Challenges to NGS Analysis of Cancer Nucleic Acids
Tumor Cellularity
Heterogeneity (Regional versus Genotypic)
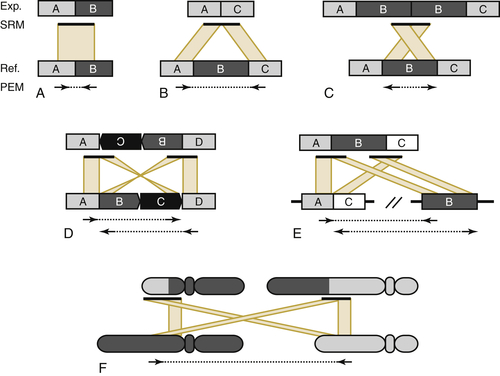
Ploidy and Copy Number Alterations in DNA
FFPE Preservation and Nucleic Acid Integrity
Applications of NGS to Study and Analyze Nucleic Acids
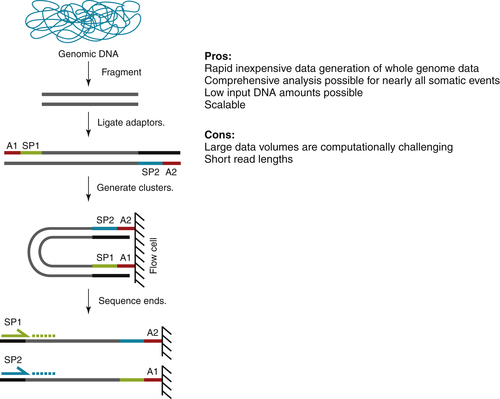
Whole-Genome Sequencing
Exome Sequencing
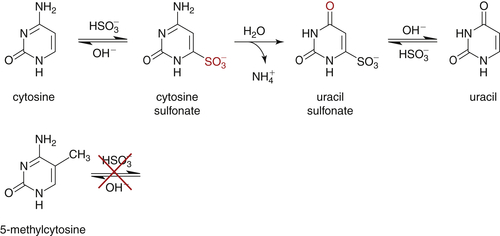
DNA Methylation
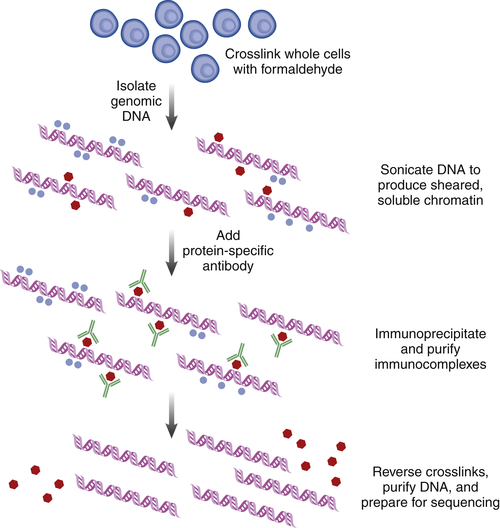
Chromatin Immunoprecipitation
Sequencing Messenger RNA (mRNA)
Sequencing Noncoding RNAs
Conclusions
1. A decade’s perspective on DNA sequencing technology . Nature . 2011 ; 470 : 198 – 203 .
2. Nucleotide sequence of bacteriophage phi X174 DNA . Nature . 1977 ; 265 : 687 – 695 .
3. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase . J Mol Biol . 1975 ; 94 : 441 – 448 .
4. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing . J Mol Biol . 1980 ; 143 : 161 – 178 .
5. DNA sequencing with chain-terminating inhibitors . Proc Natl Acad Sci U S A . 1977 ; 74 : 5463 – 5467 .
6. . Finishing the euchromatic sequence of the human genome . Nature . 2004 ; 431 : 931 – 945 .
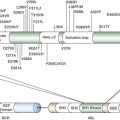
7. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia . Lancet . 1977 ; 1 : 549 – 550 .
8. The cytogenetics of acute leukaemia . Clin Haematol . 1978 ; 7 : 385 – 406 .
9. Chromosome abnormalities in leukemia . Haematol Blood Transfus . 1979 ; 23 : 43 – 52 .
10. Ph1-positive leukaemia, including chronic myelogenous leukaemia . Clin Haematol . 1980 ; 9 : 55 – 86 .
11. Identification of the constant chromosome regions involved in human hematologic malignant disease . Science . 1982 ; 216 : 749 – 751 .
12. Biological implications of consistent chromosome rearrangements in leukemia and lymphoma . Cancer Res . 1984 ; 44 : 3159 – 3168 .
13. The Philadelphia chromosome translocation. A paradigm for understanding leukemia . Cancer . 1990 ; 65 : 2178 – 2184 .
14. The role of chromosome translocations in leukemogenesis . Semin Hematol . 1999 ; 36 : 59 – 72 .
15. The consensus coding sequences of human breast and colorectal cancers . Science . 2006 ; 314 : 268 – 274 .
16. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib . Proc Natl Acad Sci U S A . 2004 ; 101 : 13306 – 13311 .
17. Cancer genome sequencing: a review . Hum Mol Genet . 2009 ; 18 : R163 – R168 .
18. Mapping short DNA sequencing reads and calling variants using mapping quality scores . Genome Res . 2008 ; 18 : 1851 – 1858 .
19. Fast and accurate long-read alignment with Burrows-Wheeler transform . Bioinformatics . 2010 ; 26 : 589 – 595 .
20. Complete Khoisan and Bantu genomes from southern Africa . Nature . 2010 ; 463 : 943 – 947 .
21. International network of cancer genome projects . Nature . 2010 ; 464 : 993 – 998 .
22. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing . Nature . 2012 ; 481 : 506 – 510 .
23. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing . N Engl J Med . 2012 ; 366 : 883 – 892 .
24. Whole-genome analysis informs breast cancer response to aromatase inhibition . Nature . 2012 ; 486 : 353 – 360 .
25. CMDS: a population-based method for identifying recurrent DNA copy number aberrations in cancer from high-resolution data . Bioinformatics . 2010 ; 26 : 464 – 469 .
26. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma . Proc Natl Acad Sci U S A . 2007 ; 104 : 20007 – 20012 .
27. The life history of 21 breast cancers . Cell . 2012 ; 149 : 994 – 1007 .
28. Biochemical analysis of damage induced in yeast by formaldehyde. I. Induction of single-strand breaks in DNA and their repair . Mutat Res . 1978 ; 50 : 181 – 193 .
29. SomaticSniper: identification of somatic point mutations in whole genome sequencing data . Bioinformatics . 2012 ; 28 : 311 – 317 .
30. Distant metastasis occurs late during the genetic evolution of pancreatic cancer . Nature . 2010 ; 467 : 1114 – 1117 .
31. Direct selection of human genomic loci by microarray hybridization . Nat Methods . 2007 ; 4 : 903 – 905 .
32. Genome-wide in situ exon capture for selective resequencing . Nat Genet . 2007 ; 39 : 1522 – 1527 .
33. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing . Nat Biotechnol . 2009 ; 27 : 182 – 189 .
34. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications . Nat Biotechnol . 2010 ; 28 : 1097 – 1105 .
35. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands . Proc Natl Acad Sci U S A . 1992 ; 89 : 1827 – 1831 .
36. Human DNA methylomes at base resolution show widespread epigenomic differences . Nature . 2009 ; 462 : 315 – 322 .
37. Chromatin profiling by directly sequencing small quantities of immunoprecipitated DNA . Nat Methods . 2010 ; 7 : 47 – 49 .
38. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals . Mol Cell . 2012 ; 47 : 810 – 822 .
39. Mapping and quantifying mammalian transcriptomes by RNA-Seq . Nat Methods . 2008 ; 5 : 621 – 628 .
40. Computational methods for transcriptome annotation and quantification using RNA-seq . Nat Methods . 2011 ; 8 : 469 – 477 .
41. Next-generation genomics: an integrative approach . Nat Rev Genet . 2010 ; 11 : 476 – 486 .
42. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review . EMBO Mol Med . 2012 ; 4 : 143 – 159 .
43. The emergence of lncRNAs in cancer biology . Cancer Discov . 2011 ; 1 : 391 – 407 .