Chapter 10 Thrombosis
Overview of Thrombosis
When the endothelial surface becomes damaged, however, release of many procoagulant proteins (especially tissue factor) and activation of platelets result in uncontrolled hemostasis at the site of vascular injury.1 As the thrombus begins to form, it recruits additional platelets to the area, leading to further platelet activation. Initially, tethering of platelets is dependent upon exposure of glycoprotein (Gp)Ib-V-IX in damaged collagen, which binds to von Willebrand factor (vWF), resulting in adhesion of platelets to the area of injury. Further recruitment of platelets is mediated through activation of the GPIIb-IIa platelet receptor, which undergoes a conformational change leading to increased affinity for fibrinogen. These events culminate with further platelet activation that results in release of many essential components for thrombus formation, including adenosine diphosphate (ADP), serotonin, and thromboxane A 2 (TxA2).
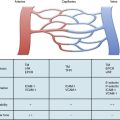
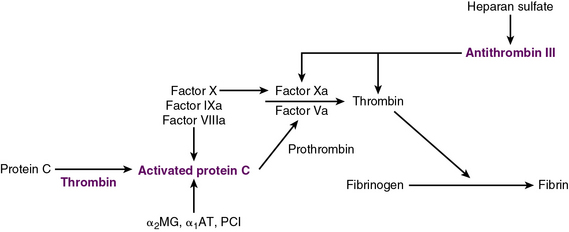
Exposure of vascular collagen also leads to activation of the normal mechanisms of hemostasis—including the coagulation cascade—through exposure of tissue factor, leading to “hemostasis in the wrong place.” The coagulation regulatory system is outlined in Figure 10-1 and discussed in detail in Chapter 5. Briefly, both the tissue factor–mediated pathway (extrinsic) and the contact-mediated pathway (instrinsic pathway) rely on activation of inactive enzyme precursors of serine proteases, which then reflexively lead to activation of another protein within the cascade. The ultimate step results in cross-linking of fibrin to stabilize a platelet plug, leading to thrombus formation. The tissue factor–initiated pathway is essential for thrombus formation. When tissue factor is released during cellular injury, factor VII is activated and complexes. This complex next activates factors X and XI. Activation of factor X is essential for conversion of prothrombin (factor II) to thrombin through the prothrombinase complex on activated platelets. This cascade of coagulation proteins is essential for hemostasis but also can have deleterious affects when it occurs unregulated, leading to unwanted thrombotic complications.
Platelets, Thrombosis, and Vascular Disease
Venous Thrombosis
Genetic risk factors associated with increased risk of VTE include mutations in factor V (Leiden) and prothrombin 20210, as well as mutations leading to deficiencies in antithrombin, protein C, and protein S. Approximately 5% of the Caucasian population has at least one mutation for factor V Leiden, and 15% to 20% of patients who present with a VTE carry the mutation.2–5 Approximately 2% of the population carry the prothrombin gene mutation, but it may be present in approximately 5% to 15% of persons with VTE.6 The population frequencies of mutations in other genes responsible for other coagulation factors (e.g., protein C) are estimated to be 1 in 500 individuals. Antithrombin III deficiency is associated with a frequency of 1 in 300 in the general population, and in 3% to 5% of those with thrombotic events. Previously it was thought that genetic mutations in genes important for methylene tetrahydrofolate reductase and hyperhomocysteinemia increased the risk of VTEs; however, recently this association has been shown to be less likely.7
One of the acquired risk factors known to be important in both venous and arterial thrombosis is acquisition of antiphospholipid antibodies, which represent a family of antibodies against phospholipids (e.g., cardiolipins) and phospholipid binding proteins (e.g., GpI β2). Mechanisms responsible for thrombosis are still speculative but may include inhibition of protein C, antithrombin, and annexin A5 expression; binding and activation of platelets; enhanced EC tissue factor expression; and activation of the complement cascade.8 Criteria for diagnosis of the associated disorder, antiphospholipid syndrome, includes the presence of both clinical events and laboratory evidence for the presence of antiphospholipid antibodies.9
Arterial Thrombosis
A primary mechanism of arterial thrombosis is rupture of atherosclerotic plaques, precipitating platelet-rich aggregates. Arterial thrombosis can have catastrophic consequences when it occurs in the coronary or carotid artery circulation. Factors that can exacerbate these types of thrombotic events include smoking, diabetes, hypertension, and hyperlipidemia. Thrombosis generally occurs when there is disruption in the hemostatic balance that results when pro- and anticoagulant molecules are at disequilibrium. Endothelial damage shifts this balance towards a more procoagulant force, leading to exposure of collagen and tissue factor. Collagen that is now exposed can activate platelets in the blood flowing through the vessel, and concomitantly thrombin is generated as the coagulation cascade is initiated in the presence of tissue factor. Genetic modifications of proteins important in coagulation can alter this process, creating a propensity to form thrombi in the arterial system (Box 10-1). These mutations affect platelet function, leading to increased propensity to aggregation.
 Box 10-1
Box 10-1Polymorphisms in the endothelial nitric oxide synthase (eNOS) gene, which is essential for NO production by ECs, have been described. The 894-G/T polymorphism I exon 7 results in a glutamate-to-aspartate change at position 298. This polymorphism is associated with increased levels of nitrogen oxides that increase risk of hypertension, MI, and stroke in patients who are homozygous for the abnormality.10,11 A unique polymorphism in the promoter of the glutathione peroxidase-3 gene has been associated with thrombotic strokes in children. Genome-wide associations have identified other loci associated with cardiovascular thrombotic disease.12
Increased levels of fibrinogen have been associated with an increased risk of MI, ischemic stroke, and peripheral artery disease.13 Age, elevated lipids, and smoking increase the risk associated with fibrinogen. β-Chain variants, such as Arg448Lys, BclI, -148 C/T, -455 G/A, and -854 G/A, with the -455 G/A polymorphism, is present in 10% to 20% of the population and is associated with a significant rise in fibrinogen levels.14 Studies, however, have not been consistent, and the association between this polymorphism and risk of arterial thrombosis is not established.15 Another site of potential polymorphisms in the fibrinogen gene is the Thr312Ala substitution in the α chain. When this polymorphism is present, the fibrin stranding is thicker, and there is increased cross-linking that may predispose to an increased thrombotic risk.16
Other potential associations between arterial thrombosis and increased risk include hyperhomocysteinemia, elevated C-reactive protein (CRP), factor VII polymorphisms, increased plasminogen activator inhibitor (PAI)-1, and platelet hyperreactivity. Wald et al. performed a meta-analysis of 72 prospective cohort studies focusing on a mutation in the methylenetetrahydrofolate reductase (MTHFR) C677T gene and the occurrence of various thrombotic events, including stroke and cardiovascular disease, and found a mild association between the mutation and risk of arterial events.17 Other factors including CRP, factor VII, and PAI-1 have shown even less promising results.14
Drugs that modulate arterial thrombosis
Use of aspirin to prevent arterial thrombosis was first established in the ISIS-2 trial in which aspirin was shown to reduce the mortality rate associated with MI. Other studies showed that aspirin resulted in a 25% relative risk reduction from all vascular-associated events, including MI and stroke, and the benefit occurred with treatment with low-dose aspirin.18 Although the half-life of aspirin is only 20 minutes, and inhibitory effects of aspirin on COX occur as quickly as 5 minutes after administration, irreversible inhibition of COX ensures that its effects are preserved for the lifespan of the platelet (7-10 days) such that COX activity does not return to normal levels until a new generation of platelets is produced. Interestingly, aspirin’s inhibitory properties appear to be most effective when exposed to weak platelet agonists (e.g., TxA, ADP), whereas exposure to stronger agonists (e.g., thrombin) leaves platelet function, as measured by aggregation, intact. For this reason, many of the essential functions of platelets, such as platelet adhesion to vWF or activation by thrombin, are not inhibited by aspirin. Aspirin resistance may explain some of the clinical failure seen with its use. Two recent meta-analyses regarding aspirin resistance have shown that laboratory evidence of unresponsiveness to aspirin may be associated with a high risk of recurrent thrombotic cardiovascular events.19,20
Another important modulator of platelet function is ADP, which acts as a weak platelet agonist through two different platelet membrane receptors, P2Y1 and P2Y12.21 When ADP stimulates the P2Y1 receptor, the platelet undergoes a shape change and Ca2 + is mobilized through activation of phospholipase C to initiate platelet aggregation in a reversible manner. The P2Y12 receptor is essential for secretion and stabilization of platelet aggregation by lowering cAMP levels.
There are two available thienopyridine derivatives that act as inhibitors of ADP-induced platelet aggregation: ticlopidine and clopidogrel. Clopidogrel is metabolized by cytochrome P450 (CYP450) into an active metabolite that irreversibly blocks the P2Y12 receptor. Clopidogrel reduces recurrent thrombotic events in patients with cardiovascular disease.22–24 The Clopidogrel vs. Aspirin in Patients at Risk of Ischemic Events (CAPRIE) trial found that clopidogrel is more effective than aspirin in reducing the risk of cardiovascular events in patients with recent MI, recent ischemic stroke, and established peripheral artery disease. Ticlopidine is also metabolized by CYP450 to an active metabolite that functions to block the PGY12 receptor. Although it functions in a similar manner to clopidogrel, it is associated with a higher degree of neutropenia and thrombotic thrombocytopenic purpura and is therefore not used as readily as clopidogrel.25,26 Other antiplatelet drugs in clinical development include ticagrelor, cangrelor, and elinogrel, all of which reversibly inhibit the P2Y12 receptor.27
These drugs work on the final common pathway of platelet aggregation to inhibit binding to fibrinogen in a similar manner to abciximab. They also appear to have anticoagulant activity because there is evidence of prolongation of the activated clotting time. These actions are thought to be regulated by inhibition of thrombin generation via tissue factor and a decrease in microparticle formation. Multiple clinical trials have shown that inhibition of GpIIb-IIIa is effective in preventing recurrent thrombotic events. Use of these drugs leads to a 35% decrease in acute ischemic events and a 26% decrease in recurrent events within 6 months. Long-term use of these drugs was shown to be efficacious in the EPILOG (Evaluation of PTCA to Improve Long-Term Outcome by c7E3 GpIIb-IIIa Receptor Blockade) trial, with a reduction in the incidence of death. Other trials have supported use of GpIIb-IIIa blockade in management of acute coronary syndromes (ACSs).28,29 Their use, however, has been reserved for high-risk circumstances such as percutaneous coronary intervention (PCI) in ACSs, owing to the recent finding of long-term benefits of ADP receptor antagonists. 30
Oral GpIIb-IIIa inhibitors have not been shown to limit cardiovascular events to date.31 This may be due to conformational changes in the GpIIb-IIIa receptor after antagonist dissociation from it. In this case, the receptor remains active and increases binding to fibrinogen and vWF, leading to a paradoxical thrombotic effect. Novel GpIIb-IIIa antagonists that do cause such conformational changes are under development. In one study, RUC-1, which is a novel compound discovered through high-throughput screening, induced partial exposure of the binding site yet still led to decreased platelet aggregation without enhanced fibrinogen binding. RUC-1 may represent a prototype molecule for these types of derivative drugs.32
Other antiplatelet agents that may inhibit thrombus formation yet preserve hemostasis so bleeding complications do not result are under investigation. One potential new therapeutic target is the GPIb-V-IX complex. Initial studies of patients who have Bernard-Soulier’s syndrome identified a deficiency of the GPIb complex. The GPIb complex is important for building a platelet bridge through vWF at areas of endothelial damage. Drugs under development act as antagonists for the GPIb-vWF interaction, including specific monoclonal antibodies, the GpIb complex antagonists isolated from snake venoms,33,34 and the Fab fragment of 6B4, which is a murine monoclonal antibody that targets human GPIb and prevents binding to vWF. Early nonhuman primate studies have suggested that thrombus formation can be attenuated using these drugs.35
Inflammation and Thrombosis
Recent evidence has clearly established a role for inflammation in the atherothrombotic process. Patients with ACSs have increased interactions between platelets and leukocytes forming detectable aggregates. The process of inflammation involves a variety of cell types, including leukocytes, ECs, and platelets. The endothelium becomes activated, and multiple cell-adhesion molecules are released, including P-selectin, which essentially has two important roles in inflammation: recruitment of proinflammatory cells and establishing signaling cascades leading to increased expression of CD11b/CD18 (MAC-1).36
Platelets are instrumental in the inflammatory aspects of atherosclerosis. Thrombin activation of platelets leads to release of many procoagulant molecules and release of inflammatory molecules, including platelet factor 4 (PF4), platelet-derived growth factor (PDGF), and RANTES, which is regulated upon activation of normal T-cell expression. In addition, platelets that have been activated in the presence of thrombin secrete CD40 ligand (CD40L). This chemoattractant is key to the recruitment of ECs, smooth muscle cells (SMCs), and macrophages. CD40 ligand also recruits a variety of proinflammatory cytokines (e.g., interleukin [IL]-1, IL-6, and IL-8) and increases expression of the adhesion molecules intercellular adhesion molecule (ICAM)-1, vascular (V)CAM-1, and P-selectin.37 CD40 ligand is also important for release of matrix metalloproteinases (MMPs), which are needed for plaque progression, neovascularization, and plaque rupture. CD40 ligand also initiates release of tissue factor, which then interacts with other cells to create a thrombogenic microenvironment. T lymphocytes orchestrate an inflammatory cascade that begins by binding to VCAM-1 through signaling regulated by interferon (IFN)-γ-inducible chemokine ligands (CXCLs), protein-10, and chemoattractant (I-TAC). Through this binding, a number of inflammatory cytokines are released, including the CD40 ligand; CD154, which leads to metalloproteinase generation; and tissue factor expression, which initiates the coagulation cascade. Mice that lack the CD40L have less atherothrombosis. Patients with ACSs have elevated levels of CD40L, and plasma levels of CD40L predict the risk of future cardiovascular events.38–40 It has been shown that elevated soluble CD40L levels can be decreased by treatment with abciximab.41
Our understanding of the role of inflammation in the thrombotic response has been expanded by many clinical studies that have shown an association between bacterial infections and increased risk of MI or stroke, although more studies are needed to prove this association.42,43 One possible mechanism is through Toll-like receptors (TLRs), which are present in platelets. Platelet activation through stimulation of TLR2 activates signaling mechanisms responsible for both thrombotic and inflammatory responses. These effects are responsible for the mechanism by which bacteria induce a proinflammatory cascade in platelets, suggesting that bacteria can directly activate platelet-dependent thrombotic responses.44 Recently this process was further refined by demonstrating that TLR2 stimulation leads to platelet activation through PI3-kinase, which is known to be important in platelet activation–associated shape change, calcium release, and granular content secretion.45
1 Alfirevic Z, Alfirevic I. Hypercoagulable state, pathophysiology, classification and epidemiology. Clin Chem Lab Med. 2010;48(Suppl 1):S15–S26.
2 Prandoni P. Acquired risk factors for venous thromboembolism in medical patients. Hematology Am Soc Hematol Educ Program. 2005:458–461.
3 Rosendaal F.R. Venous thrombosis: the role of genes, environment, and behavior. Hematology Am Soc Hematol Educ Program. 2005:1–12.
4 Cushman M. Inherited risk factors for venous thrombosis. Hematology Am Soc Hematol Educ Program. 2005:452–457.
5 Vossen C.Y., Conard J., Fontcuberta J., et al. Risk of a first venous thrombotic event in carriers of a familial thrombophilic defect. The European Prospective Cohort on Thrombophilia (EPCOT). J Thromb Haemost. 2005;3(3):459–464.
6 Kottke-Marchant K. Genetic polymorphisms associated with venous and arterial thrombosis: an overview. Arch Pathol Lab Med. 2002;126(3):295–304.
7 Ray J.G. Hyperhomocysteinemia: no longer a consideration in the management of venous thromboembolism. Curr Opin Pulm Med. 2008;14(5):369–373.
8 Lim W. Antiphospholipid antibody syndrome. Hematology Am Soc Hematol Educ Program. 2009:233–239.
9 Cohen D., Berger S.P., Steup-Beekman G.M., et al. Diagnosis and management of the antiphospholipid syndrome. BMJ. 2010;340:c2541.
10 Elbaz A., Poirier O., Moulin T., et al. Association between the Glu298Asp polymorphism in the endothelial constitutive nitric oxide synthase gene and brain infarction. The GENIC Investigators. Stroke. 2000;31(7):1634–1639.
11 Hingorani A.D., Liang C.F., Fatibene J., et al. A common variant of the endothelial nitric oxide synthase (Glu298–>Asp) is a major risk factor for coronary artery disease in the UK. Circulation. 1999;100(14):1515–1520.
12 Malarstig A., Hamsten A. Genetics of atherothrombosis and thrombophilia. Curr Atheroscler Rep. 2010;12(3):159–166.
13 Scarabin P.Y., Arveiler D., Amouyel P., et al. Plasma fibrinogen explains much of the difference in risk of coronary heart disease between France and Northern Ireland. The PRIME study. Atherosclerosis. 2003;166(1):103–109.
14 Voetsch B., Loscalzo J. Genetic determinants of arterial thrombosis. Arterioscler Thromb Vasc Biol. 2004;24(2):216–229.
15 Endler G., Mannhalter C. Polymorphisms in coagulation factor genes and their impact on arterial and venous thrombosis. Clin Chim Acta. 2003;330(1–2):31–55.
16 Standeven K.F., Grant P.J., Carter A.M., et al. Functional analysis of the fibrinogen Aalpha Thr312Ala polymorphism: effects on fibrin structure and function. Circulation. 2003;107(18):2326–2330.
17 Wald D.S., Law M., Morris J.K. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ. 2002;325(7374):1202.
18 Jneid H., Bhatt D.L. Advances in antiplatelet therapy. Expert Opin Emerg Drugs. 2003;8(2):349–363.
19 Snoep J.D., Hovens M.M., Eikenboom J.C., et al. Association of laboratory-defined aspirin resistance with a higher risk of recurrent cardiovascular events: a systematic review and meta-analysis. Arch Intern Med. 2007;167(15):1593–1599.
20 Krasopoulos G., Brister S.J., Beattie W.S., et al. Aspirin “resistance” and risk of cardiovascular morbidity: systematic review and meta-analysis. BMJ. 2008;336(7637):195–198.
21 Daniel J.L., Dangelmaier C., Jin J., et al. Role of intracellular signaling events in ADP-induced platelet aggregation. Thromb Haemost. 1999;82(4):1322–1326.
22 Yusuf S., Zhao F., Mehta S.R., et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494–502.
23 Mehta S.R., Yusuf S., Peters R.J., et al. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCI-CURE study. Lancet. 2001;358(9281):527–533.
24 Steinhubl S.R., Berger P.B., Mann J.T.3rd, et al. Early and sustained dual oral antiplatelet therapy following percutaneous coronary intervention: a randomized controlled trial. JAMA. 2002;288(19):2411–2420.
25 Michelson A.D. P2Y12 antagonism: promises and challenges. Arterioscler Thromb Vasc Biol. 2008;28(3):s33–s38.
26 Bertrand M.E., Rupprecht H.J., Urban P., et al. Double-blind study of the safety of clopidogrel with and without a loading dose in combination with aspirin compared with ticlopidine in combination with aspirin after coronary stenting: the clopidogrel aspirin stent international cooperative study (CLASSICS). Circulation. 2000;102(6):624–629.
27 Michelson A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat Rev Drug Discov. 2010;9(2):154–169.
28 van ’t Hof A.W., Valgimigli M. Defining the role of platelet glycoprotein receptor inhibitors in STEMI: focus on tirofiban. Drugs. 2009;69(1):85–100.
29 Tamhane U.U., Gurm H.S. GP IIb/IIIa inhibitors during primary percutaneous coronary intervention for STEMI: new trial and registry data. Curr Cardiol Rep. 2008;10(5):424–430.
30 Mukherjee D., Roffi M. Glycoprotein IIb/IIIa receptor inhibitors in 2008: do they still have a role? J Interv Cardiol. 2008;21(2):118–121.
31 Chew D.P., Bhatt D.L., Sapp S., et al. Increased mortality with oral platelet glycoprotein IIb/IIIa antagonists: a meta-analysis of phase III multicenter randomized trials. Circulation. 2001;103(2):201–206.
32 Blue R., Murcia M., Karan C., et al. Application of high-throughput screening to identify a novel alphaIIb-specific small-molecule inhibitor of alphaIIbbeta3-mediated platelet interaction with fibrinogen. Blood. 2008;111(3):1248–1256.
33 Chang M.C., Lin H.K., Peng H.C., et al. Antithrombotic effect of crotalin, a platelet membrane glycoprotein Ib antagonist from venom of Crotalus atrox. Blood. 1998;91(5):1582–1589.
34 Yeh C.H., Chang M.C., Peng H.C., et al. Pharmacological characterization and antithrombotic effect of agkistin, a platelet glycoprotein Ib antagonist. Br J Pharmacol. 2001;132(4):843–850.
35 Fontayne A., Meiring M., Lamprecht S., et al. The humanized anti-glycoprotein Ib monoclonal antibody h6B4-Fab is a potent and safe antithrombotic in a high shear arterial thrombosis model in baboons. Thromb Haemost. 2008;100(4):670–677.
36 Neumann F.J., Zohlnhofer D., Fakhoury L., et al. Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J Am Coll Cardiol. 1999;34(5):1420–1426.
37 Schonbeck U., Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci. 2001;58(1):4–43.
38 Aukrust P., Muller F., Ueland T., et al. Enhanced levels of soluble and membrane-bound CD40 ligand in patients with unstable angina. Possible reflection of T lymphocyte and platelet involvement in the pathogenesis of acute coronary syndromes. Circulation. 1999;100(6):614–620.
39 Schonbeck U., Varo N., Libby P., et al. Soluble CD40L and cardiovascular risk in women. Circulation. 2001;104(19):2266–2268.
40 Varo N., de Lemos J.A., Libby P., et al. Soluble CD40L: risk prediction after acute coronary syndromes. Circulation. 2003;108(9):1049–1052.
41 Freedman J.E. CD40 ligand–assessing risk instead of damage? N Engl J Med. 2003;348(12):1163–1165.
42 Fagoonee S., De Angelis C., Elia C., et al. Potential link between Helicobacter pylori and ischemic heart disease: does the bacterium elicit thrombosis? Minerva Med. 2010;101(2):121–125.
43 Stassen F.R., Vainas T., Bruggeman C.A. Infection and atherosclerosis. An alternative view on an outdated hypothesis. Pharmacol Rep. 2008;60(1):85–92.
44 Balogh S., Kiss I., Csaszar A. Toll-like receptors: link between “danger” ligands and plaque instability. Curr Drug Targets. 2009;10(6):513–518.
45 Rex S., Beaulieu L.M., Perlman D.H., et al. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb Haemost. 2009;102(1):97–110.