Chapter 15 The respiratory system
Long Cases
Asthma
Recent advances in management have included improved understanding of the importance of gene by environment interactions, and of the underlying pathophysiology of asthma. The effects of exposure to tobacco smoke, the most important environmental factor that can adversely affect the asthmatic population, are now known to be largely determined by whether children have a particular glutathione-S-transferase (GSTM1) genotype. Adverse effects (increased risk of development of asthma and wheezing) after in utero exposure to tobacco products are determined largely by the GSTM1 null genotype being present. An increased prevalence of asthma related phenotypes does not occur in children exposed in utero to tobacco smoke who possess the GSTM1+ genotype. Although several genes have been reported to be associated with various asthma phenotypes, no single gene has emerged as responsible for asthma susceptibility.
History
Symptoms
Dyspnoea, wheeze, cough, exercise tolerance (last in races?), nocturnal symptoms (cough, wheeze, wakening), morning symptoms (tightness, wheeze), use of bronchodilators, viral upper respiratory tract infections, cyanosis, syncope. Provide a detailed expansion of important or specific symptoms; for example, for cough, note duration, nature (e.g. productive/loose), frequency, timing (day/night), effects (vomiting, awakening, family disruption), sputum (amount, colour, blood), associated symptoms (fever, wheeze, shortness of breath, symptoms of allergic rhinitis, upper airway obstructive symptoms such as snoring and obstructive sleep apnoea), and responsiveness to beta-2 agonists/antibiotics.
Examination
Respiratory examination
1. Hands: tremor (beta-2 agonists).
2. Chest: deformity, increased anteroposterior diameter, Harrison’s sulcus, expansion, tracheal position, apex position, palpable pulmonary valve closure, right-ventricular overactivity, percussion, auscultation. If age appropriate, peak flow (ideally before and after nebulised salbutamol; impractical in exams).
3. ENT: ears—serous otitis media; nose—allergic rhinitis; pale, swollen, nasal mucosa, visible inferior turbinates, green/clear discharge; throat—tonsillar size, redness or exudate. Cervical nodes: lymphadenopathy.
Diagnosis and investigations
Generally a clinical diagnosis; on occasion, some investigations may be warranted.
Other investigations
Provocation inhalation challenge testing (mannitol, hypertonic saline) may be useful to confirm airway hyperresponsiveness in cases with diagnostic difficulty, recurrent cough or recurrent breathlessness or exercise-induced dyspnoea. Exercise testing may also be useful to elucidate the cause of exercise-induced dyspnoea. Tests to exclude other diagnoses that may present with cough or wheeze are as follows: (a) to exclude cystic fibrosis (CF)—sweat test, CF genotype; (b) to exclude alpha-1-antitrypsin deficiency—alpha1-antitrypsin phenotype; (c) to exclude infection—sputum microscopy and culture, nasopharyngeal aspirate for viral pathogens; (d) to exclude immune deficiency states—full blood count, T-cell subsets, immunoglobulins, including IgG subclasses, vaccine antibody responses, complement levels, HIV testing; (e) to exclude structural airway disease—bronchoscopy, endobronchial ultrasound, CT 3D reconstruction; may detect congenital tracheomalacia or bronchomalacia, especially if wheeze and hyperinflation commenced early in life, and are unresponsive to anti-asthma treatment; (f) to exclude parenchymal pathology such as bronchiectasis or congenital structural lung lesions—high-resolution CT (HRCT) scan of thorax; (g) to exclude mediastinal or vascular lesions (e.g. vascular ring)—helical CT scan or MRI with vascular reconstruction; (h) to exclude occult cardiovascular disease—electrocardiogram.
Treatment
Acute
1. Position. Sit child up, for ease of chest expansion and diaphragmatic excursion.
2. Oxygen. All children with acute severe asthma are hypoxic. Always check pulse oximetry. The aim is maximum inspired oxygen; keep the SaO2 above 90%.
3. Beta-2 agonists (e.g. salbutamol). Nebulised for severe, life-threatening asthma; for mild and moderate asthma, pMDI with spacer. For very severe asthma, continuous nebulised therapy (dose 0.3 mg/kg per hour; prevents rebound bronchospasm); for moderately severe cases, either intermittent nebulised therapy (dose 0.15 mg/kg per dose [to maximum of 5 mg] or 6–12 puffs pMDI via spacer, every 20 minutes, initially). If nebulised therapy is needed, the optimum volume of drug in the ‘acorn’ of the nebuliser is 4 mL, with the driving oxygen rate being 8 L/min; can give with ipratropium bromide (see below).
4. Intravenous beta-2 agonists. If nebulised therapy is not working, when inspiratory flow rates are very low, or the need for high-flow oxygen precludes nebuliser. An initial salbutamol bolus is followed by an infusion, incrementally increased until there is a good response. Toxicities: hypokalaemia, tachyarrhythmias, metabolic acidosis.
5. Nebulised anticholinergics. Ipratropium bromide augments the actions of beta-2 agonists. The respiratory solution concentration is 250 mcg/mL, and the dose is 0.25–1 mL every 4–8 hours; it can be given (with salbutamol) every 20 minutes for three doses initially, then continued at 4–6 hour intervals.
6. Corticosteroids (CS) (oral prednisolone, IV hydrocortisone or methylprednisolone). Used in all moderate to severe episodes; decreases morbidity. High dose for 3 days, then stop. The treatment for any longer duration should be slowly weaned, the main reason to wean is to prevent rebound in those with more persistent asthma; it is now accepted that oral steroids can be given for up to 2 weeks without the need to wean to avoid adrenal suppression. Recent studies have failed to show any additional benefit of oral steroids in preschool children with mild to moderate viral-induced wheezing.
7. Intravenous magnesium sulphate. An initial bolus of magnesium sulphate 50%: 0.1 mL/kg (50mg/kg) over 20 minutes, then an infusion of 0.06 mL/kg per hour (30 mg/kg) with serum drug level goal 1.5–2.5 mmol/L. This can be very useful if the child is not responding to intravenous salbutamol. Magnesium sulphate has been shown to be effective and safe in acute severe asthma in children. It is worth considering in a child with refractory asthma, with impending respiratory failure. It has yet to be compared directly to IV salbutamol.
9. Face mask continuous positive airway pressure (CPAP). Safer than ventilation, CPAP decreases resistance to air flow, inflates the lungs, decreases the work of the respiratory muscles and recruits the expiratory muscles. This can reverse deterioration such that children will request it once they have experienced it. Pressures of 5–10 cm H2O for 10 minutes every hour can be effective. Bronchodilators can be nebulised through the circuit. BiPAP (bilevel positive airway pressure) also may be used; this has two levels of support, a higher level during inspiration and a lower level during expiration.
10. Mechanical ventilation. A last resort. Indications are respiratory arrest, extreme fatigue or relentness hypercapnia. A treacherous path, with morbidity risks including barotrauma, gas trapping (compromised cardiac function), dysrhythmias, atelectasis and nosocomial pneumonia. Strategies to minimise these include: initial rapid sequence induction, oral intubation, sedation and paralysis; permissive hypercapnia; minimal positive end expiratory pressure (PEEP), prolonged expiratory time, low rate, limitation of peak inspiratory pressure (PIP).
Preventative
2 Inhaled corticosteroids (ICS): fluticasone propionate [FP], budesonide [BUD], beclomethasone diproprionate [BDP-HFA], ciclesonide [CIC], mometasone [MOM]
ICS are the mainstay of treatment in persistent asthma. FP, BUD and BDP are used in most countries; the newest ICS are CIC and MOM. CIC has been available in Australia since 2008; MOM is available in the UK and the USA. Concerns regarding side effects centre around hypothalamic–pitutiary–adrenal axis suppression and effects on linear growth. To evaluate the risks, the equivalent corticosteroid dosing must be appreciated, as follows, from least to most potent: BUD 200 microgram = BPD-HFA 100 microgram = FP 100 microgram = CIC 80 microgram. The most appropriate dosage is the lowest that gives symptom control. Side effects are minimal in doses below 200 mcg of FP or equivalent daily in children over 5, for periods of at least 24 months. If doses of 200 mcg or above are used, side effects may include short-term growth suppression (at 400 mcg, a decrease in linear growth of 1.5 cm per year; reversible) and adrenal suppression. Approximately 36 mcg/kg per day of BUD will cause some HPA axis suppression.
4 Leukotriene modifiers (LTMs)
There is evidence of efficacy of LTRAs as protection against exercise–induced bronchoconstriction, where it could be an alternative to SABAs; single-dose montelukast can be taken the night before, or at least 2 hours before, exercise. LTRAs can be used as steroid-sparers, and for prevention of exercise-induced bronchoconstriction, where they are superior to LABA. Well tolerated, montelukast is a chewable tablet, available in a 4 mg size (for ages 2–5) or a 5 mg size (for ages 6–15); the maximum effect is 12 hours after the dose is given. Specific side effects described include raised liver enzymes with higher than recommended dosage (zileuton), and ‘unmasking’ of eosinophilic vasculitis (Churg–Strauss disease) suppressed by steroids, becoming evident as steroids withdrawn. Recently, concerns have been raised about behavioural issues and depression with use of montelukast.
6 Combination therapies: ICS + LABAs—fluticasone propionate (FP) + salmeterol xinofoate (SX); budesonide (BUD) + eformoterol fumarate dihydrate (EFD)
Delivery methods
Spacers
1. Load with one puff pMDI at a time.
2. Allow 30 seconds (timed) for inhalation of drug from the loaded spacer.
3. Do not clean the spacer until the valve ‘clogs up’, as cleaning can cause static electricity, and the minimally charged medication particles of the pMDI ‘stick’ to the walls of the spacer. In short, ‘One puff, 30 seconds; don’t clean it’. When cleaning is needed, use detergent and leave to dry.
Optimum management for the child
The Australian National Asthma Campaign’s six-point plan is as follows:
2. Achieve best lung function.
3. Maintain best lung function—avoid trigger factors.
This plan is available on the National Asthma Council Australia website, at www.nationalasthma.org.au. In keeping with this plan remember the following points:
1. Every child should have a written asthma treatment programme, fully explained, and an appropriate crisis management plan.
2. Every child requires regular monitoring of his or her disease and its treatment, plus the complications of each, including growth (failure of linear growth can be due to undertreatment, or to overtreatment with steroids). Other treatment complications should be sought: tremor, hyperactivity (LABAs, SABAs), Cushingoid features (ICS, oral steroids), nausea, vomiting (theophylline).
3. Every child should be assessed for any inadequacy in current treatment, suggested by the following:
Common management issues
Are there any upper airway issues?
Allergic rhinitis can contribute to poor asthma control, and treating this can improve control. Ask about symptoms such as runny nose, blocked nose, sneezing, itchy eyes, runny eyes, throat clearing, hoarse voice, mouth breathing, halitosis, pain/pressure over sinuses, loss of sense of smell, coughing after first lying down at night, headaches, poor sleep or snoring. Management may include allergen avoidance and pharmacological treatment. For continuous treatment, intranasal corticosteroid (INCS) is the treatment of choice. Intranasal mometasone, fluticasone, budesonide and triamcinolone do not have a significant effect on the hypothalamic–pituitary–adrenal (HPA) axis. Treating the coexisting allergic rhinitis can improve asthma control significantly. Similarly, obstructive sleep apnoea, from whatever cause, including allergic rhinitis, can contribute to poor asthma control and successful treatment of this may aid in asthma control. See the long case approach to OSA in this chapter.
Is there adequate education of those involved?
This includes education of the child, parents and teachers:
1. The child. Should know to take beta-2 agonist before exercise.
2. The parents. Need to understand the treatment and know when to initiate more frequent treatment; how to monitor the child. Do they understand that the child should not avoid sport at school? Are they aware of the prognosis?
3. The teachers/school. Is a pMDI with spacer available to the child at all times? Do teachers appreciate the need for treatment? Do sports instructors inappropriately exclude the child from games?
Is there a problem with adherence to treatment?
Adherence to treatment in adolescence is often a problem, and there may also be parental concern about long-term corticosteroid usage, which can lead to non-adherence on their behalf. The added risk of smoking induced by peer pressure or of medication avoidance is not an uncommon management problem. Another compliance issue is the parents’ almost invariable inability to stop smoking or, in many cases, even smoke away from the child, despite constant requests. This has been documented by testing the urine of the child for cotinine (a metabolite, and anagram, of nicotine) levels before and after education about smoking. The other point to explore is the responsibility of care—whether it belongs predominantly to the patient or parent—as this is a key issue in adolescence.
Neonatal intensive care unit graduate: chronic lung disease/bronchopulmonary dysplasia (CLD/BPD)
Recent progress has been made in the understanding and management of BPD. There is now a consensus-validated description of diagnostic criteria for BPD and its severity. In the last few years, a genetic component to BPD has been identified, with establishment of a familial tendency and heritability in twin studies; this suggests that genetic factors are as important in BPD as they are in adults for hypertension, cancer or psychiatric disease. As yet, however, there are no identifiable reproducible allelic associations to the susceptibility to BPD, although ongoing research is attempting to identify specific candidate genes involved in the pathogenic pathways of BPD.
‘New’ BPD occurs in the context of multi-hit insults to the developmentally immature lung (especially under 26 weeks’ gestation), positioned between canalicular and saccular phases of lung growth (at 23–30 weeks). There is the intrinsic problem of developmental arrest of alveologenesis and vasculogenesis, with dysregulated angiogenesis, resulting in large simplified alveoli and dysmorphic lung vasculature, in addition to a premature anti-oxidant system, surfactant deficiency, and a very compliant chest wall; these intrinsic qualities increase susceptibility to the noxious effects of extrinsic problems. These extrinsic problems include mechanical ventilation and ventilator-induced lung injury (which include barotrauma [from pressure], volutrauma [from overdistension], atelectotrauma [from insufficient tidal volume], biotrauma [from infection, inflammation] and rheotrauma [from inappropriate airway flow]; these alter the integrated morphogenic programme of pulmonary development. Other inhibitors of alveolarisation and lung growth include oxygen toxicity, intrauterine, lung and systemic infections, and cytokine exposure. BPD is still the most prevalent sequel of preterm birth. Requirement for treatment with supplemental oxygen at 36 weeks’ postmenstrual age (PMA) is the commonest accepted definition of BPD at present (2010).
History
Past history
1. Pregnancy, gestation, delivery, birth weight, Apgar scores, reason for premature delivery.
2. Initial resuscitation required, when intubated, underlying respiratory diagnosis (e.g. hyaline membrane disease, meconium aspiration), duration of ventilation, continuous positive airways pressure (CPAP), oxygen requirement.
3. Complications of ventilation (e.g. air leaks, subglottic stenosis, tracheal stenosis, tube blockage), apnoeic episodes, associated problems (e.g. PDA, intraventricular haemorrhage [IVH], periventricular leucomalacia [PVL], ROP).
4. Drugs/treatments used (e.g. salbutamol, ipratropium bromide, theophylline, corticosteroids, diuretics, RSV intravenous immune globulin [RSV-Ig], palivizumab).
5. Monitoring since extubation, discharge details (e.g. age, weight, treatment).
6. Hospitalisation details (frequency, duration, usual treatment).
7. Outpatient clinics attended (where, how often, usual tests: e.g. chest X-ray, pulse oximetry).
Current status
2. Home management; for example, nasal oxygen, nebulised bronchodilators, oral theophylline, diuretic (e.g. frusemide), antibiotics.
3. Status of other systems where ex-premmies have increased risk of dysfunction; for example, ears, eyes, development, renal, cardiac, gut from necrotising enterocolitis (NEC).
Examination
Table 15.1 gives an approach to the cardiorespiratory examination (specifically) of the NICU graduate. It does not include looking for other complications of prematurity (e.g. IVH), but does include toxicities relating to ventilation and oxygen.
Table 15.1 An approach to the cardiorespiratory examination of the NICU graduate
| General observations |
| Parameters |
CCF = congestive cardiac failure; PDA = patent ductus arteriosus; ROP = retinopathy of prematurity.
Management
Oxygen is needed if the child’s PO2 in air is below 60 torrs. The aim is for the oxygen saturation (SaO2) to be between 94% and 98% in the awake and asleep phases. Aiming for higher levels may worsen lung disease, and does not necessarily improve long-term growth or development. The actual range is controversial. Neonatologists generally use a lower limit of 90%, but respiratory physicians generally use a higher range (>94%), which is probably reflective of the age of infants and hence the risk of ROP. In the early phase, a lower SaO2 is usually accepted, but upon discharge when infants are well over the corrected term of gestation, a higher minimal SaO2 is usually used. Oxygen administration is associated with increased weight gain, decreased pulmonary hypertension, decreased SIDS-like events, and decreased morbidity and mortality. Home oxygen therapy avoids prolonged and expensive hospitalisation.
At discharge from NICU, the average requirement for CLD is low-flow subnasal oxygen at 250–1000 mL/min. The median duration of oxygen requirement is 6–10 months. Weaning should occur very slowly, over about 12 weeks, guided by regular saturation monitoring. An intercurrent acute respiratory tract infection may require reinstitution of oxygen.
Obstructive airways disease and bronchodilators
These patients have obstructive lung disease, but variable airway hyperactivity, and lack uniformity in their response to drugs. Many have hypertrophied lung smooth muscle and respond to bronchodilators (even when preterm) earlier than true ‘asthmatics’. Combinations of inhaled beta-2 agonists, ipratropium bromide and inhaled corticosteroids are widely used, with variable success. Most units try these medications for at least some weeks to ascertain efficacy. Despite the common usage, there is inadequate long-term data to recommend the use of any beta agonist, or ipratropium bromide, in CLD.
Social issues
The degree of psychosocial difficulty depends to some extent on the associated problems that the child has. If there is severe neurological impairment, there is a parental separation rate approaching 50%. There tend to be three phases after the initial discharge of the child from hospital: first, a ‘euphoric’ phase, which may last about 6 weeks; then a period of despair and exhaustion, lasting from around 6 weeks to 6 months post-discharge; and finally the stage of acceptance. Another common problem is the ‘vulnerable child syndrome’, a parent–infant behaviour disorder that may include problems with feeding, difficulty separating from the mother, overindulgence and overprotection, leading to the child ‘running the household’. This may be prevented by spending more time with the parents, educating them in potential problem areas that are well recognised, by normalising the management of the baby, and by normal and appropriate discipline.
Cystic fibrosis (CF)
Genetics
Cystic fibrosis is autosomal recessive. Molecular genetics: cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator gene (CFTR); the locus is on the long arm of chromosome 7 (7q31.2), and the protein is called CFTR. Over 1525 different mutations in the CFTR gene are known; almost all are point mutations or small (1–84 base pair) deletions. The CFTR gene codes for a 1480-amino-acid integral membrane protein of 170 kDa, a cyclic-AMP-regulated chloride channel on the apical surface of epithelial cells. The deficiency of CFTR function leads to abnormal regulation of chloride channels and decides the phenotype of the epithelial cells. The most common mutation worldwide is delta-F508, where there is deletion of phenylalanine at the 508th amino acid within the CFTR protein; this accounts for 30–80% of mutant alleles, depending on the ethnic group.
There are five functional classes of mutation in CF:
• Class I mutations cause reduced or absent synthesis of CFTR, and are associated with nonsense, frameshift or splice junction mutations (e.g. G542X: 2.5% of cases; no synthesis of CFTR).
• Class II mutations cause a block in protein processing, and are associated with missense mutations and amino-acid deletions (e.g. delta-F508: 70% of cases in Australia; blocked processing of CFTR).
• Class III mutations cause a block in the regulation of the CFTR chloride channel, and are associated with missense mutations (e.g. G551D: 1.6% of cases; blocked regulation of CFTR).
• Class IV mutations cause altered conductance of CFTR chloride channel, and are associated with missense mutations (e.g. R117H: 0.3% of cases).
• Class V mutations cause decreased splicing of normal CFTR, which decreases membrane CFTR function and can act concurrently with other mutations on the same allele.
History
Current status
Respiratory disease
1. Symptoms: upper respiratory tract infection (URTI) symptoms, impaired exercise tolerance (important but relatively uncommon in children); cough frequency, severity (cough syncope extremely rare), nocturnal or exercise-induced wheeze or asthma, recent change in pattern; sputum volume, colour, blood and any recent change in these; fatigue, dyspnoea, wheeze, response to bronchodilators, peak flow pattern (limited value); need for home oxygen; chest pain; chronic sinusitis; glue ears; nasal polyps (nasal polyps produce symptoms of rhinorrhea, nasal blockage, snoring and even occasionally have protruded from the nose).
2. Infective agents: acquisition of chronic infection with Pseudomonas is an important prognostic factor, with early acquisition (under 5–6 years) of mucoid Pseudomonas being associated with increased mortality, especially in females, and increased morbidity. Also Burkholderia cepacia acquisition is important.
3. Past complications; for example, pneumothorax, moderate-to-large haemoptyses, allergic bronchopulmonary aspergillosis (ABPA).
4. Investigations; for example, sputum colonisation, chest X-ray, pulmonary function tests, overnight oximetry.
5. Home management; for example, exercise, physiotherapy (frequency, type and by whom), PEP mask; nebulised antibiotics, saline, bronchodilators, or dornase alpha; pMDIs (bronchodilators, ICS); oral antibiotic or corticosteroid; venous port access for parenteral antibiotics.
6. Future therapy plans: use of newer antibiotics, oral or nebulised; corticosteroids; consideration of lung transplant, gene therapy, experimental therapies.
Gastrointestinal disease
1. Symptoms; for example, growth, weight loss, appetite, dietary history, stool pattern (oily, pale, bulky or offensive, blood, melaena), passing wind and burping (may not be popular, but can have an impact on kids at school), abdominal pain, vomiting, haematemesis, heartburn.
2. Past complications; for example, meconium ileus or equivalent (this is now termed DIOS [distal intestinal obstruction syndrome]), rectal prolapse, jaundice, portal hypertension, fibrosing colonopathy, gastrostomy.
3. Investigations; for example, faecal fat studies, liver function tests, hepatobiliary ultrasonography or Tc-99m scintigraphy.
4. Home management; for example, pancreatic enzymes, vitamins, salt tablets.
5. Future plans; for example, gastrostomy, sclerotherapy, liver transplantation.
Past history of CF
Immunisations (especially pneumococcal, influenza vaccines).
Allergy may be an important issue, with some children requiring desensitisation.
Social history
Disease impact on patient
Treatment regime, disruption of going to hospital, recurrent/permanent loss of: normal social life, being part of peer group, being part of family, freedom, independence, normal childhood, trust, self-esteem (impaired growth, development, may have short stature, delayed puberty, gastrostomy button, intravenous access ports, offensive flatus, cough, requirement for treatment [pancreatic enzymes, vitamins, bronchodilators], decreased ability for physical activity, stamina). By around 8–12 years, aware of differences from normal peer group, and shame and embarrassment can become very prominent. There is the question of ‘Why me?’ Other issues: seeing friends or relatives with CF deteriorating in hospital, requests for transfer to adult care, compliance (reluctant to take medications in front of peers), schooling (attendance, performance, teacher awareness of disease and its treatment, peer interactions), employment prospects, limitation of activities of daily living (including sport), depression (inevitability of death), consideration of marriage, and prevention (smoking). Fertility must be discussed: What has been said and to whom? Many units normally talk with boys around 14 years of age and discuss all health-related issues, including normal sexual function, reduced ejaculate, and choices for the future with micro-aspiration sperm and IVF techniques. Smoking must be discussed. Alcohol is another discussion area for the pancreatic-sufficient children, as they have an increased risk of pancreatitis.
Disease impact on siblings
Sibling rivalry, hostility, isolation, alienation, resentment, guilt (‘survivor guilt’), concern, worry, effect of family’s financial burden, genetic counselling. There are camps for siblings (e.g. CFQ sibling camps). A useful website is www.siblingsaustralia.org.au.
Examination
The approach given in Table 15.2 assesses patients with CF for disease progression, severity and the current status of the disease. It looks particularly at clubbing, flap, chest deformity, cor pulmonale, nutrition, puberty, diabetes, chronic liver disease and hypertrophic pulmonary osteoarthropathy. Two very important signs are the cough and the sputum, and these may give a better indication of the state of the lungs than any other finding on physical examination.
| General inspection |
| Position patient: standing, with adequate exposure, for a complete examination, but sensitive to the patient’s modesty—although ideal, the patient being fully undressed (as stated in previous editions) is neither practical nor sensitive in most patients, and should not be encouraged |
| Parameters |
• Distension (poor abdominal musculature with protein–calorie malnutrition, ascites with CLD)
• Scars (e.g. gastrostomy, previous meconium ileus, hepatobiliary surgery for common bile duct stenosis, fundoplication)
• Venous access port (e.g. Infuse-A-Port)
• Prominent abdominal wall veins (CLD)
BSL = blood sugar level; CCF = congestive cardiac failure; CLD = chronic liver disease; HPOA = hypertrophic pulmonary osteoarthropathy.
Investigations
Monitoring of disease
Not many routine tests are done in CF:
1. Routine clinic visits, usually at least every 3 months: note weight, nutrition, sputum, spirometry.
2. Documentation of disease progression at intervals of 6 months to 1 year. The following may be done: chest X-ray (CXR), pulmonary function tests, liver function tests, full blood count, measurement of skin fold thickness. Vitamin levels and work-up for ABPA with total IgE can be performed annually. A high-resolution CT scan of the thorax in primary and secondary school can assess progress of bronchiectasis.
Other investigations as indicated
1. Cardiac: electrocardiography, echocardiography (right-ventricular hypertrophy) usually only done when heading for transplant.
2. Gastrointestinal: serum protein and albumin, liver function tests, coagulation studies, abdominal X-ray (meconium ileus, equivalent or peritonitis), abdominal ultrasound (gallbladder, liver), hepatobiliary Tc-99m scintigraphy (common bile duct strictures), percutaneous transhepatic cholangiogram (PTC) and endoscopic retrograde cholangiopancreatography (ERCP) to detect strictures, pancreatic function tests.
3. Other. Whether CFRD should be screened for, with an oral glucose tolerance test (GTT) in children over 10 years, is debatable. A fasting blood glucose is not particularly useful in this context, as it does not exclude diabetes and can be normal in patients with significant CFRD; a GTT is a better test. Urea and electrolytes may be used to assess salt loss, and dye studies in patients with venous access ports can be used for assessment of port function.
Management
Treatment of lung disease
The focus of CF treatment over the next decade will be the prevention of lung disease.
CF microbiology: ‘old’ established and ‘new’ emerging pathogens
The microbiology of lung pathogens dictates the usual treatment plans. Initially, the main organisms encountered are Staphylococcus aureus, Streptococcus pneumoniae, Haemophilus influenzae, P. aeruginosa non-mucoid (probably the cause of at least 30% of exacerbations in children under 2 years of age) and mucoid strains of P. aeruginosa (in >50% of CF patients). B. cepacia infection may be associated with rapidly progressive lung disease. Approximately one third of CF patients have precipitating antibodies to Aspergillus fumigatus, and two thirds have positive skin tests for this. Infection-control issues are important. Cross-infection must be minimised, avoiding the risk of transmission of B. cepacia or P. aeruginosa between infected and non-infected patients. Some clinics cohort by strain of P. aeruginosa as well.
There are now emerging newer pathogens:
MRSA (methicillin-resistant Staphylococcus aureus). This can be associated with a rapid decline in lung function. This can be treated early with clindamycin and rifampicin, given intravenously whenever admitted to hospital.
Stenotrophomonas maltophilia. This is an aerobic Gram-negative organism, aquatic, and previously classified in the genus Pseudomonas. This can be treated with oral antibiotics: trimethoprim with sulfamethoxazole (TMP-SMX).
Alcaligenes xylosoxidans.This is an aerobic Gram-negative bacillus. It can respond to piperacillin and ceftazidine.
Atypical mycobacteria. These can colonise, or actively infect. Should look for signs of disease before treating; can repeat sputum cultures, and perform chest X –ray (CXR) or CT scan of lung fields.
Fungi: Aspergillus. Around 30% CF patients may be colonised. Should look for signs of disease features of ABPA (see below), such as wheezing, positive IgE, positive skin test, and CXR consistent with ABPA.
Fungi: Scedosporium apiospermum. Poor sensitivity to antifungal drugs. Similar to Aspergillus, it is the second commonest filamentous fungus in CF patients (after Aspergillus). It is a saprophyte, obtaining nutrition by assimilating decaying organic matter. Its presence may prevent transplant.
Antibiotics
1. Sputum cultures do reflect lung flora in CF (but only in children who can produce sputum easily). Oropharyngeal cultures have poor sensitivity and positive prediction (around 55%), and better specificity and negative prediction (around 85%).
2. Choice of antibiotic for hospital treatment is usually an antipseudomonal penicillin (e.g. ticarcillin with clavulate, piperacillin) and an aminoglycoside (e.g. tobramycin), plus oral flucloxacillin. Cephalosporins such as ceftazidime, nalidixic acid derivatives and polymyxins may also be used.
3. Dose of antibiotic. Abnormal metabolism, and rapid excretion, of antibiotics occurs. ‘Double dose’ antibiotics are needed, especially aminoglycosides (e.g. tobramycin, gentamicin). Adult-dose IV antibiotics can be given from 7 years of age, in general, but this does depend on the antibiotic. As patients get older, the tobramycin dose may need to be wound back. Aminoglycosides are given as single daily doses.
Most exacerbations of chest disease are probably due to viral infections.
Chest physiotherapy
Other methods encourage independence and may be used in combination with the above. Positive expiratory pressure (PEP) masks are used by several centres. The resistance of the mask is variable; the appropriate value can be chosen by lung-function testing to document the highest increase in forced vital capacity achieved. It is usually used for 10 minutes at a time, once or twice a day. Forced expiratory technique can be taught from 8–9 years. Exercise should be encouraged from a very young age and especially encouraged to continue in the teenage years.
Allergic broncho-pulmonary aspergillosis (ABPA)
This is difficult to diagnose in CF, as the features of ABPA (CXR findings, increased serum immunoglobulins) occur in CF, except for IgE. A high IgE level is suggestive of ABPA if other features are present. ABPA can occur in progressive stages: acute stage (upper- and middle-lobe infiltrates, eosinophilia, IgE level very high, prednisolone responsiveness); remission (after 6 months; normal CXR, no prednisolone); recurrent exacerbation stage (recurrence of infiltrates, eosinophlia, IgE elevated, prednisolone responsiveness, if steroid resistant respond to omalizumab); steroid-dependent asthma stage (chronic obstructive airways disease, responds to prenisolone or inhaled steroids, or omalizumab); fibrotic stage (diffuse airway disease, with obstruction and fibrosis, variable CXR, poor response to steroids; this phase can be terminal). Treatment is stage dependent. Agents used may include: prednisolone; methylprednisolone pulses; antifungal agents (the ‘-conazoles’—itraconazole, voriconazole or posaconazole); anti-IgE agent (omalizumab); G-CSF (granulocyte colony-stimulating factor); GM-CSF (granulocyte–macrophage colony-stimulating factor); interferon; TNF (tumour necrosis factor); therapeutic bronchoscopy/surgery. For acute stage, management often includes steroids and antifungal agents for 6 months. Progress may be monitored by IgE levels. ICS may ease the asthma symptoms.
Lung transplantation
CF is the most common indication for paediatric lung transplant. CF does not re-occur in transplanted lungs. Bilateral sequential single-lung transplantation is now the operation of choice, the sole option for long-term survival of terminal patients. The mainstem bronchus and pulmonary artery are connected by end-to-end anastomoses; the two pulmonary veins from each lung are harvested intact with a patch of the left atrium of the donor, and then each left atrial patch is sewn to the recipient’s left atrium. This is performed using cardiopulmonary bypass. Living-donor lobar transplantation (LDLT) is another option, which is rarely performed because of technical and ethical complexities: it requires two donors, each undergoing a lower lobectomy to provide a right and left lower lobe to serve as right and left lungs for the recipient; for adolescents taller than 152 cm, the donors need to be tall enough to provide adequate lung tissue.
Complications fall into three groups:
1. Immediate phase (first few days post-transplant). Subgroups include the following:
2. Early phase (first few weeks post-transplant). Subgroups include the following:
3. Late phase (months post-transplant). Subgroups include the following:
Females may require transplantation earlier than males, as they have a median survival of 28 years, compared to males’ 44 years. Suggestions are that females should be assessed and transplanted at a different lung function compared with males, but this varies with different centres. The survival rate approaches 87% at 1 year, 70% at 3 years and 60% at 5 years. The three most significant factors for poor outcome are repeat transplant, mechanical ventilation at transplant and coexistent congenital heart disease. The three main causes of death are early graft dysfunction (most deaths in the first 30 days post-transplant), infection (first year post-transplant) and bronchiolitis obliterans (beyond the first year post-transplant, for cadaveric transplant recipients; rare in LDLT). Preoperatively, a 2-week assessment in hospital in necessary. The usual age has been around 17–19 years. The rate-limiting factor for this treatment is the scarcity of donor organs. To overcome this, as well as LDLT, reduction surgery and xenograft transplantation from genetically engineered porcine donors have been developed. Concurrent heart and lung transplants have been performed in patients with severe CF lung disease and significant associated cor pulmonale. A study reported in 2007, based on a retrospective cohort study of 514 children with CF on a transplant waiting list, of whom 248 had lung transplants, estimated that only five patients would clearly have improved survival from lung transplantation, inferring that lung transplantation was not necessarily associated with an improved survival in children. Some 50% of lung-transplanted patients have obstructive lung disease in 4 years. Patients can have re-transplants.
Sinonasal disease
Most children with CF have sinonasal disease, but it is underreported. On CT scan, 92–100% of CF patients have chronic rhinosinusitis, the average age of onset being 5–14 years. Nasal polyps are seen in 30–50% of CF patients, versus 0.1% of non-CF/normal children; of these in CF patients, only around 60% are symptomatic. The history for sinonasal disease may include nasal obstruction, mouth breathing, cough, nasal discharge, post-nasal drip, headache, facial pain, hyposmia, anosmia, poor sleep, snoring, limited activity, hoarseness and halitosis. Examination findings may include congested turbinates, post-pharyngeal oedema, nasal polyps, medialisation of the lateral nasal wall and craniofacial distortion. Nasal polyps can be managed medically, with oral antibiotics, nasal toilet with normal saline irrigation, irrigation with tobramycin or nasal inhalation of pulmozyme.
Treatment of gastrointestinal disease
Pancreatic insufficiency (PI)
Some patients appear refractory to PERT (i.e. stool fat output persistently above 25% of fat intake), related in some to duodenal acidity. Treatment for these patients may include gastric acid suppression with antacids, H2 receptor blockers (e.g. ranitidine), proton pump inhibitors (e.g. omeprazole) or prostaglandin analogues (e.g. misoprostol) to aid fat digestion. An alternative pancreatic enzyme preparation may be tried, as dissolution profiles may vary, or preparations may be given at the start of the meal, for earlier onset.
Nutrition
A high-energy diet is recommended. CF patients require approximately 120% of normal energy requirements because of increased energy use (due to the effort of breathing, and infection), decreased protein synthesis with acute exacerbations of chest disease, nutrient loss with malabsorption, anorexia and raised basal metabolic rate. Less than optimal nutrition may lead to poor growth, impaired respiratory function and decreased exercise tolerance.
Salt
Other gastrointestinal problems
The spectrum of gastrointestinal involvement is as follows:
1. Pancreas: insufficiency (see above); pancreatitis (in patients with pancreatic sufficiency).
2. Liver: focal biliary cirrhosis, multilobular biliary cirrhosis (complications include portal hypertension, liver failure), intrahepatic stones and sludge.
3. Gallbladder: non-functioning, stones.
4. Common bile duct: stones, stricture, rarely carcinoma.
5. Oesophagus: gastro-oesophageal reflux (increased abdominal pressure from coughing and wheezing, depressed diaphragm from hyperinflation), oesophagitis, oesophageal varices (from portal hypertension; beta blockers can reduce portal venous pressure, and risk of bleeding with varices in CF, but 1 in 5 get bronchospasm).
6. Stomach: portal gastropathy (congested stomach, with purpura in stomach, from portal hypertension).
8. Small intestine: meconium ileus (with or without the associated complications of volvulus, atresia, perforation), meconium ileus equivalent (DIOS), intussusception.
9. Large intestine: rectal prolapse, constipation with acquired megacolon, meconium plug, fibrosing colonopathy.
10. Gastrointestinal malignancy: occurs more frequently in CF, although the overall risk of cancer anywhere is no different to that for the general population.
These complications are managed by standard therapy, but some deserve particular consideration.
CF-associated liver disease (CFLD): liver and biliary tract disease
CFLD is an indolent process. Focal biliary cirrhosis occurs secondary to inspissated bile. Cirrhosis is second to lung disease as a cause of mortality in CF patients. CFLD occurs in around 15% of the CF population, cholelithiasis in 12%, cirrhosis in 10%, portal hypertension in 2–5% and clinically significant disease in 1–2%. The average age of presentation is around 10 years, with the peak incidence at 16–20 years. Once cirrhosis has developed, the duration of survival is 4–5 years. In the liver, only intrahepatic biliary epithelial cells express CFTR chloride channels. CF-associated liver disease (CFLD) is more common with PI. Management of cholestasis may include giving ursodeoxycholic acid (URSO), which mimics endogenous bile acid production. Investigations may include ultrasound, which can diagnose cirrhosis, but nuclear scans such as HIDA have largely been abandoned, as they were neither sensitive nor specific enough for precise diagnosis. CFLD has two major effects; portal hypertension (leading to varices and hypersplenism) and end-stage liver disease (ESLD). Splenic enlargement can squash the stomach and splint the diaphragm, and can cause abdominal pain via the splenic capsule being stretched or via small peripheral splenic infarcts. Management of portal hypertension may include sclerotherapy, variceal bonding, endoscopic variceal ligation, portosystemic shunting or transjugular intrahepatic portosystemic shunt placement. If progressive, irreversible hepatic insufficiency develops, liver transplantation (LTx) is the treatment of choice; LTx is a relevant management option in around 5% of CF patients. Long-term survival after LTx in CF is comparable to LTx performed for other indications (see the section on LTx in Chapter 8, Gastroenterology). Lung function can also improve after LTx. Distal common bile duct stricture, recurrent pain or biliary tree obstruction can be managed with surgical intervention: cholecystojejunostomy if the gallbladder is functioning, or choledochojejunostomy for non-visualised gallbladder, or microgallbladder.
Protracted nausea
A. Abdominal pain (functional; this is probably intestinal hypervigilence, where minor sensations become major)
S. Small bowel bacterial overgrowth/Splenic enlargement (portal hypertension)/Sinusitis
T. Treatment: prescribed drug side effects; over-the-counter drug side effects
I. Inflammatory bowel disease, IBD (especially Crohn’s disease, which is 12 times more common in CF)/Increased intracranial pressure (ICP)
 Non-prescribable (alternative/complementary) treatments
Non-prescribable (alternative/complementary) treatments
G. Gastrointestinal tract causes (five Gs): Gastrooesophageal reflux, Gastritis, Gastroparesis (diabetic), Gallstones, Giardia—to sort out which is the case, investigations can involve endoscopy and imaging; treatment may involve treating CFRD, PERT tailoring and medications (depending on the likeliest diagnosis, the possibilities include high-dose proton pump inhibitors, metronidazole, domperidone, ondansetron or even antidepressants)
Abdominal pain
G. Gastro-oesophageal reflux/oesophagitis
U. Underdosing pancreatic enzymes
R. Renal stones/Respiratory disease related: lower lobe pneumonia, muscle strain (rectus)
I. Inflamed pancreas (pancreatitis)/Inflammatory bowel disease/Intussusception
N. Non-organic/functional (e.g. grief reaction with death of friend with CF; recognising own mortality with deteriorating health—may be unrelated to CF)
G. Gallbladder/biliary related: cholelithiasis, common bile duct stricture, cholecystitis
Treatment of other complications
Cystic fibrosis related bone disease (CFR-BD)
• Hypertrophic pulmonary osteoarthropathy (HPOA) is the most common disorder (it occurs in 2–7% patients with CF). It is characterised by digital clubbing, and long-bone and joint pain (especially wrists, knees and ankles), worsened by pulmonary exacerbations. The aetiology remains unclear. Bone scan can detect HPOA early. Management includes optimising pulmonary care and aggressive treatment of exacerbations. Non-steroidal anti-inflammatory drugs (NSAIDs) may relieve discomfort.
• Kyphosis with an angle over 40° occurs in over 75% of adult female CF patients, and around one third of adult male patients. Both HPOA and kyphosis are associated with deteriorating lung function, and are seen as markers of a poor prognosis.
• Low bone mass is common, despite supplementation with vitamin D, calcium and PERT. The bone mineral density decreases and the fracture risk increases with increasing age. Osteoporosis and crush fractures can lead to significant back problems. Rib fractures are also reported. Severe bone loss occurs in transplant patients.
• CF episodic arthritis usually presents with recurrent (non-destructive) mono-articular involvement or a symmetrical polyarthritis. It can be managed by standard therapy (NSAIDs or aspirin). Glucocorticoids may be required, orally or intra-articularly. It has been observed that antibiotic therapy for lung disease can improve joint symptoms. Eruptions resembling erythema nodosum may occur in association with it.
• Rheumatoid arthritis has been reported, and treated along standard lines.
• Ciprofloxacin-induced arthralgia is treated by stopping the drug.
Is modification of current medical treatments warranted?
Alternate daily oral steroids may impact negatively on growth and bone density, and increase the risk of diabetes, although previously reported positive effects have included increased growth and appetite, decreased infection and increased well-being in some patients. Home intravenous antibiotics using venous access ports had appeared promising, and seemed as effective as hospital antibiotics, but now many studies have shown that home IV treatment may require a longer time on therapy, compared to hospital care, to achieve the same end points. Many units reserve this for patients who are very well motivated and have a lot of support. Home oxygen is useful for later-stage ‘restless’ nights and morning headache with significant oxygen desaturation overnight: this needs assessment (e.g. overnight oxygen saturation monitoring, echocardiography to detect cor pulmonale).
Prognosis
• female sex (teenage and young adult age period);
• complications, including CFRD or CFLD, or ABPA;
• abnormal chest X-ray 12 months after diagnosis;
• age of acquisition of chronic pulmonary infection with P. aeruginosa and, in particular, mucoid P. aeruginosa under 6 years;
Obstructive sleep apnoea (OSA)
Background information
Other conditions causing anatomical impingement on the airway calibre include those that decrease the bony cross-section of the lower face (sickle cell anaemia [via bone marrow hyperplasia], juvenile idiopathic arthritis [via micrognathia from temporomandibular joint involvement]), and those that cause increased airway soft tissue (adenotonsillar hypertrophy, obesity [parapharyngeal fat deposits], mucopolysaccharidoses, airway papillomatosis), collapsible upper airway tissues (Marfan; important to pick OSA, because its presence may increase risk of aortic root dilatation; other contributors to OSA in Marfan syndrome are crowded teeth, high-arched palate and narrowed jaw) or laryngeal/tracheal narrowing (laryngomalacia, subglottic stenosis). Various neurological conditions affect the muscle tone, whether upper motor neurone (cerebral palsy) or lower motor neurone (spinal muscular atrophy, myasthenia gravis, various myopathies).
C. Crouzon syndrome (FGFR 2 & 3)/Cervical fusion (Klippel–Feil syndrome)
O. Obesity/Overbite (and other dental anomalies)
M. Marfan syndrome (FBN 1)/Moebius sequence/Myelomeningocoele
P. Pfeiffer syndrome (FGFR 1 & 2)/Prader–Willi (deletion at 15 q11–q13)/Paralysed vocal cord
A. Adenotonsillar hypertrophy/Apert syndrome (FGFR 2)/Achondroplasia (FGFR 3)
C. Cerebral palsy/CHARGE syndrome (via choanal stenosis/atresia)(CHD7)/Chotzen [Saethre–Chotzen] (TWIST gene)
T. Tracheal/subglottic stenosis/Tracheomalacia/laryngomalacia
E. Extra tissue (e.g. fatty infiltration upper airway structures, storage disorders, airway papillomatosis, cystic hygroma [lymphatic malformation], nasal polyps in cystic fibrosis)
D. Down syndrome (normal-size tongue, small pharynx causing relative macroglossia)
B. Burns to head and neck/Beckwith–Wiedemann (11 p15.5) (macroglossia)
R. Rhinitis (allergic)/Robin sequence (hypoplasia of mandibular area before 9 weeks in utero) (glossoptosis)/Repaired cleft lip/palate/pharyngeal flap
E. Endocrine disease: hypothyroidism/hypopituitarism (both can cause decreased muscle tone)
A. Arthritis (juvenile idiopathic arthritis) (temporomandibular joint [TMJ] involvement causing micrognathia)/Arnold–Chiari malformation type II (including in spina bifida)
T. Treacher Collins syndrome (TCOF 1)
H. Haematological disease: sickle cell anaemia/Hallermann–Streiff syndrome (malar hypoplasia, micrognathia with hypoplastic rami and anterior displacement of TMJ, high narrow palate)
I. Inborn errors of metabolism: mucopolysaccharidoses
N. Neuromuscular weakness (spinal muscular atrophy, myasthenia gravis, myopathies)
G. Goldenhar syndrome (craniofacial microsomia; first and second branchial arch syndrome [14 q32])
Diagnosis of OSA
The following parameters are recorded on PSG (mnemonic SLEEPING OVER):
I. Intensity of respiratory effort
G. Grade snoring (frequency and/or volume)
O. Oxygen saturation (can detect brief drops in saturation by lowering averaging time to 2 seconds)
E. End-tidal CO2 (qualitative evaluation of airflow [hypopnoea causes reduction of, and apnoea causes loss/absence of, the end-tidal CO2 signal])
History
Symptoms
In school-aged children, ask about sleep-talking, insomnia, daytime fatigue, aggression, shyness, depression, low self-esteem, delayed puberty, dental problems (e.g. overcrowded teeth, malocclusion, severe enough to see a dentist/orthodontist). The increase in partial arousal parasomnias (sleepwalking, night terrors), is due to compensatory increase in slow wave sleep in response to sleep fragmentation.
Examination
Respiratory and cardiovascular examinations
Examine the chest for: asymmetry (left chest prominence with chronic increased right-ventricular activity, Beckwith–Wiedemann), deformity (Marfan), expansion, tracheal position, apex position, palpable pulmonary valve closure, right-ventricular overactivity, percussion and auscultation. Examine the cardiovascular system, noting any signs of pulmonary hypertension (single S2, P2 loud, ejection click, early diastolic murmur of pulmonary incompetence, systolic murmur of tricuspid incompetence) or cor pulmonale (signs of right-sided failure: hepatomegaly, ankle oedema).
Management
Treatments for OSA with known efficacy include the following.
Surgical procedures
1. Adenotonsillectomy (tonsillectomy and adenoidectomy, Ts & As) remains the key treatment for OSA, and leads to significant improvement in most children with OSA, with regard to breathing, sleeping and quality of life. Reports have been somewhat contradictory as to whether there may be improvements in concentration, school performance, cognitive or developmental progress. Adenotonsillectomy has been reported as reducing the need for CPAP in morbidly obese children with OSA. Adenotonsillectomy with the addition of uvulopharyngopalatoplasty (UPPP) has been used in some units for patients with Down syndrome or cerebral palsy (CP), but other units warn against UPPP, as there is a success rate of only around 50%, and complications can include velopharyngeal incompetence.
2. Other surgical therapies: for patients with life-threatening OSA (usually patients with craniofacial syndromes), then the following could be needed: mandibular distraction, maxillomandibular advancement, plus or minus tracheostomy (see below). Procedures to relieve nasal obstruction include inferior turbinate reduction, septoplasty, nasal valve surgery and rapid maxillary expansion. Procedures to relieve retroglossal obstruction include tongue reduction, genioglossus advancement and hyoid myotomy.
3. Tracheostomy is another surgical treatment that is available for selected patients with life-threatening OSA, particularly those with craniofacial anomalies (e.g. Pierre Robin sequence, CHARGE, Treacher Collins and Beckwith–Wiedemann syndromes), craniofacial and laryngeal tumours, or bilateral vocal cord paralysis.
Medical therapies
1. Intranasal corticosteroids are effective in decreasing OSA in children with moderate to severe adenoidal hypertrophy (associated with some decrease in adenoid size), those with allergic rhinitis, and those with residual postoperative OSA following adenotonsillectomy.
2. Leukotriene modifiers; the combination of intranasal budesonide and oral montelukast is effective in resolving mild residual OSA after adenotonsillectomy.
3. Weight loss is recommended as supplementary therapy for obsess children.
Short Cases
The respiratory system
‘Examine the respiratory system’ implies something different from ‘Examine the chest’. The former includes the hands (starting with the nails for clubbing), chest wall, praecordium, lungs, ears, nose, throat and regional lymph nodes. The latter includes chest wall (starting with this, not the hands), heart and lungs, as the primary focus of the examination, only then followed by more peripheral signs. The key is to do exactly what the examiners ask, and not interpret their instructions inappropriately.
Only ask for the PEFR if it would be relevant (making sure you know the appropriate expected values for that patient). Depending on the prior findings, further examination of the fundi, skin and neurological system may be required. The various findings sought are listed in Figure 15.1, and a comprehensive listing of possible signs on initial inspection is given in Table 15.3.
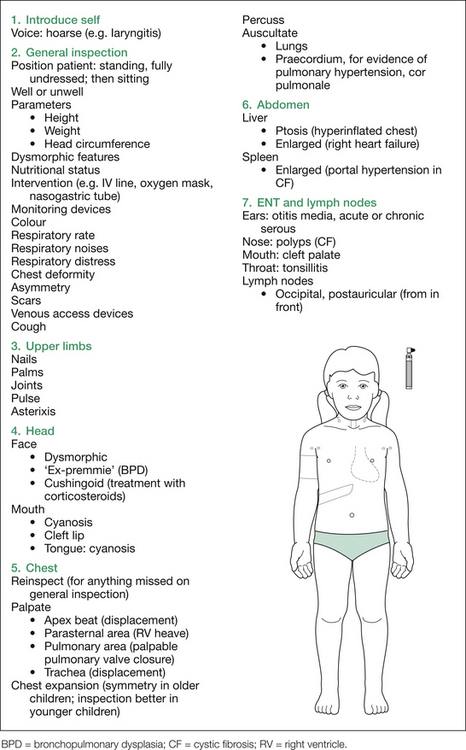
Table 15.3 Additional information: details of possible findings on respiratory examination
| Inspection |
| Dysmorphic features |
BSL = blood sugar level; CF = cystic fibrosis; HFOA = hypertrophic pulmonary osteoarthropathy; NICU = neonatal intensive care unit.
At the completion of the physical examination, the chest X-ray (CXR) can be requested; logical and succinct interpretation is expected (see the section on reading CXRs at the end of this chapter).
Stridor
Note the infant’s colour, posture (e.g. hyperextended neck with supralaryngeal problems, hypertonic spastic posturing in infants with cerebral palsy and pseudobulbar palsy), activity (paucity of movement with some neurological causes) and the degree of respiratory distress. Note the respiratory rate, tracheal tug and degree of chest recession (sternal, intercostal, substernal, subcostal or supraclavicular). Also note the following:
1. Whether the child is receiving any supports, such as nasogastric feeding (which implies at least one patent choanal opening), intravenous fluids, oxygen.
2. Whether the child has been receiving supports (which are temporarily unnecessary), such as nebulised therapy or an oropharyngeal airway on the bedside cupboard, or whether there is perinasal linear skin reddening, suggesting recent removal of tape securing a nasogastric tube.
3. Whether the child is being monitored; for example, with pulse oximetry.
Palpate the neck for any masses (e.g. lymphatic malformation [cystic hygroma]). If any masses are present, examine these in the standard manner for any lump (i.e. size, shape, consistency, pulsatility, attachments, fluctuation, transillumination, auscultation, regional lymph nodes). At this stage, if there is a neck mass, turn the child’s head to either side, then flex and extend the neck to assess if this worsens or alleviates the stridor.
Now come the least pleasant aspects of the examination:
1. Inspect the oral cavity, using a spatula and torch, looking for cleft palate and any obvious swellings or tumours (e.g. retropharyngeal or tonsillar).
2. Test the gag reflex and the suck (for bulbar dysfunction in neurological causes).
3. Palpate the tongue all the way back, to detect any mucus retention, cyst or other obstructive lesion.
4. If the condensation test suggests complete obstruction, it is now time to request a feeding tube and pass this down each nostril. Only do this at the very end of the examination, as it always upsets the child.
If a syndrome seems likely, look for other dysmorphic features.
Chest X-rays
Accurate interpretation is expected. Note the date of the X-ray and the name, to check that it is the correct film. Then note which side is labelled ‘right’ to avoid missing dextrocardia (although it should have been noted clinically), particularly in the child with clubbing and purulent sputum, who has Kartagener’s syndrome (immotile cilia/primary ciliary dyskinesia) and not CF. Note whether the film is well centred. This is the time to quickly scan the bony structures, as rotated films will show asymmetry of clavicles on the P-A view. Check the chest symmetry and note any scoliosis or rib crowding. Check that the film is well penetrated, and has been taken during a full inspiration. Expiratory films are notoriously difficult to interpret and may be quite misleading. Note the centring of the trachea and the cardiac shadow, checking for any deviation. Assess the cardiac size: the cardiac diameter is normally 50% or less of the cardiothoracic diameter (except in neonates, where it can be 60%) (see Figure 15.2). The heart may be enlarged due to pathology related to the lungs, such as cor pulmonale. The cardiac contour is then inspected, looking for evidence of the following:
1. Loss of right-heart border definition, which indicates right middle-lobe involvement.
2. Loss of apex definition, which indicates lingula involvement.
3. Increased opacification behind the heart, usually in a ‘sail’ or triangular shape, indicating left lower-lobe involvement.
4. A prominent pulmonary artery shadow, which can indicate pulmonary hypertension (PHT) .
5. A prominent right-atrial shadow, which can occur with PHT.
6. A prominent right-ventricular region, which can occur with PHT. (See Figure 15.3.)
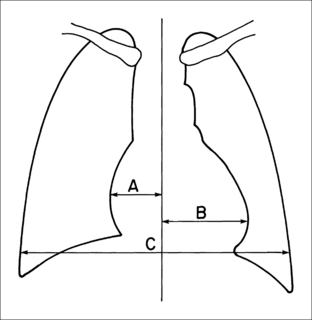
Figure 15.2 Chest roentgenography.
Diagram showing how to measure the cardiothoracic (CT) ratio from the posteroanterior view of a chest X-ray film. The CT ratio is obtained by dividing the largest horizontal diameter of the heart (A B) by the longest internal diameter of the chest (C). Myung Park 2007. Pediatric Cardiology for Practitioners, 5th edition, p. 66, Figure 4.1.
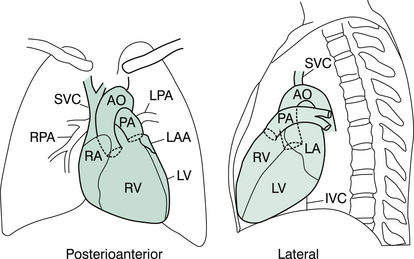
Figure 15.3 Posteroanterior and lateral projections of a normal cardiac silhouette.
Note that in the lateral projection, the right ventricle (RV) is contiguous with the lower third of the sternum and that the left ventricle (LV) normally crosses the posterior margin of the inferior vena cava (IVC) above the diaphragm. AO, aorta; LA, left atrium; LAA, left atrial appendage; LPA, left pulmonary artery; PA, pulmonary artery; RA, right atrium; RPA, right pulmonary artery; SVC, superior vena cava. Redrawn from; Myung Park 2007. Pediatric cardiology for practitioners, 5th edition, p. 66, Figure 4.2.
1. Loss of the line of the left hemidiaphragm suggests left lower-lobe involvement.
2. Loss of the line of the right hemidiaphragm suggests right lower-lobe involvement.
3. Loss of a clear costophrenic angle suggests pleural effusion (remember to examine the apices for a pleural cap).
4. The right hemidiaphragm is normally higher than the left, due to the liver; if the lungs are hyperinflated, the hemidiaphragms may be at almost the same level.
In assessing the lateral film, first note the bony structures. Note the degree of opacification of the vertebrae: normally the upper vertebrae are quite opaque, due to the overlying shoulder region, and as one progresses down the spine, the vertebral bodies appear increasingly lucent, because of the lack of overlying soft tissues in that region. Thus, if there is increased opacification of the lower vertebral area, there may be consolidation, or other process, involving the posterior segments of the affected lobe. Also note any kyphosis (rare) or rib abnormalities. Check the tracheal position followed by assessing the positions of the hemidiaphragms. Now inspect the lung fields; they are normally obscured in the postero-superior aspect (by the shoulders) and in the anteroinferior aspect (by the heart). Look for the interlobar fissures, which may delineate a collapsed or consolidated area.




