[level-membership-for-basic-science-category]
Topical and transdermal drug delivery
Adrian C. Williams
Chapter contents
Permeant properties affecting permeation
Mathematics of skin permeation
Experimental methods for studying transdermal drug delivery
Transdermal and topical preparations
Key points
• Transdermal drug delivery the skin to deliver the drug to the systemic circulation.
 between 1 and 4, and an effective daily dose of < 10 mg/day.
between 1 and 4, and an effective daily dose of < 10 mg/day.• Patches, of varying complexities, are the most common transdermal drug delivery systems.
Introduction
Topical and transdermal formulations have a long history of use. Over 2000 years ago, Greek physicians used formulations containing salt, vinegar, honey and resins to treat skin lesions and ulcers. Chinese, Egyptian and Roman medical histories describe numerous remedies applied topically as pastes and poultices.
Topical and transdermal products remain key formulations for delivering drugs not only to the skin, but also through it for systemic action. An estimated 40 million topical items are dispensed annually in England and Wales, including 16 million emollient products and 13 million topical corticosteroid preparations. In addition, many other products are dispensed for topical anaesthesia and antisepsis or for transdermal delivery, such as fentanyl patches. Additionally, ‘over-the-counter’ products are widely sought by patients and range from emollients to non-steroidal anti-inflammatory creams and gels, to treatments for warts, verrucae and fungal infections, such as athletes’ foot. Thus, pharmacists often supply topical and transdermal formulations which contain a broad variety of active ingredients; indeed it has been reported that up to 20% of all repeat prescriptions are for application to the skin. The efficacy of these products is critically dependent on biological factors, such as the integrity of the skin, on the physicochemical properties of the active ingredient and on the formulation designed to release and deliver the active into or across the skin.
Terminology
The literature occasionally contains different terminology relating to transdermal and topical drug delivery. For this Chapter, it may be helpful to clarify the following at this point:
Topical drug delivery.
The application of a formulation to the skin to treat a local disorder, i.e. the intention is to retain the active pharmaceutical ingredient (API) within the skin, for example, a locally acting hydrocortisone cream.
Transdermal drug delivery.
The application of a formulation to the skin to deliver a drug to the systemic circulation, for example, estradiol patches.
Locally acting.
The active pharmaceutical ingredient acts directly on the skin.
Regionally acting.
The active pharmaceutical ingredient acts in the area close to where the formulation is applied. This is often also described as locally acting, but here the drug does not act directly on the skin, for example topically applied ibuprofen gels to treat musculoskeletal conditions.
Permeant.
The chemical species that is moving through or into the tissue. This will be the drug, but may also be other ingredients within the formulation.
Permeation.
Movement of drug through the membrane.
Penetration.
Entry into the tissue. Penetration does not necessarily require the molecules to pass out of the tissue.
Diffusion.
Movement of molecules through a domain, from high concentration to low concentration, by random molecular movement.
Diffusivity.
This is a property of the permeant in the membrane and is a measure of how easily it will traverse through the tissue. It is expressed in units of area/time (usually cm2/h or cm2/s).
Diffusion coefficient (D).
This is the diffusion coefficient of the permeant, and is sometimes a term used interchangeably with diffusivity. As with diffusivity, its units are area/time (usually cm2/h or cm2/s).
Permeability coefficient (kp).
Describes the speed of permeant transport, given in units of distance/time (usually cm/h).
Partition coefficient (P).
This is a measure of the distribution of molecules between two phases. For transdermal delivery studies, a partition coefficient (usually expressed as a log10, hence ‘log P’) between octanol and water is often used as a guide to how well a molecule will distribute between water and stratum corneum lipids. In some texts, the symbol K is used for the partition coefficient; here, and to avoid confusion with the permeability coefficient (kp), the symbol P (also widely used in the literature) has been employed.
Partitioning.
The process of molecules distributing between two domains. In transdermal drug delivery, partitioning is generally used to describe molecular redistribution from one domain to another, such as from an aqueous domain to a lipid domain.
Flux (J).
The rate of a permeant crossing the skin (or entering the systemic circulation). It is given in units of mass/area/time (usually µg/cm2/h).
Lag time (L).
This is obtained from a permeation profile by extrapolating the steady state flux line to the time axis. Older texts have used the symbol τ whereas others have used tL for the lag time, but most modern texts use the abbreviation L.
Vehicle.
The base formulation in which the drug is applied to the skin.
Thermodynamic activity.
Used here as a measure of the ‘escaping tendency’ of a molecule from its formulation. A thermodynamic activity of 1 equates to a saturated solution, or suspension, since the molecules in a saturated solution have the greatest ‘escape tendency’.
Skin structure and function
Human skin is a highly complex multilayered organ designed to ‘keep the outside out and the insides in’. It is the largest organ of the body, comprising around 10% of the body mass and covers an area of approximately 1.8 m2 in a typical adult. As a self-repairing barrier, skin permits terrestrial life by preventing the ingress of microorganisms and chemicals whilst regulating heat and water loss from the body. Indeed, the body continually loses water and transepidermal water loss (TEWL) is in the region of 1 mg/cm2/h, but its value varies with body site and external conditions (temperature, humidity).
For drug delivery and therapy, the intact skin presents a formidable barrier and a difficult challenge to formulation scientists. The properties of the skin limit the range of active ingredients that can be delivered through the barrier to achieve therapeutic levels. However, skin can be relatively easily damaged through mechanical, chemical or microbiological assault and by radiation, such as sun damage. In these cases, drug delivery may be enhanced and could in fact lead to adverse reactions.
Structure of the skin
In terms of drug delivery, human skin can be considered as a series of layers which potentially provide a series of barriers to a molecule traversing the tissue (Fig. 39.1).
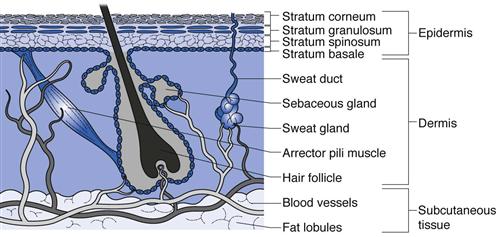
Fig. 39.1 A diagrammatical cross-section through human skin showing the different skin layers and appendages.
The subcutaneous layer
The inner subcutaneous fatty layer is typically several millimetres thick, except for some areas such as the eyelids where it is mostly absent. This subcutaneous layer of adipose tissue provides mechanical protection against physical shock, insulates the body, provides a store of high-energy molecules and carries the principal blood vessels and nerves to the skin. The subcutaneous layer is seldom an important barrier to transdermal and topical drug delivery.
The dermis
Overlying the fatty layer is the dermis, a layer typically 3–5 mm thick that is the major component of human skin. The dermis is composed of a network of mainly collagen and elastin in a mucopolysaccharide gel; essentially this combination provides an aqueous environment similar to a hydrogel. The dermis has several structures embedded within it, termed appendages, in particular nerve endings, pilosebaceous units (hair follicles and sebaceous glands) and eccrine and apocrine sweat glands (see below).
The dermis is metabolically active and requires extensive vasculature for this, as well as for regulating body temperature, for wound repair, to deliver oxygen and nutrients to the tissue and to remove waste products. The blood supply reaches to approximately 0.2 mm below the skin surface, near the dermis-epidermis boundary, and so most molecules passing through the outer layer of the skin are rapidly diluted and are carried systemically by the blood. This rich blood flow keeps the dermal concentration of most transdermally delivered drugs low, which in turn provides a concentration gradient from the outside of the body into the skin and it is this concentration gradient (more accurately, it is the chemical potential gradient) that allows drug delivery through the skin.
The epidermis
The epidermis overlies the dermis and is itself a multiple layer containing various cell types, including keratinocytes, melanocytes and Langerhans cells. Keratinocytes in the basal layer (stratum basale) undergo division and then differentiate as they migrate outwards, forming the stratum spinosum, then the stratum granulosum and finally the stratum corneum. Differentiation is complex and essentially changes the metabolically active basal cells that contain typical organelles, such as mitochondria and ribosomes, into stratum corneum that comprises anucleate flattened corneocytes packed into multiple lipid bilayers.
The stratum corneum
This outer skin layer is predominantly responsible for the barrier properties of human skin and limits drug delivery into and across the skin. The stratum corneum typically comprises only 10 to 15 cell layers and is around 10 µm thick when dry (although it can swell to several times this when wet). The stratum corneum is thinnest on the lips and eyelids and thickest on the load-bearing areas of the body such as the soles of the feet and palms of the hands. The lipid bilayers in which the keratin filled cells are embedded are uniquely different to other lipid bilayers in the body since they are comprised largely of ceramides, fatty acids, triglycerides and cholesterol/cholesterol sulphate, whilst phospholipids are largely absent. Longer chain ceramides act as ‘rivets’ connecting bilayers together and corneo-desmosomes interconnect corneocytes. The resulting structure can be likened to a brick wall (Fig. 39.1) where the keratin-filled cells act as the bricks in a mortar of multiply bilayered lipids.
In normal skin, it takes approximately 14 days for a daughter cell in the stratum basale to migrate and differentiate into a stratum corneum cell, and these cells are then retained in the stratum corneum for a further 2 weeks before they are shed.
The appendages
In terms of drug delivery, the appendages can be viewed as shunt routes or ‘short cuts’ through which molecules can pass across the stratum corneum barrier.
Specialized apocrine glands are found at specific body sites, such as the axillae and nipples, whereas eccrine glands are found over most of the body surface at a density of 100–200 glands per cm2. When stimulated by heat or emotional stress, eccrine glands secrete sweat, which is a dilute salt solution of around pH 5.
The largest appendages are the hair follicles and associated sebaceous glands which secrete sebum, composed of fatty acids, waxes and triglycerides. These lubricate the skin surface and help to maintain the skin surface pH at around 5. Skin typically has 50 to 100 hair follicles per cm2 but the load bearing areas of the soles and palms are largely devoid of these appendages.
Whilst these shunt routes offer a potential route through intact skin, the fractional area that they occupy is relatively small; for example, on the forearm, hair follicles occupy approximately 0.1% of the surface area although on the forehead this may be as much as 13%. The ducts are seldom empty, being occupied by sweat or sebum flowing out of the body which again inhibits drug delivery. However, formulators are able to target these structures, for example by delivering nano-sized drug delivery systems, such as liposomes, to the follicles in order to treat acne.
The shunt routes are important for electrical enhancement of transdermal drug delivery (iontophoresis) and also play a role in the early time course of passive drug delivery through the skin, where diffusion through the intact stratum corneum barrier has yet to reach steady state; rub a cut clove of garlic on your leg and it can be tasted within minutes, which is too fast for molecules to diffuse across the intact stratum corneum.
Transport through the skin
From the above discussion on the structure of skin it is clear that delivery of drug molecules from a topically applied formulation into the systemic circulation is complex, with numerous processes occurring and several routes of transport in operation, as illustrated in Figure 39.2.
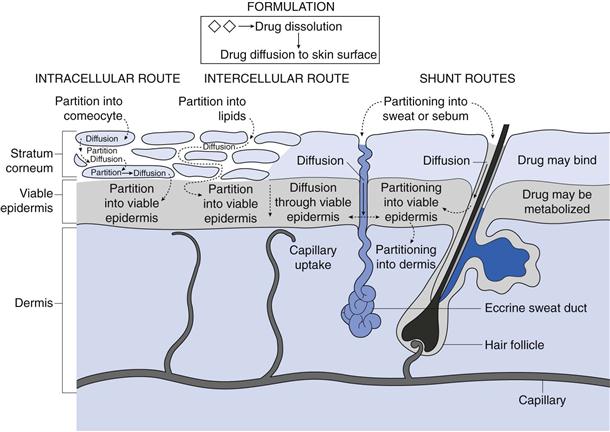
Fig. 39.2 Some of the processes occurring during transdermal drug delivery from a suspension formulation.
Initially, drug molecules must be presented to the skin surface. Consequently, if the formulation contains solid drug, then dissolution and diffusion through the formulation is the initial step in delivery. If the formulation contains dissolved drug, then as the molecules nearest to the skin surface enter the tissue these must be replaced by other molecules diffusing within the formulation towards the skin surface. Once at the outer layer of the stratum corneum, the drug molecule has three potential routes to cross the skin. Firstly it can pass via the shunt routes as described above. In this case molecules will partition into sweat or sebum before diffusing against the outflow from the glands.
More usually, the molecule encounters the intact stratum corneum ‘brick wall’ where transport can either be via an intracellular (also termed transcellular) route or transport can be intercellular.
Considering the intracellular route, the drug molecule initially partitions into a keratin-filled corneocyte, which is essentially an aqueous environment, they then diffuse through the corneocyte before partitioning into the intercellular lipid domains. For transcellular transport to continue, the molecule must then diffuse through the lipoidal region before repeatedly partitioning into and diffusing through the aqueous keratin in corneocytes and then intercellular lipids.
In contrast, the intercellular route requires the molecule to partition into the lipid bilayers between the corneocytes and then diffusion is via a tortuous route within the continuous lipid domain, i.e. following the mortar in the ‘brick wall’.
Having travelled through the stratum corneum, molecules diffuse through the lower epidermal layers before being cleared by the capillaries at the epidermal-dermal junction. During transport, there is potential for the permeant to bind to skin components such as keratin, in which case it may not reach the systemic circulation but could be sloughed off. In addition, skin is metabolically active and contains esterases, peptidases and hydrolases that can reduce the bioavailability of topically applied drugs such that, for example, only around 70% of topically applied glyceryl trinitrate (nitroglycerin) may be bioavailable.
It is important to recognize that, whilst three different routes exist for drugs to cross the skin (intercellular, transcellular, and shunt routes), for any permeant ALL three routes operate but the proportion of molecules crossing by the different routes will vary depending on the physicochemical properties of the permeant.
Permeant properties affecting permeation
Considering the processes described above, it is evident that the physicochemical properties of the permeant will control its transport into and through the skin. For both the transcellular and intercellular routes, the drug molecule has to cross the multiple lipid bilayers between the corneocytes and hence partitioning into, and diffusion through these lipid environments is essential. However, to reach the systemic circulation the molecule must also pass through the more aqueous environment of the viable epidermal cells and enter the blood. Thus, molecules which are lipophilic are usually seen as better candidates for transdermal delivery than hydrophilic compounds, but high lipophilicity is problematic for clearance.
The molecular weight of the permeant also impacts dramatically on its transport through the skin. The skin is designed to act as a barrier to external chemicals and so prevents the entry of large molecules, such as larger peptides and proteins. Not only is molecular weight an important factor in diffusion, but molecular structure (in particular hydrogen-bonding potential) can control the extent of binding to skin constituents and hence affect bioavailability. Naturally, the drug must have some solubility in the formulation and whilst transport through the stratum corneum is usually the rate limiting step in transdermal delivery, poor drug release from a formulation can occasionally limit drug transport. Finally, the effective dose of the drug must be relatively low to allow the application of appropriately sized patches/formulations.
The above processes restrict the range of drugs that can be delivered transdermally to therapeutically useful levels and some generic ‘rules of thumb’ can be used to predict whether transdermal delivery is viable for an active pharmaceutical ingredient. These include:
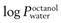 ) should be in the range 1–4, though more lipophilic molecules may be effectively delivered, such as fentanyl
) should be in the range 1–4, though more lipophilic molecules may be effectively delivered, such as fentanylIndeed, the active pharmaceutical ingredients currently used in transdermal formulations have many of the above properties; estradiol MW is 272 and it is lipophilic, with a 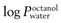 of 2.7; fentanyl MW is 336 with a log
of 2.7; fentanyl MW is 336 with a log 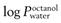 of 4.4; nicotine MW is 162 with a
of 4.4; nicotine MW is 162 with a 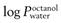 of 1.2.
of 1.2.
Mathematics of skin permeation
With such a highly complex multiple layered organ as skin, and numerous factors affecting transdermal drug delivery, it appears daunting to apply mathematical principles to describe such a complex process. However, simple mathematical principles can be used to understand the basic principles of permeation through membranes, including skin, and these assist in designing dosage forms for transdermal and topical drug delivery.
Fick’s laws of diffusion
Considering simple passive diffusion where molecules move by random motion from one region to another in the direction of decreasing concentration, then transport can be described by Fick’s First Law of Diffusion (Chapter 2):
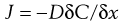 (39.1)
(39.1)
where J is the flux (the mass flow rate at which the material passes through unit area of the surface), C is the concentration of diffusing substance, x is the space co-ordinate measured normal to the section and D is the diffusion coefficient of the permeant. The negative sign demonstrates that the flux of molecules is in the direction of decreasing concentration. When a topically applied drug enters the skin, it is usually assumed that the diffusion gradient is from the outer surface into the tissue, i.e. is unidirectional.
Fick’s Second Law of Diffusion gives:
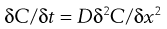 (39.2)
(39.2)
where t is time. Essentially this equation shows that the rate of change of concentration with time at a point within a diffusional field is proportional to the rate of change in the concentration gradient at that point.
The above laws assume that diffusion is through an isotropic material (i.e. one that has the same structural and diffusional properties in all directions); skin clearly is not isotropic with multiple layers, different permeation routes, etc. and indeed it is remarkable that Fickian diffusion can be used to generate valuable approximations from transdermal drug delivery data.
Experimental estimation of skin penetration
Experimentally, it is usually difficult to study transdermal drug delivery in vivo, so most researchers use in vitro protocols to mimic as closely as possible the in vivo situation. Most commonly, a membrane (for example human epidermis) is used to separate two compartments in a diffusion cell. The drug in a vehicle (for example water, buffer or in a formulation) is then applied to the uppermost skin surface (stratum corneum). This is usually termed the ‘donor’ solution. The other compartment contains a ‘receptor’ (or receiver) solution that is a good solvent for the drug, but which will not affect the skin barrier. This receptor solution thus provides essentially sink conditions (near zero concentration) of the permeant and allows a concentration gradient to exist between the donor and receptor phase, which in turn provides the driving force for diffusion across the membrane. If the cumulative mass of permeant that crosses the membrane is plotted as a function of time, then a typical permeation profile can be drawn, as illustrated in Figure 39.3.
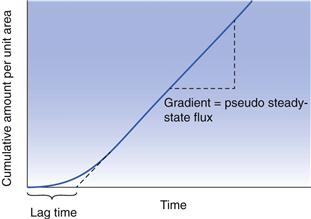
As can be seen, after sufficient time the plot approaches a straight line and from the slope we can obtain the steady state flux, dm/dt and Equation 39.2 can then be simplified to:
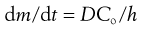 (39.3)
(39.3)
where dm/dt is the flux, usually termed J which is the cumulative mass of permeant that passes per unit area of the membrane in time t, Co is the concentration of permeant in the first layer of the membrane (at the skin surface, in contact with the donor solution) and h is the membrane thickness.
It is difficult to measure Co, the concentration of permeant in the first layer of the skin, but the concentration of the drug in the vehicle (donor solution), termed Cv, which bathes the skin membrane is usually known or can be determined easily. Differences in drug concentration between the donor solution and the first skin layer arise due to partitioning of the molecule between the membrane and donor solution so:
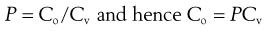 (39.4)
(39.4)
where P is the partition coefficient of the permeant between the membrane and the vehicle. Simply substituting Equation 39.4 into Equation 39.3 gives:
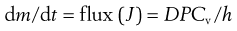 (39.5)
(39.5)
Equation 39.5 thus reiterates that the flux of a permeant through skin will be high for molecules with a high diffusion coefficient (e.g. generally having a relatively small molecular weight), will increase with increasing partitioning between the membrane and the donor solution (e.g. for lipophilic molecules) and will increase with increasing effective concentration in the donor solution (which increases the chemical potential gradient), whereas the flux through thicker membranes is reduced.
Figure 39.3 also shows that the lag time can be evaluated experimentally. The lag time (L) can be related to the diffusion coefficient by:
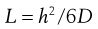 (39.6)
(39.6)
So, if the thickness of the membrane (h) is known then the diffusion coefficient can be calculated. This approach works well for relatively uniform and simple membranes such as polymers but as has been seen above, skin is far from simple. The effective thickness of the skin membrane is very difficult to estimate; if molecules traverse via the tortuous intercellular route then a simple measure of membrane thickness does not reflect the diffusional pathway. Because of this difficulty, a composite parameter, the permeability coefficient kp is often used since:
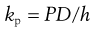 (39.7)
(39.7)
Using Equation 39.7, Equation 39.5 can then be simplified to:
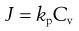 (39.8)
(39.8)
The above considers transdermal delivery where the donor solution remains essentially at the same concentration during the time-course of delivery, known as infinite dose conditions, and sink conditions prevail in the receptor solution. When a finite dose is applied, such as the application of a small amount of gel or cream, then the drug in contact with the skin surface will diminish with time. In this case, the permeation profile will typically resemble that in Figure 39.4 where flux initially increases to a maximum value (Jmax), beyond which depletion of the drug in the donor solution means that the concentration difference across the membrane, which drives diffusion, starts to fall. Finite dose profiles can be characterized by the time to maximum flux (tmax), the maximum flux (Jmax) and from the area under the curve (AUC).
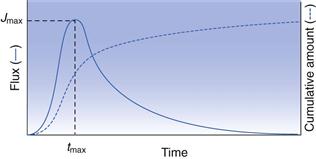
Experimental methods for studying transdermal drug delivery
When designing and optimizing transdermal and topical formulations, most researchers begin with a review of:
Various mathematical predictions have been constructed from databases of permeation experiments and relate potential flux to factors such as molecular weight, lipophilicity, aqueous solubility and hydrogen bond donor/acceptor groups. Such predictions can offer a useful guide to rule out molecules with unfavourable characteristics before undertaking time-consuming experimental studies.
In-vivo experiments
Clearly the ‘gold standard’ evaluation of transdermal and topical delivery is to apply the formulation to patients and to assess drug levels at the target site. In practice this is difficult to achieve, except in cases where a local biological response can be recorded, for instance blanching of the skin in response to vasoconstriction, such as with corticosteroids. For systemically acting drugs as well as the majority of locally acting agents, formulation design and development uses in vitro tests.
Whilst most studies use in vitro techniques, some in vivo methods are available. One option is to determine plasma levels following transdermal delivery allowing typical pharmacokinetic parameters such as Cmax and AUC to be determined, as for other routes of administration. However, for initial formulation development studies, such an approach is time consuming, expensive and may not secure regulatory approval.
Microdialysis has been used where a semi-permeable tube is inserted either underneath the skin or to a defined depth within the skin. The formulation is then applied to the skin surface and drug molecules permeating through the tissue are collected in the perfusate which is continually pumped through the probe. This technique requires specialized training for probe insertion and can present analytical challenges but offers significant advantages in assaying the permeant that has travelled through the skin barrier, i.e. it can be very valuable for assessing metabolites.
An alternative in vivo approach is to measure the loss of material from the skin surface. Whilst it is relatively easy to assay the amount of drug remaining in a formulation, typically only a small fraction of the applied dose partitions into the skin, and this approach does not define the fraction of absorbed dose that is bioavailable, i.e. unbound and active at the target sites. As an extension to this approach, tape stripping of the skin in vivo is a useful technique particularly for locally acting drugs. Essentially, a drug is applied to a defined skin area, left for a period and then that remaining at the surface is recovered. The stratum corneum is then sequentially removed using adhesive tape and the drug content in each strip is determined to build a drug profile through the tissue. As each strip can remove variable amounts of the skin, then drug levels can be normalized to protein content in each strip, reflecting the amount of stratum corneum removed. More invasive is the removal of skin biopsies, but the depth of the biopsy can be varied such that a punch biopsy can provide data for drug levels within the stratum corneum, viable epidermis, dermis and fatty layer, whilst a simple suction blister can be used to remove only the stratum corneum and viable epidermis.
Finally, non-invasive analysis of drug content in different skin strata can be determined for locally acting drugs that elicit a pharmacological or physiological response. Typical responses may be an increase in sweat secretion, vasoconstriction, vasodilation, changes to pain thresholds or changes in blood pressure that can be monitored by Laser Doppler Velocimetry.
In-vitro diffusion cells
In principle, in-vitro diffusion experiments are relatively simple, employing a two-chamber diffusion cell (examples of which are shown in Fig. 39.5) with the chambers separated by a membrane. Formulations can be applied in the donor compartment and samples then taken from the receptor compartment at intervals and the drug assayed before permeation profiles are constructed as in Figures 39.3 or 39.4.
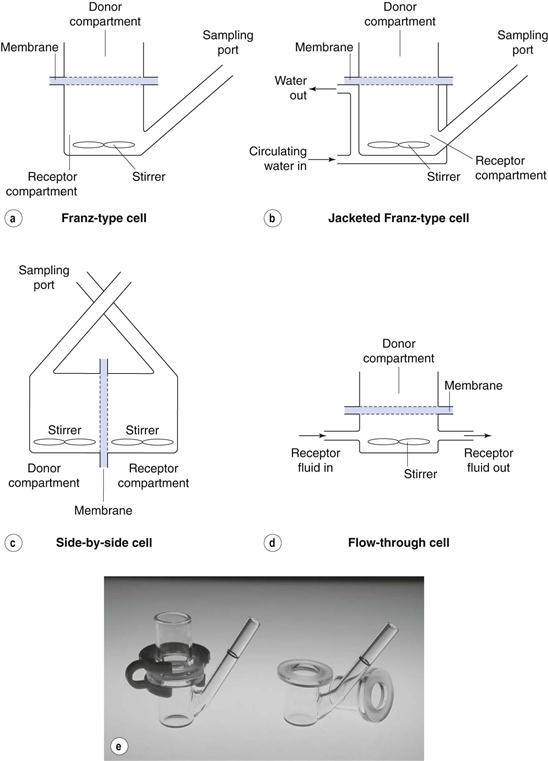
Fig. 39.5 Examples of diffusion cells commonly used in transdermal and topical drug delivery studies.
In practice, each element of the above experiment requires careful thought and consideration.
Selection of an appropriate membrane
Careful selection is essential and needs to consider the purpose of the experiment. If, for example, the purpose is to compare release from a series of formulations, or to assess stability on storage then a simple artificial (e.g. polymeric) membrane may be appropriate. If the purpose is to assess the feasibility of delivering a drug through human skin, then which skin strata to select? Most commonly, epidermal membranes (from stratum corneum to the basal membrane) are chosen since the drug is cleared from the dermal/epidermal boundary in vivo. However, if metabolism is likely to be significant then conditions must maintain enzyme activity in the tissue. Many researchers use animal skin as a substitute for human tissue due to legal constraints in some countries or lack of availability of human samples. However, it is well established that drug diffusion through some animal skins differs significantly from that through human skin.
Receptor solution
Similar detailed consideration should be given to the choice of receptor solution. This should be a good solvent for the permeant so that the drug does not violate sink conditions, usually taken to be less than 10% of saturated concentration in the receptor phase at any time. The receptor should not affect the integrity of the skin barrier, so the use solvents such as ethanol at high concentrations should be avoided since it too could diffuse ‘backwards’ from the receptor solution into, for example an aqueous solvent in the donor phase; not only could this damage the stratum corneum barrier but it could also alter the partitioning into the skin and/or affect drug release from the donor solution. A surfactant could be added but may also damage the integrity of the stratum corneum barrier. It is important to stir the receptor compartment to ensure appropriate mixing and to clear drug molecules from directly beneath the membrane.
Temperature
Diffusion is temperature dependent, so most researchers submerge their diffusion cells in a water bath or circulate water in a jacket around the cell. Typically, the aim is to maintain the skin surface temperature near 32 °C, and typically submerging the diffusion cells in a water bath at around 37 °C achieves this.
Other factors
Amongst other factors to consider is the amount of a formulation to apply to the membrane surface, selected to mimic in vivo use and so, for example, this may be either a finite dose for a locally acting cream, or an infinite dose to mimic a 7-day patch application. The choice of vehicle from which a drug may be applied must also be considered; aqueous solutions are often used but buffering may be required depending on ionization, or if solubility is poor then the solution may rapidly deplete as the drug crosses the membrane. As has been seen above, there is a lag time until pseudo-steady state permeation is reached so experimental duration needs consideration. Steady state flux is reached after approximately 2.7 times the lag time so for a permeant with a long lag time, e.g. 10 hours, steady state is not reached until 27 hours and then data need to be collected to evaluate flux. Maintaining skin integrity for extended periods is thus necessary and may require the use of an antimicrobial agent. An appropriately accurate and sensitive method needs to be established to determine the amount of permeant in the receptor solution at defined time points, and analytical interference by skin components that leach from the tissue during the experiment must be avoided. The number of replicate experiments must be considered. If using an artificial and well characterized membrane then reproducibility should be high but when a biological membrane is selected then natural variability usually dictates that a minimum of 6 replicates are needed, and best practice is to use more than one skin donor. The use of tissue from more than one source reduces potential errors arising from a damaged piece of skin, but the integrity of the membrane prior to beginning the permeation study should be verified.
Transdermal and topical preparations
A remarkably broad range of formulation options are available for topical and transdermal preparations, ranging from simple solutions and lotions, through commonly used creams (aqueous or oily), ointments, gels and patches to the less common aerosols and foams. When selecting and designing formulations, account must be taken of the physicochemical properties of the drug, such as its solubility and pKa, as described above. Equally, the formulation must be stable, the drug and excipients must be compatible and drug must be released from the dosage form following application. Importantly, the formulation should be cosmetically acceptable with a good skin feel, texture and fragrance. Ultimately, the product will be applied to human skin in vivo and so the pathophysiology of the tissue must be understood; the application of an alcoholic gel formulation to broken skin is unlikely to enhance patient compliance. Taking the above factors into account, topical and transdermal formulations aim to deliver the drug to therapeutically active levels at the target site (either local or systemic). Interestingly, it has been estimated that, for topical products such as creams and gels, typically only between 1 and 3% of the applied dose is bioavailable whereas bioavailability from patches is typically 30–70% for drugs such as buprenorphine and fentanyl.
Formulation principles
Based on the consideration of skin structure and the mathematical theory explained above, some general principles are useful to guide the selection of a dosage form and excipients.
Principle 1. Select a suitable drug molecule.
As described above, large hydrophilic molecules are poor candidates to deliver across intact skin. Ideally a drug should be moderately lipophilic (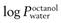 = 1–4), relatively low molecular weight (<500 Da) and be effective at low doses (<10 mg/day for transdermal delivery).
= 1–4), relatively low molecular weight (<500 Da) and be effective at low doses (<10 mg/day for transdermal delivery).
Principle 2. Release the drug.
The formulation should be designed to ensure appropriate release of the drug; this may be rapid release for a locally acting drug, or sustained and slow release for a 7-day patch. If the formulation contains a moderately lipophilic drug in a lipophilic oily base, the drug is less likely to partition out of the formulation and enter the lipophilic skin barrier than if it is applied from a more aqueous base. Essentially, the vehicle should allow some solubility of the drug but should not retain the drug by being a very good solvent (see Principle 3).
Principle 3. Use thermodynamics.
The driving force for diffusion is the chemical potential gradient across the membrane; often the term ‘concentration gradient’ is used but ‘chemical potential gradient’ is more accurate for complex membranes such as skin. Equation 39.5 described how transdermal flux is dependent on drug concentration, but in thermodynamic terms it can be shown that:
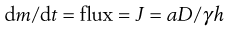 (39.9)
(39.9)
where a is the thermodynamic activity of the drug in the donor formulation and γ is the effective activity coefficient in the skin barrier. Thus, to achieve the highest flux, the drug in the vehicle should be at its maximum thermodynamic activity. By definition, a pure solid is at maximum thermodynamic activity and is given a value of 1. Thus, a saturated solution which is in equilibrium with excess solid is also at maximum thermodynamic activity and so the greatest flux can be achieved by using the drug at its solubility limit in the formulation.
For formulation development, thermodynamic activity can be considered as the ‘escape tendency’ of the drug from its vehicle; if a drug is at saturation there is a strong thermodynamic drive for it to leave the formulation, hence it will enter the skin and permeate whereas if it is present at a small fraction of its solubility limit then the drive to escape is low. In practice, a saturated system tends to be relatively unstable and so a formulation with the drug near saturation is a useful compromise.
This principle has important formulation and clinical implications. Firstly, for example, a finely divided suspension formulation with saturated drug will provide the maximum flux. If further drug is then added to the formulation, the concentration of drug in the system increases, but the thermodynamic active or ‘effective’ concentration (i.e. that of dissolved drug molecules) remains the same and so flux remains the same. Conversely, if a suspension is diluted but the drug remains saturated then flux remains the same until dilution reduces the drug to below its saturation point, and for a sub-saturated solution formulation then dilution will reduce flux. Secondly, Equation 39.9 illustrates that different formulations of any particular drug at the same thermodynamic activity will give the same flux (as long as the excipients do not modify any properties of skin). Thus, by appropriate formulation it is possible to reduce the drug loading in a topical product whilst maintaining the same thermodynamic activity and hence delivering the same amount of drug; Dioderm® contains 0.1% hydrocortisone but is clinically equivalent to 1% Hydrocortisone Cream BP as the drug is at the same thermodynamic activity in both formulations despite differences in concentration.
Principle 4. Alcohol can help.
Many effective topical and transdermal products contain low molecular weight alcohols or other volatile ingredients. Alcohols themselves can partition into skin and can provide a transient ‘reservoir’ into which the drug can partition. In addition, they may improve the diffusion coefficient of the drug in the stratum corneum. Further, whilst alcohols are typically ‘good’ solvents for most drugs, they evaporate from the skin surface when rubbed on in a finite dose application. Taking ibuprofen hydro-alcoholic gels as an example, the drug has a low aqueous solubility of less than 1 mg/mL, whereas it is readily soluble in ethanol and in a 20 : 80 w/w ethanol:water system its solubility is nearly 10 mg/mL. If a gel containing 5 mg/mL was rubbed into the skin, initially the drug is dissolved in the formulation but as the ethanol evaporates, the formulation becomes steadily more aqueous until only water remains. At this point the ibuprofen is in excess of its solubility limit, so has maximum thermodynamic activity resulting in increased delivery into the tissue (as described in Principle 3).
Principle 5. Occlusion increases delivery of most drugs.
Occlusion (covering the skin with an impermeable barrier) hydrates the skin by blocking transepidermal water loss to the external environment. The water content of the stratum corneum can rise up to 400% of the tissue’s dry weight and this increased hydration improves transdermal and topical delivery of most drugs. Some preparations require occlusion to deliver the required dose to therapeutic levels, such as EMLA cream (which contains lidocaine and prilocaine), which should be applied as a thick layer under an occlusive dressing. Alternatively occlusion can also be inadvertent, such as when applying hydrocortisone ointments or creams to treat nappy rash when tightly fitting waterproof pants can occlude the area.
Formulation options
Considering the broad range of topical and transdermal formulations ranging from simple solutions to complex multiple emulsions, these systems can be classified in numerous ways. Most commonly, formulations are described in terms of their physical properties but, as can be seen above, this in itself can be complex and changing, for example where a simple gel can lose solvent by evaporation to deposit a solid film on the skin surface. However, the scheme in Table 39.1 may be helpful when considering formulation options.
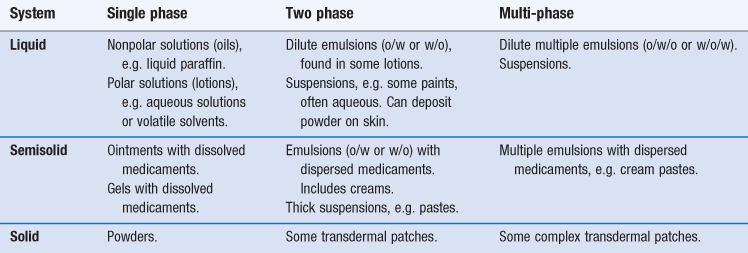
Generally, semi-solid formulations are selected for increased residence on the skin, transdermal patches for extended drug delivery through the skin and liquid formulations for a rapid short-term input of permeant into the skin. In both the clinical and cosmetic domains, skin type can affect the choice of formulation base, in that generally:
• for normal to oily skin types, gels are often preferred
In addition to skin type, the skin site to be treated can affect the selection of the vehicle thus:
However, it is mainly clinical rationale as to which formulation type is selected for topical therapy. Dependent upon the lesion type then:
A simplified decision tree to illustrate how the clinical condition can influence the choice of formulation options and formulation design is given in Figure 39.6.
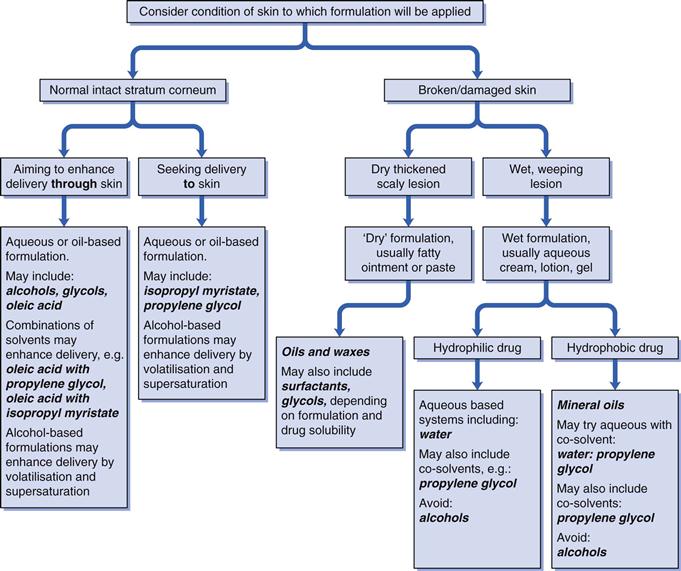
Beyond the broad nature of the vehicle, specific formulation components may be required dependent on the physicochemical properties of the drug (e.g. pKa and the need for a buffer, stability of the active and the need for an antioxidant), clinical considerations (e.g. intact or broken skin, duration of use) and to improve patient compliance. Commonly used components in topical and transdermal formulations include solvents, solubilising agents, oils, thickening agents and pH modifiers, examples of which are given in Table 39.2. It should be borne in mind that the active pharmaceutical ingredient itself may have properties that can affect the formulation, for example chlorhexidine is surface active and forms micelles.
Table 39.2
Examples of some typical components found in topical and transdermal formulations
| Component | Examples |
| Solvent | Water, propylene glycol, ethanol, isopropyl alcohol. |
| Solubilizing agent | Surfactants; anionic, e.g. sodium lauryl sulphate (SLS), cationic, e.g. cetrimide, non-ionic e.g. nonoxynol series, zwitterionic, e.g. dodecyl betaine. |
| Oils | Mineral oil, liquid or soft paraffins, silicone oils. |
| Thickening agents | Gums, celluloses, e.g. hydroxypropylmethyl cellulose (HPMC), carbomers, polyvinylpyrrolidone (PVP). |
| Preservatives | Antioxidants, e.g. ascorbic acid, butylated hydroxyanisole; Antimicrobial agents, e.g. parabens; Chelating agents, e.g. ethylenediaminetetraacetic acid (EDTA). |
| pH modifiers | Monoethanolamine, lactic acid. |
Common formulation types
Whilst Table 39.1 provides a broad classification scheme of topical and transdermal formulations, the most common systems are described in further detail below.
Liquid formulations
Liquid formulations for external application may be simple single-phase solutions using either i) an aqueous base, ii) a solvent, iii) a miscible co-solvent system (e.g. ethanol and water) or iv) an oil. Examples of single-phase solutions include soaks and paints, such as chlorhexidine solutions for skin disinfection, or lotions of malathion used to treat head lice. Liquid preparations generally have poor residence time on the skin, usually resulting in low drug delivery into the tissue and so tend to be used to treat surface conditions (e.g. disinfection). More viscous preparations can be generated to improve residence time, for example by addition of glycerol, propylene glycol or polyethylene glycol, as employed in anti-infective ear drops. When simple solutions are applied to the skin, the solvent evaporates and can cool and soothe the skin, an effect that is more pronounced from alcoholic vehicles. The evaporation of a solvent can increase the thermodynamic activity of the drug in the evaporating vehicle that can influence delivery as described in Principle 4 above.
Low-viscosity ‘thin’ oil-in-water (o/w) or water-in-oil (w/o) emulsions and suspensions are two-phase liquid systems, whereas more complex o/w/o or w/o/w ‘thin’ emulsions are multi-phase liquids. The designation between a ‘thin’ emulsion that is a liquid formulation and a semi-solid cream system is rather arbitrary, but emulsions are most widely used in creams as described below.
As with single phase solutions, solvent can evaporate from an aqueous or solvent-based suspension when applied to intact skin, so providing a cooling sensation, though clearly alcoholic-based formulations should not be applied to broken skin. As the solvent evaporates, drug delivery can be enhanced due to increased thermodynamic activity of the permeant in the vehicle (as described above). Suspensions such as calamine lotion deposit drug powder on the skin surface which improves residence time of the active on the skin surface but any deposited solid must undergo dissolution on the skin surface prior to permeation; this will limit drug absorption flux.
Semi-solid formulations
The vast majority of topically applied formulations are semi-solids. They offer good residence time on the skin. There are numerous options available to the formulator and semi-solids are generally well accepted by patients. Single phase semi-solid systems include ointments and gels in which the active ingredient is dissolved, whereas ointments or gels containing drug powder (usually microcrystalline) are two-phase semi-solids.
Ointments
Ointments are fatty preparations that are usually self-occlusive and are generally used on dry lesions. Unmedicated ointments are used as emollients to soothe, smooth and hydrate dry skin conditions.
Commonly, ointments are produced from soft, hard and liquid paraffins (or similar excipients) to generate a hydrocarbon base. The bases are highly occlusive and so prevent transepidermal water loss; this, in turn, causes the skin to hydrate and hence their usefulness in dry skin disorders. Hydration of the stratum corneum also tends to increase transdermal drug flux and so, coupled with the excellent residence time of these formulations on the skin, can provide prolonged drug delivery. However, thick greasy ointments can be difficult to spread, particularly on broken skin where they are commonly applied, and patients often find these formulations to be messy to use.
Absorption bases.
These contain an emulsifying agent that allows the formulation to soak up water or aqueous secretions whilst remaining semi-solid. Generally, they tend to contain a hydrocarbon, such as a paraffin, together with a miscible substance that is polar, such as sorbitan monooleate. This combined system can absorb up to 15% water. Thus, these formulations provide some occlusion of the skin, hydrate the stratum corneum and can be left in contact with the tissue for prolonged periods of time. Due to potential allergic reactions and sensitization, wool fats and lanolin, traditionally used in absorption bases, have largely been replaced with purified lanolin or other excipients.
Emulsifying bases.
These are similar to absorption bases but can form an oil-in-water system, for example by using a mixture of paraffins with cetostearyl alcohol and a surface active agent such as sodium lauryl sulphate (SLS) or cetrimide. These emulsifying agents generate a water-miscible ointment which can be easily washed from the skin surface, in contrast to greasy hydrocarbon bases. Anionic (e.g. SLS), cationic (e.g. cetrimide) or non-ionic (e.g. cetomacrogol) emulsifying ointments can be formulated, to ensure compatibility with any incorporated drug such that, for example, a cationic or non-ionic base would be selected for a cationic drug.
Water-soluble bases.
These are usually prepared from a mixture of water-soluble polyethylene glycols of varying molecular weights which can be blended to generate bases which soften or melt when applied to the skin surface. Water-soluble bases mix easily with skin secretions, spread well on the skin surface and can be readily washed off. However, they lose their semi-solid consistency if around 10% water is taken into the ointment and they may be incompatible with several classes of compounds including phenols and penicillin.
Gels
Gels are typically formed from a liquid phase that has been thickened with other components and may contain dissolved (single phase) or dispersed (two-phase) drug in a semi-solid system. The liquid in the gel essentially forms a continuous phase with the thickening agent enhancing viscosity by providing the porous scaffold of the gel. As with solutions or suspensions, the liquid phase may be aqueous, alcoholic, miscible blends or a non-polar solvent, and as described above, the solvent may evaporate, cooling the skin and increasing drug flux by enhancing thermodynamic activity of the drug in the evaporating vehicle.
As gelled solutions, the continuous phase allows unhindered diffusion of dissolved molecules throughout the polymer scaffold and hence drug release should be equivalent to that from a simple solution, unless the drug binds to the polymer in the gel or polymer loading generates a highly viscous gel. Numerous thickening agents are available, the selection of which is influenced by the physicochemical properties of the drug and compatibility with the solvent. Natural polymers such as carageenans or refined/synthetic polymers such as hydroxypropyl methylcellulose or Carbopols are commonly employed gelling agents.
Creams
Creams are the most common semi-solid topical dosage form and are typically two phase emulsions, either water-in-oil or oil-in-water (Chapter 27). Oily creams (w/o semi-solid emulsions) are less greasy than ointments, are easier to apply and can usually be simply washed off the skin surface, and hence are well accepted by patients. However, whilst a water-in-oil cream can deposit a protective oily layer on the surface, they tend to be less occlusive than an ointment and so may not be as beneficial in dry skin conditions. Oil-in-water creams (also called ‘washable’ or ‘vanishing’ creams) with a continuous aqueous phase are often rubbed into skin and again can provide a cooling sensation. The processes occurring during delivery from a cream are complex and difficult to define; the cream changes as it is applied, rubbed in, the continuous phase evaporates (or if oily may penetrate the skin) and the emulsion cracks. In addition, the active ingredient may be loaded in one phase but will partition between the continuous and dispersed phases or may become incorporated into micelles as the formulation collapses. Further, creams usually include an antimicrobial preservative which may have surface activity (as indeed may the medicament), a buffer, an antioxidant and fragrance materials. Considering this complexity, formulation for optimal release and delivery tends to be on an individual drug basis, taking the principles described earlier in this chapter into account.
Multi-phase semi-solid formulations
Multi-phase semi-solid formulations tend to use emulsions or multiple emulsions (o/w/o or w/o/w) with the drug dispersed in a paste. Pastes (either two- or multi-phase) are stiff preparations containing up to 50% solids, commonly in a fatty base. These preparations are useful for treating localized skin sites, as with Lassar’s paste (zinc and salicylic acid) or dithranol paste. Pastes also lay down a thick impermeable film that can be cut and used in sub blocks. Pastes tend to be less greasy than ointments as the powder may absorb some of the more mobile hydrocarbons from the fatty base.
Solid formulations
Powders.
These are seldom applied directly to the skin for drug delivery since drug dissolution is necessary prior to permeation; without the use of a solvent, skin exudates are generally unable to dissolve the applied powder
Topical sprays.
These are available to deposit powders on the skin surface but these generally incorporate a volatile solvent that dissolves some drug prior to evaporation (with consequent elevated thermodynamic activity).
Dusting powders.
These are used to reduce friction between skin surfaces, and antiseptic dusting powders are available, but do not aim to deliver drug into the skin.
Patches.
These are solid dosage forms that vary in complexity from simple two-phase to multi-phase systems. Most simply, in-situ film-forming systems are available that allow a patient to deposit a thin film on a diseased site for local therapy; these formulations contain polymers such as polyvinylpyrrolidone, polyvinyl alcohol or silicones in either an aqueous or more volatile solvent system that can be applied by spraying onto the affected area. As the solvent evaporates, the film forms. It can be un-medicated for use as a wound dressing, or may contain an antimicrobial agent to prevent infection. Also, for example, antifungal agents such as terbinafine can be incorporated to treat athletes’ foot with the spray offering simple dosing between toes.
In-situ films remain at the affected site for extended periods and then can be designed to be easily washed off or to resist water. However, as described above, drug must be in solution for absorption and hence some residual solvent or non-volatile solvent (such as an oil) may be incorporated into the formulation.
An alternate delivery device is pre-fabricated transdermal patches. These are worthy of more detailed consideration.
Transdermal delivery patches
Pre-fabricated transdermal patches are designed to deliver a constant and controlled dosage over extended periods of time for systemic therapy. They offer advantages over conventional oral dosage forms in that:
• drug administration through skin avoids the pH variations seen with gastrointestinal transit
• patches can be removed easily and quickly in cases where adverse drug reactions occur
However, due to the barrier properties of the skin, relatively few drug molecules have the appropriate physicochemical and therapeutic properties for sustained transdermal delivery. However some successful products have reached the market.
Scopolamine patches for motion sickness were first approved by the United States Food and Drug Administration (FDA) in 1979. Since then, nicotine, estradiol, fentanyl, buprenorphine, testosterone and glyceryl trinitrate patches have been commercialized. More recently, a methylphenidate transdermal patch has been developed to treat Attention Deficit Hyperactivity Disorder, the monoamine oxidase inhibitor (MAOI) selegiline has been developed in patch form for Parkinson’s disease and for major depression, whilst a rivastigmine patch is available for Alzheimer’s patients. It is notable that recent patches have aimed to meet a clinical need, for example to assist compliance with Alzheimer’s patients, rather than broadly exploring the physicochemical properties of candidate drugs.
Designs of transdermal patches
Numerous patch designs exist, some are illustrated in Figure 39.7. The simplest systems contain the drug in an adhesive, with more complexity introduced in matrix type patches and reservoir systems.
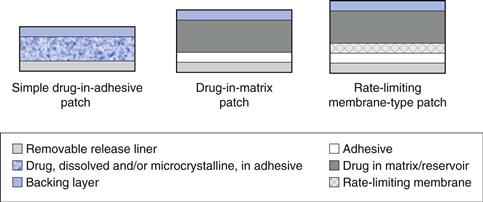
Fig. 39.7 Illustration of common patch designs.
Drug-in-adhesive patches are the simplest and most common patch design and are widely used to deliver nicotine and glyceryl trinitrate. These patches are formed by dissolving or dispersing drug within an adhesive which is then coated onto a backing layer before a release liner is applied. Drug-in-adhesive patches tend to be thinner and more flexible than other systems, but drug loading constraints can reduce the period of delivery; nicotine patches are designed for less than one day use.
Drug can be included in a separate matrix which can be formulated to increase the drug content in the system or to control drug release, allowing longer term delivery. The drug containing matrix or reservoir is often a polymeric mixture, for example polyvinylpyrrolidone and polyvinylacetate, potentially with the addition of a plasticizer such as glycerol; hydrogels may also be used as the matrix. Clearly drug released from the matrix will partition into and diffuse through the adhesive layer.
More complex rate limiting membrane systems typically contain the drug in a reservoir but with release controlled through a semi-permeable membrane. The reservoir may be liquid or more usually a gel and can be designed to contain higher drug loadings than a simple drug-in-adhesive system for prolonged delivery. More complex patch configurations based on the above are feasible, for example, multilayered drug-in-adhesive systems with a rate limiting membrane separating two adhesive layers of different drug loadings.
For all the above configurations, patches have some common components.
Removable release liner.
A liner temporarily covers the adhesive and is the layer that is removed to allow the patch to be applied to the skin. Liners are often made from polymers such as ethylene vinyl acetate, or aluminium foil, dependent on the nature of the adhesive that it covers. The liner must easily peel away from the adhesive but must be bonded firmly enough to prevent accidental removal. Liners are usually occlusive to prevent the loss of volatile patch components such as ethanol prior to use.
Adhesive.
The adhesive is a crucial component of all transdermal delivery patches and pressure sensitive adhesives (PSAs), such as acrylates, polyisobutylene (PIB) or polysiloxane adhesives, are usually used. Clearly the adhesive must:
• stick to the skin for the patch’s lifetime
• it must be non-irritating and non-allergenic as it may be in place for up to 7 days
During formulation development, considerable effort is spent testing patch wear but the presence of a drug in the adhesive can affect its properties, hence data from placebo tack, wearability and irritation studies may not truly reflect in vivo use of a medicated system.
Backing layer.
Numerous materials can be used for patch backing layers, depending on the patch design, size and length of intended use. For relatively short use small patches, an occlusive backing layer may be selected and this will hydrate the underlying skin which can improve delivery. Example materials include polyethylene or polyester films. For larger and longer term use patches, backing layers that permit some vapour transmission are preferred, such as polyvinylchloride films. In addition, the backing layer should allow multidirectional stretch and be pliable to allow the patch to move as the skin moves.
Matrix/reservoir.
A drug matrix or reservoir is usually prepared by dissolving the drug and polymers in a common solvent before adding in other excipients such as plasticizers. The viscosity of the matrix can be modified by the amounts of polymers incorporated, or by cross-linking polymers in the matrix, and can consequently be used to control diffusion of the active ingredient through the matrix to the adhesive and then on to the skin surface. Reservoirs tend not to contain gelled polymers but rather utilize a viscous liquid, such as a silicone or a cosolvent system, occasionally with ethanol into which drug is dissolved and dispersed. In these cases drug diffusion within the reservoir towards the skin surface is unhindered.
Rate-limiting membrane.
Transdermal patches were originally designed so that the patch itself controlled the rate of delivery of the active ingredient to the skin surface, and so the patch would control drug flux. In practice, it is usually the stratum corneum barrier that limits the rate of drug input into the skin and hence provides the rate limiting barrier. However, semi-permeable membranes are used to separate reservoirs from the underlying adhesive and can also be found separating multiple drug-in-adhesive layers. Membranes can be prepared from co-polymers of ethylene acetate with vinyl acetate, with or without plasticizers. As with other patch components, the rate limiting membrane must be compatible with the drug, non-toxic, stable and pliable.
Other formulations
Beyond the dosage forms described in Table 39.1, less common formulations are available for topical and transdermal drug delivery. These include:
Liposomes.
These are spherical, bilayered structures, typically between 100 and 200 nm in diameter, that can be produced from phospholipids, and which are able to encapsulate a range of different drugs either in their aqueous core or within the lipid bilayer. The properties of the liposome are determined by their size, number of bilayers and by the component lipids; often materials such as dipalmitolyphosphatidylcholine (DPPC) are used but cholesterol can be added to produce rigid vesicles whereas addition of ethanol or surfactants can produce flexible liposomes.
Whilst liposomal formulations have been developed for parenteral administration of anticancer and antifungal agents, they are not widely used for topical and transdermal drug delivery, and are predominantly found in cosmetics and long acting sunscreen formulations. The liposomes promote delivery of the active sunscreen into the stratum corneum and reduce wash off, as seen with surface deposited agents. Liposomes as drug delivery vehicles are discussed further in Chapter 45.
Foams.
Foams have been used to deliver various drugs to and through the skin, for example ibuprofen and betamethasone. Foams allow relatively easy dosing to the target site and, as dynamic systems where solvent evaporates and foams collapse on the skin surface, delivery can be enhanced beyond that seen for simple creams.
Solids or particulates.
These can be used to target drug delivery to the hair follicles. Liposomal products can also do this. The pore diameter of the follicle ranges from ~70 µm on the forehead to ~170 µm on the calf. Hair shaft diameters range from ~15 µm on the forehead to ~42 µm on the calf; these are usually filled with sebum. The volume of the follicular reservoir on the forehead has been estimated at 0.19 mm3, into which micro-particulates can be deposited to provide prolonged activity of materials such as benzoyl peroxide (which is highly insoluble), to manage acne vulgaris.
Enhancement of transdermal and topical drug delivery
The range of drugs that can be delivered transdermally to therapeutic levels is restricted due to the effective barrier provided by skin, in particular the stratum corneum, as discussed above. The factors influencing the magnitude of the permeant flux are summarized in Equation 39.5.
 (39.5)
(39.5)
Consequently, various approaches have been used to modify the above parameters by manipulating the barrier properties of the skin (increasing diffusivity), the nature of the permeant or barrier (to increase partitioning) or to increase the concentration (thermodynamic activity) of the drug in its formulation. Manipulating skin thickness is difficult, though the application site can be selected as one where the stratum corneum is relatively thin, such as the scrotum that is used to deliver testosterone. Additionally, external forces can be used effectively to circumvent the stratum corneum barrier.
Formulation manipulation
For a given drug in a defined formulation, maximum flux is achieved when the active ingredient is present at saturation (i.e. when Co, in for example Eqn 39.3, is at its maximum). However, it is feasible to generate supersaturated systems where the drug is present in excess of its solubility. This occurs when, for example, a hydro-alcoholic gel containing a poorly-water soluble compound is applied to the skin. As the alcohol (good solvent) evaporates, the drug can exceed its solubility limit in the remaining aqueous phase and so becomes supersaturated. Supersaturated states are inherently unstable and the excess drug will tend to crystallize rapidly. However, if the formulation is viscous or anti-nucleating polymers are included, then drug crystallization can be inhibited for a period of time, in which case the drug remains in a supersaturated state and provides a greater flux than can be obtained from a saturated solution. In practice, many topically applied formulations are dynamic (gels, foams, creams, etc.) and patches can contain volatile ingredients that may evaporate or partition into skin, resulting in transient supersaturated states.
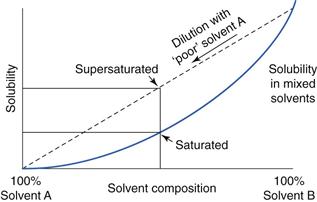
Supersaturation can also be achieved from co-solvents as shown in Figure 39.8; if solvent A is a ‘poor’ solvent for the permeant and solvent B is miscible with A but is a ‘good’ solvent, then the solubility of the drug in mixtures of A and B would follow the solid curve. If a solution of drug in solvent B is then diluted with the poor solvent A, solubility of the drug follows the dotted line and generates supersaturated systems. With time, the drug will crystallize from this supersaturated state to equilibrate with the saturation curve but crystallization can be inhibited by addition of some polymers and this provides a transient increase in drug solubility beyond saturation, hence increasing drug flux.
Other formulation strategies can provide optimal transdermal delivery using the principles described above. For example, formulations should ensure optimal drug release and encourage partitioning into the stratum corneum by using a vehicle in which the drug is only moderately soluble. The active drug should have appropriate physicochemical properties, perhaps by using a pro-drug containing a lipophilic moiety which will enhance partitioning into the lipophilic stratum corneum; ester-linked fatty acids can serve this purpose with the link then cleaved by esterases within the skin, liberating the active principle. Control over pH in the formulation of ionizable drugs is important since ions permeate less well than neutral compounds, or ionic charges can be neutralized by employing ion pairs.
Skin modification
Numerous chemicals, collectively termed penetration enhancers, interact with stratum corneum components to increase transdermal drug delivery. These enhancers act by disrupting the highly organized lipid bilayer packing through interacting with intercellular proteins, by increasing partitioning into the membrane or by a combination of these mechanisms. Ideally, penetration enhancers will be pharmacologically inert, will modify the skin barrier in a reversible manner, will be non-toxic, non-irritating, compatible with drugs and excipients and acceptable to patients (good skin ‘feel’, odourless, colourless, etc.).
The safest and most widely used penetration enhancer is water, and the transdermal flux of most drugs is greater through hydrated skin than through dry tissue. Thus, occlusion is an effective means of increasing the flux of most drugs. Ethanol and other low molecular weight alcohols that are often incorporated into topically applied formulations can also act as penetration enhancers. Ethanol can disrupt the intercellular lipid packing and so increase diffusivity through the stratum corneum but additionally can diffuse into the membrane and act as a solvent within the stratum corneum, into which drug can more easily partition.
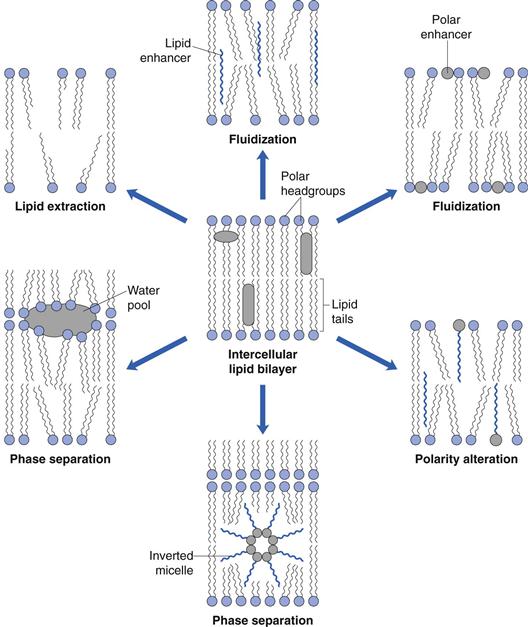
Small aprotic solvents, such as dimethylsulphoxide, can interact with lipid bilayer head groups in the stratum corneum to disrupt their close packing and facilitate drug diffusion, whereas fatty acids (e.g. oleic acid) insert along the stratum corneum lipid chains to disrupt packing. Bespoke penetration enhancers, such as Azone (1-dodecylazacyclohpetan-2-one), have been designed to possess a bulky polar head group and a lipid chain. The molecule can insert within the lipid lamellae to disrupt the endogenous stratum corneum lipid bilayers at both head and chain regions. Other commonly used excipients with enhancer activities include terpenes (fragrance agent) and surfactants. Potential mechanisms by which penetration enhancers can disrupt the lipid bilayers of the stratum corneum are illustrated in Figure 39.9.
External forces
Researchers have developed more active methods for increasing transdermal drug delivery, of which one approach is iontophoresis. An electrical potential gradient across the skin can be generated using relatively low current densities with an anode (positive electrode) and cathode (negative electrode) placed on the skin surface. A charged drug is placed under the electrode of the same polarity (e.g. anion placed under the cathode) such that when the current flows the anion is repelled by the cathode. As it attempts to migrate towards the anode it enters the skin. In addition, as applied or endogenous ions migrate from one electrode to another (such as Na+ migrating towards the cathode), water and neutral molecules can be transported along with the ions into the skin.
Devices designed to deliver liquids or particles across the skin using needleless injectors have also been developed. Using compressed gasses to propel particles or liquid at very high speed, the technology seeks to avoid needle-phobia and deliver large molecules (e.g. vaccines) up to therapeutic levels in defined skin strata. As the particles are relatively small, when fired into the skin they do not trigger the pain receptors, though the propellant gas may cause some sensation. Numerous factors can influence the efficiency of delivery from needleless injectors, ranging from the density of the particles to the thickness of the stratum corneum and the need to hold the device vertically to the skin surface to ensure that the particles penetrate to the correct depth.
Alternatively, the stratum corneum barrier can be removed or reduced to enhance transdermal delivery. Thus, dermabrasion, laser ablation or localized heating have all been used to facilitate transdermal drug delivery.
Microneedles offer significant potential benefits for delivering both small and large molecules into and through the skin. These devices can be generated from a range of substances (stainless steel, silicon, plastic, dissolvable sugars and polymers), can be hollow or intact and can vary in length from tens to hundreds of micrometers. Hundreds of microneedles can be produced in an array that is less than 1 cm2 in area. The intact microneedles will puncture the stratum corneum prior to application of a dosage form, or the drug formulation can be delivered through hollow needles. Alternatively the active drug can be administered within a dissolving microneedle array.
As the needles are small and can be designed to puncture only the depth of the stratum corneum, pain receptors deeper in the skin are not stimulated so delivery is painless. This technology is currently attracting considerable interest, particularly for delivering vaccines and it also offers potential for sampling body fluids for monitoring purposes.
Nail delivery
Despite significant anatomical differences, many of the principles described above apply to formulation design for drug delivery through nails (often termed transungual delivery). Human fingernails consist of three main structures, the outermost nail plate, the underlying nail bed and the nail matrix (Fig. 39.10).
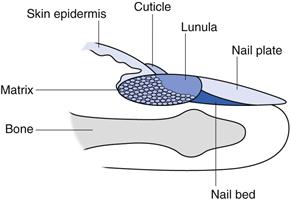
The nail matrix, sometime called the nail root, contains onychocytes, a specialized version of the keratinocytes found in stratum corneum. Much of the matrix is hidden, but the lunula, (‘the moon’) is the white crescent shaped part of the matrix most clearly visible on the thumbs.
The nail bed is an extension of the matrix and contains blood vessels and nerves.
Overlying the nail bed is the protective nail plate, a translucent keratinized layer that appears pink because of the blood vessels in the nail bed. The nail plate comprises approximately 25 layers of keratinized cells and is 0.25 to 0.6 mm thick on the fingers, whereas toenails are up to 1.3 mm thick. Nail plate growth is variable but is typically around 1 mm per month for toenails whereas fingernails grow at around 3 mm per month. The nail plate has three distinct layers, an outer dorsal layer that is dense and hard, an intermediate layer that is fibrous with fibres aligned perpendicular to the direction of growth and a thin ventral layer that connects to the nail bed. The nail plate contains between 0.1 and 1% lipids, less than that found in the skin stratum corneum, and so is a more hydrophilic barrier than skin.
Diseases of the nail plate, bed and matrix are often unsightly and cause patient distress. Onychomycosis is a common fungal infection of the nail bed or plate and affects up to 5% of the population and can cause discolouration, thickening or crumbling of the nail plate. Nail psoriasis occurs in most patients with skin psoriasis and can cause pitting of the nail, discolouration and roughening or crumbling of the nail plate.
These and other nail disorders are difficult to treat, since the nail plate provides a formidable barrier to drug delivery and so enhancement strategies have been sought. One approach is to simply abrade the nail, thinning it by filing or laser ablation or etching with acid, or creating pores using needles or microneedles. Alternatively, penetration enhancers can be included in formulations.
As with the stratum corneum, formulations can use a vehicle which encourages drug partitioning into the nail plate with ethanol and other solvents also commonly used to increase delivery from supersaturated states and by interacting with the nail. Other chemical enhancers seek to disrupt the keratin fibres that are the main constituent of nail plates. As the nail plate has relatively low lipid levels, enhancers that work in skin by disrupting the lipid bilayers are ineffective in the nail. Thus, keratolytic agents such as urea, oxidizing agents such as hydrogen peroxide and reducing agents including thioglycolic acid which disrupt disulphide bonds in keratins have been used to increase drug diffusivity through nail plates.
Antifungal agents such as terbinafine or itraconazole are commonly used orally to treat serious onychomycosis infections of the nail but can result in adverse reactions since orally delivered drugs must enter the systemic circulation before diffusing out to the infected area. Local delivery is thus clinically attractive, but limited by the nail plate barrier. Antifungal formulations of amorolifine, salicylic acid (which is keratolytic) and tioconazole for application to the nail are available as lacquers, paints and solutions. Excipients in the formulations include boric and tannic acids to etch the nail surface, alcoholic vehicles to enhance flux and, notably, patient information usually includes the direction to file the nail before application. At present, topical formulations to manage nail psoriasis are not widely available.
Bibliography
1. Bronaugh RL, Maibach HI, eds. Percutaneous Absorption. 4th edn New York: Marcel Dekker; 2005.
2. Nakashima E, Noonan PK, Benet LZ. Transdermal bioavailability and first-pass skin metabolism: A preliminary evaluation with nitroglycerin. Journal of Pharmacokinetics and Pharmacodynamics. 1987;15:423–437.
3. Otberg N, Richter H, Schaefer H, Blume-Peytavi U, Sterry W, Lademann J. Variations of hair follicle size and distribution in different body sites. Journal of Investigative Dermatology. 2004;122:14–19.
4. Smith EW, Maibach HI, eds. Percutaneous Penetration Enhancers. 2nd edn Taylor and Francis, Boca Raton, Florida: CRC Press; 2005.
5. Smith EW, Surber C, Maibach HI. Topical dermatological vehicles: a holistic approach. In: Bronaugh RL, Maibach HI, eds. Percutaneous Absorption; Drugs – Cosmetics – Mechanisms – Methodology. 3rd edn New York: Marcel Dekker Inc; 1999.
6. Touitou E, Barry BW, eds. Enhancement in Drug Delivery. Taylor and Francis, London: CRC Press; 2007.
7. Williams AC. Transdermal and Topical Drug Delivery. London: Pharmaceutical Press; 2003.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Topical and transdermal drug delivery
Adrian C. Williams
Chapter contents
Permeant properties affecting permeation
Mathematics of skin permeation
Experimental methods for studying transdermal drug delivery
Transdermal and topical preparations
Key points
• Transdermal drug delivery the skin to deliver the drug to the systemic circulation.
• Patches, of varying complexities, are the most common transdermal drug delivery systems.
Introduction
Topical and transdermal formulations have a long history of use. Over 2000 years ago, Greek physicians used formulations containing salt, vinegar, honey and resins to treat skin lesions and ulcers. Chinese, Egyptian and Roman medical histories describe numerous remedies applied topically as pastes and poultices.
Topical and transdermal products remain key formulations for delivering drugs not only to the skin, but also through it for systemic action. An estimated 40 million topical items are dispensed annually in England and Wales, including 16 million emollient products and 13 million topical corticosteroid preparations. In addition, many other products are dispensed for topical anaesthesia and antisepsis or for transdermal delivery, such as fentanyl patches. Additionally, ‘over-the-counter’ products are widely sought by patients and range from emollients to non-steroidal anti-inflammatory creams and gels, to treatments for warts, verrucae and fungal infections, such as athletes’ foot. Thus, pharmacists often supply topical and transdermal formulations which contain a broad variety of active ingredients; indeed it has been reported that up to 20% of all repeat prescriptions are for application to the skin. The efficacy of these products is critically dependent on biological factors, such as the integrity of the skin, on the physicochemical properties of the active ingredient and on the formulation designed to release and deliver the active into or across the skin.
Terminology
The literature occasionally contains different terminology relating to transdermal and topical drug delivery. For this Chapter, it may be helpful to clarify the following at this point:
Topical drug delivery.
The application of a formulation to the skin to treat a local disorder, i.e. the intention is to retain the active pharmaceutical ingredient (API) within the skin, for example, a locally acting hydrocortisone cream.
Transdermal drug delivery.
The application of a formulation to the skin to deliver a drug to the systemic circulation, for example, estradiol patches.
Locally acting.
The active pharmaceutical ingredient acts directly on the skin.
Regionally acting.
The active pharmaceutical ingredient acts in the area close to where the formulation is applied. This is often also described as locally acting, but here the drug does not act directly on the skin, for example topically applied ibuprofen gels to treat musculoskeletal conditions.
Permeant.
The chemical species that is moving through or into the tissue. This will be the drug, but may also be other ingredients within the formulation.
Permeation.
Movement of drug through the membrane.
Penetration.
Entry into the tissue. Penetration does not necessarily require the molecules to pass out of the tissue.
Diffusion.
Movement of molecules through a domain, from high concentration to low concentration, by random molecular movement.
Diffusivity.
This is a property of the permeant in the membrane and is a measure of how easily it will traverse through the tissue. It is expressed in units of area/time (usually cm2/h or cm2/s).
Diffusion coefficient (D).
This is the diffusion coefficient of the permeant, and is sometimes a term used interchangeably with diffusivity. As with diffusivity, its units are area/time (usually cm2/h or cm2/s).
Permeability coefficient (kp).
Describes the speed of permeant transport, given in units of distance/time (usually cm/h).
Partition coefficient (P).
This is a measure of the distribution of molecules between two phases. For transdermal delivery studies, a partition coefficient (usually expressed as a log10, hence ‘log P’) between octanol and water is often used as a guide to how well a molecule will distribute between water and stratum corneum lipids. In some texts, the symbol K is used for the partition coefficient; here, and to avoid confusion with the permeability coefficient (kp), the symbol P (also widely used in the literature) has been employed.
Partitioning.
The process of molecules distributing between two domains. In transdermal drug delivery, partitioning is generally used to describe molecular redistribution from one domain to another, such as from an aqueous domain to a lipid domain.
Flux (J).
The rate of a permeant crossing the skin (or entering the systemic circulation). It is given in units of mass/area/time (usually µg/cm2/h).
Lag time (L).
This is obtained from a permeation profile by extrapolating the steady state flux line to the time axis. Older texts have used the symbol τ whereas others have used tL for the lag time, but most modern texts use the abbreviation L.
Vehicle.
The base formulation in which the drug is applied to the skin.
Thermodynamic activity.
Used here as a measure of the ‘escaping tendency’ of a molecule from its formulation. A thermodynamic activity of 1 equates to a saturated solution, or suspension, since the molecules in a saturated solution have the greatest ‘escape tendency’.
Skin structure and function
Human skin is a highly complex multilayered organ designed to ‘keep the outside out and the insides in’. It is the largest organ of the body, comprising around 10% of the body mass and covers an area of approximately 1.8 m2 in a typical adult. As a self-repairing barrier, skin permits terrestrial life by preventing the ingress of microorganisms and chemicals whilst regulating heat and water loss from the body. Indeed, the body continually loses water and transepidermal water loss (TEWL) is in the region of 1 mg/cm2/h, but its value varies with body site and external conditions (temperature, humidity).
For drug delivery and therapy, the intact skin presents a formidable barrier and a difficult challenge to formulation scientists. The properties of the skin limit the range of active ingredients that can be delivered through the barrier to achieve therapeutic levels. However, skin can be relatively easily damaged through mechanical, chemical or microbiological assault and by radiation, such as sun damage. In these cases, drug delivery may be enhanced and could in fact lead to adverse reactions.
Structure of the skin
In terms of drug delivery, human skin can be considered as a series of layers which potentially provide a series of barriers to a molecule traversing the tissue (Fig. 39.1).
Fig. 39.1 A diagrammatical cross-section through human skin showing the different skin layers and appendages.
The subcutaneous layer
The inner subcutaneous fatty layer is typically several millimetres thick, except for some areas such as the eyelids where it is mostly absent. This subcutaneous layer of adipose tissue provides mechanical protection against physical shock, insulates the body, provides a store of high-energy molecules and carries the principal blood vessels and nerves to the skin. The subcutaneous layer is seldom an important barrier to transdermal and topical drug delivery.
The dermis
Overlying the fatty layer is the dermis, a layer typically 3–5 mm thick that is the major component of human skin. The dermis is composed of a network of mainly collagen and elastin in a mucopolysaccharide gel; essentially this combination provides an aqueous environment similar to a hydrogel. The dermis has several structures embedded within it, termed appendages, in particular nerve endings, pilosebaceous units (hair follicles and sebaceous glands) and eccrine and apocrine sweat glands (see below).
The dermis is metabolically active and requires extensive vasculature for this, as well as for regulating body temperature, for wound repair, to deliver oxygen and nutrients to the tissue and to remove waste products. The blood supply reaches to approximately 0.2 mm below the skin surface, near the dermis-epidermis boundary, and so most molecules passing through the outer layer of the skin are rapidly diluted and are carried systemically by the blood. This rich blood flow keeps the dermal concentration of most transdermally delivered drugs low, which in turn provides a concentration gradient from the outside of the body into the skin and it is this concentration gradient (more accurately, it is the chemical potential gradient) that allows drug delivery through the skin.
The epidermis
The epidermis overlies the dermis and is itself a multiple layer containing various cell types, including keratinocytes, melanocytes and Langerhans cells. Keratinocytes in the basal layer (stratum basale) undergo division and then differentiate as they migrate outwards, forming the stratum spinosum, then the stratum granulosum and finally the stratum corneum. Differentiation is complex and essentially changes the metabolically active basal cells that contain typical organelles, such as mitochondria and ribosomes, into stratum corneum that comprises anucleate flattened corneocytes packed into multiple lipid bilayers.
The stratum corneum
This outer skin layer is predominantly responsible for the barrier properties of human skin and limits drug delivery into and across the skin. The stratum corneum typically comprises only 10 to 15 cell layers and is around 10 µm thick when dry (although it can swell to several times this when wet). The stratum corneum is thinnest on the lips and eyelids and thickest on the load-bearing areas of the body such as the soles of the feet and palms of the hands. The lipid bilayers in which the keratin filled cells are embedded are uniquely different to other lipid bilayers in the body since they are comprised largely of ceramides, fatty acids, triglycerides and cholesterol/cholesterol sulphate, whilst phospholipids are largely absent. Longer chain ceramides act as ‘rivets’ connecting bilayers together and corneo-desmosomes interconnect corneocytes. The resulting structure can be likened to a brick wall (Fig. 39.1) where the keratin-filled cells act as the bricks in a mortar of multiply bilayered lipids.
In normal skin, it takes approximately 14 days for a daughter cell in the stratum basale to migrate and differentiate into a stratum corneum cell, and these cells are then retained in the stratum corneum for a further 2 weeks before they are shed.
The appendages
In terms of drug delivery, the appendages can be viewed as shunt routes or ‘short cuts’ through which molecules can pass across the stratum corneum barrier.
Specialized apocrine glands are found at specific body sites, such as the axillae and nipples, whereas eccrine glands are found over most of the body surface at a density of 100–200 glands per cm2. When stimulated by heat or emotional stress, eccrine glands secrete sweat, which is a dilute salt solution of around pH 5.
The largest appendages are the hair follicles and associated sebaceous glands which secrete sebum, composed of fatty acids, waxes and triglycerides. These lubricate the skin surface and help to maintain the skin surface pH at around 5. Skin typically has 50 to 100 hair follicles per cm2 but the load bearing areas of the soles and palms are largely devoid of these appendages.
Whilst these shunt routes offer a potential route through intact skin, the fractional area that they occupy is relatively small; for example, on the forearm, hair follicles occupy approximately 0.1% of the surface area although on the forehead this may be as much as 13%. The ducts are seldom empty, being occupied by sweat or sebum flowing out of the body which again inhibits drug delivery. However, formulators are able to target these structures, for example by delivering nano-sized drug delivery systems, such as liposomes, to the follicles in order to treat acne.
The shunt routes are important for electrical enhancement of transdermal drug delivery (iontophoresis) and also play a role in the early time course of passive drug delivery through the skin, where diffusion through the intact stratum corneum barrier has yet to reach steady state; rub a cut clove of garlic on your leg and it can be tasted within minutes, which is too fast for molecules to diffuse across the intact stratum corneum.
Transport through the skin
From the above discussion on the structure of skin it is clear that delivery of drug molecules from a topically applied formulation into the systemic circulation is complex, with numerous processes occurring and several routes of transport in operation, as illustrated in Figure 39.2.
Fig. 39.2 Some of the processes occurring during transdermal drug delivery from a suspension formulation.
Initially, drug molecules must be presented to the skin surface. Consequently, if the formulation contains solid drug, then dissolution and diffusion through the formulation is the initial step in delivery. If the formulation contains dissolved drug, then as the molecules nearest to the skin surface enter the tissue these must be replaced by other molecules diffusing within the formulation towards the skin surface. Once at the outer layer of the stratum corneum, the drug molecule has three potential routes to cross the skin. Firstly it can pass via the shunt routes as described above. In this case molecules will partition into sweat or sebum before diffusing against the outflow from the glands.
More usually, the molecule encounters the intact stratum corneum ‘brick wall’ where transport can either be via an intracellular (also termed transcellular) route or transport can be intercellular.
Considering the intracellular route, the drug molecule initially partitions into a keratin-filled corneocyte, which is essentially an aqueous environment, they then diffuse through the corneocyte before partitioning into the intercellular lipid domains. For transcellular transport to continue, the molecule must then diffuse through the lipoidal region before repeatedly partitioning into and diffusing through the aqueous keratin in corneocytes and then intercellular lipids.
In contrast, the intercellular route requires the molecule to partition into the lipid bilayers between the corneocytes and then diffusion is via a tortuous route within the continuous lipid domain, i.e. following the mortar in the ‘brick wall’.
Having travelled through the stratum corneum, molecules diffuse through the lower epidermal layers before being cleared by the capillaries at the epidermal-dermal junction. During transport, there is potential for the permeant to bind to skin components such as keratin, in which case it may not reach the systemic circulation but could be sloughed off. In addition, skin is metabolically active and contains esterases, peptidases and hydrolases that can reduce the bioavailability of topically applied drugs such that, for example, only around 70% of topically applied glyceryl trinitrate (nitroglycerin) may be bioavailable.
It is important to recognize that, whilst three different routes exist for drugs to cross the skin (intercellular, transcellular, and shunt routes), for any permeant ALL three routes operate but the proportion of molecules crossing by the different routes will vary depending on the physicochemical properties of the permeant.
Permeant properties affecting permeation
Considering the processes described above, it is evident that the physicochemical properties of the permeant will control its transport into and through the skin. For both the transcellular and intercellular routes, the drug molecule has to cross the multiple lipid bilayers between the corneocytes and hence partitioning into, and diffusion through these lipid environments is essential. However, to reach the systemic circulation the molecule must also pass through the more aqueous environment of the viable epidermal cells and enter the blood. Thus, molecules which are lipophilic are usually seen as better candidates for transdermal delivery than hydrophilic compounds, but high lipophilicity is problematic for clearance.
The molecular weight of the permeant also impacts dramatically on its transport through the skin. The skin is designed to act as a barrier to external chemicals and so prevents the entry of large molecules, such as larger peptides and proteins. Not only is molecular weight an important factor in diffusion, but molecular structure (in particular hydrogen-bonding potential) can control the extent of binding to skin constituents and hence affect bioavailability. Naturally, the drug must have some solubility in the formulation and whilst transport through the stratum corneum is usually the rate limiting step in transdermal delivery, poor drug release from a formulation can occasionally limit drug transport. Finally, the effective dose of the drug must be relatively low to allow the application of appropriately sized patches/formulations.
The above processes restrict the range of drugs that can be delivered transdermally to therapeutically useful levels and some generic ‘rules of thumb’ can be used to predict whether transdermal delivery is viable for an active pharmaceutical ingredient. These include:
Indeed, the active pharmaceutical ingredients currently used in transdermal formulations have many of the above properties; estradiol MW is 272 and it is lipophilic, with a of 2.7; fentanyl MW is 336 with a log of 4.4; nicotine MW is 162 with a of 1.2.
Mathematics of skin permeation
With such a highly complex multiple layered organ as skin, and numerous factors affecting transdermal drug delivery, it appears daunting to apply mathematical principles to describe such a complex process. However, simple mathematical principles can be used to understand the basic principles of permeation through membranes, including skin, and these assist in designing dosage forms for transdermal and topical drug delivery.
Fick’s laws of diffusion
Considering simple passive diffusion where molecules move by random motion from one region to another in the direction of decreasing concentration, then transport can be described by Fick’s First Law of Diffusion (Chapter 2):
(39.1)
where J is the flux (the mass flow rate at which the material passes through unit area of the surface), C is the concentration of diffusing substance, x is the space co-ordinate measured normal to the section and D is the diffusion coefficient of the permeant. The negative sign demonstrates that the flux of molecules is in the direction of decreasing concentration. When a topically applied drug enters the skin, it is usually assumed that the diffusion gradient is from the outer surface into the tissue, i.e. is unidirectional.
Fick’s Second Law of Diffusion gives:
(39.2)
where t is time. Essentially this equation shows that the rate of change of concentration with time at a point within a diffusional field is proportional to the rate of change in the concentration gradient at that point.
The above laws assume that diffusion is through an isotropic material (i.e. one that has the same structural and diffusional properties in all directions); skin clearly is not isotropic with multiple layers, different permeation routes, etc. and indeed it is remarkable that Fickian diffusion can be used to generate valuable approximations from transdermal drug delivery data.
Experimental estimation of skin penetration
Experimentally, it is usually difficult to study transdermal drug delivery in vivo, so most researchers use in vitro protocols to mimic as closely as possible the in vivo situation. Most commonly, a membrane (for example human epidermis) is used to separate two compartments in a diffusion cell. The drug in a vehicle (for example water, buffer or in a formulation) is then applied to the uppermost skin surface (stratum corneum). This is usually termed the ‘donor’ solution. The other compartment contains a ‘receptor’ (or receiver) solution that is a good solvent for the drug, but which will not affect the skin barrier. This receptor solution thus provides essentially sink conditions (near zero concentration) of the permeant and allows a concentration gradient to exist between the donor and receptor phase, which in turn provides the driving force for diffusion across the membrane. If the cumulative mass of permeant that crosses the membrane is plotted as a function of time, then a typical permeation profile can be drawn, as illustrated in Figure 39.3.
As can be seen, after sufficient time the plot approaches a straight line and from the slope we can obtain the steady state flux, dm/dt and Equation 39.2 can then be simplified to:
(39.3)
where dm/dt is the flux, usually termed J which is the cumulative mass of permeant that passes per unit area of the membrane in time t, Co is the concentration of permeant in the first layer of the membrane (at the skin surface, in contact with the donor solution) and h is the membrane thickness.
It is difficult to measure Co, the concentration of permeant in the first layer of the skin, but the concentration of the drug in the vehicle (donor solution), termed Cv, which bathes the skin membrane is usually known or can be determined easily. Differences in drug concentration between the donor solution and the first skin layer arise due to partitioning of the molecule between the membrane and donor solution so:
(39.4)
where P is the partition coefficient of the permeant between the membrane and the vehicle. Simply substituting Equation 39.4 into Equation 39.3 gives:
(39.5)
Equation 39.5 thus reiterates that the flux of a permeant through skin will be high for molecules with a high diffusion coefficient (e.g. generally having a relatively small molecular weight), will increase with increasing partitioning between the membrane and the donor solution (e.g. for lipophilic molecules) and will increase with increasing effective concentration in the donor solution (which increases the chemical potential gradient), whereas the flux through thicker membranes is reduced.
Figure 39.3 also shows that the lag time can be evaluated experimentally. The lag time (L
[/not-level-membership-for-basic-science-category]
