CHAPTER 4 The Origins of Behavior and Cognition in the Developing Brain*
Pediatricians specializing in developmental, learning, and behavioral problems have a strong interest in how the brain develops. As clinicians, pediatricians are also interested in the related topic of neural plasticity, especially how development can go pathologically “off track” and how treatment can help correct its course. We have argued1,2 that brain development can be described as a complex scaffolding of three categories of neural processes: gene-driven, experience-expectant, and experience-dependent. Gene-driven processes, which are comparatively insensitive to experience, serve to guide the migration of neurons, to target many of their synaptic connections, and to determine their differentiated functions. Experience-expectant processes correspond approximately to “sensitive periods,” developmentally timed periods of neural plasticity for which certain types of predictable experience are expected to be present for all juvenile members of a species. Not all brain development, however, is determined by gene-driven processes. Some species have a survival advantage if they can adapt to the environment or incorporate information from it. Indeed, many mammalian species have evolved specialized structures that can incorporate massive amounts of information. Because they have a long evolutionary history, the specialized systems vary across species and occur in multiple brain regions, so that there is no single “place” or “process” for learning and memory. Some types of neural plasticity have evolved to be incorporated into the developmental schedule of brain development, whereas others have evolved to serve the individual’s needs by incorporating information unique to their environment. This type of neural plasticity is termed experience-dependent, and it corresponds approximately to ordinary learning and memory: that is, encoding information that has adaptive value to an individual but is unpredictable in its timing or nature.
We emphasize a contemporary model of brain development that is derived from the study of dynamic, nonlinear systems. The dynamic systems perspective suggests that individuals use the interaction of genetic constraints and environmental information to self-organize highly complex systems (especially brains). Each organism follows a potentially unique and partly self-determined developmental path of brain assembly to the extent that the organism has unique experiences. The genetically determined restrictions (e.g., the initial cortical architecture) serve as constraints to the system, allowing the interaction of environmental information with existing neural structures to substantially organize and refine neural connections. In this chapter, which extends and amplifies earlier work by Black and colleagues,3 we review the evidence for these three processes, integrate them into a general model of brain development, provide evidence that the human brain is similarly plastic, and then apply this information to issues of children’s development and behavior.
GENE-DRIVEN PROCESSES
Gene-driven processes provide much of the basic structure of the brain and are intrinsically resistant to experience. Waddington4 described this tendency to resist deviations from predetermined pathways of development as canalization. Some of these genetically determined structures have evolved to constrain and organize experiential information, facilitating its storage in the brain in massive quantities. We now know much of the molecular biological processes of cell differentiation, neuron migration, and cell regulation and signaling. These processes are capable of building enormously complex neural structures without substantial input from the external environment. Evidence for the importance of gene-driven processes can be found in the tens of thousands of genes uniquely expressed in rat brain development.5,6 Indeed, in order to protect brain development, much of the basic organization of most nervous systems is largely impervious to experience. Neural activity that is intrinsically driven, such as that arising from the retina in utero, can play a role in these organization processes, by means of some of the mechanisms that seem also to be used later in the encoding of experience. For example, myelination of the optic nerve appears to be initially driven by spontaneous retinal activity and subsequently influenced by visually generated stimulation of the retina.7–10 Astrocytic development is also influenced by activity11 and is discussed in more detail later in this chapter. This theme of molecules and mechanisms, borrowed for other purposes in brain development or plasticity, can be found many times in this chapter.
TISSUE INDUCTION AND FORMING THE BASIC BRAIN PATTERN
Early central nervous system (CNS) development involves an ordered sequence of processes, beginning with formation of the neural plate and followed by an orderly program of further inductions.12 As in many embryological processes, brain tissue induction typically involves an organizer and its developmental target. Neural induction is familiar to physicians from the embryological process of gastrulation, in which the neuroectoderm just organizes itself. The signaling factors include activation of receptor tyrosine kinases, insulin-like growth factors and fibroblast growth factors, controlled inhibition of other signaling pathways (e.g., Noggin and Chordin), and the wingless pathway.13 A region of the neuroectoderm becomes differentiated by these signals, and its lateral edges become the neural crest and, later, the peripheral nervous system. Thus, these early signals point the tissue toward neural development or toward other ectodermal development. Many molecular mechanisms of neuronal development and organization have been preserved across species and time, in such a way that they remain remarkably similar in species as diverse as the fruit fly, amphibian, and mouse.14 We contend that some of these same mechanisms are then exploited later in development for analogous functions in critical periods and in learning and memory.
Some of the most important and unique characteristics of human brain structure appear to have evolved from relatively simple adjustments of genes controlling neuron number, modifying the rate of brain development, and regulating brain plasticity.15 Within the neural ectoderm, the spatial pattern determines much of future brain anatomy.16 The signals further differentiate the neural tube along an anteroposterior dimension and a mediolateral axis. After this point, each compartment has its own program of differentiation. The anteroposterior segments differentiate into the rhombencephalon (hindbrain), mesencephalon (midbrain), and prosencephalon (forebrain). Each of these subdivisions then follows a genetically controlled program of cell division and migration to swell into rhombomeres in the hindbrain and prosomeres in the forebrain, each of which becomes an important neural structure in the mature brain. Of course, early brain morphogenesis and control are very similar for many vertebrates. In contrast, analysis of genetic drift shows that the abnormal spindle-like microcephaly-associated (ASPM) gene, which affects overall brain size, began changing only in the past 5 million to 6 million years of human evolution to allow larger brain size.17 This recent adaptive evolution in a gene controlling brain growth is consistent with the role of key, distinctive features in human brain development: the timing of maturation and regulation of size, connectivity and plasticity.
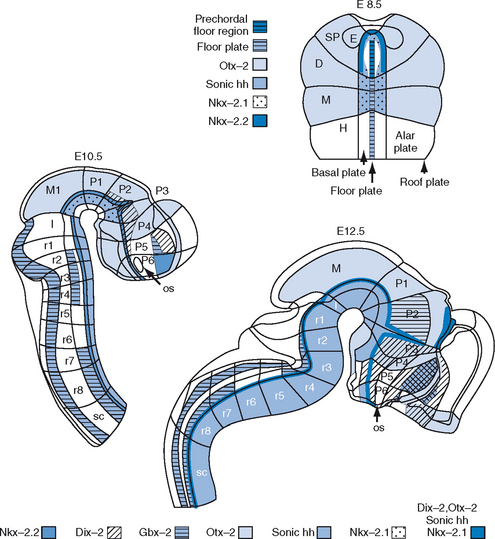
The mediolateral regionalization produces distinct tissues that are longitudinally aligned along the long axis of the CNS. Medial inductions are regulated by substances produced by axial mesodermal organizers: the notochord and the prechordal plate. These organizers are midline structures that lie underneath and produce substances such as sonic hedgehog that induce the medial neural plate to form the floor plate and basal plate. Growth factor proteins such as transforming growth factor–β mediate inductions from the lateral edge of the neural plate that are produced by the nonneural ectoderm. Lateral inductions are likely to be essential for the development of the neural crest, alar plate, and roof plate. Further patterning is determined in a checkerboard organization of brain subdivisions specified by the coordinates of anteroposterior and mediolateral location. Within the checkerboard, specific cues trigger the formation of swellings and vesicles that later become the telencephalon, eyes, and posterior pituitary gland. Although the process of regionalization subdivides the neural plate into the major brain structures, the process of morphogenesis transforms the shape of the neural plate into first a tube and then a complex tube with flexures and evaginations. As the neural plate transforms into the neural tube (neurulation), it converts the lateral-medial dimension of the neural plate into the dorsal-ventral dimension of the neural tube.14 The fusing of the neural tube is complete 26 days after conception in humans.18 The neural tube now has four ventral-to-dorsal subdivisions—the floor, basal, alar, and roof plates—each of which extends along much of the anteroposterior axis of the CNS and contributes to the distinct functional elements of the nervous system. The basal plate is the origin of the motor neurons, the alar plate is the origin of the secondary sensory neurons, and the floor plate is devoid of neurons and has several functions that are required during development. Like the notochord, the floor plate produces sonic hedgehog and is believed to serve as a secondary organizer guiding certain sensory neurons. Most of the roof plate forms the nonneuronal dorsal midline, including the choroid plexus and the pineal gland (Fig. 4-1).
HISTOGENESIS, MIGRATION, AND CELL FATE
As the neural tube organizes into regions that will become major structures (e.g., cerebrum, striatum, thalamus, cerebellum), tissue-specific genetic programs of histogenesis are begun within each region. Histogenesis can be subdivided into two general parts: neuron proliferation and differentiation.19 In general, each of these processes takes place in distinct zones within the wall of the neural tube. Proliferation takes place in the ventricular zone, which lines the inner surface of the neural tube and is adjacent to the ventricular cavity, whereas differentiation takes place largely in the mantle, which surrounds the ventricular zone. The ventricular zone cells are undifferentiated and mitotically active. Each brain region has distinct proliferation programs that regulate the rate of cell division, the number of cell divisions, and the character of cell division. Cell division can be symmetrical or asymmetrical. Symmetrical division produces cells that are identical; both the daughter cells either continue to proliferate or go on to differentiate. Asymmetrical division produces one daughter cell that differentiates and one that continues to proliferate. The regulation of these processes is integral to controlling how many cells are produced and when they are made, and local differences in replication rate give rise to the gross morphological structures of the forebrain, including the massive cerebral cortex.20
Like the ventricular zone, the subventricular zone is involved in the proliferation of brain cells, but it emerges somewhat later, between 8 to 10 weeks of gestation.21,22 In the human, migration into the telencephalic region (destined to become the cerebral cortex, hippocampus, and associated structures) begins at approximately 8 weeks of gestation, when the progenitor cells engage in asymmetrical proliferation to create postmitotic neurons.23 Proliferation ends at approximately 4.5 months of gestation, and the last cells begin their migration.23 Two waves of neurons migrate, in such a way that postmitotic neurons from the ventricular zone are first to leave, and the neurons from the subventricular zone emerge next.24 Cortical neurons migrate in an inside-out pattern, whereby neurons that developed earlier migrate to lower cortical layers, and the cells that developed later travel through and beyond previously migrated neurons for destinations in the outer cortex.25 Therefore, neurons generated in the ventricular zone take up residence in the lower layers of the cortex, and neurons that are derived from the subventricular zone become located in the outer regions of the cortex.
Although neuroanatomy texts may display a dazzling array of brain cell types, all these cells belong to only two major cell classes: neurons and glia.26 There are two major types of neurons: projection neurons, whose axons migrate to distant territories, and local circuit neurons (interneurons), whose processes ramify nearby. There are many distinct types of projection and local circuit neurons. There are two types of CNS-derived glia: astrocytes and oligodendrocytes. Astrocytes, which are believed to be derived from radial glia, probably regulate the local chemical milieu and have been shown in some cases to release neurotransmitters such as glutamate in ways that can affect neuronal activity. Oligodendrocytes produce the myelin sheaths that surround many axons; these sheaths function as insulators that increase the velocity of action potentials. As described later in the chapter, myelin appears to play key roles in regulating the plastic capacities of axons of neurons and the offset of sensitive periods for experiential organization of neuronal networks.27 Thus, these CNS-derived glial cells are increasingly seen as partners of neurons in plasticity during development and neural repair. (Another major glial type, the microglia, is derived from mesoderm and performs a phagocytic and immune system role.) Early in development, the ventricular zone contains proliferative cells that have the potential to produce both neurons and glia. In general, neurogenesis precedes gliogenesis. Most regions of the CNS can produce both neurons and astrocytic glia. Different types of neurons are generated at distinct dorsal-ventral positions in the CNS. Motor neurons in the spinal cord, for example, are generated by ventral progenitors, whereas sensory neurons are generated by dorsal progenitors. Likewise, in the telencephalon, ventral progenitors produce the motor neurons of the basal ganglia, whereas dorsal progenitors produce sensory cortical neurons.
In addition to patterning the regions of the nervous system (e.g., cerebral cortex and basal ganglia), histogenesis also regulates where cells travel (migration) and their ultimate functional fate (differentiation). The mechanisms underlying cell fate decisions in the nervous system involve both intrinsic and extrinsic signals. These signals have integral roles in regulating whether these cells continue to divide, whether they undergo symmetrical or asymmetrical division, and what lineage they will follow. Notch signaling is an example of molecular genetic control of differentiation and is mediated by Notch receptors and their ligands.28 Activation of Notch by its ligand biases a cell not to differentiate; thus, neurogenesis requires inhibition of Notch signaling.29 Notch signaling can control the rate and timing of neuron production, or it can bias progenitors toward an astrocytic fate. Notch signaling activates a complex cascade of molecular switches that culminates in altered gene expression in the differentiating cell.30 Many other types of transcription factors have important roles and serve as examples of gene-driven brain development, including the homeobox, helix-loop-helix, T-box, Winged-helix, and HMG-box families. Each of these families consists of subfamilies; for instance, key homeobox genes include Dlx, Emx, Kx, Otx, Pax, and POU, which control such processes as regional fate, cell type identity, neuronal maturation, and cell migration.16
Once neurons are generated, the next step in their differentiation is migration to the appropriate destination. Each brain region has a specific migration program. In some structures (e.g., cerebral cortex and superior colliculus), migrations are orchestrated to form layered or laminar structures. In most subcortical regions, migrations originate from nuclear structures that generally are not laminar. There are two general types of migration: radial and tangential. Radial migration is movement perpendicular to the wall of the ventricle toward the pial surface; tangential migration is movement parallel to the plane of the ventricle. Radial migration involves the interaction between elongated processes of radial glial cells and migrating immature neurons. Immature neurons are programmed to migrate to a specific location within the wall of the neural tube, where they disengage from the radial glial cell and continue to differentiate. One of the key molecules in regulating this process was identified through the analysis of the reeler mutant mouse, whose reeling behavior reflected the effects of its mutation on functional brain organization.31 In the cerebral cortex of reeler mice, later born neurons fail to migrate past their earlier born counterparts, leading to partial inversion of the usual inside out lamination. The reeler gene encodes a large secreted molecule named Reelin that appears to promote dissociation of neuroblasts from radial glia. Mouse genetic studies have implicated two low-density lipoproteins (VLDLR and ApoER2) as the receptors for the Reelin molecule. Intriguingly, this pathway appears to be significantly disturbed in the neurodevelopmental disorder of schizophrenia.32 Tangential migration of neurons has long been known to occur in the cerebellum and in the rostral migratory stream of the olfactory bulbs. Within the telencephalon, many of the γ-amino butyric acid (GABA)–based local circuit neurons are like cousins rather than like siblings, as they appear to have migrated tangentially from the basal ganglia primordial to the cerebral cortex and hippocampus.19 Progress has also been made in identifying genes that control cytoskeletal processes that are essential for migration. Several of these genes were first identified as causing neuronal migration defects in humans, including lissencephaly-1, doublecortin, and filamin.33 It is of clinical interest that a gene (DCDC2) associated with doublecortin has now been associated with heritable reading disabilities.34
NEURAL PATHWAYS AND SYNAPTIC CONNECTIONS
As the immature neurons and glia migrate from the proliferative ventricular zone to the mantle, they elaborate into more complex cellular structures. Neurons extend thin processes away from their cell body, including multiple dendrites and a single axon that can sometimes traverse long distances to find its targets. (For review, see Tessier-Lavigne and Goodman35 and Grunwald and Klein.36) The growing tip of the axon is called the growth cone. This dynamic weblike structure contains filopodia that extend and retract in multiple directions, seeking potential targets. Certain molecules can attract or repel the growing axons through their specific receptor interactions, whereas other molecules provide paths for the growing axons. Other signals provide more specific information about local branching geometry and pathways. For example, glial cells serve as guideposts for axons. Through fasciculation, late-arriving axons adhere and are bundled together with earlier axons. Molecules on the surface of the axons, some of which are related to immunoglobulins, regulate the pattern of fasciculation and later defasciculation, when the bundle separates.
As axons grow and navigate, they express receptors for guidance molecules that are expressed by neighboring cells.35,37 These processes operate as growth cones extending along specific pathways, the most well-studied of which involve crossing midline structures (commissures), such as the optic chiasm and corpus callosum. Activation of these receptors determines whether an axon grows toward or away from a target cell. At least four conserved families of guidance molecules have been identified: (1) The semaphorins, which constitute a large, 20-member family of soluble, membrane-bound molecules that elicit repulsive signals through two receptor families, neuropilins and plexins; (2) the Slit family of proteins, which consists of three members in mammals and acts through Robo receptors in commissural axons to prevent them from recrossing the midline; (3) the netrin family, whose members can be repulsive or attractive for a growth cone, depending on the receptor on the axon; and (4) the ephrin family, whose members are membrane bound and interact with two families of receptors, EphA and EphB.38 In addition to regulating axonal path finding, these same guidance molecules (i.e., semaphorins, slits, netrins, and ephrins) are involved in controlling aspects of neuron migration. Upon reaching their target, some growth cones form specialized connections with dendrites to become synapses.39 Presynaptic and postsynaptic signals induce the formation and stabilization of molecules on both sides to become specialized synaptic structures.40 On the presynaptic side, for example, synaptic vesicles filled with neurotransmitter are grouped together; on the postsynaptic side, receptor molecules are grouped together into a dense domain that is sometimes located within a dendritic protrusion called a synaptic spine.41
The wiring of complex CNS systems requires a connection of multiple cell types that are located in different positions. The wiring diagram of the visual system is an example of this process that has received a massive amount of experimental attention. The retina contains primary sensory receptor neurons (rods and cones), interneurons (amacrine, bipolar, and horizontal cells), glia, and projection neurons called retinal ganglion cells. The retinal ganglion cells extend optic nerve axons that must make several choices as they proceed to their targets. As they pass the optic chiasm, axons from the temporal retina do not cross, whereas axons from the nasal retina do cross. Intrinsic signals that distinguish nasal and temporal cells (brain factor 1 and 2 transcription factors) help guide the growing axons. Upon exiting the chiasm, the optic axons grow caudally toward their two main targets: the thalamus and the superior colliculus. Branches perpendicular to the optic tracts enter the visual center of the thalamus and form synapses with the lateral geniculate nucleus (LGN). Other optic axons continue caudally to the midbrain into the superior colliculus, where they are sorted into a retinotopic map by the ephrin family receptors.
In the LGN, the optic axons form another retinotopic map. In higher mammals, the LGN is a laminar structure, with each layer connected to only one eye. During development, however, axons from both eyes have processes extending into many LGN layers. Neuronal activity related to visual experience is required for pruning back synapses and for axonal branches to segregate into layers specific for one eye or the other. The projection neurons in the LGN send axons anteriorly into the telencephalon, in which they traverse the striatum in the internal capsule and enter the cerebral cortex. The thalamocortical axons enter the cortex while neurogenesis is still actively occurring and grow into a layer called the intermediate zone that is interposed between the proliferative zones (ventricular zone and subventricular zone) and mantle zones (cortical plate). The thalamocortical fibers then innervate specific regions of neocortex. The neocortex is subdivided into functionally distinct areas, each with its own thalamic inputs. Primary visual cortex receives LGN axons. Cortical maps from other thalamic nuclei determine primary sensory cortex (e.g., auditory cortex), whereas other regions of cerebral cortex are described as associative, because their connections are primarily to other areas of cortex. In humans, some of these associative areas are both enormous and complex, including the dorsolateral prefrontal cortex, which is involved in executive function, and the ventromedial prefrontal cortex, which is involved in complex emotional/social reasoning. Both regions have late and lengthy developmental schedules, and maldevelopment of each is implicated in numerous psychiatric disorders (see Fuster42).
EXPERIENCE-EXPECTANT DEVELOPMENT
A general process observed in many mammalian species is that a surplus of connections is produced, a large portion of which is subsequently eliminated. Evidence for overproduction and partial elimination of synapses during development has been found in many brain regions and species, including cats,43 rodents,44 monkeys,45,46 and humans.47 The overshoot in the number of synapses produced in cortical areas in many animals, including humans, has been estimated to be approximately double the number found in adults48 (see Huttenlocher49 for review). In humans, synaptic density and estimates of total synapse numbers in the visual cortex reach a peak at approximately 8 months of age, and synapse numbers decline thereafter.48 Another important finding by Huttenlocher50 is that the blooming and pruning of synapses in the frontal cortex is substantially delayed; its peak occurs during childhood. Although synapse density and absolute synapse number may differ, depending on other tissue elements, we assume for purposes of this discussion that they are equivalent. A measure that has been interpreted as reflecting synapse overproduction and loss is the volume of regions of the human cerebral cortex and other brain areas, measured by structural magnetic resonance imaging.51 Heterosynchrony (i.e., staggered developmental timing with late development of prefrontal cortex) is unique in humans among the primates.51 The clinical implications of late-developing prefrontal cortex are discussed in a later section.
The process of overproduction and selective elimination of synapses appears to be a mechanism whereby the brain is made ready to capture critical and highly reliable information from the environment. This possibility is supported by several lines of research (described in the following sections) indicating that the pruning into structured patterns of functional neural connections requires appropriate patterns of neural activity that are obtained through experience. These events occur during known critical or sensitive periods. Furthermore, the pruning appears to be driven by competitive interactions between neural connections, so that inactive neural connections are lost and connections that are most actively driven by experience are selectively maintained. “Most active” may refer to synchronous or correlated activation, such as presynaptic activity coincident with postsynaptic activity, as first proposed by Hebb,52 or some mechanism other than the mere frequency of firing. In many cases, it appears that these plastic neural systems have evolved to take advantage of information that could be “expected” for all juvenile members (i.e., it has an adaptive value for the whole species, not just individuals). In many of the experiments described in this section, investigators used interventions that disturb some aspect of the “expected” experience, leading to substantial disruptions of further development. Many patients with developmental and behavioral disorders have had similarly disturbed experiences with subsequently disrupted development.
Visual Deprivation Experiments
Studies of the effects of early visual deprivation have provided some of the strongest examples of experience inducing neural structure during development. Together, they indicate a direct link between patterns of experience-expectant visual information and patterns of neural connectivity. Experimental visual deprivation falls into two main classes. Binocular visual deprivation can be complete, depriving animals of all visual stimuli, or partial, depriving animals of patterned visual stimuli but allowing diffuse, unpatterned stimulation. This deprivation may be achieved, for example, by suturing both eyelids shut (complete deprivation) or by raising animals in complete darkness (partial deprivation). Partial deprivation reduces or distorts visual experience in some manner but allows some effect of experience on neural activity. Complete deprivation in both eyes leads to a loss in complex visuomotor learning and in the precision of neuronal response properties, but it preserves balance in eye dominance and basic perceptual skills.53 In contrast, selective deprivation in one eye during the critical period leads to a drastic reduction in its control over visual cortex neurons and behavior, whereas the nondeprived eye correspondingly gains in control. The degree of recovery from deprivation depends on the species and on the onset and duration of the deprivation period.
Binocular Deprivation
Studies of binocular deprivation have shown that appropriate visual stimulation during certain developmental stages is critical for the development of normal neural connectivity in the visual system. Dark rearing or bilateral lid closure in developing animals results in behavioral, physiological, and structural abnormalities in visual pathways.54–56 The severity and reversibility of the visual impairments are dependent on the onset and duration of the deprivation, corresponding to defined sensitive periods of a given species.57 Even short periods of early visual deprivation can result in impairments in visuomotor skills, such as visually guided placement of the forepaw in cats.58 The structural effects of dark rearing include smaller neuronal dendritic fields, reduced spine density, and reduced numbers of synapses per neuron within the visual cortex.43,59–61 In kittens, for example, developmental binocular deprivation resulted in a 40% reduction in the number of adult visual cortex synapses.43
Selective Deprivation
Experiments in selective deprivation have indicated the importance of specific types of visual experience to normal brain development. For example, kittens reared in a strobe-illuminated environment have plentiful visual pattern experience but are selectively deprived of the normal experience of movement (i.e., movement in the visual field would appear jerky or disconnected). Specific impairments in motion perception have been found in such kittens.62 These animals had visual cortical neurons that were insensitive to visual motion,63 and they exhibited impairment on visuomotor behavioral tasks that involve motion.64
Other researchers limited visual experience to specific visual patterns, or contours. Hirsch and Spinelli65 raised kittens in chambers with one eye exposed to only horizontal stripes and the other eye to only vertical stripes. Physiological recordings of visual cortical neurons from these kittens revealed that neurons were most responsive to stimuli oriented in the direction of the stripes they had experienced. These neurons also occupy twice as much of the visual cortex as neurons sensitive to stripes in nonexposed directions.66 Behaviorally, stripe-reared animals performed best on tests involving stimuli in the orientation to which they were exposed during development.67,68 Unlike dark rearing or bilateral lid closure, stripe rearing does not appear to result in an overall diminishment of neuronal size, but it does alter the orientation of the neuronal dendritic arbors.69,70 Thus, neural function appears to be determined by the pattern, in addition to the overall number, of neural connections. A related, albeit debatable clinical finding in humans who have uncorrected astigmatism in a particular orientation in one eye is reduced acuity in that axis.71
Monocular Deprivation
A great deal of information about experience-expectant processes has been learned from one particular deprivation model. In species with stereoscopic vision, including cats and monkeys, binocular regions of the cortex receive information from each eye via projections from the LGN in adjacent stripes or columns within cortical layer IV, termed ocular dominance columns. With normal experience early in development, the cortical input associated with each eye initially projects in overlapping terminal fields within layer IV. During development in normal animals, these axonal terminal fields are selectively pruned, which results in sharply defined borders between ocular dominance columns in adult animals. The neurons of this layer send convergent input to other layers, made up in large part by binocularly driven neurons.72
Studies of monocular deprivation in stereoscopic animals have shown that the formation of the ocular dominance columns is dependent on competitive interactions between the visual input from each eye.73 In monocularly deprived monkeys, the axons projecting from the deprived eye regress, whereas the axons from the experienced eye do not. This pruning back results in the thinning of the columns corresponding to the deprived eye, whereas the columns of the nondeprived eye are enlarged in relation to those of normal animals.72,74 Thus, the axonal terminals from the dominant eye appear to be selectively maintained at the expense of the inactive input of the deprived eye, in which the excess synapses are eliminated. Physiologically, the number and responsiveness of cells activated by the deprived eye are severely decreased.55 Functionally, monocular deprivation for an extended period during development results in near blindness to visual input in the deprived eye. In contrast, binocular deprivation results principally in a loss of visual acuity. Physiologically, it reduces but does not abolish the response of neurons to visual stimuli.55 It also does not prevent the formation of ocular dominance columns, although the segregation of columns is well below normal.72,75,76 Thus, in binocular deprivation, cortical input from the eyes may be partially maintained in the absence of competing information.
The physiological and anatomical effects of monocular deprivation occur fairly rapidly. Antonini and Stryker74 found that the shrinkage of geniculocortical arbors corresponding to the deprived eye was profound in cats with only 6 to 7 days of monocular deprivation, similar to that found after 33 days of deprivation. Like binocular deprivation, the recovery from the deprivation is sensitive to the time of onset and duration of the deprivation. Monocular deprivation corresponding to the sensitive period of a given species results in enduring impairments and physiological nonresponsiveness,55 whereas even very extensive deprivation in adult animals has little effect.77 In humans, early monocular deprivation resulting from congenital cataracts can have severe effects on acuity, even after treatment, whereas adults who develop cataracts in one eye show little post-treatment impairment.78 The sensitive period for the effects of monocular deprivation can be affected by prior experience. For example, the maximum sensitivity to monocular deprivation in kittens is normally during the fourth and fifth weeks after birth.79,80 Cynader and Mitchell81 found that kittens dark-reared from birth to several months of age maintain a physiological sensitivity to monocular deprivation at ages that normal kittens are insensitive. Dark-reared animals do not, however, simply show normal visual development at this later age. With binocular deprivation early in life, the ocular dominance columns of layer IV do not segregate in a fully normal pattern and do not maintain a structural sensitivity to monocular deprivation effects.75
The implication of these studies is that the sensitive period for experience effects is self-limiting; that is, as supernumerary synapses are eliminated and/or as additional synapses cease to be generated, the capacity for responding, or the experience-sensitive phase, comes to an end. Alternative models might invoke changes in the synapses that survive, leaving them immutable to further pruning or to forces acting at a distance, such as the development of local GABA-based inhibitory systems82,83 or the influence of modulatory axonal activity from other parts of the brain. There are a variety of such proposals, many with supporting data (e.g., α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA] and N-methyl-d-aspartate [NMDA] glutamate receptor distribution and subunit composition84,85), but one merits additional mention because it illustrates the interactions of neurons and glia and has been increasingly implicated in the regulation of synaptic plasticity in both development and adulthood (the latter in damaged or diseased nervous systems). It has long been known that mature axons in the brain and spinal cord show much less tendency to regrow connections after crush or transection than do seemingly equivalent axons outside of the central nervous system. Still-developing pathways in the CNS show greater flexibility. A search for the mechanism revealed an interacting series of signals typified by the molecule Nogo and its receptor. Nogo is produced by the oligodendrocytic myelin surrounding nerve cell pathways and inhibits axonal sprouting in vitro. A function blocking antibody to Nogo facilitates axonal growth after nerve injury to the adult rat spinal cord in vivo86,87 and enhances recovery of behavioral function. Work by McGee and colleagues27 has further implicated Nogo in the termination of visual sensitive periods; a mouse rendered genetically incapable of producing Nogo receptor exhibited sensitivity to monocular deprivation extending well beyond the normal age, which suggests that the Nogo mechanism limits postdevelopmental plasticity in multiple systems. Investigators have proposed involvement of various signaling pathway mechanisms of synapse stabilization and maintenance similar to those described for genotype-driven development, such as α-calcium–calmodulin kinase type II (αCaMKII) for ocular dominance maturation.88,89
Deprivation in Other Sensory Systems
Research in other sensory systems has also demonstrated experience-expectant processes. Within layer IV of the somatosensory cortex in rodents, each whisker is represented by a distinctly clustered group of neurons arranged in what have been called barrels.90 The cell bodies of these neurons form the barrel walls, and a cell-sparse region forms the barrel hollow. In adult animals, the input from each whisker (via the thalamus) terminates predominantly within the barrel hollows. Positioned to receive this input, most of the dendrites of the neurons lining the barrel wall are also oriented into the barrel hollow. This distinctive pattern of barrel walls surrounding a hollow forms postnatally, before which neurons in this region appear homogeneous. The simultaneous regression of dendrites inside the barrel walls and continued growth of dendrites in the barrel hollows mask the expected synapse overproduction and pruning back, inasmuch as the overall process is dendritic expansion.44 Were it not for the location of information provided about the structure of the barrel, this dendritic pruning in the barrel walls would be entirely masked by the simultaneous dendritic extension in the hollows.
Many rodents use their highly developed whiskers, or vibrissae, to navigate in the dark (along with heightened olfactory perception). The whisker barrel region, with its overlapping blooming and pruning of synapses, might therefore be expected to be sensitive to experience. Indeed, Glazewski and Fox91 were able to demonstrate experience-expectant plasticity in the barrel field cortex of young rats by reducing the complement of vibrissae on one side of the muzzle to a single whisker for a period of 7, 20, or 60 days. The vibrissa dominance distribution was shifted significantly toward the spared vibrissa, which gained control of more neurons in barrel cortex, whereas the deprived whiskers lost control. As the deprived whiskers grew back in, they progressively gained back some control of neurons from the spared whiskers. Whisker deprivation had the strongest effects in weanling animals and very little effect in adult rats. However, manipulations that are known to induce plasticity in sensory maps, such as the alternate trimming of individual whiskers, can cause changes in spine dynamics, even in adult animals.92 Whisker deprivation alone has been shown to alter spine dynamics over time, dramatically decreasing elimination rates in young animals but having a more subtle effect on elimination rates in adult animals.93
Humans and some other species appear to have a critical period for attachment, during which the lack of expected nurturing behavior in a timely manner disrupts subsequent emotional development. Human and monkey studies have revealed substantial effects of disrupted attachment on behavior and endocrine function, but little is known about any underlying neural plasticity. The phenomenon known as imprinting (e.g., by which newly hatched chicks learn to recognize mothers) involves both the formation of new synapses and elimination of preexisting synapses.94,95 Imprinting fits the definition of experience-expectant neural plasticity, but it is an example of social rather than perceptual development. Various primate species are differentially sensitive to maternal deprivation,96 and humans appear to be relatively sensitive. For example, rhesus monkeys raised in isolation show enduring heightened responses to stress; abnormal motor behaviors, including stereotyped movements; sexual dysfunction; eating disorders; and various extreme forms of social and emotional dysfunction.97–99 The effects of total social isolation are more severe than partial isolation, which permits visual and auditory interactions with other animals without direct physical contact. Dendritic arbors of neurons within the neocortex100 and the cerebellum101 have been found to be poorly developed in socially deprived monkeys in comparison with normal animals. Martin and associates102 found that socially deprived rhesus monkeys show a marked reduction in the dopaminergic and peptidergic innervations within the caudate-putamen, substantia nigra, and globus pallidus. In addition to evidence of reduced neuronal growth and development, socially deprived monkeys show brain abnormalities more typical of neurological disorders. However, in many of the studies just mentioned, social deprivation was confounded with broader experiential deprivation; therefore, there is still relatively little knowledge about structural brain changes specifically related to social experience.
The fragile X mental retardation syndrome is another phenomenon that might involve a different type of disruption of the systems involved in attachment and social development that leads to pathology. This syndrome is caused by impaired or blocked expression of the fragile X mental retardation protein (FMRP), which results from a triplet repeat mutation in the regulatory region of the gene. In addition to cognitive impairment and learning disabilities, fragile X retardation is often accompanied by symptoms of attention-deficit/hyperactivity disorder (ADHD) and autism.103 There is also abundant evidence for a link between the presence of the fragile X chromosome and psychiatric symptoms. Even individuals with a relatively small expansion of the cytosine-guanine-guanine repeat, who are “unaffected” cognitively, exhibit anxiety disorder (31%), bipolar disorder (23%), panic disorder (17%), and social phobia (11%).104 Men with the fragile X syndrome exhibit elevated degrees of schizoid and schizotypal features.105 “Mood lability” that does not reach criterion for bipolar diagnosis is also common in these patients.106 Investigators frequently report social problems such as “gaze avoidance” and other disturbances.
The failure to develop normal social skills through experience is a plausible origin for these disturbances, and there is reason to suspect a failure of experience-expectant developmental mechanisms in the fragile X syndrome. There are indications that, at least in the cerebral cortex, the maturation and elimination of dendrites and synapses is developmentally delayed, both in humans with fragile X syndrome, according to studies of autopsy tissue and in the knockout mouse model for the syndrome, in which the fragile X gene has been rendered nonfunctional. Dendritic spines in visual, auditory, and somatosensory cortices of humans or mice exhibit an appearance suggestive of an immature structure: longer and thinner than typically developing spines.107–111 In addition, the dendritic pruning typical in the previously described whisker barrel cortex fails to occur in the knockout mouse,112 and a similar failure of typical dendritic pruning appears in the olfactory system.113 Thus, a failure of an experience-expectant mechanism caused by an inherited genetic disorder may underlie some of the behavioral pathology described in the fragile X syndrome, a good example of an interaction, albeit a debilitating one, between genetic and experiential contributions to the development process.
FMRP is a messenger RNA (mRNA) binding protein that appears to function by binding cargo mRNAs in the nucleus and accompanying them through cytoplasmic transport to where they are ultimately translated, often in response to local, synaptically associated signaling pathways.114–117 To date, the mRNAs shown to associate with FMRPs represent a heterogeneous group of encoded proteins, although a number of them appear to be involved directly or indirectly in synaptic plasticity.118–120 Investigators have speculated that FMRP may play a role in modulating protein synthesis and its effects on the synaptic plasticity process involved in developmental information storage,117 but further work is needed to confirm these ideas. However, in view of the essential synaptic role of many of the proteins whose mRNA FMRP is hypothesized to regulate, it is possible that dysregulation of the distribution and accumulation of specific cargo mRNAs accounts for altered synaptic plasticity in these patients. For example, Postsynaptic Density-95 (PSD-95) is a developmentally and environmentally regulated scaffolding protein thought to be intimately involved with plasticity at the synapse, being one of the most highly expressed synaptic proteins whose overexpression, in turn, has dramatic effects on synaptic structure and on spine maturation and dynamics.121,122 In vivo studies in which expression of PSD-95 was tagged with photoactivatable green-fluorescent protein (paGFP) have demonstrated that the protein’s presence in the spine is dynamic, especially in younger animals, and dependent on experience.123 Although direct evidence is still lacking to tie modulation of PSD-95 to FMRP, the mRNA of PSD-95 contains a G-quartet motif thought to be a common feature of FMRP cargoes. In cultured neurons from FMRP knockout mice, metabotropic glutamate receptor (mGluR) activation–induced expression of PSD-95 appears to be deficient, and this lack of regulation could, in theory, explain many of the dendritic spine and plasticity abnormalities observed in the fragile X syndrome. However, the deficits observed in the fragile X syndrome are more likely to arise by the misregulation of a combination of mRNAs. This “cargo hypothesis” of the fragile X syndrome suggests that analysis of FMRP cargoes, in isolation and in combination, may lead not only to targets for treating fragile X symptoms but also to a potential understanding of overlap between fragile X symptoms and those of other genetically complex disorders such as autism.
EXPERIENCE-DEPENDENT DEVELOPMENT
Manipulating Environmental Complexity
In accordance with a tradition established by the well-known Berkeley group (e.g., Bennett et al124), the experimental groups are often referred to as “enriched” and “impoverished.” It is important to emphasize that these are more accurately described in terms of varying degrees of deprivation, in relation to the typical environment of feral rats. Barring considerations of stress or nutrition, we argue that rats in enriched environments experience something close to “normal” brain development and that their brains would more closely resemble those of rats raised in the wild. Although a great deal of useful information can be obtained from laboratory animals, it is important to understand that standard animals are generally overfed, understimulated, and physically out of shape.
Animals raised in complex environments have superior performance on many different types of learning tasks (reviewed by Greenough and Black2). Various studies have suggested that animals living in enriched environments may use more and different types of cues to solve tasks and may possess enhanced information-processing rates and capacities.125–128 Their superiority in complex mazes may rely, in part, on a greater familiarity with complicated spatial arrangements obtained through their rearing environment. These abilities are generalized across a wide range of other learning tests, however, which suggests that the enriched environment’s abilities do not simply reflect specific types of information gathered from the rearing environment. Rather, the brain adaptation to complex environment rearing involves changing how information is processed; that is, the rat in enriched environments appears to have learned to learn better. In the initial publication of this result, Hebb52 noted that the enriched rats learned more quickly than do their laboratory cage–reared counterparts and improved more rapidly as they were subjected to consecutive tests.
Examination of brain structure in animals in complex environments reveals a growth of neurons and synaptic connections in comparison with siblings raised in standard cages. This has been most prominently studied in the visual cortex, which shows an overall increase in thickness, volume, and weight124; an increase in dendritic branching complexity and spine density129,130; more synapses per neuron131,132; and larger synaptic contacts133 in complex-environment rats. The number of synapses in rats living in enriched environments is elevated by approximately 20% to 25% within superficial layers of the visual cortex.132 The visual cortex also shows physiological alterations, which indicates that neuronal firing in animals housed in enriched environments is enhanced in comparison with that in animals housed in standard cages.134 Very comparable anatomical data and concomitant electrophysiological alterations have been reported in cats given complex experience.135,136
The effects of environmental complexity have many different dimensions. The enriched environment’s effects on brain structure cannot be attributed to general metabolic, hormonal, or stress differences across the different rearing conditions.137 Thus, the structural brain changes may be specifically the result of altered neuronal activity and information storage. Young rats living in enriched environments have been shown to add new capillaries to the visual cortex, presumably in support of increased metabolic activity,138 and increased capillary branching alters perfusion capacity.139 Rats reared in a complex environment tend to have slower growth of skeleton and internal organs,137 as well as altered immune system responsiveness.140 Evidence that male and female rats have different responses to the complex environment in both the visual cortex and the hippocampus suggests that sex hormones have a modulatory role in the brain effects in the enriched and individual environments, at least in early postnatal development.141 Multiple brain regions have shown evidence of structural change in animals in enriched environments, including the temporal cortex,142 the striatum,143 the hippocampus,141 the superior colliculus,144 and cerebellum.101,145 Mice reared in a complex environment generate more neurons in the dentate gyrus than do those in the other conditions.146 Investigators have detected significant changes in rat cortical thickness and dendritic branching after just 4 days of enrichment.147 These effects are not limited to young animals, inasmuch as changes in neuronal dendrites and synapses in adult rats placed in the complex environment are substantial, although less so than those found in rats reared from weaning in enriched environments.148,149
Structural Effects of Learning
Although a variety of activities occur in an enriched environment, learning is clearly an important one. If learning in the enriched environment results in structural brain changes, then similar changes would be expected in animals in response to a variety of training procedures. Such studies have indeed demonstrated that major brain structure changes occur during learning. These changes have been found in the specific brain regions apparently involved in the learning. For example, training in complex mazes necessitating visuospatial memory has been found to result in increased dendritic arbors of the visual cortex in adult rats.150 When split-brain procedures were performed and unilateral occluders placed on one eye, dendrites of neurons in the monocular cortex mediating vision in the unoccluded eye showed greater growth than in the visually inexperienced hemisphere of the visual cortex.151
Training animals on motor learning tasks results in site-specific neuronal changes. Rats extensively trained to use one forelimb to reach through a tube to receive highly attractive food showed dendritic growth within the region of the cortex involved in forelimb function,152 in comparison with controls. When allowed to use only one forelimb for reaching, the dendritic arborizations of rats within the cortex opposite the trained forelimb were significantly increased in relation to the cortex opposite the untrained forelimb. Furthermore, reach training selectively alters only certain subpopulations of neurons; for example, layer II/III pyramidal neurons showed forked apical shafts.153 Reach training may produce similar results in developing animals as well. Rat pups trained to reach with one forelimb over 9 days, beginning at weaning, exhibited increased cortical thickness in the hemisphere opposite the trained limb, in comparison to the nontrained limb.154 A review and meta-analysis of more than 100 studies concluded that the neocortex tends to respond to learning with synaptogenesis, whereas the hippocampal formation tends to alter the structure of existing synapses, in accord with the roles of these two structures in persisting memory.155
A critical question is whether these training-induced brain changes result from special processes specifically involved in brain information storage or are simply an effect of increased activity within the affected brain systems; that is, do these changes reflect some generally trophic nature of experience, comparable to muscle hypertrophy with exercise, or do they correspond to changes in the brain’s “wiring diagram” that actually subserve memory? A motor learning paradigm in which rats are required to master several new complex motor coordination tasks (“acrobatic” rats) addressed this question. These animals showed increased numbers of synapses per Purkinje neuron within the cerebellum, in comparison with inactive controls.156 In contrast, animals exhibiting greater amounts of motor activity in running wheels or treadmills in which little information was learned156 or yoked-control animals that made an equivalent amount of movement in a simple straight alley157 did not show significant alterations in synaptic connections in the cerebellum. Thus, learning, and not simply the repetitive use of synapses that may occur during dull physical exercise, is selectively associated with synaptogenesis in the cerebellum. Subsequent research has reached a similar conclusion for synaptic changes in the motor cortex that arise after learning, in the same behavioral paradigm.158
Interestingly, the exercising animals did show some structural changes: The increase in density of capillaries in the involved region of cerebellum corresponded to expectations for new blood vessel development to support increased metabolic demand.156 This indicates that the brain can independently generate adaptive changes in different cellular components. When metabolic “stamina” is required, vasculature is added. When motor skills need to be learned or refined, new synapses modify neural organization. In the enriched environment, both exercise and learning effects appear to be combined.
Cerebellar synaptic changes are accompanied by functional changes in electrophysiological recording. Stimulation of parallel fibers, constituting the primary excitatory input to Purkinje cells and accounting for the bulk of the added synapses, evoked larger postsynaptic changes in acrobatic rats than in motor activity controls,159 which indicates that the training-induced synapses are functional. This effect probably also reflects increased parallel fiber input to inhibitory neurons, also evident in morphological changes of the acrobatic rats.160
We describe one example of neural plasticity and therapeutic training in an animal model of a clinical disorder, because the literature is quite extensive: There is increasing evidence that the postnatal environment strongly influences the outcome of prenatal exposure to alcohol in fetal alcohol syndrome and exposure.161 Animal models of this important developmental disorder have been carefully developed.162 Hannigan and colleagues163 found that raising rats in a complex environment greatly attenuated the behavioral effects of prenatal exposure to low to moderate levels of alcohol. Animals with fetal alcohol syndrome that were raised from weaning in isolation showed ataxia and impairments in learning spatial tests. These alcohol-induced effects were largely absent in rats with fetal alcohol syndrome raised in a complex environment. Although the investigators found no indication of rehabilitation effects on hippocampal structure, a program of forced motor skill training (similar to the “acrobatic” training described previously) nearly eliminated motor dysfunction in rats with fetal alcohol exposure and substantially increased synapse number in their cerebellar cortex.164–166 The intervention did not reverse the substantial loss of neurons in the cerebellum resulting from alcohol treatment, but the new synapses appeared to support enhanced motor performance. This work suggests that intervention focused on the skills that have been lost as a result of CNS damage can have potentially important therapeutic effects.
EVIDENCE FOR HUMAN NEURAL PLASTICITY
One kind of human experience-expectant process sensitive to selective deprivation involves perceptual mismatch from both eyes, such as when one eye deviates outward (strabismus) during early development. As in the cat and monkey studies described earlier, if the two eyes are sending competing and conflicting signals to the visual cortex during the sensitive period, the brain effectively “shuts down,” or becomes insensitive to input from, the nondominant eye. In humans, the resulting perceptual disorder, amblyopia (or “lazy eye”), results in clear perceptual deficits if surgery does not correct this visual misalignment during the critical period. The strabismus-related perceptual deficit was the first and still best-established example of human neural plasticity.167 Technological methods, such as positron-emission tomography, has demonstrated that patients with uncorrected strabismus use different areas of the visual cortex for visual processing than do normal controls.168 Although the timing, regulation, and structural changes of this sensitive period need further study, the early evidence suggests a clear parallel to the described studies of kittens with selective deprivation of vision.
Another developmental process with innate roots but nonetheless quite dependent on early experience is language acquisition.169 Although the question of whether language has an innate deep structure is still debated, it is clear that children rapidly acquire an enormous amount of vocabulary, grammar, and related information. For middle-income American families, the rate of vocabulary acquisition is directly related to the amount of verbal stimulation that the mother provides.170 There is apparently a sensitive period for acquiring the ability to discriminate speech contrasts. For example, Kuhl171 reported that before about 6 months of life, infants from English-speaking homes are able to discriminate speech contrasts from a variety of languages, including Thai, Czech, and Swedish, much the way native adult speakers are able to. However, sometime between ages 6 and 12 months, this ability is gradually lost. After this age, infants become more like adults who are most proficient in discriminating the speech contrasts from their native language,171 possibly paralleling synapse elimination as described by Huttenlocher.50 Early exposure to the native language can be interpreted as a “neural commitment” of the brain’s resources to the acoustic properties of the native language, in such a way that this dedication of resources interferes with any subsequent foreign language learning. Evidence for this commitment can be observed in both behavioral changes and in imaging data, such as magnetoencephalography.172
There exists some preliminary evidence that humans can alter brain function with extensive training, corresponding to the experience-dependent processes described previously. For example, using functional magnetic resonance imaging (fMRI) to measure regional blood flow in the brain, Karni and associates173 demonstrated increased cortical involvement after training subjects in a finger-tapping sequence. Elbert and colleagues174 showed substantial expansion of cortical involvement associated with the amount of training to play the violin. No one has yet shown directly that humans produce new synapses with this type of learning, but the fMRI changes are what would be expected if synaptogenesis were occurring in an experience-dependent process. Similarly, researchers have found that brain myelination patterns differ in musicians, depending on the ages during which training occurred175; this again emphasizes that plasticity extends beyond neurons and their synapses to glia as partners. Another provocative finding involved the relatively enlarged posterior hippocampi of London taxi drivers, who were required to encode massive amounts of spatial information for their occupation. Although synaptogenesis cannot be demonstrated directly, it is one of very few examples of structural plasticity in healthy humans.176 An indirect line of evidence indicated that healthy, well-educated subjects had greater dendritic branching of large pyramidal neurons in the language cortex than did less well-educated subjects,177 which is consistent with storage of education information in the neuropil. However, cause and effect remain obscure in this intriguing study.
Impressive findings of cortical reorganization after peripheral injury in adult humans correspond well to the previously described findings in nonhuman primates.178 For example, Ramachandran and coworkers179 examined adults who had experienced various forms of amputation, such as the forearm. One such individual experienced sensation in the limb that had, in fact, been amputated (i.e., “phantom limb phenomenon”). Ramachandran and coworkers then examined sensitivity to tactile stimulation along the regions of the face known to innervate the somatosensory cortex adjacent to the area previously innervated by the missing limb. When this region of the face was lightly stimulated, the patient reported sensation in both the face and the missing limb. Ramachandran and coworkers were eventually able to determine the degree to which the cortical surface had been reorganized to subsume the area previously occupied by the missing limb. Brain reorganization after trauma appears to be very complex, and this subject is beyond the scope of this chapter. In addition to developmental changes in the ability of adjacent tissue to reorganize,180 the patient’s learning of new behaviors is also an important component of neurobehavioral recovery.181
CLINICAL APPLICATIONS
Gene-driven processes are important in constructing a brain that is enormously complex before any effects of experience. Much has been learned about the genetic and molecular basis of histogenesis, migration, and differentiation. These complex brain structures are the foundation upon which any subsequent modification by experience is made. If a child is born with a different brain, his or her experience of the world may be dramatically different. For example, if an infant with cerebral palsy is unable to control eye and hand movements smoothly, then processes involving coordinated information from both sources will be disrupted. Even if the subsequent experience-expectant and experience-dependent processes are functionally unimpaired, the experience itself is distorted by the disorder and will not be appropriately used. Many of the most common disorders of developmental and behavioral pediatrics (e.g., autism, ADHD, mental retardation, epilepsy, and learning disabilities) originate in both neurodevelopmental alteration and distorted experience. One widely applied clinical implication is that children whose experience is distorted or impoverished by neuropathology can benefit from corrective or enabling technology to restore the quality of experience. For example, the mandate to provide hearing- or vision-impaired children with corrective devices as early as possible may be conceptualized as important in restoring the quality of their experience. Similarly, computer technology enhances the experience of children with motor problems by enabling them to better control their actions and to greatly improve their communication.182 In summary, the environment of children with disorders of brain development must be adapted to their needs; otherwise, their experience will be further distorted or impoverished, and development will go astray. As the understanding of neurodevelopmental disorders grows, the ability to provide evidence-based treatments to correct drifting developmental trajectories will also improve.
Some children with normal brains at birth may suffer from the effects of impoverishment or the poor quality of information during early development. As previously described, the effect of uncorrected, conflicting visual input caused by early strabismus is functional blindness in one eye. Similarly, the amount of verbal experience provided by a child’s mother can significantly determine the child’s vocabulary. Children’s brain development can probably be substantially influenced by the quality and the amount of their experience. Finally, the child’s early emotional relationship with a caregiver is probably important in fostering subsequent, healthy emotional development. For example, a child’s attachment to a primary caretaker is known to develop over the first 2 years of life and, perhaps most critically, between 6 and 18 months of life. Failure to develop healthy attachment relationships may ultimately prove maladaptive to both the emotional and cognitive domains (e.g., securely attached children tend to be better at problem solving than are insecurely attached children). The vulnerability of the circuits that are critically involved in emotion, emotion regulation, and memory (e.g., corticolimbic) is the likely basis for the existence of this sensitive period. Collectively, these observations support efforts to enrich experience for young children (e.g., the Head Start program). This argument is probably strongest with regard to cognitive development, but it probably also extends to other important aspects of development, such as social abilities or attachment. We suspect that neural plasticity underlies the lasting and profound effects of early experience on emotional regulation and social behavior, so that humans have a sensitive period for emotional and social development.183 It is thus important that researchers determine what brain regions may use associated experience-expectant processes and early experience to shape the brain. This knowledge may guide clinical efforts to redirect brain development in a more timely or focused manner. Advocacy and support for such interventions mandate evidence of their effectiveness.
A corollary of early and intensive intervention is that clinicians should not allow children to languish with active symptoms of their disorder. Delays in intervention may “waste” a sensitive period, making subsequent clinical intervention much more difficult and possibly leading to relatively irreversible pathology. In addition, both experience-expectant and experience-dependent mechanisms may continue to operate in various pathological states, and a child with “pathological” experience in these circumstances may very well acquire neuropathology instead of functional connections. Consider what may happen to a child’s brain structure after years of experience with auditory hallucinations, drug abuse, depression, or violence. The human brain has delayed prefrontal cortical development to adolescence, making young people prone to forms of executive function problems and emotional instability and leaving this important brain region vulnerable to damage from alcohol and drugs.184 Exposure to addictive substances can result in lasting brain changes in cortical and subcortical systems, affecting hedonic drive, developing cognitive systems, and emotional regulation.185 The effects of trauma on the developing brain are very complex. For example, Pollak and colleagues186 demonstrated that children exposed to early violence had lasting changes in their emotional system, arousal responses, and their perception of negative expressions, as if they approached the world differently than did children who had not been abused; this finding emphasizes the importance of avoiding the consequences of negative experiences during development.
Considerable work remains to be done in neuroscience, developmental psychology, and the affiliated clinical disciplines, because interactions between children, altered brain structure, and the environment are very complex.187 One example among many gene-environment interactions is the specific interaction of an adverse childhood environment with the genotype for low monoamine oxidase A activity, which results in an increased risk for conduct disorder in children188 (Fig. 4-2). In addition to the genetic factors, prenatal exposure to smoking or alcohol increases the risk of a complex neurodevelopmental disorder such as ADHD.189 Troubled or mentally ill parents serve as proximal mediators between children and the environment and can thus place a burden on children’s mental health.190 On the other hand, children with ADHD can present with such a burden of illness for parents that divorce is a common outcome.191 Developmental-behavioral pediatricians are well aware of the complexity of these interactions, because they work closely with families, social institutions, and their developing patients.
On a positive note, children, like many organisms, are resilient and can thrive in a wide variety of environments. We suggest that children are actively involved in obtaining and structuring developmentally appropriate experience. One well-known aspect of this self-structuring of experience is play, a developmental process used by many mammalian species to improve skills and learn socialization. Just as Winnicott192 proposed that a “good enough mother” can often suffice for normal emotional development, we suggest that many children can extract what they need from a “good enough environment.” Clearly, parents play an important role in facilitating a child’s experience, and their role should be considered extensively in studies of what might constitute a “good enough environment.” Society and clinicians often need only to provide “just enough” of what children require to recover and resume a healthier trajectory.193 The self-correcting and self-organizing properties of brain development may be among the most impressive themes of this chapter.
1 Black JE, Greenough WT. Induction of pattern in neural structure by experience: Implications for cognitive development. Lamb ME, Brown AL, Rogoff B, editors. Advances in Developmental Psychology. Hillsdale, NJ: Erlbaum; 1986;4:1-50.
2 Greenough WT, Black JE. Induction of brain structure by experience: Substrates for cognitive development. Gunnar M, Nelson CA, editors. Behavioral Developmental Neuroscience. Minnesota Symposia on Child Psychology. Hillsdale, NJ: Erlbaum; 1992;24:155-200.
3 Black JE, Jones TA, Nelson CA, et al. Neural plasticity. In: Alessi N, editor. The Handbook of Child and Adolescent Psychiatry, Volume 4: Varieties of Development, Section I: Developmental Neuroscience. New York: Wiley; 1998:31-53.
4 Waddington CH. Concepts of development. In: Tobach E, Aronson LR, Shaw E, editors. The Biopsychology of Development. New York: Academic Press; 1971:17-23.
5 Chaudhari N, Hahn WE. Genetic expression in the developing brain. Science. 1983;220:924-928.
6 Milner RJ, Sutcliffe JG. Gene expression in rat brain. Nucleic Acids Res. 1983;11:5497-5520.
7 Gyllensten L, Malmfors T. Myelinization of the optic nerve and its dependence on visual function-A quantitative investigation in mice. J Embryol Exp Morphol. 1963;11:255-266.
8 Demerens C, Stankoff B, Logak M, et al. Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci U S A. 1996;93:9887-9892.
9 Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361:258-260.
10 Uesaka N, Hirai S, Maruyama T, et al. Activity dependence of cortical axon branch formation: A morphological and electrophysiological study using organotypic slice cultures. J Neurosci. 2005;25:1-9.
11 Hawrylak N, Greenough WT. Monocular deprivation alters the morphology of glial fibrillary acidic protein-immunoreactive astrocytes in the rat visual cortex. Brai n Res. 1995;683:187-199.
12 Jessell TM, Sanes JR. Development. The decade of the developing brain. Curr Opin Neurobiol. 2000;10:599-611.
13 Wilson SI, Edlund T. Neural induction: Toward a unifying mechanism. Nat Neurosci. 2001;4(Suppl):1161-1168.
14 Smith JL, Schoenwolf GC. Neurulation: Coming to closure. Trends Neurosci. 1997;20:510-517.
15 Rakic P. A Small step for the cell, a giant leap for mankind: A hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383-388.
16 Wilson SW, Rubenstein JL. Induction and dorsoventral patterning of the telencephalon. Neuron. 2000;28:641-651.
17 Evans PD, Anderson JR, Vallender EJ, et al. Adaptive evolution of ASPM, a major determinant of cerebral cortical size in humans. Hum Mol Genet. 2004;13:489-494.
18 Sidman R, Rakic P. Development of the human central nervous system. In: Haymaker W, Adams RD, editors. Histology and Histopathology of the Nervous System. Springfield, IL: CC Thomas; 1982:3-143.
19 Marin O, Rubenstein JL. A long, remarkable journey: Tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780-790.
20 Caviness VSJr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: A general developmental and evolutionary model. Trends Neurosci. 1995;18:379-383.
21 Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: A review. Brain Res. 1973;62:1-35.
22 Brazel CY, Romanko MJ, Rothstein RP, et al. Roles of the mammalian subventricular zone in brain development. Prog Neurobiol. 2003;69:49-69.
23 Rakic P. Neuronal migration and contact guidance in the primate telencephalon. Postgrad Med J. 1978;54(Suppl 1):25-40.
24 Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61-83.
25 Rakic P. Neurons in rhesus monkey visual cortex: Systematic relation between time of origin and eventual disposition. Science. 1974;183:425-427.
26 Lemke G. Glial control of neuronal development. Annu Rev Neurosci. 2001;24:87-105.
27 McGee AW, Yang Y, Fischer QS, et al. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222-2226.
28 Justice NJ, Jan YN. Variations on the Notch pathway in neural development. Curr Opin Neurobiol. 2002;12:64-70.
29 Gaiano N, Fishell G. The role of Notch in promoting glial and neural stem cell fates. Annu Rev Neurosci. 2002;25:471-490.
30 Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517-530.
31 Rice DS, Curran T. Role of the Reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005-1039.
32 Eastwood SL, Harrison PJ. Interstitial white matter neurons express less Reelin and are abnormally distributed in schizophrenia: Towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol Psychiatry. 2003;8:769-831. 821
33 Gleeson JG, Walsh CA. Neuronal migration disorders: From genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352-359.
34 Meng H, Smith SD, Hager K, et al. DCDC2 is associated with reading disability and modulates neuronal development in the brain. Proc Natl Acad Sci USA. 2005;102:17053-17058.
35 Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123-1133.
36 Grunwald IC, Klein R. Axon guidance: Receptor complexes and signaling mechanisms. Curr Opin Neurobiol. 2002;12:250-259.
37 Brose K, Tessier-Lavigne M. Slit proteins: Key regulators of axon guidance, axonal branching, and cell migration. Curr Opin Neurobiol. 2000;10:95-102.
38 Kullander K, Klein R. Mechanisms and functions of Eph and Ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475-486.
39 Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791-805.
40 Benson DL, Colman DR, Huntley GW. Molecules, maps and synapse specificity. Nat Rev Neurosci. 2001;2:899-909.
41 Jan YN, Jan LY. Dendrites. Genes Dev. 2001;15:2627-2641.
42 Fuster JM. The Prefrontal Cortex: Anatomy, Physiology, and Neuropsychology of the Frontal Lobe, 3rd ed. Philadelphia: Lippincott-Williams & Wilkins, 1997.
43 Cragg BG. The development of synapses in the visual system of the cat. J Comp Neurol. 1975;160:147-166.
44 Greenough WT, Chang F-LF. Plasticity of synapse structure and pattern in the cerebral cortex. Peters A, Jones EG, editors. Cerebral Cortex. New York: Plenum Press; 1988;7:391-440.
45 Boothe RG, Greenough WT, Lund JS, et al. A quantitative investigation of spine and dendrite development of neurons in visual cortex (area 17) of Macaca nemestrina monkeys. J Comp Neurol. 1979;186:473-489.
46 Bourgeois JP, Goldman-Rakic PS, Rakic P. Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cereb Cortex. 1994;4:78-96.
47 Conel JL. The Postnatal Development of the Human Cerebral Cortex. Cambridge, MA: Harvard University Press; 1939–1967;1–8.
48 Huttenlocher PR, de Courten C. The development of synapses in striate cortex of man. Hum Neurobiol. 1987;6:1-9.
49 Huttenlocher PR. Synaptogenesis, synapse elimination, and neural plasticity in human cerebral cortex. In: Nelson CA, editor. Minnesota Symposia on Child Psychology, Volume 27: Threats to Optimal Development: Integrating Biological, Psychological, and Social Risk Factors. Hillsdale, NJ: Erlbaum; 1994:35-54.
50 Huttenlocher PR. Synaptic density in human frontal cortex-Developmental changes and effects of aging. Brain Res. 1979;163:195-205.
51 Giedd JN, Blumenthal J, Jeffries NO, et al. Brain development during childhood and adolescence: A longitudinal MRI study. Nat Neurosci. 1999;2:861-863.
52 Hebb DO. The Organization of Behavior; a Neuropsychological Theory. New York: Wiley, 1949.
53 Zablocka T, Zernicki B. Partition between stimuli slows down greatly discrimination learning in binocularly deprived cats. Behav Brain Res. 1990;36:13-19.
54 Riesen AH. Effects of visual deprivation on perceptual function and the neural substrate. In: DeAjuriaguerra J, editor. Symposium Bel Air II, Desafferentation Experimentale Et Clinique. Geneva: George & Cie; 1965:47-66.
55 Wiesel TN, Hubel DH. Comparison of the effects of unilateral and bilateral eye closure on cortical unit responses in kittens. J Neurophysiol. 1965;28:1029-1040.
56 Michalski A, Wrobel A. Correlated activity of lateral geniculate neurones in binocularly deprived cats. Acta Neurobiol Exp (Wars). 1994;54:3-10.
57 Walk RD, Gibson EJ. A comparative and analytical study of visual depth perception. Psychol Monogr. 1961;75(15):1-44.
58 Crabtree JW, Riesen AH. Effects of the duration of dark rearing on visually guided behavior in the kitten. Dev Psychobiol. 1979;12:291-303.
59 Gabbott PL, Stewart MG. Quantitative morphological effects of dark-rearing and light exposure on the synaptic connectivity of layer 4 in the rat visual cortex (area 17). Exp Brain Res. 1987;68:103-114.
60 Coleman PD, Riesen AH. Environmental effects on cortical dendritic fields. I. Rearing in the dark. J Anat. 1968;102(P t 3):363-374.
61 Valverde F. Rate and extent of recovery from dark rearing in the visual cortex of the mouse. Brain Res. 1971;33:1-11.
62 Marchand AR, Cremieux J, Amblard B. Early sensory determinants of locomotor speed in adult cats: II. Effects of strobe rearing on vestibular functions. Behav Brain Res. 1990;37:227-235.
63 Cynader M, Chernenko G. Abolition of direction selectivity in the visual cortex of the cat. Science. 1976;193:504-505.
64 Hein A, Held R, Gower EC. Development and segmentation of visually controlled movement by selective exposure during rearing. J Comp Physiol Psychol. 1970;73:181-187.
65 Hirsch HV, Spinelli DN. Visual experience modifies distribution of horizontally and vertically oriented receptive fields in cats. Science. 1970;168:869-871.
66 Sengpiel F, Stawinski P, Bonhoeffer T. Influence of experience on orientation maps in cat visual cortex. Nat Neurosci. 1999;2:727-732.
67 Corrigan JG, Carpenter DL. Early selective visual experience and pattern discrimination in hooded rats. Dev Psychobiol. 1979;12:67-72.
68 Pettigrew JD, Freeman RD. Visual experience without lines: Effect on developing cortical neurons. Science. 1973;182:599-601.
69 Coleman PD, Flood DG, Whitehead MC, et al. Spatial sampling by dendritic trees in visual cortex. Brain Res. 1981;214:1-21.
70 Tieman SB, Hirsch HV. Exposure to lines of only one orientation modifies dendritic morphology of cells in the visual cortex of the cat. J Comp Neurol. 1982;211:353-362.
71 Banks MS. Infant Refraction and Accommodation. Int Ophthalmol Clin. 1980;20:205-232.
72 LeVay S, Wiesel TN, Hubel DH. The development of ocular dominance columns in normal and visually deprived monkeys. J Comp Neurol. 1980;191:1-51.
73 Shatz CJ. Impulse activity and the patterning of connections during CNS development. Neuron. 1990;5:745-756.
74 Antonini A, Stryker MP. Rapid remodeling of axonal arbors in the visual cortex. Science. 1993;260:1819-1821.
75 Mower GD, Caplan CJ, Christen WG, et al. Dark rearing prolongs physiological but not anatomical plasticity of the cat visual cortex. J Comp Neurol. 1985;235:448-466.
76 Swindale NV. Role of visual experience in promoting segregation of eye dominance patches in the visual cortex of the cat. J Comp Neurol. 1988;267:472-488.
77 Blakemore C, Garey LJ, Vital-Durand F. The physiological effects of monocular deprivation and their reversal in the monkey’s visual cortex. J Physiol. 1978;283:223-262.
78 Bowering ER, Maurer D, Lewis TL, et al. Sensitivity in the nasal and temporal hemifields in children treated for cataract. Invest Ophthalmol Vis Sci. 1993;34:3501-3509.
79 Hubel DH, Wiesel TN. The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J Physiol. 1970;206:419-436.
80 Olson CR, Freeman RD. Monocular deprivation and recovery during sensitive period in kittens. J Neurophysiol. 1978;41:65-74.
81 Cynader M, Mitchell DE. Prolonged sensitivity to monocular deprivation in dark-reared cats. J Neurophysiol. 1980;43:1026-1040.
82 Fagiolini M, Fritschy JM, Low K, et al. Specific GABAA circuits for visual cortical plasticity. Science. 2004;303:1681-1683.
83 Fagiolini M, Hensch TK. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404:183-186.
84 Durand GM, Zukin RS. Developmental regulation of mRNAs encoding rat brain kainate/AMPA receptors: A Northern analysis study. J Neurochem. 1993;61:2239-2246.
85 Cao Z, Lickey ME, Liu L, et al. Postnatal development of Nr1, NR2A and NR2B immunoreactivity in the visual cortex of the rat. Brain Res. 2000;859:26-37.
86 Freund P, Schmidlin E, Wannier T, et al. Nogo-A-specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nat Med. 2006;12:790-792. Nat Med. 2006;12:1220. [erratum in:]
87 Liebscher T, Schnell L, Schnell D, et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol. 2005;58:706-719.
88 Taha S, Hanover JL, Silva AJ, et al. Autophosphorylation of aCaMKII is required for ocular dominance plasticity. Neuron. 2002;36:483-491.
89 Tropea D, Kreiman G, Lyckman A, et al. Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat Neurosci. 2006;9:660-668.
90 Woolsey TA, Van der Loos H. The structural organization of layer IV in the somatosensory region (SI) of mouse cerebral cortex. The description of a cortical field composed of discrete cytoarchitectonic units. Brain Res. 1970;17:205-242.
91 Glazewski S, Fox K. Time course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J Neurophysiol. 1996;75:1714-1729.
92 Holtmaat A, Wilbretcht L, Knott GW, et al. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979-983.
93 Zuo Y, Yang G, Kwon E, et al. Long-term sensory deprivation prevents dendritic spine loss in primary somatosensory cortex. Nature. 2005;436:261-265.
94 Horn G. Imprinting, learning, and memory. Behav Neurosci. 1986;100:825-832.
95 Patel SN, Rose SN, Stewart MG. Training induced dendritic spine density changes are specifically related to memory formation processes in the chick. Gallus domesticus. Brain Res. 1988;463:168-173.
96 Sackett GP, Novak MSFX, Kroeker R. Early experience effects on adaptive behavior: Theory revisited. Ment Retard Dev Disabil Res Rev. 1999;5:30-40.
97 Harlow HF, Harlow MK. The affectional systems. Schrier AM, Harlow HF, Stollnitz F, editors. Behavior of Non-Human Primates. New York: Academic Press; 1965;2:287-334.
98 Sackett GP. Prospects for research on schizophrenia. 3. Neurophysiology. Isolation-rearing in primates. Neurosci Res Progr Bull. 1972;10:388-392.
99 Bennett AJ, Lesch KP, Heils A, et al. Early experience and serotonin transporter gene variation interact to influence primate CNS function. Mol Psychiatry. 2002;7:118-122.
100 Struble RG, Riesen AH. Changes in cortical dendritic branching subsequent to partial social isolation in stumptailed monkeys. Dev Psychobiol. 1978;11:479-486.
101 Floeter MK, Greenough WT. Cerebellar plasticity: Modification of Purkinje cell structure by differential rearing in monkeys. Science. 1979;206:227-229.
102 Martin LJ, Spicer DM, Lewis MH, et al. Social deprivation of infant rhesus monkeys alters the chemo-architecture of the brain: I. Subcortical regions. J Neurosci. 1991;11:3344-3358.
103 Baumgardner TL, Reiss AL, Freund LS, et al. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics. 1995;95:744-752.
104 Franke P, Maier W, Hautzinger M, et al. Fragile X carrier females: evidence for a distinct Psychopathological Phenotype? Am J Med Genet. 1996;64:334-339.
105 Kerby DS, Dawson BL. Autistic features, personality, and adaptive behavior in males with the fragile X syndrome and no autism. Am J Ment Retard. 1994;98:455-462.
106 Sobesky WE, Hull CE, Hagerman RJ. Symptoms of schizotypal personality disorder in fragile X women. J Am Acad Child Adolesc Psychiatry. 1994;33:247-255.
107 Hinton VJ, Brown WT, Wisniewski K, et al. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41:289-294.
108 Comery TA, Harris JB, Willems PJ, et al. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401-5404.
109 Irwin SA, Patel B, Idupulapati M, et al. Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: A quantitative examination. Am J Med Genet. 2001;98:161-167.
110 Irwin SA, Idupulapati M, Gilbert ME, et al. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am J Med Genet. 2002;111:140-146.
111 Galvez R, Greenough WT. Sequence of abnormal dendritic spine development in primary somatosensory cortex of a mouse model of the fragile X mental retardation syndrome. Am J Med Genet A. 2005;135:155-160.
112 Galvez R, Gopal AR, Greenough WT. Somatosensory cortical barrel dendritic abnormalities in a mouse model of the fragile X mental retardation syndrome. Brain Res. 2003;971:83-89.
113 Galvez R, Smith RL, Greenough WT. Olfactory bulb mitral cell dendritic pruning abnormalities in a mouse model of the fragile-X mental retardation syndrome: Further support for FMRP’s involvement in dendritic development. Brain Res Dev Brain Res. 2005;157:214-216.
114 Antar LN, Afroz R, Dictenberg JB, et al. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and Fmr1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648-2655.
115 Antar LN, Dictenberg JB, Plociniak M, et al. Localization of FMRP-associated mRNA granules and requirement of micro tubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 2005;4:350-359.
116 Bagni C, Greenough WT. From mRNP trafficking to spine dysmorphogenesis: The roots of fragile X syndrome. Nat Rev Neurosci. 2005;6:376-387.
117 Grossman AW, Elisseou NM, McKinney BC, et al. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158-164.
118 Brown V, Jin P, Ceman S, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001;107:477-487.
119 Darnell JC, Jensen KB, Jin P, et al. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function.[Comment]. Cell. 2001;107:489-499.
120 Miyashiro KY, Beckel-Mitchener A, Purk TP, et al. RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron. 2003;37:417-431.
121 El-Husseini AE, Schnell E, Chetkovich DM, et al. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364-1368.
122 Chen X, Vinade L, Leapman RD, et al. Mass of the postsynaptic density and enumeration of three key molecules. Proc Natl Acad Sci U S A. 2005;102:11551-11556.
123 Gray NW, Weimer RM, Bureau I, et al. Rapid redistribution of synaptic PSD-95 in the neocortex in vivo. PLoS Biol. 2006;4:e370.
124 Bennett EL, Diamond MC, Krech D, et al. Chemical and anatomical plasticity brain. Science. 1964;146:610-619.
125 Ravizza RJ, Hershberger AC. The effect of prolonged motor restriction upon later behavior of the rat. Psychol Rec. 1966;16:73-80.
126 Thinus-Blanc C. Volume discrimination learning in golden hamsters: Effects of the structure of complex rearing cages. Dev Psychobiol. 1981;14:397-403.
127 Greenough WT, Wood WE, Madden TC. Possible memory storage differences among mice reared in environments varying in complexity. Behav Biol. 1972;7:717-722.
128 Juraska JM, Henderson C, Muller J. Differential rearing experience, gender, and radial maze performance. Dev Psychobiol. 1984;17:209-215.
129 Holloway RLJr. Dendritic branching: Some preliminary results of training and complexity in rat visual cortex. Brain Res. 1966;2:393-396.
130 Volkmar FR, Greenough WT. Rearing complexity affects branching of dendrites in the visual cortex of the rat. Science. 1972;176:1445-1447.
131 Turner AM, Greenough WT. Synapses per neuron and synaptic dimensions in occipital cortex of rats reared in complex, social, or isolation housing. Acta Stereol. 1983;2(Suppl):239-244.
132 Turner AM, Greenough WT. Differential rearing effects on rat visual cortex synapses. I. Synaptic and neuronal density and synapses per neuron. Brain Res. 1985;329:195-203.
133 Sirevaag AM, Greenough WT. Differential rearing effects on rat visual cortex synapses. II. Synaptic morphometry. Brain Res. 1985;351:215-226.
134 Greenough WT, Wang X. Altered postsynaptic response in the visual cortex in vivo of rats reared in complex environments. Abstr Soc Neurosci. 1993;19(11):19. 164
135 Beaulieu C, Colonnier M. Effect of the richness of the environment on the cat visual cortex. J Comp Neurol. 1987;266:478-494.
136 Beaulieu C, Cynader M. Effect of the richness of the environment on neurons in cat visual cortex. I. Receptive field properties. Brain Res Dev Brain Res. 1990;53:71-81.
137 Black JE, Sirevaag AM, Wallace CS, et al. Effects of complex experience on somatic growth and organ development in rats. Dev Psychobiol. 1989;22:727-752.
138 Black JE, Sirevaag AM, Greenough WT. Complex experience promotes capillary formation in young rat visual cortex. Neurosci Lett. 1987;83:351-355.
139 Swain RA, Harris AB, Wiener EC, et al. Prolonged exercise induces angiogenesis and increases cerebral blood volume in primary motor cortex of the rat. Neuroscience. 2003;117:1037-1046.
140 Kingston SG, Hoffman-Goetz L. Effect of environmental enrichment and housing density on immune system reactivity to acute exercise stress. Physiol Behav. 1996;60:145-150.
141 Juraska JM. Sex differences in dendritic response to differential experience in the rat visual cortex. Brain Res. 1984;295:27-34.
142 Greenough WT, Volkmar FR, Juraska JM. Effects of rearing complexity on dendritic branching in fronto-lateral and temporal cortex of the rat. Exp Neurol. 1973;41:371-378.
143 Comery TA, Shah R, Greenough WT. Differential rearing alters spine density on medium-sized spiny neurons in the rat corpus striatum: Evidence for association of morphological plasticity with early response gene expression. Neurobiol Learn Mem. 1995;63:217-219.
144 Fuchs JL, Montemayor M, Greenough WT. Effect of environmental complexity on size of the superior colliculus. Behav Neural Biol. 1990;54:198-203.
145 Pysh JJ, Weiss GM. Exercise during development induces an increase in Purkinje cell dendritic tree size. Science. 1979;206:230-232.
146 Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;386:493-495.
147 Wallace CS, Kilman VL, Withers GS, et al. Increases in dendritic length in occipital cortex after 4 days of differential housing in weanling rats. Behav Neural Biol. 1992;58:64-68.
148 Green EJ, Greenough WT, Schlumpf BE. Effects of complex or isolated environments on cortical dendrites of middle-aged rats. Brain Res. 1983;264:233-240.
149 Juraska JM, Greenough WT, Elliott C, et al. Plasticity in adult rat visual cortex: An examination of several cell populations after differential rearing. Behav Neural Biol. 1980;29:157-167.
150 Greenough WT, Juraska JM, Volkmar FR. Maze training effects on dendritic branching in occipital cortex of adult rats. Behav Neural Biol. 1979;26:287-297.
151 Chang FL, Greenough WT. Lateralized effects of monocular training on dendritic branching in adult split-brain rats. Brain Res. 1982;232:283-292.
152 Greenough WT, Larson JR, Withers GS. Effects of unilateral and bilateral training in a reaching task on dendritic branching of neurons in the rat motor-sensory forelimb cortex. Behav Neural Biol. 1985;44:301-314.
153 Withers GS, Greenough WT. Reach training selectively alters dendritic branching in subpopulations of layer II-III pyramids in rat motor-somatosen-sory forelimb cortex. Neuropsychologia. 1989;27:61-69.
154 Diaz E, Pinto-Hamuy T, Fernandez V. Interhemi-spheric structural asymmetry induced by a lateralized reaching task in the rat motor cortex. Eur J Neurosci. 1994;6:1235-1238.
155 Marrone DF. Ultrastructural plasticity associated with hippocampal-dependent learning: A meta-analysis. Neurobiol Learn Mem. 2006;87:361-371.
156 Black JE, Isaacs KR, Anderson BJ, et al. Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc Natl Acad Sci U S A. 1990;87:5568-5572.
157 Kleim JA, Swain RA, Czerlanis CM, et al. Learning-dependent dendritic hypertrophy of cerebellar stellate cells: Plasticity of local circuit neurons. Neurobiol Learn Mem. 1997;67:29-33.
158 Kleim JA, Lussnig E, Schwarz ER, et al. Synaptogenesis and Fos expression in the motor cortex of the adult rat after motor skill learning. J Neurosci. 1996;16:4529-4535.
159 Bendre AA, Swain RS, Wheeler BC, et al. Augmentation of cerebellar responses to parallel fiber activation following skilled motor acquisition in rats. Soc Neurosci Abstr. 1995;21:444.
160 Kleim JA, Swain RA, Armstrong KA, et al. Selective synaptic plasticity within the cerebellar cortex following complex motor skill learning. Neurobiol Learn Mem. 1998;69:274-289.
161 Brown RT, Coles CD, Smith IE, et al. Effects of prenatal alcohol exposure at school age. II. Attention and behavior. Neurotoxicol Teratol. 1991;13:369-376.
162 Gallo PV, Weinberg J. Neuromotor development and response inhibition following prenatal ethanol exposure. Neurobehav Toxicol Teratol. 1982;4:505-513.
163 Hannigan JH, Berman RF, Zajac CS. Environmental enrichment and the behavioral effects of prenatal exposure to alcohol in rats. Neurotoxicol Teratol. 1993;15:261-266.
164 Klintsova AY, Matthews JT, Goodlett CR, et al. Therapeutic motor training increases parallel fiber synapse number per Purkinje neuron in cerebellar cortex of rats given postnatal binge alcohol exposure: Preliminary report. Alcohol Clin Exp Res. 1997;21:1257-1263.
165 Klintsova AY, Cowell RM, Swain RA, et al. Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats. I. Behavioral results. Brain Res. 1998;800:48-61.
166 Klintsova AY, Scamra C, Hoffman M, et al. Therapeutic effects of complex motor training on motor performance deficits induced by neonatal binge-like alcohol exposure in rats: II. A quantitative stereological study of synaptic plasticity in female rat cerebellum. Brain Res. 2002;937:83-93.
167 Crawford ML, Harwerth RS, Smith EL, et al. Keeping an eye on the brain: The role of visual experience in monkeys and children. J Gen Psychol. 1993;120:7-19.
168 Demer JL. Positron emission tomographic studies of cortical function in human amblyopia. Neurosci Biobehav Rev. 1993;17:469-476.
169 Locke JL. Thirty years of research on developmental neurolinguistics. Pediatr Neurol. 1992;8:245-250.
170 Huttenlocher J, Haight W, Bryk A, et al. Early vocabulary growth: Relation to language input and gender. Dev Psychol. 1991;27:236-248.
171 Kuhl PK. Learning and representation in speech and language. Curr Opin Neurobiol. 1994;4:812-822.
172 Zhang Y, Kuhl PK, Imada T, et al. Effects of language experience: Neural commitment to language-specific auditory patterns. Neuroimage. 2005;26:703-720.
173 Karni A, Meyer G, Jezzard P, et al. Functional MRI evidence for adult motor cortex plasticity during motor skill learning. Nature. 1995;377:155-158.
174 Elbert T, Pantev C, Wienbruch C, et al. Increased cortical representation of the fingers of the left hand in string players. Science. 1995;270:305-307.
175 Bengtsson SL, Nagy Z, Skare S, et al. Extensive piano practicing has regionally specific effects on white matter development. Nat Neurosci. 2005;8:1148-1150.
176 Maguire EA, Gadian DG, Johnsrude IS, et al. Navigation-related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci U S A. 2000;97:4398-4403.
177 Jacobs B, Schall M, Scheibel AB. A quantitative dendritic analysis of Wernicke’s area in humans. II. Gender, hemispheric, and environmental factors. J Comp Neurol. 1993;327:97-111.
178 Weinberger NM. Dynamic regulation of receptive fields and maps in the adult sensory cortex. Annu Rev Neurosci. 1995;18:129-158.
179 Ramachandran VS, Rogers-Ramachandran D, Stewart M. Perceptual correlates of massive cortical reorganization. Science. 1992;258:1159-1160.
180 Ward NS. Functional reorganization of the cerebral motor system after stroke. Curr Opin Neurol. 2004;17:725-730.
181 Nudo RJ. Adaptive plasticity in motor cortex: Implications for rehabilitation after brain injury. J Rehabil Med. 2003;41(Suppl):7-10.
182 Merzenich MM, Jenkins WM, Johnston P, et al. Temporal processing deficits of language-learning impaired children ameliorated by training. Science. 1996;271:77-81.
183 Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549-579.
184 Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417-463.
185 Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the “dark side” of drug addiction. Nat Neurosci. 2005;8:1442-1444.
186 Pollak SD, Vardi S, Putzer Bechner AM, et al. Physically abused children’s regulation of attention in response to hostility. Child Dev. 2005;76:968-977.
187 Caspi A, Moffitt TE. Gene-environment interactions in psychiatry: Joining forces with neuroscience. Nat Rev Neurosci. 2006;7:583-590.
188 Foley DL, Eaves LJ, Wormley B, et al. Childhood adversity, monoamine oxidase a genotype, and risk for conduct disorder. Arch Gen Psychiatry. 2004;61:738-774. 4
189 Mick E, Biederman J, Faraone SV, et al. Case-control study of attention-deficit hyperactivity disorder and maternal smoking, alcohol use, and drug use during pregnancy. J Am Acad Child Adolesc Psychiatry. 2002;41:378-385.
190 Oyserman D, Bybee D, Mowbray C, et al. When mothers have serious mental health problems: parenting as a proximal mediator. J Adolesc. 2005;28:443-463.
191 Murphy KR, Barkley RA. Parents of children with attention-deficit/hyperactivity disorder: Psychological and attentional impairment. Am J Orthopsychia-try. 1996;66:93-102.
192 Winnicott DW. The family and Individual Development. London: Tavistock Press, 1965.
193 Hoghughi M, Speight AN. Good enough parenting for all children-A strategy for a healthier society. Arch Dis Child. 1998;78:293-296.