[level-membership-for-neurosurgery-category]
CHAPTER 3 The Origin of Meningiomas
INTRODUCTION
Meningiomas account for approximately 30% of all primary brain tumors constituting the largest subset of all intracranial tumors.1–3 They can occur at any age, but most commonly in middle age. Women are more likely to develop intracranial meningiomas, with a female:male ratio of nearly 2:1. Even though it is generally agreed that meningiomas are of neuroectodermal in origin and arise from the arachnoidal (meningothelial, arachnoid cap) cells based on the ultrastructural and histologic similarities between meningiomas and arachnoid cells, the cellular origins of meningiomas still have not been identified clearly.4–21 The histologic expression of diverse meningioma subtypes ranging from meningothelial to fibroblastic patterns matches well with the various non-neoplastic cells in the arachnoid villi in a similar range of meningeal to fibroblastic cells. Thus, the critical question merges at this point: Is there a universal pluripotent cell that gives rise to all different subtypes of meningiomas or does each meningioma subtype takes origin from different tumor initiating cells in various subsets of cells in the arachnoid villi? The answer to this questions still remains obscure because there is no as yet identified subset of meningioma cells with unique molecular signatures that may give rise to all meningioma subtypes.
Even though it was possible to characterize meningiomas well cytogenetically in the past several decades, they are still poorly understood and defined molecularly. Hence histopathologic grading of the tumor does not necessarily predict its clinical course, particularly in atypical meningiomas.22,23 Current findings in molecular genetics provide convincing evidence that meningiogenesis is a dynamic process whereas histopathologic grading, which reflects only a snapshot of tumor behavior, falls short in capturing the complexity of the underlying molecular dynamics of the neoplastic process.
Recently, in addition to the well known tumor suppressor NF-2 gene deletion on chromosome 22, several other genetic aberrations including the deletion of the INK4a-ARF locus have been discovered, and altered biological pathways that potentially promote tumor growth have been suggested.22,24–28
In this chapter, we report the latest findings regarding the cellular origins of meningiomas. As well known, the classically described “arachnoid-cell derived meningioma” concept is based on histopathologic and electron microscopic studies. In addition, recent molecular and genetic studies in animal models have shown that biallelic inactivation of the NF2 gene has resulted in meningioma formation, which further supports the concept of “arachoid cell-derived meningioma” at the molecular and genetic levels.29,30
HISTOLOGIC AND ULTRASTRUCTURAL SIMILARITIES BETWEEN ARACHNOID CELLS AND MENINGIOMAS
Arachnoid granulations, or arachnoid villi, are small projections of the arachnoid membrane into the superior sagittal sinus and its major tributaries, involved in the absorption process of cerebrospinal fluid (CSF). There is a general agreement that meningiomas take origin from these granulations. In 1831, Bright noticed the histologic similarities between meningioma cells and arachnoid villi cells. Cleland and Robin proposed for the first time that meningiomas derive from arachnoid cells. Soon after, Schmidt observed obvious histologic similarities between meningioma and arachnoid cells at the ultrastructural level, and with respect to cell adhesion mechanisms and the components of extracellular matrix11,13 (Table 3-1).
TABLE 3-1 Ultrastructural and histological features of non-neoplastic arachnoid cells and meningioma cells.
| Arachnoid cells | Meningioma cells | |
|---|---|---|
| Arachnoid cap cell aggregates | Psammoma bodies | Psammoma bodies |
| Polygonal arachnoid cells | Numerous junctional complexes and interdigitations | Fewer junctional complexes and interdigitations |
| Phospholipid composition | Phosphatidyl choline-multilamellar bodies to lubricate the surfaces of arachnoid cells thus facilitating the flow or absorption of CSF | Phosphatidyl serine ribbonlike rings in meningioma whorls are thought to be the precursors of psammoma bodies |
| E-cadherin expression | Localized at the intermediate junctions and anchored to cytoskeleton via intracytoplasmic microfilaments in normal arachnoid cells | Distributed along the cell borders and variations exist between the expressions of E-cadherin in different meningioma subtypes |
| Prostaglandin D2 synthase (PGDS) | Mainly localized in the rough endoplasmic reticulum of arachnoid cells and detected in higher concentrations in the core arachnoid cells suggesting that it may play role in the absorption process of CSF | The exact role of PGDS in meningioma cells is yet to be identified, besides being a candidate as a cell marker for meningiomas |
Ultrastructural Similarities
Human arachnoid villi are composed of five layers: endothelial layer, fibrous capsule, arachnoid cell layer, cap cells, and central core. The outermost layer, an endothelial lining has a pivotal role in the absorption process of CSF, and displays a number of micropinocytotic vesicles, intracytoplasmic vacuoles, and villous projections. Endothelial cells are interconnected to each other by tight junctions. The arachnoid cell layer of the villus is the direct continuation of arachnoid membrane itself. This arachnoid cell layer forms cap cell aggregates that contains calcified organelles (psammoma bodies), which are also one of the histopathologic features of meningiomas. The arachnoid cell layer contains numerous extracellular cisterns that may contain granular material and multilamellar phospholipids. These cisterns form channels from the central core into the venous lumen and are involved in the transport of CSF. In addition, polygonal arachnoid cells are tightly attached via junctional complexes that are less frequently seen in meningioma cells.13 Several studies in the literature revealed that syncytial areas of meningiomas and normal arachnoid villi are similar ultrastructurally; however, the ultrastructure of the meningioma cells are less organized and display fewer interdigitations.11,13,31
Yamashima and colleagues investigated two forms of phospholipids in arachnoid villi and meningiomas: phosphatidyl choline and phosphatidyl serine. Human arachnoid villi display multilamellar bodies that are similar to pulmonary surfactant and are assumed to lubricate the surfaces of arachoid cells thus facilitating the flow or absorption of CSF. Conversely, phosphatidyl serine appeared as ribbonlike rings in meningioma whorls that are thought to be the precursors of psammoma bodies.32
Cell Adhesion Mechanisms
Cadherins
Epithelial (E)-cadherin is a transmembrane glycoprotein and functions in cell–cell adhesion via β-catenin that indirectly binds E-cadherin to actin filaments. This results in strong adhesive forces between the adjacent arachnoid cells in arachnoid villi, thus enabling individual arachnoid cells to undergo conformational changes during CSF absorption.33,34
Apart from their involvement in CSF absorption process in arachnoid villi, cadherins have profound roles in embryogenesis, normal tissue growth, and maintenance of the tumor cell nest. Shimoyama and colleagues reported that E-cadherin is expressed in all epithelial tissues and cancer cells, loss of which may contribute to the invasiveness of cancer cells. Interestingly, most meningiomas display en block growth, compressing the surrounding brain without infiltration. This growth pattern can be partly explained by the expression of E-cadherins, particularly in syncytial and transitional types of meningiomas. Several experimental studies reported an inverse correlation between the invasiveness of meningiomas and the expression of E-cadherins. Further, variations exist between the expressions of E-cadherin in different meningioma subtypes: It is expressed diffusely in syncytial type, less in transitional type, and not expressed in the fibroblastic type. This variation in the expression of E-cadherins in meningioma types correlates with the proposed corresponding cell types in arachnoid villi. Tohma and colleagues33 claimed that meningiomas may derive from arachnoid cells or fibroblasts (fibrous capsule) in the arachnoid villi rather than a single uniform cell based on the expression pattern of E-cadherin in different meningioma types. It is noteworthy that whereas fibrous capsule and the fibroblastic type of meningiomas do not express E-cadherin, the rest of the layers of arachnoid villi (cap cell cluster, arachnoid layer, and core arachnoid cells), and the proposed corresponding meningioma types do express E-cadherins. Tohma and colleagues also demonstrated ultrastructurally that E-cadherins are distributed along the cell borders in meningioma cells whereas they are localized at the intermediate junctions and anchored to cytoskeleton via intracytoplasmic microfilaments in normal arachnoid cells.33 This change in the distribution of E-cadherin was thought to be at the receptor level, rendering the E-cadherin inactive and thus resulting in more arbitrary architecture and the increased motility of embryonic and meningioma cells.
Prostaglandin D2 synthase
Prostaglandin D2 synthase (PGDS or β-trace) is an enzyme playing a role in the synthesis of prostaglandin D2 in the central nervous system (CNS).9,35 The function of PGDS in arachnoid and meningioma cells was reported in detail by Yamashima and colleagues in 1997. This study demonstrated that PGDS is localized mainly in the rough endoplasmic reticulum of arachnoid cells and detected in higher concentrations in the core arachnoid cells, suggesting that it may play role in the CSF absorption process.9
Extracellular Matrix
It is also reported that the two basic subtypes—meningothelial and fibroblastic meningiomas—display common ultrastructural features of intermediate filaments such as vimentin, interdigitations, and desmosomes.11,36 Bellon and colleagues37 have shown extracellular deposition of type 4 collagen in the transitional type. Likewise, McComb and Bigner38 demonstrated the fibrillar distribution of the laminin in the transitional and the fibroblastic types but not in the meningothelial type. Further, Kubota and colleagues39 found that whereas type I, III, and IV collagens and laminin occurred diffusely in between the tumors cells of fibroblastic types, these extracellular matrix proteins were detected in the fibrous septum of the meningothelial type that separates the clusters of tumor cells. In addition, Rutka and colleagues demonstrated that cultured meningioma cells express type I and III procollagens, type IV collagen, and laminin independent of the histologic subtypes.40
ORIGINS OF MENINGIOMAS AT THE GENETIC AND MOLECULAR LEVELS
The majority of meningiomas occur spontaneously or in association with the inherited autosomal dominant disorder, neurofibromatosis 2 (NF2). Mutation of the neurofibromatosis 2 (NF2) gene on chromosome 22q12 is an early aberration in meningioma tumorigenesis and individuals with NF2 are at significantly elevated risk for developing meningiomas.10,26,41–43 Mutations in the both alleles of NF2 tumor suppressor gene result in the loss of merlin protein, which is thought to play a central role in regulating leptomeningeal cell proliferation. Biallelic inactivation of the NF2 gene is the initial and most common genetic defect present in at least 50% (30% to 70%) of spontaneous meningiomas.26,29,30,42 In addition, recent studies have identified merlin inactivation through calpain-mediated proteolysis or aberrant methylation in the 5′ region of the NF2 gene in the remaining approximately 50% of meningiomas.7,8,44 Merlin is a radixin-like protein that is localized underneath the cell membrane and is implicated in the control of cell membrane–cytoskeletal interaction. Merlin acts on linking cell membrane proteins and actin filaments, thus leading to contact inhibition of normal cell growth. Notably, meningioma cells exhibit weak immunostaining for merlin compared to non-neoplastic arachnoid cells. Recent studies have reported that merlin-deficient meningioma cells are prone to develop cytoskeletal and cell contact defects, altered cell morphology, and delayed cellular apoptosis.41,44,45
Another protein called 4.1 B belonging to the same protein superfamily as merlin was localized in DAL1 gene locus on chromosome 18p11.32. Although several studies have demonstrated the loss of 4.1 B protein expressions in up to 76% of cases, no genetic or epigenetic change on the DAL1 gene locus itself could be identified.46 Similarly, no mutations have been identified in the genes coding for ezrin, radixin, and moesin which are structural relatives of merlin.26,27,47
Atypical and malignant meningiomas have more intricate genetic aberrations with losses of the G1–S phase cell cycle checkpoint regulators, CDKN2A and CDKN2B, and p14ARF on chromosome 9p contributing to more aggressive meningioma phenotypes.25,28 Recently, Kalamarides and colleagues showed that there is a synergy of NF2 and p16Ink4a mutations in the natural history of meningioma development in mice with biallelic inactivation of NF2.30 In this study, authors investigated that additional loss of the p16Ink4a locus increased the frequency of meningioma and meningothelial proliferation in NF2 knockout mice regardless of the tumor grade. Likewise, Kalamarides and colleagues developed an animal model earlier. In this animal model, they targeted Cre recombinase to the leptomeninges of NF2 knockout mice by adenoviral delivery.29 Consequently, these mice developed a range of meningioma subtypes mimicking human meningiomas; hence the authors concluded that NF2 biallelic gene activation in arachnoid cells is rate-limiting for meningioma development in the mouse.
However, in contrast to spontaneous meningiomas, radiation-induced meningiomas express fewer NF2 mutations or losses on chromosome 22. Radiation-induced meningiomas arise in fewer than 1% of irradiated patients and tend to be multifocal and more aggressive, possibly due to additional chromosomal losses on 1p, 6q, and 7p.26,41
The correlation between the aforementioned genetic aberrations and the corresponding alterations in molecular pathways during meningiomagenesis presents a challenge. Several studies have been investigating the global gene expression profiles of meningiomas with the goal of providing more insight into the molecular biology of these neoplasms. A recent study by Lal and colleagues has demonstrated that meningiomas of all three histopathologic grades can be divided into two main subgroups—low-proliferative and high-proliferative meningiomas—based on the global gene profiles and underlying molecular mechanisms.11 The results of this study redefined the grade II meningiomas as either grade I or grade III based on their gene-expression patterns. In this study, gains and losses of chromosomes were described, but no gene amplifications were found in 23 meningioma specimens studied. The frequency of chromosome losses in descending order was chromosome 22, 14q, and 1p. Aberrations have also been detected on chromosomes 3p, 6q, 10, 14q, and 18, and gains on chromosome 1q. The study also claimed that alterations in the transforming growth factor-β (TGF-β) pathway may contribute to the anaplasia of grade III meningiomas, as a striking difference in the number of aberrations in genes that regulate TGF-β pathway was observed between the grade I and grade III meningiomas.
THE MENINGIOMA STEM CELL CONCEPT AND ITS IMPLICATIONS IN THE ORIGIN OF MENINGIOMAS
Stem cells can be described as self-renewing, omnipotent cells that may eventually form various cell types with multilineage differentiation. Likewise, the concept of “cancer stem cells” denotes cancer cells with stem cell–like features that are responsible for tumor initiation, tumor renewal, and resistance to antineoplastic medications. Initially, this concept took its origin from the obvious similarities between the self-renewal mechanisms of stem cells and cancer cells derived from leukemia, multiple myeloma, and breast cancer. More than a decade ago, Singh and colleagues presented striking evidence regarding the existence of cancer stem cells in medulloblastomas and gliomas.48 This study demonstrated that CD133+ cancer cells had the potential to form clusters of cells resembling neurospheres with self-renewal and differentiation abilities. The authors posted that the origin of cancer initiating cells might be a normal CD133 expressing neural stem cell, because CD133, a neural stem cell surface marker, was also detected in the normal human fetal brain. Several more recent studies have also revealed similar findings suggestive of a linkage between the normal neurogenesis and carcinogenesis.
The idea of a “meningioma stem cell” is the extension of the cancer stem cell concept in other various solid tumors.47
Preliminary Results Supporting the Concept of Meningioma Stem Cells
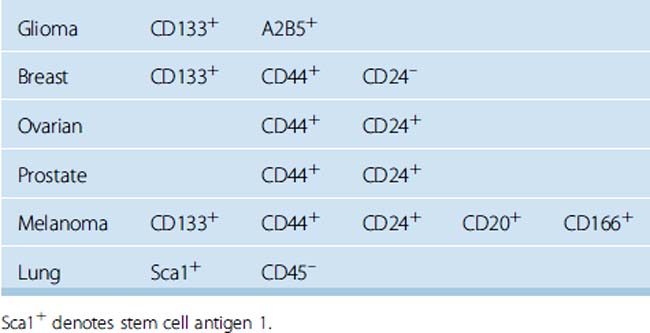
Considering the cell surface markers of tumor-initiating cells, or, in other words, “cancer stem cells,” several transmembrane glycoproteins including CD24, CD34, CD44, CD133, and CD166 were studied in cultured meningioma cells in neural stem cell medium and on paraffin-embedded tissue sections obtained from grade I to III meningiomas. In addition, double staining with the proliferation marker Ki-67 was performed with each of the aforementioned cell surface markers to show the co-localization of Ki-67 staining with any of the stem cell markers. We observed consistent co-localization of CD133 and CD44 with the nuclear proliferation marker Ki-67 in vivo and in vitro. These findings suggested that meningioma stem cells may arise from CD133+ CD44+ CD24– CD166– meningioma cells. Further, CD133+CD44+ CD24–CD166– meningioma cell populations had significantly longer survival times and increased proliferation rates in vitro. In the literature we reviewed, breast cancer stem cells displayed similar expression of surface markers as CD133+ CD44+ CD24– (Table 3-2).
Expression of CD44
CD44 is a widely distributed cell surface marker and cell adhesion molecule. The insertion of alternatively spliced exons into the CD44 mRNA creates various isoforms of CD44, each involved in diverse biologic functions. Suzuki and colleagues demonstrated the differential expression of CD44 in various meningioma subtypes.49 In this study, only the secretory meningiomas appeared to express variant forms of CD44, favoring tumor cell differentiation to epithelial type, whereas meningothelial, fibrous, and malignant meningiomas express the standard form of CD44. Further, several other studies in the literature revealed convincing evidence that the overexpression of CD44 was often associated with increased migration ability and anaplasia in meningioma cells.49,50 Sainio and colleagues demonstrated the co-localization of NF2 gene–encoded merlin protein with CD44 and noted the interaction of CD44 and cytoskeleton via ezrin, radixin, and moesin proteins which are structurally related to merlin protein. Similarly, Morrison and colleagues presented additional evidence regarding the role of merlin-mediated contact inhibition of cell growth through interactions with CD44 in schwannoma cell lines.
[1] CBTRUS. Statistical report: Primary Brains Tumors in the United States, 1998–2002. Published by the Central Brain Tumor Registry of the United States, 2005.
[2] Claus E.B., Bondy M.L., Schildkraut J.M., Wiemels J.L., Wrensch M., Black P.M. Epidemiology of intracranial meningioma. Neurosurgery. 2005;57(6):1088-1095. discussion 1088–1095
[3] Black P.M. Meningiomas. Neurosurgery. 1993;32(4):643-657.
[4] Vandenabeele F., Creemers J., Lambrichts I. Ultrastructure of the human spinal arachnoid mater and dura mater. J Anat. 1996;189(Pt 2):417-430.
[5] Nicholas D.S., Weller R.O. The fine anatomy of the human spinal meninges. A light and scanning electron microscopy study. J Neurosurg. 1988;69(2):276-282.
[6] Weller R.O. Microscopic morphology and histology of the human meninges. Morphologie. 2005;89(284):22-34.
[7] Kaneko T., Yamashima T., Tohma Y., et al. Calpain-dependent proteolysis of merlin occurs by oxidative stress in meningiomas: a novel hypothesis of tumorigenesis. Cancer. 2001;92(10):2662-2672.
[8] Kimura Y., Koga H., Araki N., et al. The involvement of calpain-dependent proteolysis of the tumor suppressor NF2 (merlin) in schwannomas and meningiomas. Nat Med. 1998;4(8):915-922.
[9] Yamashima T., Sakuda K., Tohma Y., et al. Prostaglandin D synthase (beta-trace) in human arachnoid and meningioma cells: roles as a cell marker or in cerebrospinal fluid absorption, tumorigenesis, and calcification process. J Neurosci. 1997;17(7):2376-2382.
[10] Sakuda K., Kohda Y., Matsumoto T., et al. Expression of NF2 gene product merlin in arachnoid villi and meningiomas. Noshuyo Byori. 1996;13(2):145-148.
[11] Yamashima T. On arachnoid villi and meningiomas: functional implication of ultrastructure, cell adhesion mechanisms, and extracellular matrix composition. Pathol Oncol Res. 1996;2(3):144-149.
[12] Yamashima T., Tohma Y., Nitta H., et al. Synthesis of multilamellar phospholipids in meningioma cells. Noshuyo Byori. 1994;11(1):1-6.
[13] Yamashima T., Kida S., Yamamoto S. Ultrastructural comparison of arachnoid villi and meningiomas in man. Mod Pathol. 1988;1(3):224-234.
[14] Schachenmayr W., Friede R.L. The origin of subdural neomembranes. I. Fine structure of the dura-arachnoid interface in man. Am J Pathol. 1978;92(1):53-68.
[15] Yamashima T., Friede R.L. [Light and electron microscopic studies on the subdural space, the subarachnoid space and the arachnoid membrane]. Neurol Med Chir (Tokyo). 1984;24(10):737-746.
[16] Kida S., Yamashima T., Kubota T., Ito H., Yamamoto S. A light and electron microscopic and immunohistochemical study of human arachnoid villi. J Neurosurg. 1988;69(3):429-435.
[17] Haines D.E., Frederickson R.G. The meninges. In: Al-Mefty O., editor. Meningiomas. New York: Raven Press; 1991:9-25.
[18] Lopes C., Mair W.G.P. Ultrastructure of the arachnoid membrane in man. Acta Neuropathol (Berlin). 1974;28:167-173.
[19] Alcolado R., Weller R.O., Parrish E.P., Garrod D. The cranial arachnoid and pia mater in man: anatomical and ultrastructural observations. Neuropathol Appl Neurobiol. 1988;14(1):1-17.
[20] Tripathi R.C. Ultrastructure of the arachnoid mater in relation to outflow of cerebrospinal fluid. A new concept. Lancet. 1973;819:8-11.
[21] Anderson D.R. Ultrastructure of meningeal sheaths. Arch Opthalmol. 1969;82:659-674.
[22] Kleihues P., Louis D.N., Scheithauer B.W., et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61(3):215-225. discussion 226–229
[23] Kleihues P., Burger P.C., Scheithauer B.W. The new WHO classification of brain tumours. Brain Pathol. 1993;3(3):255-268.
[24] Wrobel G., Roerig P., Kokocinski F., et al. Microarray-based gene expression profiling of benign, atypical and anaplastic meningiomas identifies novel genes associated with meningioma progression. Int J Cancer. 2005;114(2):249-256.
[25] Perry A., Banerjee R., Lohse C.M., Kleinschmidt-DeMasters B.K., Scheithauer B.W. A role for chromosome 9p21 deletions in the malignant progression of meningiomas and the prognosis of anaplastic meningiomas. Brain Pathol. 2002;12(2):183-190.
[26] Simon M., Boström J.P., Hartmann C. Molecular genetics of meningiomas: from basic research to potential clinical applications. Neurosurgery. 2007;60(5):787-798. discussion 787–798
[27] Simon M., Park T.W., Köster G., et al. Alterations of INK4a(p16–p14arf)/INK4b(p15) expression and telomerase activation in meningioma progression. J Neurooncol. 2001;55(3):149-158.
[28] Weber R.G., Boström J., Wolter M., et al. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci USA. 1997;94(26):14719-14724.
[29] Kalamarides M., Stemmer-Rachamimov A.O., Takahashi M., et al. Natural history of meningioma development in mice reveals: a synergy of Nf2 and p16(Ink4a) mutations. Brain Pathol. 2008;18(1):62-70.
[30] Kalamarides M., Niwa-Kawakita M., Leblois H., et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes Dev. 2002;16(9):1060-1065.
[31] Yamashima T., Kida S., Kubota T., Yamamoto S. The origin of psammoma bodies in the human arachnoid villi. Acta Neuropathol. 1986;71(1–2):19-25.
[32] Yamashima T., Yamashita J. Histological, ultrastructural and chromatographical discrimination of phospholipids in meningiomas. Acta Neuropathol. 1990;80(3):255-259.
[33] Tohma Y., Yamashima T., Yamashita J. Immunohistochemical localization of cell adhesion molecule epithelial cadherin in human arachnoid villi and meningiomas. Cancer Res. 1992;52(7):1981-1987.
[34] Brunner E.C., Romeike B.F., Jung M., Comtesse N., Meese E. Altered expression of beta-catenin/E-cadherin in meningiomas. Histopathology. 2006;49(2):178-187.
[35] Kawashima M., Suzuki S.O., Yamashima T., Fukui M., Iwaki T. Prostaglandin D synthase (beta-trace) in meningeal hemangiopericytoma. Mod Pathol. 2001;14(3):197-201.
[36] Nitta H., Yamashima T., Yamashita J., Kubota T. An ultrastructural and immunohistochemical study of extracellular matrix in meningiomas. Histol Histopathol. 1990;5(3):267-274.
[37] Bellon G., Caulet T., Cam Y., et al. Immunohistochemical localisation of macromolecules of the basement membrane and extracellular matrix of human gliomas and meningiomas. Act Neuropathol. 1985;66(3):245-252.
[38] McComb R.D., Bigner D.D. Immunolocalization of laminin in neoplasms of the central and peripheral nervous systems. J Neuropathol Exp Neurol. 1985;44(3):242-253.
[39] Kubota T., Yamashima T., Hasegawa M., Kida S., Hayashi M., Yamamoto S. Formation of psammoma bodies in meningocytic whorls. Ultrastructural study and analysis of calcified material. Acta Neuropathol. 1986;70(3–4):262-268.
[40] Rutka J.T., Giblin J., Dougherty D.V., McCulloch J.R., DeArmond S.J., Rosenblum M.L. An ultrastructural and immunocytochemical analysis of leptomeningeal and meningioma cultures. J Neuropathol Exp Neurol. 1986;45(3):285-303.
[41] Perry A., Gutmann D.H., Reifenberger G. Molecular pathogenesis of meningiomas. Neuro-oncol. 2004;70(2):183-202.
[42] Giovannini M., Robanus-Maandag E., van der Valk M., et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14(13):1617-1630.
[43] Riemenschneider M.J., Perry A., Reifenberger G. Histological classification and molecular genetics of meningiomas. Lancet Neurol. 2006;12(5):1045-1054. Erratum in:Lancet Neurol. 2007;6(2):105.
[44] Kimura Y., Koga H., Araki N., et al. The involvement of calpain-dependent proteolysis of the tumor suppressor NF2 (merlin) in schwannomas and meningiomas. Nat Med. 1998;4(8):915-922.
[45] McClatchey A.I., Giovannini M. Membrane organization and tumorigenesis—the NF2 tumor suppressor, Merlin. Genes Dev. 2005;19(19):2265-2277.
[46] Robb V.A., Li W., Gascard P., Perry A., Mohandas N., Gutmann D.H. Identification of a third Protein 4.1 tumor suppressor, Protein 4.1R, in meningioma pathogenesis. Neurobiol Dis. 2003;13(3):191-202.
[47] Sauvageot C.M., Kesari S., Stiles C.D. Molecular pathogenesis of adult brain tumors and the role of stem cells. Neurol Clin. 2007;25(4):891-924. vii
[48] Singh S.K., Clarke I.D., Terasaki M., et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821-5828.
[49] Suzuki S.O., Iwaki T., Kitamoto T., Mizoguchi M., Fukui M., Tateishi J. Differential expression of CD44 variants among meningioma subtypes. Clin Mol Pathol. 1996;49(3):M140-M146.
[50] Rooprai H.K., Liyanage K., King A., Davies D., Martin K., Pilkington G.J. CD44 expression in human meningiomas: an immunocytochemical, immunohistochemical and flow cytometric analysis. Int J Oncol. 1999;14(5):855-860.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
CHAPTER 3 The Origin of Meningiomas
INTRODUCTION
Meningiomas account for approximately 30% of all primary brain tumors constituting the largest subset of all intracranial tumors.1–3 They can occur at any age, but most commonly in middle age. Women are more likely to develop intracranial meningiomas, with a female:male ratio of nearly 2:1. Even though it is generally agreed that meningiomas are of neuroectodermal in origin and arise from the arachnoidal (meningothelial, arachnoid cap) cells based on the ultrastructural and histologic similarities between meningiomas and arachnoid cells, the cellular origins of meningiomas still have not been identified clearly.4–21 The histologic expression of diverse meningioma subtypes ranging from meningothelial to fibroblastic patterns matches well with the various non-neoplastic cells in the arachnoid villi in a similar range of meningeal to fibroblastic cells. Thus, the critical question merges at this point: Is there a universal pluripotent cell that gives rise to all different subtypes of meningiomas or does each meningioma subtype takes origin from different tumor initiating cells in various subsets of cells in the arachnoid villi? The answer to this questions still remains obscure because there is no as yet identified subset of meningioma cells with unique molecular signatures that may give rise to all meningioma subtypes.
Even though it was possible to characterize meningiomas well cytogenetically in the past several decades, they are still poorly understood and defined molecularly. Hence histopathologic grading of the tumor does not necessarily predict its clinical course, particularly in atypical meningiomas.22,23 Current findings in molecular genetics provide convincing evidence that meningiogenesis is a dynamic process whereas histopathologic grading, which reflects only a snapshot of tumor behavior, falls short in capturing the complexity of the underlying molecular dynamics of the neoplastic process.
Recently, in addition to the well known tumor suppressor NF-2 gene deletion on chromosome 22, several other genetic aberrations including the deletion of the INK4a-ARF locus have been discovered, and altered biological pathways that potentially promote tumor growth have been suggested.22,24–28
In this chapter, we report the latest findings regarding the cellular origins of meningiomas. As well known, the classically described “arachnoid-cell derived meningioma” concept is based on histopathologic and electron microscopic studies. In addition, recent molecular and genetic studies in animal models have shown that biallelic inactivation of the NF2 gene has resulted in meningioma formation, which further supports the concept of “arachoid cell-derived meningioma” at the molecular and genetic levels.29,30
HISTOLOGIC AND ULTRASTRUCTURAL SIMILARITIES BETWEEN ARACHNOID CELLS AND MENINGIOMAS
Arachnoid granulations, or arachnoid villi, are small projections of the arachnoid membrane into the superior sagittal sinus and its major tributaries, involved in the absorption process of cerebrospinal fluid (CSF). There is a general agreement that meningiomas take origin from these granulations. In 1831, Bright noticed the histologic similarities between meningioma cells and arachnoid villi cells. Cleland and Robin proposed for the first time that meningiomas derive from arachnoid cells. Soon after, Schmidt observed obvious histologic similarities between meningioma and arachnoid cells at the ultrastructural level, and with respect to cell adhesion mechanisms and the components of extracellular matrix11,13 (Table 3-1).
TABLE 3-1 Ultrastructural and histological features of non-neoplastic arachnoid cells and meningioma cells.
| Arachnoid cells | Meningioma cells | |
|---|---|---|
| Arachnoid cap cell aggregates | Psammoma bodies | Psammoma bodies |
| Polygonal arachnoid cells | Numerous junctional complexes and interdigitations | Fewer junctional complexes and interdigitations |
| Phospholipid composition | Phosphatidyl choline-multilamellar bodies to lubricate the surfaces of arachnoid cells thus facilitating the flow or absorption of CSF | Phosphatidyl serine ribbonlike rings in meningioma whorls are thought to be the precursors of psammoma bodies |
| E-cadherin expression | Localized at the intermediate junctions and anchored to cytoskeleton via intracytoplasmic microfilaments in normal arachnoid cells | Distributed along the cell borders and variations exist between the expressions of E-cadherin in different meningioma subtypes |
| Prostaglandin D2 synthase (PGDS) | Mainly localized in the rough endoplasmic reticulum of arachnoid cells and detected in higher concentrations in the core arachnoid cells suggesting that it may play role in the absorption process of CSF | The exact role of PGDS in meningioma cells is yet to be identified, besides being a candidate as a cell marker for meningiomas |
Ultrastructural Similarities
Human arachnoid villi are composed of five layers: endothelial layer, fibrous capsule, arachnoid cell layer, cap cells, and central core. The outermost layer, an endothelial lining has a pivotal role in the absorption process of CSF, and displays a number of micropinocytotic vesicles, intracytoplasmic vacuoles, and villous projections. Endothelial cells are interconnected to each other by tight junctions. The arachnoid cell layer of the villus is the direct continuation of arachnoid membrane itself. This arachnoid cell layer forms cap cell aggregates that contains calcified organelles (psammoma bodies), which are also one of the histopathologic features of meningiomas. The arachnoid cell layer contains numerous extracellular cisterns that may contain granular material and multilamellar phospholipids. These cisterns form channels from the central core into the venous lumen and are involved in the transport of CSF. In addition, polygonal arachnoid cells are tightly attached via junctional complexes that are less frequently seen in meningioma cells.13 Several studies in the literature revealed that syncytial areas of meningiomas and normal arachnoid villi are similar ultrastructurally; however, the ultrastructure of the meningioma cells are less organized and display fewer interdigitations.11,13,31
Yamashima and colleagues investigated two forms of phospholipids in arachnoid villi and meningiomas: phosphatidyl choline and phosphatidyl serine. Human arachnoid villi display multilamellar bodies that are similar to pulmonary surfactant and are assumed to lubricate the surfaces of arachoid cells thus facilitating the flow or absorption of CSF. Conversely, phosphatidyl serine appeared as ribbonlike rings in meningioma whorls that are thought to be the precursors of psammoma bodies.32
Cell Adhesion Mechanisms
Cadherins
Epithelial (E)-cadherin is a transmembrane glycoprotein and functions in cell–cell adhesion via β-catenin that indirectly binds E-cadherin to actin filaments. This results in strong adhesive forces between the adjacent arachnoid cells in arachnoid villi, thus enabling individual arachnoid cells to undergo conformational changes during CSF absorption.33,34
Apart from their involvement in CSF absorption process in arachnoid villi, cadherins have profound roles in embryogenesis, normal tissue growth, and maintenance of the tumor cell nest. Shimoyama and colleagues reported that E-cadherin is expressed in all epithelial tissues and cancer cells, loss of which may contribute to the invasiveness of cancer cells. Interestingly, most meningiomas display en block growth, compressing the surrounding brain without infiltration. This growth pattern can be partly explained by the expression of E-cadherins, particularly in syncytial and transitional types of meningiomas. Several experimental studies reported an inverse correlation between the invasiveness of meningiomas and the expression of E-cadherins. Further, variations exist between the expressions of E-cadherin in different meningioma subtypes: It is expressed diffusely in syncytial type, less in transitional type, and not expressed in the fibroblastic type. This variation in the expression of E-cadherins in meningioma types correlates with the proposed corresponding cell types in arachnoid villi. Tohma and colleagues33 claimed that meningiomas may derive from arachnoid cells or fibroblasts (fibrous capsule) in the arachnoid villi rather than a single uniform cell based on the expression pattern of E-cadherin in different meningioma types. It is noteworthy that whereas fibrous capsule and the fibroblastic type of meningiomas do not express E-cadherin, the rest of the layers of arachnoid villi (cap cell cluster, arachnoid layer, and core arachnoid cells), and the proposed corresponding meningioma types do express E-cadherins. Tohma and colleagues also demonstrated ultrastructurally that E-cadherins are distributed along the cell borders in meningioma cells whereas they are localized at the intermediate junctions and anchored to cytoskeleton via intracytoplasmic microfilaments in normal arachnoid cells.33 This change in the distribution of E-cadherin was thought to be at the receptor level, rendering the E-cadherin inactive and thus resulting in more arbitrary architecture and the increased motility of embryonic and meningioma cells.
Prostaglandin D2 synthase
Prostaglandin D2 synthase (PGDS or β-trace) is an enzyme playing a role in the synthesis of prostaglandin D2 in the central nervous system (CNS).9,35 The function of PGDS in arachnoid and meningioma cells was reported in detail by Yamashima and colleagues in 1997. This study demonstrated that PGDS is localized mainly in the rough endoplasmic reticulum of arachnoid cells and detected in higher concentrations in the core arachnoid cells, suggesting that it may play role in the CSF absorption process.9
Extracellular Matrix
It is also reported that the two basic subtypes—meningothelial and fibroblastic meningiomas—display common ultrastructural features of intermediate filaments such as vimentin, interdigitations, and desmosomes.11,36 Bellon and colleagues37
[/not-level-membership-for-neurosurgery-category]
