[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Figure 33-1 The Field Cancerization Theory is defined as the presence of one or more mucosal areas consisting of epithelial cells with cancer-associated genetic or epigenetic alterations. A precursor field (light blue) is monoclonal but does not show invasive growth or metastatic behavior, which are the hallmarks of an invasive carcinoma (dark blue). A field is preneoplastic by definition; it may or may not have histological alterations characteristic of dysplasia. 4 A leukoplakia is the clinical manifestation of a field, though most fields are clinically invisible. Additional genetic changes are needed to transform a field into a carcinoma. The field and primary tumor share genetic alterations and have a common clonal origin. Clinically, a field may be the source of local recurrences, second field tumors, and second primary tumors after surgical resection of the initial carcinoma. These legions are distinguished on the basis of their distance from the index tumor, or the time interval after which they develop. A local recurrence (lower center) arises from residual tumor cells and is less than 2 cm away from, and/or occurs within 3 years of, the primary tumor. A second primary tumor (lower left) is more than 2 cm away from, and/or occurs more than 3 years after, the primary tumor. Tumors that arise from a contiguous portion of the same field that gave rise to the original primary tumor have been described as second field tumors (lower right). 4 Studies attempting to identify specific genetic characteristics that determine the risk of a field developing into cancer have shown that genetic changes at chromosome 9p, decreased cytokeratin 4 expression, and decreased cornulin expression are potential biomarkers. 69,70 Leukoplakia studies have demonstrated that the presence and number of genetic changes, typically chromosome 9p loss, chromosome 3p loss, and chromosome 17p loss, are associated with the risk of progression. 4 At far right, the TP63/NOTCH1 expression gradient of normal epithelium is illustrated. Perturbation of this gradient is believed to be a component of precancerous fields and invasive HNSCC legions. The normal process of squamous differentiation in mucosa is controlled in part by TP63 and NOTCH1. TP63 is expressed in keratinocytes of the basal layer, where it maintains their proliferative potential and regulates expression of basal keratins. Expression of NOTCH1 results in terminal differentiation of cells in the spinous and granular layers expressing alternative keratins. 44,46–48,52,53 (Adapted from C. René Leemans, Boudewijn J. M. Braakhuis, and Ruud H. Brakenhoff. (2010). The molecular biology of head and neck cancer. Nature Reviews Cancer. doi:10.1038/nrc2982.)
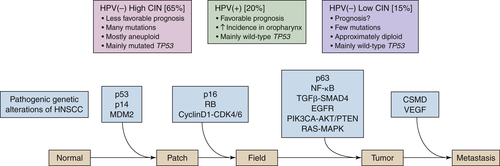
Figure 33-2 A hypothetical model of HNSCC development depicting the genetic alterations implicated in the process by current data. The model is a generalization and thus is varyingly accurate among subtypes of HNSCC. Three steps are critical in this model: A progenitor or adult stem cell acquires one (or more) genetic alterations, usually including an alteration of p53, and forms a patch containing clonal, genetically altered daughter cells. Then, by escaping normal growth control and/or gaining growth advantage, this clonal patch develops into an expanding field. Eventually, through a further accumulation of genetic alterations, a subclone in the field evolves into an invasive cancer and progresses to metastasis. Both aneuploidy and the accumulation of cancer-associated genetic changes in fields are linked to the risk of malignant progression. In addition, the three main clinicopathologic subtypes of HNSCC are depicted: HPV(+) HNSCC, HPV(–) HNSCC with many numerical genetic changes (high CIN), and HPV(–) HNSCC with few genetic changes (low CIN). 4 Although drawn as distinct steps for the purpose of illustration, the actual order of acquisition of distinct alterations is not known at this time. CDK, Cyclin-dependent kinase; CSMD, CUB and SUSHI multiple domain protein; NF-κB, nuclear factor-κB; PIK3CA, phosphoinositide-3 kinase subunit-α; TGFβ, transforming growth factor-β; VEGF, vascular endothelial growth factor. (Adapted from C. René Leemans, Boudewijn J. M. Braakhuis, and Ruud H. Brakenhoff. (2010). The molecular biology of head and neck cancer. Nature Reviews Cancer. doi:10.1038/nrc2982.)
Table 33-1
Common Genetic Alterations in HNSCC
| Gene ∗ |
Description |
Mutated (Activating/Missense/Inactivating) |
| TP53 a,b |
Transcription factor |
53% (NA/28%/25%) |
| CCND1 b |
Cell cycle activator (Cyclin D1) |
25% (25%/NA/NA) |
| CDKN2A a,b |
Cell cycle inhibitor/p53 activator |
21% (NA/3%/18%) |
| NOTCH1 a,b, ∗ |
Receptor/transcription factor |
15% (NA/8%/8%) |
| CSMD3 a,b |
Putative adhesion factor |
13% (NA/12%/1%) |
| USH2A a,b |
Basement membrane protein |
12% (NA/11%/1%) |
| PIK3CA a,b, ∗ |
PI3 kinase catalytic subunit |
10% (7%/7%/NA) |
| PRDM9 b,∗ |
Histone methyltransferase |
8% (NA/8%/1%) |
| COL22A1 b, ∗ |
Pro-apoptotic effector |
8% (NA/7%/2%) |
| RIMS2 b, ∗ |
Putative synaptic vesicle regulator |
8% (NA/8%/NA) |
| ZFHX4 b |
Zinc finger homeodomain |
8% (NA/8%/NA) |
| MLL2 b, ∗ |
Histone methyltransferase |
8% (NA/4%/4%) |
| NAV3 b |
Axonal/cytoskeleton guide |
8% (NA/7%/1%) |
| CASP8 a.b |
Pro-apoptotic proteolyase |
7% (NA/2%/5%) |
| TP63 a,b |
Transcription factor |
7% (NA/5%/1%) |
| NSD1 b |
Histone methyltransferase |
7% (NA/3%/4%) |
| EGFR b |
Growth factor Rtk |
7% (7%/NA/NA) |
| PTEN b |
Lipid phosphatase—PI3K inhibitor |
6% (NA/3%/3%) |
| FBXW7 a, ∗ |
Ubiquitin ligase |
5% (NA/3%/2%) |
| HRAS a,b,∗ |
RTK signaling protein |
4% (4%/4%/NA) |
| IRF6 b |
Interferon regulatory factor |
4% (NA/3%/1%) |
| NOTCH2 b |
Receptor/transcription factor |
4% (NA/3%/1%) |
| NOTCH3 b, ∗ |
Receptor/transcription factor |
4% (NA/2%/2%) |
Displays the percentage of samples in the aAgrawal et al. 15 and bStranksy et al. 16 exome sequencing studies with at least one mutant allele or copy number loss/amplification of a given gene. It also identifies the percentage of each respective mutation that is activating (known activating mutation or copy number amplification), missense, and inactivating (nonsense, splice site, frame shift, insertion/deletion, or copy number loss).
∗ Mutations present in at least one HPV(+) tumor.
Molecular Pathogenesis of HNSCC: Interfacing Genomic Pathways
Conceptually, there are six major hallmarks that define the current understanding of a cancerous cellular phenotype: sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis.
17 Research to date indicates that the altered oncogenes and tumor suppressors of HNSCC act primarily in functional pathways known to largely determine cellular proliferation, cell survival, squamous epithelial differentiation, and invasion and metastasis. Individual genes may function in more than one pathway, and the pathways themselves interface with, and influence, each other, as depicted in
Figure 33-3 . The following sections discuss some of the most common gene alterations believed to contribute to HNSCC oncogenesis in the context of these pathways.
Cell Cycle and Proliferation: TP53/CDKN2A/RB/CCND1/TERT
Though oncogenes have been identified, the high mutation rates seen in most cases of HNSCC, along with the recent exome sequencing data, suggest that loss-of-function mutations in tumor suppressor genes represent the predominant genetic pathology observed in HNSCC.
4,15,16 Foremost among these tumor suppressors is the
TP53 gene product: p53. A nuclear phosphoprotein that signals through its key
downstream partner, cyclin-dependent kinase inhibitor 1 (p21), p53 can influence both the G
1 and G
2 checkpoints of the cell cycle, although it is traditionally thought of as being the primary G
2 checkpoint regulator. Canonically, in response to DNA damage, p53 activation inhibits cell cycle progression and prevents apoptosis, allowing the cell time to repair the damaged DNA. If the DNA damage cannot be repaired, apoptosis ensues. Loss of p53 function allows cells with damaged DNA to proliferate freely, resulting in the accumulation of potentially oncogenic mutations in the genome of affected cells.
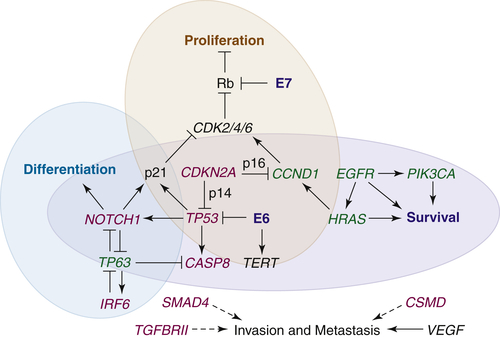
Figure 33-3 The interfacing genetic alterations of putative oncogenes (green), tumor suppressors (red), and signaling pathways that mediate the hallmarks of HNSCC. Loss of TP53 and CDKN2A, either through mutation or expression of the HPV E6 and E7 proteins (blue), along with amplification of CCND1 favors survival and permits proliferation through the increased activity of cyclin-dependent kinases (CDKs) and loss of p53-dependent apoptosis. Although intact differentiation programs and alternative apoptotic programs may restrict abnormal cell cycling for a time, loss of NOTCH1 and/or abnormal expression of TP63, along with the acquisition of alterations in other survival genes, such as CASP8, PIK3CA, and EGFR, remove additional barriers to tumor cell proliferation and survival. Upregulation of pro-angiogenic genes permits the growth of tumors, and the loss of cell adhesion genes allows for the release of cells from the mucosal lining. Invasion through the basement membrane is promoted by TGFβ-SMAD signaling, the loss of which initially contributes to tumorigenesis, and whose later reactivation drives metastasis. Several genes and signaling pathways, including TP53, TP63, and NOTCH1, contribute to more than one hallmark by influencing each other’s expression and/or activity.
Mutation of
TP53 is the earliest and most frequent mutation event observed in HPV-negative HNSCC. Occurring in more than 50% of cases, mutation is significantly associated with decreased survival.
18 The majority of
TP53 mutations are found in exons 5 through 9, the DNA binding region, with mutations at several specific codons known to be associated with tobacco exposure.
7 In a large portion of HNSCC without somatic
TP53 mutations, the activity of p53 is compromised by other mechanisms, including E6 expression in HPV(+) cancers, which inactivates p53, and overexpression and/or amplification of
MDM2, which promotes the degradation of p53.
Chromosomal loss of 9p21 has been reported in 70% to 80% of dysplastic oral mucosa legions progressing to HNSCC.
19 This illustrates an interface between pathways, as the
CDKN2A locus is found within 9p21. Two
CDKN2A protein products, p16
INK4A and p14
ARF/INK4B, are involved in cell cycle regulation. Specifically, p14
ARF/INK4B is known to downregulate
MDM2, thereby regulating p53 levels.
20 In all, p53 function is believed to be downregulated through one or more mechanisms in at least 80% of HNSCC.
4 A second
CDKN2A transcript, p16
INK4A, implicates the Retinoblastoma (
RB) pathway, which is the primary G
1 checkpoint regulator, in HNSCC. p16
INK4A inhibits the Cyclin D1/Cyclin-Dependent Kinase (CDK) complex, which normally functions to inactivate
RB-encoded pocket proteins via phosphorylation, allowing for the dissociation and activation of Elongation Factor-2 and subsequent entry into S phase. Inactive, phosphorylated
RB pocket proteins are unable to block the G
1-to-S phase transition in the setting of p16
INK4A loss.
20 In addition to chromosomal loss of 9p21, recent studies have demonstrated
CDKN2A mutations in approximately 7% of tumors and copy number losses in another 20% to 30%.
15,16 The mechanism of p16
INK4A loss has been shown to be of prognostic value in oral SCC: epigenetic silencing was found to be associated with higher recurrence rates, and deletion with increased rates of nodal metastases.
21 Analogous to the inhibition of p53 by HPV E6 expression, E7 expression in HPV(+) HNSCC inactivates the
RB pathway by binding RB1. Because E7 expression can inhibit the
RB pathway, there is less selective pressure for p16
INK4A loss in HPV(+) HNSCC. As a result, immunohistochemical staining for p16
INK4A is used clinically in establishing the HPV status of HNSCC, along with polymerase chain reaction (PCR)-based methods.
22 Further evidence of the important role played by the
RB pathway in HNSCC is that the commonly found amplification of 11q13, which contains
CCND1, in combination with other potential mechanisms, results in the overexpression of Cyclin D1 in up to 80% of HPV(–) tumors.
4 Intriguingly,
CDKN2A loss and
CCND1 gain, though seemingly redundant mechanisms to evade the G
1 checkpoint, are not mutually exclusive events in HNSCC. Both occur frequently and remain under investigation as independent and synergistic markers of poor prognosis.
23 Cyclin D1 has been found to sequester certain
CDK inhibitors and to bind transcription factors such as PPARγ, and various DNA repair proteins such as Rad51.
24 It remains to be established whether any of these interactions contributes to a noncanonical consequence of
CCND1 functional loss in HNSCC.
Finally, the role of telomerase in promoting limitless replicative potential must be considered. The activity of telomerase (TERT) is detectable by immunostaining in approximately 80% of HNSCC cases analyzed. In most in vitro HNSCC models, TERT activity is necessary for immortalization of cell lines. However, keratinocytes may elongate their telomeres in a TERT-independent fashion, and
TERT (5p15.33) is not known to be frequently gained or amplified in HNSCC. Although the exact role of
TERT is still unclear, it remains a candidate cancer gene in HNSCC.
4 Apoptosis and Survival: EGFR/RAS-MAPK/PIK3CA-AKT/CASP8
Cell cycle alterations, reduced immunogenicity, promotion of angiogenesis, and inhibition of apoptosis are just some of the many mechanisms underlying the enhanced survival of HNSCC. These cancerous traits are generated by genetic and epigenetic alterations in several pathways. Of particular importance in HNSCC are the receptor tyrosine kinase (RTK)-based signaling pathways. The class 1a phosphatidyl-inositol-3 kinases (PI3K) are heterodimers coupled to RTKs, such as the EGFR, or adaptor molecules. The PI3K-AKT kinase (AKT) signaling pathway mediates resistance to apoptosis and survival. Activated PI3K generates the lipid second-messenger phosphatidylinositol-3,4,5-P3 (PIP3), which serves to activate AKT. AKT is a serine/threonine kinase that, when activated, phosphorylates many downstream transcription factors, apoptosis inhibitors, cell cycle inhibitors, and other proteins, ultimately promoting cell survival and proliferation. This pathway is held in check by the action of the tumor suppressor phosphate and tensin homologue (PTEN), which dephosphorylates PIP3, thereby deactivating AKT. If PTEN activity is compromised, PI3K-AKT signaling can be irreversibly activated by RTK stimulation.
25 Inactivating
PTEN mutations have been reported in approximately 10% of HNSCC, PTEN expression is undetectable in nearly 30% of tongue cancers, and loss of heterozygosity of the
PTEN locus has been observed in up to 40% of HNSCC.
26 Furthermore, recent evidence suggests that loss of even a single
PTEN allele can contribute to tumorigenesis.
27 Three different “hotspot” activating mutations have been reported in
PI3KCA, which codes for the catalytic subunit of PI3K.
28 Notably, the frequency of
PI3KCA mutations is higher in HPV(+) HNSCC, suggesting a possible interaction between the PI3K pathway and the E6/E7 proteins of HPV. This has been suggested to be contributory to the development of invasive SCC in cervical cancer.
15,16,29 The PI3K-AKT axis is of consequence therapeutically in HNSCC as well, with numerous targeted inhibitors now in clinical trials.
29,30 RAS family GTPases (HRAS, KRAS, and NRAS) are molecular switches that cycle between two conformational states: an active GTP-bound form, and an inactive GDP-bound form. The first RAS effector pathway to be identified was the RAS-RAF-MEK-MAPK pathway. The pathway is a common and essential element of mitogenic signaling driven by RTKs, resulting in a diverse array of cellular responses. RAF proteins are serine/threonine kinases that bind to the effector region of RAS-GTP. This interaction induces translocation of the protein to the plasma membrane. There, RAF proteins are activated and phosphorylated by different protein kinases. Active RAF phosphorylates MEK that, in turn, phosphorylates and activates MAPK. Activated MAPK serves as the terminal effector of the pathway, influencing cellular growth, differentiation, inflammation, apoptosis, and senescence. Mutant RAS, in which it assumes a permanently active conformation, is a well-established oncogene, found in approximately 25% of human tumors.
31 HNSCC is unique in that
HRAS mutations, being found in 3% to 5% of HNSCC, are more prevalent than
KRAS or
NRAS mutations.
15,16 These
HRAS mutations are known to be associated with HNSCC in smokers, and in mouse models exposed to chemical carcinogens.
32 The exact contribution of the
HRAS mutations to oncogenesis has yet to be elucidated in HNSCC. The RAS-MAPK and PI3K-ATK pathways interact directly and indirectly, through multiple intermediates.
31 In addition,
HRAS mutations have been detected in HPV(+) tumors, allowing for the possibility of cooperation with oncogenic viral proteins.
15,16 Recent in vitro evidence suggests that even a single
HRAS mutation, in the background of HPV and
MYC alteration, can contribute to tumorigenesis.
33 Although the success of therapies targeting RAS proteins has been limited to date, several attempts to target their downstream effectors have shown promising results in preclinical models.
34 RTKs lie upstream of both the RAS-MAPK and PI3K-ATK pathways. Most importantly for HNSCC, is
EGFR (7p12), which codes for the prototypical ErbB family Type I RTK. Signaling through EGFR represents another interface between pathways, as it is involved in a variety of cellular processes, including survival and differentiation. EGFR has an extracellular ligand-binding domain, a transmembrane portion, and an intracellular kinase domain with five autophosphorylation sites. Ligand binding by EGFR monomers drives homodimerization or heterodimerization with another RTK, resulting in the initiation of downstream survival and proliferation signaling pathways. Two important and well-studied pathways activated by EGFR ligand binding are the RAS-MAPK and PI3K-ATK pathways. These independent cascades converge via the ultimate upregulation of Cyclin D1. Furthermore, when bound to EGF, EGFR itself can translocate to the nucleus, where it acts as a transcription factor for several genes including
CCND1, and as a co-activator for other transcription factor proteins, such as the STAT proteins.
4 EGFR is expressed in most epithelial tissues, and its dysregulation has been repeatedly shown to contribute to epithelial oncogenesis. In HNSCC, EGFR expression levels are nearly ubiquitously elevated in tumor and tumor-adjacent tissue compared to corresponding normal mucosa. Higher expression levels and copy number gain correlate with decreased survival but have not been highly indicative of improved response to EGFR-directed therapy. There are three FDA-approved EGFR targeting agents in clinical use: gefitinib and erlotinib, both TKIs, and cetuximab, a monoclonal antibody against EGFR, which is the only agent approved for use in HNSCC. All have shown modest efficacy as monotherapies to date, with EGFR-targeted therapies being effective in about 20% of patients in large multicenter trials, generally in combination with radiation and/or chemotherapy.
35 Expression of EGFRvIII, an
EGFR allele harboring a large in-frame deletion of exons 2 through 7, can confer resistance to anti-EGFR therapy. The prevalence of the EGFRvIII variant remains controversial in HNSCC, with various studies reporting its expression to be present in anywhere from 0% to 42% of the tumors assayed.
36–39 Investigations into EGFRvIII mechanism(s) of oncogenesis continue, as therapies specifically directed against EGFRvIII have shown promise in glioblastoma and may be applicable in refractory HNSCC.
39 Another genetic alteration, reported in some cases of HNSCC, that is believed to contribute to anti-EGFR therapy resistance is mutation or amplification of the
MET gene, which codes for another RTK.
40,41 MET has been implicated as a cancer gene in HNSCC that influences cell growth, motility, and angiogenesis.
4 This, too, may be of particular clinical consequence because there are both monoclonal antibodies and small-molecule inhibitors, FDA-approved in other cancers, with the ability to inhibit MET kinase activity.
42,43
Finally, in addition to the growth factor signaling pathways that indirectly influence apoptosis, recent studies in HNSCC have found alterations directly within the apoptosis cascade itself.
CASP8, a proteolyase responsible for initiating the caspase cascade that drives apoptosis, was found to be mutated in 8% of HNSCC by exome sequencing;
BCL2, which prevents apoptosis, has been observed to be overexpressed in some HNSCC cell lines, usually coincident with the underexpression of p63.
16,44 Differentiation and Mesenchymal Transition: NOTCH/TP63
Many of the expression profile studies in HNSCC contain a large number of genes that are thought to reflect the process of epithelial-to-mesenchymal transition (EMT), especially profiles of metastatic HNSCC. EMT is a biological process, wherein cells change from an epithelial phenotype to a mesenchymal-like phenotype. Because epithelial cells do not possess the cellular plasticity for metastatic dissemination, this process is a common occurrence in cancer cells.
4 TP63 codes for p63, a p53-related transcription factor that, via its target genes, regulates differentiation in stratified epithelium, lineage specification, and subsequently proliferative potential. Mice lacking
TP63 undergo total failure of epidermal maturation.
45,46 In normally differentiated mature epithelium,
TP63 expression is present as a gradient. The highest level occurs in the basal epithelial cells, where it serves to antagonize
NOTCH1 expression. Rising superficially through the strata,
TP63 levels decrease and
NOTCH1 levels increase, driving terminal differentiation of the epithelial cell type (see
Figure 33-1). In dysplastic mucosa, this patterning is lost, and
TP63 expression is evident throughout all layers of the epithelium. In addition,
TP63 overexpression and/or amplification are seen in the majority of HNSCC, and mutations were found in approximately 7% of tumors by exome sequencing.
15,16,47 An isoform of
TP63, ΔNp63, known to contribute to cell survival, senescence suppression, and growth factor signaling, was also found to be specifically upregulated in HNSCC.
16,44 Another recent finding that emerged from the HNSCC exome sequencing studies is the discovery of
NOTCH family mutations in 15% to 20% of tumors, with most being present in
NOTCH1 (12% to 15%).
15,16 NOTCH signaling has been shown to influence cell survival, self-renewal capacity, and cell cycle exit, in addition to driving epithelial differentiation in concert with p63 and other signaling pathways. Ligands on adjacent cells bind to
the NOTCH receptor, resulting in the cleavage of intracellular portions of the receptor that subsequently translocate to the nucleus and drive the transcription of NOTCH target genes.
48 Overactivation of this pathway is believed to be tumorigenic in diffuse large B-cell lymphoma, T-cell acute lymphoblastic leukemia, and chronic lymphocytic leukemia. In those hematologic malignancies, translocations and activating mutations within NOTCH receptor genes have been observed.
49–51 In contrast, the observed
NOTCH mutations in HNSCC are believed to be largely inactivating, loss-of-function–type mutations, suggesting a possible tumor suppressor role.
15,52 The exact role of NOTCH signaling in HNSCC remains to be elucidated and is likely tissue and/or context dependent, as has been observed in mouse models of epidermal and hematopoietic malignancies.
53,54 Invasion and Metastasis: MMP/TGFβ-SMAD/NFκB/CSMD/VEGF
Like many cancers, HNSCC tumors metastasize primarily to the regional lymph nodes. The number of lymph node metastases in the neck, in more distant tissues, and the presence of extranodal spread are important prognostic factors, predictive of disseminated disease and survival. Although expression profile signatures of primary tumors that are predictive of metastasis have been identified, attempts to elucidate the mechanisms driving HNSCC metastasis are preliminary and in some cases conflicting.
4 Metastasis is a multifaceted process that ultimately results in a primary tumor “seeding” a distant anatomical site in the body. It involves several steps, one of which is invasion via the degradation of the extracellular matrix surrounding the primary tumor, in order to gain access to other areas of the body via the bloodstream or lymph system. Many studies have investigated the involvement of the matrix metalloproteinases (MMPs), which facilitate the degradation of the extracellular matrix. To date, strong associations have not been found, and treatments targeting MMPs have not achieved appreciable success in HNSCC.
55 In the context of invasion, transforming growth factor β pathway (TGFβ), which normally functions to inhibit growth, has been implicated in HNSCC. TGFβ ligands bind to the receptors TGFBR1 and TGFBR2, resulting in phosphorylation of TGFBR1, which then activates the proteins SMAD2 and SMAD3. A SMAD complex is formed with the addition of SMAD4. This complex enters the nucleus and binds transcription factors, co-activators, and co-repressors, which modulate the expression of TGFβ target genes. Several of these are known to suppress cell proliferation, such as the cell cycle inhibitors cyclin-dependent kinase inhibitor 2. In addition, the TGFβ pathway has been identified as a key player in the EMT process.
56 The TGFβ pathway has been implicated in HNSCC—most commonly through 18q deletion, which contains
SMAD2,
SMAD3,
SMAD4, and the
TGFBRII gene.
4 A recent mouse model found that conditional deletion of
SMAD4 in the head and neck epithelium was sufficient to generate invasive HNSCC. The loss of
SMAD4 expression in these animals correlated with increased expression of
TGFBRI and increased activation of SMAD3, and the Fanconi anemia DNA repair pathway was found to be downregulated.
57 Previous studies reported missense mutations in
TGFBRII in primary HNSCC, as well as mutations in
SMAD2 and
SMAD4 in some HNSCC cell lines.
58,59 The prevalence of such mutations, however, has been called into question by recent exome sequencing studies, which did not report any in the 106 tumors that were fully analyzed.
15,16 Recently, it has been demonstrated that reduced activity of the TGFβ pathway correlates with increased NF-κB signaling in HNSCC. Though alterations in TGFβ and NF-κB signaling have long been implicated in cancer, the exact mechanism(s) of their interaction, as well as their independent and/or cooperative contributions to invasion and metastasis in HNSCC, still need to be precisely elucidated.
60,61 Tumors require blood vessels in order to grow to sizes larger than a few millimeters in diameter. These vessels facilitate nutrient and oxygen delivery, as well as metabolic by-product disposal. The exploitation of neo-angiogenesis, usually by producing angiogenic factors, is common to all solid tumors. Of the many inducers of angiogenesis, the strongest is vascular endothelial growth factor (VEGF). Many studies have linked VEGF expression to HNSCC prognosis, including a meta-analysis that found a significantly increased risk of 1.88, as well as an association with VEGF expression and metastasis to lymph nodes.
4 Although these data suggest a link between VEGF expression and outcome, neither VEGF nor EGFR expression has thus far been found to be a better prognostic indicator than HPV status.
62 Both of the recent exome sequencing studies reported mutations in
CSMD3, a putative adhesion factor that maps to 8p23 along with its family member,
CSMD1.
CSMD1 is a putative tumor suppressor, the loss of which is associated with high tumor grade and poor prognosis in other cancers, and whose role in HNSCC remains under investigation.
15,16,63 Though functional studies have not yet been performed, these mutations may underlie a mechanism permitting the dissociation of cells from an otherwise cohesive sheet of cancerous epithelium, ultimately allowing for migration and metastasis of HNSCC tumors.
Future Directions
The elucidation of molecular pathways responsible for the pathogenesis of HNSCC has already led to the development and implementation of several targeted therapies.
25,30,35 Given the heterogeneity of this disease, future efforts will be required to identify effective targets in specific patient populations. One potential target may be miRNAs: several preliminary studies have identified a set of 66 commonly deregulated miRNAs in HNSCC that may in fact contribute to many of the cancerous phenotypes observed in this disease and are predictive of metastasis.
64,65 A recent study reports distinct miRNA expression profiles in HNSCC dependent on HPV status.
66 Similarly, epigenetic studies have identified promoter hypermethylation patterns that correspond to HPV status, as well as a series of changes that correspond to the development of cancer from a precancerous plaque.
67,68 The continuation of these early investigative efforts, in molecular markers of prognosis and treatment response, will be vital to our ability to generate tumor-tailored therapies for individual patients.
References
1. Duvvuri U. , Myers J.N. Cancer of the head and neck is the sixth most common cancer worldwide . Curr Probl Surg . 2009 ; 46 : 114 – 117 .
2. Ferlay J. , Shin H.R. , Bray F. , Forman D. , Mathers C. , Parkin D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008 . Int J Cancer . 2010 ; 127 : 2893 – 2917 .
3. American Cancer Society . Cancer Facts and Figures 2012 . Surveillance Research ; 2012 : Retrieved from: http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/cancer-facts-figures-2012 .
4. Leemans C.R. , Braakhuis B.J. , Brakenhoff R.H. The molecular biology of head and neck cancer . Nat Rev Cancer . 2011 ; 11 : 9 – 22 .
5. Human papillomavirus-associated cancers—United States, 2004-2008 . MMWR Morb Mortal Wkly Rep . 2012 ; 61 : 258 – 261 .
6. Ang K.K. , Harris J. , Wheeler R. et al. Human papillomavirus and survival of patients with oropharyngeal cancer . N Engl J Med . 2010 ; 363 : 24 – 35 .
7. Cardesa A. , Nadal A. Carcinoma of the head and neck in the HPV era . Acta Dermatovenerol Alp Panonica Adriat . 2011 ; 20 : 161 – 173 .
8. Park J.W. , Pitot H.C. , Strati K. et al. Deficiencies in the Fanconi anemia DNA damage response pathway increase sensitivity to HPV-associated head and neck cancer . Cancer Res . 2010 ; 70 : 9959 – 9968 .
9. Okano M. , Gross T.G. Acute or chronic life-threatening diseases associated with Epstein-Barr virus infection . Am J Med Sci . 2012 ; 343 : 483 – 489 .
10. Edge S.B.B.D. , Compton C.C. et al. AJCC Cancer Staging Manual . 7th ed. New York, NY : Springer Verlag ; 2009 .
11. Bonner J.A. , Harari P.M. , Giralt J. et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck . N Engl J Med . 2006 ; 354 : 567 – 578 .
12. Vermorken J.B. , Mesia R. , Rivera F. et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer . N Engl J Med . 2008 ; 359 : 1116 – 1127 .
13. Roesch-Ely M. , Nees M. , Karsai S. et al. Proteomic analysis reveals successive aberrations in protein expression from healthy mucosa to invasive head and neck cancer . Oncogene . 2007 ; 26 : 54 – 64 .
14. Olivier M. , Hollstein M. , Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use . Cold Spring Harb Perspect Biol . 2010 ; 2 a001008 .
15. Agrawal N. , Frederick M.J. , Pickering C.R. et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1 . Science . 2011 ; 333 : 1154 – 1157 .
16. Stransky N. , Egloff A.M. , Tward A.D. et al. The mutational landscape of head and neck squamous cell carcinoma . Science . 2011 ; 333 : 1157 – 1160 .
17. Hanahan D. , Weinberg R.A. Hallmarks of cancer: the next generation . Cell . 2011 ; 144 : 646 – 674 .
18. Poeta M.L. , Manola J. , Goldwasser M.A. et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck . N Engl J Med . 2007 ; 357 : 2552 – 2561 .
19. Molinolo A.A. , Amornphimoltham P. , Squarize C.H. , Castilho R.M. , Patel V. , Gutkind J.S. Dysregulated molecular networks in head and neck carcinogenesis . Oral Oncol . 2009 ; 45 : 324 – 334 .
20. Vousden K.H. , Lane D.P. p53 in health and disease . Nat Rev Mol Cell Biol . 2007 ; 8 : 275 – 283 .
21. Sailasree R. , Abhilash A. , Sathyan K.M. , Nalinakumari K.R. , Thomas S. , Kannan S. Differential roles of p16INK4A and p14ARF genes in prognosis of oral carcinoma . Cancer Epidemiol Biomarkers Prev . 2008 ; 17 : 414 – 420 .
22. Pannone G. , Rodolico V. , Santoro A. et al. Evaluation of a combined triple method to detect causative HPV in oral and oropharyngeal squamous cell carcinomas: p16 immunohistochemistry, consensus PCR HPV-DNA, and in situ hybridization . Infect Agent Cancer . 2012 ; 7 : 4 .
23. Higuchi E. , Oridate N. , Homma A. et al. Prognostic significance of cyclin D1 and p16 in patients with intermediate-risk head and neck squamous cell carcinoma treated with docetaxel and concurrent radiotherapy . Head Neck . 2007 ; 29 : 940 – 947 .
24. Musgrove E.A. , Caldon C.E. , Barraclough J. , Stone A. , Sutherland R.L. Cyclin D as a therapeutic target in cancer . Nat Rev Cancer . 2011 ; 11 : 558 – 572 .
25. Engelman J.A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations . Nat Rev Cancer . 2009 ; 9 : 550 – 562 .
26. Bian Y. , Hall B. , Sun Z.J. et al. Loss of TGF-βsignaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation . Oncogene . 2012 ; 31 3322-3232 .
27. Berger A.H. , Knudson A.G. , Pandolfi P.P. A continuum model for tumour suppression . Nature . 2011 ; 476 : 163 – 169 .
28. Murugan A.K. , Hong N.T. , Fukui Y. , Munirajan A.K. , Tsuchida N. Oncogenic mutations of the PIK3CA gene in head and neck squamous cell carcinomas . Int J Oncol . 2008 ; 32 : 101 – 111 .
29. Henken F.E. , Banerjee N.S. , Snijders P.J. et al. PIK3CA-mediated PI3-kinase signalling is essential for HPV-induced transformation in vitro . Mol Cancer . 2011 ; 10 : 71 .
30. Kundu S.K. , Nestor M. Targeted therapy in head and neck cancer . Tumour Biol . 2012 ; 33 : 707 – 721 .
31. Castellano E. , Downward J. RAS interaction with PI3K: more than just another effector pathway . Genes Cancer . 2011 ; 2 : 261 – 274 .
32. Lu S.L. , Herrington H. , Wang X.J. Mouse models for human head and neck squamous cell carcinomas . Head Neck . 2006 ; 28 : 945 – 954 .
33. Narisawa-Saito M. , Inagawa Y. , Yoshimatsu Y. et al. A critical role of MYC for transformation of human cells by HPV16 E6E7 and oncogenic HRAS . Carcinogenesis . 2012 ; 33 : 910 – 917 .
34. Baines A.T. , Xu D. , Der C.J. Inhibition of Ras for cancer treatment: the search continues . Future Med Chem . 2011 ; 3 : 1787 – 1808 .
35. Sharafinski M.E. , Ferris R.L. , Ferrone S. , Grandis J.R. Epidermal growth factor receptor targeted therapy of squamous cell carcinoma of the head and neck . Head Neck . 2010 ; 32 : 1412 – 1421 .
36. Hama T. , Yuza Y. , Saito Y. et al. Prognostic significance of epidermal growth factor receptor phosphorylation and mutation in head and neck squamous cell carcinoma . Oncologist . 2009 ; 14 : 900 – 908 .
37. Sok J.C. , Coppelli F.M. , Thomas S.M. et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting . Clin Cancer Res . 2006 ; 12 : 5064 – 5073 .
38. Szabo B. , Nelhubel G.A. , Karpati A. et al. Clinical significance of genetic alterations and expression of epidermal growth factor receptor (EGFR) in head and neck squamous cell carcinomas . Oral Oncol . 2011 ; 47 : 487 – 496 .
39. Wheeler S.E. , Morariu E.M. , Bednash J.S. et al. Lyn kinase mediates cell motility and tumor growth in EGFRvIII-expressing head and neck cancer . Clin Cancer Res . 2012 ; 18 : 2850 – 2860 .
40. Seiwert T.Y. , Jagadeeswaran R. , Faoro L. et al. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma . Cancer Res . 2009 ; 69 : 3021 – 3031 .
41. Sequist L.V. , Waltman B.A. , Dias-Santagata D. et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors . Sci Transl Med . 2011 ; 3 75ra26 .
42. Kwak E.L. , Bang Y.J. , Camidge D.R. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer . N Engl J Med . 2010 ; 363 : 1693 – 1703 .
43. Pacchiana G. , Chiriaco C. , Stella M.C. et al. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody . J Biol Chem . 2010 ; 285 : 36149 – 36157 .
44. Rocco J.W. , Leong C.O. , Kuperwasser N. , DeYoung M.P. , Ellisen L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis . Cancer Cell. J . 2006 ; 9 : 45 – 56 .
45. Mills A.A. , Zheng B. , Wang X.J. , Vogel H. , Roop D.R. , Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis . Nature . 1999 ; 398 : 708 – 713 .
46. Yang A. , Schweitzer R. , Sun D. et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development . Nature . 1999 ; 398 : 714 – 718 .
47. Yugawa T. , Narisawa-Saito M. , Yoshimatsu Y. et al. DeltaNp63alpha repression of the Notch1 gene supports the proliferative capacity of normal human keratinocytes and cervical cancer cells . Cancer Res . 2010 ; 70 : 4034 – 4044 .
48. Dang T.P. Notch, apoptosis and cancer . Adv Exp Med Biol . 2012 ; 727 : 199 – 209 .
49. Lee S.Y. , Kumano K. , Nakazaki K. et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma . Cancer Sci . 2009 ; 100 : 920 – 926 .
50. Puente X.S. , Pinyol M. , Quesada V. et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia . Nature . 2011 ; 475 : 101 – 105 .
51. Weng A.P. , Ferrando A.A. , Lee W. et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia . Science . 2004 ; 306 : 269 – 271 .
52. Dotto G.P. Notch tumor suppressor function . Oncogene . 2008 ; 27 : 5115 – 5123 .
53. Nicolas M. , Wolfer A. , Raj K. et al. Notch1 functions as a tumor suppressor in mouse skin . Nat Genet . 2003 ; 33 : 416 – 421 .
54. Pear W.S. , Aster J.C. , Scott M.L. et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles . J Exp Med . 1996 ; 183 : 2283 – 2291 .
55. Rosenthal E.L. , Matrisian L.M. Matrix metalloproteases in head and neck cancer . Head Neck . 2006 ; 28 : 639 – 648 .
56. Ikushima H. , Miyazono K. TGFbeta signalling: a complex web in cancer progression . Nat Rev Cancer . 2010 ; 10 : 415 – 424 .
57. Bornstein S. , White R. , Malkoski S. et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation . J Clin Invest . 2009 ; 119 : 3408 – 3419 .
58. Qiu W. , Schonleben F. , Li X. , Su G.H. Disruption of transforming growth factor beta-Smad signaling pathway in head and neck squamous cell carcinoma as evidenced by mutations of SMAD2 and SMAD4 . Cancer Lett . 2007 ; 245 : 163 – 170 .
59. Wang D. , Song H. , Evans J.A. , Lang J.C. , Schuller D.E. , Weghorst C.M. Mutation and downregulation of the transforming growth factor beta type II receptor gene in primary squamous cell carcinomas of the head and neck . Carcinogenesis . 1997 ; 18 : 2285 – 2290 .
60. Cohen J. , Chen Z. , Lu S.L. et al. Attenuated transforming growth factor beta signaling promotes nuclear factor-kappaB activation in head and neck cancer . Cancer Res . 2009 ; 69 : 3415 – 3424 .
61. Freudlsperger C. , Bian Y. , Contag Wise S. et al. TGF-β and NF-κB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers . Oncogene . 2013 ; 32 : 1549 – 1559 .
62. Fei J. , Hong A. , Dobbins T.A. et al. Prognostic significance of vascular endothelial growth factor in squamous cell carcinomas of the tonsil in relation to human papillomavirus status and epidermal growth factor receptor . Ann Surg Oncol . 2009 ; 16 : 2908 – 2917 .
63. Kamal M. , Shaaban A.M. , Zhang L. et al. Loss of CSMD1 expression is associated with high tumour grade and poor survival in invasive ductal breast carcinoma . Breast Cancer Res Treat . 2010 ; 121 : 555 – 563 .
64. Babu J.M. , Prathibha R. , Jijith V.S. , Hariharan R. , Pillai M.R. A miR-centric view of head and neck cancers . Biochim Biophys Acta . 2011 ; 1816 : 67 – 72 .
65. Chen Z. , Jin Y. , Yu D. et al. Down-regulation of the microRNA-99 family members in head and neck squamous cell carcinoma . Oral Oncol . 2012 ; 48 : 686 – 691 .
66. Lajer C.B. , Garnaes E. , Friis-Hansen L. et al. The role of miRNAs in human papilloma virus (HPV)-associated cancers: bridging between HPV-related head and neck cancer and cervical cancer . Br J Cancer . 2012 ; 106 : 1526 – 1534 .
67. Worsham M.J. , Stephen J.K. , Chen K.M. et al. Delineating an epigenetic continuum in head and neck cancer . Cancer Lett . 2012 [Epub ahead of print] .
68. Sartor M.A. , Dolinoy D.C. , Jones T.R. et al. Genome-wide methylation and expression differences in HPV(+) and HPV(-) squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis . Epigenetics . 2011 ; 6 : 777 – 787 .
69. Graveland A.P. , Golusinski P.J. , Buijze M. et al. Loss of heterozygosity at 9p and p53 immunopositivity in surgical margins predict local relapse in head and neck squamous cell carcinoma . Int J Cancer . 2011 ; 128 : 1852 – 1859 .
70. Schaaij-Visser T.B. , Graveland A.P. , Gauci S. et al. Differential proteomics identifies protein biomarkers that predict local relapse of head and neck squamous cell carcinomas . Clin Cancer Res . 2009 ; 15 : 7666 – 7675 .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Figure 33-1 The Field Cancerization Theory is defined as the presence of one or more mucosal areas consisting of epithelial cells with cancer-associated genetic or epigenetic alterations. A precursor field (light blue) is monoclonal but does not show invasive growth or metastatic behavior, which are the hallmarks of an invasive carcinoma (dark blue). A field is preneoplastic by definition; it may or may not have histological alterations characteristic of dysplasia. 4 A leukoplakia is the clinical manifestation of a field, though most fields are clinically invisible. Additional genetic changes are needed to transform a field into a carcinoma. The field and primary tumor share genetic alterations and have a common clonal origin. Clinically, a field may be the source of local recurrences, second field tumors, and second primary tumors after surgical resection of the initial carcinoma. These legions are distinguished on the basis of their distance from the index tumor, or the time interval after which they develop. A local recurrence (lower center) arises from residual tumor cells and is less than 2 cm away from, and/or occurs within 3 years of, the primary tumor. A second primary tumor (lower left) is more than 2 cm away from, and/or occurs more than 3 years after, the primary tumor. Tumors that arise from a contiguous portion of the same field that gave rise to the original primary tumor have been described as second field tumors (lower right). 4 Studies attempting to identify specific genetic characteristics that determine the risk of a field developing into cancer have shown that genetic changes at chromosome 9p, decreased cytokeratin 4 expression, and decreased cornulin expression are potential biomarkers. 69,70 Leukoplakia studies have demonstrated that the presence and number of genetic changes, typically chromosome 9p loss, chromosome 3p loss, and chromosome 17p loss, are associated with the risk of progression. 4 At far right, the TP63/NOTCH1 expression gradient of normal epithelium is illustrated. Perturbation of this gradient is believed to be a component of precancerous fields and invasive HNSCC legions. The normal process of squamous differentiation in mucosa is controlled in part by TP63 and NOTCH1. TP63 is expressed in keratinocytes of the basal layer, where it maintains their proliferative potential and regulates expression of basal keratins. Expression of NOTCH1 results in terminal differentiation of cells in the spinous and granular layers expressing alternative keratins. 44,46–48,52,53 (Adapted from C. René Leemans, Boudewijn J. M. Braakhuis, and Ruud H. Brakenhoff. (2010). The molecular biology of head and neck cancer. Nature Reviews Cancer. doi:10.1038/nrc2982.)
Figure 33-2 A hypothetical model of HNSCC development depicting the genetic alterations implicated in the process by current data. The model is a generalization and thus is varyingly accurate among subtypes of HNSCC. Three steps are critical in this model: A progenitor or adult stem cell acquires one (or more) genetic alterations, usually including an alteration of p53, and forms a patch containing clonal, genetically altered daughter cells. Then, by escaping normal growth control and/or gaining growth advantage, this clonal patch develops into an expanding field. Eventually, through a further accumulation of genetic alterations, a subclone in the field evolves into an invasive cancer and progresses to metastasis. Both aneuploidy and the accumulation of cancer-associated genetic changes in fields are linked to the risk of malignant progression. In addition, the three main clinicopathologic subtypes of HNSCC are depicted: HPV(+) HNSCC, HPV(–) HNSCC with many numerical genetic changes (high CIN), and HPV(–) HNSCC with few genetic changes (low CIN). 4 Although drawn as distinct steps for the purpose of illustration, the actual order of acquisition of distinct alterations is not known at this time. CDK, Cyclin-dependent kinase; CSMD, CUB and SUSHI multiple domain protein; NF-κB, nuclear factor-κB; PIK3CA, phosphoinositide-3 kinase subunit-α; TGFβ, transforming growth factor-β; VEGF, vascular endothelial growth factor. (Adapted from C. René Leemans, Boudewijn J. M. Braakhuis, and Ruud H. Brakenhoff. (2010). The molecular biology of head and neck cancer. Nature Reviews Cancer. doi:10.1038/nrc2982.)
Table 33-1
Common Genetic Alterations in HNSCC
| Gene ∗ |
Description |
Mutated (Activating/Missense/Inactivating) |
| TP53 a,b |
Transcription factor |
53% (NA/28%/25%) |
| CCND1 b |
Cell cycle activator (Cyclin D1) |
25% (25%/NA/NA) |
| CDKN2A a,b |
Cell cycle inhibitor/p53 activator |
21% (NA/3%/18%) |
| NOTCH1 a,b, ∗ |
Receptor/transcription factor |
15% (NA/8%/8%) |
| CSMD3 a,b |
Putative adhesion factor |
13% (NA/12%/1%) |
| USH2A a,b |
Basement membrane protein |
12% (NA/11%/1%) |
| PIK3CA a,b, ∗ |
PI3 kinase catalytic subunit |
10% (7%/7%/NA) |
| PRDM9 b,∗ |
Histone methyltransferase |
8% (NA/8%/1%) |
| COL22A1 b, ∗ |
Pro-apoptotic effector |
8% (NA/7%/2%) |
| RIMS2 b, ∗ |
Putative synaptic vesicle regulator |
8% (NA/8%/NA) |
| ZFHX4 b |
Zinc finger homeodomain |
8% (NA/8%/NA) |
| MLL2 b, ∗ |
Histone methyltransferase |
8% (NA/4%/4%) |
| NAV3 b |
Axonal/cytoskeleton guide |
8% (NA/7%/1%) |
| CASP8 a.b |
Pro-apoptotic proteolyase |
7% (NA/2%/5%) |
| TP63 a,b |
Transcription factor |
7% (NA/5%/1%) |
| NSD1 b |
Histone methyltransferase |
7% (NA/3%/4%) |
| EGFR b |
Growth factor Rtk |
7% (7%/NA/NA) |
| PTEN b |
Lipid phosphatase—PI3K inhibitor |
6% (NA/3%/3%) |
| FBXW7 a, ∗ |
Ubiquitin ligase |
5% (NA/3%/2%) |
| HRAS a,b,∗ |
RTK signaling protein |
4% (4%/4%/NA) |
| IRF6 b |
Interferon regulatory factor |
4% (NA/3%/1%) |
| NOTCH2 b |
Receptor/transcription factor |
4% (NA/3%/1%) |
| NOTCH3 b, ∗ |
Receptor/transcription factor |
4% (NA/2%/2%) |
Displays the percentage of samples in the aAgrawal et al. 15 and bStranksy et al. 16 exome sequencing studies with at least one mutant allele or copy number loss/amplification of a given gene. It also identifies the percentage of each respective mutation that is activating (known activating mutation or copy number amplification), missense, and inactivating (nonsense, splice site, frame shift, insertion/deletion, or copy number loss).
∗ Mutations present in at least one HPV(+) tumor.
Molecular Pathogenesis of HNSCC: Interfacing Genomic Pathways
Conceptually, there are six major hallmarks that define the current understanding of a cancerous cellular phenotype: sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis.
17 Research to date indicates that the altered oncogenes and tumor suppressors of HNSCC act primarily in functional pathways known to largely determine cellular proliferation, cell survival, squamous epithelial differentiation, and invasion and metastasis. Individual genes may function in more than one pathway, and the pathways themselves interface with, and influence, each other, as depicted in
Figure 33-3 . The following sections discuss some of the most common gene alterations believed to contribute to HNSCC oncogenesis in the context of these pathways.
Cell Cycle and Proliferation: TP53/CDKN2A/RB/CCND1/TERT
Though oncogenes have been identified, the high mutation rates seen in most cases of HNSCC, along with the recent exome sequencing data, suggest that loss-of-function mutations in tumor suppressor genes represent the predominant genetic pathology observed in HNSCC.
4,15,16 Foremost among these tumor suppressors is the
TP53 gene product: p53. A nuclear phosphoprotein that signals through its key
downstream partner, cyclin-dependent kinase inhibitor 1 (p21), p53 can influence both the G
1 and G
2 checkpoints of the cell cycle, although it is traditionally thought of as being the primary G
2 checkpoint regulator. Canonically, in response to DNA damage, p53 activation inhibits cell cycle progression and prevents apoptosis, allowing the cell time to repair the damaged DNA. If the DNA damage cannot be repaired, apoptosis ensues. Loss of p53 function allows cells with damaged DNA to proliferate freely, resulting in the accumulation of potentially oncogenic mutations in the genome of affected cells.
Figure 33-3 The interfacing genetic alterations of putative oncogenes (green), tumor suppressors (red), and signaling pathways that mediate the hallmarks of HNSCC. Loss of TP53 and CDKN2A, either through mutation or expression of the HPV E6 and E7 proteins (blue), along with amplification of CCND1 favors survival and permits proliferation through the increased activity of cyclin-dependent kinases (CDKs) and loss of p53-dependent apoptosis. Although intact differentiation programs and alternative apoptotic programs may restrict abnormal cell cycling for a time, loss of NOTCH1 and/or abnormal expression of TP63, along with the acquisition of alterations in other survival genes, such as CASP8, PIK3CA, and EGFR, remove additional barriers to tumor cell proliferation and survival. Upregulation of pro-angiogenic genes permits the growth of tumors, and the loss of cell adhesion genes allows for the release of cells from the mucosal lining. Invasion through the basement membrane is promoted by TGFβ-SMAD signaling, the loss of which initially contributes to tumorigenesis, and whose later reactivation drives metastasis. Several genes and signaling pathways, including TP53, TP63, and NOTCH1, contribute to more than one hallmark by influencing each other’s expression and/or activity.
Mutation of
TP53 is the earliest and most frequent mutation event observed in HPV-negative HNSCC. Occurring in more than 50% of cases, mutation is significantly associated with decreased survival.
18 The majority of
TP53 mutations are found in exons 5 through 9, the DNA binding region, with mutations at several specific codons known to be associated with tobacco exposure.
7 In a large portion of HNSCC without somatic
TP53 mutations, the activity of p53 is compromised by other mechanisms, including E6 expression in HPV(+) cancers, which inactivates p53, and overexpression and/or amplification of
MDM2, which promotes the degradation of p53.
Chromosomal loss of 9p21 has been reported in 70% to 80% of dysplastic oral mucosa legions progressing to HNSCC.
19 This illustrates an interface between pathways, as the
CDKN2A locus is found within 9p21. Two
CDKN2A protein products, p16
INK4A and p14
ARF/INK4B, are involved in cell cycle regulation. Specifically, p14
ARF/INK4B is known to downregulate
MDM2, thereby regulating p53 levels.
20 In all, p53 function is believed to be downregulated through one or more mechanisms in at least 80% of HNSCC.
4 A second
CDKN2A transcript, p16
INK4A, implicates the Retinoblastoma (
RB) pathway, which is the primary G
1 checkpoint regulator, in HNSCC. p16
INK4A inhibits the Cyclin D1/Cyclin-Dependent Kinase (CDK) complex, which normally functions to inactivate
RB-encoded pocket proteins via phosphorylation, allowing for the dissociation and activation of Elongation Factor-2 and subsequent entry into S phase. Inactive, phosphorylated
RB pocket proteins are unable to block the G
1-to-S phase transition in the setting of p16
INK4A loss.
20 In addition to chromosomal loss of 9p21, recent studies have demonstrated
CDKN2A mutations in approximately 7% of tumors and copy number losses in another 20% to 30%.
15,16 The mechanism of p16
INK4A loss has been shown to be of prognostic value in oral SCC: epigenetic silencing was found to be associated with higher recurrence rates, and deletion with increased rates of nodal metastases.
21 Analogous to the inhibition of p53 by HPV E6 expression, E7 expression in HPV(+) HNSCC inactivates the
RB pathway by binding RB1. Because E7 expression can inhibit the
RB pathway, there is less selective pressure for p16
INK4A loss in HPV(+) HNSCC. As a result, immunohistochemical staining for p16
INK4A is used clinically in establishing the HPV status of HNSCC, along with polymerase chain reaction (PCR)-based methods.
22 Further evidence of the important role played by the
RB pathway in HNSCC is that the commonly found amplification of 11q13, which contains
CCND1, in combination with other potential mechanisms, results in the overexpression of Cyclin D1 in up to 80% of HPV(–) tumors.
4 Intriguingly,
CDKN2A loss and
CCND1 gain, though seemingly redundant mechanisms to evade the G
1 checkpoint, are not mutually exclusive events in HNSCC. Both occur frequently and remain under investigation as independent and synergistic markers of poor prognosis.
23 Cyclin D1 has been found to sequester certain
CDK inhibitors and to bind transcription factors such as PPARγ, and various DNA repair proteins such as Rad51.
24 It remains to be established whether any of these interactions contributes to a noncanonical consequence of
CCND1 functional loss in HNSCC.
Finally, the role of telomerase in promoting limitless replicative potential must be considered. The activity of telomerase (TERT) is detectable by immunostaining in approximately 80% of HNSCC cases analyzed. In most in vitro HNSCC models, TERT activity is necessary for immortalization of cell lines. However, keratinocytes may elongate their telomeres in a TERT-independent fashion, and
TERT (5p15.33) is not known to be frequently gained or amplified in HNSCC. Although the exact role of
TERT is still unclear, it remains a candidate cancer gene in HNSCC.
4 Apoptosis and Survival: EGFR/RAS-MAPK/PIK3CA-AKT/CASP8
Cell cycle alterations, reduced immunogenicity, promotion of angiogenesis, and inhibition of apoptosis are just some of the many mechanisms underlying the enhanced survival of HNSCC. These cancerous traits are generated by genetic and epigenetic alterations in several pathways. Of particular importance in HNSCC are the receptor tyrosine kinase (RTK)-based signaling pathways. The class 1a phosphatidyl-inositol-3 kinases (PI3K) are heterodimers coupled to RTKs, such as the EGFR, or adaptor molecules. The PI3K-AKT kinase (AKT) signaling pathway mediates resistance to apoptosis and survival. Activated PI3K generates the lipid second-messenger phosphatidylinositol-3,4,5-P3 (PIP3), which serves to activate AKT. AKT is a serine/threonine kinase that, when activated, phosphorylates many downstream transcription factors, apoptosis inhibitors, cell cycle inhibitors, and other proteins, ultimately promoting cell survival and proliferation. This pathway is held in check by the action of the tumor suppressor phosphate and tensin homologue (PTEN), which dephosphorylates PIP3, thereby deactivating AKT. If PTEN activity is compromised, PI3K-AKT signaling can be irreversibly activated by RTK stimulation.
25 Inactivating
PTEN mutations have been reported in approximately 10% of HNSCC, PTEN expression is undetectable in nearly 30% of tongue cancers, and loss of heterozygosity of the
PTEN locus has been observed in up to 40% of HNSCC.
26 Furthermore, recent evidence suggests that loss of even a single
PTEN allele can contribute to tumorigenesis.
27 Three different “hotspot” activating mutations have been reported in
PI3KCA, which codes for the catalytic subunit of PI3K.
28 Notably, the frequency of
PI3KCA mutations is higher in HPV(+) HNSCC, suggesting a possible interaction between the PI3K pathway and the E6/E7 proteins of HPV. This has been suggested to be contributory to the development of invasive SCC in cervical cancer.
15,16,29 The PI3K-AKT axis is of consequence therapeutically in HNSCC as well, with numerous targeted inhibitors now in clinical trials.
29,30 RAS family GTPases (HRAS, KRAS, and NRAS) are molecular switches that cycle between two conformational states: an active GTP-bound form, and an inactive GDP-bound form. The first RAS effector pathway to be identified was the RAS-RAF-MEK-MAPK pathway. The pathway is a common and essential element of mitogenic signaling driven by RTKs, resulting in a diverse array of cellular responses. RAF proteins are serine/threonine kinases that bind to the effector region of RAS-GTP. This interaction induces translocation of the protein to the plasma membrane. There, RAF proteins are activated and phosphorylated by different protein kinases. Active RAF phosphorylates MEK that, in turn, phosphorylates and activates MAPK. Activated MAPK serves as the terminal effector of the pathway, influencing cellular growth, differentiation, inflammation, apoptosis, and senescence. Mutant RAS, in which it assumes a permanently active conformation, is a well-established oncogene, found in approximately 25% of human tumors.
31 HNSCC is unique in that
HRAS mutations, being found in 3% to 5% of HNSCC, are more prevalent than
KRAS or
NRAS mutations.
15,16 These
HRAS mutations are known to be associated with HNSCC in smokers, and in mouse models exposed to chemical carcinogens.
32 The exact contribution of the
HRAS mutations to oncogenesis has yet to be elucidated in HNSCC. The RAS-MAPK and PI3K-ATK pathways interact directly and indirectly, through multiple intermediates.
31 In addition,
HRAS mutations have been detected in HPV(+) tumors, allowing for the possibility of cooperation with oncogenic viral proteins.
15,16 Recent in vitro evidence suggests that even a single
HRAS mutation, in the background of HPV and
MYC alteration, can contribute to tumorigenesis.
33 Although the success of therapies targeting RAS proteins has been limited to date, several attempts to target their downstream effectors have shown promising results in preclinical models.
34 RTKs lie upstream of both the RAS-MAPK and PI3K-ATK pathways. Most importantly for HNSCC, is
EGFR (7p12), which codes for the prototypical ErbB family Type I RTK. Signaling through EGFR represents another interface between pathways, as it is involved in a variety of cellular processes, including survival and differentiation. EGFR has an extracellular ligand-binding domain, a transmembrane portion, and an intracellular kinase domain with five autophosphorylation sites. Ligand binding by EGFR monomers drives homodimerization or heterodimerization with another RTK, resulting in the initiation of downstream survival and proliferation signaling pathways. Two important and well-studied pathways activated by EGFR ligand binding are the RAS-MAPK and PI3K-ATK pathways. These independent cascades converge via the ultimate upregulation of Cyclin D1. Furthermore, when bound to EGF, EGFR itself can translocate to the nucleus, where it acts as a transcription factor for several genes including
CCND1, and as a co-activator for other transcription factor proteins, such as the STAT proteins.
4 EGFR is expressed in most epithelial tissues, and its dysregulation has been repeatedly shown to contribute to epithelial oncogenesis. In HNSCC, EGFR expression levels are nearly ubiquitously elevated in tumor and tumor-adjacent tissue compared to corresponding normal mucosa. Higher expression levels and copy number gain correlate with decreased survival but have not been highly indicative of improved response to EGFR-directed therapy. There are three FDA-approved EGFR targeting agents in clinical use: gefitinib and erlotinib, both TKIs, and cetuximab, a monoclonal antibody against EGFR, which is the only agent approved for use in HNSCC. All have shown modest efficacy as monotherapies to date, with EGFR-targeted therapies being effective in about 20% of patients in large multicenter trials, generally in combination with radiation and/or chemotherapy.
35 Expression of EGFRvIII, an
EGFR allele harboring a large in-frame deletion of exons 2 through 7, can confer resistance to anti-EGFR therapy. The prevalence of the EGFRvIII variant remains controversial in HNSCC, with various studies reporting its expression to be present in anywhere from 0% to 42% of the tumors assayed.
36–39 Investigations into EGFRvIII mechanism(s) of oncogenesis continue, as therapies specifically directed against EGFRvIII have shown promise in glioblastoma and may be applicable in refractory HNSCC.
39 Another genetic alteration, reported in some cases of HNSCC, that is believed to contribute to anti-EGFR therapy resistance is mutation or amplification of the
MET gene, which codes for another RTK.
40,41 MET has been implicated as a cancer gene in HNSCC that influences cell growth, motility, and angiogenesis.
4 This, too, may be of particular clinical consequence because there are both monoclonal antibodies and small-molecule inhibitors, FDA-approved in other cancers, with the ability to inhibit MET kinase activity.
42,43
Finally, in addition to the growth factor signaling pathways that indirectly influence apoptosis, recent studies in HNSCC have found alterations directly within the apoptosis cascade itself.
CASP8, a proteolyase responsible for initiating the caspase cascade that drives apoptosis, was found to be mutated in 8% of HNSCC by exome sequencing;
BCL2, which prevents apoptosis, has been observed to be overexpressed in some HNSCC cell lines, usually coincident with the underexpression of p63.
16,44 Differentiation and Mesenchymal Transition: NOTCH/TP63
Many of the expression profile studies in HNSCC contain a large number of genes that are thought to reflect the process of epithelial-to-mesenchymal transition (EMT), especially profiles of metastatic HNSCC. EMT is a biological process, wherein cells change from an epithelial phenotype to a mesenchymal-like phenotype. Because epithelial cells do not possess the cellular plasticity for metastatic dissemination, this process is a common occurrence in cancer cells.
4 TP63 codes for p63, a p53-related transcription factor that, via its target genes, regulates differentiation in stratified epithelium, lineage specification, and subsequently proliferative potential. Mice lacking
TP63 undergo total failure of epidermal maturation.
45,46 In normally differentiated mature epithelium,
TP63 expression is present as a gradient. The highest level occurs in the basal epithelial cells, where it serves to antagonize
NOTCH1 expression. Rising superficially through the strata,
TP63 levels decrease and
NOTCH1 levels increase, driving terminal differentiation of the epithelial cell type (see
Figure 33-1). In dysplastic mucosa, this patterning is lost, and
TP63 expression is evident throughout all layers of the epithelium. In addition,
TP63 overexpression and/or amplification are seen in the majority of HNSCC, and mutations were found in approximately 7% of tumors by exome sequencing.
15,16,47 An isoform of
TP63, ΔNp63, known to contribute to cell survival, senescence suppression, and growth factor signaling, was also found to be specifically upregulated in HNSCC.
16,44 Another recent finding that emerged from the HNSCC exome sequencing studies is the discovery of
NOTCH family mutations in 15% to 20% of tumors, with most being present in
NOTCH1 (12% to 15%).
15,16 NOTCH signaling has been shown to influence cell survival, self-renewal capacity, and cell cycle exit, in addition to driving epithelial differentiation in concert with p63 and other signaling pathways. Ligands on adjacent cells bind to
the NOTCH receptor, resulting in the cleavage of intracellular portions of the receptor that subsequently translocate to the nucleus and drive the transcription of NOTCH target genes.
48 Overactivation of this pathway is believed to be tumorigenic in diffuse large B-cell lymphoma, T-cell acute lymphoblastic leukemia, and chronic lymphocytic leukemia. In those hematologic malignancies, translocations and activating mutations within NOTCH receptor genes have been observed.
49–51 In contrast, the observed
NOTCH mutations in HNSCC are believed to be largely inactivating, loss-of-function–type mutations, suggesting a possible tumor suppressor role.
15,52 The exact role of NOTCH signaling in HNSCC remains to be elucidated and is likely tissue and/or context dependent, as has been observed in mouse models of epidermal and hematopoietic malignancies.
53,54 Invasion and Metastasis: MMP/TGFβ-SMAD/NFκB/CSMD/VEGF
Like many cancers, HNSCC tumors metastasize primarily to the regional lymph nodes. The number of lymph node metastases in the neck, in more distant tissues, and the presence of extranodal spread are important prognostic factors, predictive of disseminated disease and survival. Although expression profile signatures of primary tumors that are predictive of metastasis have been identified, attempts to elucidate the mechanisms driving HNSCC metastasis are preliminary and in some cases conflicting.
4 Metastasis is a multifaceted process that ultimately results in a primary tumor “seeding” a distant anatomical site in the body. It involves several steps, one of which is invasion via the degradation of the extracellular matrix surrounding the primary tumor, in order to gain access to other areas of the body via the bloodstream or lymph system. Many studies have investigated the involvement of the matrix metalloproteinases (MMPs), which facilitate the degradation of the extracellular matrix. To date, strong associations have not been found, and treatments targeting MMPs have not achieved appreciable success in HNSCC.
55
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading.
Learn more here
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
The Molecular Basis of Cancer Expert Consult - Online
