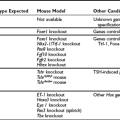
Thyroid-Stimulating Hormone Receptor Mutations
Gain-of-Function Mutations
From what we know of thyroid cell physiology, it is easy to predict the phenotypes associated with gain-of-function mutations of the cyclic adenosine monophosphate (cAMP)-dependent regulatory cascade. Two observations provide pertinent models of this situation. Transgenic mice made to ectopically express the adenosine A2a receptor in their thyroid display severe hyperthyroidism associated with thyroid hyperplasia.1 Because the A2a adenosine receptor is coupled to Gs and displays constitutive activity as a result of continuous stimulation by ambient adenosine, this model closely mimics the situation expected for a gain-of-function germline mutation of the TSH receptor. Patients with the McCune-Albright syndrome are mosaic for mutations in Gs (gsp mutations), which also leads to constitutive stimulation of adenylyl cyclase.2 Hyperfunctioning thyroid adenomas develop in these patients from cells harboring the mutation, which makes them a model for gain-of-function somatic mutations of the TSH receptor. A transgenic model in which gsp mutations are targeted for expression in the mouse thyroid has been constructed. Although with a less dramatic phenotype, this represents a pertinent model for a gain of function in the cAMP regulatory cascade.3
Familial Nonautoimmune Hyperthyroidism or Hereditary Toxic Thyroid Hyperplasia
The major cause of hyperthyroidism in adults is Graves’ disease, in which an autoimmune reaction is mounted against the thyroid gland and autoantibodies are produced that recognize and stimulate the TSH receptor. This origin may explain why the initial description by the group of Leclère of a family showing segregation of thyrotoxicosis as an autosomal dominant trait in the absence of signs of autoimmunity was met with skepticism.4 Reinvestigation of this family together with that of another family from Reims (France) identified two mutations of the TSH receptor gene that segregated in perfect linkage with the disease.5 A series of additional families have been studied since, almost systematically with a different mutation of the TSH receptor gene6–19 (Fig. 20-1). (For comprehensive lists of activating mutations, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.) The functional characteristics of these mutant receptors confirm that they are constitutively stimulated (see below). Hereditary toxic thyroid hyperplasia (HTTH), sometimes called Leclère’s disease, is characterized by the following clinical characteristics: autosomal dominant transmission; hyperthyroidism with a variable age of onset (from infancy to adulthood, even within a given family); hyperplastic goiter of variable size, but with steady growth; and absence of clinical or biological stigmata of autoimmunity. An observation common to cases to date is the need for drastic ablative therapy (surgery or radioiodine) to control the disease once the patient has become hyperthyroid. The autonomous nature of the thyroid tissue from these patients has been elegantly demonstrated by grafting in nude mice.20 In contrast to tissue from patients with Graves’ disease, HTTH cells continue to grow in the absence of stimulation by TSH or thyroid-stimulating antibody.
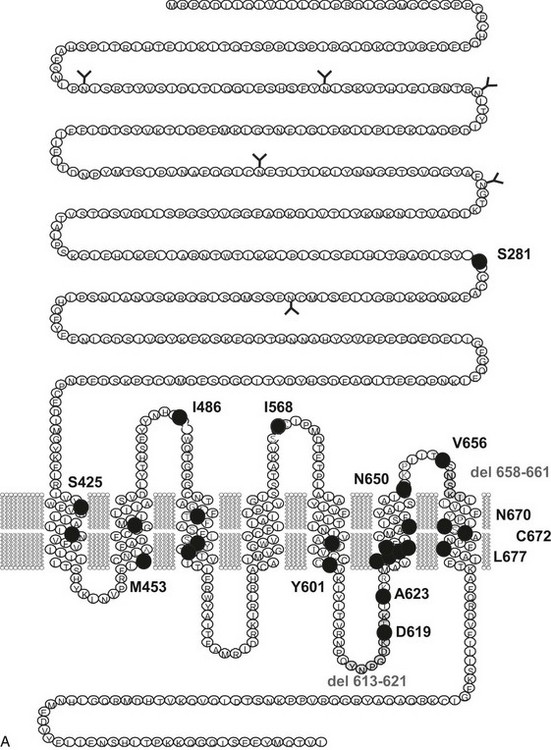
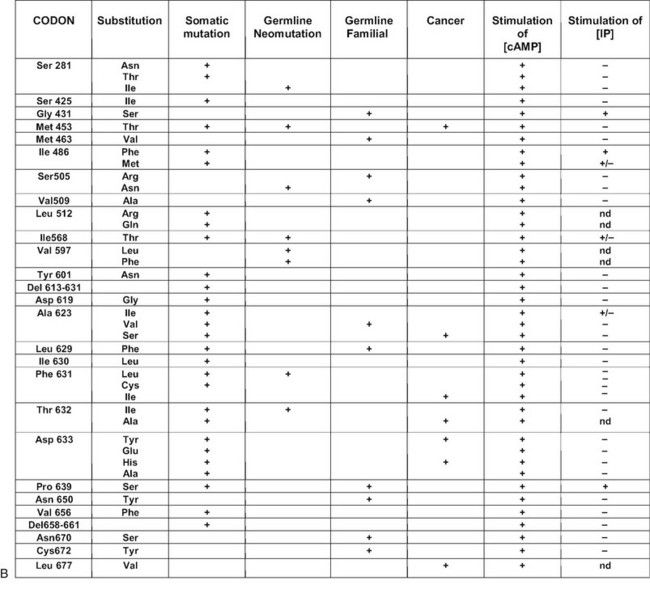
FIGURE 20-1 Schematic representation of the thyroid-stimulating hormone (TSH) receptor. A, The locations of known activating mutations are indicated.B, The nature of the mutations is indicated along with their origins (somatic, germline sporadic, germline familial, cancer) and effects on intracellular regulatory cascades. (For a complete list of activating mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.)
The prevalence of hereditary toxic thyroid hyperplasia is difficult to estimate at the present time. It is likely that many cases have been (and still are) mistaken for Graves’ disease. This may be explained by the high frequency of thyroid autoantibodies (antithyroglobulin, antithyroperoxidase) in the general population. It is expected that wider knowledge of the existence of the disease will lead to better diagnosis. This is not a purely academic problem, in that presymptomatic diagnosis in children of affected families may prevent the developmental or psychological complications associated with infantile or juvenile hyperthyroidism. A country-wide screening for the condition has been performed in Denmark. It was found in 1 out of 121 patients with juvenile thyrotoxicosis (0.8%; 95% confidence interval [CI]: 0.02% to 4.6%), which corresponds to 1 in 17 patients with presumed nonautoimmune juvenile thyrotoxicosis (6%; 95% CI: 0.15% to 28.69%).21
Sporadic Toxic Thyroid Hyperplasia
Cases of toxic thyroid hyperplasia have been described in children of unaffected parents.7,22–29 Conspicuously, congenital hyperthyroidism was present in most of the patients and required aggressive treatment. Mutations of one TSH receptor allele were identified in the children but were absent in the parents. Because paternity was confirmed by minisatellite or microsatellite testing, these cases qualify as true neomutations. When the amino acid substitutions implicated in hereditary and sporadic cases are compared, for the majority, they do not overlap (see Fig. 20-1). Although most of the sporadic cases harbor mutations that are also found in toxic adenomas, most of the hereditary cases have “private” mutations. Analysis of the functional characteristics of the individual mutant receptors in COS cells and the clinical course of individual patients suggests an explanation for this observation: “sporadic” mutations seem to have a stronger activating effect than “hereditary” mutations do. From their severe phenotypes, it is likely that newborns with neomutations would not have survived if not treated efficiently. On the contrary, from inspection of the available pedigrees, it seems that the milder phenotype of patients with hereditary mutations has only a limited effect on reproductive fitness. The fact that hereditary mutations are rarely observed in toxic adenomas is compatible with the suggestion that they would cause extremely slow tissue growth and, accordingly, would rarely cause thyrotoxicosis, if somatic. There is, however, no a priori reason for neomutations to cause stronger gain of function than hereditary mutations. Accordingly, an activating mutation of the TSH receptor gene has been found in a 6-month-old child with subclinical hyperthyroidism who presents with weight loss as the initial symptom.30
Somatic Mutations: Autonomous Toxic Adenomas
Soon after mutations of Gsα had been found in adenomas of the pituitary somatotrophs,31 similar mutations (also called gsp mutations) were found in some toxic thyroid adenomas and follicular carcinomas.32–35 The mutated residues (Arg201, Glu227) are homologous to those found mutated in the ras proto-oncogenes, that is, the mutations decrease the endogenous guanine triphosphatase activity of the G protein, thereby resulting in a constitutively active molecule.
Toxic adenoma was found to be a fruitful source of somatic mutations activating the TSH receptor, probably because the phenotype is very conspicuous and easy to diagnose. Whereas mutations are distributed all along the serpentine portion of the receptor36–42 and even in the extracellular aminoterminal domain,43 there is clearly a hotspot at the cytoplasmic side of the sixth transmembrane segment (see Fig. 20-1). (For a complete list of TSH receptor gene mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.) The clustering reflects the pivotal role of this portion of the molecule in activation mechanisms.44–49
Despite some dispute about the prevalence of TSH receptor mutations in toxic adenomas, which may be due to different origins among patients50 or different sensitivities of methods, the current consensus is that activating mutations of the TSH receptor are the major cause of solitary toxic adenomas and account for about 60% to 80% of cases.36,42,51–53 Contrary to initial suggestions, the same percentage of TSH receptor mutation is observed in Japan, an iodine-sufficient country with low prevalence of toxic adenomas.54 In some patients with multinodular goiter and two zones of autonomy at scintigraphy, a different mutation of the TSH receptor was identified in each nodule.55–58 This finding indicates that the pathophysiologic mechanism responsible for solitary toxic adenomas can be at work on a background of multinodular goiter. In agreement with this notion, activating mutations of the TSH receptor have been identified in hyperfunctioning areas of multinodular goiter.57,59–61 The independent occurrence of two activating mutations in a patient may seem highly improbable at first. However, the multiplicity of the possible amino acid targets for activating mutations within the TSH receptor makes this event less unlikely. It is also possible that a mutagenic environment is created in glands exposed to chronic stimulation by TSH in which H2O2 is produced.62,63 Finally, involvement of TSH receptor mutations in thyroid cancer has been implicated in a limited proportion of follicular thyroid carcinomas.61,64–69
Structure-Function Relationships of The TSH Receptor, as Deduced from Activating Mutations
Most of the activating mutations of the TSH receptor have been studied by transient expression in COS or HEK293T cells. There is no guarantee that the mutants will function in an identical way in these artificial systems as they do in thyrocytes. Arguments have been obtained for such cell-type specific effects.70 In thyrocytes, a better relation has been observed between adenylyl cyclase stimulation and differentiation than with growth.70 However, the built-in amplification associated with transfection of constructs in COS or HEK 293T cells makes it possible to detect even slight increases in constitutive activity of TSH receptor mutants. An important observation has been that the wild-type receptor itself displays significant constitutive activity.38,71 This characteristic is not unique to the TSH receptor, but it is interesting to note that it is not shared by its close relatives, the luteinizing hormone/chorionic gonadotropin (LH/CG) receptor and the follitropin receptor (FSH). The effect of activating mutations accordingly must be interpreted in terms of “increase in constitutive activity.” Most receptor mutants found in toxic adenomas and/or toxic thyroid hyperplasia share common characteristics: (1) they increase the constitutive activity of the receptor toward stimulation of adenylyl cyclase; (2) with a few notable exceptions (see Fig. 20-1),72 they do not display constitutive activity toward the inositol phosphate/diacylglycerol pathway; (3) their expression at the cell surface is decreased (from slightly to severely); (4) most but not all of them keep responding to TSH for stimulation of cAMP and inositol phosphate generation, with a tendency to do so at decreased median effective concentrations; and (5) they bind 125I-labeled bovine TSH with an apparent affinity higher than that of the wild-type receptor.
No direct relationship has been found between the level of cAMP achieved by different mutants in transfected COS cells and their level of expression at the cell membrane,73 which means that individual mutants have widely different “specific constitutive activity” (measured as the stimulation of cAMP accumulation/receptor number at the cell surface). Although this specific activity may tell us something about the mechanisms of receptor activation, it is not a measure of the actual phenotypic effect of the mutation in vivo. Indeed, one of the relatively mild mutations, observed up until now only in a family with HTTH (Cys672Tyr), is among the strongest according to this criterion. It would be logical to expect the best correlation to be found between the phenotype and the actual level of cAMP achieved, irrespective of the level of receptor expression. However, differences between the effects of the mutants in transfected COS or HEK293 cells and thyrocytes in vivo may render these correlations a difficult exercise.70
According to a current model of G protein–coupled receptor (GPCR) activation, the receptor would exist under at least two interconverting conformations: R (silent conformations) and R* (the active forms).44 The unliganded receptor would shuttle between both forms, the equilibrium being in favor of R. Binding of the ligand to the slit between the transmembrane segments (for biogenic amines) and/or the residues of the N-terminal segment or extracellular loops (for neuropeptides) is believed to stabilize the R* conformations. The resulting R-to-R* transition is supposed to involve a conformational change that modifies the relative position of transmembrane helices, which in turn would translate into conformational changes in the cytoplasmic domains interacting with trimeric G proteins. Seminal studies with the adrenergic receptor α1b have shown that a variety of amino acid substitutions in the C-terminal portion of the third intracellular loop lead to their constitutive activation.74 The observation that all amino acid substitutions at Ala293 were effective in activating the receptor led to the concept that the silent form of GPCRs would be submitted to a structural constraint requiring the wild-type primary structure of the third intracellular loop. This constraint could be released by a wide spectrum of amino acid substitutions in this segment.44,75
The precise effects of individual mutations in structural terms are beginning to emerge from analogies with the limited list of GPCRs whose tridimensional structure has been solved.76
Familial Gestational Hyperthyroidism
Some degree of stimulation of the thyroid gland by human chorionic gonadotropin (hCG) is commonly observed during early pregnancy. It is usually responsible for a decrease in serum thyrotropin with an increase in the concentration of free thyroxine (T4), which remains within the normal range (for references, see 77). When concentrations of hCG are abnormally high, as in molar pregnancy, true hyperthyroidism may ensue. The pathophysiologic mechanism is believed to be promiscuous stimulation of the TSH receptor by excess hCG, as suggested by the rough direct or inverse relation between serum hCG and free T4 or TSH concentrations, respectively.77 A convincing rationale is provided by the close structural relationships and evolutionary origins of the glycoprotein hormones and their receptors.78
A new syndrome was described in 1998 in a family with dominant transmission of hyperthyroidism limited to pregnancy.79 The proposita and her mother had severe thyrotoxicosis together with hyperemesis gravidarum during the course of each of their pregnancies. When nonpregnant, they were clinically and biologically euthyroid. Both patients were heterozygous for a Lys183Arg mutation in the extracellular aminoterminal domain of the TSH receptor. When tested by transient transfection in COS cells, the mutant receptor displayed normal characteristics toward TSH. However, a convincing explanation for the phenotype was provided in that it showed higher sensitivity to stimulation by hCG than the wild-type TSH receptor.
The amino acid substitution responsible in these patients for promiscuous stimulation of the TSH receptor by hCG is surprisingly conservative. Also surprising is the observation that residue 183 is a lysine in both the TSH and LH/CG receptors. When placed on the three-dimensional structure of the hormone-binding domain of the TSH receptor,80 residue 183 belongs to one of the β sheets that constitute the surface of interaction with the hormones. Detailed analysis of the effect of the K183R mutation by site-directed mutagenesis indicated that any amino acid substitution at this position confers a slight increase in stability to the illegitimate hCG/TSH receptor complex.81 This increase in stability would be enough to cause signal transduction by the hCG concentrations achieved in pregnancy, but not by the LH concentrations observed after menopause. Indeed, the mother of the proposita remained euthyroid after menopause. This finding is compatible with a relatively modest gain in function of the Lys183Arg mutant upon stimulation by hCG.
In contrast to other mammals, humans and primates rely on CG for maintenance of the corpus luteum in early pregnancy. The frequent partial suppression of TSH observed at peak hCG levels during normal pregnancy indicates that evolution has selected physiologic mechanisms operating very close to the border of thyrotoxicosis. This finding may provide a rationale for the observation that in comparison with other species, the glycoprotein hormones of primates display lower biological activity because of positive evolutionary selection of specific amino acid substitutions in their α subunits.78 Up to now, no spontaneous mutation has been identified that would increase the bioactivity of hCG. An interesting parallel may be drawn between familial gestational hyperthyroidism and cases of spontaneous ovarian hyperstimulation syndrome (sOHSS).82,83 In sOHSS, mutations of the FSH receptor gene render the receptor abnormally sensitive to hCG. The result is recurrent hyperstimulation of the ovary, on the occasion of each pregnancy.
Loss-of-Function Mutations
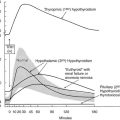
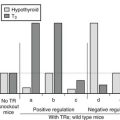
Loss-of-function mutations in the TSH receptor gene are expected to cause a syndrome of “resistance to TSH.” The expected phenotype is likely to resemble that of patients with mutations in TSH itself. These mutations have been described early because of the prior availability of information on TSH α and β genes.84 Mouse models of resistance to TSH are available as natural (hyt/hyt mouse)85 or experimental TSH receptor mutant lines.86,87 It is interesting to note and contrary to the situation in humans (see below) that the thyroid of newborn TSH receptor knockout mice is of normal size. As expected, the homozygote animals displayed profound hypothyroidism. Their thyroids did not express the sodium-iodide symporter but showed significant (noniodinated) thyroglobulin production. From this information, one would expect patients with two TSH receptor–mutated alleles to exhibit a degree of hypothyroidism in accordance with the extent of loss of function, going from mild, compensated hypothyroidism to profound neonatal hypothyroidism with absent iodide trapping. Heterozygous carriers are expected to be normal or to display minimal increase in plasma TSH (see below).
Clinical Cases with The Mutations Identified
A few patients with convincing resistance to TSH had been described before molecular genetics permitted identification of the mutations.88,89 The first cases described in molecular terms involved euthyroid siblings with elevated TSH.90 Sequencing of the TSH receptor gene identified a different mutation in each allele of the affected individuals, which made them compound heterozygotes. The substitutions occurred in the extracellular aminoterminal portion of the receptor (maternal allele, Pro162Ala; paternal allele, Ile167Asn). The functional characteristics of the mutant receptors showed that the paternal allele was virtually completely nonfunctional, whereas the maternal allele displayed an increase in the median effective TSH concentration for stimulation of cAMP production. Current knowledge of the tridimensional structure of part of the ectodomain of the receptor80,91 allows one to establish structure-function relationships for mutations affecting this portion of the receptor.92
A large number of familial cases with loss-of-function mutations of the TSH receptor have been identified in the course of screening programs for congenital hypothyroidism93–106 (Fig. 20-2). (For a complete list of TSH receptor gene mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.) Some of the patients displayed the usual criteria for congenital hypothyroidism, including high TSH, low free T4, and undetectable trapping of 99Tc. In some cases, plasma thyroglobulin levels were normal or high. Patients can be compound heterozygotes for complete loss-of-function mutations,94 or they may be homozygotes, born to consanguineous93 or apparently unrelated parents.100 In agreement with the phenotype of knockout mice with homozygous invalidation of the TSH receptor, patients with complete loss of function of the receptor display an in-place thyroid with completely absent iodide or 99Tc trapping. However, in contrast to the situation in mice, the gland is hypoplastic. Activation of the cAMP pathway, although dispensable for the anatomic development of the gland and thyroglobulin production, thus is absolutely required for expression of the sodium-iodide-symporter (NIS) gene and, at least in humans, for normal growth of the tissue during fetal life. This explains how in the absence of thyroglobulin measurements or expert echography, loss-of-function mutations of the TSH receptor may easily be misdiagnosed as thyroid agenesis.
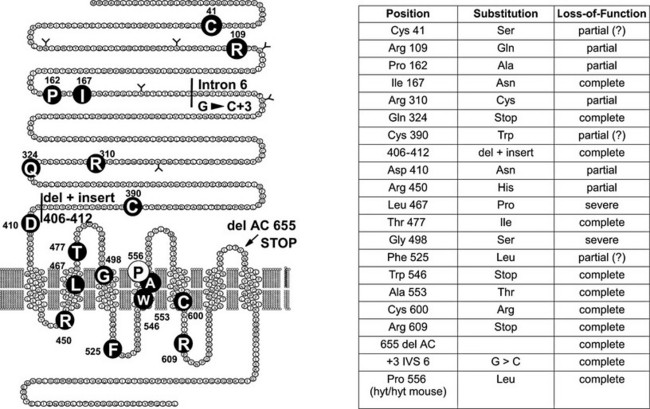
FIGURE 20-2 Loss-of-function mutations of the thyroid-stimulating hormone (TSH) receptor. The locations of known loss-of-function mutations are indicated (left) together with their nature and functional characteristics. (For a complete list of loss of function mutations with their functional characteristics, see http://gris.ulb.ac.be/ and http://www.ssfa-gphr.de/main/ssfa.php.)
In the heterozygous state, complete loss-of-function mutation of the TSH receptor is a cause of moderate hyperthyrotropinemia (subclinical hypothyroidism), segregating as an autosomal dominant trait.107 Finally, it must be stressed that an autosomal dominant form of partial resistance to TSH has been demonstrated in families in which linkage to the TSH receptor gene has been excluded.108 A locus has been identified on chromosome 15q25.3–26.1, but the gene responsible for this phenotype has not been identified yet.109
Polymorphisms
A series of single-nucleotide polymorphisms affecting the coding sequence have been identified in the TSH receptor gene. After the initial suggestion that some of these (D36H, P52T, D727E) would be associated with susceptibility to autoimmune thyroid disease,110–112 the current consensus is that they represent neutral alleles with no pathophysiologic significance.113–116 However, a genome-wide study involving a large cohort of patients recently demonstrated an association between noncoding single nucleotide polymorphisms (SNPs) at the TSH receptor gene locus and Graves’disease.117,118 The genetic substratum responsible for this association is still under study.117
One polymorphic residue deserves special mention: position 601 was found to be a tyrosine or a histidine in the two initial reports of TSH receptor cloning.119,120 Characterization of the two alleles by transfection in COS cells indicated interesting functional differences: the Y601 allele displayed readily detectable constitutive activity, whereas H601 was completely silent; the Y601 allele responded to stimulation by TSH by activating the adenylyl cyclase– and phospholipase C–dependent regulatory cascades, when the H601 allele was active only on the cAMP pathway.121 The Y601 allele is by far the most frequent among all populations tested, and a Y601N mutation was found in a toxic adenoma. Characterization of the mutant demonstrated increased constitutive activation of the cAMP regulatory cascade,121 making the 601 residue an interesting target for structure-function studies.
References
1. Ledent, C, Dumont, JE, Vassart, G, et al. Thyroid expression of an A2 adenosine receptor transgene induces thyroid hyperplasia and hyperthyroidism. EMBO J. 1992;11:537–542.
2. Weinstein, LS, Yu, S, Warner, DR, et al. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705.
3. Michiels, FM, Caillou, B, Talbot, M, et al. Oncogenic potential of guanine nucleotide stimulatory factor alpha subunit in thyroid glands of transgenic mice. Proc Natl Acad Sci U S A. 1994;91:10488–10492.
4. Thomas, JS, Leclere, J, Hartemann, P, et al. Familial hyperthyroidism without evidence of autoimmunity. Acta Endocrinol Copenh. 1982;100:512–518.
5. Duprez, L, Parma, J, Van Sande, J, et al. Germline mutations in the thyrotropin receptor gene cause non autoimmune autosomal dominant hyperthyroidism. Nat Genet. 1994;7:396–401.
6. Tonacchera, M, Van Sande, J, Cetani, F, et al. Functional characteristics of three new germline mutations of the thyrotropin receptor gene causing autosomal dominant toxic thyroid hyperplasia. J Clin Endocrinol Metab. 1996;81:547–554.
7. Fuhrer, D, Wonerow, P, Willgerodt, H, et al. Identification of a new thyrotropin receptor germline mutation (Leu629Phe) in a family with neonatal onset of autosomal dominant nonautoimmune hyperthyroidism. J Clin Endocrinol Metab. 1997;82:4234–4238.
8. Fuhrer, D, Warner, J, Sequeira, M, et al. Novel TSHR germline mutation (Met463Val) masquerading as Graves’ disease in a large Welsh kindred with hyperthyroidism. Thyroid. 2000;10:1035–1041.
9. Biebermann, H, Schoneberg, T, Hess, C, et al. The first activating TSH receptor mutation in transmembrane domain 1 identified in a family with nonautoimmune hyperthyroidism. J Clin Endocrinol Metab. 2001;86:4429–4433.
10. Arturi, F, Chiefari, E, Tumino, S, et al. Similarities and differences in the phenotype of members of an Italian family with hereditary non-autoimmune hyperthyroidism associated with an activating TSH receptor germline mutation. J Endocrinol Invest. 2002;25:696–701.
11. Alberti, L, Proverbio, MC, Costagliola, S, et al. A novel germline mutation in the TSH receptor gene causes non-autoimmune autosomal dominant hyperthyroidism. Eur J Endocrinol. 2001;145:249–254.
12. Akcurin, S, Turkkahraman, D, Tysoe, C, et al. A family with a novel TSH receptor activating germline mutation (p.Ala485Val). Eur J Pediatr. 2008;167:1231–1237.
13. Gozu, HI, Mueller, S, Bircan, R, et al. A new silent germline mutation of the TSH receptor: coexpression in a hyperthyroid family member with a second activating somatic mutation. Thyroid. 2008;18:499–508.
14. Liu, Z, Sun, Y, Dong, Q, et al. A novel TSHR gene mutation (Ile691Phe) in a Chinese family causing autosomal dominant non-autoimmune hyperthyroidism. J Hum Genet. 2008;53:475–478.
15. Ferrara, AM, Capalbo, D, Rossi, G, et al. A new case of familial nonautoimmune hyperthyroidism caused by the M463V mutation in the TSH receptor with anticipation of the disease across generations: a possible role of iodine supplementation. Thyroid. 2007;17:677–680.
16. Nishihara, E, Nagayama, Y, Amino, N, et al. A novel thyrotropin receptor germline mutation (Asp617Tyr) causing hereditary hyperthyroidism. Endocr J. 2007;54:927–934.
17. Nwosu, BU, Gourgiotis, L, Gershengorn, MC, et al. A novel activating mutation in transmembrane helix 6 of the thyrotropin receptor as cause of hereditary nonautoimmune hyperthyroidism. Thyroid. 2006;16:505–512.
18. Claus, M, Maier, J, Paschke, R, et al. Novel thyrotropin receptor germline mutation (Ile568Val) in a Saxonian family with hereditary nonautoimmune hyperthyroidism. Thyroid. 2005;15:1089–1094.
19. Vaidya, B, Campbell, V, Tripp, JH, et al. Premature birth and low birth weight associated with nonautoimmune hyperthyroidism due to an activating thyrotropin receptor gene mutation. Clin Endocrinol (Oxf). 2004;60:711–718.
20. Leclere, J, Béné, MC, Duprez, A, et al. Behavior of thyroid tissue from patients with Graves’ disease in nude mice. J Clin Endocrinol Metab. 1984;59:175–177.
21. Lavard, L, Jacobsen, BB, Perrild, H, et al. Prevalence of germline mutations in the TSH receptor gene as a cause of juvenile thyrotoxicosis. Acta Paediatr. 2004;93:1192–1194.
22. Holzapfel, HP, Wonerow, P, von Petrykowski, W, et al. Sporadic congenital hyperthyroidism due to a spontaneous germline mutation in the thyrotropin receptor gene. J Clin Endocrinol Metab. 1997;82:3879–3884.
23. Kopp, P, Van Sande, J, Parma, J, et al. Congenital non-autoimmune hyperthyroidism caused by a neomutation in the thyrotropin receptor gene. N Engl J Med. 1995;332:150–154.
24. Kopp, P, Jameson, JL, Roe, TF. Congenital nonautoimmune hyperthyroidism in a nonidentical twin caused by a sporadic germline mutation in the thyrotropin receptor gene. Thyroid. 1997;7:765–770.
25. Gruters, A, Schoneberg, T, Biebermann, H, et al. Severe congenital hyperthyroidism caused by a germ-line neo mutation in the extracellular portion of the thyrotropin receptor. J Clin Endocrinol Metab. 1998;83:1431–1436.
26. Bircan, R, Miehle, K, Mladenova, G, et al. Multiple relapses of hyperthyroidism after thyroid surgeries in a patient with long term follow-up of sporadic non-autoimmune hyperthyroidism. Exp Clin Endocrinol Diabetes. 2008;116:341–346.
27. Watkins, MG, Dejkhamron, P, Huo, J, et al. Persistent neonatal thyrotoxicosis in a neonate secondary to a rare thyroid-stimulating hormone receptor activating mutation: case report and literature review. Endocr Pract. 2008;14:479–483.
28. Fricke-Otto, S, Pfarr, N, Muhlenberg, R, et al. Mild congenital primary hypothyroidism in a Turkish family caused by a homozygous missense thyrotropin receptor (TSHR) gene mutation (A593 V). Exp Clin Endocrinol Diabetes. 2005;113:582–585.
29. Nishihara, E, Fukata, S, Hishinuma, A, et al. Sporadic congenital hyperthyroidism due to a germline mutation in the thyrotropin receptor gene (Leu 512 Gln) in a Japanese patient. Endocr J. 2006;53:735–740.
30. Pohlenz, J, Pfarr, N, Kruger, S, et al. Subclinical hyperthyroidism due to a thyrotropin receptor (TSHR) gene mutation (S505R). Acta Paediatr. 2006;95:1685–1687.
31. Landis, CA, Masters, SB, Spada, A, et al. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–696.
32. Goretzki, PE, Lyons, J, Stacy Phipps, S, et al. Mutational activation of RAS and GSP oncogenes in differentiated thyroid cancer and their biological implications. World J Surg. 1992;16:576–581.
33. Lyons, J, Landis, CA, Harsh, G, et al. Two G protein oncogenes in human endocrine tumors. Science. 1990;249:655–659.
34. O’Sullivan, C, Barton, CM, Staddon, SL, et al. Activating point mutations of the gsp oncogene in human thyroid adenomas. Mol Carcinog. 1991;4:345–349.
35. Suarez, HG, du Villard, JA, Caillou, B, et al. GSP mutations in human thyroid tumours. Oncogene. 1991;6:677–679.
36. Fuhrer, D, Holzapfel, HP, Wonerow, P, et al. Somatic mutations in the thyrotropin receptor gene and not in the Gs alpha protein gene in 31 toxic thyroid nodules. J Clin Endocrinol Metab. 1997;82:3885–3891.
37. Holzapfel HP, Scherbaum WA, Paschke R: Identification of two different somatic TSH receptor mutations in the same patient with hyperthyroidism due to multifocal thyroid autonomy. International Congress of Endocrinology San Francisco, 1996, Abstract 1:P2–P945, 1996.
38. Parma, J, Duprez, L, Van Sande, J, et al. Somatic mutations in the thyrotropin receptor gene cause hyperfunctioning thyroid adenomas. Nature. 1993;365:649–651.
39. Paschke, R, Tonacchera, M, Van Sande, J, et al. Identification and functional characterization of two new somatic mutations causing constitutive activation of the TSH receptor in hyperfunctioning autonomous adenomas of the thyroid. J Clin Endocrinol Metab. 1994;79:1785–1789.
40. Porcellini, A, Ciullo, I, Laviola, L, et al. Novel mutations of thyrotropin receptor gene in thyroid hyperfunctioning adenomas. J Clin Endocrinol Metab. 1994;79:657–661.
41. Wonerow, P, Schoneberg, T, Schultz, G, et al. Deletions in the third intracellular loop of the thyrotropin receptor. A new mechanism for constitutive activation. J Biol Chem. 1998;273:7900–7905.
42. Parma, J, Duprez, L, Van Sande, J, et al. Diversity and prevalence of somatic mutations in the TSH receptor and Gs alpha genes as a cause of toxic thyroid adenomas. J Clin Endocrinol Metab. 1997;82:2695–2701.
43. Duprez, L, Parma, J, Costagliola, S, et al. Constitutive activation of the TSH receptor by spontaneous mutations affecting the N-terminal extracellular domain. FEBS Lett. 1997;409:469–474.
44. Gether, U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21:90–113.
45. Govaerts, C, Lefort, A, Costagliola, S, et al. A conserved ASN in TM7 is an on/off switch in the activation of the TSH receptor. J Biol Chem. 2001;276:22991–22999.
46. Neumann, S, Krause, G, Chey, S, et al. A free carboxylate oxygen in the side chain of position 674 in transmembrane domain 7 is necessary for TSH receptor activation. Mol Endocrinol. 2001;15:1294–1305.
47. Jaeschke, H, Kleinau, G, Sontheimer, J, et al. Preferences of transmembrane helices for cooperative amplification of G(alpha)s and G (alpha)q signaling of the thyrotropin receptor. Cell Mol Life Sci. 2008;65:4028–4038.
48. Kleinau, G, Claus, M, Jaeschke, H, et al. Contacts between extracellular loop two and transmembrane helix six determine basal activity of the thyroid-stimulating hormone receptor. J Biol Chem. 2007;282:518–525.
49. Vassart, G, Pardo, L, Costagliola, S. A molecular dissection of the glycoprotein hormone receptors. Trends Biochem Sci. 2004;29:119–126.
50. Takeshita, A, Nagayama, Y, Yokoyama, N, et al. Rarity of oncogenic mutations in the thyrotropin receptor of autonomously functioning thyroid nodules in Japan. J Clin Endocrinol Metab. 1995;80:2607–2611.
51. Georgopoulos, NA, Sykiotis, GP, Sgourou, A, et al. Autonomously functioning thyroid nodules in a former iodine-deficient area commonly harbor gain-of-function mutations in the thyrotropin signaling pathway. Eur J Endocrinol. 2003;149:287–292.
52. Trulzsch, B, Krohn, K, Wonerow, P, et al. Detection of thyroid-stimulating hormone receptor and Gs alpha mutations: in 75 toxic thyroid nodules by denaturing gradient gel electrophoresis. J Mol Med. 2001;78:684–691.
53. Palos-Paz, F, Perez-Guerra, O, Cameselle-Teijeiro, J, et al. Prevalence of mutations in TSHR, GNAS, PRKAR1A and RAS genes in a large series of toxic thyroid adenomas from Galicia, an iodine-deficient area in NW Spain. Eur J Endocrinol. 2008;159:623–631.
54. Vanvooren, V, Uchino, S, Duprez, L, et al. Oncogenic mutations in the thyrotropin receptor of autonomously functioning thyroid nodules in the Japanese population. Eur J Endocrinol. 2002;147:287–291.
55. Duprez, L, Hermans, J, Van Sande, J, et al. Two autonomous nodules of a patient with multinodular goiter harbor different activating mutations of the thyrotropin receptor gene. J Clin Endocrinol Metab. 1997;82:306–308.
56. Holzapfel, HP, Fuhrer, D, Wonerow, P, et al. Identification of constitutively activating somatic thyrotropin receptor mutations in a subset of toxic multinodular goiters. J Clin Endocrinol Metab. 1997;82:4229–4233.
57. Tonacchera, M, Vitti, P, Agretti, P, et al. Activating thyrotropin receptor mutations in histologically heterogeneous hyperfunctioning nodules of multinodular goiter. Thyroid. 1998;8:559–564.
58. Tonacchera, M, Chiovato, L, Pinchera, A, et al. Hyperfunctioning thyroid nodules in toxic multinodular goiter share activating thyrotropin receptor mutations with solitary toxic adenoma. J Clin Endocrinol Metab. 1998;83:492–498.
59. Tonacchera, M, Agretti, P, Chiovato, L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab. 2000;85:2270–2274.
60. Georgopoulos, NA, Sykiotis, GP, Sgourou, A, et al. Autonomously functioning thyroid nodules in a former iodine-deficient area commonly harbor gain-of-function mutations in the thyrotropin signaling pathway. Eur J Endocrinol. 2003;149:287–292.
61. Gozu, H, Avsar, M, Bircan, R, et al. Mutations in the thyrotropin receptor signal transduction pathway in the hyperfunctioning thyroid nodules from multinodular goiters: a study in the Turkish population. Endocr J. 2005;52:577–585.
62. Krohn, K, Fuhrer, D, Bayer, Y, et al. Molecular pathogenesis of euthyroid and toxic multinodular goiter. Endocr Rev. 2005;26:504–524.
63. Maier, J, van Steeg, H, van Oostrom, C, et al. Deoxyribonucleic acid damage and spontaneous mutagenesis in the thyroid gland of rats and mice. Endocrinology. 2006;147:3391–3397.
64. Russo, D, Arturi, F, Schlumberger, M, et al. Activating mutations of the TSH receptor in differentiated thyroid carcinomas. Oncogene. 1995;11:1907–1911.
65. Spambalg, D, Sharifi, N, Elisei, R, et al. Structural studies of the TSH receptor and Gsa in human thyroid cancers: low prevalence of mutations predicts infrequent involvement in malignant transformation. J Clin Endocrinol Metab. 1996;81:3898–3901.
66. Camacho, P, Gordon, D, Chiefari, E, et al. A Phe 486 thyrotropin receptor mutation in an autonomously functioning follicular carcinoma that was causing hyperthyroidism. Thyroid. 2000;10:1009–1012.
67. Fuhrer, D, Tannapfel, A, Sabri, O, et al. Two somatic TSH receptor mutations in a patient with toxic metastasising follicular thyroid carcinoma and non-functional lung metastases. Endocr Relat Cancer. 2003;10:591–600.
68. Mircescu, H, Parma, J, Huot, C, et al. Hyperfunctioning malignant thyroid nodule in an 11-year-old girl: pathologic and molecular studies. J Pediatr. 2000;137:585–587.
69. Niepomniszcze, H, Suarez, H, Pitoia, F, et al. Follicular carcinoma presenting as autonomous functioning thyroid nodule and containing an activating mutation of the TSH receptor (T620I) and a mutation of the Ki-RAS (G12C) genes. Thyroid. 2006;16:497–503.
70. Fuhrer, D, Lewis, MD, Alkhafaji, F, et al. Biological activity of activating thyroid-stimulating hormone receptor mutants depends on the cellular context. Endocrinology. 2003;144:4018–4030.
71. Kosugi, S, Okajima, F, Ban, T, et al. Mutation of Alanine 623 in the third cytoplasmic loop of the rat TSH receptor results in a loss in the phosphoinositide but not cAMP signal induced by TSH and receptor autoantibodies. J Biol Chem. 1992;267:24153–24156.
72. Parma, J, Van Sande, J, Swillens, S, et al. Somatic mutations causing constitutive activity of the thyrotropin receptor are the major cause of hyperfunctioning thyroid adenomas: identification of additional mutations activating both the cyclic adenosine 3′,5′-monophosphate and inositol phosphate-Ca2+ cascades. Mol Endocrinol. 1995;9:725–733.
73. Van Sande, J, Parma, J, Tonacchera, M, et al. Somatic and germline mutations of the TSH receptor gene in thyroid diseases. J Clin Endocrinol Metab. 1995;80:2577–2585.
74. Kjelsberg, MA, Cotecchia, S, Ostrowski, J, et al. Constitutive activation of the alpha 1B-adrenergic receptor by all amino acid substitutions at a single site, Evidence for a region which constrains receptor activation. J Biol Chem. 1992;267:1430–1433.
75. Lefkowitz, RJ, Cotecchia, S, Samama, P, et al. Constitutive activity of receptors coupled to guanine nucleotide regulatory proteins. TiPS. 1994;14:303–307.
76. Mustafi, D, Palczewski, K. Topology of class A G protein-coupled receptors: insights gained from crystal structures of rhodopsins, adrenergic and adenosine receptors. Mol Pharmacol. 2009;75:1–12.
77. Glinoer, D. The regulation of thyroid function in pregnancy: pathways of endocrine adaptation from physiology to pathology. Endocr Rev. 1997;18:404–433.
78. Grossmann, M, Weintraub, BD, Szkudlinski, MW. Novel insights into the molecular mechanisms of human thyrotropin action: structural, physiological, and therapeutic implications for the glycoprotein hormone family. Endocr Rev. 1997;18:476–501.
79. Rodien, P, Bremont, C, Sanson, ML, et al. Familial gestational hyperthyroidism caused by a mutant thyrotropin receptor hypersensitive to human chorionic gonadotropin. N Engl J Med. 1998;339:1823–1826.
80. Sanders, J, Chirgadze, DY, Sanders, P, et al. Crystal structure of the TSH receptor in complex with a thyroid-stimulating autoantibody. Thyroid. 2007;17:395–410.
81. Smits, G, Govaerts, C, Nubourgh, I, et al. Lysine 183 and glutamic acid 157 of the thyrotropin receptor: two interacting residues with a key role in determining specificity towards TSH and hCG. Mol Endocrinol. 2002;16:722–735.
82. Smits, G, Olatunbosun, O, Delbaere, A, et al. Ovarian hyperstimulation syndrome due to a mutation in the follicle-stimulating hormone receptor. N Engl J Med. 2003;349:760–766.
83. Vasseur, C, Rodien, P, Beau, I, et al. A chorionic gonadotropin-sensitive mutation in the follicle-stimulating hormone receptor as a cause of familial gestational spontaneous ovarian hyperstimulation syndrome. N Engl J Med. 2003;349:753–759.
84. Szkudlinski, MW, Fremont, V, Ronin, C, et al. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev. 2002;82:473–502.
85. Stein, SA, Oates, EL, Hall, CR, et al. Identification of a point mutation in the thyrotropin receptor of the hyt/hyt hypothyroid mouse. Mol Endocrinol. 1994;8:129–138.
86. Marians, RC, Ng, L, Blair, HC, et al. Defining thyrotropin-dependent and -independent steps of thyroid hormone synthesis by using thyrotropin receptor-null mice. Proc Natl Acad Sci U S A. 2002;99:15776–15781.
87. Postiglione, MP, Parlato, R, Rodriguez-Mallon, A, et al. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci U S A. 2002;99:15462–15467.
88. Codaccioni, JL, Carayon, P, Michel Bechet, M, et al. Congenital hypothyroidism associated with thyrotropin unresponsiveness and thyroid cell membrane alterations. J Clin Endocrinol Metab. 1980;50:932–937.
89. Stanbury, JB, Rocmans, P, Buhler, UK, et al. Congenital hypothyroidism with impaired thyroid response to thyrotropin. N Engl J Med. 1968;279:1132–1136.
90. Sunthornthepvarakul, T, Gottschalk, ME, Hayashi, Y, et al. Resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N Engl J Med. 1995;332:155–160.
91. Smits, G, Campillo, M, Govaerts, C, et al. Glycoprotein hormone receptors: determinants in leucine-rich repeats responsible for ligand specificity. EMBO J. 2003;22:2692–2703.
92. Kleinau, G, Krause, G. Thyrotropin and homologous glycoprotein hormone receptors: structural and functional aspects of extracellular signaling mechanisms. Endocr Rev. 2009;30:133–151.
93. Abramowicz, MJ, Duprez, L, Parma, J, et al. Familial congenital hypothyroidism due to inactivating mutation of the thyrotropin receptor causing profound hypoplasia of the thyroid gland. J Clin Invest. 1997;99:3018–3024.
94. Biebermann, H, Schoneberg, T, Krude, H, et al. Mutations of the human thyrotropin receptor gene causing thyroid hypoplasia and persistent congenital hypothyroidism. J Clin Endocrinol Metab. 1997;82:3471–3480.
95. Clifton-Bligh, RJ, Gregory, JW, Ludgate, M, et al. Two novel mutations in the thyrotropin (TSH) receptor gene in a child with resistance to TSH. J Clin Endocrinol Metab. 1997;82:1094–1100.
96. De Roux, N, Misrahi, M, Brauner, R, et al. Four families with loss of function mutations of the thyrotropin receptor. J Clin Endocrinol Metab. 1996;81:4229–4235.
97. Gagne, N, Parma, J, Deal, C, et al. Apparent congenital athyreosis contrasting with normal plasma thyroglobulin levels and associated with inactivating mutations in the thyrotropin receptor gene: are athyreosis and ectopic thyroid distinct entities? J Clin Endocrinol Metab. 1998;83:1771–1775.
98. Park, SM, Clifton-Bligh, RJ, Betts, P, et al. Congenital hypothyroidism and apparent athyreosis with compound heterozygosity or compensated hypothyroidism with probable hemizygosity for inactivating mutations of the TSH receptor. Clin Endocrinol (Oxf). 2004;60:220–227.
99. Bretones, P, Duprez, L, Parma, J, et al. A familial case of congenital hypothyroidism caused by a homozygous mutation of the thyrotropin receptor gene. Thyroid. 2001;11:977–980.
100. Jordan, N, Williams, N, Gregory, JW, et al. The W546X mutation of the thyrotropin receptor gene: potential major contributor to thyroid dysfunction in a Caucasian population. J Clin Endocrinol Metab. 2003;88:1002–1005.
101. Tonacchera, M, Agretti, P, Pinchera, A, et al. Congenital hypothyroidism with impaired thyroid response to thyrotropin (TSH) and absent circulating thyroglobulin: evidence for a new inactivating mutation of the TSH receptor gene. J Clin Endocrinol Metab. 2000;85:1001–1008.
102. Sura-Trueba, S, Aumas, C, Carre, A, et al. An inactivating mutation within the first extracellular loop of the thyrotropin receptor impedes normal posttranslational maturation of the extracellular domain. Endocrinology. 2009;150:1043–1050.
103. Yuan, ZF, Mao, HQ, Luo, YF, et al. Thyrotropin receptor and thyroid transcription factor-1 genes variant in Chinese children with congenital hypothyroidism. Endocr J. 2008;55:415–423.
104. Kanda, K, Mizuno, H, Sugiyama, Y, et al. Clinical significance of heterozygous carriers associated with compensated hypothyroidism in R450H, a common inactivating mutation of the thyrotropin receptor gene in Japanese. Endocrine. 2006;30:383–388.
105. Tsunekawa, K, Onigata, K, Morimura, T, et al. Identification and functional analysis of novel inactivating thyrotropin receptor mutations in patients with thyrotropin resistance. Thyroid. 2006;16:471–479.
106. Tenenbaum-Rakover, Y, Grasberger, H, Mamanasiri, S, et al. Loss-of-function mutations in the thyrotropin receptor gene as a major determinant of hyperthyrotropinemia in a consanguineous community. J Clin Endocrinol Metab. 2009;94:1706–1712.
107. Alberti, L, Proverbio, MC, Costagliola, S, et al. Germline mutations of TSH receptor gene as cause of nonautoimmune subclinical hypothyroidism. J Clin Endocrinol Metab. 2002;87:2549–2555.
108. Xie, J, Pannain, S, Pohlenz, J, et al. Resistance to thyrotropin (TSH) in three families is not associated with mutations in the TSH receptor or TSH [see comments]. J Clin Endocrinol Metab. 1997;82:3933–3940.
109. Grasberger, H, Vaxillaire, M, Pannain, S, et al. Identification of a locus for nongoitrous congenital hypothyroidism on chromosome 15q25.3–26.1. Hum Genet. 2005;118:348–355.
110. Bohr, UR, Behr, M, Loos, U. A heritable point mutation in an extracellular domain of the TSH receptor involved in the interaction with Graves’ immunoglobulins. Biochim Biophys Acta. 1993;1216:504–508.
111. Bahn, RS, Dutton, CM, Heufelder, AE, et al. A genomic point mutation in the extracellular domain of the thyrotropin receptor in patients with Graves’ ophthalmopathy. J Clin Endocrinol Metab. 1994;78:256–260.
112. Gabriel, EM, Bergert, ER, Grant, CS, et al. Germline polymorphism of codon 727 of human thyroid-stimulating hormone receptor is associated with toxic multinodular goiter. J Clin Endocrinol Metab. 1999;84:3328–3335.
113. Ban, Y, Greenberg, DA, Concepcion, ES, et al. A germline single nucleotide polymorphism at the intracellular domain of the human thyrotropin receptor does not have a major effect on the development of Graves’ disease. Thyroid. 2002;12:1079–1083.
114. Muhlberg, T, Herrmann, K, Joba, W, et al. Lack of association of nonautoimmune hyperfunctioning thyroid disorders and a germline polymorphism of codon 727 of the human thyrotropin receptor in a European Caucasian population. J Clin Endocrinol Metab. 2000;85:2640–2643.
115. Simanainen, J, Kinch, A, Westermark, K, et al. Analysis of mutations in exon 1 of the human thyrotropin receptor gene: high frequency of the D36H and P52T polymorphic variants. Thyroid. 1999;9:7–11.
116. Ho, SC, Goh, SS, Khoo, DH. Association of Graves’ disease with intragenic polymorphism of the thyrotropin receptor gene in a cohort of Singapore patients of multi-ethnic origins. Thyroid. 2003;13:523–528.
117. Brand, OJ, Barrett, JC, Simmonds, MJ, et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves’ disease (GD). Hum Mol Genet. 2009;18:1704–1713.
118. Burton, PR, Clayton, DG, Cardon, LR, et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat Genet. 2007;39:1329–1337.
119. Libert, F, Lefort, A, Gerard, C, et al. Cloning, sequencing and expression of the human thyrotropin (TSH) receptor: evidence for binding of autoantibodies. Biochem Biophys Res Commun. 1989;165:1250–1255.
120. Nagayama, Y, Kaufman, KD, Seto, P, et al. Molecular cloning, sequence and functional expression of the cDNA for the human thyrotropin receptor. Biochem Biophys Res Commun. 1989;165:1184–1190.
121. Arseven, OK, Wilkes, WP, Jameson, JL, et al. Substitutions of tyrosine 601 in the human thyrotropin receptor result in increase or loss of basal activation of the cyclic adenosine monophosphate pathway and disrupt coupling to Gq/11. Thyroid. 2000;10:3–10.