[level-membership-for-pediatrics-category]
Chapter 672 The Neck
672.1 Torticollis
Torticollis is a symptom rather than a diagnosis, and describes the clinical findings of tilting of the head to the right or left side in combination with rotation of the head to the opposite side. Congenital muscular torticollis (CMT) is the most common etiology, but a variety of other conditions result in torticollis, and a detailed workup is often required to rule out other diagnoses in patients who lack the characteristic features of CMT (approximately 20%) (Fig. 672-1). The differential diagnosis includes trauma (clavicle fracture or brachial plexopathy), tumors or malformations of the central nervous system, ocular disorders, congenital bony abnormalities (Klippel-Feil syndrome), inflammatory conditions, and other diagnoses such as atlantoaxial rotatory displacement or Sandifer syndrome (Table 672-1).
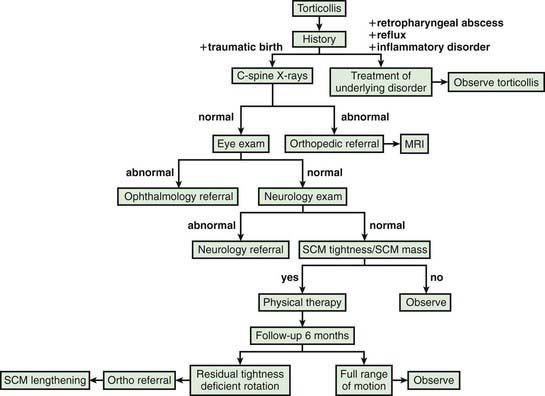
Figure 672-1 Algorithm for evaluation of muscular torticollis. SCM, sternocleidomastoid muscle.
(From Do TT: Congenital muscular torticollis: current concepts and review of treatment, Curr Opin Pediatr 18:26–29, 2006.)
Table 672-1 DIFFERENTIAL DIAGNOSIS OF TORTICOLLIS
CONGENITAL
ACQUIRED
INFLAMMATION
NEUROLOGIC
OTHER
Cheng JC, Tang SP, Chen TM, et al. The clinical presentation and outcome of treatment of congenital muscular torticollis in infants—a study of 1086 cases. J Pediatr Surg. 2000;35:1091-1096.
Cheng JC, Wong MW, Tang SP, et al. Clinical determinants of the outcome of manual stretching in the treatment of congenital muscular torticollis in infants: a prospective study of eight hundred and twenty-one cases. J Bone Joint Surg Am. 2001;83:679-687.
Collins A, Jankovic J. Botulinum toxin injection for congenital muscular torticollis presenting in children and adults. Neurology. 2006;67:1083-1085.
Do TT. Congenital muscular torticollis: current concepts and review of treatment. Curr Opinion Pediatr. 2006;18:26-29.
Herman MJ. Torticollis in infants and children: common and unusual causes. Instr Course Lect. 2006;55:647-653.
Ohman A, Nilsson S, Lagerkvist AL, et al. Are infants with torticollis at risk of a delay in early milestones compared to a control group of healthy infants? Dev Med Child Neurol. 2009;51(7):545-550.
Pang D, Li V. Atlantoaxial rotatory fixation: part III of a prospective study of the clinical manifestation, diagnosis, management, and outcome of children with atlantoaxial rotatory fixation. Neurosurgery. 2005;57:952-972.
Rosman NP, Douglass LM, Sharif UM, et al. The neurology of benign paroxysmal torticollis of infancy: report of 10 new cases and review of the literature. J Child Neurol. 2009;24:155-160.
Rubio AS, Griffet JR, Caci H, et al. The moulded baby syndrome: incidence and risk factors regarding 1001 neonates. Eur J Pediatr. 2009;168:605-611.
Shim JS, Jang HP. Operative treatment of congenital torticollis. J Bone Joint Surg Br. 2008;90:934-939.
Snyder EM, Coley BD. Limited value of plain radiographs in infant torticollis. Pediatrics. 2006;118:e1779-e1784.
Stellwagen L, Hubbard E, Chambers C, et al. Torticollis, facial asymmetry and plagiocephaly in normal newborns. Arch Dis Child. 2008;93:827-831.
Yu CC, Wong FH, Lo LJ, et al. Craniofacial deformity in patients with uncorrected congenital muscular torticollis: an assessment from three-domernsional computed tomographic imaging. Plast Reconstr Surg. 2004;113:24-33.
van Vlimmeren LA, Helders PJ, van Adrichem LN, et al. Torticollis and plagiocephaly in infancy: therapeutic strategies. Pediatr Rehabil. 2006;9:40-46.
von Heideken J, Green DW, Burke SW, et al. The relationship between developmental dysplasia of the hip and congenital muscular torticollis. J Pediatr Orthop. 2006;26:805-808.
672.2 Klippel-Feil Syndrome
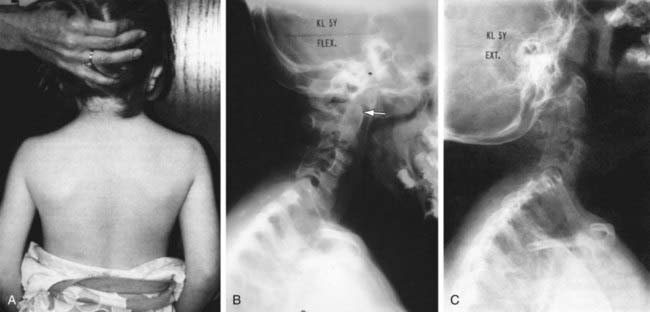
Klippel-Feil syndrome involves a congenital fusion (failure of segmentation) of one or more cervical motion segments, and the clinical triad of short neck, low hairline, and restriction of neck motion is seen in only about half of the patients. Most patients have associated congenital anomalies at the craniocervical junction (occiput-C2), the subaxial spine (below C2), or both (Fig. 672-2). A familial gene locus on the long arm of chromosome 8 has been identified. Abnormalities in other organ systems must be ruled out. Associated anomalies have been identified in the genitourininary system (30-40%; unilateral renal agenesis, duplicated collecting systems, horseshoe kidney), auditory system, heart, neural axis, and the musculoskeletal system (Sprengel deformity in one third, scoliosis). Congenital cervical fusions or anomalies are also commonly seen in patients with Goldenhar syndrome, Mohr syndrome, VACTERL syndrome (vertebral anomalies, anal atresia, cardiovascular anomalies, tracheoesophageal fistula, esophageal atresia, renal and/or radial anomalies, limb defects), and fetal alcohol syndrome.
Hensinger RN, Lang JE, MacEwen GD. Klippel-Feil syndrome: a constellation of associated anomalies. J Bone Joint Surg Am. 1974;56:1246-1253.
Pizzutillo PD, Woods M, Nicholson L, et al. Risk factors in Klippel-Feil syndrome. Spine. 1994;19:2110-2116.
Samartzis D, Kalluri P, Herman J, et al. The role of congenitally fused cervical segments upon the space available for the cord and associated symptoms in Klippel-Feil patients. Spine. 2008;33:1442-1450.
672.3 Cervical Anomalies and Instabilities
Anomalies of the craniovertebral junction and/or the lower cervical spine may be seen in isolation or in association with other conditions such as genetic syndromes and skeletal dysplasias. Congenital anomalies can result from a mutation in the homeobox genes. Coexisting abnormalities in other organ systems (renal, cardiac, intraspinal) must be ruled out. The true incidence is unknown, because many of these anomalies remain asymptomatic and undiagnosed. A subset of these anomalies, whether symptomatic or asymptomatic, places the patient at risk of neurologic injury due to either cervical instability or spinal stenosis. There seems to be no difference between syndromic and nonsyndromic anomalies when considering symptoms, findings, or treatment. Cervical instabilities may be associated with certain congenital anomalies, or with a variety of other conditions predisposing to excessive laxity or mobility (connective tissue disorders, metabolic diseases). The host of cervical anomalies and instabilities are categorized in Table 672-2.
Table 672-2 CAUSES OF PEDIATRIC CERVICAL INSTABILITY
CONGENITAL
Vertebral (Bony Anomalies)
Ligamentous or Combined Anomalies
Found at birth as an element of somatogenic aberration
Syndromic Disorders
ACQUIRED
The most common anomaly of the lower cervical spine (subaxial) is Klippel-Feil syndrome, and congenital fusions between vertebrae may also be seen in up to 50% of patients with fetal alcohol syndrome. Instabilities in this region are post-traumatic or develop from the abnormal stress distribution in the setting on congenital cervical fusions (Chapter 672.2).
Down Syndrome
Ligamentous hyperlaxity is a characteristic feature of Down syndrome and can result in hypermobility or instability at the occipitoatlantal or the atlantoaxial joints (Chapter 76). Hypermobility or instability at C1-C2 is found in up to 40% of children with Down syndrome, with occipitoatlantal hypermobility in up to 61%. These patients can also have coexisting congenital or developmental anomalies of the cervical spine such as occipitalization of the atlas, atlantal arch hypoplasia, basilar invagination, os odontoideum, and odontoid hypoplasia. All patients require screening by history and physical examination (at regular intervals) and at least a single series of cervical spinal radiographs, including a lateral view in flexion and extension. The goal is to establish whether patients have normal mobility (normal radiographic parameters), hypermobility (excessive translation but not expected to be at neurologic risk), or instability (high risk of neurologic deterioration). Although the specific recommendations vary between states, both clinical and radiographic screenings are required before participation in Special Olympics.
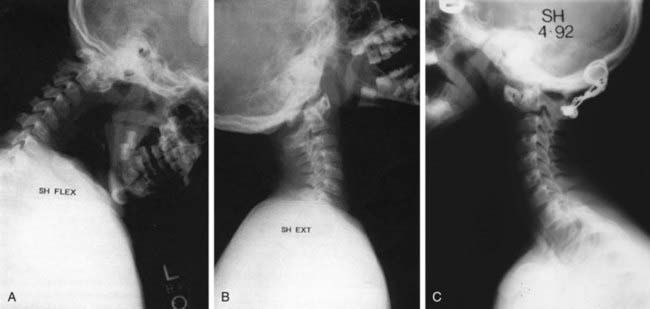
The clinical diagnosis of neurologic dysfunction may be challenging in this population of patients, and subtle findings such as decreased exercise tolerance and gait abnormalities (increased tripping or falling) may be the earliest signs of myelopathy. Although formal neurologic testing may be impossible, clonus and hyperreflexia may be identified on physical examination. Imaging studies are commonly used to diagnose and follow patients with hypermobility or instability. With regard to the atlantoaxial joint, the atlanto-dens interval (ADI) is measured as the space between the dens and the anterior ring of C1 (ADI) on lateral radiographs in neutral, flexion, and extension (Fig. 672-3). A normal ADI in children with Down syndrome is <4.5 mm. Hypermobility is diagnosed with an ADI between 4.5 and 10 mm, and an ADI >10 mm represents instability and carries a significant risk of neurologic injury. An MRI with flexion and extension, usually performed under supervision, helps to further evaluate instability and neurologic compression. Although hypermobility at the occipitoatlantal joint is present in >50% of children with Down syndrome, most patients do not develop instability or neurologic symptoms. The relationships at this articulation are difficult to measure reliably on plain radiographs, and a dynamic MRI can help to clarify the significance of any questionable radiographic findings. Involvement of the subaxial spine is less common and is typically encountered in the adult population of patients with Down syndrome. Degenerative changes and/or instability can result in pain, radiculopathy, and myelopathy.
22q11.2 Deletion Syndrome
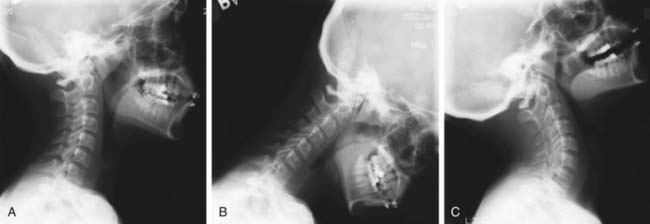
The chromosome abnormality deletion of 22q11.2 is a common genetic syndrome and encompasses a wide spectrum of abnormalities including cardiac, palate, and immunologic anomalies. At least one developmental anomaly of the occiput or cervical spine is noted in all patients (Fig. 672-4). The occipital variations observed include platybasia and basilar impression. Atlas variations include dysmorphic shape, open posterior arch, and occipitalization, and axis variations include a dysmorphic dens. A range of cervical vertebral fusions is noted in these patients, most commonly at C2 and C3. Increased segmental motion is observed on dynamic imaging in >50% of patients, often at more than one level. All patients should have screening radiographs, and many require follow-up for the cervical spine.
Drummond DS, Hosalkar HS. Treatment of cervical instability. In: Clark CR, editor. The cervical spine: the Cervical Spine Research Society editorial committee. ed 4. Philadelphia: Lippincott Williams & Wilkins; 2005:427-447.
Gholve PA, Hosalkar HS, Ricchetti ET, et al. Occipitalization of the atlas in children. Morphologic classification, associations, and clinical relevance. J Bone Joint Surg Am. 2007;89:571-578.
Guille JT, Sherk HH. Congenital osseous anomalies of the upper and lower cervical spine in children. J Bone Joint Surg Am. 2002;84:277-288.
Hosalkar HS, Sankar WN, Wills BP, et al. Congenital osseous anomalies of the upper cervical spine. J Bone Joint Surg Am. 2008;90:337-348.
Menezes AH. Craniovertebral junction database analysis: incidence, classification, presentation, and treatment algorithms. Childs Nerv Syst. 2008;24:1101-1118.
Menezes AH, Vogel TW. Specific entities affecting the craniocervical region: syndromes affecting the craniocervical junction. Childs Nerv Syst. 2008;24:1155-1163.
Ricchetti ET, States L, Hosalkar HS, et al. Radiographic study of the upper cervical spine in the 22q11.2 deletion syndrome. J Bone Joint Surg Am. 2004;86:1751-1760.
Sankar WN, Wills BP, Dormans JP, et al. Os odontoideum revisited: the case for a multifactorial etiology. Spine. 2006;31:979-984.
Segal LS, Drummond DS, Zarotti RM, et al. Complications of posterior arthrodesis of the cervical spine in patients who have Down syndrome. J Bore Joint Surg [Am]. 1991;73(10):1547-1554.
Stevens CA, Pearce RG, Burton EM. Familial odontoid hypoplasia. Am J Med Genet Part A. 2009;149:1290-1292.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Chapter 672 The Neck
672.1 Torticollis
Torticollis is a symptom rather than a diagnosis, and describes the clinical findings of tilting of the head to the right or left side in combination with rotation of the head to the opposite side. Congenital muscular torticollis (CMT) is the most common etiology, but a variety of other conditions result in torticollis, and a detailed workup is often required to rule out other diagnoses in patients who lack the characteristic features of CMT (approximately 20%) (Fig. 672-1). The differential diagnosis includes trauma (clavicle fracture or brachial plexopathy), tumors or malformations of the central nervous system, ocular disorders, congenital bony abnormalities (Klippel-Feil syndrome), inflammatory conditions, and other diagnoses such as atlantoaxial rotatory displacement or Sandifer syndrome (Table 672-1).
Figure 672-1 Algorithm for evaluation of muscular torticollis. SCM, sternocleidomastoid muscle.
(From Do TT: Congenital muscular torticollis: current concepts and review of treatment, Curr Opin Pediatr 18:26–29, 2006.)
Table 672-1 DIFFERENTIAL DIAGNOSIS OF TORTICOLLIS
CONGENITAL
ACQUIRED
INFLAMMATION
NEUROLOGIC
OTHER
Paroxysmal torticollis of infancy
[/not-level-membership-for-pediatrics-category]
