[level-membership-for-pathology-category]
Chapter 6 Responses to cellular injury
CELLULAR INJURY



Cell survival depends upon several factors: a constant supply of energy, intact plasma membrane, biologically safe and effective function of generic and specific cellular activities, genomic integrity, controlled cell division, and internal homeostatic mechanisms. Cell death may result from significant disturbance of these factors. However, cell replication proceeds in a human body at a rate of c. 10 000 new cells per second; so, although eventually some will be lost to the environment via the skin or gut surfaces, many will inevitably need to be deleted. Thus, cell death is a normal physiological process as well as a reaction to injury. Similarly, failure or poor regulation of death processes may underlie some diseases.
Causative agents and processes
A wide range of possible agents or circumstances result in cellular injury (Fig. 6.1). These could be categorised according to the nature of the injurious agent, the cellular target, the pattern of cellular reaction or mode of cell death. The sequence of agent, target and mode will be uniform, but some injurious agents have variable effects depending on concentration, duration or other contributory influences such as co-existent disease. Some examples are given in Table 6.1. Major types of cellular injury include:
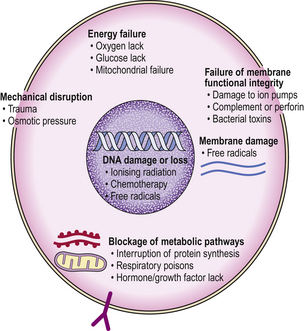
Fig. 6.1 Mechanisms of cellular injury. Different agents can injure the various structural and functional components of the cell. Some cells with specific function are selectively prone to certain types of injury.
Table 6.1 Examples of causes of cellular injury and their mode of action
| Example agent | Mode of action |
|---|---|
| Trauma (e.g. road traffic accident) | Mechanical disruption of tissue |
| Carbon monoxide inhalation | Prevents oxygen transport |
| Contact with strong acid | Coagulates tissue proteins |
| Paracetamol overdose | Metabolites bind to liver cell proteins and lipoproteins |
| Bacterial infections | Toxins and enzymes |
| Ionising radiation (e.g. X-rays) | Damage to DNA |
Physical agents
Most physical agents cause passive cell destruction by gross membrane disruption or catastrophic functional impairment. Trauma and thermal injury cause cell death by disrupting cells and denaturing proteins, and also cause local vascular thrombosis with consequent tissue ischaemia or infarction (Ch. 8). Freezing damages cells mechanically because their membranes are perforated by ice crystals. Missile injury combines the effects of trauma and heat; much energy is dissipated into tissues around the track. Blast injuries are the result of shearing forces, where structures of differing density and mobility are moved with respect to one another; traumatic amputation is a gross example. Microwaves (wavelengths in the range from 1 mm to 1 m) cause thermal injury. Laser light falls into two broad categories: relatively low energy produces tissue heating, with coagulation for example; higher energy light breaks intramolecular bonds by a photochemical reaction, and effectively vaporises tissue. Ionising radiation is considered on p. 115.
Chemical and biological agents
Cells may be injured by contact with drugs and other chemicals; the latter may include enzymes and toxins secreted by micro-organisms. This category of agents can give rise to the full range of modes of death.
Drugs and poisons
Many naturally occurring and synthetic chemicals cause cellular injury; the effect is usually dose-related, but in a few instances the effect is exacerbated by constitutional factors.
Some are highly toxic systemic metabolic poisons, while others exert their damage locally; the latter includes caustic liquids applied to skin or mucous membranes, or gases that injure the lung. Furthermore, some substances produce one effect locally and another systemically. For example, some drugs are potentially caustic, and care needs to be taken to avoid extravasation into soft tissues when giving them by intravenous injection.
Caustic agents cause rapid local cell death due to their extreme alkalinity or acidity, in addition to having a corrosive effect on the tissue by digesting proteins.
Infectious organisms
The mechanisms of tissue damage produced by infectious organisms are varied, but with many bacteria it is their metabolic products or secretions that are harmful (Ch. 3). Thus, the host cells receive a chemical insult that may be toxic to their metabolism or membrane integrity. The mode of cell death generally induces an acute inflammatory response, which may be damaging to adjacent cells; organisms that do this are called pyogenic. In contrast, bacterial endotoxin (lipopolysaccharide) induces apoptosis with different pathological consequences. Intracellular agents such as viruses often result in the physical rupture of infected cells, but with some viruses such as hepatitis B (Ch. 16) local tissue damage may result from host immune reactions. Therefore, the cellular response to injury caused by infections will depend on a combination of the damage inflicted directly by the agent and indirectly as a result of the host response to the agent.
Blockage of metabolic pathways
Cell injury may result from specific interference with intracellular metabolism, effected usually by relative or total blockage of one or more pathways.
Cellular respiration
Prevention of oxygen utilisation results in the death of many cells due to loss of their principal energy source. Cyanide ions act in this way by binding to cytochrome oxidase and thus interrupting oxygen utilisation. Cells with higher metabolic requirements for oxygen (e.g. cardiac myocytes) are most vulnerable.
Glucose deprivation
Glucose is another important metabolite and source of energy. Some cells, cerebral neurones for example, are highly dependent. In diabetes mellitus there is inadequate utilisation of glucose due to an absolute or relative lack of insulin.
Protein synthesis
Cell function and viability will also be compromised if protein synthesis is blocked at the translational level because there is a constant requirement to replace enzymes and structural proteins. Ricin, a potent toxin from the castor oil plant, acts in this manner at the ribosomal level. Many antibiotics, such as streptomycin, chloramphenicol and tetracycline, act by interfering with protein synthesis, although toxic effects by this mechanism are fortunately rare.
Loss of growth factor or hormonal influence
Many cells rely on growth factors for their survival. Typically, these bind to growth factor receptors spanning the plasma membrane, triggering an intracellular cascade, often via a tyrosine kinase. This pathway can fail or be blocked at many points including growth factor deficit, receptor loss or blockade, or tyrosine kinase inhibitor (e.g. imatinib) (Ch. 4); affected cells may undergo apoptosis. Similar consequences can follow hormone withdrawal, as either a physiological response or part of a disease process. If widespread in an organ, it will shrink (atrophy).
Ischaemia and reperfusion injury
Impaired blood flow (Ch. 8) causes inadequate oxygen delivery to cells. Mitochondrial production of ATP will cease, and anaerobic glycolysis will result in acidosis due to the accumulation of lactate. The acidosis promotes calcium influx. Cells in different organs vary widely in their vulnerability to oxygen deprivation; those with high metabolic activity such as cortical neurones and cardiac myocytes will be most affected.
When the blood supply is restored, the oxygen results in a burst of mitochondrial activity and excessive release of reactive oxygen species (free radicals).
Free radicals
Free radicals are atoms or groups of atoms with an unpaired electron (symbolised by a superscript dot); they avidly form chemical bonds. They are highly reactive, chemically unstable, generally present only at low concentrations, and tend to participate in or initiate chain reactions.
Free radicals can be generated by two principal mechanisms:
The consequences of free radical formation include the following:
The clinicopathological events involving free radicals include:
Cells irreversibly damaged by free radicals are deleted, generally by apoptosis.
Failure of membrane integrity
Cell membrane damage is an important mode of cellular injury for which there are several possible mechanisms:
Cell membrane damage is one of the consequences of complement activation (Ch. 9); some of the end products of the complement cascade have cytolytic activity. Another effector of cytolysis is perforin, a mediator of lymphocyte cytotoxicity that causes damage to the cell membrane of the target cells such as those infected by viruses. Incidental membrane tears or perforations can be repaired very quickly, so do not necessarily result in cell death.
Intramembrane channels permit the controlled entry and exit of specific ions. Blockage of these channels is sometimes used therapeutically. For example, verapamil is a calcium channel blocker used in the treatment of hypertension and ischaemic heart disease. Used in inappropriate circumstances or at high dosage, however, the calcium channel blockage may have toxic effects.
Membrane ion pumps that are responsible for maintaining intracellular homeostasis, for example calcium, potassium and sodium concentrations within cells, are dependent on an adequate supply of ATP. Any chemical agents that deplete ATP, either by interfering with mitochondrial oxidative phosphorylation or by consuming ATP in their metabolism, will compromise the integrity of the membrane pumps and expose the cell to the risk of lysis. The Na/K ATPase in cell membranes can be directly inhibited by ouabain. Failure of membrane ion pumps frequently results in cell swelling, also called oncosis or hydropic change (see below), which may progress to cell death.
Just as disastrous for the cell is biochemical alteration of the lipoprotein bilayer forming the cell membrane. This can result from reactions with either the phospholipid or protein moieties. Membrane phospholipids may be altered through peroxidation by reactive oxygen species and by phospholipases. If the membrane damage results in lysosome permeability, release of its contents precipitates further cell damage or death. Membrane proteins may be altered by cross-linking induced by free radicals.
DNA damage or loss
Damage to DNA results primarily from reactive oxygen species attack, for example following ionising radiation (p. 117). Damage may not be evident immediately; dividing cells are more susceptible. Cell populations that are constantly dividing (i.e. labile cells such as intestinal epithelium and haemopoietic cells) are soon affected by a dose of radiation sufficient to alter their DNA. Other cell populations may require a growth or metabolic stimulus before the DNA damage is revealed. Since non-lethal DNA damage may be inherited by daughter cells, a clone of transformed cells with abnormal growth characteristics may be formed; this is the process of neoplastic transformation that results in tumours (Ch. 11).
Normal erythrocytes are particularly sensitive to injury because they lack a nucleus and, therefore, the means for inducing repair mechanisms. This will also be the fate of any cell in which the nucleus is severely damaged, or when mitosis is attempted but its completion is blocked. The latter is the result of DNA strand breaks or cross-linkages; ionising radiation and some cytotoxic drugs used in cancer therapy have this effect. Damaged cells are deleted, usually by apoptosis.
Cellular appearances following injury
The agents and mechanisms mentioned above cause a variety of histological abnormalities, although very few are specific for each agent. Two patterns of sublethal cellular alteration seen fairly commonly are hydropic change and fatty change.
Hydropic change (oncosis)
In hydropic change the cytoplasm becomes pale and swollen due to accumulation of fluid. Hydropic change generally results from disturbances of metabolism such as hypoxia or chemical poisoning. These changes are reversible, although they may herald irreversible damage if the causal injury is persistent.
Fatty change (steatosis)
Vacuolation of cells is due often to the accumulation of lipid droplets as a result of a disturbance to ribosomal function and uncoupling of lipid from protein metabolism. The liver is commonly affected in this way by several causes, such as hypoxia, alcohol or diabetes. Moderate degrees of fatty change are reversible, but severe fatty change may not be.
Mechanisms of cell death
There are two distinct mechanisms by which cells die: necrosis and apoptosis. However, there are also other cellular deaths combining features of both these processes. Discussion of cell death is further complicated by a lack of uniform nomenclature; some authors use the term necrosis to denote cell death by any cellular mechanism, but more often it is used to describe a specific mechanism. Although there are usually particular triggers for one process or another, there are some situations where apoptosis follows a lower dose or shorter duration of insult while necrosis occurs above that threshold. Mechanisms of cell death involve defined metabolic pathways. Consequently, cell death processes may be amenable to therapeutic interventions.
Necrosis









Necrosis is characterised by bioenergetic failure and loss of plasma membrane integrity. The ischaemia–reperfusion model has been the focus of much resear Ch. Failure of ATP production renders plasma membrane ion pumps ineffective with resulting loss of homeostasis, influx of water, oncosis, lysis and cell death, but in many circumstances this sequence may be an oversimplification.
Anaerobic conditions result in acidosis, thus promoting calcium inflow. Calcium uptake by mitochondria eventually exceeds their storage capacity, and contributes to disruption of the inner membrane (mitochondrial permeability transition); ATP production ceases and contents leak into the cytosol. This mitochondrial sequence is particularly exacerbated, if not initiated, by reperfusion causing a burst of reactive oxygen species production.
DNA damage, for example by free radicals or alkylating agents, initiates repair sequences including activation of the nuclear enzyme poly (ADP-ribose) polymerase (PARP). In proliferating cells, as they are dependent on glycolysis, this leads to NAD depletion and thus ATP depletion and consequently necrosis.
Falling ATP levels can open plasma membrane channel-mediated calcium uptake (death channels); large rises in cytosol calcium activate calcium-dependent proteases or lead on to mitochondrial permeability transition. In contrast, free radical damage to endoplasmic reticulum allows calcium stores to leak into the cytosol; smaller rises in calcium tend to cause apoptosis rather than necrosis.
Free radical damage to lysosomal membranes releases proteases, such as cathepsins which damage other membranes and can cause cell death. By a similar mechanism, binding of tumour necrosis factor to its cell surface receptor stimulates excessive mitochondrial reactive oxygen species with the results noted above and hence necrosis.
All these pathways eventually lead to rupture of the plasma membrane and spillage of cell contents, but this is not the end of the sequence. Some of the contents released are immunostimulatory, for example heat shock proteins and purine metabolites. These provoke the inflammatory response (Ch. 10), which paves the way for repair.
Several distinct morphological types of necrosis are recognised:
The type of tissue and nature of the causative agent determine the type of necrosis.
Necrosis must be distinguished from apoptosis, in which cell death results from a different mechanism. The cell membrane remains intact and there is no inflammatory reaction.
Coagulative necrosis
Coagulative necrosis is the commonest form of necrosis and can occur in most organs. Following devitalisation, the cells retain their outline as their proteins coagulate and metabolic activity ceases. The gross appearance will depend partly on the cause of cell death, and in particular on any vascular alteration such as dilatation or cessation of flow. Initially, the tissue texture will be normal or firm, but later it may become soft as a result of digestion by macrophages. This can have disastrous consequences in necrosis of the myocardium following infarction, as there is a risk of ventricular rupture (Ch. 13).
Microscopic examination of an area of necrosis shows a variable appearance depending on the duration. In the first few hours, there will be no discernable abnormality. Subsequently, there will be progressive loss of nuclear staining until it ceases to be haematoxyphilic; this is accompanied by loss of cytoplasmic detail (Fig. 6.2). The collagenous stroma is more resistant to dissolution. The result is that, histologically, the tissue retains a faint outline of its structure until such time as the damaged area is removed by phagocytosis (or sloughed off a surface), and is then repaired or regenerated. The presence of necrotic tissue usually evokes an inflammatory response; this is independent of the initiating cause of the necrosis.
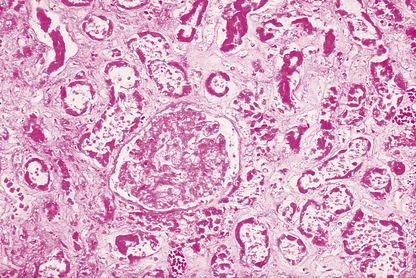
Fig. 6.2 Necrosis. Histology of part of a kidney deprived of its blood supply by an arterial embolus (Ch. 8). This is an example of coagulative necrosis. Cellular and nuclear detail has been lost. The ghost outline of a glomerulus can be seen in the centre, with remnants of tubules elsewhere.
Colliquative necrosis
Colliquative necrosis occurs in the brain because of its lack of any substantial supporting stroma; thus, necrotic neural tissue may totally liquefy. There will be a glial reaction around the periphery, and the site of necrosis will be marked eventually by a cyst.
Caseous necrosis
Tuberculosis is characterised by caseous necrosis, a pattern of necrosis in which the dead tissue is structureless. Histological examination shows an amorphous eosinophilic area stippled by haematoxyphilic nuclear debris. Although not confined to tuberculosis, nor invariably present, caseation in a biopsy should always raise the possibility of tuberculosis.
Gangrene
Gangrene is necrosis with putrefaction of the tissues, sometimes as a result of the action of certain bacteria, notably clostridia. The affected tissues appear black because of the deposition of iron sulphide from degraded haemoglobin. Thus, ischaemic necrosis of the distal part of a limb may proceed to gangrene if complicated by an appropriate infection. As clostridia are very common in the bowel, intestinal necrosis is particularly liable to proceed to gangrene; it can occur as a complication of appendicitis, or incarceration of a hernia if the blood supply is impeded. These are examples of ‘wet’ gangrene. In contrast, ‘dry’ gangrene is usually seen in the toes, as a result of gradual arterial or small vessel obstruction in atherosclerosis or diabetes mellitus, respectively. In time, a line of demarcation develops between the gangrenous and adjacent viable tissues.
In contrast to the above, primary infection with certain bacteria or combinations of bacteria may result in similar putrefactive necrosis. Gas gangrene is the result of infection by Clostridium perfringens, while synergistic gangrene follows infection by combinations of organisms, such as Bacteroides and Borrelia vincenti.
Fibrinoid necrosis
In the context of malignant hypertension (Ch. 13), arterioles are under such pressure that there is necrosis of the smooth muscle wall. This allows seepage of plasma into the media with consequent deposition of fibrin. The appearance is termed fibrinoid necrosis. With haematoxylin and eosin staining, the vessel wall is a homogeneous bright red. Fibrinoid necrosis is sometimes a misnomer because the element of necrosis is inconspicuous or absent. Nevertheless, the histological appearance is distinctive and its close resemblance to necrotic tissue perpetuates the name of this lesion.
Fat necrosis
Following trauma to adipose tissue, the release of intracellular fat elicits a brisk inflammatory response, with polymorphs and macrophages phagocytosing the fat, proceeding eventually to fibrosis. The result may be a palpable mass, particularly at a superficial site such as the breast.
In acute pancreatitis, there is release of pancreatic lipase (Ch. 16). As a result, fat cells have their stored fat split into fatty acids, which then combine with calcium to precipitate out as white soaps. In severe cases, hypocalcaemia can ensue.
Apoptosis




Apoptosis is quite different from necrosis (Table 6.2); indeed, it includes suppression of necrosis. It is an energy-dependent process for deletion of unwanted individual cells. Apoptosis is involved in morphogenesis (Ch. 5), and is the mechanism for controlling organ size. Unwanted or defective cells also undergo apoptosis; thus, lymphocyte proliferation in germinal centres and the thymus is followed by apoptosis of unwanted cells. Factors controlling apoptosis include substances outside the cell and internal metabolic pathways:
Table 6.2 Comparison of cell death by apoptosis and necrosis
| Feature | Apoptosis | Necrosis |
|---|---|---|
| Induction | May be induced by physiological or pathological stimuli | Invariably due to pathological injury |
| Extent | Single cells | Cell groups |
| Biochemical events | Energy-dependent fragmentation of DNA by endogenous endonucleases | Energy failure |
| Impairment or cessation of ion homeostasis | ||
| Lysosomes intact | Lysosomes leak lytic enzymes | |
| Cell membrane integrity | Maintained | Lost |
| Morphology | Cell shrinkage and fragmentation to form apoptotic bodies with dense chromatin | Cell swelling and lysis |
| Inflammatory response | None | Usual |
| Fate of dead cells | Ingested (phagocytosed) by neighbouring cells | Ingested (phagocytosed) by neutrophil polymorphs and macrophages |
| Outcome | Cell elimination | Defence, and preparation for repair |
Exposure to inducers or withdrawal of inhibitors acts via the bcl-2 protein family, which then inhibit or activate the death pathway, resulting in activation of initiator and executioner caspases. Alternatively, activation of the plasma membrane receptor Fas (CD95) by its ligand bypasses bcl-2 to activate other caspases (Fig. 6.3). These result in degradation of the cytoskeletal framework, fragmentation of DNA and loss of mitochondrial function. The nucleus shrinks (pyknosis) and fragments (karyorrhexis). The cell shrinks, retaining an intact plasma membrane (Fig. 6.4), but alteration of this membrane rapidly induces phagocytosis. Dead cells not phagocytosed fragment into smaller membrane-bound apoptotic bodies. There is no inflammatory reaction to apoptotic cells, probably because the cell membrane is intact. Various diseases are associated with reduced or increased apoptosis.
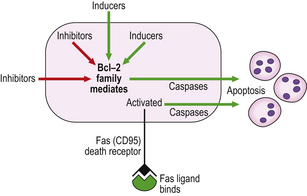
Fig. 6.3 Mechanisms of apoptosis. Apoptosis results from activation of caspases triggered either by the bcl-2 family or by the binding of Fas ligand to its receptor.
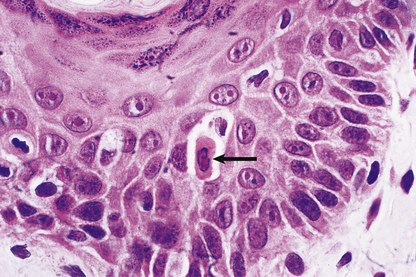
Fig. 6.4 Apoptosis. Histology of skin from a case of graft-versus-host disease (Ch. 9) in which there is individual cell death (arrowed) in the epidermis as a result of immune injury.
Reduced apoptosis
The product of the p53 gene checks the integrity of the genome before mitosis; defective cells are switched to apoptosis instead. In contrast, bcl-2 protein inhibits apoptosis. Therefore, loss of p53 function or excess bcl-2 expression may result in failure of initiation of apoptosis with resulting cell accumulation; both these defects are important in neoplasia (Ch. 11). Autoimmune disease (Ch. 9) might reflect failure of induction of apoptosis in lymphoid cells directed against host antigens; in systemic lupus erythematosus, alterations are reported in the Fas lymphocyte receptor. Some viruses enhance their survival by inhibiting apoptosis of cells they infect, and latent infection by Epstein–Barr virus upregulates bcl-2.
Increased apoptosis
Diseases in which increased apoptosis is probably important include acquired immune deficiency syndrome (AIDS), neurodegenerative disorders and anaemia of chronic disorders (Ch. 23). In AIDS, human immunodeficiency virus proteins may activate CD4 on uninfected T-helper lymphocytes, inducing apoptosis with resulting immunodepletion. Apoptosis is the usual mode of cell death in exposure to ionising radiation (p. 117).
Autophagy
Autophagy is another cellular response to stress, such as deficiency of nutrients or growth factor-mediated effects, or organelle damage. Cell components are isolated into intracellular vacuoles and then processed through to lysosomes. Although generally a means of staving off cell death, it may progress to cell death if the stimulus is more severe, or the cell metabolic pathways may switch to apoptosis.
Patterns of cell death in systematic pathology
The clinical value of knowing the metabolic pathways to cell death lies in the potential to modify them by increasing or decreasing cell survival as appropriate by targeting cell death or cell survival pathways. Thus, exposure to minor degrees of hypoxia has a protective effect in subsequent severe hypoxia; this is called preconditioning. Diseases such as myocardial infarction and stroke are major causes of morbidity, so any intervention improving cell survival could have major benefits. Solid organ transplantation includes an episode of graft ischaemia and reperfusion, so reduction in harm to the graft may be achievable. In contrast, increasing cell kill in cancer treatment is beneficial. In recognition of the complexity of pathways in necrosis, the phrase ‘programmed cell necrosis’ has been suggested as a balance to the established phrase ‘programmed cell death’ (apoptosis).
The outline of necrosis and apoptosis above treats these as particular events in particular circumstances; the reality of disease is often more complex. For example, myocardial ischaemia and reperfusion is characterised by necrosis, but probably has an element of apoptosis in marginally affected tissues. Acute lung injury (adult respiratory distress syndrome) results in widespread alveolar damage following a wide range of circumstances (Ch. 14); thus the precise pathway to cell death varies between Gram-positive sepsis, Gram-negative sepsis, trauma, oxygen toxicity and so on, and includes combinations of necrosis, oncosis, apoptosis and caspase-independent cell death. Treatment strategies will presumably need to be tailored to the precise circumstances; at present generic approaches, such as blocking pro-inflammatory cytokines like tumour necrosis factor, give limited success.
REPAIR AND REGENERATION
 Cells can be divided into labile, stable or permanent populations; only labile and stable cells can be replaced if lost
Cells can be divided into labile, stable or permanent populations; only labile and stable cells can be replaced if lost


The ultimate consequences of injury depends on many factors. Most important is the capability of cells to replicate, replacing those that are lost, coupled with the ability to rebuild complex architectural structures.
Structures such as intestinal villi depending largely on the epithelium for their shape can be rebuilt. However, complex structures such as the renal glomeruli cannot be reconstructed if destroyed.
Cell renewal
Cells in adult individuals are classified according to their potential for renewal (Ch. 5):
Stem cells
Cells lost through injury or normal senescence are replaced from the stem cell pool present in many labile and stable populations. When stem cells undergo mitotic division, one of the daughter cells progresses along a differentiation pathway according to the needs and functional state of the tissue; the other daughter cell retains the stem cell characteristics. Stem cells are a minority population in many tissues and are often located in discrete compartments: in the epidermis, stem cells are in the basal layer immediately adjacent to the basement membrane, in the hair follicles and sebaceous glands; in intestinal mucosa, the stem cells are near the bottom of the crypts. The liver has an equivalent population of progenitor cells, lying between hepatocytes and bile ducts.
There also seems to be a separate pool of stem cells available in the bone marrow; these haemopoietic stem cells are able to seed into other organs and differentiate locally into the appropriate tissue.
The ability of a tissue to regenerate may be dependent on the integrity of the stem cell population. Stem cells are particularly vulnerable to radiation injury; this can result either in their loss, thus impairing the regenerative ability of the tissue, or in mutations propagated to daughter cells with the risk of neoplastic transformation.
Complete restitution
Loss of part of a labile cell population can be completely restored. For example, consider the result of a minor skin abrasion (Fig. 6.5). The epidermis is lost over a limited area, but at the margins of the lesion there remain cells that can multiply to cover the defect. In addition, the base of the lesion probably transects the neck of sweat glands and hair follicles; cells from here can also proliferate and contribute to healing. At first, cells proliferate and spread out as a thin sheet until the defect is covered. When they form a confluent layer, the stimulus to proliferate is switched off; this is referred to as contact inhibition, and controls both growth and movement. Once in place, the epidermis is rebuilt from the base upwards until it is indistinguishable from normal. This whole process is called healing.
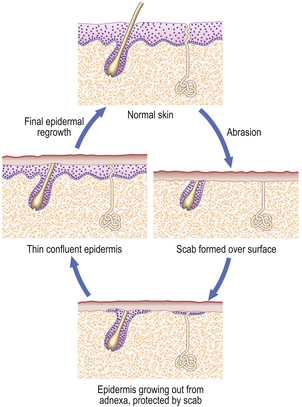
Fig. 6.5 Healing of a minor skin abrasion. The scab, a layer of fibrin, protects the epidermis as it grows to cover the defect. The scab is then shed and the skin is restored to normal.
Contact inhibition of growth and of movement are important control mechanisms in normal cells. In neoplasia (Ch. 11) these control mechanisms are lost, allowing the continued proliferation of tumour cells.
The contribution of adnexal gland cells to regeneration is made use of in plastic surgery when using split skin grafts. The whole of the epidermis is removed and positioned as the donor graft, but the necks of adnexa are left in place to generate a replacement at the donor site.
Organisation


Organisation is the process whereby specialised tissues are repaired by the formation of mature fibrovascular connective tissue. Granulation tissue is formed in the early stages, often on a scaffold of fibrin, and any dead tissue is removed by phagocytes such as neutrophil polymorphs and macrophages. The granulation tissue contracts and gradually accumulates collagen to form the scar, which then undergoes remodelling.
Organisation is a common consequence of pneumonia (Ch. 14). The alveolar exudate becomes organised. Organisation also occurs when tissue dies as a result of cessation of its blood supply (an infarct). In all instances, the organised area is firmer than normal, and often shrunken or puckered.
Granulation tissue




When specialised or complex tissue is destroyed, it cannot be reconstructed. A stereotyped response then follows—a process known as repair. Capillary endothelial cells proliferate and grow into the area to be repaired; initially they are solid buds but soon they open into vascular channels. The vessels are arranged as a series of loops arching into the damaged area. Simultaneously, fibroblasts are stimulated to divide and to secrete collagen and other matrix components. They also acquire bundles of muscle filaments and attachments to adjacent cells. These modified cells are called myofibroblasts and display features and functions of both fibroblasts and smooth muscle cells. As well as secreting a collagen framework, they play a fundamental role in wound contraction. This combination of capillary loops and myofibroblasts is known as granulation tissue. (The name derives from the appearance of the base of a skin ulcer. When the repair process is observed, the capillary loops are just visible and impart a granular texture.) Excessive granulation tissue protruding from a surface is called proud flesh. Granulation tissue must not be confused with a granuloma (an aggregate of epithelioid histiocytes).
Wound contraction and scarring
Wound contraction is important for reducing the volume of tissue for repair; the tissue defect may be reduced by 80%. It results from the contraction of myofibroblasts in the granulation tissue. These are attached to each other and to the adjacent matrix components, so that granulation tissue as a whole contracts and indraws the surrounding tissues. Collagen is secreted and forms a scar, replacing the lost specialised tissues. Infection and associated inflammation are liable to increase scarring.
Although wound contraction serves a very useful function, it can also lead to problems. If the tissue damage is circumferential around the lumen of a tube such as the gut, subsequent contraction may cause stenosis (narrowing) or obstruction due to a stricture. Similar tissue distortion resulting in permanent shortening of a muscle is called a contracture. Similarly, burns to the skin can be followed by considerable contraction, with resulting cosmetic damage and often impaired mobility.
Outcome of injuries in different tissues
Having considered the general principles of healing and repair, the particular outcome of injuries to a variety of tissues will be considered.
Skin
The process of healing of a skin wound depends on the size of the defect.
Incised wound: healing by first intention
An incision, such as that made by a surgical scalpel, causes very little damage to tissues on either side of the cut. If the two sides of the wound are brought together accurately, then healing can proceed with the minimum of delay or difficulty (Fig. 6.6). Obviously, some small blood vessels will have been cut, but these will be occluded by thrombosis, and close apposition of wound edges will help. Fibrin deposited locally will then bind the two sides. Coagulated blood on the surface forms the scab and helps to keep the wound clean. This join is very weak, but is formed rapidly and is a framework for the next stage. It is important that it is not disrupted; sutures, sticking plaster or other means of mechanical support are invaluable aids. Over the next few days, capillaries proliferate sufficiently to bridge the tiny gap, and fibroblasts secrete collagen as they migrate into the fibrin network. If the sides of the wound are very close, then such migration is minimal, as would be the amount of collagen and vascular proliferation required. By about 10 days, the strength of the repair is sufficient to enable removal of sutures. The only residual defect will be the failure to reconstruct the elastic network in the dermis.
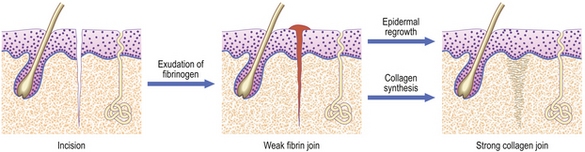
While these changes are proceeding in the dermis, the basal epidermal cells proliferate to spread over any gap. If the edges of the wound are gaping, then the epidermal cells will creep down the sides. Eventually, when the wound is healed, these cells will usually stop growing and be resorbed, but occasionally they will remain and grow to form a keratin-filled cyst (implantation dermoid).
Tissue loss: healing by second intention
When there is tissue loss or some other reason why the wound margins are not apposed, then another mechanism is necessary for repair. For example, if there is haemorrhage (persistent bleeding) locally, this will keep the sides apart and prevent healing by first intention; infection similarly compromises healing. The response will be characterised by:
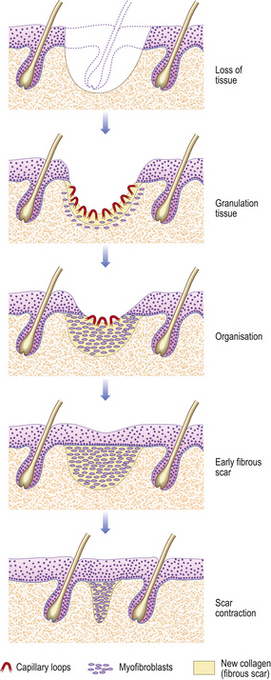
Fig. 6.7 Skin wound repaired by second intention. The tissue defect becomes filled with granulation tissue, which eventually contracts, leaving a small scar.
The timescale depends on the size of the defect, as this determines the amount of granulation tissue to be generated and the area to cover with epithelium. Quite large expanses of tissue can be removed if necessary, and the defect left to heal by second intention. The final cosmetic result depends on how much tissue loss there has been, as this affects the amount of scarring.
Keloid nodules
Dermal injury is sometimes followed by excessive fibroblast proliferation and collagen production. This phenomenon is genetically determined, and is particularly prevalent among blacks. A mass several centimetres across may follow surgery or injury, particularly burns.
Mechanism of skin healing and repair
Healing and repair involve a complex interplay of cytokines (Ch. 5). There is considerable complexity and redundancy in this system, with the same cell producing many cytokines, and most cytokines having many functions. The initiating signals are probably hypoxia together with the release of growth factors from platelet degranulation. These trigger the production of numerous cytokines such as epidermal growth factors (EGFs) and keratinocyte growth factor (KGF) from platelets, macrophages and dermal fibroblasts, to stimulate keratinocyte proliferation and mobility. Keratinocytes and macrophages produce vascular endothelial growth factor (VEGF), inducing new blood vessel formation (angiogenesis). Platelet-derived growth factor (PDGF) from platelets, macrophages and keratinocytes facilitates the local accumulation and activation of macrophages, proliferation of fibroblasts and matrix production. Control of myofibroblasts and collagen formation is partly influenced by transforming growth factor-beta (TGF-beta); abnormalities can result in hypertrophic scars or keloid.
Failure to regenerate structures such as skin adnexal glands and hair is a post-natal problem. Formation of these complex cellular configurations is controlled by a small number of homeotic (patterning) genes which then control the necessary growth and differentiation genes. Damage to fetal skin is healed completely, but in the adult the homeotic genes are not activated, necessitating an imperfect repair. Adult epidermis is capable of responding to produce hairs and so forth, but the wounded dermis fails to produce the required signals.
Peritoneum
The healing of a peritoneal defect may follow two distinct alternative patterns. If it is sutured or contaminated with fibrin or foreign material such as glove powder, then local ischaemia or platelet granules initiate the cytokine networks seen in skin healing (and elsewhere) with resulting angiogenesis. Contact with another surface, such as serosa of the intestine, can then result in adhesion. In time a band of collagen is laid down which may subsequently cause bowel obstruction.
However, a peritoneal defect that is simply cleaned and left unsutured will heal by a different mechanism. Perivascular connective tissue cells in the base of the defect proliferate to fill it, while any debris is removed by macrophages. When this is complete, the surface cells differentiate into new mesothelial lining cells, rather than awaiting migration over the surface from the surviving mesothelial cells at the margin (Fig. 6.8). Healing is thus rapid and complete, irrespective of the size of the defect, with very little risk of forming adhesions.
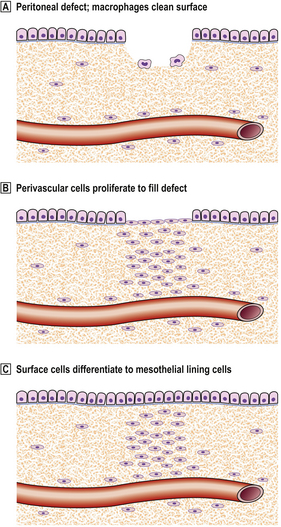
Gastrointestinal tract
The fate of an intestinal injury depends upon its depth.
Mucosal erosions
An erosion is defined as loss of part of the thickness of the mucosa. Viable epithelial cells are immediately adjacent to the defect and proliferate rapidly to regenerate the mucosa. Such an erosion can be re-covered in a matter of hours, provided that the cause has been removed. Notwithstanding this remarkable speed of recovery, it is possible for a patient to lose much blood from multiple gastric erosions before they heal. If endoscopy to identify the cause of haematemesis is delayed, the erosions may no longer be present, and thus escape detection.
Mucosal ulceration
Ulceration is loss of the full thickness of the mucosa, and often the defect goes much deeper to penetrate the muscularis propria; further complications are discussed in Chapter 15. The principles of repair have been outlined above. Destroyed muscle cannot be regenerated, and the mucosa must be replaced from the margins. The outcome of mucosal ulceration is discussed below with reference to a gastric ulcer, but colonic ulcers show similar features. Damaged blood vessels will have bled and the surface will become covered by a layer of fibrin. Macrophages then remove any dead tissue by phagocytosis. Meanwhile, granulation tissue is produced in the ulcer base, as capillaries and myofibroblasts proliferate. Also, the mucosa will begin to regenerate at the margins and spread out on to the floor of the ulcer.
If the cause persists, the ulcer becomes chronic and there is oscillation between further ulceration and repair, possibly resulting in considerable destruction of the gastric wall. If healing ever proceeds far enough, the fibrous scar tissue that has replaced muscle will contract, with distortion of the stomach and possible obstruction. Any larger arteries that lie in the path of the advancing ulceration are at risk of rupture, with resulting haemorrhage. However, there may be a zone of inflammation around the ulceration, and if this abuts the vessel it results in a reactive proliferation of the vascular intima. This feature is referred to as endarteritis obliterans because of the obliteration of the lumen (it has nothing to do with end arteries).
Bone




Fracture healing
Immediately after the fracture there will be haemorrhage within the bone from ruptured vessels in the marrow cavity, and also around the bone in relation to the periosteum. A haematoma at the fracture site facilitates repair by providing a foundation for the growth of cells (Fig. 6.9). There will also be devitalised fragments of bone, and probable soft tissue damage nearby. Thus, the initial phases of repair involve removal of necrotic tissue and organisation of the haematoma. In the latter, the capillaries will be accompanied by fibroblasts and osteoblasts. These deposit bone in an irregularly woven pattern. The mass of new bone, sometimes with islands of cartilage, is called callus; that within the medullary cavity is internal callus, while that at the periosteum is external callus. The latter is helpful as a splint, although it will need to be resorbed eventually. Woven bone is subsequently replaced by more orderly, lamellar bone; this in turn is gradually remodelled according to the direction of mechanical stress.
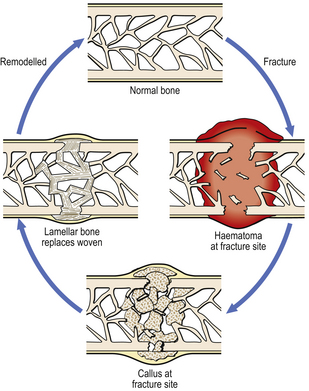
Problems with fracture healing
Several factors can delay, or even arrest, the repair of a fracture:
Movement between the two ends, apart from causing pain, also results in excessive callus and prevents or slows down tissue union. Persistent movement prevents bone formation, and collagen is laid down instead to give fibrous union; this results in a false joint at the fracture site. Movement of a lesser degree leads to excessive callus which takes longer to be resorbed and may impinge on adjacent structures.
Interposed soft tissues between the broken ends delay healing at least until they are removed, and there is an increased risk of non-union.
Gross misalignment also slows the rate of healing and will prevent a good functional result, leading to increased risk of degenerative disease (osteoarthrosis) in adjacent joints.
Infection at the fracture site will delay healing and there is the additional risk of chronic osteomyelitis. Infection is more likely if the skin over the fracture is broken; this is referred to as a compound fracture.
If the bone broken was weakened by disease, the break is called a pathological fracture. Pathological fracture may be the result of a primary disorder of bone, or the secondary involvement of bone by some other condition, such as metastatic carcinoma. In most instances, a pathological fracture will heal satisfactorily, but sometimes treatment of the underlying cause will be required first.
Liver
Hepatocytes, a stable cell population, have excellent regenerative capacity. In some circumstances, hepatic regeneration comes from liver progenitor cells rather than hepatocytes; bone marrow-derived stem cells are a third option. The hepatic architecture, however, cannot be satisfactorily reconstructed if severely damaged. Consequently, conditions that result only in hepatocyte loss may be followed by complete restitution, whereas damage destroying both the hepatocytes and architecture may not. In the latter situation, the imbalance between hepatocyte regeneration and failure to reconstruct the architecture may proceed to cirrhosis (Fig. 6.10).
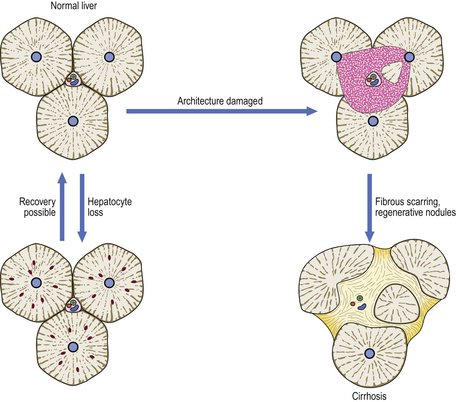
Fig. 6.10 Consequences of liver injury depending on extent of tissue damage. Loss of only scattered liver cells, or even small groups, can be restored without architectural disturbance. However, if there is confluent loss of liver cells and architectural damage, the liver heals by scarring and nodular regeneration of liver cells, resulting in cirrhosis.
Kidney
The kidney is similar to the liver with respect to tissue injury, in that it has an epithelium that can be regenerated but an architecture that cannot. Loss of tubular epithelium following an ischaemic episode or exposure to toxins may result in renal failure, but in general there is sufficient surviving epithelium to repopulate the tubules and enable normal renal function to return. Inflammatory or other damage resulting in destruction of the glomerulus is likely to be permanent or result in glomerular scarring, with loss of filtration capacity. Similarly, interstitial inflammation is liable to proceed to fibrosis and, thus, impaired reabsorption from tubules.
Muscle
Cardiac muscle fibres and smooth muscle cells are permanent cells; vascular smooth muscle may be different, in that new vessels can be formed. This means that damaged muscle is replaced by scar tissue. However, if the contractile proteins only are lost, then it is possible to synthesise new ones within the old endomysium. Voluntary muscle has a limited capacity for regeneration from satellite cells.
Neural tissue

 Peripheral nerves show Wallerian degeneration (Ch. 26) distal to trauma; variable recovery depending on alignment and continuity
Peripheral nerves show Wallerian degeneration (Ch. 26) distal to trauma; variable recovery depending on alignment and continuity
Even though recent evidence suggests that adult nerve cells may have a low replicative capacity, there is no effective regeneration of neurones in the central nervous system. Glial cells, however, may proliferate in response to injury, a process referred to as gliosis.
Peripheral nerve damage affects axons and their supporting structures, such as Schwann cells. If there is transection of the nerve, axons degenerate proximally for a distance of about one or two nodes; distally, there is Wallerian degeneration followed by proliferation of Schwann cells in anticipation of axonal regrowth. If there is good realignment of the cut ends, the axons may regrow down their previous channels (now occupied by proliferated Schwann cells); however, full functional recovery is unusual. When there is poor alignment or amputation of the nerve, the cut ends of the axons still proliferate, but in a disordered manner, to produce a tangled mass of axons and stroma called an amputation neuroma. Sometimes, these are painful and require removal.
Modifying influences







The description of tissue injury and repair given above applies to an otherwise healthy adult. However, various factors can impair healing and repair:
Age
Early in life, cellular injury is likely to impair or prevent the normal growth and development of an organ. Organogenesis is at risk if there is impaired function, differentiation or migration of the precursor cells. For example, rubella infection or thalidomide administration in early pregnancy can cause congenital abnormalities; therapeutic doses of radiation are associated with microcephaly and mental retardation.
Similar considerations apply to childhood, in that there may be growth disturbance following tissue damage. For example, the distal pulmonary airways may be permanently damaged by severe infection or mechanical stress, as in whooping cough. High doses of radiation will result in loss of replicating cells and in local failure to grow; the affected area will then be smaller in proportion to the rest of the body. On the other hand, wound healing proceeds rapidly in healthy children, and fractures unite more quickly than in adults.
The physiology of ageing is complex (Ch. 12); one characteristic is a reduced ability to repair damaged tissues. Connective tissues become less elastic, renal function diminishes, bones weaken and cerebral neurones are lost. Consequently, a more substantial effect from the same insult occurs when compared with that in a younger adult. Wound healing is often delayed in old age because of ischaemia or other significant disease.
Disorders of nutrition
Wound healing is profoundly influenced by the ability to synthesise protein and collagen. The latter is dependent on vitamin C for the hydroxylation of proline as a step in collagen synthesis. Scurvy (vitamin C deficiency) leads to wound healing of greatly reduced strength; capillaries are also fragile and thus haemorrhages occur.
Protein malnutrition, whether due to dietary deficiency or the consequence of protein loss, also impairs wound healing. In addition, severe malnutrition impairs the response to infection which may then proceed to a fatal outcome. For example, measles is generally a transient problem in well-nourished children, but is frequently fatal in the malnourished.
Neoplastic disorders
In advanced malignant neoplastic disease with widely disseminated tumours, or gastrointestinal symptoms such as dysphagia, the patient is malnourished. However, a catabolic state with profound weight loss may be an early feature of some cancers. Such patients have impaired healing, and this may compromise the recovery from attempted surgical removal of the lesion.
There may also be evidence of impaired healing in the vicinity of the tumour. Skin stretched over a superficial tumour will often break down and ulcerate, and it is necessary to treat the tumour to promote healing of the ulcer. A pathological fracture of bone through a metastatic tumour may not heal unless the tumour is dealt with first.
Cushing’s syndrome and steroid therapy
Excessive circulating corticosteroids, whether they result from tumour or from therapeutic administration, have two effects on tissue injury.
Diabetes mellitus and immunosuppression
Both diabetes mellitus (Ch. 7) and immunosuppression (Ch. 9) increase susceptibility to infection by low-virulence organisms, and increase the risk of tissue damage. Normal healing responses are possible, although they may be impaired by continuing infection. Diabetes may affect polymorph function, and may also result in occlusion of small blood vessels and cause neuropathy. There also seems to be a direct effect on keratinocytes, reducing their motility, and also that of myofibroblasts, both of which delay healing.
Vascular disturbance
An adequate vascular supply is essential for normal cellular function. An impaired supply can result in ischaemia or infarction (Ch. 8). Note that an adequate supply for resting tissue may prove inadequate if the demand increases. For example, in coronary artery disease, the blood flow may be sufficient for the resting state, but not for exertion when the cardiac output increases. The deficit of oxygen may then result in tissue damage.
Another effect of a reduced vascular supply is impaired healing. This occurs because hypoxia and reduced local nutrition result in poorer tissue regrowth or repair.
Denervation
An intact nerve supply supports the structural and functional integrity of many tissues. In addition, nerves have a role in mediating the inflammatory response as part of the host mechanism for limiting the effects of injury. Denervated tissues may become severely damaged, probably through a combination of unresponsiveness to repeated minor trauma, and lack of pain of intercurrent infection or inflammation. Thus, patients with conditions such as peripheral neuropathy or leprosy may develop foot ulcers (neuropathic ulcers). A neuropathic joint (Charcot’s joint) may be damaged unwittingly and progressively beyond repair.
INJURY DUE TO IONISING RADIATION





Radiation is generally perceived by the public as harmful. In the European Union it is now mandatory that medical practitioners using radiation for investigating or treating patients know about radiation protection. This section deals with certain aspects of this, particularly in relation to:
Definition and sources
Radiation of medical importance is largely restricted to that which causes the formation of ions on interaction with matter (ionising radiation). The exception to this is some ultraviolet light. Ionising radiation includes:
Electromagnetic radiation
Only part of the electromagnetic spectrum produces ionising events. The production of ions requires a photon of high energy and thus of short wavelength, in practice shorter than that of ultraviolet light. If the photon is emitted by a machine, the radiation is called an X-ray. If it is emitted as a result of the disintegration of an unstable atom, it is referred to as a gamma ray. It follows that X-rays can be switched on and off, while gamma ray emission is continuous so protection requires a physical barrier.
Particulate radiation
As well as photons, certain subatomic particles may also produce ionisation. These include alpha particles (helium nuclei), beta particles (electrons) and neutrons. The distinction between beta particles and electrons is the same as that between gamma rays and X-rays: beta particles are produced through the process of radioactive decay, whereas electrons are a structural component of atoms that may be artificially projected as a beam.
Units of dose
Various units have been used for measuring radiation. The current unit of absorbed dose is the gray (Gy)—1 joule of radiation energy deposited in 1kg of matter— and is the usual measure of therapeutic radiation when a uniform type of radiation is administered to a specified tissue.
However, different forms of radiation vary in the distribution of energy deposited in tissues, hence the biological effect. Alpha particles, having a high linear energy transfer (LET), deposit a large amount of energy over a short distance, so are about 20 times more damaging than beta particles or X-rays, which have low LET. Tissues also differ in their sensitivity (Table 6.3). Therefore, when subjects are exposed to a mixture of different forms of radiation to several tissues, it is useful to make mathematical corrections for comparative purposes, and express the result as the effective dose equivalent, measured in sieverts (Sv) (Fig. 6.11).
Table 6.3 Relative sensitivities of different tissues to harmful effects of ionising radiation
| Tissue | Factor |
|---|---|
| Gonads | 0.25 |
| Breasts | 0.15 |
| Haemopoietic tissue | 0.12 |
| Lungs | 0.12 |
| Liver | 0.06 |
| Thyroid | 0.03 |
| Bone | 0.03 |
| Other organs (total) | 0.24 |
| Total for body = 1.00 | |
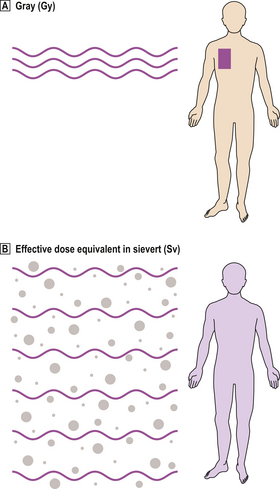
Fig. 6.11 Units of dose of radiation.  The gray (Gy) is a measure of absorbed dose.
The gray (Gy) is a measure of absorbed dose.  The sievert (Sv) measures the radiation dose corrected for different types of radiation and the differing sensitivities of tissues to them.
The sievert (Sv) measures the radiation dose corrected for different types of radiation and the differing sensitivities of tissues to them.
Another relevant unit is a measure of the rate of disintegration of unstable atoms; 1 becquerel (Bq) is one emission per second. The becquerel is not itself a measure of dose, because it expresses only a rate of disintegration irrespective of the nature or energy of the products of disintegration. However, for any particular atom the latter is known, so the dose can be calculated.
Background radiation
Everyone is exposed to background radiation from their environment. In the UK the average annual dose is 2.7mSv, which comes from:
Over 90% of the artificial component is from medical usage, such as diagnostic X-rays and nuclear medicine. The amount has increased recently, reflecting greater use of CT scans. (Note that magnetic resonance imaging does not use ionising radiation.) The natural component is made up from cosmic, terrestrial, airborne and food sources. The most locally variable among these is airborne radiation, which derives mainly from radon and radon daughters; these diffuse out of the ground and are commoner in certain types of rock, such as granite. In the UK, there are 100-fold differences from one place to another. Some draught-proofed homes in areas of high natural airborne radiation accumulate radon to concentrations exceeding acceptable industrial limits, thereby placing occupants at risk of lung disease from irradiation.
Mode of action
When radiation passes through tissue, any collisions within it will be randomly distributed amongst its components. However, it seems that direct damage as a result of ionisation of proteins or membranes does not make a major contribution to the biological end result. Water is the most prevalent molecule, and following ionisation several types of short-lived but highly reactive radicals are formed such as H• and hydroxyl radical OH•. In a well-oxygenated cell, oxygen radicals will also be formed, e.g. hydroperoxyl radical, HO2• and superoxide radical O2•−.
These radicals then interact with macromolecules, of which the most significant are membrane lipids and DNA.
DNA damage
The types of radiation-induced DNA damage include:
Breakage of the DNA strand (Fig. 6.12) is a common result of radiation. When only one strand is broken, repair can generally be accomplished accurately. Double-strand breaks may be irreparable because there is no template. Also, multiple double-strand breaks may rejoin incorrectly, resulting in chromosome translocation or inversion.
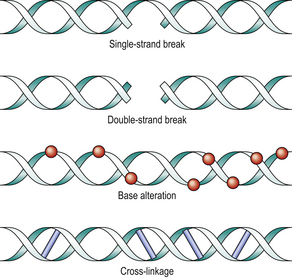
Fig. 6.12 DNA damage by radiation. Single-strand breaks can be reconstituted by DNA repair enzymes, because the complementary strand forms a template. The other injuries are less easily remedied. Cross-linkage causes reproductive death.
Base alterations are also frequent, such that the DNA strand no longer transcribes correctly (mutation). The result may be unreadable (nonsense mutation) or may read incorrectly (missense mutation).
DNA strand cross-linking occurs when radiation-induced reactive oxygen species cause linkage between the complementary strands, resulting in an inability to separate and thus to make a new copy. DNA replication is therefore blocked. DNA damage of these types is not caused solely by radiation; it is also the mechanism of action of some chemotherapy. For example, alkylating agents cause cross-linkage and platinum-based drugs cause strand breaks.
The consequences of DNA damage depend on its nature and extent, and on the results of any attempts at repair. The mean lethal dose per cell is in the range 0.5–2Gy. Most double-strand breaks are repaired promptly, but some result in misrepair or failure to repair. Cells affected in this way are described as having ‘reproductive death’; the combination of genetic instability and lethal mutations results in cell death after two or three mitotic cycles. A much smaller proportion of cells die immediately by apoptosis or necrosis.
There are several DNA repair enzyme systems, sufficient for incidental strand breaks. Some people have defective DNA repair, so are more susceptible to ionising radiation or ultraviolet light. Loss of function mutations of the ATM gene impair excision repair of double-strand breaks, and explain the enhanced radiation sensitivity of patients with ataxia telangiectasia. Similarly, the mutated ERCC6 gene is the defect in xeroderma pigmentosum, in which there is extreme skin sensitivity to sunlight, causing tumours.
Effects on tissues
Despite knowledge of the molecular events following irradiation, there is continuing debate about how these are translated into the observed tissue responses. The immediate physico-chemical events and consequent biomolecular damage are over in a few milliseconds; the varied outcomes are manifest in hours to years.
DNA damage may have three possible consequences:
The dose given will influence this outcome, as will the radiosensitivity of the cell. Tissue and organ changes will reflect the overall reactions in the component parts. Tissue consequences are usually divided into early tissue reactions or deterministic effects, which are predictable according to the dose received, and later stochastic effects, where only the probability is related to the dose. Thus, cataract and skin erythema (tissue reactions) will not occur below a certain threshold dose, while in contrast there is no dose threshold below which there is no probability of cancer (a stochastic effect).
Early effects
Early effects of radiation are generally the result of cell killing and the interruption of successful mitotic activity. Hierarchical cell organisations, such as the bone marrow or gut epithelium, which have a dividing stem cell population and daughter cells of finite life expectancy, will show the most pronounced effects. In essence, the supply of functioning differentiated cells is cut off or suspended. In addition, there is vascular endothelial damage, resulting in fluid and protein leakage rather like that of the inflammatory response (Ch. 10).
Late effects
Late effects of radiation are the result of several factors, and the contribution of each is contentious. Vascular endothelial cell loss will result in exposure of the underlying collagen. This will prompt platelet adherence and thrombosis, which is subsequently incorporated into the vessel wall and is associated with the intimal proliferation of endarteritis obliterans. A possible result of this is long-term vascular insufficiency with consequent atrophy and fibrosis.
However, the observed atrophy may simply be a function of continuing cell loss over a long period of time, reflecting an inherently slow rate of proliferation of cells in the tissue concerned. If this is the case, the vascular alterations are part of the late effects of radiation, but not the cause of the atrophy.
The cellular alterations induced by radiation are permanent. The limits of tissue tolerance cannot be exceeded even if many years have elapsed. In addition to the effects mentioned above, radiation-induced mutation of the genome causes an increased risk of neoplastic transformation (see below).
Bone marrow
Haemopoietic marrow is a hierarchical tissue that maintains the blood concentration of functional cells of limited life-span by a constant high rate of mitotic activity. The effect of radiation is to suspend renewal of all cell lines. Subsequent blood counts will fall at a rate corresponding to the physiological survival of cells; granulocytes will diminish after a few days but erythrocytes survive much longer.
The ultimate outcome will depend on the dose received, and will vary from complete recovery to death from marrow failure (unless a marrow transplant is successful). In the long-term survivor, there is a risk of leukaemia. Localised heavy radiation will not alter the blood count, but it will result in local loss of haemopoiesis and fibrosis of the marrow cavity.
Intestine
The surface epithelial lining of the small intestine is renewed every 24–48 hours. A significant dose of radiation will therefore result in loss of protective and absorptive functions over a similar timescale; diarrhoea and the risk of infection then follow. If a high dose is given to a localised region, the mucosa will regrow, although often with a less specialised cell type, and with the probability of mutations in the remaining cells. The muscle coat will also have been damaged, and there is the risk of granulation tissue causing a stricture later.
Skin
The changes in the skin reflect its composition from epithelium, connective tissue and blood vessels. Epidermis will suffer the consequences of cessation of mitosis, with desquamation and hair loss. Provided enough stem cells survive, hair will regrow, and any defects in epidermal coverage can be re-epithelialised. The regenerated epidermis will lack rete ridges and adnexa. Damage to keratinocytes and melanocytes results in melanin deposition in the dermis where it is picked up by phagocytic cells; these tend to remain in the skin and result in local hyperpigmentation (post-inflammatory pigmentation). Some fibroblasts in the dermis will be killed, while others are at risk of an inability to divide, or to function correctly. As a consequence, the dermis is thinned, and histology shows bizarre, enlarged fibroblast nuclei.
The vessels show various changes depending on their size. Endothelial cell loss or damage is the probable underlying factor. Small and thin-walled vessels will leak fluid and proteins, and mimic the inflammatory response; in the long term, they can be permanently dilated and tortuous (telangiectatic). Larger vessels develop intimal proliferation and may permanently impair blood flow.
In summary, the skin is at first reddened with desquamation, and subsequently shows pigmentation. Later, it is thinned with telangiectasia; if damage is too severe, it will break down and ulcerate (radionecrosis).
Gonads
Germ cells are very radiosensitive, and permanent sterility can follow relatively low doses. Also of great significance is the possibility of mutation in germ cells, which could result in passing on defects to the next generation; this is a teratogenic effect. However, although this has been demonstrated experimentally in mice, no studies of radiated populations have proven a teratogenic effect in humans.
Lung
Ionising radiation is one of several agents that can damage alveoli, culminating in fibrosis (Ch. 14). Inhaled radioactive materials induce pulmonary tumours.
Whole body irradiation
Whole body irradiation can be the result of accidental or therapeutic exposure. The consequences can mostly be predicted (Fig. 6.13). At very high doses, death occurs rapidly with convulsions due to cerebral injury. At lower doses, the clinical picture is dominated in the first few days by gastrointestinal problems, and later by bone marrow suppression; either may prove fatal. In the long term, there is the risk of neoplasia.
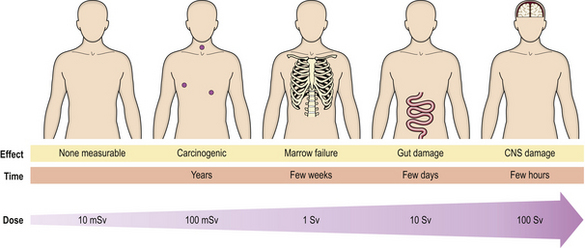
Fig. 6.13 Consequences of whole body irradiation. As the dose increases, so do the severity and immediacy of the effects.
Therapeutic usage of total body irradiation is mainly for the deliberate ablation of the bone marrow prior to transplantation of marrow, using either stored marrow from the patient or marrow from another donor.
Ionising radiation and tumours
There is no doubt that ionising radiation causes tumours (Ch. 11). This is now firmly established for relatively high doses, but with low-dose radiation some uncertainty remains.
There is a roughly linear relationship between the dose received and the incidence of tumours. The mechanism is incompletely understood, but the fundamental event is mutation of the host cell DNA; it is unlikely that a single point mutation is sufficient and more probably many will be present. As the radiation dose increases, so a greater number of cells will be lethally irradiated, thus reducing the number surviving and at risk of neoplastic transformation.
The dose–response information comes from several sources, including animal experiments and observations on patients or populations exposed to radiation. Thus, children who received radiation of the thyroid gland show an incidence of tumours corresponding to the dose received. Occupational exposure to radon gas in mines also shows a correlation with the risk of lung tumours. For a given dose, the risk of neoplasia varies between tissues (Table 6.4).
Table 6.4 Relative lifetime risk of fatal cancer from a standard dose of ionising radiation
| Tissue | Risk factor (Sv–1) |
|---|---|
| Lung | 1 in 80 |
| Female breast | 1 in 90 |
| Haemopoietic tissue | 1 in 360 |
| Bone | 1 in 2000 |
| Thyroid | 1 in 4000 |
| Other organs (total) | 1 in 43 |
| Total for body = 1 in 20 | |
Common to all these observations is a time delay between exposure to radiation and development of the tumour. Studies of Japanese survivors of the atomic bombs show significant numbers of cases of leukaemia by about 6 years, with a mean delay of 12.5 years and thereafter a decreasing incidence. For solid cancers, however, the mean delay was 25 years with a continuing increased incidence in these people four decades later; in total, there have now been many more solid cancers than leukaemias (Fig. 6.14).
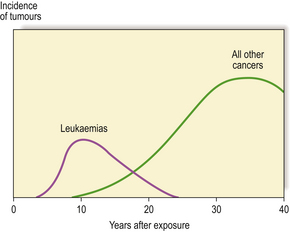
Fig. 6.14 Tumours in atom bomb survivors. There is a latent interval between exposure to radiation and detection of the tumours. This is relatively short for leukaemias, but up to several decades for solid tumours.
Regarding low doses (less than 100mSv), it is more difficult to be sure if the radiation is carcinogenic because the anticipated number of tumours would be so small compared with the overall number of tumours in the exposed population. However, more recent studies of a cohort of 100 000 Japanese atomic bomb survivors exposed to low doses suggest that linear extrapolation with no minimum threshold gives a reasonable fit with observed cancers. By way of illustration, a single CT scan of the abdomen can result in a dose of about 15mSv to the digestive tract which, for a 40 year old, may result in a lifetime risk of 0.02% of death from digestive tract cancer. However, estimates of the risk of cancer in this dose range may be two or three times too high or too low. Children may be at a greater risk than adults for any given dose, an effect compounded by their projected longer survival at risk.
Ultraviolet light
Ultraviolet light has three wavelength classes:
UVB is associated with sunburn and can also cause skin tumours; although not ionising, it damages DNA by inducing pyrimidine dimers and strand linkage. UVB is also immunosuppressive. UVA probably induces non-dimer damage, and also inhibits DNA repair processes. The tumours produced are basal cell and squamous cell carcinomas, and malignant melanomas. Melanin pigmentation, itself induced by ultraviolet light, is protective against these effects.
UVC is very toxic and is used in germicide lamps. However, solar radiation in this range is filtered out by the ozone layer.
Principles of radiation protection
In view of the risk of harm from ionising radiation, it is important that it is used safely and only when there are no suitable alternatives. The International Commission on Radiological Protection (ICRP) has published recommendations with three central requirements:
In view of the risk of harm from ionising radiation, it must be used in a discriminating manner that takes alternatives into account. In the European Union, the Ionising Radiation (Medical Exposure) Regulations (IRMER) require the doctor to consider whether a procedure, or an investigation, involving radiation is justifiable in each and every circumstance.
The second requirement is sometimes referred to as the ALARA principle. This emphasises that doses should be ‘as low as reasonably achievable, not simply kept below dose limits’.
Therapeutic radiation: radiotherapy



Like most effective treatments in medicine, radiotherapy carries certain risks of undesirable or unpredictable side-effects. This means that it tends to be reserved for serious or life-threatening conditions, or palliation of incurable diseases. The most common effect required from radiation is the ability to kill cells; this is used in the treatment of tumours. Sometimes, the object is to induce fibrosis or vascular occlusion, as in the treatment of vascular malformations.
Radiation may be given with the intention of cure, generally for a tumour. Usually, the aim is to give as high a dose as possible to the tumour, while producing the least possible damage to adjacent normal tissues. Some tumours are relatively radiosensitive, so that the therapeutic margin between tumour cell kill and tissue damage is wide; but in others the normal tissue tolerance is the limiting factor.
Basal cell carcinoma of the skin is very common and often managed by radiotherapy, although local excision is also effective. Squamous carcinoma of the larynx is usually irradiated in the first instance, because this preserves voice production. Squamous carcinoma of the uterine cervix is a tumour that may be managed by primary surgery or radiotherapy (Fig. 6.15). Localised malignant lymphoma is often irradiated, whereas generalised lymphoma is treated by chemotherapy. Metastatic seminoma of testis in para-aortic lymph nodes is usually irradiated, illustrating that radical treatment is possible even when metastases are present. Seminoma is an example of a very radiosensitive tumour. Carcinoma of the breast is usually treated by surgery, but as local recurrence is a risk patients often proceed to post-operative radical radiotherapy.
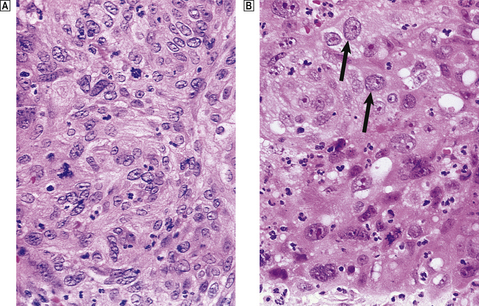
Fig. 6.15 Effect of radiation on carcinoma of the cervix.  Before irradiation.
Before irradiation.  One week after high-dose irradiation, showing injury to the tumour cells (note bloating of arrowed nuclei) and the induced inflammatory reaction.
One week after high-dose irradiation, showing injury to the tumour cells (note bloating of arrowed nuclei) and the induced inflammatory reaction.
Even when there is no hope of cure, there is much that can be done to relieve symptoms. Palliative radiotherapy is often given to treat metastatic tumour deposits, such as painful bone metastases.
Fractionation
A higher dose of radiation may be given without increasing side-effects if it is divided into a number of fractions and given on different days. In practice, it is common to treat on only 5 days a week, so that a dose divided into 25 fractions would be given over 5 weeks. Each treatment fraction induces tissue damage, but is followed by attempts at repair. Normal cells included in the treated tissue volume are better able to repair effectively than are neoplastic cells. Consequently, there is a differential cell killing of more tumour cells than normal cells. Modern radiotherapy equipment and planning techniques allow a high degree of conformation of the radiated volume to the tumour itself, with less normal tissue included in the field. Palliative treatment is often a lower dose given as fewer larger fractions, in order to get a more rapid response.
Response modifiers
In addition to the benefits of fractionating treatment, there has been considerable interest in modifying the tissue response to radiation. There are often conflicting interests, in that an increased sensitivity is required in the tumour and increased resistance in the normal tissue.
The most common reason for reduced sensitivity in a tumour is a low oxygen tension. The probable explanation is the central role of oxygen radicals in mediating the biological impact of ionising radiation; a lower oxygen concentration means, quite simply, that fewer oxygen radicals are generated. Many tumours have a poorly developed vascular network, resulting in hypoxic areas; patients are also often anaemic, which increases the problem. Blood transfusion can correct anaemia.
Radiosensitisers are drugs that diffuse into tissues and, by mimicking the effect of oxygen, enhance the response. Although they are under investigation, none is in current use. The nearest equivalent is the use of psoralens to enhance the efficacy of ultraviolet light in the management of psoriasis (Ch. 24). Hyperbaric oxygen and hyperthermia have both been used, but neither has passed into routine practice. Regarding reducing the harmful effects of radiation, research has been conducted into compounds that inhibit apoptosis, or reduce a damaging inflammatory response following irradiation.
Complications of radiotherapy
Irrespective of the part of the body treated, nausea and vomiting are very common side-effects of radiotherapy. The mechanism is not understood, but it is more likely to occur when large volumes of tissue are treated.
Major and minor salivary glands are liable to undergo permanent atrophy after irradiation. If treatment has been given from both sides of the body then this can result in a troublesome dry mouth.
Depending on the type of radiation, the skin will receive a proportion of any dose given to any internal target. Certain techniques result in skin-sparing, and radionecrosis is unlikely unless the skin itself is the target of irradiation. However, skin reactions are very common and range from the expected acute inflammatory phases to residual pigmentation. All these phenomena will be strictly delineated by the margins of the treatment field with its straight edges (Fig. 6.16). They can thus be distinguished from other diseases.

Fig. 6.16 Skin erythema due to therapeutic irradiation. Skin erythema is an immediate tissue reaction to radiation.
Fibrosis is a late manifestation in irradiated tissue and will also be restricted to the treated field (Fig. 6.17). Most treatment techniques take care to avoid clinical consequences from such fibrosis, but occasionally an individual patient will show an excessive reaction, such as a stricture of the bowel. Sometimes, fibrosis is the desired objective of therapy, as in the treatment of intracerebral vascular malformations by inducing scarring.
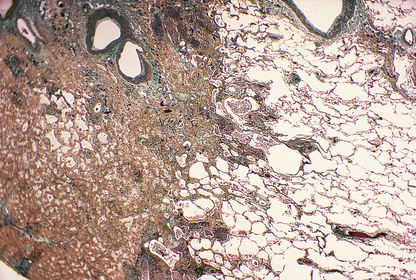
Fig. 6.17 Histology of lung fibrosis due to therapeutic irradiation. Note the abrupt demarcation between the solid scarred lung (left) and the adjacent normally aerated lung (right); this is due to the sharp cut-off at the edge of the irradiated field, to minimise the extent of damage to adjacent structures.
Commonly confused conditions and entities relating to cellular injury
| Commonly confused | Distinction and explanation |
|---|---|
| Apoptosis and necrosis | Both are modes of cell death. Apoptosis is an active process involving single-cell death occurring in normal (e.g. embryogenesis) and abnormal situations; the cell membrane is intact and no inflammatory reaction is elicited. Necrosis is a response to injury, almost always pathological and involving groups of cells; cell membrane integrity is lost and an inflammatory and repair response is common. |
| Coagulative and colliquative necrosis | In coagulative necrosis the cells are dead but the tissue architecture is often preserved in the early stages; the tissue then softens and eventually heals by fibrosis and scarring. Colliquative (or liquefactive) necrosis occurs characteristically in the brain; the tissue liquefies and heals by cyst formation. |
| Granuloma and granulation tissue | A granuloma is an aggregate of epithelioid histiocytes and a feature of some specific chronic inflammatory disorders. Granulation tissue is an important component of healing and comprises small blood vessels in a connective tissue matrix with myofibroblasts. |
| Stenosis and stricture | Stenosis most often refers to narrowing of an orifice (e.g. pylorus), aperture or valve, whereas a stricture is usually used to describe a narrowed tube (e.g. intestine). |
| First and second intentionhealing | Healing by first intention occurs when there has been no significant loss of tissue (e.g. a clean surgical incision). When there has been a significant loss of tissue (e.g. by trauma) the defect is filled initially by granulation tissue before further healing; this is second intention. |
| X-rays and gamma rays | Both have similar physical properties, but X-rays are produced by a machine and their production can be controlled by a switch, whereas gamma rays are produced by radioactive decay and protection from them can be achieved only by a barrier. |
Bomford C.K., Kunkler I.H.. Walter and Miller’s textbook of radiotherapy, 6th edn.. Edinburgh: Churchill Livingstone; 2002.
Brenner D.J., Hall E.J.. Computed tomography—an increasing source of radiation exposure. New England Journal of Medicine. 2007;357:2277-2284.
Fajardo L.P., Berthrong M., Anderson R.E.. Radiation pathology. Oxford,: Oxford University Press; 2001. Ch 1
Foley-Comer A.J., Herrick S.E., Al Mishlab T., et al. Evidence for incorporation of free-floating mesothelial cells as a mechanism of serosal healing. Journal of Cell Science. 2002;115:1383-1389.
Fuchs E.. Skin stem cells: rising to the surface. Journal of Cell Biology. 2008;180:273-284.
Gabbiani G.. The myofibroblast in wound healing and fibrocontractive diseases. Journal of Pathology. 2003;200:500-503.
Letai A.G. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nature Reviews. Cancer. 2008;8:121-132
Maiuri M.C., Zalckvar E., Kimchi A., Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature Reviews. Molecular Cell Biology. 2007;8:741-752
Metz C.N.. Fibrocytes: a unique cell population implicated in wound healing. Cellular and Molecular Life Sciences. 2003;60:1342-1350.
Price P., Sikora K.. Treatment of cancer, 4th edn. London: Chapman and Hall; 2002.
Taylor R.C., Cullen S.P., Martin S.J. Apoptosis: controlled demolition at the cellular level. Nature Reviews Molecular Cell Biology. 2008;9:231-241
Zong W.X., Thompson C.B.. Necrotic death as a cell fate. Genes and Development. 2006;20:1-15.
Health Protection Agency, radiation division (National Radiological Protection Board): http://www.hpa.org.uk/radiation
Health Risks from Exposure to Low Levels of Ionizing Radiation: BEIR VII Phase 2 (Free Executive Summary): http://www.nap.edu/catalog/11340.html
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 6 Responses to cellular injury
CELLULAR INJURY
Cell survival depends upon several factors: a constant supply of energy, intact plasma membrane, biologically safe and effective function of generic and specific cellular activities, genomic integrity, controlled cell division, and internal homeostatic mechanisms. Cell death may result from significant disturbance of these factors. However, cell replication proceeds in a human body at a rate of c. 10 000 new cells per second; so, although eventually some will be lost to the environment via the skin or gut surfaces, many will inevitably need to be deleted. Thus, cell death is a normal physiological process as well as a reaction to injury. Similarly, failure or poor regulation of death processes may underlie some diseases.
Causative agents and processes
A wide range of possible agents or circumstances result in cellular injury (Fig. 6.1). These could be categorised according to the nature of the injurious agent, the cellular target, the pattern of cellular reaction or mode of cell death. The sequence of agent, target and mode will be uniform, but some injurious agents have variable effects depending on concentration, duration or other contributory influences such as co-existent disease. Some examples are given in Table 6.1. Major types of cellular injury include:
Fig. 6.1 Mechanisms of cellular injury. Different agents can injure the various structural and functional components of the cell. Some cells with specific function are selectively prone to certain types of injury.
Table 6.1 Examples of causes of cellular injury and their mode of action
| Example agent | Mode of action |
|---|---|
| Trauma (e.g. road traffic accident) | Mechanical disruption of tissue |
| Carbon monoxide inhalation | Prevents oxygen transport |
| Contact with strong acid | Coagulates tissue proteins |
| Paracetamol overdose | Metabolites bind to liver cell proteins and lipoproteins |
| Bacterial infections | Toxins and enzymes |
| Ionising radiation (e.g. X-rays) | Damage to DNA |
Physical agents
Most physical agents cause passive cell destruction by gross membrane disruption or catastrophic functional impairment. Trauma and thermal injury cause cell death by disrupting cells and denaturing proteins, and also cause local vascular thrombosis with consequent tissue ischaemia or infarction (Ch. 8). Freezing damages cells mechanically because their membranes are perforated by ice crystals. Missile injury combines the effects of trauma and heat; much energy is dissipated into tissues around the track. Blast injuries are the result of shearing forces, where structures of differing density and mobility are moved with respect to one another; traumatic amputation is a gross example. Microwaves (wavelengths in the range from 1 mm to 1 m) cause thermal injury. Laser light falls into two broad categories: relatively low energy produces tissue heating, with coagulation for example; higher energy light breaks intramolecular bonds by a photochemical reaction, and effectively vaporises tissue. Ionising radiation is considered on p. 115.
Chemical and biological agents
Cells may be injured by contact with drugs and other chemicals; the latter may include enzymes and toxins secreted by micro-organisms. This category of agents can give rise to the full range of modes of death.
Drugs and poisons
Many naturally occurring and synthetic chemicals cause cellular injury; the effect is usually dose-related, but in a few instances the effect is exacerbated by constitutional factors.
Some are highly toxic systemic metabolic poisons, while others exert their damage locally; the latter includes caustic liquids applied to skin or mucous membranes, or gases that injure the lung. Furthermore, some substances produce one effect locally and another systemically. For example, some drugs are potentially caustic, and care needs to be taken to avoid extravasation into soft tissues when giving them by intravenous injection.
Caustic agents cause rapid local cell death due to their extreme alkalinity or acidity, in addition to having a corrosive effect on the tissue by digesting proteins.
Infectious organisms
The mechanisms of tissue damage produced by infectious organisms are varied, but with many bacteria it is their metabolic products or secretions that are harmful (Ch. 3). Thus, the host cells receive a chemical insult that may be toxic to their metabolism or membrane integrity. The mode of cell death generally induces an acute inflammatory response, which may be damaging to adjacent cells; organisms that do this are called pyogenic. In contrast, bacterial endotoxin (lipopolysaccharide) induces apoptosis with different pathological consequences. Intracellular agents such as viruses often result in the physical rupture of infected cells, but with some viruses such as hepatitis B (Ch. 16) local tissue damage may result from host immune reactions. Therefore, the cellular response to injury caused by infections will depend on a combination of the damage inflicted directly by the agent and indirectly as a result of the host response to the agent.
Blockage of metabolic pathways
Cell injury may result from specific interference with intracellular metabolism, effected usually by relative or total blockage of one or more pathways.
Cellular respiration
Prevention of oxygen utilisation results in the death of many cells due to loss of their principal energy source. Cyanide ions act in this way by binding to cytochrome oxidase and thus interrupting oxygen utilisation. Cells with higher metabolic requirements for oxygen (e.g. cardiac myocytes) are most vulnerable.
Glucose deprivation
Glucose is another important metabolite and source of energy. Some cells, cerebral neurones for example, are highly dependent. In diabetes mellitus there is inadequate utilisation of glucose due to an absolute or relative lack of insulin.
Protein synthesis
Cell function and viability will also be compromised if protein synthesis is blocked at the translational level because there is a constant requirement to replace enzymes and structural proteins. Ricin, a potent toxin from the castor oil plant, acts in this manner at the ribosomal level. Many antibiotics, such as streptomycin, chloramphenicol and tetracycline, act by interfering with protein synthesis, although toxic effects by this mechanism are fortunately rare.
Loss of growth factor or hormonal influence
Many cells rely on growth factors for their survival. Typically, these bind to growth factor receptors spanning the plasma membrane, triggering an intracellular cascade, often via a tyrosine kinase. This pathway can fail or be blocked at many points including growth factor deficit, receptor loss or blockade, or tyrosine kinase inhibitor (e.g. imatinib) (Ch. 4); affected cells may undergo apoptosis. Similar consequences can follow hormone withdrawal, as either a physiological response or part of a disease process. If widespread in an organ, it will shrink (atrophy).
Ischaemia and reperfusion injury
Impaired blood flow (Ch. 8) causes inadequate oxygen delivery to cells. Mitochondrial production of ATP will cease, and anaerobic glycolysis will result in acidosis due to the accumulation of lactate. The acidosis promotes calcium influx. Cells in different organs vary widely in their vulnerability to oxygen deprivation; those with high metabolic activity such as cortical neurones and cardiac myocytes will be most affected.
When the blood supply is restored, the oxygen results in a burst of mitochondrial activity and excessive release of reactive oxygen species (free radicals).
Free radicals
Free radicals are atoms or groups of atoms with an unpaired electron (symbolised by a superscript dot); they avidly form chemical bonds. They are highly reactive, chemically unstable, generally present only at low concentrations, and tend to participate in or initiate chain reactions.
Free radicals can be generated by two principal mechanisms:
The consequences of free radical formation include the following:
The clinicopathological events involving free radicals include:
Cells irreversibly damaged by free radicals are deleted, generally by apoptosis.
Failure of membrane integrity
Cell membrane damage is an important mode of cellular injury for which there are several possible mechanisms:
Cell membrane damage is one of the consequences of complement activation (Ch. 9); some of the end products of the complement cascade have cytolytic activity. Another effector of cytolysis is perforin, a mediator of lymphocyte cytotoxicity that causes damage to the cell membrane of the target cells such as those infected by viruses. Incidental membrane tears or perforations can be repaired very quickly, so do not necessarily result in cell death.
Intramembrane channels permit the controlled entry and exit of specific ions. Blockage of these channels is sometimes used therapeutically. For example, verapamil is a calcium channel blocker used in the treatment of hypertension and ischaemic heart disease. Used in inappropriate circumstances or at high dosage, however, the calcium channel blockage may have toxic effects.
Membrane ion pumps that are responsible for maintaining intracellular homeostasis, for example calcium, potassium and sodium concentrations within cells, are dependent on an adequate supply of ATP. Any chemical agents that deplete ATP, either by interfering with mitochondrial oxidative phosphorylation or by consuming ATP in their metabolism, will compromise the integrity of the membrane pumps and expose the cell to the risk of lysis. The Na/K ATPase in cell membranes can be directly inhibited by ouabain. Failure of membrane ion pumps frequently results in cell swelling, also called oncosis or hydropic change (see below), which may progress to cell death.
Just as disastrous for the cell is biochemical alteration of the lipoprotein bilayer forming the cell membrane. This can result from reactions with either the phospholipid or protein moieties. Membrane phospholipids may be altered through peroxidation by reactive oxygen species and by phospholipases. If the membrane damage results in lysosome permeability, release of its contents precipitates further cell damage or death. Membrane proteins may be altered by cross-linking induced by free radicals.
DNA damage or loss
Damage to DNA results primarily from reactive oxygen species attack, for example following ionising radiation (p. 117). Damage may not be evident immediately; dividing cells are more susceptible. Cell populations that are constantly dividing (i.e. labile cells such as intestinal epithelium and haemopoietic cells) are soon affected by a dose of radiation sufficient to alter their DNA. Other cell populations may require a growth or metabolic stimulus before the DNA damage is revealed. Since non-lethal DNA damage may be inherited by daughter cells, a clone of transformed cells with abnormal growth characteristics may be formed; this is the process of neoplastic transformation that results in tumours (Ch. 11).
Normal erythrocytes are particularly sensitive to injury because they lack a nucleus and, therefore, the means for inducing repair mechanisms. This will also be the fate of any cell in which the nucleus is severely damaged, or when mitosis is attempted but its completion is blocked. The latter is the result of DNA strand breaks or cross-linkages; ionising radiation and some cytotoxic drugs used in cancer therapy have this effect. Damaged cells are deleted, usually by apoptosis.
Cellular appearances following injury
The agents and mechanisms mentioned above cause a variety of histological abnormalities, although very few are specific for each agent. Two patterns of sublethal cellular alteration seen fairly commonly are hydropic change and fatty change.
Hydropic change (oncosis)
In hydropic change the cytoplasm becomes pale and swollen due to accumulation of fluid. Hydropic change generally results from disturbances of metabolism such as hypoxia or chemical poisoning. These changes are reversible, although they may herald irreversible damage if the causal injury is persistent.
Fatty change (steatosis)
Vacuolation of cells is due often to the accumulation of lipid droplets as a result of a disturbance to ribosomal function and uncoupling of lipid from protein metabolism. The liver is commonly affected in this way by several causes, such as hypoxia, alcohol or diabetes. Moderate degrees of fatty change are reversible, but severe fatty change may not be.
Mechanisms of cell death
There are two distinct mechanisms by which cells die: necrosis and apoptosis. However, there are also other cellular deaths combining features of both these processes. Discussion of cell death is further complicated by a lack of uniform nomenclature; some authors use the term necrosis to denote cell death by any cellular mechanism, but more often it is used to describe a specific mechanism. Although there are usually particular triggers for one process or another, there are some situations where apoptosis follows a lower dose or shorter duration of insult while necrosis occurs above that threshold. Mechanisms of cell death involve defined metabolic pathways. Consequently, cell death processes may be amenable to therapeutic interventions.
Necrosis
Necrosis is characterised by bioenergetic failure and loss of plasma membrane integrity. The ischaemia–reperfusion model has been the focus of much resear Ch. Failure of ATP production renders plasma membrane ion pumps ineffective with resulting loss of homeostasis, influx of water, oncosis, lysis and cell death, but in many circumstances this sequence may be an oversimplification.
Anaerobic conditions result in acidosis, thus promoting calcium inflow. Calcium uptake by mitochondria eventually exceeds their storage capacity, and contributes to disruption of the inner membrane (mitochondrial permeability transition); ATP production ceases and contents leak into the cytosol. This mitochondrial sequence is particularly exacerbated, if not initiated, by reperfusion causing a burst of reactive oxygen species production.
DNA damage, for example by free radicals or alkylating agents, initiates repair sequences including activation of the nuclear enzyme poly (ADP-ribose) polymerase (PARP). In proliferating cells, as they are dependent on glycolysis, this leads to NAD depletion and thus ATP depletion and consequently necrosis.
Falling ATP levels can open plasma membrane channel-mediated calcium uptake (death channels); large rises in cytosol calcium activate calcium-dependent proteases or lead on to mitochondrial permeability transition. In contrast, free radical damage to endoplasmic reticulum allows calcium stores to leak into the cytosol; smaller rises in calcium tend to cause apoptosis rather than necrosis.
Free radical damage to lysosomal membranes releases proteases, such as cathepsins which damage other membranes and can cause cell death. By a similar mechanism, binding of tumour necrosis factor to its cell surface receptor stimulates excessive mitochondrial reactive oxygen species with the results noted above and hence necrosis.
All these pathways eventually lead to rupture of the plasma membrane and spillage of cell contents, but this is not the end of the sequence. Some of the contents released are immunostimulatory, for example heat shock proteins and purine metabolites. These provoke the inflammatory response (Ch. 10), which paves the way for repair.
Several distinct morphological types of necrosis are recognised:
The type of tissue and nature of the causative agent determine the type of necrosis.
Necrosis must be distinguished from apoptosis, in which cell death results from a different mechanism. The cell membrane remains intact and there is no inflammatory reaction.
Coagulative necrosis
Coagulative necrosis is the commonest form of necrosis and can occur in most organs. Following devitalisation, the cells retain their outline as their proteins coagulate and metabolic activity ceases. The gross appearance will depend partly on the cause of cell death, and in particular on any vascular alteration such as dilatation or cessation of flow. Initially, the tissue texture will be normal or firm, but later it may become soft as a result of digestion by macrophages. This can have disastrous consequences in necrosis of the myocardium following infarction, as there is a risk of ventricular rupture (Ch. 13).
Microscopic examination of an area of necrosis shows a variable appearance depending on the duration. In the first few hours, there will be no discernable abnormality. Subsequently, there will be progressive loss of nuclear staining until it ceases to be haematoxyphilic; this is accompanied by loss of cytoplasmic detail (Fig. 6.2). The collagenous stroma is more resistant to dissolution. The result is that, histologically, the tissue retains a faint outline of its structure until such time as the damaged area is removed by phagocytosis (or sloughed off a surface), and is then repaired or regenerated. The presence of necrotic tissue usually evokes an inflammatory response; this is independent of the initiating cause of the necrosis.
Fig. 6.2 Necrosis. Histology of part of a kidney deprived of its blood supply by an arterial embolus (Ch. 8). This is an example of coagulative necrosis. Cellular and nuclear detail has been lost. The ghost outline of a glomerulus can be seen in the centre, with remnants of tubules elsewhere.
Colliquative necrosis
Colliquative necrosis occurs in the brain because of its lack of any substantial supporting stroma; thus, necrotic neural tissue may totally liquefy. There will be a glial reaction around the periphery, and the site of necrosis will be marked eventually by a cyst.
Caseous necrosis
Tuberculosis is characterised by caseous necrosis, a pattern of necrosis in which the dead tissue is structureless. Histological examination shows an amorphous eosinophilic area stippled by haematoxyphilic nuclear debris. Although not confined to tuberculosis, nor invariably present, caseation in a biopsy should always raise the possibility of tuberculosis.
Gangrene
Gangrene is necrosis with putrefaction of the tissues, sometimes as a result of the action of certain bacteria, notably clostridia. The affected tissues appear black because of the deposition of iron sulphide from degraded haemoglobin. Thus, ischaemic necrosis of the distal part of a limb may proceed to gangrene if complicated by an appropriate infection. As clostridia are very common in the bowel, intestinal necrosis is particularly liable to proceed to gangrene; it can occur as a complication of appendicitis, or incarceration of a hernia if the blood supply is impeded. These are examples of ‘wet’ gangrene. In contrast, ‘dry’ gangrene is usually seen in the toes, as a result of gradual arterial or small vessel obstruction in atherosclerosis or diabetes mellitus, respectively. In time, a line of demarcation develops between the gangrenous and adjacent viable tissues.
In contrast to the above, primary infection with certain bacteria or combinations of bacteria may result in similar putrefactive necrosis. Gas gangrene is the result of infection by Clostridium perfringens, while synergistic gangrene follows infection by combinations of organisms, such as Bacteroides and Borrelia vincenti.
Fibrinoid necrosis
In the context of malignant hypertension (Ch. 13), arterioles are under such pressure that there is necrosis of the smooth muscle wall. This allows seepage of plasma into the media with consequent deposition of fibrin. The appearance is termed fibrinoid necrosis. With haematoxylin and eosin staining, the vessel wall is a homogeneous bright red. Fibrinoid necrosis is sometimes a misnomer because the element of necrosis is inconspicuous or absent. Nevertheless, the histological appearance is distinctive and its close resemblance to necrotic tissue perpetuates the name of this lesion.
Fat necrosis
Following trauma to adipose tissue, the release of intracellular fat elicits a brisk inflammatory response, with polymorphs and macrophages phagocytosing the fat, proceeding eventually to fibrosis. The result may be a palpable mass, particularly at a superficial site such as the breast.
In acute pancreatitis, there is release of pancreatic lipase (Ch. 16). As a result, fat cells have their stored fat split into fatty acids, which then combine with calcium to precipitate out as white soaps. In severe cases, hypocalcaemia can ensue.
Apoptosis
Apoptosis is quite different from necrosis (Table 6.2); indeed, it includes suppression of necrosis. It is an energy-dependent process for deletion of unwanted individual cells. Apoptosis is involved in morphogenesis (Ch. 5), and is the mechanism for controlling organ size. Unwanted or defective cells also undergo apoptosis; thus, lymphocyte proliferation in germinal centres and the thymus is followed by apoptosis of unwanted cells. Factors controlling apoptosis include substances outside the cell and internal metabolic pathways:
Table 6.2 Comparison of cell death by apoptosis and necrosis
| Feature | Apoptosis | Necrosis |
|---|---|---|
| Induction | May be induced by physiological or pathological stimuli | Invariably due to pathological injury |
| Extent | Single cells | Cell groups |
| Biochemical events | Energy-dependent fragmentation of DNA by endogenous endonucleases | Energy failure |
| Impairment or cessation of ion homeostasis | ||
| Lysosomes intact | Lysosomes leak lytic enzymes | |
| Cell membrane integrity | Maintained | Lost |
| Morphology | Cell shrinkage and fragmentation to form apoptotic bodies with dense chromatin | Cell swelling and lysis |
| Inflammatory response | None | Usual |
| Fate of dead cells | Ingested (phagocytosed) by neighbouring cells | Ingested (phagocytosed) by neutrophil polymorphs and macrophages |
| Outcome | Cell elimination | Defence, and preparation for repair |
Exposure to inducers or withdrawal of inhibitors acts via the bcl-2 protein family, which then inhibit or activate the death pathway, resulting in activation of initiator and executioner caspases. Alternatively, activation of the plasma membrane receptor Fas (CD95) by its ligand bypasses bcl-2 to activate other caspases (Fig. 6.3). These result in degradation of the cytoskeletal framework, fragmentation of DNA and loss of mitochondrial function. The nucleus shrinks (pyknosis) and fragments (karyorrhexis). The cell shrinks, retaining an intact plasma membrane (Fig. 6.4), but alteration of this membrane rapidly induces phagocytosis. Dead cells not phagocytosed fragment into smaller membrane-bound apoptotic bodies. There is no inflammatory reaction to apoptotic cells, probably because the cell membrane is intact. Various diseases are associated with reduced or increased apoptosis.
Fig. 6.3 Mechanisms of apoptosis. Apoptosis results from activation of caspases triggered either by the bcl-2 family or by the binding of Fas ligand to its receptor.
Fig. 6.4 Apoptosis. Histology of skin from a case of graft-versus-host disease (Ch. 9) in which there is individual cell death (arrowed) in the epidermis as a result of immune injury.
Reduced apoptosis
The product of the p53 gene checks the integrity of the genome before mitosis; defective cells are switched to apoptosis instead. In contrast, bcl-2 protein inhibits apoptosis. Therefore, loss of p53 function or excess bcl-2 expression may result in failure of initiation of apoptosis with resulting cell accumulation; both these defects are important in neoplasia (Ch. 11). Autoimmune disease (Ch. 9) might reflect failure of induction of apoptosis in lymphoid cells directed against host antigens; in systemic lupus erythematosus, alterations are reported in the Fas lymphocyte receptor. Some viruses enhance their survival by inhibiting apoptosis of cells they infect, and latent infection by Epstein–Barr virus upregulates bcl-2.
Increased apoptosis
Diseases in which increased apoptosis is probably important include acquired immune deficiency syndrome (AIDS), neurodegenerative disorders and anaemia of chronic disorders (Ch. 23). In AIDS, human immunodeficiency virus proteins may activate CD4 on uninfected T-helper lymphocytes, inducing apoptosis with resulting immunodepletion. Apoptosis is the usual mode of cell death in exposure to ionising radiation (p. 117).
Autophagy
[/not-level-membership-for-pathology-category]
