Chapter 85 The Porphyrias
The Heme Biosynthetic Pathway
Heme is required for a variety of hemoproteins such as hemoglobin, myoglobin, respiratory cytochromes, and cytochrome P450 enzymes (CYPs). It is believed that the 8 enzymes in the pathway for heme biosynthesis are active in all tissues. Hemoglobin synthesis in erythroid precursor cells accounts for about 85% of daily heme synthesis in humans. Hepatocytes account for most of the rest, primarily for synthesis of CYPs, which are especially abundant in the liver endoplasmic reticulum (ER), and turn over more rapidly than many other hemoproteins, such as the mitochondrial respiratory cytochromes. As shown in Figure 85-1, pathway intermediates are the porphyrin precursors δ-aminolevulinic acid (ALA, also known as 5-aminolevulinic acid) and porphobilinogen (PBG), and porphyrins (mostly in their reduced forms, known as porphyrinogens). At least in humans, these intermediates do not accumulate in significant amounts under normal conditions or have important physiologic functions.
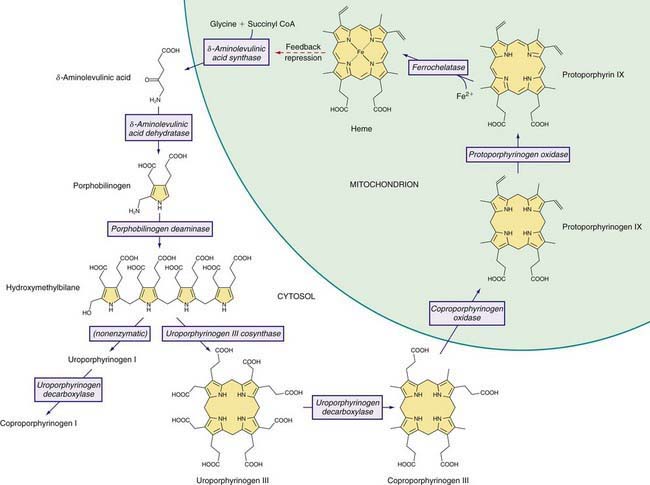
A deficiency of each enzyme in the pathway is associated with a specific porphyria (Table 85-1). The 1st enzyme, ALA synthase (ALAS), occurs in 2 forms. An erythroid specific form, termed ALAS2, is deficient in X-linked sideroblastic anemia, due to mutations of the ALAS2 gene on chromosome Xp11.2. Gain of function mutations of ALAS2 due to deletions in the last exon have been found in a variant form of erythropoietic protoporphyria (EPP). The housekeeping or ubiquitous form of this enzyme, termed ALAS1, is found in all tissues including liver, and its gene is located on chromosome 3p21.1. Disease-related mutations of ALAS1 have not been described.
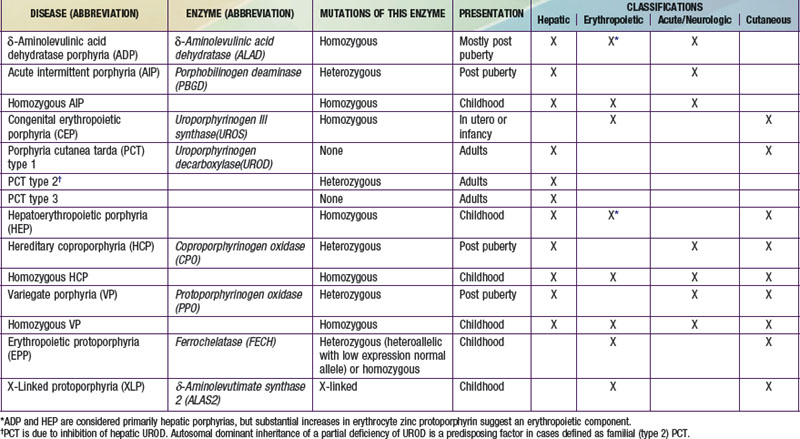
Table 85-1 THE HUMAN PORPHYRIAS: MUTATIONS, TIME OF PRESENTATION, AND TISSUE- AND SYMPTOM-BASED CLASSIFICATIONS
Regulation of heme synthesis differs in the 2 major heme-forming tissues. Liver heme biosynthesis is primary controlled by ALAS1. Synthesis of ALAS1 in liver is regulated by a “free” heme pool (see Fig. 85-1), which can be augmented by newly synthesized heme or by existing heme released from hemoproteins and destined for breakdown to biliverdin by heme oxygenase.
Intermediates of the heme biosynthetic pathway are efficiently converted to heme and, normally, only small amounts of the intermediates are excreted. Some may undergo chemical modifications before excretion. Whereas the porphyrin precursors ALA and PBG are colorless, nonfluorescent, and largely excreted unchanged in urine, PBG may degrade to colored products such as the brownish pigment called porphobilin or spontaneously polymerize to uroporphyrins. Porphyrins are red in color and display bright red fluorescence when exposed to long wavelength ultraviolet light. Porphyrinogens, which are colorless and nonfluorescent, are the reduced form of porphyrins, and when they accumulate are readily autoxidized to the corresponding porphyrins when outside the cell. Only the type III isomers of uroporphyrinogen and coproporphyrinogen are converted to heme (see Fig. 85-1).
Classification and Diagnosis of Porphyrias
Two classification schemes reflect either the underlying pathophysiology or clinical features, and both are useful for diagnosis and treatment (see Table 85-1). In hepatic and erythropoietic porphyrias, the source of excess production of porphyrin precursors and porphyrins is the liver and bone marrow, respectively. Acute porphyrias cause neurologic symptoms that are associated with increases of 1 or both of the porphyrin precursors ALA and PBG. In the cutaneous porphyrias, photosensitivity results from transport of porphyrins in blood from the liver or bone marrow to the skin. Dual porphyria refers to the very rare cases of porphyria with deficiencies of 2 different heme pathway enzymes.
It is notable that acute intermittent porphyria (AIP), porphyria cutanea tarda (PCT), and erythropoietic protoporphyria (EPP), the 3 most common porphyrias, are very different in clinical presentation, precipitating factors, methods of diagnosis, and effective therapy (Table 85-2). Two of the 4 acute porphyrias, hereditary coproporphyria (HCP) and variegate porphyria (VP), can also cause lesions indistinguishable from PCT (see Table 85-1). Congenital erythropoietic porphyria (CEP) causes more severe blistering lesions, often with secondary infection and mutilation. EPP is distinct from the other cutaneous porphyrias in causing nonblistering photosensitivity that occurs acutely after sun exposure. EPP is also the most common porphyria to become manifest before puberty.
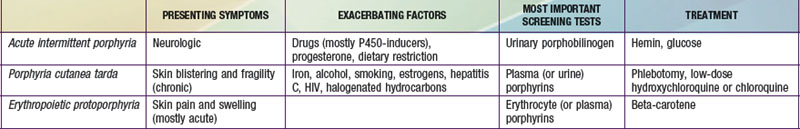
First-Line Laboratory Diagnostic Testing
δ-Aminolevulinic Acid Dehydratase Porphyria (ADP)
Pathology and Pathogenesis
ALAD catalyzes the condensation of 2 molecules of ALA to form the pyrrole PBG (see Fig. 85-1). The enzyme is subject to inhibition by a number of exogenous and endogenous chemicals. ALAD is the principal lead-binding protein in erythrocytes, and lead can displace the zinc atoms of the enzyme. Inhibition of erythrocyte ALAD activity is a sensitive index of lead exposure.
Acute Intermittent Porphyria (AIP)
Etiology
AIP results from the deficient activity of the housekeeping form of PBG deaminase (PBGD). This enzyme is also known as hydroxymethylbilane (HMB) synthase; the prior term uroporphyrinogen I synthase is obsolete. PBGD catalyzes the deamination and head-to-tail condensation of 4 PBG molecules to form the linear tetrapyrrole, HMB (also known as preuroporphyrinogen; see Fig. 85-1). A unique dipyrromethane cofactor binds the pyrrole intermediates at the catalytic site until 6 pyrroles (including the dipyrrole cofactor) are assembled in a linear fashion, after which the tetrapyrrole HMB is released. The apo-deaminase generates the dipyrrole cofactor to form the holo-deaminase, and this occurs more readily from HMB than from PBG. Indeed, high concentrations of PBG may inhibit formation of the holo-deaminase. The product HMB can cyclize nonenzymatically to form nonphysiologic uroporphyrinogen I, but in the presence of the next enzyme in the pathway is more rapidly cyclized to form uroporphyrinogen III.
Pathology and Pathogenesis
Drugs that are unsafe in acute porphyrias (Table 85-3) include those having the capacity to induce hepatic ALAS1, which is closely associated with induction of CYPs. Some chemicals (e.g., griseofulvin) can increase heme turnover by promoting the destruction of specific CYPs to form an inhibitor (e.g., N-methyl protoporphyrin) of ferrochelatase (FECH, the final enzyme in the pathway). Sulfonamide antibiotics are harmful but apparently not inducers of hepatic heme synthesis. Ethanol and other alcohols are inducers of ALAS1 and some CYPs.
Table 85-3 DRUGS REGARDED AS UNSAFE AND SAFE IN ACUTE PORPHYRIAS
| UNSAFE | SAFE |
|---|---|
| Barbiturates | Narcotic analgesics |
| Sulfonamide antibiotics* | Aspirin |
| Meprobamate* (also mebutamate,* tybutamate*) | Acetaminophen |
| Carisoprodol* | Phenothiazines |
| Glutethimide* | Penicillin and derivatives |
| Methyprylon | Streptomycin |
| Ethchlorvynol* | Glucocorticoids |
| Mephenytoin | Bromides |
| Phenytoin* | Insulin |
| Succinimides | Atropine |
| Carbamazepine* | Cimetidine |
| Clonazepam | Ranitidine† |
| Primidone* | Acetaminophen (paracetamol) |
| Valproic acid* | Acetazolamide |
| Pyrazolones (aminopyrine, antipyrine) | Allopurinol |
| Griseofulvin* | Amiloride |
| Ergots | Bethanidine |
| Metoclopramide* | Bumetanide |
| Rifampin* | Cimetidine |
| Pyrazinamide* | Coumarins |
| Diclofenac* | Fluoxetine |
| Progesterone and synthetic progestins* | Gabapentin |
| Danazol* | Gentamicin |
| Alcohol | Guanethidine |
| ACE inhibitors (especially enalapril) | Ofloxacin |
| Calcium channel blockers (especially nifedipine) | Propranolol |
| Ketoconazole | Succinylcholine |
| Rifampin | Tetracycline |
This partial listing does not include all available information about drug safety in acute porphyrias. Other sources should be consulted for drugs not listed here.
* Porphyria is listed as a contraindication, warning, precaution, or adverse effect in U.S. labeling for these drugs. Estrogens are also listed as harmful in porphyria, but have been implicated as harmful in acute porphyrias mostly based only on experience with estrogen-progestin combinations. While estrogens can exacerbate PCT, there is little evidence they are harmful in the acute porphyrias.
† Porphyria is listed as a precaution in U.S. labeling for this drug. However, this drug is regarded as safe by other sources.
Diagnosis and Differential Diagnosis
An increased urinary PBG establishes that a patient has 1 of the 3 most common acute porphyrias (see Table 85-2). Measuring PBG in serum is preferred when there is coexistent severe renal disease but is less sensitive when renal function is normal. Measurement of urinary ALA is less sensitive than PBG and also less specific but will detect ADP, the fourth type of acute porphyria. Erythrocyte PBGD activity is decreased in most AIP patients and helps confirm the diagnosis in a patient with high PBG. A normal enzyme activity in erythrocytes does not exclude AIP.
Treatment
Hemin
Hemin* is available for intravenous administration in the United States as a lyophilized hematin preparation (Panhematin, Lundbeck). Degradation products begin to form as soon as the lyophilized product is reconstituted with sterile water, and these are responsible for phlebitis at the site of infusion and a transient anticoagulant effect. Loss of venous access due to phlebitis is common after repeated administration. Stabilization of lyophilized hematin by reconstitution with 30% human albumin can prevent these adverse effects, and is recommended, especially if a peripheral vein is used for the infusion. Uncommon side effects of hemin include fever, aching, malaise, hemolysis, anaphylaxis, and circulatory collapse. Heme arginate, a more stable hemin preparation, is available in Europe and South Africa.
General and Supportive Measures
Drugs that may exacerbate porphyrias (see Table 85-3) should be discontinued whenever possible, and other precipitating factors identified. Hospitalization is warranted, except for mild attacks, for treatment of severe pain, nausea, and vomiting; for administration of hemin and fluids; and for monitoring vital capacity, nutritional status, neurologic function, and electrolytes. Pain usually requires a narcotic analgesic; there is low risk for addiction after recovery from the acute attack. Ondansetron or a phenothiazine such as chlorpromazine is needed for nausea, vomiting, anxiety, and restlessness. Chloral hydrate or low doses of short-acting benzodiazepines can be given for restlessness or insomnia. β-Adrenergic blocking agents may be useful during acute attacks to control tachycardia and hypertension, but may be hazardous in patients with hypovolemia and incipient cardiac failure.
Safe and Unsafe Drugs
Patients often do well with avoidance of harmful drugs. Some drugs known or strongly suspected to be harmful or safe in the acute porphyrias are listed in Table 85-3. Updated and more extensive listings are available on an interactive website of the European Porphyria Initiative (www.porphyria-europe.com) and from the American Porphyria Foundation (www.porphyriafoundation.com). Information regarding safety is lacking for many drugs, especially for those recently introduced, and sometimes opinions are conflicting.
Congenital Erythropoietic Porphyria (CEP)
Pathology and Pathogenesis
UROS, which is markedly deficient in CEP, catalyzes inversion of pyrrole ring D of HMB (the pyrrole ring shown on the right end of the molecule in Fig. 85-1) and rapid cyclization of the linear tetrapyrrole to form uroporphyrinogen III. This enzyme is also termed uroporphyrinogen III cosynthase. The human enzyme is a monomer. The gene for the enzyme is found on chromosome 10q25.3→q26.3, and contains 10 exons. Erythroid and housekeeping transcripts are generated by alternative promoters but encode the same enzyme.
Clinical Manifestations
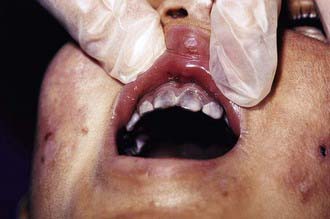
The most characteristic presentation is reddish urine or pink staining of diapers by urine or meconium shortly after birth (Fig. 85-2). With sun exposure, severe blistering lesions appear on exposed areas of skin on the face and hands, and have been termed hydroa aestivale because they are more severe with greater sunlight exposure during summer (Fig. 85-3). Vesicles and bullae, as well as friability, hypertrichosis, scarring, thickening, and areas of hypopigmentation and hyperpigmentation are very similar to those seen in PCT but usually much more severe. Infection and scarring sometimes cause loss of facial features and fingers and damage to the cornea, ears, and nails. Porphyrins are deposited in dentine and bone in utero. Reddish-brown teeth in normal light, an appearance termed erythrodontia, display reddish fluorescence under long-wave ultraviolet light (Fig. 85-4). An unaffected child born to a mother with CEP may have erythrodontia. Hemolysis and splenomegaly are common in CEP. Bone marrow compensation may be adequate, especially in milder cases. Patients with severe phenotypes, however, are often transfusion-dependent. Splenomegaly may contribute to the anemia and cause leukopenia and thrombocytopenia, which may be complicated by significant bleeding. Neuropathic symptoms are absent, and there is no sensitivity to drugs, hormones, and carbohydrate restriction. The liver may be damaged by iron overload or hepatitis acquired from blood transfusions.
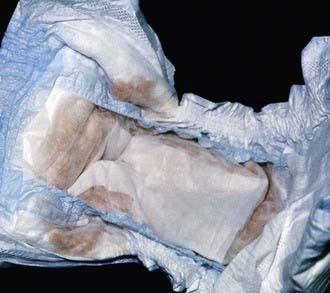
Figure 85-2 Congenital erythropoietic porphyria. The diaper of an affected baby demonstrates the red color of urine.
(From Paller AS, Macini AJ: Hurwitz clinical pediatric dermatology, ed 3, Philadelphia, 2006, Elsevier Saunders, p 517.)
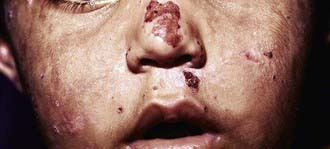
Figure 85-3 Congenital erythropoietic porphyria. Vesicles, bullae, and crusts on sun-exposed areas.
(From Paller AS, Macini AJ: Hurwitz clinical pediatric dermatology, ed 3, Philadelphia, 2006, Elsevier Saunders, p 517.)
Porphyria Cutanea Tarda (PCT)
This is the most common and readily treated human porphyria (see Table 85-2). It occurs in mid or late adult life, and is rare in children. Previous terms include symptomatic porphyria, PCT symptomatica, and idiosyncratic porphyria. The underlying cause is a liver-specific, acquired deficiency of uroporphyrinogen decarboxylase (UROD) with contributions by several types of genetic factors. UROD mutations are found in familial PCT. HEP, the homozygous form of familial PCT, usually has a more severe presentation in childhood, resembling CEP clinically.
Etiology
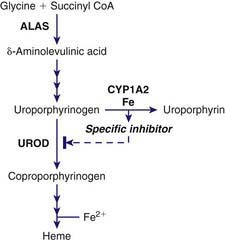
PCT is due to a reduction of hepatic UROD to 20% of normal activity or less. An inhibitor of hepatic UROD has been characterized as uroporphomethene, which is derived from partial oxidation of the enzyme substrate uroporphyrinogen. CYPs such as CYP1A2, as well as iron, are involved in its formation (Fig. 85-5). Although the enzyme is inhibited, the amount of hepatic enzyme protein measured immunochemically remains at its genetically determined level.
UROD catalyzes the decarboxylation of the 4 acetic acid side chains of uroporphyrinogen (an octacarboxyl porphyrinogen) to form coproporphyrinogen (a tetracarboxyl porphyrinogen) (see Fig. 85-1). The enzyme reaction occurs in a sequential, clockwise fashion, with the intermediate formation of hepta-, hexa-, and pentacarboxyl porphyrinogens. Uroporphyrinogen III, as compared with other uroporphyrinogen isomers, is the preferred substrate. Human UROD is a dimer with the 2 active site clefts juxtaposed. The UROD gene is on chromosome 1p34 and contains 10 exons, with only 1 promoter. Therefore, the gene is transcribed as a single mRNA in all tissues.
Pathology and Pathogenesis
CYPs, especially CYP1a2, can catalyze the oxidation of uroporphyrinogen to uroporphyrin. This uroporphyrinogen oxidase (URO-OX) activity is enhanced by iron, and leads to formation of a UROD inhibitor (see Fig. 85-5). CYP1a2 seems essential for development of uroporphyria in rodents, because experimental uroporphyria does not develop in CYP1a2 knockout mice.
Susceptibility Factors
Smoking and Cytochrome P450 Enzymes
Smoking has not been extensively studied as a susceptibility factor but is commonly associated with alcohol use in PCT. It may act to induce hepatic CYPs and oxidative stress. Hepatic CYPs are thought to be important in oxidizing uroporphyrinogen and generating a UROD inhibitor (see Fig. 85-5). Genetic polymorphisms of CYP1a2 and 1A1 have been implicated in human PCT. The frequency of an inducible CYP1a2 genotype was more common in PCT patients than in controls in several studies.
Clinical Manifestations
 of a normal tablet, twice weekly), which can be continued until plasma or urine porphyrins are normalized. There is at least some risk of retinopathy, which may be lower with hydroxychloroquine. Prospective treatment trials comparing this treatment with phlebotomy are lacking. Low-dose chloroquine was reportedly not effective in patients homozygous for the C282Y mutation in the HFE gene. The mechanism of action of 4-aminoquinolines in PCT is not known but is quite specific, because these drugs are not useful in other porphyrias.
of a normal tablet, twice weekly), which can be continued until plasma or urine porphyrins are normalized. There is at least some risk of retinopathy, which may be lower with hydroxychloroquine. Prospective treatment trials comparing this treatment with phlebotomy are lacking. Low-dose chloroquine was reportedly not effective in patients homozygous for the C282Y mutation in the HFE gene. The mechanism of action of 4-aminoquinolines in PCT is not known but is quite specific, because these drugs are not useful in other porphyrias.Hepatoerythropoietic Porphyria
Hereditary Coproporphyria (HCP)
Etiology
A partial (50%) deficiency in CPO activity has been found in all cells studied from patients with HCP. A much more profound deficiency is found in homozygous cases. Human CPO is a homodimer composed of 39-kd subunits, and contains no metals or prosthetic groups. The enzyme requires molecular oxygen, and is localized in the mitochondrial intermembrane space. A single active site on the enzyme catalyzes the oxidative decarboxylation of 2 of the 4 proprionic acid groups of coproporphyrinogen III to form the 2 vinyl groups at positions 2 and 4, on rings A and B, respectively, of protoporphyrinogen IX (see Fig. 85-1). Most of the intermediate tricarboxyl porphyrinogen, termed harderoporphyrinogen, is not released before undergoing the 2nd decarboxylation to protoporphyrinogen IX. Coproporphyrinogen I is not a substrate for this enzyme.
Variegate Porphyria (VP)
Etiology
Human PPO is a homodimer that contains FAD and is localized to the cytosolic side of the inner mitochondrial membrane. Membrane-binding domains may be docked onto human FECH, the next enzyme in the pathway, which is embedded in the opposite side of the membrane. PPO catalyzes the oxidation of protoporphyrinogen IX to protoporphyrin IX by the removal of 6 hydrogen atoms (see Fig. 85-1). The enzyme requires molecular oxygen. The substrate is readily oxidized nonenzymatically to protoporphyrin under aerobic conditions, or if exported into the cytosol. PPO is highly specific for protoporphyrinogen IX, and is inhibited by tetrapyrroles such as heme, biliverdin, and bilirubin and by certain herbicides that cause protoporphyrin to accumulate and induce phototoxicity in plants. Inhibition by bilirubin may account for decreased PPO activity in Gilbert disease.
Erythropoietic Protoporphyria (EPP)
Etiology
FECH, the enzyme that is deficient in EPP, catalyzes the final step in heme synthesis, which is insertion of ferrous iron (Fe2+) into protoporphyrin IX (see Fig. 85-1). The enzyme is also termed heme synthetase or protoheme ferrolyase. The human enzyme is a dimer, and each homodimer contains a [2Fe-2S] cluster, which may have a role in bridging homodimers. FECH is found in the mitochondrial inner membrane where its active site faces the mitochondrial matrix. It may be associated with complex I of the mitochondrial electron transport chain, and the ferrous iron substrate may be produced upon nicotinamide adenine dinucleotide (NADH) oxidation. FECH is specific for the reduced form of iron, but can utilize other metals such as Zn2+ and Co2+ and other dicarboxyl porphyrins. Accumulation of free protoporphyrin rather than zinc protoporphyrin in EPP indicates that formation of the latter is dependent on FECH activity in vivo.
Epidemiology
EPP is the 3rd most common porphyria, although its prevalence is not precisely known (see Table 85-2). It is described mostly in white people, but occurs in other races. The IVS3-48T>C splice variant is common in whites and East Asians but rare in Africans, which would be predictive of a lower disease prevalence in populations of African origin. EPP is the most common porphyria in children, but is often not diagnosed until adult life.
Anderson KE, Sassa S, Bishop DF, et al. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias, ed 8. Scriver CR, Beaudet AL, Sly WS, et al, editors. The metabolic and molecular basis of inherited disease, vol II. McGraw-Hill, New York, 2001;2991-3062.
Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924-934.
Sarkany RP. Making sense of the porphyrias. Photodermatol Photoimmunol Photomed. 2008;24:102-108.
Seth AK, Badminton MN, Mirza D, et al. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13:1219-1227.
Akagi R, Kato N, Inoue R, et al. δ-Aminolevulinate dehydratase (ALAD) porphyria: the first case in North America with two novel ALAD mutations. Mol Genet Metab. 2006;87:329-336.
Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439-450.
Solis C, Martinez-Bermejo A, Naidich TP, et al. Acute intermittent porphyria: studies of the severe homozygous dominant disease provide insights into the neurologic attacks in acute porphyrias. Arch Neurol. 2004;61:1764-1770.
Congenital Erythropoietic Porphyria
Dupuis-Girod S, Akkari V, Ged C, et al. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease). Eur J Pediatr. 2005;164:104-107.
Hallai N, Anstey A, Mendelsohn S, et al. Pregnancy in a patient with congenital erythropoietic porphyria. N Engl J Med. 2007;357:622-623.
Phillips JD, Steensma DP, Pulsipher MA, et al. Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood. 2007;109:2618-2621.
Ajioka RS, Phillips JD, Weiss RB, et al. Down-regulation of hepcidin in porphyria cutanea tarda. Blood. 2008;112:4723-4728.
Phillips JD, Bergonia HA, Reilly CA, et al. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci U S A. 2007;104:5079-5084.
Remenyik E, Lecha M, Badenas C, et al. Childhood-onset mild cutaneous porphyria with compound heterozygotic mutations in the uroporphyrinogen decarboxylase gene. Clin Exp Dermatol. 2008;33:602-605.
Goodwin RG, Kell WJ, Laidler P, et al. Photosensitivity and acute liver injury in myeloproliferative disorder secondary to late-onset protoporphyria caused by deletion of a ferrochelatase gene in hematopoietic cells. Blood. 2006;107:60-62.
Harms J, Lautenschlager S, Minder CE, et al. An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N Engl J Med. 2009;360:306-307.
Holme SA, Anstey AV, Finlay AY, et al. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574-581.
Holme SA, Whatley SD, Roberts AG, et al. Seasonal palmar keratoderma in erythropoietic protoporphyria indicates autosomal recessive inheritance. J Invest Dermatol. 2009;129:599-605.
Holme SA, Worwood M, Anstey AV, et al. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007;110:4108-4110.
Minder EI, Schneider-Yin X, Steurer J, et al. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009;55:84-97.
Rand EB, Bunin N, Cochran W, et al. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006;118:e1896-1899.
Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408-414.
* Hemin is the generic name for all heme preparations used for intravenous administration. Hemin is also a chemical term that refers to the oxidized (ferric) form of heme (iron protoporphyrin IX), and is usually isolated as hemin chloride. In alkaline solution, the chloride is replaced by the hydroxyl ion, forming hydroxyheme, or hematin.





