Chapter 9 The Life-Threatening Epilepsies of Childhood and Their Treatment
Introduction
Lennox-Gastaut Syndrome
DIAGNOSIS AND ETIOLOGY2
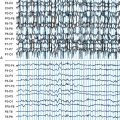
LGS develops between 2 and 8 years of age. It is a combination of various seizure types, mostly tonic and/or atonic seizures (usually called “drop-attacks” for their risk of falls and recurrent injury) and atypical absences, sometimes appearing as status epilepticus. On electroencephalogram (EEG), bilateral slow spike waves (< 2.5 Hz), generalized with bifrontal predominance, and bursts of rapid (10 Hz) rhythms during slow sleep (often corresponding to subclinical seizures) are most important for diagnosis. Psychomotor delay is present in 90% of cases, with slow behavior and frontal disorders as predominant symptoms. LGS can be cryptogenic in children with a previously normal development or symptomatic of congenital or acquired brain anomalies. Other types of epilepsy, especially IS, may precede LGS. Even if development was delayed prior to the onset of LGS, further mental deterioration is the rule due to persistently high rates of seizures together with interictal abnormal activity. Treating epilepsy has therefore ever been a key point in LGS.
TREATMENT
The open antiepileptic drugs (AEDs), the most used in LGS (valproate, benzodiazepines, and phenytoin), as well as ACTH and steroids, or the nondrug treatments, including ketogenic diet, corpus callosotomy, or vagal nerve stimulator, all remain poorly beneficial.2 Felbamate was the first of the new drugs shown to be effective in LGS by using the methodology of double-blind placebo-controlled adjunctive therapy in a randomized controlled trial (RCT).3 The risk of medullar aplasia and hepatotoxicity let the drug be proposed as the third line of treatment in severe cases. Then lamotrigine showed 33% of the lamotrigine group and 16% of the placebo group experienced a more than a 50% reduction in the frequency of all major seizures, including drop attacks. Global evaluations of patients’ functioning in terms of speech, language, and attention were significantly improved in the lamotrigine group.4 The efficacy of topiramate was also demonstrated for tonic seizures, and drop attacks at 3 months5 and maintained at long term in more than 50% of the patients.6 More recently, patients on rufinamide experienced a significant reduction in total seizure frequency and in drop attacks compared to patients on placebo, with 43% of responders for drop attacks compared to 17% on placebo.7
PARTICULAR ISSUES OF LGS REGARDING AED TRIALS
Several types of seizures coexist in LGS; each of them may be present in types of epilepsy that are not LGS. Clinical trials in LGS should therefore include EEG features in diagnosis criteria and several seizure types as efficacy endpoints to select the drug appropriate for the epileptic syndrome, as opposed to the current approach, which is based on the seizure type. LGS provided the first attempt to perform a trial dedicated to a specific syndrome, using felbamate in 1993.3
LGS is a recognized distinct medical condition, but one of the more challenging aspects of LGS is the distinction from other childhood epilepsies that might mimic either the EEG or clinical pattern. For example, trying to differentiate LGS from MAE may be highly difficult based on age of onset, seizure type (especially in LGS forms with myoclonic jerks and MAE forms with tonic seizures), and slow spike-and-wave patterns on EEG.8 Only one feature is pathognomonic of LGS, the bursts of rapid (10 Hz) rhythms during slow sleep. However, it has not been required as diagnostic criteria for trials until now, mainly because it needed sleep EEG, which could not be systematically performed. As a result, among the patients included in the LGS trials, some likely matched more closely with MAE than with LGS. Further trials should ideally include this EEG feature, at least for stratifying the analysis on a subgroup of “pure” LGS patients.
Infantile Spasms
DIAGNOSIS AND ETIOLOGY9
IS usually occurs between 3 and 8 months of age, with a peak at 4 months. More and more cases have been reported with onset later than 1 year, which seem to be considered separately. Diagnosis lies on the classical triad, which associates a specific type of seizures (epileptic spasms), a specific EEG pattern (hypsarrhythmia), and psychomotor deterioration. In fact, one of the three components may be lacking or be atypical. Epileptic spasms consist of axial contraction in flexion or extension, in clusters that correspond to a large slow wave or to flattening with rapid rhythms on EEG. Spasms may be clinically subtle or even subclinical, particularly at onset of the disease or when they are incompletely controlled, thus making EEG a necessary tool to prove their complete disappearance. The features of spasms may be either symmetric or asymmetric. Video recording is often required for detailed analysis because asymmetry of spasms or a focal discharge combined with them are arguments for a focal cortical lesion to be at the origin of IS. Psychomotor deterioration is usually rapid from epilepsy onset, affecting head control, reaching for objects, defect in visual and auditive attention, and eye–hand coordination.10,11 It is important to assess the status of psychomotor development (normal or abnormal) before onset because it carries prognostic implications, with a better outcome for patients without regression of eye tracking.
Although about 30% of IS present with normal MRI, the so-called cryptogenic/idiopathic IS, a large panel of cerebral lesions can be the cause in the remaining cases, including cortical malformations (such as agyria-pachygyria, hemimegalencephaly or focal cortical dysplasia), sequelae of pre-, per-, or postnatal anoxic-ischemia (such as periventricular leukomalacia of premature infants, porencephaly, or sequelae of subdural hematoma), infection of the central nervous system (such as meningitis or encephalitis), neurocutaneous syndromes (such as tuberous sclerosis or neurofibromatosis), chromosome disorders (such as Down syndrome or mutations in ARX, STK9 or Kir6.2 genes),12 or inherited metabolic disorders (such as pyridoxine dependency, Menkes disease, or mitochondrial disease due to NARP mutation). As a result, specific etiologic treatment (pyridoxine, surgery) is rarely possible, although it must be actively evaluated.
MEDICAL TREATMENT9
Most AEDs are usually inefficient in IS, and most of them were tested through open trials (valproate, benzodiazepines, piridoxine, lamotrigine, topiramate, felbamate, zonisamide, ketogenic diet, and thyrotropin-releasing hormone). The two major therapeutic approaches consist of hormonal treatment (ACTH and steroids) and vigabatrin. Considering an evidence-based approach, vigabatrin is less effective as a hormonal treatment at short term (2 weeks), but is as effective at 1-year follow-up.13,14 They both have side effects, although they are different in potential severity; vigabatrin may induce bilateral restriction of the peripheral visual field in around 20% of cases, whereas hormonal therapy carries a mortality rate up to 5%. However, a benefit with epilepsy seems to be associated with a mental benefit over the long term in IS: developmental and socialization outcome is favorably influenced by the initial and rapid control of spasms with vigabatrin in tuberous sclerosis15 and with steroids in cryptogenic and even symptomatic cases.16
Vigabatrin demonstrated its efficacy on IS as a first-line monotherapy at doses superior to 100 mg/kg/day compared to placebo or to low doses.17,18 Overall, more than one-third of children can be expected to have complete resolution of spasms, but the response rate mainly depends on etiology; it reached 90% in infants with tuberous sclerosis19 and 54% in patients with a variety of conditions other than tuberous sclerosis.13 In cryptogenic cases, the success rate may reach 100% when adding ACTH to patients not responding to vigabatrin monotherapy.20 The most preoccupying side effect of vigabatrin is its retinal toxicity, which induces visual field constriction. Although asymptomatic in most pediatric cases, rather less frequent in children (around 20%) than in adults (around 30%),21 and never reported in children who received less than 15 months of vigabatrin exposure,22 there is still no validated means to detect such a visual field defect before the age of 6 to 8 years.
PARTICULAR ISSUES OF IS REGARDING AED TRIALS
The risk of rapid cognitive decline allows a reduction in the duration of the evaluation period in double-blind conditions. Given the high rate of seizures, it was successfully limited to a maximum of 2 weeks in the two last RCTs in IS.13,17
Both epilepsy and its etiology contribute to the prognosis of IS. Epilepsy adds its own severity to this status, not only through the frequent seizures, but also through the interictal paroxysmal. The treatment of IS should therefore have two goals: to control seizures and to control hypsarrhythmia. Complete cessation of spasms is mandatory; the decrease of seizure rate by over 50%, which is how responders are usually defined in trials, is not adequate in IS trials. One should also consider the interictal EEG activity as a surrogate marker.
Treatment strategy in IS depends on both the local use and the drug availability in various countries. Regarding vigabatrin, the question that remains is how long treatment should be continued; in Down syndrome, 6-month control of spasms was followed by no relapse after vigabatrin discontinuation, whereas a devastating relapse was possible until the age of 5 years in tuberous sclerosis.23
Dravet Syndrome
DIAGNOSIS AND ETIOLOGY
The first seizures occur between 2 and 9 months of age.24 They are tonic-clonic or clonic seizures, either generalized or affecting alternatively one side of the body, often prolonged, resulting in recurrent status epilepticus and often provoked by fever. Children typically have a normal perinatal history and initially present with normal psychomotor development, normal neurological examination, and normal EEG between seizures. The pattern changes from the second year on: tonic-clonic or clonic seizures persist with the same characteristics, but additional myoclonia, atypical absences, and partial seizures occur; generalized spike and waves are observed during sleep; patients develop ataxia, hyperactivity, and mental retardation; and EEG shows spontaneous generalized spike waves and polyspike waves, as well as a slowing down of the background activity. Borderline forms were identified by Japanese authors with potential later onset, no later myoclonia, better cognitive outcome, and less pharmacoresistance.25 The relationships with the typical form previously described are still to be established.
Nonsense mutations of SCN1a gene, which codes for the α1 subunit of voltage-dependant sodium channel, have been identified in up to 70% of patients, including some microdeletions and duplications, which require more sophisticated procedure.26,27,28
TREATMENT
Most authors agree that valproate and benzodiazepines may decrease the frequency and duration of afebrile convulsive seizures, but the effect is only moderate. Some investigators associate phenobarbital, bromide, or phenytoin, depending on the country, usually with unsatisfactory results. Paradoxically, lamotrigine and carbamazepine can aggravate seizures and should be avoided.29
By contrast, two new drugs are helpful for treating patients with DS, stiripentol and topiramate. Stiripentol efficacy was shown in one open and then two randomized placebo-controlled trials, independently conducted in France and Italy in children with DS and receiving concomitant therapy with clobazam (CLB) and valproic acid (VPA).30,31 Despite a relatively small sample size in both trials (41 and 23 patients), 71% and 67% of patients, respectively, were responders on STP against 5% and 9%, respectively, on placebo. Tolerability was acceptable provided the dose of medication was diminished, because STP inhibits the cytochrome p450 (CYP) system in the liver, resulting in an increased plasma concentration of concomitant AED, particularly clobazam, mainly through CYP 2C19.31,32 In the long term, the frequency and duration of seizures remained significantly reduced, as was the number of episodes of convulsive status epilepticus.33
Topiramate has not been as extensively studied in DS, and only data from three open-labeled trials are available, with 55% of responders in two of them.34,35 Side effects are mainly related to rapid dosage titration and, to some extent, to the association with valproate (such as apathy and elevated blood ammonia levels). The association of topiramate to stiripentol does not need any particular adaptation of dosages and may be helpful and well tolerated in patients unsatisfactorily controlled with stiripentol.36
Preliminary open reports are also emerging using levetiracetam as adjunctive therapy with encouraging results.37
PARTICULAR ISSUES OF DRAVET SYNDROME REGARDING AED TRIALS
The severity of Dravet syndrome is due to its unfavorable prognosis for both mental and vital status. The risk of sudden unexpected death in epilepsy (SUDEP) is among the highest within all epilepsy syndromes (about 15% compared to 5%). The psychomotor development of almost all affected children is poor, evolving from a normal status before the beginning of the disease to a severe mental retardation around the age of 4 to 5 years. Epileptic seizures, and especially the number of status epilepticus in the first years of life, are likely to carry some responsibility in the mental retardation,38 so that any drug that could decrease their frequency, as STP does, may be beneficial as soon as the diagnosis is confirmed.
Dravet syndrome is a relatively easily identifiable disease, and diagnosis can be performed based on electroclinical criteria in a significant proportion of cases as early as 6 months of age. A score of early diagnosis is currently under validation in Japan.39 Thus, there is no reason to restrict the further trials to infants over the age of 1 year.
A rational approach of therapeutics could be possible in the future for Dravet syndrome in light of the defect described on a sodium channel gene in these patients: AEDs working mainly by blocking sodium channels (such as carbamazepine, phenytoin, and lamotrigine) are likely not to be effective and even to worsen seizures, whereas broad-spectrum AEDs (like valproate, benzodiazepines, and topiramate) are likely to be effective.40
Continuous Spike Waves During Sleep
DIAGNOSIS AND ETIOLOGY41
Onset is during childhood around 2 to 10 years of age. Seizures are rare, simple, or complex partial and atypical absences and drop attacks. The crucial feature is the regression of intellectual abilities, close to a CSWS pattern. All cognitive functions may be involved; some patients with verbal agnosia behave like deaf children, thus resulting in Landau-Kleffner syndrome, whereas others exhibit an acquired frontal syndrome42 with behavioral changes that may mimic psychosis or dementia. Negative myoclonus43 and oro-buccofacial apraxia have also been reported. Deterioration may be missed if the child presented with previous psychomotor delay, and CSWS diagnosis was therefore overlooked. Sleep EEG constantly shows bilateral continuous high-frequency spike wave activity (1.5 to 5 Hz). The proportion of EEG tracing with spikes is over 85% during slow sleep (but it may be around 50% in Landau-Kleffner syndrome).44 Focal activity is usually detected during awake EEG, in accordance with the particular functions involved.
Etiology comprises both cryptogenic and symptomatic cases, the later mainly resulting from pre- or perinatal anoxic-ischemia or unilateral polymicrogyria.45
TREATMENT41
Benzodiazepines (BZ) in monotherapy and ethosuximide may be effective in some cryptogenic cases, whereas carbamazepine may worsen the condition, and barbiturates, lamotrigine, and even valproate may be inefficient. Steroids have a favorable and lasting effect and are justified when BZ are not efficient (most often in symptomatic patients), provided corticotherapy will be administered for at least 1 year to avoid possible relapse. Preliminary encouraging reports using open topiramate and levetiracetam46 need to be confirmed.
Conclusion
Contrary to what has been shown in adults with epilepsy, the early choice of an adapted treatment is a key point in infants and children with EE.47 However, they remain “therapeutic orphans,” although they represent the most frequent and deleterious disorders in the field of epilepsy.48 EE do not exist in adults and therefore require specific trials. Rather than apply the guidelines for AED trials in adults, the design and the methodology of trials in EE need to be adapted to the particulars of these epilepsy syndromes (i.e., rare and age-related diseases with rapid and severe cognitive deterioration).
1. Chiron C, Dulac O, Pons G. Antiepileptic drug development in children: considerations for a revisited strategy. Drugs. 2008;68:17-25.
2. Genton P, Dravet C. Lennox-Gastaut syndrome. In: Engel JJr, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. 2nd. Philadelphia: Lippincott Williams & Wilkins; 2008:2417-2428.
3. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox-Gastaut syndrome). The Felbamate Study Group in Lennox-Gastaut Syndrome. N Engl J Med. 1993;328:29-33.
4. Motte J, Trevathan E, Arvidsson JF, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalized seizures associated with the Lennox-Gastaut syndrome. Lamictal Lennox-Gastaut Study Group. N Engl J Med. 1997;337:1807-1812.
5. Sachdeo RC, Glauser TA, Ritter F, Reife R, Lim P, Pledger G. A double-blind, randomized trial of topiramate in Lennox-Gastaut syndrome. Topiramate YL Study Group. Neurology. 1999;52:1882-1887.
6. Glauser TA, Levisohn PM, Ritter F, Sachdeo RC. Topiramate in Lennox-Gastaut syndrome: open-label treatment of patients completing a randomized controlled trial. Topiramate YL Study Group. Epilepsia. 2000;41(Suppl 1):S86-S90.
7. Arroyo S. Rufinamide. Neurotherapeutics. 2007;4:155-162.
8. Kaminska A, Ickowicz A, Plouin P, Bru MF, Dellatolas G, Dulac O. Delineation of cryptogenic Lennox-Gastaut syndrome and myoclonic astatic epilepsy using multiple correspondence analysis. Epilepsy Res. 1999;36:15-29.
9. Dulac O, Dalla BB, Chiron C. West syndrome. In: Engel JJr, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. 2nd. Philadelphia: Lippincott Williams & Wilkins; 2008:2329-2336.
10. Guzzetta F, Frisone MF, Ricci D, Rando T, Guzzetta A. Development of visual attention in West syndrome. Epilepsia. 2002;43:757-763.
11. Rando T, Baranello G, Ricci D, et al. Cognitive competence at the onset of West syndrome: correlation with EEG patterns and visual function. Dev Med Child Neurol. 2005;47:760-765.
12. Bahi-Buisson N, Eisermann M, Nivot S, et al. Infantile spasms as an epileptic feature of DEND syndrome associated with an activating mutation in the potassium adenosine triphosphate (ATP) channel, Kir6.2. J Child Neurol. 2007;22:1147-1150.
13. Lux AL, Edwards SW, Hancock E, et al. The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet. 2004;364:1773-1778.
14. Lux AL, Edwards SW, Hancock E, et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol. 2005;4:712-717.
15. Jambaque I, Chiron C, Dumas C, Mumford J, Dulac O. Mental and behavioural outcome of infantile epilepsy treated by vigabatrin in tuberous sclerosis patients. Epilepsy Res. 2000;38:151-160.
16. Riikonen R. Long-term outcome of patients with West syndrome. Brain Dev. 2001;23:683-687.
17. Appleton RE, Peters AC, Mumford JP, Shaw DE. Randomised, placebo-controlled study of vigabatrin as first-line treatment of infantile spasms. Epilepsia. 1999;40:1627-1633.
18. Elterman RD, Shields WD, Mansfield KA, Nakagawa J. Randomized trial of vigabatrin in patients with infantile spasms. Neurology. 2001;57:1416-1421.
19. Chiron C, Dumas C, Jambaque I, Mumford J, Dulac O. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Res. 1997;26:389-395.
20. Granstrom ML, Gaily E, Liukkonen E. Treatment of infantile spasms: results of a population-based study with vigabatrin as the first drug for spasms. Epilepsia. 1999;40:950-957.
21. Wild JM, Ahn HS, Baulac M, et al. Vigabatrin and epilepsy: lessons learned. Epilepsia. 2007;48:1318-1327.
22. Vanhatalo S, Nousiainen I, Eriksson K, et al. Visual field constriction in 91 Finnish children treated with vigabatrin. Epilepsia. 2002;43:748-756.
23. Kroll-Seger J, Kaminska A, Moutard ML, et al. Severe relapse of epilepsy after vigabatrin withdrawal: for how long should we treat symptomatic infantile spasms? Epilepsia. 2007;48:612-613.
24. Dravet C, Bureau M. Severe myoclonic epilepsy in infancy (Dravet syndrome). In: Engel JJr, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. 2nd. Philadelphia: Lippincott Williams & Wilkins; 2008:2337-2342.
25. Oguni H, Hayashi K, Oguni M, et al. Treatment of severe myoclonic epilepsy in infants with bromide and its borderline variant. Epilepsia. 1994;35:1140-1145.
26. Claes L, Del Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327-1332.
27. Sugawara T, Mazaki-Miyazaki E, Fukushima K, et al. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology. 2002;58:1122-1124.
28. Marini C, Temudo T, Ferrari AR, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia. 2007;48:1678-1685.
29. Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O. Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia. 1998;39:508-512.
30. Perez J, Chiron C, Musial C, et al. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia. 1999;40:1618-1626.
31. Chiron C, Marchand MC, Tran A, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000;356:1638-1642.
32. Giraud C, Treluyer JM, Rey E, et al. In vitro and in vivo inhibitory effect of stiripentol on clobazam metabolism. Drug Metab Dispos. 2006;34:608-611.
33. Thanh TN, Chiron C, Dellatolas G, et al. Efficacité et tolérance à long terme du stiripentol dans le traitement de l’épilepsie myoclonique sévère du nourrisson (syndrome de Dravet). Arch Pediatr. 2002;9:1120-1127.
34. Nieto-Barrera M, Candau R, Nieto-Jimenez M, Correa A, del Portal LR. Topiramate in the treatment of severe myoclonic epilepsy in infancy. Seizure. 2000;9:590-594.
35. Coppola G, Capovilla G, Montagnini A, et al. Topiramate as add-on drug in severe myoclonic epilepsy in infancy: an Italian multicenter open trial. Epilepsy Res. 2002;49:45-48.
36. Kroll-Seger J, Portilla P, Dulac O, Chiron C. Topiramate in the treatment of highly refractory patients with Dravet syndrome. Neuropediatrics. 2006;37:325-329.
37. Striano P, Coppola G, Pezella M, et al. An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology. 2007;69:250-254.
38. Nabbout R, Gennaro E, Dalla BB, et al. Spectrum of SCN1A mutations in severe myoclonic epilepsy of infancy. Neurology. 2003;60:1961-1967.
39. Hattori H, Ouchida M, Ono J, et al. A screening test for the prediction of Dravet syndrome before one year of age. Epilepsia. 2007;11:1-8.
40. Ceulemans B, Boel M, Claes L, et al. Severe myoclonic epilepsy in infancy: toward an optimal treatment. J Child Neurol. 2004;19:516-521.
41. Smith MC. Landau-Kleffner Syndrome and CSWS. In: Engel JJr, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. 2nd. Philadelphia: Lippincott Williams & Wilkins; 2008:2429-2436.
42. Roulet E, Davidoff V, Despland PA, Deonna T. Mental and behavioural deterioration of children with epilepsy and CSWS: acquired epileptic frontal syndrome. Dev Med Child Neurol. 1993;35:661-674.
43. Guerrini R, Dravet C, Genton P, et al. Epileptic negative myoclonus. Neurology. 1993;43:1078-1083.
44. Veggiotti P, Beccaria F, Guerrini R, Capovilla G, Lanzi G. Continuous spike-and-wave activity during slow-wave sleep: syndrome or EEG pattern? Epilepsia. 1999;40:1593-1601.
45. Guerrini R, Genton P, Bureau M, et al. Multilobar polymicrogyria, intractable drop attack seizures, and sleep-related electrical status epilepticus. Neurology. 1998;51:504-512.
46. Aeby A, Poznanski N, Verheulpen D, Wetzburger C, Van BP. Levetiracetam efficacy in epileptic syndromes with continuous spikes and waves during slow sleep: experience in 12 cases. Epilepsia. 2005;46:1937-1942.
47. Arroyo S, Brodie MJ, Avanzini G, et al. Is refractory epilepsy preventable? Epilepsia. 2002;43:437-444.
48. Trevathan E. Antiepileptic drug development for “therapeutic orphans.”. Epilepsia. 2003;44(Suppl 7):19-25.