CHAPTER 15 Autism Spectrum Disorders
Before the 1990s, autism was thought to be a rare disorder with a dismal prognosis whose victims rarely achieved independent living and enduring relationships. Instead, most affected adults lived with their parents or in state institutions.1 Most individuals in whom autism as diagnosed were nonverbal and assumed to have some degree of mental retardation even when standardized measurements of intelligence were not available. Treatment programs, if existent, were usually housed in facilities serving segregated populations. There were no published autism guidelines, and there was little interest in research outside a relatively small circle of dedicated investigators. The media, lay public, and members of Congress were generally not even aware of the term autism, much less concerned about it.
The 1990 decade was proclaimed “the decade of the brain,”2 partly because of advances in neuroimaging and expanding knowledge about how the central nervous system worked. However, the 1990s might also be called the “the decade of autism” because of the rapidly expanding body of knowledge in the field. The 1990s also marked a period of significantly heightened public awareness, attributable at least in part to the 1988 release of the Academy Award–wining movie “Rainman.” Although some advocates expressed concern about how autism was portrayed, the movie nevertheless brought autism to public awareness. Public attention has remained high because autism has been and continues to be engulfed in a sea of controversy. Concerns about a possible “epidemic” relating to vaccines and toxins and the media’s frenzy regarding miraculous cures (auditory integration therapy, facilitated communication, secretin injections, and mercury chelation) have made autism a household term. Furthermore the media, motivated by dedicated advocates, has played an important role in capturing the attention of Congress, which in turn has resulted in autism-specific activities mandated by national legislation: the Children’s Health Act of 2000,3 the New Freedom Initiative of 2001,4 and the Combating Autism Act of 2005.5
Because of mandated federal support, autism “centers of excellence” emerged and contributed to a rapidly expanding body of knowledge. Thus, interest in autism within professional circles paralleled that of the lay public; this attention is illustrated by the exponential growth of training activities and published articles during the 1990s. Whereas before 2000 professional organizations such as the American Academy of Pediatrics (AAP) offered no stand-alone course in autism at its national conferences, since then autism has been at the top of the AAP list of “hot topics” and now consistently appears as a topic on conference agendas. Similarly, approximately 3000 autism-related articles were published in scientific peer-reviewed journals between 1943 (when it was first described by Kanner6) and 1990, whereas more than 4000 appeared in the 1990s alone.7 Beginning at the end of the 20th century, the first policy statements and practice guidelines were published.8–13 In spite of this, the cause of autism is still not known, and there is no known cure.





It is impossible to discuss the voluminous literature surrounding the autistic spectrum disorders (ASDs) in a single chapter. The ambitious reader is referred to Handbook of Autism and Pervasive Developmental Disorders (Volumes 1 and 2),14 Neurobiology of Autism,15 Autism Spectrum Disorders in Children,16 and Autism Spectrum Disorders.17
TERMINOLOGY
Terminology, definitions, and diagnostic criteria have changed over the years. The concept of “autism” before the 1990s apparently represented only a small proportion of ASDs. Although frequently used by European investigators in the 1990s, the term autistic spectrum disorder did not become popular in the United States until about 2000.18 It has become an “umbrella term” that includes three of the five pervasive developmental disorders (PDDs) listed in the most recent revisions of The Diagnostic and Statistic Manual of Mental Disorders (DSM) of the American Psychiatric Association19,20 and the Diagnostic and Statistical Manual for Primary Care, Child and Adolescent Version21: autistic disorder, Asperger disorder (referred to as Asperger syndrome in this chapter), and pervasive developmental disorder, not otherwise specified (PDD-NOS). The remaining two PDDs, Rett syndrome and childhood disintegrative disorder, are not discussed in this chapter.
The ASDs are neurodevelopmental conditions characterized by one or a combination of the following: significant social skill deficits, both qualitative and quantitative language abnormalities, restricted interests, and repetitive motor mannerisms. Although ASDs appear to have a strong genetic basis,22,23 the precise cause is unknown; thus, there is no pathognomonic physical sign or laboratory test. Instead, the diagnosis is made by determining the presence of characteristic developmental and behavioral criteria described in the fourth edition of the DSM (DSM-IV)19 or in the text revision (DSM-IV-TR).20 Many clinicians use a standardized evaluation tool that operationalizes the DSM criteria. Nevertheless, considerable subjectivity still exists in making this diagnosis, largely because of the wide range of symptoms included within the scope of the spectrum.
Autism Disorder
Autism was first described in the third edition of the DSM (DSM-III) in 198024 as “Infantile Autism”; the current term, “Autistic Disorder” replaced “Infantile Autism” in the revised version of the DSM-III (DSM-III-R) in 1987.25 Although clinical patterns vary in regard to severity, age at onset, underlying cognitive deficits, and other features, diagnosis of “classic” autistic disorder is dependent on the presence of at least half (six) of the criteria (Table 15-1).20 Symptoms in at least one of these areas must have been present before the age of 3.

TABLE 15-1 Diagnostic Criteria for 299.00 Autism Disorder
Rights were not granted to include this table in electronic media. Please refer to the printed book.
Reprinted with permission from the Diagnostic and Statistical Manual of Mental Disorders, 4th ed, Text Revision. Washington, DC: American Psychiatric Association, 2000, p 75.
Asperger Syndrome
Asperger syndrome is characterized by the same impairment in social interaction and restricted interests as in autistic disorder; however, language skills are relatively normal (defined as use of single words by age 2 years and phrases by 3 years) (Table 15-2).20 Later language is characterized by pragmatic deficits (problems with the social use of language). In addition, cognitive and adaptive skills are normal. Depending on the child’s age, it is sometimes quite challenging to distinguish between children with Asperger syndrome and children with autistic disorder and normal intelligence. Because of this, controversy exists as to whether Asperger syndrome represents a high-functioning form of autism or a separate entity.26,27 Children with Asperger syndrome are usually not recognized until after 4 years of age, when social interactions with peers in preschool settings become a concern.

TABLE 15-2 Diagnostic Criteria for 299.80 Asperger Disorder*
Rights were not granted to include this table in electronic media. Please refer to the printed book.
Reprinted with permission from the Diagnostic and Statistical Manual of Mental Disorders, 4th ed, Text Revision. Washington, DC: American Psychiatric Association, 2000, p 75.
Pervasive Developmental Disorder, Not Otherwise Specified
Pervasive Developmental Disorder, Not Otherwise Specified is a subthreshold term that is used when a child demonstrates some but not all of the criteria necessary to make a diagnosis of one of the specific PDDs. Unfortunately, there was an error in the DSM-IV text19: It stated that PDD-NOS should be used when there was an “impairment in… social interaction” or in verbal and nonverbal skills. This allowed clinicians to apply the PDD-NOS label in the absence of social skill deficits, thus broadening the definition of PDD-NOS and causing loss of specificity. This error was corrected in the DSM-IV-TR.20 The PDD-NOS label is reserved for persons who demonstrate “severe and pervasive impairment in the development of reciprocal social interaction” and either communication deficits or restricted interests/repetitive behaviors. Confusion still exists regarding the actual number of criteria that are necessary to apply the PPD-NOS label; by convention, at least two but not more than five should be present. PDD-NOS also includes “atypical autism,” which refers to persons with at least one feature that is dissonant with traditional autism, such as later onset or absence of stereotypies.28,29
Throughout this chapter, ASD refers to all three disorders as a group. When a specific ASD is discussed, the appropriate term is used. Autism is used in reference to older literature published before the concept of a spectrum of autistic disorders emerged. A broad spectrum does appear to exist, although its external and internal boundaries are hazy.30,31 Family studies have shown that the entire spectrum may be expressed in the same pedigree. Sometimes the term broader autism phenotype is used for individuals with isolated social deficits, particularly in the context of extended-family relatives of probands with autism.32,33
HISTORY
In 1943, Leo Kanner, a psychiatrist at the Johns Hopkins University School of Medicine, first defined autism as it is known today.6 About the same time, Hans Asperger, an Austrian pediatrician, unaware of Kanner’s work, published an article in German34 describing four children who demonstrated symptoms similar to those described by Kanner with the exception of better verbal and cognitive skills. Asperger syndrome escaped recognition until it was popularized by Wing’s translation into English.35,36 In 1978, Rutter37 published the first set of “essential criteria” for autism. These were incorporated into the next edition of the DSM (DSM-III),24 and autism became recognized as a separate entity within the newly created category of Pervasive Developmental Disorders.
New criteria were developed for the DSM-III-R,25 published in 1987, and were criticized for being too inclusive and thus promoting overidentification of autistic disorder.38 The criteria still in use today were published in 1994 in the DSM-IV19; Asperger syndrome criteria appeared for the first time in this version. The DSM-IV Autistic Disorder criteria were the result of years of analyses to reduce the overinclusiveness of DSM-III-R. Furthermore, collaboration with European groups working on the manual for the revised International Classification of Diseases, 10th edition (ICD-10),39 promoted conformity between the two classification systems. Studies have revealed that the DSM-IV criteria have better specificity (0.87) than do DSM-III-R criteria.40 Criteria for autistic disorder and Asperger syndrome have not changed in the DSM-IV-TR.20
Although Kanner initially hypothesized that autism was an inborn, biological condition,41 misconceptions based on psychodynamic theory soon became prevalent. Probably the most important one was the mistaken concept that autism might be caused by cold and unnurturing parents (“the refrigerator theory”). Bettelheim42 promoted this concept in his book, The Empty Fortress: Infantile Autism and the Birth of Self. The refrigerator theory remained popular until the 1960s when Rimland hypothesized a neurological cause.43 This hypothesis was later supported by the demonstration of neuropathological abnormalities on magnetic resonance imaging (MRI)44 and documentation of a high rate of coexisting seizures.45
The science of ASD has advanced a great deal. Collaborative research centers and multidisciplinary diagnostic teams proliferated during the late 1990s and continue to do so with even greater momentum in the new millennium. Since the 1990s, there have been a number of rapid developments: the debut of the first screening tools, the development of evaluation tools that operationalize DSM-IV criteria, neuropathic studies that revealed an early prenatal onset, identification of multiple genetic susceptibility genes, recognition of the relative importance of social skill deficits in defining ASDs, and evidence that early and appropriate intervention is effective in improving outcomes.7,13
PREVALENCE
The apparent dramatic rise in prevalence of ASDs has become a focus for parent advocacy groups and the media and may well be one of the most controversial topics in the field of autism (Table 15-3). More than 30 studies documented an apparent increase in prevalence of these diagnoses.46–55c In 2000, the Centers for Disease Control and Prevention organized the Autism and Developmental Disabilities Monitoring (ADDM) Network, a multisite, records-based surveillance program, to study the prevalence of ASDs. The ADDM Network employs systematic screening of developmental evaluation records for autistic behaviors rather than depending on a medical or educational diagnostic label of an ASD. In 2007, the ADDM Network reported ASD rates ranging from 1 in 303 to 1 in 94 8-year-old children for two time periods (2000, 2002) in a total of 14 sites in the United States; the average rate was 1 in 150 or 6.6 per 1000 8-year-olds.56 Studies varied in methods, definition, and case ascertainment strategies, but overall there appeared to have been up to a 10-fold increase worldwide since the 1950s.
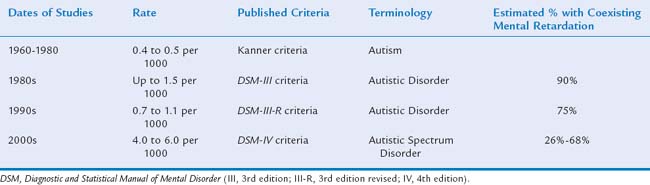
Several factors complicate the interpretation these data, making it very difficult to discern whether there has been a true rise in prevalence or simply an apparent one. Most investigators have demonstrated that the apparent rise in prevalence is attributable, at least in part, to changing broader criteria and increased public and professional awareness.51,54,57 This is supported by a greater increase in the numbers of milder cases (i.e., PDD-NOS and Asperger syndrome). Other factors contributing to the apparent rise include the emergence of screening tools in the 1990s, which resulted in improved ascertainment, and the development of better diagnostic tools that can more reliably identify children at younger ages. The media (e.g., the National Broadcasting Company’s Autism Speaks Campaign, April 2006) and advocacy groups have been successful in raising public awareness so that parents are now recognizing ASD symptoms in their children and voicing their concerns earlier to physicians. There has also been an increase in recognition of ASDs among children who have disorders unrelated to ASD, such as Down syndrome58 and the syndrome of coloboma, heart anomaly, choanal atresia, retardation, genital lesions, and ear abnormalities (CHARGE association).59 Autistic features can also be detected in some children with congenital sensory disorders, especially when severe vision and/or hearing deficits are not detected and intervention is not implemented early.60,61
Finally, public policies have also made a significant effect on reported prevalence rates obtained from public administrative data. The Education of All Handicapped Children Act (Public Law 94-142) of 197562 made schools accessible to children with some disabilities. Many children with more severe disabilities, such as those with both autism and severe mental retardation, continued to live in segregated state institutions. Later laws such as the Americans with Disabilities Act of 199063 promoted closure of institutions; which caused more children with disabilities to live at home and attend community schools. Autism first became a diagnostic category for which children could receive services with passage of the Individuals with Disabilities Education Act (IDEA) in 1991.64 Before 1991, children with autism were most likely to be served under categories established by older laws, such as “mental retardation,” “learning disabled,” “speech delayed,” “or emotionally disturbed.”65 Since the passage of IDEA,64 school eligibility diagnoses have usually conformed to medical diagnoses of an ASD. This phenomenon of “diagnostic substitution” may account for a substantial proportion of the apparent rise in prevalence; however, educational administrative data may not be completely reliable.66–71 IDEA amendments72,73 have made supplementary services available to children with ASD that are not available to children with other disabilities, such as “year-around school.” When criteria for ASD are marginal, professionals may be tempted to apply the label in order to secure these supplementary services, which often also provide additional support for the parents. Such strategies tend to inflate the “prevalence” when values are obtained solely from educational sources.
The reasons for the reported 10-fold rise in prevalence of ASD remain controversial.50,51,74–82 However, there is broad agreement that more boys than girls are consistently found to be affected with ASD; sex ratios range from 2 : 1 to 4 : 1.50,53,79,83–86 The male : female ratio is even higher for high-functioning autism and Asperger syndrome, ranging from 6 : 1 to 15 : 1.87 A 2006 study, in which most affected children (53.3%) had normal intelligence, demonstrated a male : female ratio of 9 : 1.55
ETIOLOGY
ASDs are now believed to be biologically based neurodevelopmental disorders that are highly heritable.88 Because of the wide phenotypic spectrum, most experts believe that many genes are involved.89 In a minority of cases, ASDs may be associated with a medical condition or a known syndrome characterized by dysmorphic features and some degree of comorbid mental retardation.47,48 Although ASDs are believed to be mainly genetic in origin, the lack of 100% concordance in monozygotic twins indicates that environmental factors may modulate the phenotypic expression.22,88 Thus it has become increasingly apparent that the cause is multifactorial, with a variety of genetic and, to a lesser extent, environmental factors playing a role.
Genetic Underpinnings
Studies of twins have revealed concordance rates of classic autism in 60% of monozygotic pairs and in 0% to 3% of dizygotic pairs.22,88,90 When the broader phenotype was taken into consideration, the rates were 60% to 92% and 0% to 10%, respectively. In addition, family studies have demonstrated a rapid decrease in prevalence among first, second, and third-degree relatives. Using these data, Bailey and colleagues22 calculated that the predisposition for autism was more than 90% heritable, with multiple interacting genetic influences and strong family clustering.91 In spite of these strong genetic underpinnings, the exact cause or causes are still unknown. The task has been daunting because of genetic complexity and phenotypic variation. First, ASDs appear to be complex heritable disorders involving multiple genes; estimates based on family studies range from 5 to 20 genes.89 Each gene or gene combination may result in somewhat different subtype but often with overlapping behavioral phenotypes. The number of genes contributing to the disorder and the relative prevalence of each will increase or decrease the probability of identifying the cause; that is, success is more likely if there are relatively few genes that are somewhat common than if there are many genes that are rarer.91 A second factor making gene identification more challenging is the variability in the ASD phenotype. The wide spectrum of symptoms sometimes promotes inclusion of participants in a study with various ASDs, sometimes even including individuals with disorders falsely categorized to be in the spectrum. This imprecise diagnosis contaminates the study group and makes identification of a unifying etiological agent elusive.92,93
Two major strategies have been used in the search for the ASD susceptibility genes: targeted cytogenetic studies and whole genome screens of families of children with ASD.91,94 The first strategy depends on developing a hypothesis regarding the pathogenesis of ASD, focusing on one or more potential candidate genes and testing them genetically for an association with ASD. Candidate genes in ASD include, among others, those that appear to play a role in brain development (e.g., cerebellar Purkinje cell proliferation)15,95 or in neurotransmitter function (e.g., serotonin transmitter).96 The second strategy entails an indirect method and does not require investigators to make assumptions regarding the mechanism of inheritance. Instead, families with multiple members demonstrating an ASD (multiplex families) are studied to identify recurring DNA markers (breakpoints, translocations, duplications, and deletions) present in affected, but not in unaffected, members. Unfortunately, progress has been limited because the phenotypic endpoints of ASD are not well defined. Changes in DSM criteria and inconsistency in ascertainment strategies, resulting in a hazy delineation between “affected” and “unaffected” family members, contaminate outcomes and challenge interpretation of results. This phenotypic heterogeneity has challenged molecular searches for the ASD gene or genes in spite of several genomewide screens (International Molecular Genetic Study of Autism Consortium [IMGSAC]) and multicenter collaborative efforts since the 1980s.93,97–99 The results are very enlightening; however, rather than providing conclusions based on replicated findings from multiple labs, these studies have often produced confusion and uncertainty as more susceptibility loci are described. Although at least one autism-linked abnormality has been found on almost every chromosome, few sites have been identified with any frequency. Table 15-4 provides a summary of some of the more consistent findings; however, the reader is advised to consult more extensive reviews.7,92,100–104 Large study samples with pooled data from multiple populations (maximizing homogeneous samples) are needed to confirm the validity of reported candidate genes and susceptibility sites in ASD.93
TABLE 15-4 Selected Autism Spectrum Disorder (ASD) Genetic Markers
|
The high male : female ratio and the discovery of genes for both fragile X syndrome and Rett syndrome on the X-chromosome made it a plausible target.116,362,553 Investigators have targeted a variety of possible roles for the X-chromosome in ASD:
Skewed X-chromosome inactivation is known to occur in X-linked mental retardation carriers554 and to be responsible for most of the phenotypic variability seen in Rett syndrome.362 X-chromosome inactivation patterns in female patients with ASD and controls (from the AGRE database) were studied to determine whether skewness might account for expression of possible autism genes on the X-chromosome.553 Indeed, statistically greater skewness was found in those with classic Autism disorder than in controls (33% vs. 11%). Furthermore, of 10 asymptomatic mothers of Autism daughters demonstrating skewness, 5 also had highly skewed X-chromosome inactivation; of the mothers of the four control daughters showing skewness, none showed skewed inactivation. These results warrant further study to determine the possibility of skewed X-chromosome inactivation and/or X-linked candidate genes in the etiology of ASD in both male and female patients.
An epigenetic phenomenon similar to the one occurring in Rett syndrome has been proposed because of overlapping clinical presentations (i.e., stereotypies and regression in social and communication skills) and the discovery that a few individuals with ASD also demonstrated Rett syndrome–like mutations.362,363,361a A mutation in a regulator gene on the X-chromosome may cause the inappropriate activation or inactivation of otherwise normal genes that affect brain development and, in turn result in ASD. Between 2% and 7% of children with Angelman syndrome and a rare child with ASD also have been found to have MECP2 mutations.124,361a
Imprinting on the X-chromosome has been offered as a possible explanation for the high male : female ratio. Investigations of girls with either Turner syndrome or partial deletions of the X-chromosome revealed an increase risk for social skill deficits similar to those seen in ASD.97,100,555,556 Paternally rather than maternally derived deletions were more strongly associated with poor social cognition. Thus, it appears the paternal X-chromosome is important for development of this skill, and because boys do not receive X-chromosomes from their fathers, they might be at higher risk for social deficits as a result of imprinting (parental origin effect).
Isolated findings of Xp22 deletions or duplications have been reported in a few individuals with ASD.557
Genome screens have found a linkage to the Xq13-21 region that contains the genes that code for neuroligin, a cell-adhesion molecule that is thought to be involved with synaptogenesis.558
Mutations of the angiotensin II receptor gene on the Xq22-23 region have been implicated in one of the X-linked mental retardation syndromes in which almost 20% also meet criteria for ASD.559
A site on 2q, which appears to contain a susceptibility gene for autism and language delay (2q37), was identified in several studies, including two IMGSAC screens92,96,97,559; however, a more recent screen entailing a different database failed to confirm this.93
In a genome screen of pooled data from two countries, the 3p24-26 region emerged as the most promising.93 This locus contains the oxytocin receptor gene (OXTR). The possible link between ASD and oxytocin regulation of social behavior has been noted since the mid-1990s,96,561,562 and oxytocin receptors have been found throughout the limbic system. Social deficits in oxytocin knockout mice563 reduced oxytocin plasma levels,564 and some evidence that synthetic oxytocin ameliorated repetitive behaviors in adults with ASD565 have all pointed toward a contributing role for oxytocin.
Genome screens of this chromosome have resulted in the most consistent findings.97,98,566 Researchers have postulated a susceptibility site, called the AUTSI locus (7q31-33), where mutations in one or more of the involved genes can potentially increase the risk of an ASD.198 The RELN (7q22-33) gene and its secretory glycoprotein, reelin, appear to play a role in migration and cell lamination in the brain, especially in the cerebellum, where some of the most consistent neuropathological abnormalities occur.198,567 Other genes also occur at this site and may play a role. The FOXP2 gene (7q31-35) appears to play a role in the embryonic development of neural pathways involved in the acquisition of expressive language. Several mutations have been found in patients with speech disorders.568 Finally, the WNT2 gene on chromosome 7 appears to play a role in social skills.569
A variety of cytogenetic abnormalities occur at the 15q11-13 locus (duplications, deletions, translocations). In regard to ASD, 1% to 4% of study cohorts may demonstrate a duplication, usually maternally derived, at this site.100,122,123,570,571 A “chromosome 15 phenotype” has begun to emerge that is characterized by hypotonia, joint laxity, global (especially motor) developmental delays, seizures, speech delay, social deficits, stereotypies, and a variable pattern of mild facial dysmorphisms.123 Other abnormalities (deletions) also occur at the site and produce either Angelman or Prader-Willie syndrome, depending on the parent of origin. A GABA receptor gene (coding for a neurotransmitter highly implicated in ASD) also occurs at this site.92,100
Because serotonin is pivotal during brain development213,572 and platelet serotonin is one of the most common laboratory abnormalities in children with ASD,96,573,574 the serotonin transporter gene (17q11-12) has become a popular target for study. A susceptibility site at 17p12-q21 was noted to be the second most promising one in a comprehensive genome screen involving two databases from different countries.93
Terminal deletions at 22q13 have been associated with hypotonia, developmental delay, autistic-like behavior, and subtle physical features (ear anomalies, short nose, smooth philtrum, and full lips).366 Another study reported that 14% of patients with a confirmed microdeletion of 22q11.2 also met criteria for an ASD.367 Two other well-known syndromes (velocardiofacial [Shprintzen] syndrome and DiGeorge syndrome) are also associated with microdeletions at this site.113,575
Older IMGSAC studies have consistently revealed possible sites on 2q, 7q, and 17q; a more recent combined analysis of two primary genome scans from AGRE (United States) and Finnish populations (314 autism-affected families) revealed the best loci to be 3p24-26 and 17p12-q21 with additional promising sites at 1p12-q25, 4q21-31; 5p15-q12; 6q14-21, 7q33-36; 8q22-24; and 19p13-q13.93
|
AGRE, Autism Genetic Resource Exchange; GABA, γ-amino butyric acid; IMGSAC, International Molecular Genetic Study of Autism Consortium.
Table 15-4 describes findings of genetic investigations that have, for the most part, targeted etiological possibilities for “idiopathic ASD,” which represents most cases of ASD. Although the literature provides multiple systems for characterizing ASDs, it is perhaps most helpful in a discussion of etiology to subtype ASDs as either idiopathic or secondary.103 For the purposes of this discussion, patients with idiopathic ASD are those who do not have a coexisting associated medical condition or syndrome known to cause an ASD. Most individuals with ASD (perhaps almost all of those with Asperger syndrome) have the idiopathic subtype. Children with idiopathic ASD demonstrate variable behavioral phenotypes, are less likely to have coexisting mental retardation, and do not have dysmorphic features heralding a recognizable syndrome. Nevertheless, twin and family studies have revealed that idiopathic ASD is very heritable, with a recurrence rate of 3% to 7%,22 although phenotypic expression may be modified by other variables.92
Patients with “secondary” ASD are children with a known identifiable syndrome or medical disorder believed to play an etiological role in ASD; this occurs in only 2% to 10% of cases.23,66,103–113 In a meta-analysis of 23 epidemiological studies, Chakrabarti and Fombonne47 reported that a recognizable condition was identified in an average of 6% of those with a confirmed ASD. The rate of coexisting mental retardation was 26%, the lowest reported prevalence to date. The presence of severe mental retardation, especially when associated with dysmorphic features, increases the likelihood of identifying a genetic etiology.10,104,110,114 Genetic syndromes associated with ASD and coexisting mental retardation include




Of these four entities, the fragile X syndrome is the most common known genetic cause for the autistic phenotype, present in 1% to 5% of children with ASD, whereas 30% to 50% of those with genetically confirmed fragile X syndrome demonstrate some characteristics of ASD.117,126 The presence of a known disorder does not automatically imply causation. A few children with genetic syndromes characterized by features quite different from ASD may also meet full DSM criteria. For example, investigators have reported that that 6% to 7% of children with Down syndrome (usually characterized by relatively good social skills and obvious physical stigmata)58,127,128 and almost 50% of children with the CHARGE association59,129 meet criteria for a diagnosis of either autistic disorder or PDD-NOS. Children with severe congenital sensory impairments (visual and/or auditory) are also at risk for the development of symptoms consistent with ASD, especially when appropriate early intervention is not provided.60,61 Advancing paternal age has been shown to be associated with an increased risk of ASD possibly due to de novo spontaneous mutations and/or alterations in genetic imprinting.129a
A variety of immunological abnormalities in T cells, immunoglobulins, and anti–brain autoantibodies have all been reported in retrospective case studies,130–134 but systematic studies have confirmed neither their existence nor their relevance.102,135 Prospective studies have revealed that, except for a few individuals with recurrent infections, healthy children with ASD generally have normal immune function.135 Epidemiological data revealed clustering of autoimmune disorders in ASD families; however there was no increase in autoimmune disorders of the central nervous system and the patients with ASD did not themselves exhibit autoimmune disorders.136 Food allergies have also been implicated to play an etiological role in a few case reports,137,138 but, again, this has not been confirmed with rigorous studies.12,102,139
Environmental Factors
Regardless of the mechanism, a review of studies published since the 1950s reveals convincing evidence that most cases of ASD result from genetic factors with possible interacting environmental factors.22,92,140,141 Environmental influences may represent a “second hit” or “trigger” phenomenon; that is, they may modulate/stimulate preexisting genetic factors to result in the manifestation of ASD in an individual child.
Environmental factors should have their greatest effect during the prenatal period, especially early in gestation, because the developmental brain abnormalities associated with ASD occur during the first and second trimesters.141–144 Factors already identified to play a role include maternal rubella145 or cytomegalovirus infections146,147 and treatment with valproate148,149 or thalidomide.144,150 An isolated report150 indicated that fetal exposure to thalidomide during days 20 to 24 of gestational age was associated with ASD symptoms; later exposure resulted in the more characteristic limb abnormalities but no ASD characteristics. Nelson and associates151 reported increased cord blood levels of brain-derived neurotrophic factor and other neurotrophins in newborns in whom ASD was later diagnosed, which may have implications regarding the mechanism of the characteristic early brain overgrowth. Some investigators feel that fetal toxin exposure might be implicated by studies that have demonstrated higher rates of ASD in offspring of mothers who resided in urban settings during pregnancy152,153; however, other factors such as better access to diagnostic services in urban areas may be operative.
PERINATAL
Perinatal events have also been investigated, but findings have not been consistent and need replication.154–157 Among other factors, the strongest associations have been with threatened abortion and advanced maternal age.158 An association has been suggested between full-term neonatal encephalopathy and later diagnoses of ASD.159 In one study, ASD were diagnosed in 5% of survivors, which represented an almost sixfold increase in comparison with matched controls. This association, if replicated and confirmed, may represent a genetically derived predisposition making the infants vulnerable to both encephalopathy and ASD or a causative mechanism.
POSTNATAL
Postnatal causes of ASD are less likely possibilities. Moderate environmental deprivation has not been found to play an etiological role in ASD.152,160 One study of Romanian orphanages revealed that severe environmental deprivation could lead to something resembling at least “quasi-autism”; however, most symptoms disappeared if the children were adopted into nurturing homes during early childhood.161 A causal association between the measles, mumps, and rubella (MMR) vaccine and the development of ASD was proposed in a controversial case series study of 12 children with autistic regression and colitis.162 The onset of autistic behavior reportedly occurred shortly after receipt of the MMR vaccination. The Institute of Medicine,163 the AAP,164 the British Medical Research Council,165 and the Cochrane Collaboration166 reviewed epidemiological studies and other published and unpublished evidence and concluded that there is no evidence of a causal association. In 2004, 10 of the 13 authors of the original article stated in a retraction that they did not believe the data supported the conclusions regarding a possible causal relationship.167 Most recently, a multisite Collaborative Programs of Excellence in Autism study of 351 children with ASD failed to reveal any evidence of a causal association between ASD symptoms and the MMR vaccine.168
Questions have been raised repeatedly80,81,153 about the possible effects of environmental mercury exposure and mercury-containing vaccines on the development of ASD and other developmental disabilities. From large data sets from the United States, Sweden, and Denmark, no consistent association has been found between vaccines containing thimerosal (the mercury-based preservative) and ASD or other neurodevelopmental outcomes.169–171 Statements that the neuropathological and clinical presentations of ASD and mercury poisoning are similar were also refuted by Nelson and Bauman.172 Although scientific evidence170 of no association between vaccines (specifically MMR and thimerosal- or mercury-containing vaccines) and ASD continues to accumulate, many parents, as well as some professionals, remain unconvinced of the validity of the evidence.81a In a best seller (Evidence of Harm, 2005), Kirby82 hypothesized that various governmental agencies and medical organizations have conspired to cover up the possible harmful effects of thimerosal. The disbelief in the scientific evidence regarding mercury and vaccines remains one of the most challenging public heath problems faced by pediatricians in the United States today.
Although the preceding discussion reveals the wide variety of coexisting conditions known to be associated with ASD, a thorough etiological investigation in the individual child with ASD rarely identifies a known cause, especially in the absence of mental retardation, dysmorphic features, a positive family history, and/or positive results of a focal neurological examination.10,12,47,104,107 Multicentered collaborative studies are needed and are currently being designed to systematically evaluate children with ASD for comorbid conditions.173 The inclusion of controls with mental retardation and/or other disabilities is important in determining whether the conditions are unique to ASD or equally prevalent among children with significant neurodevelopmental disabilities in general.
NEUROLOGICAL CORRELATES
A neurobiological basis for autism was first suspected in the 1970s when it was noted that approximately one third of persons with autism had epilepsy.174 The neurological basis was further supported by evidence that autism was associated with tuberous sclerosis, a neurocutaneous disorder.118 Since that time, a growing body of evidence has revealed that brain growth and organization are different between normal children and those with ASD and that the onset of these abnormalities begins early in prenatal brain development, in some cases as early as days 20 to 24 of gestation.15,95,144,150,175 Information regarding brain development, cytoarchitecture, and functioning has been accessed through a variety of methods, including evoked potentials, electroencephalography (EEG) and magnetoencephalography, structural and functional MRI (sMRI and fMRI), positron emission tomography, neurotransmitter levels, neuropsychological testing, and postmortem examination of brain tissue. Many findings have been nonspecific and inconsistent; neuroimaging studies sometimes reveal results that are disparate from those of microscopic tissue examinations. Study samples have been characterized by a wide spectrum of behavioral heterogeneity and IQ scores. The presence or absence of coexisting mental retardation has often confounded the interpretation of reports and has been responsible for conflicting results. The highly variable and complex ASD phenotype, coupled with changing DSM criteria, has prevented the recognition of a cohesive neurological mechanism to explain all core symptoms. Research has been further impeded by the dearth of available specimens, lack of an animal model, and logistical challenges in studying children who are nonverbal with behavioral challenges.
Although individuals have been evaluated with clinical neuroimaging since the 1960s, the first rigorous research-quality neuroimaging studies did not begin to appear in the literature until the 1980s.176 The first systematic postmortem study of tissue in an individual with known autism was reported in 1985.177 In 1988, the first MRI abnormality (hypoplasia of the cerebellum) was reported.44 Since then, studies describing neurophysiological and neuropathological abnormalities, as well as theories regarding their effect on behavior and learning, have mushroomed. For a more thorough review of the neurological aspects of ASD, the reader is referred to related chapters by Volkmar and colleagues14 (see also Anderson and Hoshino178) and reviews by Bauman and Kemper15 and Polleux and Lauder.179 In 2006, the neurological findings supported by the greatest degree of evidence included the following:







Although early investigators occasionally reported slightly enlarged ventricles, the changes were therapeutically insignificant.176 Studies of the cingulated gyrus, basal ganglia, thalamus, and brainstem are fewer in number and less conclusive. There are many additional findings and theories, but these are not as well studied, more controversial, or confounded by comorbid medical conditions and/or intellectual deficits.
Seizures and Electroencephalographic Data
Electrophysiological studies were among the earliest used to probe differences in the autistic brain and provided the first evidence that autism was a neurological rather than a behavioral disorder. Abnormal electroencephalographic results are more prevalent in autism than is frank epilepsy (50% vs. 30%, respectively)176,180 and reflect the disruption of balance between inhibition and excitation, which in turn can negatively affect attention and sensory processing.179,181
Brain Growth
Kanner6 himself noted that 5 of his 11 original patients had large heads, but it was not until the1990s that brain size/volume in ASD was systematically studied through both indirect methods (occipitofrontal circumference measurements) and direct methods (sMRI and postmortem examinations). Although these early studies revealed increased brain size182 and volume,183 the mechanisms and changes in growth velocity over the developmental period in children were not recognized until the late 1990s. Head size does not appear to be large at birth; in fact, it is sometimes reported as somewhat smaller than average.184 Acceleration of growth, as evidenced by serial occipitofrontal circumference measurements and confirmed with volumetric studies, begins to occur around 6 months and peaks between 2 and 4 years of age, thus occurring in concert with (or even before) the appearance of ASD symptoms.185–188 Approximately one third of children with ASD meet criteria for macrocephaly, and 90% have greater than average brain volumes.185 Growth appears to level off in late childhood in the majority; thus, brain volume is not significantly different from that in controls by age 12 years, although macrocephaly (as measured by occipitofrontal circumference) may persist.186,189,190 Furthermore, increased cortical folding resulting in abnormal gyral patterns, reflecting increased volume, has been noted in affected children but not adolescents or adults.191 These findings appear very consistent, especially when study subjects are matched for intelligence. The main contribution to increased size lies in nonuniform changes in the hippocampus, amygdala, and cerebral white matter and, less consistently, gray matter.185,192–194 Although several theories have been proposed for the abnormal growth pattern (e.g., increased neurogenesis, glial cell proliferation, abnormal myelin, and/or decreased apoptosis/pruning), it seems most likely that it is the outer radiate zone, related to intrahemispheric synaptogenesis and connectivity, that matures postnatally and produces the rapid growth in ASD. Interestingly, parents of children with ASD can have macrocephaly in the absence of ASD symptoms. Thus, macrocephaly might be caused by a susceptibility gene that works in concert with other genes to produce ASD.
Although there are numerous published sMRI studies, no consistent abnormalities have been reported with regard to the gross anatomy or growth of other brain structures such as the brainstem, basal ganglia, and cerebellum.44,182,183,192,193 Several studies have consistently shown impaired growth (caused by hypoplasia, not atrophy) in the body and posterior regions of the corpus callosum. Only one study has correlated these anatomical findings with deficits in interhemispheric cognitive tasks.195 Rather than a focal neurological abnormality, ASD seems to be characterized by abnormalities of neural distribution and connectivity with excessive intrahemispheric connectivity and deficient interhemispheric connections (corpus callosum).176
Cerebellar Purkinje Cells
Although comparisons of sMRI and volumetric studies of the cerebellum have been controversial, one of the most consistent findings over time has been the marked decrease in Purkinje cells noted in postmortem microscopic studies.15,95,196 The absence of empty baskets suggests that the process is one of hypoplasia rather than atrophy after a noxious event, but some authorities disagree.197 Reductions in reelin may contribute abnormal regulation of neuronal layering and microscopic abnormalities found in the cerebellum.198 The absence of glial hyperplasia indicates the pathological process occurs early in brain development, before the time when the brain is able to initiate a reaction to neuronal injury. Furthermore, the number of olivary neurons is preserved, which provides additional evidence that the process must occur before weeks 28 to 30 of gestation. After this time, tight neuronal unions form between the two areas, and cell loss in the cerebellum would prompt an obligatory retrograde cell loss in the ascending olivary neurons.15,95,143,175 Although it has long been known that the cerebellum played a role in motor learning, modulation, and coordination, there is growing evidence that it also plays a role in verbal processing, affective behavior, and shifting of attention.199 Bauman and Kemper175 suggested that early in embryological life, the climbing olivary neurons might form primitive unions with collateral cells in the lamina dessicans (which disappears at 28 to 30 weeks) of the cerebellar peduncles. These neural units are not as efficient as the primary pathways and cease to function after a brief period. An intriguing question is whether this process may contribute to “autistic regression.”
Decreased Cell Size, Increased Cell Number, and Increased Packing Density in Limbic Structures
Investigators have targeted the limbic system because it plays an important role in social behavior/cognition (amygdala) and associative social memory, especially relationships among the emotional aspects of an experience (hippocampus). Postmortem microscopic studies have consistently revealed abnormalities in cell number and size and packing density in both the amygdala and hippocampus. However, sMRI and volumetric data have been inconsistent, diverse, and often contradictory.193,200,201
Abnormal Minicolumns in the Cerebral Cortex
Abnormal cortical minicolumns (defined as the most basic unit of neural organization) have been added to the growing number of neuropathological abnormalities found in ASD. Although Bailey and colleagues202 described several abnormalities of pyramidal neuronal migration (ectopic neurons in white matter zones, misoriented apical dendrites, and disorganized cellular layers) in the superior temporal gyrus. Casanova and coworkers203 more recently introduced “minicolumn” terminology into ASD literature. Minicolumns in some areas of the autistic frontal cortex were found to be smaller (representing an underdeveloped system) and had abnormal patterning. Both findings are consistent with deviant processes that occur very early in the second trimester. These anatomical abnormalities may result in deficient neuronal “insulation” and serve as the structural basis for increased neuronal “cross-talk” and overstimulation. This, in turn, may cause the sensory gating and processing difficulties found in some individuals with ASD.203,204 Other autistic symptoms then might be explained by a disregulation of axonal outgrowth, dendritic arborization, and synaptic connectivity.179 Abnormalities described in cortical frontostriatal circuits may be associated with ritualistic and repetitive behaviors.204 Volumetric sMRI studies of cortical systems serving language functions have revealed the absence of the usual left hemispheric hypertrophy (representing left brain dominance and language specialization). Instead of a larger left hemisphere (specifically, the Wernicke receptive language processing area), the planum temporale volumes were equal in subjects with ASD.205 Furthermore, decreased gray matter in the left inferior prefrontal gyrus or Broca’s area (expressive language center) resulted in actual reversal of the typical hemispheric asymmetry (left larger than right) in language-impaired subjects with ASD.206
Hypoactivity in the Fusiform Gyrus during Face Recognition Tasks
Functional neuroimaging techniques, primarily positron emission tomography and fMRI, have confirmed clinical and neuroanatomical data depicting ASD as a disorder characterized by uneven rather than generalized deficits.207 The most consistent fMRI finding has been hypoactivity in the fusiform gyrus, particularly in the fusiform facial area, confirming the clinical impression that deficits in facial recognition are characteristic of ASD.208,209 fMRI has also demonstrated associated deficits in related areas of the “social brain,” such as the amygdala, which plays a critical role in emotional arousal and integration of emotional data.210 Persons with ASD appear to be less motivated to look at faces or to follow the point of conversational partners.208 Computerized eye tracking techniques have also revealed that they pay less attention to faces and more attention to inanimate details in the background.209,211,212 When they do look at the face, they target the mouth rather than the eyes. Because oral expressions provide less information about emotional states than the eyes, persons with ASD often fail to detect meaningful social information during interactions.208
Neurochemical Testing
Neurochemical abnormalities may also be present in children with ASD.178 Increased levels of 5-hydroxytryptamine in whole blood, chiefly platelets, has been a fairly consistent finding. Although 5-hydroxytryptamine is an important neurotransmitter for brain development and modulation of sleep, mood, body temperature, appetite, and hormone release, no consistent abnormalities have been found in central nervous system levels. Age-related differences in serotonin synthesis capacity have also been demonstrated between children with autism and nonautistic controls.213
In conclusion, it is widely accepted that ASD is a “neurodevelopmental disorder,” although the specific underlying abnormalities have not been identified. ASD may actually represent a disorder of neural distribution rather than frank structural abnormalities.176 A project to create the first ever atlas of the autistic brain at several ages is well under way.214 Newer functional brain studies have provided some intriguing links between the neuroanatomical substrate and characteristic clinical features. Well-designed studies with participants matched for IQ levels and with the most sophisticated technology (e.g., diffusion tensor imaging) are needed to unravel the mystery of anatomical differences and white matter “connectivity” and associated discrepancies in learning and neuropsychological functioning. Such studies may provide valuable information that can be used to design, implement, and evaluate new and effective intervention strategies.
CLINICAL SIGNS
Although emphasis has historically been placed on language deficits, they are not specific to ASD and are commonly also the presenting feature of children with mental retardation, hearing loss, and communication disorders. Stereotypies may be obvious and easily recognized, but they also occur in other conditions, primarily severe mental retardation and blindness. Furthermore, they often do not appear until after 3 years of age,215 and some forms (e.g., hand flapping) can be normal in certain situations (e.g., in an excited toddler). Thus, language deficits and stereotypies do not clearly distinguish ASD from other childhood disorders. During the 1990s, it became apparent that the social deficits, specifically those relating to “social communication,” were the most consistent and characteristic symptoms of ASD. The diagnosis of classic autistic disorder currently requires that at least one criterion be met in each of the language and restricted interests domains and that two criteria be met in the social skills domain.19,20 Several early recognizable social communication deficits (e.g., joint attention) appear to be fairly specific for ASD. The severity of these deficits varies significantly from patient to patient, thus creating diagnostic challenges.
Most parents first become concerned about their children’s development when they are between 15 and 18 months of age216,217; their first concerns usually focus on speech delays. Indeed, this has been the historical hallmark of ASD and will probably continue to be so because these deficits are easily recognized. However, with heightened public awareness about the early signs that occur before development of vocal speech, parents are beginning to voice concerns about more subtle receptive language skills (the child’s not responding to his or her name being called) and social skills (e.g., decreased eye contact, unusual attachments to objects, not caring whether parent is nearby). Studies have demonstrated that symptoms can appear before 1 year of age, although these may be subtle.218 Some infants appear to develop normally until approximately the second year of life, when they demonstrate regression in speech and social skills, withdraw, and become indifferent to their surroundings.219,220 Subtle abnormalities in social communication may be evident on careful examination of 1-year-old birthday video recordings.221–223 Expanded discussions can be found in chapters and reviews dedicated solely to early clinical characteristics.224–227
Social Skills Deficits
As noted previously, abnormalities in social communication skills are the most unique and consistent findings in infants with ASD. They appear earlier than identifiable speech deficits but are often more subtle. Children with ASD universally demonstrate deficits in social relatedness, defined as the inherent drive to connect with others and share complementary emotional states.228 Children with other types of disabilities (e.g., mental retardation, sensory disabilities) still attempt to connect with others: Those with hearing deficits compensate with eye gaze and gestures, and those with vision deficits compensate with voice and touch. Children with ASD do not appear to seek this connectedness; they are usually content with being alone, rarely make eye contact or bids for others’ attention with gestures or vocalizations, and react little to praise or bids for attention from others. In later years, they have difficulty in sharing the emotional state of others in cooperative games and group settings and have few, if any, friends.
DEFICITS IN JOINT ATTENTION AND SHARING OF INTERESTS
One of the most distinguishing characteristics of very young children with ASD is a deficit in “joint attention.”229–235 Joint attention is the desire coupled with the ability (facial expressions, gestures and/or speech) to draw another’s attention to objects, events, or other persons simply for the enjoyment of sharing experiences. Like other developmental skills, joint attention appears to develop in graduated stages, usually between 8 and 16 months. At approximately 8 months of age, a typically developing infant may participate in “gaze monitoring”: that is, when a parent looks away (e.g., to check the time), the infant follows the parent’s gaze and looks in the same direction. This social skill should be differentiated from simple auditory orienting, in which both the infant and the parent are stimulated by an environmental stimulus (e.g., clock alarm) at the same time. Children begin to “follow a point” at about 10 to 12 months of age. If a parent points in the direction of an interesting object or event and says, “Look,” the typically developing child looks in the direction that the parent is pointing. Upon seeing the object/event, the child looks back at the parent and smiles, frowns, or shows fear, whichever emotion is appropriate to the situation. An infant with ASD does not follow a point even when a parent tries repeatedly, calling the child’s name in a loud voice or with physical prompts such as touching the child’s shoulder before pointing.218
At 14 to 16 months of age, the typically developing child begins to point simply to “comment” about, or “share,” an interesting object/event (protodeclarative pointing). As the child points, he or she looks alternatively between the object/event of interest and the caregiver. The same triad exists (child, caregiver, object), but the goal is reversed. It is the shared social experience, not the object, that the child seeks. Children with ASD consistently fail to point to “comment” at age-appropriate times. If and when they do start to point, they are less likely to show positive affect and connectedness during the act. About the same time, typical children also begin to “show” items of interest to parents. This bid for attention is distinct from asking for help (e.g., bringing a jar of bubbles to the parent as a request to open it). Mastery of joint attention appears to be a reliable predictor of functional language development.13,236–238
The ability to disengage and shift focus of attention from one stimulus to a novel one is a very basic skill that can be measured in normally developing 6-month-olds. The inability to shift attention was proposed as a characteristic deficit in autism that possibly contributed to deficits in joint attention, inasmuch as it relies on shifting attention between an object/event and a partner.44,199 Multicenter studies of infant siblings of older children with ASD have revealed that the inability to shift one’s attention (from parent to object of interest and back to parent again) is measurable and perhaps the first reliable sign of ASD.218
POOR SOCIAL AND EMOTIONAL RECIPROCITY
One of the earliest developmental milestones is the ability to orient to social stimuli—in particular, turning to respond to one’s own name.239 At about 8 to 10 months of age, most children turn preferentially when their name is called. Like children with hearing impairments, those with ASD often fail to orient to their name. In fact, an early concern of parents of children later diagnosed with ASD is about their infant’s hearing. Parents are often puzzled because such children seem to attend to environmental sounds better than to human voices.240 Retrospective studies of 1-year-old birthday video recordings in children who later received diagnoses of ASD have demonstrated that failure to orient to one’s name being called is one of the most consistent deficits in affected children at that age.221,222. Reciprocal social interaction includes ongoing back-and-forth bids for attention and social interactions with multiple emotional expressions, sounds, and other gestures. Social referencing241 is the ability to recognize the emotional states of others as they respond to various stimuli. When faced with a novel situation, a normal infant might look to his or her caregiver for an indication of delight, anger, or fear in her facial expression. His or her facial expression will then usually mimic the caregiver’s, although he or she may not fully understand the situation. A child with ASD engages in less social referencing and less imitation.242
DIFFICULTY IN MAKING AND KEEPING FRIENDS
Because children with ASD lack the fundamental social skill building blocks described previously, they are less likely to develop appropriate peer relationships. They may have few or no friends, and they tend to relate better with either much younger children or adults. These relationships, when present, usually evolve around the child’s own special interests. Later developing skills may be deficient and also impair friendships. Many authorities believe that impaired central coherence is a basic characteristic of children with ASD, especially older ones. Central coherence is the ability to interpret stimuli in a relatively global way, taking context into account.243,244 Persons with ASD tend to focus on parts and to make less use of context; their processing is more piecemeal. They have difficulty integrating component features into a cohesive unit and seeing the “big picture.” Although central coherence is not a true social skill, deficits in central coherence can impair social interactions, because this type of information processing is very different from that of typically developing peers. Theory-of-mind skills enable a person to take the perspective of another person and are based on the realization that others have thoughts and emotions that are independent from one’s own.245–247 Theory-of-mind skills include the ability to infer states of mind on the basis of external behavior. This inability to take the perspectives of other people is another impediment to forming and maintaining friendships. Although the deficit itself is not unique to patients with ASD, the degree of the deficit is much more severe than has been noted in other disorders. Because of deficits in perspective taking, children with ASD have difficulties with social-emotional behaviors such as empathy, sharing, and comforting. It is now generally accepted that in the course of typical development, children have a sense of the mental states of others by 4 years of age.248,249 Although not helpful in the early diagnosis of autistic disorder, lack of theory-of-mind skills is critical in the early diagnosis of later recognized Asperger syndrome. Unlike deficits in joint attention, theory-of-mind deficits are not specific for ASD; similar findings can be seen in children with cognitive impairments and are consistent with their general level of developmental functioning.250
Communication Deficits
Most children in whom autistic disorder and PPD-NOS are later diagnosed present initially with “speech delay,” although this trend is slowly changing as parents become more aware of social milestones and sense that something is wrong before the child is 18 months old.230,249 Although lack of or severe deficits in speech without any effort to compensate with gestures has long been thought to be characteristic of autistic disorder, more children who now receive diagnoses of autistic disorder do have some speech. Vocabulary deficits are often the focus of concern, but there are typically earlier communication deficits that, if detected, could promote earlier diagnosis.241,249
 Lack of the alternating to-and-fro pattern of vocalizations between baby and parent that usually occurs at approximately 5 months (i.e., babies with ASD usually continue vocalizing without regard for the parent’s speech).
Lack of the alternating to-and-fro pattern of vocalizations between baby and parent that usually occurs at approximately 5 months (i.e., babies with ASD usually continue vocalizing without regard for the parent’s speech).





Parents are often unaware of these deficits unless the milestones are brought to their attention.
Approximately 25% to 30% of children with autistic disorder and PDD-NOS begin to say words at 12 to 18 months but then stop using them. “Autistic regression” characteristically takes place between 15 and 24 months of age after the child has mastered 5 to 10 words.219,251 Many such children become completely nonverbal and cease to gesture (wave, point, and so forth). Although this regression is seemingly dramatic, some parents are able to rationalize the regression and attribute the loss of skills to a family event such as the birth of a new sibling or a move to a new house. Home videos recorded before the onset of regression have revealed that, in at least some children, mild delays and subtle early signs were present before the apparent regression, although there is a subset of children who were apparently normal.168,220
Some children use “pop-up words”: that is, words that are verbalized inconsistently and with no apparent communicative intent. These words are said out of context for a short period of time (days or weeks) and then, as suddenly as they might pop up for no apparent reason, they also disappear.17,238,249 On occasion, these utterances may be phrases or entire sentences, also said out of context. Spontaneously uttered pop-up words should be distinguished from “echolalia.” Echolalia, sometimes called “parroting” by lay individuals, is the repetition of another’s speech. It is classified as “immediate” when the child repeats another’s words right after they are heard or as “delayed” when repeated at distant time later. Normal children pass through a brief developmental stage (“vocabulary burst stage”) in which they imitate other’s speech, particularly the last one or two words of a sentence. Autistic echolalia can persist throughout the lifespan with little or no apparent communicative function. It often occurs long after the utterance (delayed echolalia) and is also qualitatively different in that the utterances are more exact, have a monotone quality and consist of larger verbal “chunks” (e.g., TV advertisement jingles, video re-enactments, or nursery rhymes). These verbalizations often exceed the child’s functional language skills and may be misinterpreted as “advanced” when in fact the child has difficulty following a simple one-step command. Some children with ASD become quite obsessed with labeling colors, shapes, numbers, and letters of the alphabet, and yet they cannot point to them on request or incorporate the labels into functional language. Later they may develop hyperlexia or advanced oral reading without corresponding comprehension skills.
Children with Asperger syndrome may have mild or very limited speech delays and thus escape recognition until around 3 to 4 years of age, when their inability to make friends becomes a concern. Although unnoticed, language development is atypical in that children are often quite verbal about some subjects (usually things or events), but unable to express simple feelings or recognize the feelings and viewpoints of others. Speech may be fluent but limited to only a few topics, typically those that hold a strong, all-consuming interest for the child. It can also be overly formal (pedantic), a reason why they are sometimes described as “little professors.”247 Children with Asperger syndrome also have deficits in the social use of language (pragmatics): how to choose a topic of conversation; understanding and producing appropriate tempo, facial expression, and body language during conversation; turn-taking; recognizing when the partner has lost interest in a topic; knowing when to start, sustain, and end a conversation on the basis of listener cues; knowing when and how to repair a communication breakdown; and using the appropriate degree of formality and politeness. Language may seem odd, pedantic, self-centered, and not listener-responsive and often results in a monotone monologue. These children may demonstrate unique delivery of speech (prosody) in regard to intonation, volume, rhythm, pitch, and personal space, and they tend to disregard listener needs. Children with Asperger syndrome and high-functioning autism have difficulty with abstract reasoning and with discussion of thoughts and opinions of others. Inability to discern and judge the conversational intents of others, especially when their conversation includes words or phrases with ambiguous meanings impairs their ability to understand metaphors, humor, sarcasm, teasing, metaphors, irony, lies, jokes, faux pas, and deception.245,246
Play Skill Deficits
Play has many attributes. It can be sensorimotor, functional, constructive, pretend, or imaginary. Play can take place in isolation of, in parallel with, or through interaction with other children. It can also be pathological (i.e., ritualistic play). Mastery of pretend play, especially during interaction with others, builds on both communication and social skills. Lack of or significantly delayed pretend play, coupled with persistent sensorimotor and/or ritualistic play, is very characteristic of ASD and serves as a discriminating feature of both screening and evaluation ASD tools. Some children with severe ASD may never progress past the sensorimotor play stage. They mouth, twirl, bang, and manipulate objects in a stereotypic or ritualistic manner. Often they prefer to play with common objects (string, sticks, rocks, or ballpoint pens) rather than store-bought toys. One exception is puzzles, especially shape-matching ones and computerized “puzzle games.” These represent a form of “constructive play” (i.e., using objects in combination to create a product).241 Children with ASD are often content to play alone for hours, requiring little attention or supervision. Often this “play” is either constructive (puzzles, computer games, and blocks), ritualistic (lining objects up or sorting/matching shapes or colors), or sensorimotor (mouthing, banging, twirling) in nature. Children with ASD may seem to enjoy chase games and roughhousing, but it is actually the games’ sensorimotor aspects rather than the social aspects that the child enjoys. Even when symbolic play does develop, it may not advance to more sophisticated social play such as role-playing with a peer. These children have trouble interacting in groups and cooperating in the social rules of games. Often they are left out, ignored, and at high risk of being bullied by peers.
Atypical Behaviors
Children with ASD often manifest repetitive, nonfunctional, atypical behaviors or stereotypies (e.g., hand flapping, finger movements, rocking, twirling).19,20,227 Although most such behaviors are harmless by themselves, they are problematic in that they may prevent the child from accomplishing tasks and learning new skills and may interfere with inclusion in natural environments with typically developing peers. Although stereotypies are very distinctive and obvious, they are not specific to children with ASD. Children with severe mental retardation and/or severe visual deficits also commonly demonstrate stereotypies. Even normal toddlers, especially before the onset of fluent language, may flap their arms briefly when they are excited or frustrated. True autistic stereotypies often do not appear until after age 3 years and may include some behaviors such as habitual toe walking and/or sensory stereotypies (persistent sniffing and licking of nonfood items) that are less common in children with other disorders.
Children with ASD, especially those with cognitive deficits, may demonstrate various forms of self-injurious behavior.252 Such behaviors (e.g., head banging, skin picking, eye poking, hand biting) represent a class of stereotypies that, unlike those described previously, may cause bodily harm. Reasons for self-injurious behavior include those that may cause any child, with or without ASD, to display inappropriate behavior. These may include frustration during unsuccessful communication attempts to procure a desired object, protest against transitions, anxiety in new environments, boredom, pain, depression, fatigue, and sleep deprivation. In rare cases, it may result from an endogenous neurochemical abnormality in dopa, serotonin, opioid, or γ-amino butyric acid neural transmitter systems and/or a part of a behavioral phenotype associated with a known genetic syndrome such as the finger and lip biting characteristic in Lesch-Nyhan syndrome.
Additional Clinical Features That Are Common but Are Not Core Features
MENTAL RETARDATION
In the past, cognitive deficits were thought to be extremely common in autism, and in fact, in most studies published before 1990, investigators reported the estimated prevalence of coexisting mental retardation as 90%.83 Reviews and guidelines published from the late 1990s generally reported the prevalence as approximately 75%.8,10–12,92,114 This statistic dropped with more recent prevalence studies,52,53 with a low of 26% in England47,48 and 47% in the United States.55–55b Better ascertainment of children with milder disorders, improved professional training, and more effective strategies/tools for evaluating cognitive abilities in children with ASD have all been cited as possible reasons for the decreasing prevalence of coexisting mental retardation.
SPLINTER AND SAVANT SKILLS
A unique characteristic of ASD is the unevenness of skills. Abilities may be significantly delayed in many areas of development but advanced in others, often because of exceptional memory, calculation, music, or art abilities.253 These advanced skills are often called splinter skills when they serve little or no purpose in day-to-day life and do not improve the ultimate prognosis. For some patients, they may lead to a career that provides financial independence and even widespread recognition254,255 and thus may be called “savant skills.”
ABNORMAL SENSORY PROCESSING
Children with ASD may demonstrate deficits in multisensory integration and processing.256 They may demonstrate simultaneous hyposensitivities and hypersensitivities for different stimuli even within the same sensory modality.257 Although a child may seem not to hear his or her name being called, he or she is annoyed by the sound of dripping water in a distant room. In the visual modality, a child may explore toys while holding them very close to his or her eyes (as if visually impaired) and yet be exceptionally sensitive to the subtle flickering of fluorescent lights. Children with ASD may have oral aversions and/or overall “tactile defensiveness” to soft touch but no apparent response to injuries and other painful stimuli. The dichotomy may arise from an abnormal arousal level or an abnormal sensory gating system.
MOTOR ABNORMALITIES
In addition to the peculiar motor stereotypies that serve as a defining characteristic of the ASD, some affected children also demonstrate poor coordination and even frank delays, usually in the context of global developmental delay (GDD) or severe mental retardation. Others actually appear to have advanced motor skills; still others may have deficits in praxis (the planning, execution, and sequencing of movements).228 Apraxia (severe deficits) and dyspraxia (milder deficits) affect the imitation of speech, facial expressions, play, and/or motor patterns of the extremities. Some investigators believe that, although not a defining characteristic by DSM or ICD-10 standards, motor clumsiness is a distinguishing characteristic of Asperger syndrome.101,258 Some children with ASD may appear to be “hyperactive” and “motor-driven” with an exterior focus of attention, whereas others may be hypoactive and withdrawn and move little.257
IDENTIFICATION AND DIAGNOSIS
Screening
The importance of screening for ASD has been emphasized because early identification allows early intervention that can potentially improve outcome and also leads to etiological investigation and counseling with regard to recurrence risk.13 Although the clinical practice of developmental-behavioral pediatricians is more likely to involve comprehensive diagnostic evaluations than screening, training of general pediatricians and other primary health care providers in effective autism-specific screening strategies has become a primary obligation. Developmental-behavioral pediatricians may also be in a position to train or advise early intervention multidisciplinary teams with regard to screening for ASD.
Historically, the initial concerns of parents of children who later received diagnoses of ASD were dismissed, and diagnosis and intervention were therefore delayed.216,217,259 In spite of increased public and professional awareness, the diagnosis of ASD is still often delayed. In a 2006 Centers for Disease Control and Prevention Atlanta-based study, Wiggins and colleagues55 reported that the average age at the first documented ASD diagnosis was 60 months (range, 17 to 105 months). The average age at diagnosis was significantly younger (i.e., 41 months) in children with overall impairments. An average delay of 13 months occurred between the first evaluation by a qualified professional and the first ASD diagnosis. To address these ongoing challenges, the AAP now recommends administering a standardized autism-specific screening tool at the 18-month evaluation260 and, perhaps additionally at the 24-month health supervision visits261,262 and at any age when ASD concerns are raised spontaneously by parents or as a result of clinicians’ observations or surveillance questions about social, communicative, and play behaviors.
ASD-specific screening tools are sometimes described as level 1 or level 2 screens.263 Level 1 screening measures are administered to all children and are designed to differentiate children at risk for an ASD from the general population, especially those with typical development. Level 2 screening measures are more often used in settings such as early intervention programs or developmental clinics that serve children with a variety of developmental problems; they help differentiate children at risk for ASD from those at risk for other developmental disorders such as mental retardation or specific language impairment. Level 2 screening tools generally require more time and training to administer, score, and interpret than do level 1 measures, and there is considerable overlap between the concept of a level 2 screening tool and that of a diagnostic instrument.263,264 Level 2 screening measures may be used as part of a diagnostic evaluation, but they should not be used in isolation to make a diagnosis. It is important for developmental-behavioral pediatricians to be familiar with the array of ASD screening tools available in order to train primary care providers and to conduct or assist with advanced level 2 screening.
Properties of some level 1 and level 2 ASD screening tools designed for use with very young children are reviewed in Table 15-5.263,265–276 Several level 1 tools, such as the Checklist for Autism in Toddlers (CHAT) and the Modified Checklist for Autism in Toddlers (M-CHAT), are available to the clinician at no cost. Wong and associates276 translated the M-CHAT into Chinese, modified the response choices and the scoring system, and combined it with the five observational items from the CHAT to form the CHAT-23. The Screening Tool for Autism in Two-Year-Olds (STAT) is an interactive measure developed for use as a level 2 screening measure in children between the ages of 24 and 36 months; investigation of its utility with younger and older children is under way.263 Completion of a training workshop is required before use of the STAT. Tools such as the Autism Behavior Checklist,277 the Childhood Autism Rating Scale (CARS),278–281 the Gilliam Autism Rating Scale (GARS),282 and the Social Communication Questionnaire283 can be used to screen for risk of ASD over a wide age range, including the preschool age group (see Table 15-5). Significant concerns about the psychometric properties of the Autism Behavior Checklist, GARS, and Pervasive Developmental Disorders Screening Test–II have been raised.263,284,285 The Social Communication Questionnaire was derived from the Autism Diagnostic Interview–Revised and has fairly strong psychometric properties.283,286 The Social Communication Questionnaire is recommended for use in children older than 4 years and is currently being evaluated to determine the most appropriate cutoff scores for 2- and 3-year-old children.
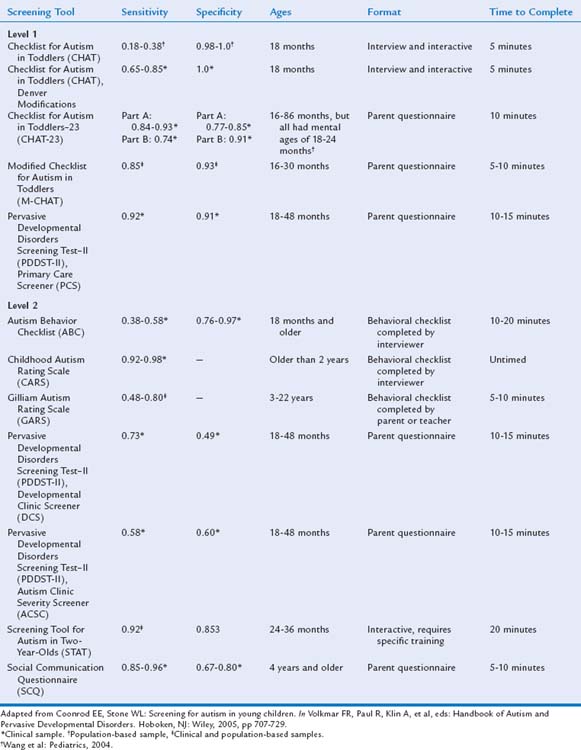
Many of the ASD-specific screening measures are currently being revised or further evaluated, and new tools are being developed to address some of the weaknesses of existing instruments. Attempts are being made to design instruments capable of detecting ASDs at younger ages. For example, the Early Screening for Autism287,288 is being developed as a level 1 ASD screen for 14-month-old children, and the Systematic Observation of Red Flags for Autism Spectrum Disorders in Young Children,289 which is based on the Communication and Symbolic Behavior Scales Developmental Profile Behavior Sample, may be a valuable level 2 screening tool for use in the second year of life.
Comprehensive Evaluation
Because there can be a long waiting period for a subspecialty evaluation, the developmental-behavioral pediatrician or clinic staff should ensure at the time of the initial referral that the child has already been referred for an audiological evaluation and to an early intervention program or a school program (depending on the child’s age) by the primary care provider.260,262 If the child has not, then this should be done immediately so that intervention strategies can be implemented in a timely manner. Immediately available services should address the child’s individual pattern of developmental deficits; strategies can be revised to be more ASD-specific, if necessary, after the definitive diagnosis is made.
For a comprehensive discussion regarding the rationale and strategies for accomplishing these tasks, the reader is referred to published neurology guidelines10; pediatric guidelines11,12; the American Speech and Hearing Association Position Statement290 and Technical Report291; and chapters in two books, Autism Spectrum Disorders: A Transactional Developmental Perspective17 and Handbook of Autism and Pervasive Developmental Disorders14 particularly Chapter 20 (on medical workup), 21 (on diagnostic instruments), 29 (on evaluation components), 30 (on communication assessment), 31 (on behavior assessment), and 32 (on sensorimotor assessment).
COMPREHENSIVE HISTORY
A comprehensive evaluation should begin with thorough health (including neonatal events),159 family, developmental, emotional, and behavioral histories. A three-generation family history especially targeting ASD and its broader phenotype—mental retardation, language delays, and psychiatric and learning disorders—can be very helpful in guiding the etiological workup. A review of systems may be more challenging with the nonverbal child who cannot vocalize symptoms (e.g., depression) or localize pain (e.g., abdominal). Conditions that occur with a higher frequency in children with ASD (e.g., seizures292) should be targeted, as should conditions (e.g., lead toxicity) that result from predisposing ASD-related behaviors (e.g., prolonged mouthing or pica).293 Symptoms of other DSM conditions (e.g., anxiety, obsessive-compulsive disorder, and/or attention-deficit/hyperactivity [ADHD] disorder) may overlap with those of ASD, making diagnosis more difficult. ASD-specific surveys and behavioral interview tools (see discussion in the section “Determination of the Presence or Absence of DSM-IV-TR Criteria”) may assist the developmental-behavioral pediatrician in obtaining pertinent developmental, behavioral and emotional information relating to DSM criteria.
THOROUGH PHYSICAL EXAMINATION
Although a thorough physical examination may be challenging, it is important in identifying syndromes and medical conditions known to be associated with ASD. Measurement of head circumference, a neurodevelopmental examination, and a meticulous search for dysmorphic features, including a Wood’s lamp examination for early subtle neurocutaneous lesions, are especially relevant components of the examination. As previously noted, approximately one third of affected children meet criteria for macrocephaly.184–188 Gillberg and Coleman294 found posteriorly rotated ears in 30%, but this finding has not been replicated in subsequent studies. Mild hypotonia has been noted in some children with idiopathic ASD.295 Positive findings are important in determining the type and extent of an etiological workup; however, because most children have “idiopathic ASD,” the physical examination is chiefly noteworthy for its lack of positive findings.
DEVELOPMENTAL AND/OR PSYCHOMETRIC EVALUATION
Young children with ASD, especially those with high-functioning autism, often demonstrate a significant discrepancy among domains; that is, motor, cognitive and adaptive skills are generally more advanced than language and social (in particular, joint attention) skills.235,296–298 Individual scores for each domain that reveal the child’s relative strengths and deficits are often more valuable than a summarized score in describing the child and in planning intervention. Often multiple tools are needed to address all domains. High “outlier” scores may represent splinter skills that do not necessarily reflect the child’s ability to function in natural settings.7,235 In fact, the discrepancy between scores on standardized tests and real-life functioning is a relatively common finding.
Although the Bayley Scales of Infant Development–II has excellent properties and has been the most widely used developmental assessment tool299 in infant research and many clinical settings, it has not been as helpful in the evaluation of ASD. The Mental Development Index score summarizes nonverbal problem-solving, language, and social skills into a single numerical value, thus obscuring the discrepancies that characterize the profile scatter in ASD.235 The newer Bayley scales, third edition,300 offers promise because they do provide individual scores; however, further study is needed in this population. The Mullen Scales of Early Learning301 have often been used in research endeavors because individual domain scores are possible; however, additional tools are needed to assess adaptive and social skills. Deficits in adaptive ability are critical in the diagnosis of comorbid GDD/mental retardation, and deficits in social skills in relation to general functioning are critical in the diagnosis of ASD.20,37,235 Voigt and associates297 used the Cognitive Adaptive Test/Clinical Linguistic and Auditory Milestone Scale (CAT-CLAMS)303–305 to illustrate discrepant scores in children with ASD, although this measure is not regarded as a comprehensive evaluation tool. All study children demonstrated significant discrepancies in that the developmental quotient for visual-motor problem-solving (CAT) skills averaged 36.4 points above that for language (CLAMS) skills (SD = 15.9; p < 0.00001). In general, the higher the general level of functioning (overall developmental quotient) was, the more significant was the discrepancy (p < 0.0001). A more comprehensive language-specific tool may be helpful with fluent children in order to assess both the quantitative and qualitative differences characteristic of high-functioning autism and Asperger syndrome. Although some rating scales and checklists have been developed to facilitate the assessment process,306 the speech and language pathologist must draw from clinical expertise to detect and describe samples of atypical language such as echolalia, pronoun reversal, pop-up words, neologisms, and pragmatic deficits.290,291 A more comprehensive measure of sensorimotor function may be helpful in supplementing the information gained from a neurological examination.307
Measurement of abilities across all domains can be more challenging and time consuming in older, higher functioning children. The menu of instruments is more extensive, and some knowledge of the child’s verbal abilities is necessary to make appropriate choices.235 Regardless of the tool or tools chosen, subtest scores are again often more helpful than the composite score in making a diagnosis and in planning intervention. Although the Wechsler Preschool and Primary Scale of Intelligence and the Wechsler Intelligence Scale for Children (depending on the child’s age) are “gold standards” for assessment of intelligence, the behavioral challenges and language deficits characteristic of this population may preclude their use. Thus, the clinician must be flexible and able to quickly and skillfully transition to alternative strategies and/or tools while, at the same time, maintaining the child’s interest and attention. The Leiter Scales of Nonverbal Intelligence308 can be helpful in children who have little speech and/or who are noncompliant with tasks that require pointing responses.309–311 This tool uses manipulatives that seem to foster cooperation in children who are otherwise difficult to test; however, it measures only nonverbal skills, and thus the resulting IQ score may not represent the child’s ability to problem solve in real-life situations. The original Leiter scales contained relatively few items in each age category, and some stimulus drawings are now outdated; in addition, the IQ score is calculated. The revised Leiter scales312 are an attempt to ameliorate these disadvantages; however, rather than a manipulative paradigm, it uses the more standard pointing response, which decreases its utility in some difficult-to-test children. Distinguishing between children with severe GDD/mental retardation and stereotypies from those with primary ASD and coexisting GDD/mental retardation can be challenging especially in children with mental ages less than 18 months who are unable to participate optimally in standardized testing. The Pervasive Developmental Disorder in Mental Retardation Scale (PDD-MRS) was developed to assist in this task, but a standardized measure of cognition is necessary to implement the tool.313 Although not a routine component of the evaluation, a more extensive battery of neuropsychological tests can sometimes be helpful in the evaluation of additional deficits (e.g., executive function, central coherence, theory of mind, memory, and shifting of attention) that are characteristic of older and higher-functioning children with ASD.
Knowing whether a child has coexisting mental retardation is critical in determining the type and extent of the etiological search, the optimal school placement, and eligibility for additional financial and support services. In addition to an IQ score below 70 to 75, a measurement of adaptive ability is necessary.314 Adaptive skills can be assessed with the Vineland Adaptive Behavior Scales,315 a semistructured interview technique that addresses motor, social, communication, and daily living skills. Several versions exist for use in different settings. Children with ASD usually demonstrate relative strengths in daily living and motor skills in comparison with communication and social skills.235,316 New norms have been developed for specific application to persons with ASD.317 In addition, a new version, the Vineland Social Emotional Early Childhood Scales,318 contains more early-emerging social skills applicable to ASD. Although not as informative as direct testing of actual skills, the composite Vineland Adaptive Behavior Scales score can be compared with the social and communication subscale scores to demonstrate whether there is a discrepancy between general functioning and these skills.235,319,320
DETERMINATION OF THE PRESENCE OR ABSENCE OF DSM-IV-TR CRITERIA
Since the 1980s, the DSM-IV and DSM-IV-TR criteria have served as the “gold standard” for the diagnosis of an ASD. However, very young children who later receive diagnoses of autistic disorder may not demonstrate full DSM criteria. For example, “failure to form age-appropriate peer relationships” is really not applicable in very young children. In addition, in a preverbal child, it is difficult to demonstrate abnormal conversational skills and stereotypic language. As noted previously, ritualistic behaviors, a need for routines, and stereotypies are often not present or at least not recognized in children younger than 3 years. Thus, even children appearing to have severe autism may not meet full criteria at very young ages. Instead, they usually receive the “subthreshold” provisional diagnosis of PDD-NOS or “speech delay with autistic tendencies” and then, if additional signs appear and full criteria are met, the diagnosis is revised to one of the ASDs. Realizing this diagnostic dilemma, especially because earlier diagnosis is being promoted, Stone215 recommended a modified DSM strategy for children younger than 3 years whereby the clinician consider only three DSM criteria from the social skills domain and one from the communication domain:
Although the DSM criteria are still regarded as the “gold standard,” a significant amount of subjectivity exists when they are used alone, especially when clinicians are inexperienced. In addition, observation of the child during a brief unstructured encounter may fail to reveal DSM-related deficits (e.g., joint attention and/or pretend play) but amplify atypical behaviors (i.e., stereotypies associated with boredom and lack of stimulation in a typical examination room). Ideally, an evaluation should include observation of the child in free play to determine whether he or she engages in spontaneous bids for joint attention, imitative play, and/or engagement with the parents, coupled with a more structured session in which the tester attempts to elicit DSM-related behaviors by using a standardized format.235
Standardized tools have been developed to assist the clinician in operationalizing DSM criteria and in making the diagnosis of ASD. Although several tools have been in existence since the 1980s, a 2006 Atlanta study revealed that only 30% of practitioners actually used one to make the ASD diagnosis.55 A few of the more widely used tools are briefly discussed in the next section; however, the reader is advised to refer to much more detailed descriptions of these tools and their properties in excellent reviews.114,249,264,322,323 Knowledge of the child’s verbal skills is often important in choosing the appropriate ASD-specific tool (e.g., the most appropriate Autism Diagnostic Observation Schedule [ADOS] module). Tools include those that support a DSM-IV diagnosis and are used in clinical settings, or those that are considered “gold standard” diagnostic instruments and are required for NIH-funded research endeavors.
Standardized Tools That Support a Clinical DSM-IV-TR Diagnosis
“Gold Standard” Diagnostic Tools Necessary for Research Endeavors
Knowledge of the child’s overall developmental or mental age is important for interpreting results, because some of these ASD tools tend to overidentify ASD in children with severe GDD/mental retardation.264 For a more detailed discussion of these and additional tools, the reader is referred to the multidisciplinary panel review114 and practice parameter10 and to Volkmar and colleagues.14
SPECIFIC ASSESSMENTS FOR ASPERGER SYNDROME
Diagnosis of Asperger syndrome usually occurs after 4 years of age. Deficits in social skills without accompanying language delays often go unnoticed until children attend school and demonstrate difficulties in classroom activities with peers. For this reason, school personnel, rather than parents or pediatricians, often initiate the Asperger syndrome evaluation. Targeted level 2 screening when symptoms are recognized, rather than universal screening, has been suggested by published guidelines10–12,114; this remains the current suggestion.262 Teachers, parents, and, depending on the age, the students themselves may be asked to complete an Asperger syndrome checklist as a first step in the evaluation process. Although many checklists are currently available for level 2 screening, none is ideal. Of the level 2 tools, the Australian Scale for Asperger Syndrome is perhaps the most popular, mainly because it is easily accessed333 (see www.aspergersyndrome.org). However, it has not been standardized, and the Web site reports low specificity. Like many of the other level 2 surveys, it queries the parents/teachers about abnormalities in social, emotional, communication, cognitive, and movement skills and the presence of unusual interests and rigid routines/rituals. Campbell334 evaluated five of the rating scales. The scales reviewed included the Asperger Syndrome Diagnostic Scale,335 Autism Spectrum Screening Questionnaire,336 Childhood Asperger Syndrome Test,337 Gilliam Asperger’s Disorder Scale,338 and Krug Asperger’s Disorder Index.339 All five measures fell short of current standards, but the Krug measure showed the strongest properties. None of these surveys should ever be used in isolation; however, they can be helpful as a component of a multidisciplinary evaluation.
Pediatric and neurology guidelines did not address the comprehensive evaluation of a child with Asperger syndrome.10–12,114 Some guidance can be found in two other consensus-driven protocols: the California Practice Guidelines for children older than 6 years323 and the ASHA guidelines.290,291 Developing a consensus statement may be more challenging for several reasons: (1) There remains controversy about whether Asperger syndrome is a distinct entity versus a subtype of high-functioning autism27; (2) there are several sets of criteria for Asperger syndrome36,340; (3) because these children’s skills are more complex and differentiated, a wider variety of tests is necessary to quantify the skills323; and (4) most instruments lack sensitivity in identifying more subtle social communication deficits.290,291
For these reasons, the process necessary to make an accurate diagnosis of Asperger syndrome is challenging and almost always involves a team approach. The Autism Diagnostic Interview–Revised and Module 3 or 4 of the ADOS are ideally suited for higher functioning verbal children such as those with Asperger syndrome. In addition to historical aspects of the child’s early development (particularly language development), a complete battery of standardized tests is needed to differentiate Asperger syndrome from learning disorders with overlapping characteristics (e.g., nonverbal learning disability, pragmatic disorder, semantic-pragmatic language disorder, hyperlexia). Asperger syndrome is even less often associated with a known medical condition than is autistic disorder18; nevertheless, a number of mental health conditions (i.e., schizophrenia, schizoid personality, anxiety disorder, obsessive-compulsive disorder, oppositional defiant disorder, and selective/elective mutism) may mimic Asperger syndrome or coexist with it. Thus, a physician with expertise in ASD and mental health conditions should be involved in the diagnostic process. Table 15-6 lists similar and contrasting characteristics of some of these learning and mental health disorders.
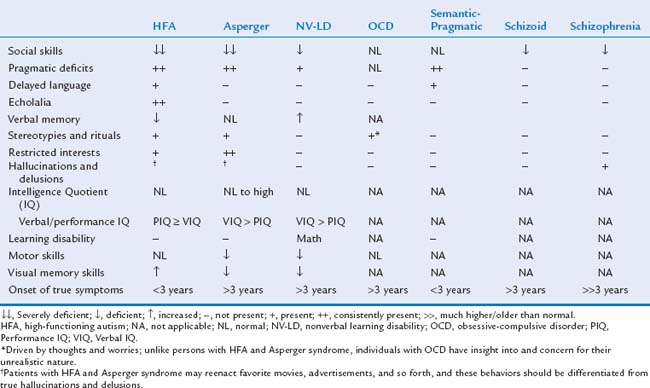
ASSESSMENT OF FAMILY FUNCTIONING AND AVAILABLE RESOURCES
An important part of the comprehensive evaluation of a child with a suspected ASD is the assessment of the family. It should begin with an assessment of the parents’ current understanding of ASD in order to determine both the family’s ability to advocate effectively for their child and the appropriate level of training activities needed. Sometimes parents may need assistance in evaluating the information they already have, especially if it is not peer reviewed and has been accessed from the Internet. The developmental-behavioral pediatrician should also assess the family’s coping strategies, resources (financial, childcare, health insurance), and support systems, including family, friends, neighbors, and religious and local community agencies. On the basis of these considerations, the developmental-behavioral pediatrician or, when available, a team social worker or nurse coordinator can formulate a family support plan.341–343 Referrals to local or Internet-based sources of information, advocacy, and support should be provided. Sleep deprivation, depression, physical well-being, and emotional conditions resulting from stress are more likely to occur in members of families with children with ASD.344–347 These problems should be considered and referrals made when appropriate.
LABORATORY INVESTIGATION
The final challenge in the evaluation of ASD, and perhaps the most controversial, is determining the extent of the etiological “search.” Published etiological yields vary and generally are more highly correlated with the presence or absence of coexisting mental retardation rather than with ASD itself. The majority of reports describe finding an underlying cause in 2% to 10% of patients.47,48,104,107–110,348 These yields are lower than the 10% to 81% reported in studies of GDD/mental retardation without coexisting ASD.349,350 Unfortunately, the extent and sophistication of the laboratory investigations vary a great deal among ASD-specific studies and even within the same study. Patient factors such as the presence of coexisting GDD/mental retardation, dysmorphic features, and/or a positive family history are often not addressed. Some investigators report a “positive yield,” whereas, in fact, the identified abnormality is nonspecific, is not related to a known autism-related etiology, and does not affect counseling and/or management (e.g., delayed mylinization on MRI). In other studies, positive test results indicate coexisting conditions that, although they may be common in children with ASD (e.g., gastrointestinal disorders), are not known to play an etiological role. Thus, a positive laboratory test result does not necessarily mean a positive “yield.”
The emergence of new technology and the lack of overall consensus among subspecialty expert panels presents a formidable challenge when clinicians attempt to develop a consistent search strategy for children with ASD, especially those with comorbid GDD/mental retardation. The American College of Medical Genetics349,351 and the American Academy of Neurology and Child Neurology Society (AAN-CNS)350 have published guidelines for the evaluation of children with GDD/mental retardation that are based on a considerable body of evidence in this population; however, their recommendations are somewhat discordant. Additional recommendations are anticipated in the future from the AAP Committee on Genetics because they will be based on the availability of newer technology (Brad Schaefer, personal communication, March 2006). In the period 2000 to 2001, the first ASD guidelines were published both by the AAN-CNS10 and by the AAP11,12 Because there was some overlap of the authoring panels, there is much agreement between the two documents. Although implied, neither of the ASD guidelines specifically addresses the laboratory investigation of children with high functioning autism or Asperger syndrome. Prevalence studies suggest that the yield is extremely low in the absence of GDD/mental retardation and perhaps even lower in Asperger syndrome.36,47,104,107,352,353 Etiological yield tends to be highest in isolated GDD/mental retardation (10% to 81%), moderate in ASD with coexisting GDD/mental retardation (2% to 10%), and lowest in isolated ASD with normal intelligence (<5%).
In view of comorbidity, discordant guidelines, limitations in current evidence, and emerging technology, we support a practical approach such as a tiered strategy. The AAP Committees on Children with Disabilities and Genetics are in the process of developing ASD guidelines that will be published at a future date and may deviate somewhat from the following recommendations. The proposed strategy advocates for informed clinical judgment and decision making that is based on the presence or absence of coexistent GDD/mental retardation and/or clinical indicators.
Proposed Strategy for a Tiered Etiological Search
LEVEL 1 SEARCH
All children with language delays, including those with ASD, should have an audiology evaluation. Published ASD-specific guidelines for young children presenting with symptoms of ASD reinforce this recommendation.10–12,114,262 School-based hearing screening may be adequate in older children with Asperger syndrome who have no significant language deficits, especially in the absence of any academic concerns.
LEVEL 2 SEARCH
The workup of a child with ASD with coexisting GDD/mental retardation should be guided by published guidelines for both conditions. As noted previously, the etiological yield rates are typically lower in children with both ASD and GDD/mental retardation than in those with isolated GDD/mental retardation, especially those with severe GDD. For children who have both conditions, ASD guidelines recommend (1) a high-resolution karyotype and (2) a molecular study for the fragile X syndrome, unless the child has a known etiology to explain the GDD/mental retardation or presents with characteristics of a disorder that can be confirmed by a specific targeted laboratory test (e.g., MECP2 in a girl with classic Rett symptoms).10–12,114,262 These guidelines also suggest that the tests just described may be helpful in the absence of GDD/mental retardation if there is a family history of mental retardation (especially mental retardation caused by the fragile X syndrome), but it is recognized that the yield will be extremely low. The presence of more than two dysmorphic features increases the likelihood of a positive yield; however, this finding is more highly correlated with GDD/mental retardation than with ASD.294,350,354 The relative importance of the fragile X syndrome in the cause of ASD has been somewhat controversial and, depending on the study, the prevalence of a positive DNA assay varies between 1.6% and 16%.355 In a review of 40 studies,356 identical pooled proportions of the fragile X syndrome were found in boys with autism and in boys with isolated mental retardation; this raises concern about the relative causative role of the fragile X syndrome in ASD. If the karyotype and fragile X test are negative (as most are), but a recognizable cause is still suspected, then the clinician should consider level 3 tests and/or consultation with a clinical geneticist and/or neurologist, depending on indicators from the history and/or physical examination.
Although the 2003 AAN-CNS practice parameter350 recommends additional “screening” laboratory tests (e.g., MRI, subtelomere fluorescent in situ hybridization [FISH] studies) for all children with GDD/mental retardation, it has not been endorsed by the AAP for GDD/mental retardation or ASD. Published genetic GDD/mental retardation guidelines have recommended fewer “screening” tests and a more targeted approach that is based on clinical judgment.349,351,354 Although more sophisticated and effective screening techniques may be on the horizon, there are currently no existing data to support more extensive laboratory evaluation in children with ASD. As the apparent prevalence of coexisting GDD/mental retardation continues to decrease, the need to consider GDD/mental retardation guidelines will occur less often.47,55–55b,348
LEVEL 3 SEARCH
A geneticist can assist with an extended pedigree history and a more meticulous dysmorphic examination. Expensive, more sophisticated laboratory tests may require the assistance of a geneticist for authorization (especially when they are not yet clinically available) and interpretation. When the child’s evaluation includes profound mental retardation, consanguinity, a complex family history, or a vague dysmorphic gestalt, the geneticist may order subtelomere testing.356b Yields for subtelomere studies are extremely low in those with mild mental retardation and usually zero in studies that target individuals with ASD.104,112 The geneticist may order single-locus studies (e.g., 15q or 22q) or newer macroarray FISH studies; however, the latter are expensive and may not be clinically available in all settings. When specific indicators are present, the geneticist is also helpful in ordering and interpreting the level 3 targeted tests listed in nos. 1 to 9 below.
A child neurologist can be very helpful in assisting with the evaluation of a child with regression, seizures, or specific neurological examination findings. The neurologist is able to address certain aspects of the history and physical examination in more detail and thus may identify indicators for level 3 laboratory studies. A high index of suspicion and meticulous monitoring is recommended to identify subtle seizures.10–12,114,350 Some neurologists may recommend “screening” EEG and MRI. Although nonspecific abnormalities have been found in the majority of children, the significance of these abnormalities is not clear, treatment has not been of any proven value,357 and there is currently insufficient evidence to recommend or discourage the use of screening EEG.358,359 Furthermore, MRI has not been recommended for all children with isolated macrocephaly associated with idiopathic ASD.10,321
The clinician (developmental-behavioral pediatrician, geneticist, neurologist, neurodevelopmental disabilities specialist, or other qualified specialist) should pursue level 3 tests when clinical findings characteristic of one or more specific disorders become apparent. Identification of these clinical indicators depends, in part, on the thoroughness of the history and physical examination. The following recommendations for a targeted etiological search in the individual child are relatively consistent with the original ASD guidelines.10–12,114 Level 3 testing should be individualized according to the following specific clinical findings:
 Because Rett mutations can sometimes occur in boys (especially those with Klinefelter syndrome), MECP2 assays should also be considered in boys who present with regressive ASD. Universal screening as part of a research protocol has revealed that MECP2 mutations are present in a few ASD children without evidence of the Rett phenotype; if these findings are replicated, universal screening of all children with ASD may be recommended in the future.361a,362,363
Because Rett mutations can sometimes occur in boys (especially those with Klinefelter syndrome), MECP2 assays should also be considered in boys who present with regressive ASD. Universal screening as part of a research protocol has revealed that MECP2 mutations are present in a few ASD children without evidence of the Rett phenotype; if these findings are replicated, universal screening of all children with ASD may be recommended in the future.361a,362,363 Because an association has been found between Angelman syndrome and assisted reproductive techniques,364 methylation testing for Angelman syndrome should be considered if children conceived by assisted reproductive techniques present with ASD features, because some overlap with the Angelman phenotype.
Because an association has been found between Angelman syndrome and assisted reproductive techniques,364 methylation testing for Angelman syndrome should be considered if children conceived by assisted reproductive techniques present with ASD features, because some overlap with the Angelman phenotype. Because an association between ASD and a mild variant of Smith-Lemli-Opitz syndrome has been described, 7-dehydrocholesterol testing might be considered when a child with ASD presents with hypotonia and syndactyly of the second and third toes. This suggestion is important for genetic counseling because Smith-Lemli-Opitz syndrome is an autosomal recessive disorder.365
Because an association between ASD and a mild variant of Smith-Lemli-Opitz syndrome has been described, 7-dehydrocholesterol testing might be considered when a child with ASD presents with hypotonia and syndactyly of the second and third toes. This suggestion is important for genetic counseling because Smith-Lemli-Opitz syndrome is an autosomal recessive disorder.365Reviews18 continue to support the recommendations published in previous guidelines10–12,114 that the following tests are not indicated in the typical workup of a child with ASD: hair analysis for trace elements, vitamin levels, celiac antibodies, immunological workup, allergy testing, intestinal permeability studies, stool analysis, urinary peptide measurements, thyroid function tests, or erythrocyte glutathione perioxidase level measurements.
Patients enrolled in one of several multicenter research studies may also undergo a number of additional tests (e.g., functional imaging, immunological studies, fibroblast karyotypes, neuroligan gene testing, mitochondrial gene sequencing, and additional metabolic studies)173 that may not currently be universally available or clinically indicated in most children with idiopathic ASD, especially those with Asperger syndrome and high-functioning autism. Insurers rarely pay for such tests, evidence does not support doing these routinely, and the results may promote unrealistic expectations. As information regarding genetic markers for ASD expands and technology continues to become more sophisticated, the yield of complex genetic and laboratory investigations may increase in children with idiopathic ASD and eventually become clinically useful as part of the routine ASD workup. Currently the most promising probes are those that target 15q duplications, 22q deletions, and abnormalities on the X-chromosome and chromosomes 2, 3, 7, and 17. Some investigators have already suggested screening FISH for the 15q and 22q abnormalities111 (Brad Schaefer, personal communication, March 2006); however, more evidence is needed before this becomes standard of care. Testing for 22q deletions, may be particularly important for genetic counseling purposes because about half of the cases in one study were caused by balanced translocations.366–367
SUMMARY
In conclusion, developmental-behavioral pediatricians play an important role in teaching primary care providers to recognize the early signs of ASD, conduct ongoing developmental surveillancem and use an ASD-specific screening tool at the 18- and 24-month well-child visits. They should also encourage primary care providers to listen to parents’ concerns relating to language and social skills and to act on them. Such action should include simultaneous referrals for audiological testing, an appropriate intervention program, and a pediatric subspecialist/developmental team with expertise in the evaluation of children with ASD. Depending on the clinical presentation, referrals to neurology and/or genetics may also be indicated. Either acting alone or as a member of a developmental assessment team, the developmental-behavioral pediatrician should evaluate the child’s health, developmental, and behavioral status; apply DSM-IV-TR criteria, preferably through the use of a standardized ASD-specific evaluation tool; and decide on the extent of the etiological laboratory workup, sometimes with guidance from genetic and/or neurology colleagues. The entire process should be done in a collaborative manner and communicated with a concise summary report. The developmental-behavioral pediatrician should interpret the results of the clinical and laboratory evaluations with the parents in an unhurried, sensitive, and compassionate manner that is culturally appropriate. The evaluation process should include appropriate follow-up because genetic and neuroimaging technology is constantly evolving, manifestations of the DSM criteria may evolve with development, and children may present with additional comorbid symptoms and/or challenging behaviors at any point in their lives.
MANAGEMENT
Educational Interventions
In the late1960s and the 1970s, when the psychogenic theory of causation faded and Section 504 of the Rehabilitation Act of 1973 (Public Law 93-112)368 and the Education of All Handicapped Children Act of 1975 (Public Law 94-142)62 mandated appropriate public education for all children with disabilities, education of the child began to replace psychodynamic treatment of the parents as the primary intervention for autism. Education has been defined as fostering of acquisition of skills and knowledge to assist a child in developing independence and personal responsibility; it encompasses not only academic learning but also socialization, adaptive skills, communication, amelioration of interfering behaviors, and generalization of abilities across multiple environments.13 Teachers, behavior therapists, speech and language therapists, occupational therapists, paraprofessional staff, parents, and other experts commonly play key roles in the education of children with ASD. Physicians and other clinicians are often in a position to guide families to empirically supported practices and help them evaluate the appropriateness of the educational services that are being offered.
PROGRAMS FOR PRESCHOOL- AND EARLY SCHOOL-AGED CHILDREN
Since the 1980s, autism education research and program development have focused disproportionately on very young children as a result of earlier identification and evidence that early intensive intervention may result in substantially better outcomes.369 Model programs have been described,13,370,371 and selected examples are summarized in Table 15-7. Although these programs may differ in philosophy and relative emphasis on particular strategies, they share many common goals, and there is a growing consensus that important principles and components of effective early childhood intervention for children for with ASD include the following13,369,372,373,374:
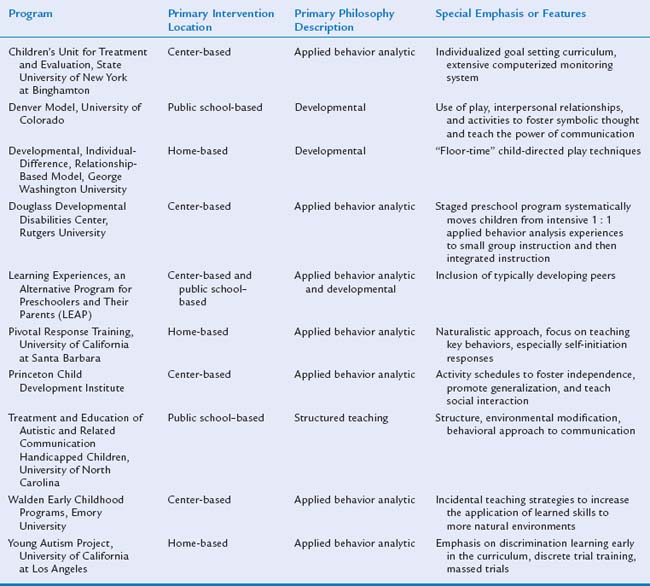
PROGRAMS FOR OLDER CHILDREN
Some model programs, such as the Princeton Child Development Institute375 and the Treatment and Education of Autistic and Related Communication- Handicapped Children (TEACCH) program,376 provide programming throughout childhood and into adulthood. More commonly, the focus of specialized programs is on early childhood; there have been few evaluations of comprehensive educational programs for older children and adolescents with ASD. There is empirical support for the use of certain strategies, especially those based on applied behavior analysis (ABA), across all age groups to increase and maintain desirable adaptive behaviors, reduce interfering maladaptive behaviors or narrow the conditions in which they occur, teach new skills, and generalize behaviors to new environments or situations.377–379
The specific goals and objectives in the Individualized Education Plan and the supports required to achieve them should be the driving force behind decisions regarding the most appropriate, least restrictive educational settings. Appropriate setting may range from self-contained special education classrooms to regular classrooms. Often a mix of specialized and inclusion experience is appropriate. Even high-functioning students with ASD often require accommodations and other supports such as provision of explicit directions, modification of classroom and homework assignments, organizational supports, access to a computer and word processing software for writing tasks, and social communication skills training. When a paraprofessional aide is assigned, it is important that there is an infrastructure of expertise and support for the child beyond the immediate presence of the aide.380 The specific duties of the aide should be outlined, the strategies to be used should be defined, and the aide should receive adequate training.
SPECIFIC STRATEGIES
A variety of specific methods are used in educational programs for children with ASD. Detailed reviews of intervention strategies to enhance communication13,377,381–387 and reduce interfering maladaptive behaviors377,388,389 are available. A few specific strategies are briefly reviewed as follows because of their importance, based on empirical support in the literature or popularity.
Applied Behavior Analysis
Highly structured comprehensive early intervention programs for children with ASD such as the Young Autism Project at the University of California, Los Angeles,390,391 rely heavily on discrete trial training methods, but this is only one of dozens of techniques used within the realm of ABA. Discrete trial training methods are very useful in establishing learning readiness by teaching foundation skills such as attention, compliance, imitation, and discrimination learning, as well as a variety of other skills. However, discrete trial training has been criticized because of problems with generalization of learned behaviors to spontaneous use in natural environments and because the highly structured teaching environment is not representative of natural adult-child interactions. Traditional ABA techniques have been modified to address these issues. Naturalistic behavioral interventions such as incidental teaching and natural language paradigm/pivotal response training are more child centered and take place in more loosely structured environments.379
Functional behavior analysis, or functional assessment, is an important aspect of ABA-based treatment of unwanted behaviors. Most problem behaviors serve an adaptive function of some type and are reinforced by their consequences, such as (1) attainment of adult attention; (2) attainment of a desired object, activity, or sensation; or (3) escape from an undesired situation or demand. Functional assessment is a rigorous, empirically based method of gathering information that can be used to maximize the effectiveness and efficiency of behavioral support interventions.392 It includes formulating a clear description of the problem behavior (including frequency and intensity); identifying the antecedents, consequences, and other environmental factors that maintain the behavior; developing hypotheses specifying the motivating function of the behavior; and collecting direct observational data to test the hypothesis. Functional analysis is also useful in identifying antecedents and consequences that are associated with increased frequency of desirable behaviors so that these can be used to evoke new adaptive behaviors.
Empirically validated behavioral methods for the treatment of problem behaviors include antecedent interventions, consequence-based interventions, and some classical or respondent conditioning procedures.377 Antecedent interventions focus on preventing the occurrence of problem behaviors and include strategies such as (1) providing optimal levels of environmental stimulation, predictable schedules and routines, favorable staffing patterns, antecedent physical exercise, personal choices, and balanced task difficulty and (2) utilizing errorless learning, behavioral momentum, stimulus change, functional communication training, social skills training, and self-management procedures. Adaptive skill acquisition is a vital component of antecedent interventions. Consequence-based interventions include interruption and redirection, reinforcement-based interventions, extinction procedures, and sometimes noncontingent reinforcement and punishment procedures. Classical conditioning procedures sometimes used in clinical practice include desensitization and relaxation techniques.
Speech and Language Therapy
Individuals with ASD have deficits in social communication, and treatment by a speech and language pathologist is almost always warranted. Most children with ASD can develop useful speech. Chronological age, lack of typical prerequisite skills, failure to benefit from previous language intervention, and lack of discrepancy between language and IQ scores should not preclude speech and language services.290,291 Speech and language therapists are likely to be most effective when they train and work in close collaboration with teachers, support personnel, families, and the child’s peers to promote functional communication in natural settings throughout the day; traditional, low-intensity pull-out service delivery models are often ineffective.
Augmentative and alternative communication modalities, including gestures, sign language, and picture communication programs, often are effective in enhancing communication.381,384 The picture exchange communication system393,394 is widely used. The picture exchange communication system method incorporates ABA and developmental-pragmatic principles, by which the child is taught to initiate a picture request and persist with the communication until the partner responds. There is also evidence that some nonverbal individuals with ASD benefit from exposure to voice output communication devices.384 Introduction of augmentative and alternative communication systems to nonverbal children with ASD does not keep them from learning to talk, and there is some evidence that they may be more stimulated to learn speech if they already understand something about symbolic communication.393,395
Social Skills Instruction
There is some objective evidence to support various approaches to teaching social skills.385–387,396–399,404 Joint attention training may be especially beneficial in young, preverbal children with ASD, because joint attention behaviors precede and are predictive of social language development.400,401 Families can facilitate joint attention and other reciprocal social interaction experiences throughout the day in the course of the child’s regular activities. Examples of these techniques are described in the AAP parent booklet Understanding Autism Spectrum Disorders402 (pp 27 to 30).
A social skills curriculum should focus on responding to the social overtures of other children and adults, initiating social behavior, minimizing stereotyped perseverative behavior while using a flexible and varied repertoire of responses, and self-managing new and established skills.369 Social skills groups, social stories, visual cuing, social games, video modeling, scripts, peer-mediated techniques, and play and leisure curricula are supported primarily by descriptive and anecdotal literature, but the quantity and quality of research is increasing.369,373,398 Relationship Development Intervention focuses on activities that elicit interactive behaviors with the goal of engaging the child in a social relationship so that he or she discovers the value of positive interpersonal activity and becomes more motivated to learn the skills necessary to sustain these relationships.403. However, the evidence of efficacy of Relationship Development Intervention is anecdotal; published empirical scientific research is lacking at this time. A number of social skills curricula and guidelines are available for use in school programs and at home.369,396,404
Sensory Integration Therapy
Sensory integration therapy is often used alone or as part of a broader program of occupational therapy for children with ASD. The goal of sensory integration therapy is not to teach specific skills or behaviors but to remediate deficits in neurological processing and integration of sensory information to allow the child to interact with the environment in a more adaptive manner. Unusual sensory responses are common in children with ASD, but there is no strong evidence that these symptoms differentiate ASD from other developmental disorders, and the efficacy of sensory integration therapy has not been objectively demonstrated.405–407 Available studies are plagued by methodological limitations, but proponents have noted that higher quality sensory integration research is forthcoming.408
COMPARATIVE EFFICACY OF EDUCATIONAL INTERVENTIONS
All treatments, including educational interventions, should be based on sound theoretical constructs, rigorous methods, and empirical studies of efficacy.373 Proponents of behavior-analytic approaches have been the most active in using scientific methods to evaluate their work, and most research studies of comprehensive treatment programs that meet minimal scientific standards involve treatment of preschoolers with behavioral approaches.374 The effectiveness of ABA-based intervention in autism is well documented, primarily through five decades of research through single-subject methods77,388,378,409 but also in controlled studies of comprehensive early intensive behavioral interventions.390,410–415
There is limited empirical evidence of efficacy for non-ABA early intervention models such as the Denver model,416–418 TEACCH,376,419–422 and the responsive teaching curriculum developed by Mahoney and colleagues.423,424 Greenspan and Wieder’s developmental, individual-difference, relationship-based model (“Floor Time”) is supported in the literature only by an unblinded review of case records with comparison to an inappropriate control group and use of a questionable primary outcome measure425 and a descriptive follow-up study of 8% of the original group of patients.426
Although categorization by philosophical orientation (e.g., behavior analytic, developmental, structured teaching) has some meaning, there is also overlap among the different approaches. For example, contemporary comprehensive behavioral curricula borrow from ideas that were introduced from a developmental or cognitive orientation (such as addressing joint attention, reciprocal imitation, symbolic play, and theory of mind and using indirect language stimulation and contingent imitation techniques), and in developmental models such as the Denver model and the structured teaching approach of the TEACCH program, behavioral techniques are used to fulfill their curriculum goals.369,379
Most educational programs available to young children with ASD are based in their communities, and often an “eclectic” treatment approach is used. This approach draws from a combination of methods, including ABA methods such as discrete trial training, TEACCH-based procedures, speech and language therapy (with or without the picture exchange communication system or related augmentative or alternative communication strategies), sensory integration therapy, and typical preschool activities. In three studies in which intensive ABA programs (25 to 40 hours per week) were compared with equally intensive “eclectic” approaches, results have suggested that the ABA programs were significantly more effective.410,411,427 Although the groups of children achieved very similar scores on key dependent measures before treatment began, parent-determined rather than random assignment to treatment group was a limitation of these studies. Additional comparisons of educational treatment approaches are warranted.
PROGRAM FOR ADOLESCENTS: TRANSITION ISSUES
At some point, depending on the adolescent’s cognitive level, communication and social skills, and comorbid behavior challenges, the adolescent with ASD may decide to move out from the family home into the community. The school transition process, if properly executed, should help in facilitating this. Skills necessary for independent living should be taught to the degree possible with the abilities of the individual. A growing body of literature addresses sexuality education for individuals with developmental disabilities,428–430 as well as training regarding leisure skills. During the final school years, assessments should be conducted to determine whether the individual is capable of living independently or whether some degree of supervision and daily support, such as that typically provided in government subsidized group homes, is needed. As with decisions regarding educational placement settings, the least restrictive environment should be sought in order to ensure maximum possible autonomy and self-determination.
Some comprehensive programs, such as the Princeton Child Development Institute and TEACCH, have curricula for adults with ASD that extend many of the same approaches offered for children while increasing the emphasis on self-care and community-living skills. A self-determination program designed specifically for individuals with ASD who want to have more control over financial and other important decisions has been described,431 and the emphasis on self-determination will probably continue to grow.
Medical Aspects of Habilitation
GENETIC COUNSELING
Genetic counseling regarding recurrence risk in siblings is very important even when the etiological workup results are negative. Parents need to understand that such results do not mean that future siblings are without risk; the recurrence risk is approximately 5% to 6% (range, 2% to 8%) in idiopathic ASD.92,432 The prevalence of abnormality in siblings is even higher, perhaps 20%, when the broader phenotype or milder constellation of similar social, communication, and behavioral abnormalities is considered.432 If there are already two siblings with ASD in a family, it is likely that the recurrence risk for strictly defined ASD in subsequent pregnancies is well above 8% and may approach 25%, but there is insufficient evidence to be more precise.432 It is important to discuss the recurrence risk promptly after diagnosis in order to provide parents with the opportunity to consider this information before they conceive another child.92 If a specific cause is determined, the recurrence risk may be lower or higher than the risk in idiopathic ASD, depending on the syndrome or other etiological condition identified. In addition, in some conditions (e.g., fragile X syndrome), prenatal diagnosis may be possible.
SEIZURES
The reported prevalence of epilepsy among individuals with ASD ranges from 11% to 39%.292 Coexisting severe mental retardation and motor deficits are associated with a high prevalence of seizures (42%),433 whereas the prevalence of seizures is only 6% to 8% in children with ASD but without severe mental retardation, motor deficit, associated etiological medical disorder, or positive family history of epilepsy.433,434 The prevalence of epilepsy is also higher in studies that include adolescents and young adults because the onset of epilepsy in ASD has two peaks: one before age 5 and another in adolescence. Anticonvulsant treatment in children with ASD is based on the same criteria that are used for other children with epilepsy, including accurate diagnosis of the particular seizure type.
Epileptiform abnormalities on EEG are more common than full-blown epilepsy in children with ASD; reported frequencies range from 10% to 72%.358 Some studies have suggested that epileptiform abnormalities on EEG219 and/or epilepsy435 is more common in the subgroup of children with ASD who have a history of regression, whereas other studies have not demonstrated this association.357,436 Autistic regression with epileptiform findings on EEG has been compared by analogy to Landau-Kleffner syndrome and electrical status epilepticus in sleep, but there are important differences between these conditions, and the treatment implications are unclear.180,292 Whether subclinical seizures have adverse effects on language, cognition, and behavior is debated, and there is no evidence-based recommendation for treatment of children with ASD and epileptiform findings on EEG, with or without regression.180
GASTROINTESTINAL PROBLEMS
The prevalence of gastrointestinal problems is unclear, because most investigators have not examined representative groups of children with ASD in comparison with appropriate controls.437,438 In some published studies, researchers have found that gastrointestinal problems such as chronic constipation or diarrhea occur in 46% to 70% of children with ASD,439–441 whereas lower rates, in the range of 9% to 24%, have been reported in several other studies.442–445
In children with ASD undergoing endoscopy, high rates of lymphoid nodular hyperplasia and often histologically subtle esophagitis, gastritis, duodenitis, and colitis have been described, and preliminary evidence suggests that some immunohistochemical features may be unique to inflammation associated with ASD.437,446,447 The existing literature does not support routine specialized gastroenterological testing for asymptomatic children with ASD.437 However, if a child with ASD presents with symptoms such as chronic or recurrent abdominal pain, vomiting, diarrhea, or constipation, it is reasonable to evaluate the gastrointestinal tract. Occult gastrointestinal discomfort should also be considered in a child presenting with outbursts of aggression or self-injury.
Standard acid-inhibiting therapy for the symptomatic child with reflux esophagitis or gastritis is probably warranted, as is treatment of identified Helicobacter pylori infection, although there are no data specific to ASD. The role of immune modulators for mucosal inflammation associated with ASD has not been studied, although there are anecdotal reports of response in the literature.447 Radiographic evidence of constipation has been found to be more common in children with ASD than in controls with abdominal pain (36% vs. 10%),448 and effective management may provide some benefit.
PSYCHOPHARMACOLOGY
Surveys indicate that approximately 45% of children and adolescents449–451 and up to 75% of adults452 with ASD are treated with psychotropic medication. Older age, poorer adaptive skills and social competence, and higher levels of challenging behavior are associated with likelihood of medication use.451
The Decision to Initiate Medical Treatment
Before initiating a trial of psychotropic medication, it is important to search for medical factors that may be causing or exacerbating the maladaptive behaviors. Recognition and treatment of medical conditions may eliminate the need for psychopharmacological agents in some cases. For example, in the case of an acute onset or exacerbation of aggressive or self-injurious behavior, an occult source of pain such as otitis media, otitis externa, pharyngitis, sinusitis, dental abscess, constipation, urinary tract infection, fracture, headache, esophagitis, gastritis, colitis, or allergic rhinitis may be identified and treated. When behavioral deterioration is temporally related to menstrual cycles in an adolescent girl, use of an analgesic or an oral or injectable contraceptive may be helpful. Obstructive sleep apnea may contribute to behavioral deterioration and may be ameliorated by weight reduction, tonsillectomy and adenoidectomy, or continuous positive airway pressure. Extreme food selectivity has the potential to lead to malnutrition or vitamin or mineral deficiencies; however, most evaluations of nutritional status in children with ASD suggest that despite dietary selectivity, malnutrition is uncommon.437,453 Although the prevalence in ASD is unknown, pica related to iron or zinc deficiency may respond to supplementation.
After treatable medical causes and modifiable environmental factors have been ruled out, a therapeutic trial of medication may be considered if the behavioral symptoms are causing significant impairment in functioning. In some cases, a coexisting disorder such as major depression, bipolar disorder, or an anxiety disorder can be reasonably diagnosed, and the patient can be treated with the medications that are useful for treating these conditions in otherwise typically developing children and adolescents. Modifications of diagnostic criteria may be necessary to account for clinical presentations of psychiatric conditions in individuals with developmental disabilities.454,455 In other cases, clinicians opt to treat interfering maladaptive behaviors in the absence of a clear comorbid psychiatric diagnosis.
Choice of Medication
The evidence regarding the efficacy of psychopharmacological interventions in ASD has been detailed in reviews.139,456–458 Although most psychotropic medications have been used in children with ASD, there is currently insufficient literature to establish a consensus-based, evidence-based approach to pharmacological management. Fortunately, there has been an increase in publication of randomized, double-blind, placebo-controlled clinical trials to guide clinical practice. A summary of selected target symptoms, potential psychiatric diagnoses, and medication options is provided in Table 15-8.
TABLE 15-8 Selected Medication Options for Common Target Symptoms or Comorbid Diagnoses in Children with Autism Spectrum Disorder
| Target Symptom Cluster | Possible Comorbid Diagnosis | Suggested Medications |
|---|---|---|
| Repetitive behavior, behavioral rigidity, obsessive-compulsive symptoms | Obsessive-compulsive disorder |
SSRI, selective serotonin reuptake inhibitor.
Adapted from Myers SM, Challman TD: Psychopharmacology: An approach to management in autism and intellectual disabilities. In Accardo PJ, ed: Capute & Accardo’s Neurodevelopmental Disabilities in Infancy and Childhood, 3rd ed. Baltimore: Paul H. Brookes, in press.
STIMULANTS, α2 AGONISTS, AND ATOMOXETINE
Although early studies of the effects of stimulants yielded negative results, more recent double-blind, placebo-controlled trials of methylphenidate have demonstrated improvement in hyperactivity, impulsivity, and inattention in children with ASD.459–461 Methylphenidate is effective in some children with ASD, but the response rate is lower than in children with isolated ADHD, adverse effects are more frequent, and it is unclear whether the results can be generalized to other stimulants.461,462 Two very small, double-blind, placebo-controlled trials have documented modest benefits of clonidine in reducing hyperarousal symptoms, including hyperactivity, irritability and outbursts, impulsivity, and repetitive behaviors in children with ASD.463,464 A retrospective record review study revealed that open-label treatment with guanfacine was effective in 19 (24%) of 80 children with ASD.465 Patients without mental retardation were more likely to show improvement in target symptoms, including hyperactivity, inattention, insomnia, and tics. Atomoxetine has not been evaluated in controlled trials in children with ASD, but a retrospective study suggested that it might be effective for ADHD-like symptoms in this population.466
ATYPICAL ANTIPSYCHOTIC AGENTS
Atypical antipsychotic medications currently available in the United States include clozapine, risperidone, olanzapine, quetiapine, ziprasidone, and aripiprazole. There is evidence that atypical antipsychotics are efficacious in the treatment of children and adolescents with severe disruptive behaviors associated with intellectual disability.467–469 in addition to those with psychosis, bipolar disorder, Tourette syndrome, and, potentially, conduct disorder and severe ADHD.470 Two large, multisite, randomized controlled trials have confirmed the short-term efficacy of risperidone for severe disruptive behaviors such as tantrums, aggression, and self-injurious behavior in youths with ASD.471–474 Results of two open-label studies, each with a double-blind discontinuation component, have suggested long-term benefits and tolerance.475,476 The effect of risperidone on core symptoms of ASD is less dramatic. The Research Units on Pediatric Psychopharmacology Autism Network trial revealed that treatment with risperidone improved the restricted, repetitive, and stereotyped patterns of interests and behavior but did not significantly affect impairments in social interaction or communication.473
SELECTIVE SEROTONIN REUPTAKE INHIBITORS
Selective serotonin reuptake inhibitors (SSRIs) include fluoxetine, sertraline, fluvoxamine, paroxetine, citalopram, and escitalopram. Double-blind, placebo-controlled trials have demonstrated efficacy of fluoxetine477 and fluvoxamine478,479 in the treatment of repetitive and other maladaptive behaviors in patients with ASD. Open-label trials of various serotonin reuptake inhibitors in children and adults with ASD report response rates in the range of 50% to 75% but are susceptible to publication bias and other shortcomings of uncontrolled studies.480 Improvements in target symptoms, including repetitive behaviors, irritability, depressive symptoms, tantrums, anxiety, aggression, difficulty with transitions, social interaction, and language, have been reported.477–480
ANTICONVULSANT MOOD STABILIZERS AND LITHIUM
In small open-label trials, valproate,481 levetiracetam,482 and topiramate482a were effective in reducing symptoms such as aggression, impulsivity, hyperactivity, conduct problems, and mood lability in children with ASDs. In an open-label valproate trial481 and several case reports,483,484 improvements in language and social skills were also described. A small double-blind, placebo-controlled trial485 demonstrated significant improvement in repetitive behavior in children with ASDs who were treated with valproate. Valproate may also be associated with significant improvement in electrencephalographic recordings and subjective clinical status.486 Uvebrant and Bauziene487 reported a decrease in “autistic symptoms” in 8 of 13 patients treated with lamotrigine for intractable epilepsy, regardless of efficacy in controlling the seizures. However, in a double-blind, placebo-controlled study, Belsito and coworkers488 did not find significant differences between lamotrigine and placebo. Several case reports describe children with ASD and atypical bipolar disorder or mania who responded well to open-label treatment with lithium.489–491
General Clinical Principles of Medication Management
Principles to guide the approach to psychopharmacological management of ASD in clinical practice have been proposed by several authors.139,457,458 It is important to identify the coexisting psychiatric diagnosis or formulate a hypothesis regarding the origin of the target behaviors in order to select the medication that might be most effective (see Table 15-8). In addition, medication-specific issues, such as side effects, preparations available, dosing schedules, cost, and monitoring requirements should be considered. Potential benefits and side effects should be explained, informed consent obtained, baseline data regarding behaviors and somatic complaints collected, and potential strategies for dealing with treatment failure or partial response reviewed.
It is important to have some quantifiable means of assessing the efficacy of the medication and to obtain input from a variety of sources, such as parents, teachers, therapists, and aides. Consistent use of validated, treatment-sensitive rating scales and medication side effect scales is desirable. A wide variety of outcome measures have been used in research trials and in clinical practice to measure maladaptive behavior treatment effects.492 Among the most common are the Clinical Global Impression scale, Aberrant Behavior Checklist, and Nisonger Child Behavior Rating Form.
In general, only one medication change should be made at a time in order to be able to judge the treatment effect. It is usually best to begin with a low dose and gradually titrate upwards to the target effect to minimize the risk of treatment-emergent adverse events. If a particular medication is found to be ineffective after appropriate titration (i.e., adequate dose and duration to expect therapeutic effect) or associated with intolerable side effects, the medication should be discontinued. Gradual tapering may be warranted, depending on the medication. This is particularly important with certain agents (e.g., antipsychotics) because of the risk of withdrawal dyskinesias.
SLEEP DISTURBANCE
Sleep problems are common in children and adolescents with ASD at all levels of cognitive functioning.493–497 Sleep problems are associated with family distress and may have significant impact on daytime functioning and quality of life in children with ASD. Behavioral interventions, including sleep hygiene measures, restriction of daytime sleep, positive bedtime routines, and extinction procedures, are often effective.493,498,499
Relatively little empirical information regarding pharmacological management of sleep problems in children with ASD or other developmental disabilities is available. Recommendations are typically based on case reports and open-label trials, extrapolation from the adult literature, and expert consensus.498 There is some evidence of abnormality of melatonin regulation in ASD,500,501 and melatonin may be effective for improving sleep onset in children with ASD, as well as in children with other developmental disabilities, although there have been few randomized placebo-controlled trials.502–504 Antihistamines, α2 agonists, benzodiazepines, chloral hydrate, trazodone, and newer non-benzodiazepine hypnotics such as zolpidem and zaleplon are also sometimes utilized to treat pediatric insomnia.498 In some cases, other conditions or symptoms such as epilepsy, depression, anxiety, or aggressive outbursts warrant pharmacological treatment, and an agent that may also assist with sleep can be chosen.493
COMPLEMENTARY AND ALTERNATIVE MEDICINE
Complementary and alternative medicine (CAM) use is common in children with ASD449,505 Detailed reviews of general CAM issues in developmental disabilities are available in this volume (see Chapter 8E) and elsewhere,506 and ASD-specific CAM reviews have also been published.506a,507 The boundary between therapies considered conventional and those viewed as complementary and alternative is often poorly delineated, and a therapy initially considered to fall within the realm of CAM may later be supported by sufficient evidence to become part of standard, “conventional” practice.
CAM therapies used to treat ASD have been categorized as biological or nonbiological.507 Nonbiological interventions include treatments such as auditory integration training, behavioral optometry, craniosacral manipulation, dolphin-assisted therapy, and facilitated communication. Biological therapies include immunoregulatory interventions (e.g., dietary restriction of food allergens, intravenous immune globulin, and antiviral agents), detoxification therapies (e.g., chelation), gastrointestinal treatments (e.g., digestive enzymes, antifungal agents, probiotics, “yeast-free diet,” gluten-free/casein-free diet, and vancomycin), and supplemental therapies purported to act by modulating neurotransmission (e.g., vitamin A, vitamin C, vitamin B6 and magnesium, folic acid, folinic acid, vitamin B12, dimethylglycine and trimethylglycine, carnosine, omega-3 fatty acids, inositol, and various minerals).506a,507
For most of these CAM interventions, there is little or no scientific evidence of efficacy. Some interventions have been appropriately studied and their efficacy or validity has been disproved. For example, more than a dozen randomized, double-blind, placebo-controlled trials involving more than 700 patients have demonstrated that secretin is not an effective treatment for ASD, and it should not be used for this purpose.508,509 There is also a substantial body of research indicating that facilitated communication lacks credibility.510–512 This technique is used by a trained facilitator to provide physical support to a nonverbal person’s hand or arm while that person uses a computer keyboard or other device to spell. The evidence suggests that the communications produced actually originate from the facilitator.513,514
Evidence-based recommendations regarding additional CAM therapies are not possible because they have been evaluated inadequately as a result of methodological flaws, insufficient numbers of patients, or lack of replication. The most recent and most appropriately designed trials have demonstrated no significant benefit of dimethylglycine,515,516 vitamin B6 and magnesium,517,518 or auditory integration training.519,520 Both positive520a and negative521,522 results have been described in small, methodologically flawed studies of intravenous immune globulin. Although the gluten-free/casein-free diet is popular, there is little evidence to support or refute this intervention. In 2004, a Cochrane review found that meaningful conclusions could not be drawn from the existing literature523 and the only double-blind clinical trial subsequently published demonstrated no statistically significant differences between the patients following the gluten-free/casein-free diet and the control groups.524 Larger double-blind, placebo-controlled challenge studies are in progress.507 Many popular interventions such as chelation of heavy metals, hyperbaric oxygen, antifungal agents to decrease yeast overgrowth, antiviral agents to modulate the immune system, and omega-3 fatty acids to modulate intracellular second messengers or cellular membranes have not yet been studied in ASD; their popularity is based on unproven theories and anecdotes.
Health care practitioners who diagnose ASDs and treat children with with these conditions should recognize that many of their patients will use nonstandard therapies. It is wise and practical to become somewhat knowledgeable about CAM therapies, inquire about current and past CAM use, provide balanced information and advice about treatment options, identify risks or potential harmful effects, avoid becoming defensive or dismissing CAM in ways that convey a lack of sensitivity or concern, maintain open communication, and continue to work with families even if there is disagreement about treatment choices525 (see Chapter 8E). It is also essential to evaluate the scientific merits of specific therapies critically and share this information with families, educate families about how to evaluate information and recognize pseudoscience, and insist that studies of CAM be held to the same scientific and ethical standards as all clinical research.507,525a
Family Support
Management should focus not only on the child but also on the family. It has been recognized that parents, who were once viewed as the cause of their child’s ASD, actually play a key role in effective treatment.13 A child with an ASD has a substantial effect on the family. Although most family members recognize benefits of living with their child with an ASD, such as finding greater meaning in their own lives, experiencing delight in the child’s accomplishments, and feeling enhanced empathy for others,526 parents of children with ASD experience more stress and depression than do parents of children who are typically developing or who have certain other disabilities.527–529 Supporting the family and ensuring its emotional and physical health is an extremely important aspect of overall management of ASD.
PARENT/CAREGIVER SUPPORT
Physicians and other professionals can provide support to parents by educating them about ASD, providing anticipatory guidance and training, involving them as cotherapists, assisting them in obtaining access to resources, providing emotional support through traditional strategies such as empathic listening and talking through problems, and assisting them in advocating for their child’s needs.526 In some cases, referral of parents for counseling or other appropriate mental health services may be required. These families need ongoing support, with the specific constellation of needs varying through the family life cycle.
One of the chief strategies in helping families raise children with ASD is providing them with access to needed ongoing supports, as well as additional services during critical periods and/or crises. Natural supports include spouses, extended family members, neighbors, religious institutions, and friends who can help with caregiving and who can provide psychological and emotional support. Informal supports include social networking with other families of children with ASD and community agencies that provide training, respite, social events, and recreational activities. Formal supports include publicly funded, state-administrated programs such as early intervention, special education, vocational and residential/living services, respite services, Medicaid, In-Home and Community-Based Waiver Services, Supplemental Security Income (SSI) benefits, and other financial subsidies. The breadth and depth of services vary, even within the same state or region. Few services exist in many rural areas, and public programs may have long waiting lists.
SIBLING SUPPORT
The impact of ASD on a family is not limited to the parents or primary caregivers. Depression and psychosocial problems are also common in siblings of children with ASD,530,531 who often are plagued by questions and concerns about the reason for their sibling’s disability, how to cope with embarrassment or the reaction of peers to the sibling’s behavior, and what the future holds with regard to their role in long-term care.526 Family financial difficulties and lack of knowledge about the disability may exacerbate adjustment problems in the unaffected sibling, and the relationship may be influenced by a number of other external factors, including the availability of support services, especially respite services.
Sibling support groups offer the opportunity to learn important information and skills while sharing experiences and connecting with other siblings of children with ASD.526 Although the research on support groups for siblings of children with disabilities is difficult to interpret because of study design problems and inconsistent outcome effects on sibling adjustment, these groups have generally been evaluated positively by participating siblings and parents.526,532
FINANCIAL SUPPORT AND RELATED ISSUES
Several publicly funded programs provide financial assistance (e.g., SSI benefits, Food Stamps, Medicaid, and In-Home and Community-Based Waiver Services). Access to some public benefits, such as SSI, depends on financial need. Because eligibility depends on the financial status of the family, need-based supports such as SSI can be lost, if, for example, a well-meaning family member bequeaths the child a monetary gift. However, the government has established rules allowing assets to be held in trust for SSI and Medicaid recipients through a Supplemental Needs or Special Needs Trust. The Special Needs Trust can be used to fund needs that go beyond the bare necessities of food, clothing, and shelter provided by SSI and the medical supports and services covered by Medicaid. The laws governing such trusts are complex, and the help of an attorney knowledgeable in special needs planning is usually required. More information about Special Needs Trust can be found at http://www.nichcy.org/pubs/outprint/nd18.pdf.
PROGNOSIS
Although prognosis is one of the parents’ most pressing concerns at the time of diagnosis, it is dependent on many things and cannot usually be predicted during infancy or early childhood, especially in children younger than 3 years.533 Important early predictors include functional play skills, responsiveness to others’ bids for joint attention, and the frequency of requesting behaviors.534 In fact, several variables are especially important: cognitive abilities, level of adaptive functioning, severity of autistic symptoms, and acquisition of functional language by age 5 years.452,535–543 Throughout the lifespan, these variables interact. The prognosis for any given child depends on his or her place on the spectrum for each of these interacting trajectories.
Prognosis for independence as an adult may be better correlated with level of cognitive-adaptive functioning than with the severity of autistic symptoms. Children with normal intelligence and adaptive functioning and milder autistic symptoms generally have better outcomes; conversely, those with mental retardation and severe autistic symptoms usually have the worst prognosis. Those with normal cognitive-adaptive skills but severe autistic symptoms general do better than those with mental retardation and mild autistic symptoms. Coplan and colleagues538,541,544 reaffirmed the contribution of intelligence rather than degree of atypicality (autistic symptoms) to better outcomes, although among children with normal intelligence, the degree of atypicality is important in determining prognosis. It generally appears that approximately one third of autistic children with normal intelligence and functional language tend to improve with time to the extent that they are able to participate fully in the community.
Persons with Asperger syndrome may have better outcomes than those with other disorders on the autistic spectrum, perhaps because of their normal intelligence. One study evaluated short-term outcomes in 46 children with high-functioning autism and 20 with Asperger syndrome. All were 4 to 6 years old and had normal intelligence. Tests of cognition, language, and behavior at onset and 2 years later revealed that the children with Asperger syndrome had better social skills and fewer autistic symptoms than did the children with high-functioning autism. Some of the latter group became verbally fluent and demonstrated posttest scores indistinguishable from those of the groups with Asperger syndrome. On the other hand, an adult outcome study revealed that although those with Asperger syndrome tend to have a greater likelihood of earning a college degree, the college education did not significantly affect employment or marital status.543,545
Additional factors associated with poorer outcomes include no joint attention by 4 years and no functional speech by 5 years of age13; seizures, especially when onset occurred during adolescence; coexisting medical (e.g., tuberous sclerosis) or psychiatric (e.g., schizophrenia) disorders; and extreme “aloofness” with very little interaction with others. Female gender tends to be a risk factor for poorer prognosis because of a higher incidence of mental retardation and a lower incidence of savant skills. Factors associated with better outcomes include early identification resulting in early enrollment in appropriate intervention programs,13,546 and inclusion in regular educational and community settings with typically developing peers.131
Although all of these factors have statistical significance in ASD in general, each individual is affected by a myriad of variables in the course of her or his lifetime that will help shape her or his future. A good outcome generally means that the child matures into an adult who is able to live independently and be gainfully employed. Most, if not all, such individuals retain some degree of social skill deficit. Many choose highly technical occupations that require few social interactions and gravitate to social circles that include individuals with similar characteristics. As the generation of individuals who received diagnoses early and enrolled in effective intervention programs mature and reach adulthood, more of them are marrying and having families. There is a growing number of published autobiographies and biographies now available that describe success stories, often to the credit of understanding and supportive parents and spouses.547–551
Life expectancy is reduced in persons with ASD associated with severe mental retardation because of seizures, respiratory disease, and accidents; however, even those with mild mental retardation were found to have shorter lifespans, generally because of accidents such as suffocation and drowning, as well as seizures.552
FUTURE DIRECTIONS
The developmental-behavioral pediatrician is ideally suited to train and support primary care providers, within the context of the medical home, in surveillance, screening, and management of children with ASD. The developmental-behavioral pediatrician is often the most experienced pediatric subspecialist in the health field and thus should provide leadership for a comprehensive evaluation within the context of a team or by coordinating independent evaluations by subspecialists, psychologists, and therapists. The developmental-behavioral pediatrician’s services can be valuable in the management of both the coexisting medical conditions and the challenging behavioral issues associated with ASD; in assisting intervention and school personnel with developing individualized plans for effective interventions; and in training parents, caregivers, and nonmedical professionals. On the basis of their expertise and involvement in clinical care of children with ASD, developmental-behavioral pediatricians, among others, can guide clinical research so that hypotheses tested have a significant effect on the quality of life for children with ASD, their families, and/or the professionals who care for them. It is an exciting field, one in which the fund of knowledge is mushrooming on a daily basis. It is hoped that in the near future, new research findings will uncover the causes and point to interventions that will have greater efficacy than do those available currently. Continued research may help to resolve controversial issues and clarify treatment and prognostic conundrums. It is hoped that autism “specialists” across all disciplines can unite with families as one voice in the interest of children with ASD.





