both demonstrated significant improvement in long-term kidney function, with no evidence of dysfunction when the hyperfractionated dose was limited to 9.8 to 10 Gy.209,210 Two recent dose-effect modeling studies have demonstrated that nephropathy is unlikely after a biologically equivalent total dose of 16 Gy (calculated using a linear quadratic model, with corrections for dose rate and an α/β of 2.5 Gy) and that fractionating TBI and delivering a low dose rate (<0.10 Gy/min) prevent kidney dysfunction.195,211 Monitoring blood chemistry and counts, as well as blood pressure and urine studies are advised given the prevalence of kidney disease after HSCT. Although there are no proven therapies to treat HSCT-related nephropathy, medical therapies (antihypertensives, corticosteroids), plasma exchange, hemodialysis and renal transplant, and are possible options for management (see Hingorani201 for review). The angiotensin-converting enzyme inhibitor captopril is known to prevent chronic nephropathy from diabetes212 and was been hypothesized to reduce the risk of radiation nephropathy from TBI in preclinical studies.213,214 A small RCT of captopril to mitigate chronic renal failure in patients undergoing 12 Gy of fractionated TBI (with kidney dose of 9.8 Gy) prior to HSCT revealed less nephropathy; however, this was not statistically significant.215
Primary hypothyroidism is the most common endocrinopathy following TBI with an incidence of approximately 25% after fractionated therapy.216 The incidence is significantly higher in series of single-dose TBI and is known to increase with longer follow-up. It has been suggested that adults have a lower risk of hypothyroidism than children and that some patients develop only transient hypothyroidism. This condition is easily managed with thyroid hormone replacement. Following TBI, central hypothyroidism caused by thyrotropin (thyroid-stimulating hormone) deficiency is rare, as is deficiency of corticotropin (adrenocorticotrophic hormone), and hypogonadotropic hypogonadism.
Impaired growth and loss of adult height has been described as a consequence of TBI during childhood. This effect is multifactorial because radiation can cause poor nutritional conditions, skeletal dysplasia, and also temporary or permanent growth hormone (GH) deficiency. The effects of TBI on growth are inversely related to patient age at the time of treatment. Both the total dose and dose per fraction of TBI have shown to be related to the risk of growth impairment. These factors are also important when considering GH replacement for children proven to have deficiency. Sanders et al. observed that GH replacement was not effective after single-dose TBI or when TBI was delivered after age 10 years.217
Typical TBI regimens allow preservation of testicular Leydig cell function and therefore normal testosterone levels. However, almost all males will develop azoospermia as a result of germ cell irradiation during TBI. Although male patients are universally counseled to expect infertility, there are rare reports of spermatogenic activity in men many years after exposure to TBI during childhood. Age plays an even more important role in female fertility following TBI. Although approximately half of prepubescent girls who receive TBI will undergo spontaneous puberty, many will experience premature menopause in their twenties or thirties.218 Ovarian failure is expected in virtually all females receiving TBI after age 10 years. For the small percentage of women who become pregnant after TBI, some investigators have reported an increased risk for spontaneous abortion, whereas others do not.
Other endocrine and metabolic complications of HSCT include insulin resistance, lipid disorders and hypertension. Both type 1 and type 2 diabetes mellitus are associated with TBI.219The possible mechanisms involved are active areas of investigation.
Declines in neuropsychiatric function following TBI have been observed, but are subtle because of relatively low doses and modern fractionation. A recent prospective study at St. Jude tracked 158 children who had undergone HSCT, of whom 80 had received full-dose fractionated TBI. Patients were given a battery of neurocognitive examinations at 1, 3, and 5 years after transplant. Although TBI was associated with a 3-point decline in IQ compared with non-TBI patients, this difference was small compared with the 20 point difference observed between children from high and low socioeconomic groups.220
Beyond organ-specific toxicities, the risk of developing SMNs is increased in patients that have undergone HSCT. Typically, three groups of SMNs after HSCT are described, and follow a distinct pattern of development (Fig. 15-5):221 MDS and AML, post-transplant lymphoproliferative disorder (PTLD), and solid tumors.222 There are multiple risk factors that may predispose to development of a SMN after HSCT, including but not limited to genetic aberrations, pre-HSCT therapy, conditioning regimen, graft source and processing, post-transplant immunosuppression, and GVHD (for review see DeVita et al.38); only the association of TBI with SMNs is discussed here.
Secondary MDS or AML after HSCT has been reported to occur at rates between 1.1% at 20 months to 24.3% at 43 months. The World Health Organization classifies secondary MDS and AML as alkylating agent– or radiation-related, typically occurring 4 to 7 years after treatment, or topoisomerase-II inhibitor–related, typically occurring 6 months to 5 years after treatment.223 Several studies have attempted to quantify the risk of developing secondary MDS or AML after undergoing TBI, with conflicting results. Studies from research groups from Minnesota,224 Newcastle,225 the City of Hope,226 and France227 have demonstrated no increase in rates of MDS or AML after myeloablative HSCT using TBI. Other studies from Nebraska,228 the European Group for Bone Marrow Transplantation (EBMT),229 Paris,230 Barcelona,231
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
