CHAPTER 80 THE LEUKODYSTROPHIES
The leukodystrophies are now defined as disorders that (1) have a known or presumptive genetic cause; (2) have a progressive clinical course; (3) involve predominantly and usually confluently central nervous system (CNS) white matter; and (4) are characterized by a primary lesion of myelin or myelinating cells. This is the definition recommended by Powers1 and conforms to that proposed by Bielschowsky and Henneberg, who introduced this name in 1928.2 These diseases must be distinguished from abnormalities of myelin caused by infectious or toxic agents, disturbances of blood flow, anoxia or asphyxia, trauma, or genetic disorders in which damage to myelin or myelinating cells is a secondary event. The understanding, definition, and classification have been enhanced greatly by the combined use of neuroimaging and genetic techniques. Magnetic resonance imaging (MRI) is a powerful tool for the definition and categorization of leukoencephalopathies of unknown origin.3,4 For instance, MRI pattern analysis, combined with clinical data, led to the recognition that vanishing white matter disease5 (also referred to as childhood ataxia with diffuse central nervous hypomyelination)6 and megalencephalic leukoencephalopathy with subcortical cysts (MLC)7 are distinct clinical entities. Rapid application of positional cloning led to the identification of the gene defects.8–10 Although conventional MRI techniques have been of great value for the definition and classification of the leukodystrophies, these are now being supplemented by proton magnetic resonance spectroscopy (MRS), diffusion tensor imaging, and magnetization transfer techniques. One review summarized these novel techniques, as well as the current understanding of the alterations of myelin structure and function that underlie the neuroimaging abnormalities.11
Although these techniques represent important advances, van der Knaap cautioned against “diagnostic euphoria,”3 because for up to 50% of children with white matter abnormalities evident on MRI, no specific diagnosis could be established despite repeated MRI and extensive laboratory investigations.11 The 12 leukodystrophies in which the gene defect has been defined are the primary focus of this chapter.
X-LINKED ADRENOLEUKODYSTROPHY
Other names12 by which this disease is known include adrenomyeloneuropathy, Schilder’s disease, Addison’s disease with diffuse sclerosis, encephalitis periaxialis diffusa, Bronzekrankheit, and skelosierende encephalomyelitis.
Male Patients
Female Patients
Approximately 50% of women heterozygous for the adrenoleukodystrophy gene develop pure adrenomyeloneuropathy in middle age or later.16 The onset is later, and progression is slower than in male patients, but disability may be severe, necessitating the use of a wheelchair. Inflammatory brain disease17 and clinically or biochemically demonstrable adrenal insufficiency18 are rare in this disorder. They are present in about 1% of women heterozygous for the adrenoleukodystrophy gene.
Pathology
Inflammatory Cerebral Phenotype
In the inflammatory cerebral forms, there is loss of myelin, which is confluent and usually symmetrical with caudorostral progression.19 The periventricular and central white matter are involved. Cavitation and calcification may be seen. Arcuate fibers are relatively spared. In the most common form with posterior manifestation, the posterior cingulum, corpus callosum, fornix, hippocampal commissure, posterior limb of the internal capsule, and optic systems are typically involved. The cerebellar white matter exhibits similar but milder changes. In about 15% of affected patients, the cerebral pathology begins in the frontal white matter and advances rostrocaudally. Histopathological study reveals marked loss of myelin and, to a lesser degree, axons and a loss of oligodendrocytes in association with hypertrophic reactive astrocytosis. The pathology of the inflammatory cerebral lesions can be subdivided into three zones, which are also demonstrable in imaging studies.20 Zone 1, the advancing active edge of myelin loss, includes areas of intense perivascular inflammation and accumulation of lipid-laden macrophages. In Zone 2, there are large perivascular collections of mononuclear cells, particularly lymphocytes and mostly CD8 cytotoxic cells,21 which are highly characteristic in the area of early breakdown. In Zone 3, there are gliosis, loss of myelin, and variable loss of axons. In the inflammatory cerebral forms, there is also secondary corticospinal tract degeneration extending down through the peduncles, basis pontis, medullary pyramid, and spinal cord.
Pure Adrenomyeloneuropathy
The spinal cord bears the brunt of the disease process in men with pure adrenomyeloneuropathy and also in neurologically symptomatic women heterozygous for the adrenoleukodystrophy gene. Loss of myelinated axons and a milder loss of oligodendrocytes, but with little or no inflammatory response, are observed in the long ascending and descending tracts of the spinal cord.22 The pattern of fiber loss is consistent with a distal axonopathy in that the greatest losses are observed in the distal segments: that is, the cervical region for the ascending fasciculus gracilis and the low thoracic and lumbar segments for the descending corticospinal tract. The number of dorsal root ganglion cells is not reduced. This is indicative of axonal pathology and not neuronal damage as the primary event.23 Mitochondria are abnormal and contain lipid inclusions.24 Peripheral nerve lesions in adrenomyeloneuropathy are variable and mild in comparison with those of the myelopathy. Sural and peroneal nerves have displayed loss of large- and small-diameter myelinated fibers, endoneurial fibrosis, and thin myelin sheaths.25,26 Neurophysiological studies in 18 patients suggest that primary axonal pathology (present in 67%) is the principal abnormality. Only 9% of the patients fulfilled the electrodiagnostic criteria for primary demyelination.27
Adrenal Cortex and Testis
Adrenocortical cells become ballooned as a result of the accumulation of lamellae, lamellar lipid-laden cells, and fine lipid clefts. The striated material, which contains cholesterol esters esterified with very-long-chain fatty acids (VLCFAs), appears to lead to cell dysfunction, atrophy, and death.28 In fetuses affected by X-linked adrenoleukodystrophy (X-ALD), the fetal adrenal zone is already severely involved.29 In the testes, lamellae and lamellar lipid-laden cells are present in the interstitial cells of Leydig and their precursors. Degenerative changes in the seminiferous tubules and Sertoli cells are observed in adrenomyeloneuropathy30 and may eventually lead to azoospermia.31
Neuroimaging
Inflammatory Cerebral Phenotypes
The most common MRI abnormality in the childhood inflammatory phenotype is a pattern that involves the parieto-occipital white matter most severely. It is present in about 80% of affected patients.32–34 Arcuate fibers are relatively spared. After administration of gadolinium-diethylenetriamine pentaacetic acid contrast material, a rim enhancement can be seen surrounding the most severely affected area. Three zones can often be identified.32 The outer advancing zone is in the process of active demyelination without inflammation. Inflammation is present in the second zone. The third zone is completely demyelinated and gliotic, and cavitation and calcification may be present. Demyelination advances in the frontal direction. Structures that are affected relatively early are the lateral and medial geniculate bodies and, in some cases, the lateral-inferior part of the thalamus, the posterior limb of the internal capsule, and the external capsule. In the brainstem, there is involvement of the occipitoparietotemporopontine and pyramidal tracts, the brachia of the inferior colliculus and of the superior colliculus, and the lateral lemniscus. In approximately 15% of patients, the process starts in the frontal area with concomitant involvement of the rostrum and genu of the corpus callosum and of the anterior limb of the internal capsule,32,34 and then it spreads caudally. On occasion, the initial lesions are in the pons and cerebellum.35 The initial lesions may be asymmetrical and mistaken for a brain tumor.36
Proton MRS abnormalities precede changes demonstrable by conventional MRI37–39 and can be predictive of outcome; they aid in the selection of patients for hematopoietic cell transplantation (HCT) therapy and in the evaluation of therapies.40 Reduction of N-acetyl aspartate levels is the earliest change; increases in the choline peaks are correlated with the extent of the demyelinative process.
Adrenomyeloneuropathy
Conventional brain MRI usually appears normal in patients with “pure” adrenomyeloneuropathy. Abnormalities may be seen in the corticospinal and frontopontine tracts and have been interpreted as centripetal extension of the distal axonopathy.34,41 Brain MRS studies in patients with pure adrenomyeloneuropathy have revealed evidence of diffuse axonal pathology.42 Studies of the spinal cord with conventional MRI have demonstrated diffuse atrophy mainly in the lower thoracic cord.41 The magnetization transfer MRI appears to be a sensitive method for the quantitation of structural abnormalities in adrenomyeloneuropathy.43
Genetics
The mode of inheritance is usually classified as X-linked recessive, but the fact that 50% of heterozygous women are neurologically symptomatic indicates that a designation as X-linked is more accurate, as proposed by Dobyns and associates.44 The disease incidence is estimated at 1 per 17,000 and appears to be the same in all ethnic groups.45 The defective gene has been mapped to Xq28.46 The gene codes for a peroxisomal membrane protein, adrenoleukodystrophy protein (ALDP).47 ALDP is a member of the adenosine triphosphate-binding cassette (ABC) transporter superfamily.48 Forty-eight mammalian ABC transporters are estimated to exist. They transport a variety of substrates, including ions, sugars, amino acids, proteins, and lipids.49 Four mammalian peroxisomal ABC transporters have been identified and are designed as subgroup ABCD. The gene deficient in X-ALD has been assigned the name ABCD1. ABCD2 may also be relevant to X-ALD (see section on therapy). ABCD2, which has been mapped to 12q11, codes for the adrenoleukodystrophy-related protein ALDR, which has 66% homology with ALDP50 and can substitute for some of the functions of ALDP.51,52
More than 500 mutations in ALDP have been identified in patients with X-ALD53 and are updated in the X-linked Adrenoleukodystrophy Database website (www.x-ald.nl). Fifty-seven percent of the mutations are nonrecurrent, and many are unique to a kindred. Of all the mutations, 55.9% were found to be missense mutations, 27.1% nonsense mutations, 3.9% small in-frame amino acid insertions or deletions, and another 3.9% large deletions of one or more exons.53 No correlation between phenotype and genotype has been demonstrated. A single mutation may be associated with a wide range of phenotypes.54 It is common for widely varying phenotypes to co-occur in the same family. The action of a modifier gene has been postulated.55,56
Diagnosis
Definitive diagnosis of X-ALD depends on the demonstration of biochemical abnormalities and on results of mutation analysis. Demonstration of characteristic abnormalities in the levels of saturated VLCFAs in plasma is the most frequently used test.57 Statistically significant increases in the levels of hexacosanoic acid (C26:0) and in the ratios of C26:0 to C24:0 and to C22:0 are present in more than 99% of male patients with adrenoleukodystrophy and in approximately 85% of women heterozygous for the X-ALD gene.58 Increases in VLCFA levels in patients with other peroxisomal disorders, such as Zellweger’s syndrome, neonatal adrenoleukodystrophy, and infantile Refsum’s disease,59 can usually be distinguished easily on the basis of clinical manifestation and tests of the other peroxisomal functions. Patients on the ketogenic diet may produce false-positive results60; VLCFA levels are also increased in cultured skin fibroblast,61 and study of cultured fibroblasts can be helpful when results of plasma samples are equivocal. VLCFA levels are also increased in red blood cell membranes,16,62 in leukocytes,63 and in cultured amniocytes64 and chorionic villus cells.65
The plasma VLCFA assay57 is highly reliable for the identification of affected male patients. Abnormalities are already present on the day of birth.57 There are two reports of false-negative test results in a total of three male patients with X-ALD,66,67 but this has not observed in more than 100 affected male patients.57 In contrast, false-negative results occur in 15% to 20% of women heterozygous for the X-ALD gene. Because of this, mutation analysis is much preferred for the determination of carrier status in women at risk for X-ALD. More than 500 pathogenic mutations in the ABCD2 gene have been identified in patients with X-ALD53 and are updated in the X-linked Adrenoleukodystrophy Database website. Reliable methods for their identification in affected men and in heterozygous women have been published.68 The first step is to define the nature of the mutation in affected male family members or in a family member who is an obligate carrier. Once the mutation is found in a nuclear family member or a more distant relative, it can be determined whether this mutation exists in lymphoblasts or cultured fibroblasts of the at-risk person. VLCFA analysis in cultured amniocytes or chorion villus samples is a valuable technique for the identification of affected fetuses.65 However, false-negative results have been reported,69,70 and confirmation by mutation analysis should be performed when the nature of the mutation has been defined in an affected male patient or obligate heterozygote family member.
Pathogenesis
Role and Pathogenesis of Very-Long-Chain Fatty Acid Excess
The abnormal accumulation of saturated VLCFAs is the principal biochemical abnormality in X-ALD.14,71 There is considerable evidence that this contributes to pathogenesis, according to studies in model membranes72 and in cultured human adrenal cells73 and correlative studies in postmortem brain74 and adrenal tissue.75 Excess saturated VLCFAs are normally oxidized in the peroxisome.76 This process is impaired in cultured skin fibroblasts, leukocytes, and amniocytes of patients with X-ALD.77 The defect in X-ALD was localized to the first step: namely, the formation of the coenzyme ester of VLCFA acids.78,79 This reaction is catalyzed by VLCFA-coenzyme A synthetase (VLCS).80 VLCS structure and levels are normal in patients with X-ALD and in the X-ALD animal model.81–83 ALDP, the protein that is deficient in X-ALD, is required for the oxidation of VLCFA in fibroblasts of patients with X-ALD.51,52,84 ALDP has no homology with VLCS; as already noted, it is a member of the ABC transporter superfamily.48 The mechanism by which ALDP influences VLCFA metabolism is complex and not yet understood.85 Adding to the complexity are findings of several earlier86 and more recent studies87 that suggest that VLCFA synthesis is also increased in X-ALD.
Pathogenesis of Pure Adrenomyeloneuropathy
The studies of Powers and associates established that the primary defect in pure adrenomyeloneuropathy is a noninflammatory axonopathy that affects most severely the distal portions of the dorsal columns and corticospinal tracts in the spinal cord.22,23 The demyelination appears to be secondary to the axonal damage. The mechanism responsible for the axonal dysfunction has not been defined. It is postulated that the VLCFA excess destabilizes the axonal membrane or impairs the function of trophic factors.23 Mitochondrial abnormalities23 and oxidative stress88 have been proposed as contributing to pathogenesis. The pathology and clinical features in the mouse model of X-ALD resemble those of adrenomyeloneuropathy,89 and further studies in this model may increase understanding of the pathogenesis of adrenomyeloneuropathy.
Pathogenesis of Inflammatory Cerebral Forms of X-Linked Adrenoleukodystrophy
The most generally accepted concept for the pathogenesis of the inflammatory response is that the accumulation of VLCFA has an adverse effect on myelin and oligodendrocyte stability and function, which renders them vulnerable to various other adverse events (a “second hit”), which in turn initiates a destruction cascade that results in the death of oligodendrocytes and rapid destruction of myelin.90 Cytokines, such as tumor necrosis factor α, play an important role.91 Apoptosis of oligodendrocyte has been reported.92 Ito and colleagues21 demonstrated that most of the perivascular cells that accumulate in the active demyelinating lesions are CD8 cytotoxic cells. They concluded that oligodendrocyte death was lytic/cytotoxic rather than apoptotic. There was strong CD1 immunoreactivity in astrocytes and microglia. CD1 molecules are antigen-presenting glycoproteins that, unlike the major histocompatibility complex proteins, can present self-lipid antigens to T-cells.93 This has led to the intriguing hypothesis that VLCFA-containing complex lipids, which are present in brain tissue from patients with X-ALD but not in normal brain tissue, act as triggers for the autoimmune response in patients with the inflammatory adrenoleukodystrophy phenotype. Asheuer and associates94 demonstrated decreased expressions of an ABCD transporter gene (ABCD4) and a VLCS (BG1) in patients with X-ALD and their susceptibility to the inflammatory phenotype. This is the first demonstration of a possible genotype-phenotype correlation in X-ALD.
Therapy
Steroid Replacement Therapy
The importance of evaluating adrenal function and providing appropriate replacement therapy cannot be overemphasized. Steroid replacement therapy in general does not alter neurological progression, except possibly in some patients with adrenomyeloneuropathy,95 but it can improve strength and well-being and may be lifesaving. Most male patients with X-ALD have increased plasma adrenocortical hormone and impairment of cortisol responsiveness to a 0.25-mg intravenous dose96 of cosyntropic (Cotrosyn) after 60 minutes. The authors and colleagues recommend that at least one of these tests be performed yearly. Isolated measurements of plasma cortisol levels are insufficient and may lead to the false conclusion that adrenal insufficiency has been ruled out. Glucocorticoid dose requirements are generally the same as those used for other forms of primary adrenal insufficiency.97 To mimic diurnal rhythms of physiological cortisol secretion, adult patients receive 25 mg of cortisone acetate or 20 mg of hydrocortisone in the early morning and a smaller second dose, 12.5 or 10 mg, respectively, in the late afternoon. The dosage in children is 5 to 10 mg per 24 hours. Not all require mineralocorticoid replacement. When postural hypotension, hyponatremia, or hyperkalemia does persist despite adequate glucocorticoid therapy, fludrocortisone, 0.05 mg to 0.1 mg/day, is prescribed.
Hematopoietic Cell Transplantation
HCT is an important therapeutic modality for the cerebral inflammatory forms of X-ALD. Either bone marrow cells or umbilical cord cells are used.98 Aubourg and coauthors99 were the first to report stabilization and reversal of neurological manifestation in an 8-year-old boy who had early evidence of cerebral involvement. This patient was healthy 10 years later (P. Aubourg, personal communication, July 2000). Shapiro and associates100 reported long-term stabilization in patients who had received bone marrow cells. Peters and colleagues98 provided follow-up on 126 patients with X-ALD who received HCT. This study provided important guidelines in regard to the selection of patients for HCT. The overall 5-year survival rate was 56%, compared with an estimated 45% rate among untreated patients. The rate of transplantation-related mortality was 14%. Outcome with respect to mortality, morbidity, and quality of life was unsatisfactory for patients who were already severely ill at the time of HCT, and the procedure is not recommended for them. In contrast, the 5-year survival rate was 92% in a group of patients whose neurological involvement (performance IQ > 80) and neuroradiological involvement (Loes MRI score < 9, range = 0-34)101 were still relatively mild, and the procedure is recommended for them. Peters and colleagues98 provided a more detailed discussion of the indications for HCT. At this time, the procedure is not recommended for asymptomatic patients with normal MRI findings or for those with pure adrenomyeloneuropathy. The mechanism of the beneficial effect of bone marrow cell transplantation is incompletely understood.14
Dietary Therapy with Lorenzo’s Oil
Lorenzo’s oil is a 4:1 mixture of glyceryl trioleate and glyceryl trierucate. It has the remarkable biochemical effect of normalizing the plasma levels of VLCFA in patients with X-ALD within 4 weeks, probably by inhibiting endogenous VLCFA synthesis.102 Investigators reported that administration of Lorenzo’s oil to asymptomatic boys who had normal brain MRI findings significantly reduced their risk of developing childhood cerebral X-ALD.103 It was recommended that asymptomatic patients with normal brain MRI findings who are younger than 8 years be placed on a carefully supervised program of Lorenzo’s oil and dietary therapy. Adrenal function and brain MRI appearance must be monitored at 6-month intervals. Lorenzo’s oil does not alter significantly the neurological progression in patients with X-ALD who already have evidence of inflammatory cerebral involvement104,105 and in adrenomyeloneuropathy.106 However, results of a single cohort study suggested that it may slow the progression of adrenomyeloneuropathy in patients who did not have evidence of cerebral involvement.107 A double-blind placebo-controlled study of Lorenzo’s oil therapy in men and women with pure adrenomyeloneuropathy is now in progress.
Other pharmacological therapeutic approaches have shown therapeutic potential in preclinical studies and preliminary clinical trials. These include 4-phenylbutyrate,51 lovastatin,108,109 and lovastatin with arginine butyrate.110
Gene replacement therapy is under active investigation. Preclinical studies have shown encouraging results.111,112
METACHROMATIC LEUKODYSTROPHY
Clinical Manifestations
There are late infantile, juvenile, and adult manifestations. In about 75% of patients, the late infantile and juvenile forms are equally divided in frequency. The other 25% have the adult form.113
Late Infantile Form
This form manifests between 6 months and 4 years of age and has been subdivided into four clinical stages.114 In clinical stage I, there is hypotonia of the legs or of all four limbs. If the child is walking, the gait becomes unsteady. Deep tendons are diminished or absent. This stage lasts for a few months to more than a year. In stage II, the child can no longer stand and shows mental regression. Speech deteriorates as a result of dysarthria and aphasia. Nystagmus, ataxia, and truncal signs develop, and muscle tone in the legs is increased. This stage lasts only a few months. In stage III, the flaccid paresis is superseded by spastic tetraplegia with pathological reflexes and extensor plantar reflexes, and the child becomes bedridden. Feeding difficulties and episodes of airway obstruction occur. About 25% of patients develop epileptic seizures. In stage IV, the patients enter a decerebrate state and lose all contact with their surroundings. Death usually occurs about 5 years after onset of clinical symptoms.
Adult Form
Onset of adult MLD has been reported at varying ages up to age 63.115 Patients show gradual decline in intellectual abilities. They become emotionally labile and show memory deficits, disorganized thinking, behavioral abnormalities, or psychiatric symptoms such as hallucinations or delusions.116 Clumsiness of movement and urinary and sometimes fecal incontinence are also present. A progressive spastic paresis of the arms and legs develops, with increased tendon reflexes and extensor plantar reflexes. Ataxia and extrapyramidal symptoms and dystonia may be present, as well as optic atrophy and signs of bulbar dysfunction. Peripheral neuropathy is often absent.117
Pathology
The pathology of MLD in the nervous system is characterized primarily by demyelination and deposits of metachromatic granules in the central and peripheral nervous systems. High-resolution electron microscopy of the storage granules revealed that the inclusions are surrounded by a membrane, which suggests that they are located in the lysosome.115 The central white matter is reduced in amount, is firm, and in severely affected regions may show cavitation of spongy degeneration. The subarcuate fibers are usually spared. There is moderate to severe loss of myelin sheaths. The intrafascicular oligodendrocytes are reduced in number. There is a striking accumulation of metachromatic granules in macrophages that are prominent in perivascular spaces and also in oligodendrocytes and in neurons. Immunocytochemical studies have shown that the accumulated material in neurons and glial cells contains sulfatide.118 The cerebellum is atrophic with severe demyelination, prominent gliosis, storage granules, and a reduction of Purkinje cells. The peripheral nervous system shows segmental demyelination. Metachromatic granules are present in Schwann cells and in endoneurial macrophages.
Neuroimaging
MRI reveals periventricular white matter abnormalities with a symmetrical distribution.119 In juvenile and adult cases, there is preferential frontal lobe involvement. The arcuate fibers are relatively spared. The corpus callosum is invariably affected. The posterior limb of the internal may be involved. Brainstem lesions in the pyramidal tract may be observed, and the cerebellar white matter may be involved. Contrast enhancement is usually absent; this helps differentiate MLD from X-ALD, in which enhancement is often prominent. Diffusion-weighted images vary with the stage of the illness. Oguz and associates reported that diffusion was restricted in the early stage and increased in the later stage.120
MRI in MLD often exhibits radially oriented hypointense stripes in T2-weighted hyperintense areas. Postmortem studies in which neuroimaging patterns were correlated precisely with histopathological findings revealed that the stripes corresponded to perivenular zones in which there was relative preservation of myelin, together with the accumulation of lipid-laden glial cells.121
Genetics
The mode of inheritance is autosomal recessive. The incidence of late infantile MLD varies in different countries.115 MLD appears to be most common in northern Sweden, where the incidence is estimated to be 1 per 40,000 among some groups, and among Arabs living in Israel. In France and in Germany, the incidence was estimated to be 1 per 130,000 to 1 per 170,000.
The defective gene has been mapped to locus 22q13. It codes for arylsulfatase A, which catalyzes the desulfation of 3-sulfogalactosyl-containing glycolipids. Four such glycolipids have been identified.115 Of these, galactosylceramide-3-sulfate is present in the highest concentration and most relevant to MLD.
Metachromatic Leukodystrophy-Causing Mutations
Sixty-three MLD-causing mutations have been identified. The mutations can be subdivided into two general categories: the null alleles, in which the mutation does not allow synthesis of any function, and the R-alleles, in which there are low amounts of residual enzyme activity. Null alleles tend to be associated with the late infantile phenotype, R-alleles with the adult phenotype, and heterozygosity for null and R-alleles with the juvenile phenotype. However, there is considerable phenotypical variability of severity when patients with the identical genotype are compared.122
Saposin B Deficiency
A small number of patients with clinical features that resemble those of MLD have a deficiency of saposin B, the sulfatide activator protein. These patients have increased levels of sulfatide in tissues and urine, but arylsulfatase A activity is normal. Nine cases have been reported and include patients with late infantile, juvenile, and adult onset.123 The molecular defects involve homoallelic point mutations that destroy a glycosylation site.
Multiple Sulfatase Deficiency
In multiple sulfatase deficiency, the catalytic activity of 12 sulfatases, including arylsulfatase A, is impaired. More than 50 cases have been described.124 The clinical manifestation resembles that of classic MLD but with certain additional features such as mildly coarse features, dysostosis multiplex, stiff joints, ichthyosis, hydrocephalus, deafness, and enlarged liver, attributable to defects in one of the other sulfatases. The gene defect leads to reduced activity of a post-translational system that generates an α-formyl glycine residue from a thiol group of an active site cysteine that is conserved in all members of the mammalian sulfatase family.
Diagnosis
The laboratory diagnosis of MLD depends on the demonstration of decreased arylsulfatase A activity and increased excretion of sulfatide in urine. The arylsulfatase A assay is performed in peripheral blood leukocytes or cultured fibroblasts.125 The major problems in the enzymatic diagnosis is the frequent occurrence of the clinically benign pseudodeficiency alleles, which reduce arylsulfatase A activity. Measurement of urinary sulfatide excretion is key to the distinction between MLD and pseudodeficiency. This excretion is markedly increased in patients with MLD and normal in patients with pseudodeficiency or in persons heterozygous for the MLD-causing mutation.126 MLD and pseudodeficiency can also be distinguished by the sulfatide loading assay.127 Patients with multiple sulfatase are distinguished by demonstration of deficient activity of arylsulfatase B and C, in addition to the defect of arylsulfatase A. Patients with saposin B deficiency have normal arylsulfatase A activity, but their sulfatide loading value and urinary sulfatide excretion are increased. Mutation analysis is of great value for the identification of persons heterozygous for the MLD-causing mutation. It detects the pseudodeficiency. However, it must be kept in mind that alleles bearing the relatively common pseudodeficiency polymorphism may, in addition, contain an MLD-causing mutation.
Pathogenesis
The accumulation of sulfatide, secondary to the deficiency arylsulfatase A, is the principal biochemical abnormality in MLD. The sulfatides accumulate mainly within the lysosome. The sulfatide accumulation is noted in several extraneural tissues, such as the gallbladder, liver, kidneys, pancreas, adrenal cortex, and sweat glands, but it leads to pathological changes only in the gallbladder.
The demyelination that occurs in MLD appears to be secondary to sulfatide-induced changes within the cells responsible for myelin maintenance: namely, the oligodendrocytes in the CNS and the Schwann cells in the peripheral nervous system.129 Changes in the subcellular organelles of these cells are observed before any morphological abnormalities in the myelin sheaths associated with them are detectable.115 Investigators who used immunocytochemical techniques have demonstrated the accumulation of sulfatides in neurons and glial cells in arylsulfatase A-deficient mice.118 The degeneration of neurons and glia is enhanced and accelerated by the secretion of proinflammatory cytokines by monocytes.130
Other pathogenetic mechanisms that may contribute are impaired myelin stability secondary to its abnormal biochemical composition131 and the abnormal accumulation of sulfogalactosylsphingosine (lysosulfatide).132 Lysosulfatide has been shown to inhibit the activity of protein kinase C and cytochrome oxidase at concentrations far below those found in tissues of patients with MLD.132,133
Therapy
Bone marrow transplantation is under investigation as a therapy for MLD. It has not been effective in symptomatic patients with the late infantile form of the disease and may even accelerate the progression of the disease.134 Several reports suggest that bone marrow transplantation arrests the progression in patients with the juvenile or adultonset MLD phenotype.135–137 Evaluation of these results is difficult because of the relatively short follow-up and the variability of progression in untreated patients. Several presymptomatic patients have undergone transplantation.138,139 Longer follow-up is necessary to assess effectiveness.
Therapeutic studies in the mouse model of MLD are highly encouraging. Ex vivo administration of genetic modification of hematopoietic stem cells led to a remarkable correction of neuropathological changes.140 Matzner and coauthors reported the unexpected and surprising findings that intravenous administration of purified arylsulfatase A improved nervous system pathology and function.141
GLOBOID LEUKODYSTROPHY
Clinical Features
The infantile form of the disease is most common. Hagberg and associates subdivided the course of the disease into three stages.142 In stage I, the child, apparently normal during the first few months after birth, becomes hyperirritable and hypersensitive to auditory and tactile stimuli, and there is some stiffness of the limbs. Slight regression of psychomotor development, feeding difficulties, and vomiting may be observed. Early peripheral nervous system manifestations are common.143 In stage II, there is rapid deterioration. There is marked hypertonicity, with extended and crossed legs, flexed arms, and arched back. Optic atrophy and sluggish pupillary reflexes are common. Stage III is the “burnt-out” stage, sometimes reached within few weeks or months. The child is in a decerebrate state, is blind, and has no contact with his or her surroundings. Patients rarely survive for more than 2 years.
Juvenile- and adultonset cases are well documented. The juvenile cases have been subdivided into the late infantile (early childhood)-onset form, which begins at ages 6 months to 3 years, and the late childhood-onset form, which begins at ages 3 to 8 years.144 The early childhood form progresses rapidly, with death approximately 2 years after onset. The late childhood cases manifest with loss of vision and with hemiparesis, ataxia, and psychomotor regression; death occurs 10 months to 7 years after onset. The number of reports of adultonset cases is increasing. Progressive paraparesis and tetraparesis with demyelination in the spinal cord are often the main abnormalities. One female patient developed slowly progressive paraparesis at 38 years of age.145 Other cases were misdiagnosed as amyotrophic lateral sclerosis.146
Pathology
The pathology is confined mainly to the nervous system.147 Postmortem examination reveals that the brain is small and atrophic, with shrunken gyri and widened sulci. The major histopathological changes are demyelination, gliosis, and the presence of unique macrophages, the globoid cells in the white matter. The subarcuate fibers tend to be spared. The phylogenetically newer fiber tracts are usually more involved in the demyelinating process. In the spinal cord, the pyramidal tracts are more affected than are the dorsal columns. The oligodendroglial cell population is severely diminished in the areas of demyelination. Globoid cells are clustered around blood vessels and may be multinucleated. They contain tubular inclusions that have morphological similarities to the inclusions of pure galactosylceramide.148 Secondary axonal degeneration is a consistent finding. The peripheral nerves are commonly affected. They show endoneurial fibrosis, proliferation of fibroblasts, and segmental demyelination. Inclusions similar to those in the globoid cells in the brain are found in the cytoplasm of histiocytes, macrophages, and Schwann cells.
Neuroimaging
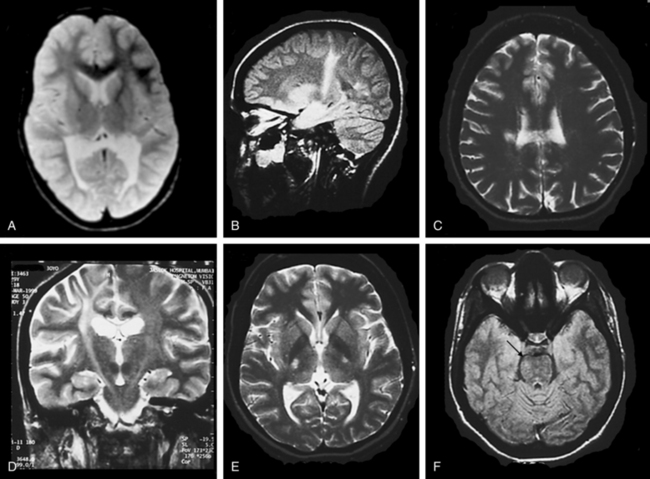
In the early stages, computed tomographic changes may be more evident than MRI changes. In stage I, computed tomographic scans may be normal or exhibit symmetrical increased density in the thalami, the corona radiata, and the posterior limb of the internal capsule and basal ganglia.149 MRI confirms the presence of white matter abnormalities with relative sparing of arcuate fibers in infantile-onset GLD (Fig. 80-1). The posterior limb of the internal capsule, corpus callosum, cerebellar white matter, and brain tracts are also involved. In cases of adultonset GLD, both computed tomography and MRI reveal predominant involvement of the occipital periventricular white matter with extension in the parietal and temporal direction and associated involvement of the splenium of the corpus callosum, a pattern that resembles that seen in X-ALD. In adultonset cases, symmetrical high-signal intensity lesions may be evident on T2-weighted MRI in frontoparietal white matter, the centrum semiovale, and the posterior limb of the internal capsule, with sparing of the periventricular white matter (see Fig. 80-1).145 MRS in patients with infantile-onset GLD revealed pronounced elevation of myoinositol- and choline-containing compounds, which reflected gliosis and demyelination. N-acetyl aspartate levels were decreased, which reflected neuroaxonal loss.150
Genetics
The mode of inheritance is autosomal recessive. The gene has been mapped to loci 14q24.3-32.1. It codes for galactocerebroside (psychosine) β-galactosidase. The incidence is estimated to be 1 per 100,000 in the United States but appears to be higher in the Scandinavian countries. More than 60 mutations have been identified.147,151 Although there is no consistent correlation between genotype and phenotype,152 some associations have been identified. The 502T/del mutation accounts for 50% of mutant alleles in The Netherlands and 75% in Scandinavian countries. The mutation probably occurred initially in Sweden and traveled from there throughout Europe, Asia, and the United States. This 30-kilobase deletion eliminates all of the coding region of one of the subunits of the enzyme, and patients who are homozygous for this deletion have the severe infantile-onset phenotype. Two other mutations, C1538T and A1652T, account for about 10% to 15% of mutant alleles of infant patients with European ancestry. The G809A mutation is common in patients with adultonset GLD. One copy of the G809A mutation is probably enough to explain a mild phenotype in heterozygous patients whose other allele has a mutation that usually is associated with a severe phenotype.147 A series of polymorphic changes in the gene that occur on the same allele as disease-causing mutations have been identified; they influence the enzyme activity and clinical manifestations in persons who are heterozygous for GLD-causing mutations.153
Diagnosis
Assays of GalC activity in peripheral leukocytes or cultured fibroblasts is the most reliable method for diagnosis.154 Leukocyte and cultured fibroblast results are equally reliable. Wenger and colleagues147 listed citations for validated techniques that use natural substrates. The same assay in cultured amniotic cells or in biopsy specimens of chorionic villi is used for prenatal diagnosis, but it is valuable for measuring the GalC enzyme activity in the carrier parents beforehand, because some carriers have enzyme levels that are sufficiently low to characterize them as homozygous affected. When this is the case, special care must be taken in the interpretation of the results of the prenatal study, and the results can be complemented by mutation analysis. The wide range of enzyme activity in normal individuals and in carriers, which results mainly from polymorphic amino acid changes that cause a wide range in the values tested, limits the reliability of the enzyme assay for carrier identification. In families in which the mutations have been defined, carrier status is determined by mutation analysis. An important diagnostic development is the progress that has been made toward mass neonatal screening for lysosomal disorders, including GLD,155–157 Neurophysiological studies, such as motor conduction velocity and visual and auditory evoked responses are abnormal in most patients with GLD,158 but it is important to realize that they may be normal in patients with the adultonset phenotypes.159
Pathogenesis
The globoid cell, the hallmark pathological feature of GLD, is the consequence of the accumulation of galactosylceramide. Galactosylceramide is unique among sphingolipids in its ability to elicit the globoid cell reaction when injected into normal rat brain.160 Even though the galactosylceramide thus plays a role in some aspects of the pathogenesis of GLD, there is increasingly compelling evidence that it is the accumulation of psychosine (sphingosine-galactose) that is the principal pathogenetic factor. This pathogenetic mechanism, now referred to as the psychosine hypothesis, was first proposed by Miyatake and Suzuki161 and reexamined in detail 25 years later by Suzuki.162 GalC, the gene product that is deficient in GLD, catalyzes the degradation of both galactosylceramide and psychosine. Psychosine is present in very low concentration in normal brain tissue, but its concentration in GLD white matter is increased 100-fold.147
Psychosine with its free amino group is highly cytotoxic. Its effect is mediated by caspase activation.163 Psychosine-induced apoptosis is a mouse oligodendrocyte precursor line mediated by caspase activity. Oligodendrocytes are selectively destroyed because psychosine formation occurs primarily in these cells. The molecular mechanisms of psychosine-induced cell death have been clarified.164 Psychosine-induced apoptosis in human oligodendrocyte cell line studies in animal models, particularly the canine model and the twitcher mouse, have been of great value for the studies of pathogenesis of GLD and its therapy.165
Therapy
HCT and bone marrow transplantation for GLD are under intense investigation. HCT was of benefit in four patients with adultonset GLD: CNS deterioration was reversed, MRI findings improved in three patients, and levels in cerebrospinal fluid (CSF) diminished.166 The effect of HCT in patients with infantile-onset GLD and in asymptomatic infants with GLD has been evaluated.167 Although the symptomatic boys with infantile GLD experienced no benefit, there was strong evidence of a beneficial effect in 11 asymptomatic newborns who received umbilical cord blood transplants at 12 to 44 days after birth. At the time of follow-up (median age, 3.0 years) they had age-appropriate cognitive function and receptive language skills, and serial MRI studies demonstrated progressive central myelination. There is no doubt the procedure has a profound effect with regard to mortality and early development, but there were mild-to-moderate delays in expressive language and gross motor function. Additional follow-up is needed to assess the long-range outcome. Ethical aspects of this therapeutic approach were discussed in an editorial.168
Another approach, therapy aimed to reduce the concentrations of substrate, is being tested in experimental animals.169 Studies of transplantation of neural cells and direct infection of the brain with viral vector containing GalC, are in progress in experimental animals.
PELIZAEUS-MERZBACHER DISEASE
Pelizaeus-Merzbacher disease is known as several entities: classic Pelizaeus-Merzbacher disease,170 connatal Pelizaeus-Merzbacher disease,171,172 X-linked spastic paraparesis type 2 (SPG2),173,174 proteolipid protein (PLP) null syndrome, and pure spastic paraplegia.
Clinical Features
Classic Pelizaeus-Merzbacher Disease
This disorder is characterized by abnormal eye movements (horizontal or rotatory) with onset during the first months of life; psychomotor deterioration before 2 years of age; and the appearance of bilateral pyramidal tract signs, dystonia, and often ataxia during the first years of life. Laryngeal stridor and optic atrophy are frequent.170
Connatal Pelizaeus-Merzbacher Disease
This disorder is characterized by nystagmus at birth, pharyngeal weakness, stridor, hypotonia, severe spasticity, and seizures.174
Complicated Spastic Paraplegia
This disorder is characterized by nystagmus, ataxia, and spastic gait, but little or no cognitive impairment.174
Pathology
Features include lack of myelin in all parts of the CNS. Islets of preserved myelin around blood vessels in deeper parts of brain produce the so-called tigroid pattern. Axons are relatively preserved. Oligodendrocytes are reduced in number, as is the number of cytoplasmic processes. A length-dependent axonal degeneration is also present.173
Neuroimaging
MRI reveals an arrest of myelination. In some cases, no myelin appears to be present; in other cases, myelin is present in parts of the brainstem, cerebellar white matter, the posterior limb of the internal capsule, the thalamus, and the globus pallidus. The pattern described is normal for a neonate or an infant in the first few months of life, although not normal at the age of the patient. The white matter may appear speckled, reflecting the tigroid pattern seen pathologically. There also is evidence of neuroaxonal injury, based on the demonstration of significant and widespread reduction in brain N-acetyl aspartate levels.176
Genetics
The mode of inheritance is X-linked recessive. Some women heterozygous for Pelizaeus-Merzbacher disease develop neurological progressive paraparesis.175,177,178 The gene maps to locus Xq22 and codes for PLP. This protein makes up approximately 50% of myelin protein weight. It is composed of four helices that span the cell membrane. Portions of the protein chain extend into the extracellular space, where they have a homophilic interaction with PLP molecules in adjacent spirals of the cell membrane.179 This region is visualized as the intraperiod line on electron microscopy.180
Duplication of the region surrounding locus Xq22 accounts for the majority, perhaps up to 70%, of the mutations in PLP.181 These duplications arise from sister chromatid exchange. Striking variations in the breakpoints and in the size of the duplication occur.182,183 Most patients with mutations have the classic phenotype182; some have the connatal form,184 and others have the mild SPG2 phenotype. Deletions of the PLP locus occur only rarely.185,186 Patients in whom no PLP protein product is formed, referred to as null mutations, tend to have a mild phenotype with relatively mild spastic paraparesis, which progressed during adolescence and (unlike that in classic Pelizaeus-Merzbacher disease) was associated with demyelinating peripheral neuropathy.173 About 20% of patients have point mutations at the PLP locus that alter the amino acid sequence of the PLP/DM20 proteins. Approximately 100 distinct mutations have been discovered to date. The majority of abnormal PLP/DM20 proteins result in severe phenotypes through a toxic gain of function, whereas the remainder result in a milder form that is associated with loss of function.
Diagnosis
Although the classic phenotype has a relatively distinct clinical manifestation,170 the connatal form can be confused with motor neuron disease or spinal muscular atrophy. The mildest forms merge clinically with the syndromes of SPG2. MRI is essential for the evaluation of individuals with clinical signs and symptoms of classic Pelizaeus-Merzbacher disease or SPG2. Virtually all patients with Pelizaeus-Merzbacher disease eventually have MRI findings consistent with a leukodystrophy.187,188
A search for proteolipid protein 1 (PLP1) gene duplication is the most efficient initial screening test. Both interphase fluorescent in situ hybridization and quantitative polymerase chain reaction should be used.182 If neither method yields a positive result, direct sequencing of the PLP1 gene should be performed.189 Prenatal diagnosis has been accomplished.190
Pathogenesis
Because mutations in which no PLP1 protein is synthesized lead to the mildest disease, it has been postulated that toxic effects are caused either by overexpression of the gene in patients with duplications of the normal gene or by the presence of mutant genes. In patients with the gene duplications, excessive amounts of PLP1 have been shown to accumulate in the late endosomal and lysosomal compartments. PLP1 normally is associated with cholesterol and other lipids to form “lipid rafts” that traffic through the Golgi compartment. In rodent models of Pelizaeus-Merzbacher disease in which there is an excess of PLP1, these lipid rafts are shunted to the late endosomal and lysosomal compartments, effectively draining myelin lipids from the Golgi compartment.191 In patients with mutations in the PLP1 coding region, the differences in clinical severity appear to be related to differential effects of the mutation on protein folding and trafficking.192 These unfolded proteins accumulate in the endoplasmic reticulum, and this leads to the unfolded protein response.193,194 This response, which involves the increased expression of unfolded protein response effector genes, protects the cell against metabolic stress to preserve or reestablish homeostasis. In cells in which homeostasis cannot be maintained, there often is apoptosis by activation of caspase cascades.195 This pathogenetic mechanism is presumed to be operative in oligodendrocytes and neurons. The understanding of the pathogenesis of Pelizaeus-Merzbacher disease has been aided greatly by studies in animal models of the disease.196 These include the “jimpy” mouse and the “jimpy-msd” mouse, in which the disorder resembles classic Pelizaeus-Merzbacher disease; the “jimpy-4j” mouse, in which the disorder resembles connatal Pelizaeus-Merzbacher disease; and the “rumpshaker” mouse, which is a model of SPG2. The myelin-deficient rat also provides a model of connatal Pelizaeus-Merzbacher disease. Because rats are larger animals, this model is well suited for neurophysiological studies of pathogenesis and for the study of therapeutic interventions.197,198
CANAVAN’S DISEASE
Clinical Features
Age at onset is most commonly between 3 and 6 months. In the patients with early onset, mental changes are severe, and the clinical phenomena of nervous system maturation are not observed.199–203 Instead, there is increasing difficulty in feeding, progressive lethargy, and increasing stiffness of the limbs. When the disease begins later in infancy, normal development of social, motor, and visual responses is seen at first, but this is lost as the disease advances. Hypotonia is present early in the course of the disease and is followed by increased tone that begins in the legs and produces a posture of extension. Optic atrophy occurs in a large proportion of cases. Macrocephaly (>90th percentile) was documented in 54 of 59 children; 51 of 58 had nystagmus, 38 of 60 had epilepsy, and optic atrophy was present in 17 of 60.202 Although cases with later onset have been reported,204,205 only two cases have been biochemically confirmed,206 and in these patients, onset of symptoms occurred by 2 years of age. Although most patients die during the first decade, 14 of the 60 patients in Traeger and Rapin’s series202 were alive in the second or third decade, albeit all severely disabled.
Pathology
The majority of affected infants show an increase in brain size and weight. In formalin-fixed sections, the white matter is soft, gelatinous, and darker than normal; enlargement of ventricles increases as the disease advances. The most striking microscopic change is the widespread vacuolization, which characteristically involves the lower layer of the cerebral cortex and the subcortical white matter; the more central zones are relatively or entirely spared. There is a widespread lack of myelin, which involves the area of spongy degeneration and extends far beyond them. In the areas of spongy degeneration, as well as in the demyelinated zones, oligodendrocytes are preserved. Axis cylinders in the areas of demyelination are diminished but not absent. Throughout the cerebral cortex and basal ganglia, there are conspicuous increases in the number and size of protoplasmic astrocytes.201 Ultrastructural studies have shown that the vacuoles in the subcortical white matter lie within the myelin sheaths, between split lamellae of the myelin spirals. The split was noted between the major dense lines.207 Some of the membranes were focally ruptured. The vacuoles communicated through these ruptured membranes into the widened extracellular spaces. Cell membranes of protoplasmic astrocytes were also disrupted. The mitochondria within the astrocytes displayed enormous elongations and contained distended and distorted cristae.
Neuroimaging
In all stages of the disease, MRI shows the most severe abnormalities in the subcortical white matter of the cerebrum and cerebellum. Central white matter structures such as the periventricular rim of white matter, internal capsule, corpus callosum, and brainstem are preserved longer. After several years, cerebral atrophy with enlargement of the ventricles ensues.208 The most characteristic neuroimaging abnormality is the accumulation of N-acetyl aspartic acid, demonstrable by proton spectroscopy.209
Genetics
The mode of inheritance is autosomal recessive. The defective gene maps to chromosome 17pter and has been isolated.210 It codes for aspartyl acylase, which hydrolyzes N-acetyl aspartate to aspartate and acetate. Canavan’s disease occurs most frequently in the Ashkenazi Jewish population, in which carrier frequency is 1 per 38.211 Two mutations are most common among Jewish patients: a missense mutation in codon 285 with substitution of glutamic acid to alanine, which accounted for 86.3% of mutations in 104 alleles, and a nonsense mutation on codon 231, tyrosine to stop codon, which was found in 13.4%. Together, these two mutations accounted for 97% of all the alleles in Jewish patients with Canavan’s disease. In non-Jewish patients, the mutations are different and more diverse: the most common is in codon 305, a missense mutation substituting alanine to glutamic acid. This mutation was observed in 35.7% of 70 alleles from 35 unrelated non-Jewish patients. Fifteen other mutations accounted for 24 mutant alleles.
Diagnosis
Definitive diagnosis is achieved by measurement of levels of N-acetyl aspartic acid in urine. It is essential that the isotope dilution assay procedure be used.212,213 N-acetylneuraminic acid (NANA) levels in Canavan’s disease are 80 to 120 times higher than control levels. NANA assay in amniotic fluid enables prenatal diagnosis.214 Further studies of this assay showed that although significantly increased levels of amniotic fluid NANA were reliable markers, moderate increases should be interpreted with caution and could lead to false-negative results.215 Measurement of aspartoacylase activity is not reliable for postnatal or prenatal diagnosis.214,216 It is recommended strongly that, whenever possible, it be combined with mutation analysis in amniocytes or trophoblasts.217,218 Mutation analysis is a key diagnostic procedure.211 It is particularly so in the Ashkenazi Jewish populations, in which two common mutations account for all the mutations that have been encountered. The American College of Obstetrics and Gynecology has recommended that molecular carrier screening be offered to Ashkenazi Jewish couples.219
Pathogenesis
The key defect in Canavan’s disease is the deficiency of N-acyl-L-aspartate aminohydrolase,220 which hydrolyses NANA to L-aspartate and acetate. NANA is found only in the nervous system, in which its normal concentration, 6 to 7μmol/g, is second only to that of glutamic acid in the free amino acid pool. NANA is synthesized in neuronal mitochondria by the enzyme aspartate N-acyltransferase, whereas the catabolic enzyme N-acyl-L-aspartate is present mainly in oligodendrocytes,221 and its concentration is even higher in Canavan’s disease.
Three pathogenetic mechanisms for Canavan’s disease have been proposed: (1) Madhavarao and colleagues221 demonstrated that levels of acetate in the brain and the synthesis of myelin lipids are reduced significantly in the mouse model of Canavan’s disease and also in a patient with Canavan’s disease, and they proposed that this is a consequence of an impaired acetate supply in the oligodendrocyte secondary to the N-acyl-L-aspartate deficiency. (2) Levels of N-acetyl aspartate glutamate may be increased in Canavan’s disease and may interfere with the function of the N-methyl-D-aspartate receptor.222 (3) Because of its high concentration, NANA may act as an organic osmolyte that normally removes excess water from neurons by acting as a molecular water pump, and this function may be deficient in Canavan’s disease.223
Two animal models of Canavan’s disease have been developed. Aspartoacylase gene knockout in the mouse leads to reproduction of the clinical, pathological, radiological, and biochemical defects of Canavan’s disease in humans.224,225
Therapy
There is no specific therapy for Canavan’s disease. It was the first neurodegenerative disorder to be treated by gene therapy.226 A nonviral lipid-entrapped, polycation-condensed delivery system in conjunction with adeno-associated virus-based plasmid containing recombinant N-acyl-L-aspartate was administered by intraventricular injection to two patients with Canavan’s disease. The procedure was well tolerated. Fifteen additional patients were treated.227 There were no definitive changes in clinical course.
The availability of animal models of Canavan’s disease has made it possible to test gene transfer therapy in these models. Stereotactic delivery of adeno-associated virus 2-mediated N-acyl-L-aspartate to “tremor rats” reduced brain NANA levels and was associated with some improvement in motor function.228 On the basis of the hypothesis that acetate deficiency in Canavan’s disease limits synthesis of myelin lipids,221 a trial of dietary supplementation with acetate or acetate precursors has been proposed.221
ALEXANDER’S DISEASE
Clinical Features
Early infantile, juvenile, and adultonset forms have been described. The early infantile-onset form manifests at approximately 6 months with rapidly progressive neurological and mental retardation, in association with macrocephaly spasticity and seizure, and leads to death in the second or third year.229,230 Among patients with the juvenile-onset form, macrocephaly was present in only 27%, and progressive cognitive defects occurred in 60%. In the adultonset form, ataxia, eye movement disturbances, and bulbar and pseudobulbar symptoms231 predominate. Palatal myoclonus,232 autonomic disturbances, and sleep disturbances have also been reported.233
Pathology
In the early infantile-onset form, the brain is enlarged with a normal convolutional pattern. The white matter is discolored, soft, and swollen in the frontal region. Light microscopy reveals that the main abnormalities are the abundance of Rosenthal fibers and demyelination of the white matter (Rosenthal fibers are eosinophilic intracytoplasm filamentous cytoplasmic fibers within astrocytes).234 The major chemical components of Rosenthal fibers are glial fibrillary acidic protein, the small heat-shock proteins α-crystalline and hsp27, and ubiquitin.235 Rosenthal fibers have a tendency to accumulate at interfaces with mesodermal tissue that is underneath the pia mater and around the blood vessels. They are most prominent in the frontal and parietal regions and much less so in the occipital lobes. The basal ganglia are also involved. The tracts and nuclei of the brainstem nuclei are involved heavily from the midbrain to lower medulla; the cerebellar hemispheres, only slightly.229,236
Neuroimaging
On the basis of the study of 5 patients with the infantile-onset form and 14 with the juvenile-onset form, van der Knaap and colleagues237 proposed five MRI criteria for the diagnosis of Alexander disease: (1) extensive cerebral white matter T2-weighted abnormalities with a frontal preponderance; (2) the presence of a periventricular rim of decreased T2-weighted signal intensity but with increased T1-weighted signal intensity in the same region; (3) abnormalities in the basal ganglia and thalami, in the form of either elevated signal intensity or atrophy and either elevated or decreased signal intensity on T2-weighted images; (4) brainstem abnormalities, particularly those involving the midbrain and medulla; and (5) contrast enhancement involving one or more of the following structures: ventricular lining, periventricular rim of tissue, white matter of the frontal lobes, optic chiasm, fornix, basal ganglia, thalamus, dentate nucleus, and brainstem structures (Fig. 80-2).
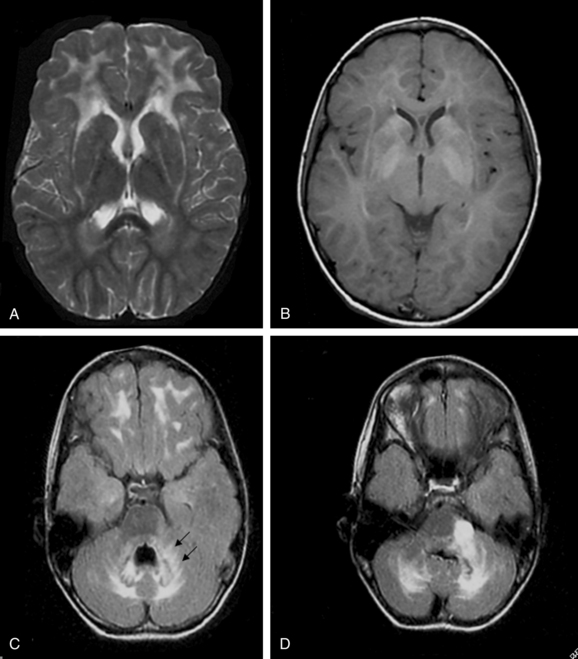
The authors concluded that the imaging pattern in infantile and juvenile-onset cases was sufficiently specific to allow the in vivo MRI diagnosis of Alexander’s disease and that brain biopsy is necessary only in atypical cases. However, in a later study of 10 patients with later onset and atypical features, the same group of investigators reported that this typical pattern was not found.238 In these patients, late-onset disease, ataxia, bulbar symptoms, and eye movement disturbances dominated the clinical findings.231,233 The MRI revealed predominantly posterior lesions, including multiple tumor-like brainstem lesions.
Genetics
The mode of inheritance is autosomal dominant. The abnormal gene has been mapped to locus 17q21. It codes for the glial fibrillary acid protein. One hundred and two pathogenic mutations have been identified.239–241 In one study, 72 patients had the infantile-onset phenotype, 22 had the juvenile-onset phenotype, 7 had the adultonset phenotype, and 1 was asymptomatic. All of the cases of the infantile phenotype, 21 of those with the juvenile phenotype, and 4 of those with the adult phenotype were sporadic. Five pedigrees with more than one affected member have been reported. All of the 16 affected persons in these pedigrees had the adult or the juvenile phenotype.
Diagnosis
In the patients with the typical infantile phenotype, the clinical features, combined with brain MRI findings,237 are sufficiently specific to establish the diagnosis. However, in juvenile and adultonset cases, mutation analysis239 is required for diagnosis. In these cases, the range of phenotypical expression and MRI abnormalities is wider than had been recognized in the past. It is recommended that mutation analysis be performed not only in the affected persons but also in their parents. This makes it possible to distinguish between sporadic cases (the majority) and familial cases. In familial cases, mutation analysis should be performed in at-risk members. Most of the familial cases have the adultonset phenotype, and their identification is required for genetic counseling.
Pathogenesis
Glial fibrillary acidic protein is one of the intermediate filament proteins, which also includes the keratins, vimentin, desmin, peripherin, and nestin.242 Intermediate filament contains α-helical polypeptides that are linked and intertwined to form protofilaments. There is a great deal of evidence that the mutations in Alexander’s disease have a dominant negative effect: namely, that the abnormal glial fibrillary acidic protein allele product in astrocytes causes secondary dysfunction of oligodendrocytes and neurons.243 In support of this is the fact that no null mutations have been identified in patients with Alexander’s disease (which suggests that these would be lethal) and that glial fibrillary acidic protein knockout genes in mice cause only subtle pathological changes.243
CEREBROTENDINOUS XANTHOMATOSIS
Another name by which this condition is known is forme cérébrale de la cholestérinose généralisée.245
Clinical Features
Clinical features develop slowly and may manifest irregularly in varying combinations. The most common features are xanthomas in the Achilles tendon, neurological dysfunction, and cataracts. As patients grow older, they develop progressive coronary arteriosclerosis and osteoporosis that predisposes to bone fractures.246 The CNS manifestations are gradual deterioration of intellectual function, which progresses to dementia; behavior and psychiatric manifestations; pyramidal tract signs; spastic paraplegia; ataxia; dysarthria; and nystagmus. Epileptic seizures occur in half of the symptomatic subjects. Peripheral neuropathy is also present. Chronic diarrhea is a frequent symptom. Some specific clinical points have been highlighted; for example, the spinal phenotype (without xanthomas) may be misdiagnosed as multiple sclerosis.247 In children, cataracts may be the first manifestation of cerebrotendinous xanthomatosis,248 and xanthomata may be absent in patients with severe neurological involvement.249
Neuroimaging
The most important and earliest MRI abnormalities occur in the cerebellum. On T2-weighted images, there are high-intensity lesions in the dentate nuclei and cerebellar hemispheres.4,214 Atrophy of cerebellar folia and symmetrical lesions in the corticospinal tracts, the medial lemniscus in the brainstem, and the inferior olive may occur. In the supratentorial region, ill-defined slight signal changes are seen in the periventricular region.
Genetics
The mode of inheritance is autosomal recessive. Several hundred cases have been reported from many countries, particularly The Netherlands, Italy, Israel, and Japan. The gene maps to locus 2q33ter. It codes for a sterol 27-hydroxylase (CYP27A).250 More than 20 mutations have been defined.251
Diagnosis
Although the clinical manifestations and MRI findings of cerebrotendinous xanthomatosis are rather characteristic, laboratory diagnosis is required for definitive diagnosis. Laboratory techniques also enable presymptomatic diagnosis and early initiation of therapy. Demonstration of abnormally increased levels of cholestanol through precise methods such as selected ion monitoring252 is a key diagnostic step.251 A screening method that detects increased levels of 7α-hydroxylated bile acids in urine is a valuable technique.253 Mutation analysis in at-risk relatives250 of known patients is recommended highly for presymptomatic identification of affected persons and for genetic counseling.
Pathogenesis
The primary defect in cerebrotendinous xanthomatosis is the deficiency of C27-steroid hydroxylase, which leads to defective bile acid synthesis and the accumulation of a variety of metabolites with the C27-steroid side chains. There are series of complex metabolic and pathological consequences.251 The major clinical manifestations are caused by generalized accumulation of cholestanol and cholesterol in nearly every tissue, including the CNS. Menkes and associates demonstrated the accumulation of cholestanol in the brains of patients with cerebrotendinous xanthomatosis in 1968.254 Cholesterol synthesis is greatly increased, because in the absence of bile acids, the normal bile acid feedback inhibition does not take place. Cholestanol is normally produced from cholesterol in small quantities by a four-step pathway. In cerebrotendinous xanthomatosis, C27-steroids that cannot be converted into bile acids may be shunted into this pathway.251 The C27-steroid hydroxylase that is deficient in cerebrotendinous xanthomatosis normally may also act on the C25-steroid hydroxylase that is involved in vitamin D metabolism, and this may contribute to osteoporosis. C27-steroid hydroxylase also functions to transport steroids out of cells, and a defect in this mechanism may contribute to the premature atherosclerosis in cerebrotendinous xanthomatosis.
Therapy
There is compelling evidence that oral administration of chenodeoxycholic acid in a dosage of 750 mg/day benefits patients with cerebrotendinous xanthomatosis. Chenodeoxycholic acid is one of the bile acids produced in normal persons, but it is not produced in patients with cerebrotendinous xanthomatosis because of the enzymatic defect. Chenodeoxycholic acid administration reduces the levels of cholestanol in plasma and CSF255,256 and was shown to reduce the excretion of urinary bile alcohols257 and bile alcohol glucuronides.258 It has been reported to improve somatosensory and motor evoked potentials,259 visual and brainstem auditory evoked responses,260 brain MRI abnormalities,261 and osteoporosis.262,263 There are several reports that it stopped or slowed neurological progression.256,264 Berginer and coauthors255 reported improvements in cerebellar function, behavioral disturbances, and seizure control after 1 year of chenodeoxycholic acid therapy. Beneficial effects are most evident if therapy is started when deficits are slight. Biochemical and DNA screening of at-risk relatives now enables identification of asymptomatic persons with cerebrotendinous xanthomatosis. It is possible that chenodeoxycholic acid therapy will reduce or prevent later disability.
Addition of lovastatin or simvastatin to the chenodeoxycholic acid regimen may further decrease cholestanol levels,265–267 but additional clinical benefits have not been demonstrated.268 Reduction of cholestanol levels can also be achieved with low-density lipoprotein apheresis,269 but this relatively invasive therapy, which would need to be administered repeatedly and is the subject of controversy,270,271 should be considered only in patients who have not responded to pharmacological therapy.
SJÖGREN-LARSSON SYNDROME
Another name by which this condition is known is fatty aldehyde dehydrogenase deficiency.
Clinical Features
The three cardinal features are ichthyosis, mental retardation, and spastic diplegia or tetraplegia.272 The ichthyosis is already present at birth or in the neonatal period. It is mild to moderate in severity and generalized in distribution. The face is mildly involved or spared. Neurological symptoms vary considerably but become evident within the first 3 years of life. Spastic diplegia is much more common than tetraplegia. Many patients never gain the ability to walk, and many who do walk require leg braces. The degree of mental retardation tends to be correlated with the severity of spasticity. In a study of Swedish patients, two thirds had an IQ of less than 50. Of enzymatically confirmed cases, 13% showed no mental retardation.273 Most patients have speech deficits of various types, including delayed speech and dysarthria. Two thirds have an associated seizure disorder. Patients with Sjögren-Larsson syndrome generally do not show neurological regression, and most survive well into adulthood. Abnormalities of the retina are frequent. The most consistent findings are glistening white dots in the foveal and perifoveal regions.274
Pathology
The nervous system shows a widely distributed loss of myelin, most prominent in the centrum semiovale, pyramidal tracts, and frontal lobes.275 There is considerable variation in the degree of myelin deficiency. Ballooning of myelin sheaths has been noted in the areas of myelin loss and near blood vessels. There is significant gliosis and proliferation of astrocytes. Gray matter is much less affected. The loss of neurons and axons tends to occur in areas where myelin loss has become extensive and appears to be secondary to myelin loss.
Neuroimaging
A study of 18 patients with Sjögren-Larsson syndrome demonstrated characteristic changes.276 MRI revealed a zone of abnormally high signal intensity in the periventricular high matter on T2-weighted images. The abnormality was present already at 2 years of age and remained relatively stable after that. The subarcuate fibers, cerebellum, and corpus callosum were relatively spared. The gray matter was normal. Mild cerebral atrophy was found in most patients older than 10 years. MRS revealed a prominent and narrow resonance at 1.3ppm, at which protons of methylene groups resonate. The nature of the lipids that give rise to this peak has not been defined. This peak was present in the white matter but not in the gray matter. The total N-acetyl aspartate level was normal, which was consistent with the relative preservation of neurons; creatine, choline, and inositol levels were increased, which was consistent with the gliosis and myelin damage.
Genetics
The mode of inheritance is autosomal recessive. Sjögren-Larsson syndrome is a rare disorder, first reported and apparently most common in Sweden, where the frequency is 0.4 per 100,000. By 2001,273 more than 200 patients had been recognized in all parts of the world. The gene maps to locus 17p11.2. It codes for fatty aldehyde dehydrogenase.277 Mutations of the gene were first identified by De Laurenzi and associates.278 Seventy-two mutations in the gene have been defined.279
Diagnosis
Diagnosis is suggested by the clinical triad of ichthyosis, mental retardation, and spasticity and is aided by brain MRI and MRS studies276 and the presence of glistening spots in the retina.274 Definitive diagnosis depends on demonstration of fatty aldehyde dehydrogenase deficiency in cultured skin fibroblasts.280,281 This assay also enables prenatal diagnosis.282 Van den Brink and associates reported an alternative enzymatic assay based on demonstration of impaired phytol degradation in cultured fibroblasts of Sjögren-Larsson syndrome patients.283 DNA-based diagnosis is feasible278 and is being applied increasingly for heterozygote identification and prenatal diagnosis.
Pathogenesis
The levels of essential polyunsaturated fatty acids in plasma are reduced.285
Treatment
Treatment is symptomatic. Topical administration of keratolytic agents and the systemic administration of short-acting retinoids has ameliorated the ichthyosis. A variety of fat-modified diets, such as those in which long-chain fatty acids are replaced by medium-chain fatty acids, and the administration of polyunsaturated fatty acids have been used without definitive favorable effects.273 Haug and Braun-Falco286 reported that adeno-associated virus vectors restored fatty aldehyde dehydrogenase deficiency in fibroblast cell lines from patients with Sjögren-Larsson syndrome, which suggests that gene therapy may eventually become an option.
VANISHING WHITE MATTER DISEASE5
Other names by which this condition is known include childhood ataxia with central hypomyelination6 and myelinopathia centralis diffusa.287 Allelic conditions include Cree eukoencephalopathy288 and leukodystrophy in patients with ovarian dysgenesis.289
Clinical Features
Classic Form
The onset is in the juvenile period (3 to 10 years of age). Ataxia and spasticity are present, and intellect is relatively preserved. The course is chronic and progressive with episodes of deterioration, which may include coma, precipitated by minor injuries, fever, or fright.290
Severe Infantile Form, Cree Leukoencephalopathy
The onset is at ages 3 to 9 months. This form is characterized by hypotonia, seizures, spasticity, hyperventilation, blindness, developmental regression, and death by 21 months.291,292
Adolescent or Adult Forms
This form is characterized by progressive ataxia and spasticity and by mildly to moderately impaired mental capacity.293 Age of onset typically is 10 to 21 years. Adultonset cases may manifest with dementia and psychiatric symptoms.294 The disorder may be associated with ovarian dysgenesis in women.289
Pathology
Extensive degeneration of white matter with cystic degeneration is a feature. The cortex is preserved. There is increased density and apoptosis of oligodendrocytes.295 Astrocyte structure is abnormal.288
Genetics
The basic defect involves the eukaryotic translation initiation factor eIF2β. eIF2β is a complex consisting of five subunits, eIF2β1 to eIF2β5, encoded on loci 3q27, 14q24, 1q34.1, 2q23.3, and 12q24.3. Mutations of eIF2β5 (3q27) are most common. In one series, 14 of 16 mutations were missense,8 and most involved nonconserved amino acids. The R113H mutations in eIF2β5 and the E213G mutations in eIF2β2 appear to be associated with milder phenotypes.296,297
Pathogenesis
eIF2β plays a fundamental role in the initiation of translation and was summarized by van der Knaap and colleagues.8 The first step of the translation process is that a complex of eIf2-guanosine triphosphate (GTP) and methionyl-transfer RNA binds to the ribosome; eIf2 leaves the ribosome as eIF-guanosine diphosphate. In order to bind another methionyl-transfer RNA, eIf2 must be reactivated by exchange of guanosine diphosphate for guanosine triphosphate. This reaction is catalyzed by eIF2β. Under a variety of stress conditions, protein synthesis is decreased. This response is a protective mechanism. The down regulation is induced by the rapid expression of a specific set of proteins called heat shock proteins. In the absence or impaired function of eIF2β, these key regulatory mechanisms cannot take place. The processes that lead to the specific defects in vanishing white matter disease are not yet clear. Apoptosis of oligodendrocytes295 and impairment of astrocyte generation288 have been demonstrated. The nearly instantaneous neurological worsening after a frightful event that has been reported by Vermeulen and associates290 provided hints about the speed and complexity of these control mechanisms.
MEGALENCEPHALIC LEUKOENCEPHALOPATHY WITH SUBCORTICAL CYSTS
Other names by which MLC is known include leukoencephalopathy with swelling and a discrepantly mild clinical course,7 van der Knaap’s megalencephalic leukoencephalopathy,298 and leukoencephalopathy with megalencephaly and mild clinical course.299
Clinical Features
Macrocephaly is present at birth or, more frequently, develops during the first year of life.7,298–301 After the first year of life, the head growth rate normalizes. The first clinical symptom is delay in walking. Walking is often unstable, and the child falls frequently.4 After an interval of several years, there is slow deterioration of motor function, with the development of ataxia. Signs of pyramidal tract dysfunction are late and minor. Most affected children become wheelchair dependent at the end of the first decade or during the second decade of life. Mental deterioration is late and mild. Speech becomes increasingly dysarthric, and dysphagia may develop. Some patients have dystonia and athetosis. Almost all patients have epilepsy from early on; it is usually controlled easily with medication.300 Minor head trauma may induce temporary degeneration.4
Pathology
The cerebral white matter exhibits status spongiosus with innumerable vacuoles.301 The white matter shows intense fibrillary astrogliosis. Electron microscopy reveals splitting of the myelin sheaths at the intraperiod line with intramyelinic vacuole formation. There is no evidence of axonal degeneration or loss. The cerebral cortex is normal.
Neuroimaging
The cerebral hemispherical white matter is diffusely abnormal and swollen. The swelling is most marked during the first year of life, with obliteration of peripheral CSF spaces and narrowing of ventricles (Fig. 80-3). The external and extreme capsules are prominently involved. The central white matter structures are relatively spared.4 Cysts are present almost invariably in the anterior temporal regions and often also in the frontal and parietal subcortical regions (see Fig. 80-3). They enlarge with age and may become very large. Only a few infants and children with MLC lack cysts. MRS shows reduction of all signals, which is indicative of high water content.
Genetics
The mode of inheritance is autosomal recessive. It is a rare disorder but relatively prevalent among Turkish people and in a certain Asian-Indian community, the Agrawal ethnic group. The defective gene has been mapped to locus 22qter.10 It encodes a protein of unknown function with eight transmembrane domains and is now referred to as MLC1. It is highly conserved throughout evolution. MLC1 mutations have been demonstrated in 70% of patients with MLC. Twelve mutations have been defined.8–10
Diagnosis
Diagnosis of MLC is based on the characteristic clinical, MRI, and MRS findings. Mutations in the MLC1 gene have been identified in 70% of the patients.4,8–10
Pathogenesis
Within the brain, MCL1 protein is expressed in astrocytic endfeet in the perivascular, subependymal, and subpial regions.4,302 This localization suggests a possible role in a transport process across the blood-brain barrier.303 Of interest is that water transport proteins such as aquaporin-4 display a similar localization.304
MEMBRANOUS LIPODYSTROPHY
Clinical Features
The clinical course is divided into four stages:305 (1) The latent stage extends into adulthood; (2) the osseous stage begins in the third decade and is characterized by fractures after minor injuries, cystic rarefaction, metaphyseal cysts, and replacement of cancellous bone by cysts; (3) The neuropsychiatric stage, which begins in the third or fourth decade, is characterized by progressive dementia with predominately frontal syndrome and by agnosia, apraxia, and loss of social inhibition; and (4) the dementia stage is characterized by cachexia, seizures, and death between 35 and 45 years of age.
Palatal myoclonus, dysarthria, nystagmus and ataxic gait have been reported in patients in stage 3.306 One 56-year-old patient had an unusually benign course, with bone lesions for 16 years but no neurological or psychiatric disturbances.307
Pathology
In the bone lesion, bone and hematopoietic tissue are replaced by cysts originating from adipocytes. They contain membrane membranes that are autofluorescent and carbohydrate, phospholipids, and fatty acid crystals. Periodic acid-Schiff stain of these cysts yields positive results.305,308
Brain weight is reduced. There is atrophy of the superior frontal gyri. Deep frontal white matter is shrunken with grayish discoloration, and basal ganglia are reduced in size. Neocortical cytoarchitecture is generally preserved; there are no pathological inclusions. There is severe loss of axons and myelin, accompanied by scattered axonal spheroids and widespread activation of microglia. Vascular alterations are present in the deep frontal and white matter. These alterations affect scattered small arterioles and capillaries and consist of concentric thickening of the vascular wall with narrowing or obliteration of lumen.309
Neuroimaging
MRI shows bilateral T2-weighted hyperintensity and areas of cavitation in the periventricular region. Subarcuate fibers are spared. There is frontotemporal atrophy. Cerebellum and brainstem are atrophic. Computed tomography revealed calcification in the dentate nucleus and putamen.306,310
Genetics
The mode of inheritance is autosomal recessive. The disorder is rare. By 1997, it had been observed in fewer than 150 patients.305 Most cases occur in two widely separated population groups: the Japanese population and the Finnish population in northern Scandinavia. The estimated population frequency in the Finns is 2 × 10−6. The defective gene maps to 19q13.1. It codes for TYRObp,311 a transmembrane protein that is a key activating signal in natural killer cells.312 Loss-of-function mutations have been identified in both Japanese and Finnish patients.
Pathogenesis
The defect of TYRObp is suggestive of a link between the bone and CNS lesions. This gene is expressed in microglial cells313 and may also be expressed in osteoclasts. The detailed pathogenetic mechanisms are still unclear. The glial proliferation in white matter and the TYRObp expression patterns suggest that microglia contribute to the pathogenesis of CNS abnormalities. It is not known whether the abnormalities in small blood vessels in white matter play a primary role in pathogenesis or are a secondary event.309
RIBOSE-5-PHOSPHATE ISOMERASE DEFICIENCY
The patient’s mental age is estimated at 2.6 to 4.8 years.314 MRI showed increased T2-weighted signal in cerebral hemispheres involving subarcuate fibers, but there was partial sparing of periventricular white matter. Some vermal atrophy was present. No signal abnormality was detected in the basal ganglia, cerebral cortex, brainstem, or cerebellum. MRS demonstrated several peaks between 3.5 and 4.0ppm, later identified as arabitol and ribitol. The mode of inheritance is autosomal recessive. The gene maps to locus 2p11.2. It codes for ribose-5-phosphate isomerase (Enzyme Commission number 5.3.1.6), which catalyzes conversion of ribulose-5-phosphate to ribose-5-phosphate. A mutation in this isomerase was demonstrated in the proband, and the mother was heterozygous for this mutation. Diagnosis is based on demonstration of increased levels of arabitol and ribitol on brain MRS and in urine, plasma, and CSF315 and on demonstration of defective activity of ribose-5-phosphate isomerase.316 Polyol accumulation is postulated to lead to polyneuropathy, possibly secondary to defective Na+/K+-adenosine triphosphatase regulation as a result of myoinositol depletion.316 There is no specific therapy.
LEUKODYSTROPHIES IN WHICH THE GENETIC DEFECT IS NOT YET DEFINED
Table 80-1 lists the disorders that are probably distinct leukodystrophies but whose genetic defects are not yet defined.
TABLE 80-1 Leukodystrophies in Which the Gene Defect Is Not Defined
| Disease | Reference(s) |
|---|---|
| Leukoencephalopathy with brainstem involvement and high lactate | 317, 318 |
| Cystic leukoencephalopathies without megalencephaly | 319, 320 |
| Familial orthochromatic leukodystrophy | 321, 322 |
| Hereditary leukoencephalopathy with spheroids | 323, 324 |
| Other leukodystrophies in children | 325, 326 |
| Leukodystrophies in adults | 327–332 |
| Hereditary leukodystrophy and palmoplantar keratoderma: a newly recognized disorder with increased skin collagen content | 333 |
| Oculodentodigital dysplasia with cerebral white matter abnormalities in a two-generation family | 334 |
DISORDERS THAT RESEMBLE LEUKODYSTROPHIES BUT IN WHICH THE PRIMARY DEFECT DOES NOT AFFECT MYELIN OR MYELINATING CELLS
These disorders are listed in Table 80-2.
TABLE 80-2 Disorders That Resemble Leukodystrophies in Which the Primary Defect Does Not Involve Myelin
| Disease | Reference(s) |
|---|---|
| Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) | 335, 336 |
| Binswanger’s microangiopathic leukoencephalopathy; leukoaraiosis | 337 |
| Leukoencephalopathy associated with mitochondrial disorders | 338–340 |
| Leukodystrophy in disorders of mucopolysaccharide metabolism | 341 |
| Leukodystrophy in disorders of amino acid metabolism | 342 |
| Leukodystrophy in inborn errors of single carbon transfer pathway | 343 |
| Leukodystrophy and muscular dystrophy | 344–346 |
| Posterior leukoencephalopathy | 347 |
SUMMARY AND CONCLUSIONS
Barkovich AJ. Magnetic resonance techniques in the assessment of myelin and myelination. J Inherit Metab Dis. 2005;28:311-343.
Di Rocco M, Biancheri R, Rossi A, et al. Genetic disorders affecting white matter in the pediatric age. Am J Med Genet B Neu-ropsychiatr Genet. 2004;129:85-93.
Lazzarini RA. Myelin Biology and Disorders. Amsterdam: Elsevier, 2004.
van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders, 3rd ed. Berlin: Springer, 2005.
van der Knaap MS. Magnetic resonance in childhood white-matter disorders. Dev Med Child Neurol. 2001;43:705-712.
1 Powers JM. Leukodystrophies: overview and classification. In: Lazzarini RA, editor. Myelin Biology and Disorders. London: Elsevier; 2004:663-690.
2 Bielschowsky M, Henneberg R. Ueber familiare diffuse Sklerose (Leukodystrophia cerebri progressive hereditaria). J Psychol Neurol (Lpz). 1928;36:131-181.
3 van der Knaap MS, Breiter SN, Naidu S, et al. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology. 1999;213:121-133.
4 van der Knaap MS, Valk J. Magnetic Resonance of Myelin, Myelination, and Myelin Disorders, 2nd ed. Berlin: Springer-Verlag, 2005.
5 van der Knaap MS, Barth PG, Gabreels FJ, et al. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48:845-855.
6 Schiffmann R, Moller JR, Trapp BD, et al. Childhood ataxia with diffuse central nervous system hypomyelination. Ann Neurol. 1994;35:331-340.
7 van der Knaap MS, Barth PG, Stroink H, et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol. 1995;37:324-334.
8 van der Knaap MS, Leegwater PA, Konst AA, et al. Mutations in each of the five subunits of translation initiation factor eIF2β can cause leukoencephalopathy with vanishing white matter. Ann Neurol. 2002;51:264-270.
9 Leegwater PA, Yuan BQ, van der Steen J, et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68:831-838.
10 Topcu M, Gartioux C, Ribierre F, et al. Vacuoliting megalencephalic leukoencephalopathy with subcortical cysts, mapped to chromosome 22qtel. Am J Hum Genet. 2000;66:733-739.
11 Barkovich AJ. Magnetic resonance techniques in the assessment of myelin and myelination. J Inherit Metab Dis. 2005;28:311-343.
12 Moser HW. Adrenoleukodystrophies. In: Lazzarini R, editor. Myelin Biology and Disorders. New York: Academic Press; 2003:807-839.
13 Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120:1485-1508.
14 Moser HW, Smith KD, Watkins PA, et al. X-linked adrenoleukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3257-3301.
15 Kurihara M, Kumagai K, Yagishita S, et al. Adrenoleuko-myeloneuropathy presenting as cerebellar ataxia in a young child: a probable variant of adrenoleukodystrophy. Brain Dev. 1993;15:377-380.
16 Tsuji S, Suzuki M, Ariga T, et al. Abnormality of long-chain fatty acids in erythrocyte membrane sphingomyelin from patients with adrenoleukodystrophy. J Neurochem. 1981;36:1046-1049.
17 Fatemi A, Barker PB, Ulug AM, et al. MRI and proton MRSI in women heterozygous for X-linked adrenoleukodystrophy. Neurology. 2003;60:1301-1307.
18 el-Deiry SS, Naidu S, Blevins LS, et al. Assessment of adrenal function in women heterozygous for adrenoleukodystrophy. J Clin Endocrinol Metab. 1997;82:856-860.
19 Schaumburg HH, Powers JM, Raine CS, et al. Adrenoleukodystrophy. A clinical and pathological study of 17 cases. Arch Neurol. 1975;32:577-591.
20 van der Knaap MS, Valk J. MR of adrenoleukodystrophy: histopathologic correlations. AJNR Am J Neuroradiol. 1989;10:S12-S14.
21 Ito M, Blumberg BM, Mock DJ, et al. Potential environmental and host participants in the early white matter lesion of adrenoleukodystrophy: morphologic evidence for CD8 cytotoxic T cells, cytolysis of oligodendrocytes, and CD1-mediated lipid antigen presentation. J Neuropathol Exp Neurol. 2001;60:1004-1019.
22 Powers JM, DeCiero DP, Ito M, et al. Adrenomyeloneuropathy: a neuropathologic review featuring its noninflammatory myelopathy. J Neuropathol Exp Neurol. 2000;59:89-102.
23 Powers JM, DeCiero DP, Cox C, et al. The dorsal root ganglia in adrenomyeloneuropathy: neuronal atrophy and abnormal mitochondria. J Neuropathol Exp Neurol. 2001;60:493-501.
24 Jansen GA, Wanders RJ, Watkins PA, et al. Phytanoyl-coenzyme A hydroxylase deficiency—the enzyme defect in Refsum’s disease. N Engl J Med. 1997;337:133-134.
25 Julien JJ, Vallat JM, Vital C, et al. Adrenomyeloneuropathy: demonstration of inclusions at the level of the peripheral nerve. Eur Neurol. 1981;20:367-373.
26 Martin JJ, Ceuterick C, Libert J. Skin and conjunctival nerve biopsies in adrenoleukodystrophy and its variants. Ann Neurol. 1980;8:291-295.
27 van Geel BM, Koelman JH, Barth PG, et al. Peripheral nerve abnormalities in adrenomyeloneuropathy: a clinical and electrodiagnostic study. Neurology. 1996;46:112-118.
28 Powers JM, Schaumburg HH, Johnson AB, et al. A correlative study of the adrenal cortex in adrenoleukodystrophy—evidence for a fatal intoxication with very long chain saturated fatty acids. Invest Cell Pathol. 1980;3:353-376.
29 Powers JM, Moser HW, Moser AB, et al. Fetal adrenoleukodystrophy: the significance of pathologic lesions in adrenal gland and testis. Hum Pathol. 1982;13:1013-1019.
30 Powers JM, Schaumburg HH. The testis in adrenoleukodystrophy. Am J Pathol. 1981;102:90-98.
31 Brown FR3rd, Van Duyn MA, Moser AB, et al. Adrenoleukodystrophy: effects of dietary restriction of very long chain fatty acids and of administration of carnitine and clofibrate on clinical status and plasma fatty acids. Johns Hopkins Med J. 1982;151:164-172.
32 van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders, 3rd ed. Berlin: Springer, 2005.
33 Kumar AJ, Rosenbaum AE, Naidu S, et al. Adrenoleukodystrophy: correlating MR imaging with CT. Radiology. 1987;165:497-504.
34 Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61:369-374.
35 Tateishi J, Sato Y, Suetsugu M, et al. Adrenoleukodystrophy with olivopontocerebellar atrophy-like lesions. Clin Neuropathol. 1986;5:34-39.
36 Afifi AK, Menezes AH, Reed LA, et al. Atypical presentation of X-linked childhood adrenoleukodystrophy with an unusual magnetic resonance imaging pattern. J Child Neurol. 1996;11:497-499.
37 Kruse B, Barker PB, van Zijl PC, et al. Multislice proton magnetic resonance spectroscopic imaging in X-linked adrenoleukodystrophy. Ann Neurol. 1994;36:595-608.
38 Eichler FS, Barker PB, Cox C, et al. Proton MR spectroscopic imaging predicts lesion progression on MRI in X-linked adrenoleukodystrophy. Neurology. 2002;58:901-907.
39 Oz G, Tkac I, Charnas LR, et al. Assessment of adrenoleukodystrophy lesions by high field MRS in non-sedated pediatric patients. Neurology. 2005;64:434-441.
40 Moser HW, Barker PB. Magnetic resonance spectroscopy: a new guide for the therapy of adrenoleukodystrophy. Neurology. 2005;64:406-407.
41 Kumar AJ, Kohler W, Kruse B, et al. MR findings in adultonset adrenoleukodystrophy. AJNR Am J Neuroradiol. 1995;16:1227-1237.
42 Dubey P, Fatemi A, Barker PB, et al. Spectroscopic evidence of cerebral axonopathy in patients with “pure” adrenomyeloneuropathy. Neurology. 2005;64:304-310.
43 Fatemi A, Smith SA, Dubey P, et al. Magnetization transfer MRI demonstrates spinal cord pathology in adrenomyeloneuropathy. Neurology. 2005;64:1739-1745.
44 Dobyns WB, Filauro A, Tomson BN, et al. Inheritance of most X-linked traits is not dominant or recessive, just X-linked. Am J Med Genet A. 2004;129:136-143.
45 Bezman L, Moser AB, Raymond GV, et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49:512-517.
46 Migeon BR, Moser HW, Moser AB, et al. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci U S A. 1981;78:5066-5070.
47 Mosser J, Douar AM, Sarde CO, et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726-730.
48 Dean M, Hamon Y, Chimini G. The human ATP-binding cassette (ABC) transporter superfamily. J Lipid Res. 2001;42:1007-1017.
49 Higgins CF. ABC transporters: from microorganisms to man. Annu Rev Cell Biol. 1992;8:67-113.
50 Lombard-Platet G, Savary S, Sarde CO, et al. A close relative of the adrenoleukodystrophy (ALD) gene codes for a peroxisomal protein with a specific expression pattern. Proc Natl Acad Sci U S A. 1996;93:1265-1269.
51 Kemp S, Wei HM, Lu JF, et al. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy. Nat Med. 1998;4:1261-1268.
52 Braiterman LT, Watkins PA, Moser AB, et al. Peroxisomal very long chain fatty acid beta-oxidation activity is determined by the level of adrenodeukodystrophy protein (ALDP) expression. Mol Genet Metab. 1999;66:91-99.
53 Kemp S, Pujol A, Waterham HR, et al. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat. 2001;18:499-515.
54 Berger J, Molzer B, Fae I, et al. X-linked adrenoleukodystrophy (ALD): a novel mutation of the ALD gene in 6 members of a family presenting with 5 different phenotypes. Biochem Biophys Res Commun. 1994;205:1638-1643.
55 Maestri NE, Beaty TH. Predictions of a 2-locus model for disease heterogeneity: application to adrenoleukodystrophy. Am J Med Genet. 1992;44:576-582.
56 Smith KD, Kemp S, Braiterman LT, et al. X-linked adrenoleukodystrophy: genes, mutations, and phenotypes. Neurochem Res. 1999;24:521-535.
57 Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol. 1999;45:100-110.
58 Moser HW, Moser AE, Trojak JE, et al. Identification of female carriers of adrenoleukodystrophy. J Pediatr. 1983;103:54-59.
59 Moser AB, Rasmussen M, Naidu S, et al. Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups. J Pediatr. 1995;127:13-22.
60 Theda C, Woody RC, Naidu S, et al. Increased very long chain fatty acids in patients on a ketogenic diet: a cause of diagnostic confusion. J Pediatr. 1993;122:724-726.
61 Moser HW, Moser AB, Kawamura N, et al. Adrenoleukodystrophy: elevated C26 fatty acid in cultured skin fibroblasts. Ann Neurol. 1980;7:542-549.
62 Antoku Y, Sakai T, Tsukamoto K, et al. A simple diagnostic method of adrenoleukodystrophy: total fatty acid analysis of erythrocyte membranes. Clin Chim Acta. 1987;169:121-125.
63 Molzer B, Bernheimer H, Heller R, et al. Detection of adrenoleukodystrophy by increased C26:0 fatty acid levels in leukocytes. Clin Chim Acta. 1982;125:299-305.
64 Moser HW, Moser AB, Powers JM, et al. The prenatal diagnosis of adrenoleukodystrophy. Demonstration of increased hexacosanoic acid levels in cultured amniocytes and fetal adrenal gland. Pediatr Res. 1982;16:172-175.
65 Moser AB, Moser HW. The prenatal diagnosis of X-linked adrenoleukodystrophy. Prenat Diagn. 1999;19:46-48.
66 Kennedy CR, Allen JT, Fensom AH, et al. X-linked adrenoleukodystrophy with non-diagnostic plasma very long chain fatty acids. J Neurol Neurosurg Psychiatry. 1994;57:759-761.
67 Wanders RJ, van Roermund CW, Lageweg W, et al. X-linked adrenoleukodystrophy: biochemical diagnosis and enzyme defect. J Inherit Metab Dis. 1992;15:634-644.
68 Boehm CD, Cutting GR, Lachtermacher MB, et al. Accurate DNA-based diagnostic and carrier testing for X-linked adrenoleukodystrophy. Mol Genet Metab. 1999;66:128-136.
69 Gray RG, Green A, Cole T, et al. A misdiagnosis of X-linked adrenoleukodystrophy in cultured chorionic villus cells by the measurement of very long chain fatty acids. Prenat Diagn. 1995;15:486-490.
70 Carey WF, Poulos A, Sharp P, et al. Pitfalls in the prenatal diagnosis of peroxisomal β-oxidation defects by chorionic villus sampling. Prenat Diagn. 1994;14:813-819.
71 Igarashi M, Schaumburg HH, Powers J, et al. Fatty acid abnormality in adrenoleukodystrophy. J Neurochem. 1976;26:851-860.
72 Ho JK, Moser H, Kishimoto Y, et al. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest. 1995;96:1455-1463.
73 Whitcomb RW, Linehan WM, Knazek RA. Effects of long-chain, saturated fatty acids on membrane microviscosity and adrenocorticotropin responsiveness of human adrenocortical cells in vitro. J Clin Invest. 1988;81:185-188.
74 Paintlia AS, Gilg AG, Khan M, et al. Correlation of very long chain fatty acids accumulation and inflammatory disease progression in childhood ALD: implications for potential therapies. Neurobiol Dis. 2003;14:425-439.
75 Powers JM, Schaumburg HH, Johnson AB, et al. A correlative study of the adrenal cortex in adrenoleukodystrophy—evidence for a fatal intoxication with very long chain saturated fatty acids. Invest Cell Pathol. 1980;3:353-376.
76 Singh I, Moser AE, Goldfischer S, et al. Lignoceric acid is oxidized in the peroxisome: implications for the Zellweger cerebro-hepato-renal syndrome and adrenoleukodystrophy. Proc Natl Acad Sci U S A. 1984;81:4203-4207.
77 Singh I, Moser AE, Moser HW, et al. Adrenoleukodystrophy: impaired oxidation of very long chain fatty acids in white blood cells, cultured skin fibroblasts, and amniocytes. Pediatr Res. 1984;18:286-290.
78 Lazo O, Contreras M, Hashmi M, et al. Peroxisomal ligno-ceroyl-CoA ligase deficiency in childhood adrenoleukodystrophy and adrenomyeloneuropathy. Proc Natl Acad Sci U S A. 1988;85:7647-7651.
79 Wanders RJ, van Roermund CW, van Wijland MJ, et al. Direct demonstration that the deficient oxidation of very long chain fatty acids in X-linked adrenoleukodystrophy is due to an impaired ability of peroxisomes to activate very long chain fatty acids. Biochem Biophys Res Commun. 1988;153:618-624.
80 Uchiyama A, Aoyama T, Kamijo K, et al. Molecular cloning of cDNA encoding rat very long-chain acyl–CoA synthetase. J Biol Chem. 1996;271:30360-30365.
81 Heinzer AK, Watkins PA, Lu JF, et al. A very long chain acyl–CoA synthetase–deficient mouse and its relevance to X-linked adrenoleukodystrophy. Hum Mol Genet. 2003;12:1145-1154.
82 Heinzer AK, Kemp S, Lu JF, et al. Mouse very long-chain acyl–CoA synthetase in X-linked adrenoleukodystrophy. J Biol Chem. 2002;277:28765-28773.
83 Heinzer AK, McGuinness MC, Lu JF, et al. Mouse models and genetic modifiers in X-linked adrenoleukodystrophy. Adv Exp Med Biol. 2003;544:75-93.
84 Braiterman LT, Zheng S, Watkins PA, et al. Suppression of peroxisomal membrane protein defects by peroxisomal ATP binding cassette (ABC) proteins. Hum Mol Genet. 1998;7:239-247.
85 McGuinness MC, Lu JF, Zhang HP, et al. Role of ALDP (ABCD1) and mitochondria in X-linked adrenoleukodystrophy. Mol Cell Biol. 2003;23:744-753.
86 Tsuji S, Sano T, Ariga T, et al. Increased synthesis of hexacosanoic acid (C23:0) by cultured skin fibroblasts from patients with adrenoleukodystrophy (ALD) and adrenomyeloneuropathy (AMN). J Biochem (Tokyo). 1981;90:1233-1236.
87 Kemp S, Valianpour F, Denis S, et al. Elongation of very long-chain fatty acids is enhanced in X-linked adrenoleukodystrophy. Mol Genet Metab. 2005;84:144-151.
88 Di Biase A, Di Benedetto R, Fiorentini C, et al. Free radical release in C6 glial cells enriched in hexacosanoic acid: implication for X-linked adrenoleukodystrophy pathogenesis. Neurochem Int. 2004;44:215-221.
89 Pujol A, Hindelang C, Callizot N, et al. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet. 2002;11:499-505.
90 Dubois-Dalcq M, Feigenbaum V, Aubourg P. The neurobiology of X-linked adrenoleukodystrophy, a demyelinating peroxisomal disorder. Trends Neurosci. 1999;22:4-12.
91 Powers JM, Liu Y, Moser AB, et al. The inflammatory myelinopathy of adrenoleukodystrophy: cells, effector molecules, and pathogenetic implications. J Neuropathol Exp Neurol. 1992;51:630-643.
92 Feigenbaum V, Gelot A, Casanova P, et al. Apoptosis in the central nervous system of cerebral adrenoleukodystrophy patients. Neurobiol Dis. 2000;7:600-612.
93 Moody DB, Besra GS, Wilson IA, et al. The molecular basis of CD1-mediated presentation of lipid antigens. Immunol Rev. 1999;172:285-296.
94 Asheuer M, Bieche I, Laurendeau I, et al. Decreased expression of ABCD4 and BG1 genes early in the pathogenesis of X-linked adrenoleukodystrophy. Hum Mol Genet. 2005;14:1293-1303.
95 Zhang LX, Bakshi R, Fine E, et al. Clinical and electrophysiological improvement of adrenomyeloneuropathy with steroid treatment. J Neurol Neurosurg Psychiatry. 2003;74:822-823.
96 Dubey P, Raymond G, Moser AB, et al. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long chain fatty acid screening. J Pediatr. 2005;146:528-532.
97 Moser HW, Bergin A, Naidu S, Ladenson PW. Adrenoleukodystrophy. Endocrinol Metab Clin North Am. 1991;20:297-318.
98 Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881-888.
99 Aubourg P, Blanche S, Jambaque I, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med. 1990;322:1860-1866.
100 Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713-718.
101 Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15:1761-1766.
102 Rizzo WB, Leshner RT, Odone A, et al. Dietary erucic acid therapy for X-linked adrenoleukodystrophy. Neurology. 1989;39:1415-1422.
103 Moser HW, Raymond GV, Lu SE, et al. Follow-up of 89 asymptomatic adrenoleukodystrophy patients treated with Lorenzo’s oil. Arch Neurol. 2005;62:1073-1080.
104 van Geel BM, Assies J, Haverkort EB, et al. Progression of abnormalities in adrenomyeloneuropathy and neurologically asymptomatic X-linked adrenoleukodystrophy despite treatment with “Lorenzo’s oil.”. J Neurol Neurosurg Psychiatry. 1999;67:290-299.
105 Uziel G, Bertini E, Bardelli P, et al. Experience on therapy of adrenoleukodystrophy and adrenomyeloneuropathy. Dev Neurosci. 1991;13:274-279.
106 Aubourg P, Adamsbaum C, Lavallard-Rousseau MC, et al. A two-year trial of oleic and erucic acids (“Lorenzo’s oil”) as treatment for adrenomyeloneuropathy. N Engl J Med. 1993;329:745-752.
107 Koehler W, Sokolowski P. Clinical phenotypes, diagnosis and treatment of adulthood X-linked adrenoleukodystrophy. In: Berger J, Köhler W, Stöckler-Ipsiroglu S, editors. Understanding and Treating Adrenoleukodystrophy: Present State and Future Perspectives. Heilbronn: Verlag; 2005:28-59.
108 Singh I, Pahan K, Khan M. Lovastatin and sodium phenylacetate normalize the levels of very long chain fatty acids in skin fibroblasts of X-adrenoleukodystrophy. FEBS Lett. 1998;426:342-346.
109 Weinhofer I, Forss-Petter S, Zigman M, et al. Cholesterol regulates ABCD2 expression: implications for the therapy of X-linked adrenoleukodystrophy. Hum Mol Genet. 2002;11:2701-2708.
110 McGovern MM, Wasserstein MP, Aron A, et al. Biochemical effect of intravenous arginine butyrate in X-linked adrenoleukodystrophy. J Pediatr. 2003;142:709-713.
111 Flavigny E, Sanhaj A, Aubourg P, et al. Retroviral-mediated adrenoleukodystrophy-related gene transfer corrects very long chain fatty acid metabolism in adrenoleukodystrophy fibroblasts: implications for therapy. FEBS Lett. 1999;448:261-264.
112 Benhamida S, Pflumio F, Dubart-Kupperschmitt A, et al. Transduced CD34+ cells from adrenoleukodystrophy patients with HIV-derived vector mediate long-term engraftment of NOD/SCID mice. Mol Ther. 2003;7:317-324.
113 Baumann M, Korenke GC, Weddige-Diedrichs A, et al. Haematopoietic stem cell transplantation in 12 patients with cerebral X-linked adrenoleukodystrophy. Eur J Pediatr. 2003;162:6-14.
114 Hagberg B. Clinical symptoms, signs, and tests in metachromatic leukodystrophy. In: Folch-Pi J, Bauer H, editors. Brain Lipids and Lipoproteins and the Leukodystrophies. Amsterdam: Elsevier; 1963:134.
115 von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3695-3724.
116 Kumperscak HG, Paschke E, Gradisnik P, et al. Adult metachromatic leukodystrophy: disorganized schizophrenia-like symptoms and postpartum depression in 2 sisters. J Psychiatry Neurosci. 2005;30:33-36.
117 Marcao AM, Wiest R, Schindler K, et al. Adult onset metachromatic leukodystrophy without electroclinical peripheral nervous system involvement: a new mutation in the ARSA gene. Arch Neurol. 2005;62:309-313.
118 Molander-Melin M, Pernber Z, Franken S, et al. Accumulation of sulfatide in neuronal and glial cells of arylsulfatase A deficient mice. J Neurocytol. 2004;33:417-427.
119 Holtzman NA. Expanding newborn screening: how good is the evidence? JAMA. 2003;290:2606-2608.
120 Oguz KK, Anlar B, Senbil N, et al. Diffusion-weighted imaging findings in juvenile metachromatic leukodystrophy. Neuropediatrics. 2004;35:279-282.
121 van der Voorn JP, Pouwels PJ, Kamphorst W, et al. Histopathologic correlates of radial stripes on MR images in lysosomal storage disorders. AJNR Am J Neuroradiol. 2005;26:442-446.
122 Gieselmann V, Zlotogora J, Harris A, et al. Molecular genetics of metachromatic leukodystrophy. Hum Mutat. 1994;4:233-242.
123 Sandhoff K, Kolter T, Harzer H. Sphingolipid activator proteins. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3371-3388.
124 Hopwood JJ, Ballabio A. Multiple sulfatase deficiency and the nature of the sulfatase family. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3725-3732.
125 Baum H, Dodgson KS, Spencer B. The assay of arylsul-phatases A and B in human urine. Clin Chim Acta. 1959;4:453-455.
126 Strasberg PM, Warren I, Skomorowski MA, et al. HPLC analysis of urinary sulfatide: an aid in the diagnosis of metachromatic leukodystrophy. Clin Biochem. 1985;18:92-97.
127 Kihara H, Ho CK, Fluharty AL, et al. Prenatal diagnosis of metachromatic leukodystrophy in a family with pseudo arylsulfatase A deficiency by the cerebroside sulfate loading test. Pediatr Res. 1980;14:224-227.
128 Gieselmann V, Polten A, Kreysing J, et al. Arylsulfatase A pseudodeficiency: loss of a polyadenylylation signal and N-glycosylation site. Proc Natl Acad Sci U S A. 1989;86:9436-9440.
129 Gieselmann V, Franken S, Klein D, et al. Metachromatic leukodystrophy: consequences of sulphatide accumulation. Acta Paediatr Suppl. 2003;92:74-79.
130 Proia RL, Wu YP. Blood to brain to the rescue. J Clin Invest. 2004;113:1108-1110.
131 Ginsberg L, Gershfeld NL. Membrane bilayer instability and the pathogenesis of disorders of myelin. Neurosci Lett. 1991;130:133-136.
132 Toda K, Kobayashi T, Goto I, et al. Lysosulfatide (sulfogalac-tosylsphingosine) accumulation in tissues from patients with metachromatic leukodystrophy. J Neurochem. 1990;55:1585-1591.
133 Hannun YA, Bell RM. Lysosphingolipids inhibit protein kinase C: implications for the sphingolipidoses. Science. 1987;235:670-674.
134 Krivit W, Lockman LA, Watkins PA, et al. The future for treatment by bone marrow transplantation for adrenoleukodystrophy, metachromatic leukodystrophy, globoid cell leukodystrophy and Hurler syndrome. J Inherit Metab Dis. 1995;18:398-412.
135 Guffon N, Souillet G, Maire I, et al. Juvenile metachromatic leukodystrophy: neurological outcome two years after bone marrow transplantation. J Inherit Metab Dis. 1995;18:159-161.
136 Krivit W, Lipton ME, Lockman LA, et al. Prevention of deterioration in metachromatic leukodystrophy by bone marrow transplantation. Am J Med Sci. 1987;294:80-85.
137 Dhuna A, Toro C, Torres F, et al. Longitudinal neurophysiologic studies in a patient with metachromatic leukodystrophy following bone marrow transplantation. Arch Neurol. 1992;49:1088-1092.
138 Malm G, Ringden O, Winiarski J, et al. Clinical outcome in four children with metachromatic leukodystrophy treated by bone marrow transplantation. Bone Marrow Transplant. 1996;17:1003-1008.
139 Pridjian G, Humbert J, Willis J, et al. Presymptomatic late-infantile metachromatic leukodystrophy treated with bone marrow transplantation. J Pediatr. 1994;125:755-758.
140 Biffi A, De Palma M, Quattrini A, et al. Correction of metachromatic leukodystrophy in the mouse model by transplantation of genetically modified hematopoietic stem cells. J Clin Invest. 2004;113:1118-1129.
141 Matzner U, Herbst E, Hedayati KK, et al. Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Hum Mol Genet. 2005;14:1139-1152.
142 Hagberg B, Kollberg H, Sourander P, et al. Infantile globoid cell leucodystrophy (Krabbe’s disease). A clinical and genetic study of 32 Swedish cases 1953–1967. Neuropadiatrie. 1969;1:74-88.
143 Korn-Lubetzki I, Dor-Wollman T, Soffer D, et al. Early peripheral nervous system manifestations of infantile Krabbe disease. Pediatr Neurol. 2003;28:115-118.
144 Loonen MC, Van Diggelen OP, Janse HC, et al. Late-onset globoid cell leucodystrophy (Krabbe’s disease). Clinical and genetic delineation of two forms and their relation to the early-infantile form. Neuropediatrics. 1985;16:137-142.
145 Satoh JI, Tokumoto H, Kurohara K, et al. Adultonset Krabbe disease with homozygous T1853C mutation in the galacto-cerebrosidase gene. Unusual MRI findings of corticospinal tract demyelination. Neurology. 1997;49:1392-1399.
146 Henderson RD, MacMillan JC, Bradfield JM. Adult onset Krabbe disease may mimic motor neurone disease. J Clin Neurosci. 2003;10:638-639.
147 Wenger DA, Suzuki K, Suzuki Y, et al. Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease). In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:3669-3694.
148 Yunis EJ, Lee RE. Tubules of globoid leukodystrophy: a right-handed helix. Science. 1970;169:64-66.
149 van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders, 3rd ed. Berlin: Springer, 2005.
150 Brockmann K, Dechent P, Wilken B, et al. Proton MRS profile of cerebral metabolic abnormalities in Krabbe disease. Neurology. 2003;60:819-825.
151 Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat. 1997;10:268-279.
152 Fu L, Inui K, Nishigaki T, et al. Molecular heterogeneity of Krabbe disease. J Inherit Metab Dis. 1999;22:155-162.
153 Harzer K, Knoblich R, Rolfs A, et al. Residual galactosyl-sphingosine (psychosine) β-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin Chim Acta. 2002;317:77-84.
154 Suzuki Y, Suzuki K. Krabbe’s globoid cell leukodystrophy: deficiency of glactocerebrosidase in serum, leukocytes, and fibroblasts. Science. 1971;171:73-75.
155 Meikle PJ, Ranieri E, Simonsen H, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics. 2004;114:909-916.
156 Li Y, Scott CR, Chamoles NA, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50:1785-1796.
157 Li Y, Brockmann K, Turecek F, et al. Tandem mass spectrometry for the direct assay of enzymes in dried blood spots: application to newborn screening for Krabbe disease. Clin Chem. 2004;50:638-640.
158 Husain AM, Altuwaijri M, Aldosari M. Krabbe disease: neurophysiologic studies and MRI correlations. Neurology. 2004;63:617-620.
159 Grewal RP, Petronas N, Barton NW. Late onset globoid cell leukodystrophy. J Neurol Neurosurg Psychiatry. 1991;54:1011-1012.
160 Austin JH, Lehfeldt D. Studies in globoid (Krabbe) leukodystrophy. 3. Significance of experimentally-produced globoid-like elements in rat white matter and spleen. J Neuropathol Exp Neurol. 1965;24:265-289.
161 Miyatake T, Suzuki K. Globoid cell leukodystrophy: additional deficiency of psychosine galactosidase. Biochem Biophys Res Commun. 1972;48:539-543.
162 Suzuki K. Twenty five years of the “psychosine hypothesis:” a personal perspective of its history and present status. Neurochem Res. 1998;23:251-259.
163 Zaka M, Wenger DA. Psychosine-induced apoptosis in a mouse oligodendrocyte progenitor cell line is mediated by caspase activation. Neurosci Lett. 2004;358:205-209.
164 Haq E, Giri S, Singh I, et al. Molecular mechanism of psychosine-induced cell death in human oligodendrocyte cell line. J Neurochem. 2003;86:1428-1440.
165 Suzuki K. Models of Krabbe disease. In: Lazzarini RA, editor. Myelin Biology and Disorders. San Diego, CA: Elsevier; 2004:1101-1113.
166 Krivit W, Shapiro EG, Peters C, et al. Hematopoietic stemcell transplantation in globoid-cell leukodystrophy. N Engl J Med. 1998;338:1119-1126.
167 Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease. N Engl J Med. 2005;352:2069-2081.
168 Weinberg KI. Early use of drastic therapy. N Engl J Med. 2005;352:2124-2126.
169 Biswas S, Biesiada H, Williams TD, et al. Substrate reduction intervention by L-cycloserine in twitcher mice (globoid cell leukodystrophy) on a B6;CAST/Ei background. Neurosci Lett. 2003;347:33-36.
170 Boulloche J, Aicardi J. Pelizaeus-Merzbacher disease: clinical and nosological study. J Child Neurol. 1986;1:233-239.
171 Kaye EM, Doll RF, Natowicz MR, et al. Pelizaeus-Merzbacher disease presenting as spinal muscular atrophy: clinical and molecular studies. Ann Neurol. 1994;36:916-919.
172 Seitelberger F. [Merzbacher-Pelizaeus disease; clinicoanatomic studies of its position among the diffuse scleroses]. Wien Z Nervenheilkd Grenzgeb. 1954;9:228-289.
173 Garbern JY, Yool DA, Moore GJ, et al. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551-561.
174 Hudson LD, Garbern JY. Pelizaeus-Merzbacher. In: Lazzarini RE, editor. Myelin Biology and Disorders. New York: Academic Press; 2003:867-885.
175 Inoue K, Tanaka H, Scaglia F, et al. Compensating for central nervous system dysmyelination: females with a proteolipid protein gene duplication and sustained clinical improvement. Ann Neurol. 2001;50:747-754.
176 Bonavita S, Schiffmann R, Moore DF, et al. Evidence for neuroaxonal injury in patients with proteolipid protein gene mutations. Neurology. 2001;56:785-788.
177 Hodes ME, DeMyer WE, Pratt VM, et al. Girl with signs of Pelizaeus-Merzbacher disease heterozygous for a mutation in exon 2 of the proteolipid protein gene. Am J Med Genet. 1995;55:397-401.
178 Nance MA, Boyadjiev S, Pratt VM, et al. Adultonset neurodegenerative disorder due to proteolipid protein gene mutation in the mother of a man with Pelizaeus-Merzbacher disease. Neurology. 1996;47:1333-1335.
179 Weimbs T, Stoffel W. Proteolipid protein (PLP) of CNS myelin: positions of free, disulfide-bonded, and fatty acid thioesterlinked cysteine residues and implications for the membrane topology of PLP. Biochemistry. 1992;31:12289-12296.
180 Knapp PE. Proteolipid protein: is it more than just a structural component of myelin? Dev Neurosci. 1996;18:297-308.
181 Mimault C, Giraud G, Courtois V, et al. Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher disease: duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. The Clinical European Network on Brain Dysmyelinating Disease. Am J Hum Genet. 1999;65:360-369.
182 Inoue K, Osaka H, Imaizumi K, et al. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Ann Neurol. 1999;45:624-632.
183 Woodward K, Kendall E, Vetrie D, et al. Pelizaeus-Merzbacher disease: identification of Xq22 proteolipid-protein duplications and characterization of breakpoints by interphase FISH. Am J Hum Genet. 1998;63:207-217.
184 Ellis D, Malcolm S. Proteolipid protein gene dosage effect in Pelizaeus-Merzbacher disease. Nat Genet. 1994;6:333-334.
185 Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet. 2002;3:199-242.
186 Raskind WH, Williams CA, Hudson LD, et al. Complete deletion of the proteolipid protein gene (PLP) in a family with X-linked Pelizaeus-Merzbacher disease. Am J Hum Genet. 1991;49:1355-1360.
187 Cambi F, Tartaglino L, Lublin F, et al. X-linked pure familial spastic paraparesis. Characterization of a large kindred with magnetic resonance imaging studies. Arch Neurol. 1995;52:665-669.
188 Hodes ME, Zimmerman AW, Aydanian A, et al. Different mutations in the same codon of the proteolipid protein gene, PLP, may help in correlating genotype with phenotype in Pelizaeus-Merzbacher disease/X-linked spastic paraplegia (PMD/SPG2). Am J Med Genet. 1999;82:132-139.
189 Hobson GM, Davis AP, Stowell NC, et al. Mutations in non-coding regions of the proteolipid protein gene in Pelizaeus-Merzbacher disease. Neurology. 2000;55:1089-1096.
190 Inoue K, Kanai M, Tanabe Y, et al. Prenatal interphase FISH diagnosis of PLP1 duplication associated with Pelizaeus-Merzbacher disease. Prenat Diagn. 2001;21:1133-1136.
191 Simons M, Kramer EM, Macchi P, et al. Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J Cell Biol. 2002;157:327-336.
192 Southwood CM, Garbern J, Jiang W, et al. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585-596.
193 Kozutsumi Y, Segal M, Normington K, et al. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332:462-464.
194 Ma Y, Hendershot LM. The unfolding tale of the unfolded protein response. Cell. 2001;107:827-830.
195 Harding HP, Calfon M, Urano F, et al. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575-599.
196 Knave AM, Griffith IR. Models of Pelizaeus-Merzbacher disease. In: Lazzarini RAE, editor. Myelin Disorders and Classification. London: Elsevier; 2004:1125-1142.
197 Duncan ID, Nadon NL, Hoffman RL, et al. Oligodendrocyte survival and function in the long-lived strain of the myelin deficient rat. J Neurocytol. 1995;24:745-762.
198 Brustle O, Jones KN, Learish RD, et al. Embryonic stem cell–derived glial precursors: a source of myelinating transplants. Science. 1999;285:754-756.
199 Bogaert L, Bertrand L. Sur une idiotie familiale avec degenescence spongieuse nevraxe—note preliminaire. Acta Neurol Belg. 1949;49:572-587.
200 Bogaert L, Bertrand I. Spongy Degeneration of the Brain in Infancy. Amsterdam: North-Holland, 1967.
201 Banker BQ, Victor ML. Spongy degeneration of the brain in infancy. In: Goodman RM, Motulsky AG, editors. Genetic Diseases Among Ashkenazi Jews. New York: Raven Press; 1979:201-216.
202 Traeger EC, Rapin I. The clinical course of Canavan disease. Pediatr Neurol. 1998;18:207-212.
203 Hogan GR, Richardson EPJr. Spongy degeneration of the nervous system (Canavan’s disease). Pediatrics. 1965;35:284-294.
204 Jellinger K, Seitelberger F. Juvenile form of spongy degeneration of the CNS. Acta Neuropathol (Berl). 1969;13:276-281.
205 Goodhue WWJr, Couch RD, Namiki H. Spongy degeneration of the CNS: an instance of the rare juvenile form. Arch Neurol. 1979;36:481-484.
206 Toft PB, Geiss-Holtorff R, Rolland MO, et al. Magnetic resonance imaging in juvenile Canavan disease. Eur J Pediatr. 1993;152:750-753.
207 Adachi M, Wallace BJ, Schneck L, Volk BW. Fine structure of spongy degeneration of the central nervous system (van Bogaert and Bertrand type). J Neuropathol Exp Neurol. 1966;25:598-616.
208 van der Knaap MS, Valk J, editors. Canavan’s disease. In Magnetic Resonance of Myelin, Myelination, and Myelin Disorders. 2nd ed. Berlin: Springer-Verlag; 1995:216-220.
209 Grodd W, Krageloh-Mann I, Petersen D, et al. In vivo assessment of N-acetylaspartate in brain in spongy degeneration (Canavan’s disease) by proton spectroscopy. Lancet. 1990;336:437-438.
210 Kaul R, Gao GP, Balamurugan K, et al. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet. 1993;5:118-123.
211 Kaul R, Gao GP, Aloya M, et al. Canavan disease: mutations among Jewish and non-Jewish patients. Am J Hum Genet. 1994;55:34-41.
212 Kelley RI, Stamas JN. Quantification of N-acetyl-L-aspartic acid in urine by isotope dilution gas chromatography-mass spectrometry. J Inherit Metab Dis. 1992;15:97-104.
213 Jakobs C, ten Brink HJ, Langelaar SA, et al. Stable isotope dilution analysis of N-acetylaspartic acid in CSF, blood, urine and amniotic fluid: accurate postnatal diagnosis and the potential for prenatal diagnosis of Canavan disease. J Inherit Metab Dis. 1991;14:653-660.
214 Bennett MJ, Gibson KM, Sherwood WG, et al. Reliable prenatal diagnosis of Canavan disease (aspartoacylase deficiency): comparison of enzymatic and metabolite analysis. J Inherit Metab Dis. 1993;16:831-836.
215 Besley GT, Elpeleg ON, Shaag A, et al. Prenatal diagnosis of Canavan disease—problems and dilemmas. J Inherit Metab Dis. 1999;22:263-266.
216 Rolland MO, Mandon G, Bernard A, et al. Unreliable verification of prenatal diagnosis of Canavan disease: aspartoacylase activity in deficient and normal fetal skin fibroblasts. J Inherit Metab Dis. 1994;17:748.
217 Elpeleg ON, Shaag A, Anikster Y, et al. Prenatal detection of Canavan disease (aspartoacylase deficiency) by DNA analysis. J Inherit Metab Dis. 1994;17:664-666.
218 Matalon R, Michals-Matalon K. Prenatal diagnosis of Canavan disease. Prenat Diagn. 1999;19:669-670.
219 ACOG committee opinion. Screening for Canavan disease. Number 212, November 1998. Committee on Genetics. American College of Obstetricians and Gynecologists. Int J Gynaecol Obstet. 1999;65:91-92.
220 Matalon R, Michals K, Sebesta D, et al. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am J Med Genet. 1988;29:463-471.
221 Madhavarao CN, Arun P, Moffett JR, et al. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci U S A. 2005;102:5221-5226.
222 Burlina AP, Ferrari V, Divry P, et al. N-acetylaspartylglutamate in Canavan disease: an adverse effector? Eur J Pediatr. 1999;158:406-409.
223 Baslow MH. Brain N-acetylaspartate as a molecular water pump and its role in the etiology of Canavan disease: a mechanistic explanation. J Mol Neurosci. 2003;21:185-190.
224 Matalon R, Rady PL, Platt KA, et al. Knockout mouse for Canavan disease: a model for gene transfer to the central nervous system. J Gene Med. 2000;2:165-175.
225 Kitada K, Akimitsu T, Shigematsu Y, et al. Accumulation of N-acetyl-L-aspartate in the brain of the tremor rat, a mutant exhibiting absence-like seizure and spongiform degeneration in the central nervous system. J Neurochem. 2000;74:2512-2519.
226 Leone P, Janson CG, Bilaniuk L, et al. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann Neurol. 2000;48:27-38.
227 Fink DJ. Gene therapy for Canavan disease? Ann Neurol. 2000;48:9-10.
228 McPhee SW, Francis J, Janson CG, et al. Effects of AAV-2–mediated aspartoacylase gene transfer in the tremor rat model of Canavan disease. Brain Res Mol Brain Res. 2005;135:112-121.
229 Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydro-cephalic infant. Brain. 1949;72:373-381.
230 Pridmore CL, Baraitser M, Harding B, et al. Alexander’s disease: clues to diagnosis. J Child Neurol. 1993;8:134-144.
231 Martidis A, Yee RD, Azzarelli B, et al. Neuro-ophthalmic, radiographic, and pathologic manifestations of adultonset Alexander disease. Arch Ophthalmol. 1999;117:265-267.
232 Thyagarajan D, Chataway T, Li R, et al. Dominantly-inherited adultonset leukodystrophy with palatal tremor caused by a mutation in the glial fibrillary acidic protein gene. Mov Disord. 2004;19:1244-1248.
233 Stumpf E, Masson H, Duquette A, et al. Adult Alexander disease with autosomal dominant transmission: a distinct entity caused by mutation in the glial fibrillary acid protein gene. Arch Neurol. 2003;60:1307-1312.
234 Hallervorden J. [The development of the myelin sheath and Rosenthal’s fibers]. Dtsch Z Nervenheilkd. 1961;181:547-580.
235 Goldman JE, Corbin E. Rosenthal fibers contain ubiquitinated αB-crystallin. Am J Pathol. 1991;139:933-938.
236 Friede RL. Alexander’s disease. Arch Neurol. 1964;11:414-422.
237 van der Knaap MS, Naidu S, Breiter SN, et al. Alexander disease: diagnosis with MR imaging. AJNR Am J Neuroradiol. 2001;22:541-552.
238 van der Knaap MS, Salomons GS, Li R, et al. Unusual variants of Alexander disease. Ann Neurol. 2005;57:327-338. [erratum in Ann Neurol 2005; 58:172].
239 Brenner M, Johnson AB, Boespflug-Tanguy O, et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117-120.
240 Rodriguez D, Gauthier F, Bertini E, et al. Infantile Alexander disease: spectrum of GFAP mutations and genotype-phenotype correlation. Am J Hum Genet. 2001;69:1134-1140.
241 Li R, Johnson AB, Salomons G, et al. Glial fibrillary acidic protein mutations in infantile, juvenile and adult forms of Alexander disease. Ann Neurol. 2005;57:310-326.
242 Fuchs E. The cytoskeleton and disease: genetic disorders of intermediate filaments. Annu Rev Genet. 1996;30:197-231.
243 Li R, Messing A, Goldman JE, et al. GFAP mutations in Alexander disease. Int J Dev Neurosci. 2002;20:259-268.
244 Stevenson M. Therapeutic potential of RNA interference. N Engl J Med. 2004;351:1772-1777.
245 Bogaert L, Scherer H, Epstein E. Une Forme Cerebrale de la Cholesterinose Generalisee. Paris: Masson et Cie, 1937.
246 Berginer VM, Salen G, Patel Y. Cerebrotendinous xanthomatosis. In: Rosenberg RN, Prusiner SB, de Mauro S, et al, editors. Molecular and Genetic Basis of Neurologic and Psychiatric Disease. Philadelphia: Elsevier; 2003:575-580.
247 Bartholdi D, Zumsteg D, Verrips A, et al. Spinal phenotype of cerebrotendinous xanthomatosis—a pitfall in the diagnosis of multiple sclerosis. J Neurol. 2004;251:105-107.
248 Wevers RA, Cruysberg JR, Van Heijst AF, et al. Paediatric cerebrotendinous xanthomatosis. J Inherit Metab Dis. 1992;15:374-376.
249 Gilad R, Lampl Y, Lev D, et al. Cerebrotendinous xanthomatosis without xanthomas. Clin Genet. 1999;56:405-406.
250 Cali JJ, Hsieh CL, Francke U, et al. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem. 1991;266:7779-7783.
251 Bjorkhem I, Boberg KM, Lettersdorf Y. Inborn errors in bile acid biosynthesis and storage of sterols other than cholesterol. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:2961-2988.
252 Seyama Y, Ichikawa K, Yamakawa T. Quantitative determination of cholestanol in plasma with mass fragmentography. Biochemical diagnosis of cerebrotendinous xanthomatosis. J Biochem (Tokyo). 1976;80:223-228.
253 Koopman BJ, van der Molen JC, Wolthers BG, et al. Screening for cerebrotendinous xanthomatosis by using an enzymatic assay for 7α-hydroxylated steroids in urine. Clin Chem. 1987;33:142-143.
254 Menkes JH, Schimschock JR, Swanson PD. Cerebrotendinous xanthomatosis. The storage of cholestanol within the nervous system. Arch Neurol. 1968;19:47-53.
255 Berginer VM, Salen G, Shefer S. Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med. 1984;311:1649-1652.
256 Federico A, Dotti MT. Cerebrotendinous xanthomatosis: clinical manifestations, diagnostic criteria, pathogenesis, and therapy. J Child Neurol. 2003;18:633-638.
257 Wolthers BG, van der Molen JC, Walrecht H, et al. Reduction of urinary bile alcohol excretion and serum cholestanol in patients with cerebrotendinous xanthomatosis after oral administration of deoxycholic acid. Clin Chim Acta. 1990;193:113-118.
258 Batta AK, Salen G, Shefer S, et al. Increased plasma bile alcohol glucuronides in patients with cerebrotendinous xanthomatosis: effect of chenodeoxycholic acid. J Lipid Res. 1987;28:1006-1012.
259 Mondelli M, Sicurelli F, Scarpini C, et al. Cerebrotendinous xanthomatosis: 11-year treatment with chenodeoxycholic acid in five patients. An electrophysiological study. J Neurol Sci. 2001;190:29-33.
260 Pedley TA, Emerson RG, Warner CL, et al. Treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. Ann Neurol. 1985;18:517-518.
261 Swanson PD, Cromwell LD. Magnetic resonance imaging in cerebrotendinous xanthomatosis. Neurology. 1986;36:124-126.
262 Berginer VM, Shany S, Alkalay D, et al. Osteoporosis and increased bone fractures in cerebrotendinous xanthomatosis. Metabolism. 1993;42:69-74.
263 Federico A, Dotti MT, Lore F, et al. Cerebrotendinous xanthomatosis: pathophysiological study on bone metabolism. J Neurol Sci. 1993;115:67-70.
264 van Heijst AF, Verrips A, Wevers RA, et al. Treatment and follow-up of children with cerebrotendinous xanthomatosis. Eur J Pediatr. 1998;157:313-316.
265 Lewis B, Mitchell WD, Marenah CB, et al. Cerebrotendinous xanthomatosis: biochemical response to inhibition of cholesterol synthesis. Br Med J (Clin Res Ed). 1983;287:21-22.
266 Nakamura T, Matsuzawa Y, Takemura K, et al. Combined treatment with chenodeoxycholic acid and pravastatin improves plasma cholestanol levels associated with marked regression of tendon xanthomas in cerebrotendinous xanthomatosis. Metabolism. 1991;40:741-746.
267 Salen G, Batta AK, Tint GS, et al. Comparative effects of lovastatin and chenodeoxycholic acid on plasma cholestanol levels and abnormal bile acid metabolism in cerebrotendinous xanthomatosis. Metabolism. 1994;43:1018-1022.
268 Dotti MT, Lutjohann D, von Bergmann K, et al. Normalisation of serum cholestanol concentration in a patient with cerebrotendinous xanthomatosis by combined treatment with chenodeoxycholic acid, simvastatin and LDL apheresis. Neurol Sci. 2004;25:185-191.
269 Mimura Y, Kuriyama M, Tokimura Y, et al. Treatment of cerebrotendinous xanthomatosis with low-density lipoprotein (LDL)–apheresis. J Neurol Sci. 1993;114:227-230.
270 Berginer VM, Salen G. LDL-apheresis cannot be recommended for treatment of cerebrotendinous xanthomatosis. J Neurol Sci. 1994;121:229-232.
271 Kuriyama M, Mimura Y. LDL-apheresis in cerebrotendinous xanthomatosis: reply to letter. J Neurol Sci. 1994;121:231-232.
272 Sjögren T, Larsson T. Oligophrenia in combination with congenital ichthyosis and spastic disorders; a clinical and genetic study. Acta Psychiatr Neurol Scand. 1957;32:1-112.
273 Rizzo WB. Sjögren-Larsson syndrome: fatty aldehyde dehydrogenase deficiency. In: Scriver CR, Beaudet AL, Sly WS, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001:2239-2258.
274 Jagell S, Polland W, Sandgren O. Specific changes in the fundus typical for the Sjögren-Larsson syndrome. An ophthalmological study of 35 patients. Acta Ophthalmol (Copenh). 1980;58:321-330.
275 McLennan JE, Gilles FH, Robb RM. Neuropathological correlation in Sjögren-Larsson syndrome. Oligophrenia, ichthyosis and spasticity. Brain. 1974;97:693-708.
276 Willemsen MA, van der Graaf M, van der Knaap MS, et al. MR imaging and proton MR spectroscopic studies in Sjögren-Larsson syndrome: characterization of the leukoencephalopathy. AJNR Am J Neuroradiol. 2004;25:649-657.
277 Rizzo WB, Dammann AL, Craft DA. Sjögren-Larsson syndrome. Impaired fatty alcohol oxidation in cultured fibroblasts due to deficient fatty alcohol:nicotinamide adenine dinucleotide oxidoreductase activity. J Clin Invest. 1988;81:738-744.
278 De Laurenzi V, Rogers GR, Hamrock DJ, et al. Sjögren-Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52-57.
279 Rizzo WB, Carney G. Sjögren-Larsson syndrome: diversity of mutations and polymorphisms in the fatty aldehyde dehydrogenase gene (ALDH3A2). Hum Mutat. 2005;26:1-10.
280 Rizzo WB, Craft DA. Sjögren-Larsson syndrome. Deficient activity of the fatty aldehyde dehydrogenase component of fatty alcohol:NAD+ oxidoreductase in cultured fibroblasts. J Clin Invest. 1991;88:1643-1648.
281 Kelson TL, Craft DA, Rizzo WB. Carrier detection for Sjögren-Larsson syndrome. J Inherit Metab Dis. 1992;15:105-111.
282 Rizzo WB, Craft DA, Kelson TL, et al. Prenatal diagnosis of Sjögren-Larsson syndrome using enzymatic methods. Prenat Diagn. 1994;14:577-581.
283 van den Brink DM, van Miert JM, Wanders RJ. Assay for Sjögren-Larsson syndrome based on a deficiency of phytol degradation. Clin Chem. 2005;51:240-242.
284 Willemsen MA, de Jong JG, van Domburg PH, et al. Defective inactivation of leukotriene B4 in patients with Sjögren-Larsson syndrome. J Pediatr. 2000;136:258-260.
285 Hernell O, Holmgren G, Jagell SF, et al. Suspected faulty essential fatty acid metabolism in Sjögren-Larsson syndrome. Pediatr Res. 1982;16:45-49.
286 Haug S, Braun-Falco M. Adeno-associated virus vectors are able to restore fatty aldehyde dehydrogenase-deficiency. Implications for gene therapy in Sjögren-Larsson syndrome. Arch Dermatol Res. 2005;296:568-572.
287 Hanefeld F, Holzbach U, Kruse B, et al. Diffuse white matter disease in three children: an encephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy. Neuropediatrics. 1993;24:244-248.
288 Dietrich J, Lacagnina M, Gass D, et al. EIF2β5 mutations compromise GFAP+ astrocyte generation in vanishing white matter leukodystrophy. Nat Med. 2005;11:277-283.
289 Schiffmann R, Tedeschi G, Kinkel RP, et al. Leukodystrophy in patients with ovarian dysgenesis. Ann Neurol. 1997;41:654-661.
290 Vermeulen G, Seidl R, Mercimek-Mahmutoglu S, et al. Fright is a provoking factor in vanishing white matter disease. Ann Neurol. 2005;57:560-563.
291 Fogli A, Wong K, Eymard-Pierre E, et al. Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2β5 locus. Ann Neurol. 2002;52:506-510.
292 Ainsworth C. Molecular medicine: lost in translation. Nature. 2005;435:556-558.
293 van der Knaap MS, Kamphorst W, Barth PG, et al. Phenotypic variation in leukoencephalopathy with vanishing white matter. Neurology. 1998;51:540-547.
294 Ohtake H, Shimohata T, Terajima K, et al. Adultonset leukoencephalopathy with vanishing white matter with a missense mutation in EIF2β5. Neurology. 2004;62:1601-1603.
295 Van Haren K, van der Voorn JP, Peterson DR, et al. The life and death of oligodendrocytes in vanishing white matter disease. J Neuropathol Exp Neurol. 2004;63:618-630.
296 Fogli A, Schiffmann R, Bertini E, et al. The effect of genotype on the natural history of eIF2β-related leukodystrophies. Neurology. 2004;62:1509-1517.
297 van der Knaap MS, Leegwater PA, van Berkel CG, et al. Arg113His mutation in eIF2βε as cause of leukoencephalopathy in adults. Neurology. 2004;62:1598-1600.
298 Besenski N, Bosnjak V, Cop S, et al. Neuroimaging and clinically distinctive features in van der Knaap megalencephalic leukoencephalopathy. Int J Neuroradiol. 1997;3:244-249.
299 Goutieres F, Boulloche J, Bourgeois M, et al. Leukoencephalopathy, megalencephaly, and mild clinical course. A recently individualized familial leukodystrophy. Report on five new cases. J Child Neurol. 1996;11:439-444.
300 Higuchi Y, Hattori H, Tsuji M, et al. Partial seizures in leukoencephalopathy with swelling and a discrepantly mild clinical course. Brain Dev. 2000;22:387-389.
301 van der Knaap MS, Barth PG, Vrensen GF, et al. Histopathology of an infantile-onset spongiform leukoencephalopathy with a discrepantly mild clinical course. Acta Neuropathol (Berl). 1996;92:206-212.
302 Boor PKI, de Groot K, Waisfisz Q, et al. MCL1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol. 2005;64:412-419.
303 Schmitt A, Gofferje V, Weber M, et al. The brain-specific protein MLC1 implicated in megalencephalic leukoencephalopathy with subcortical cysts is expressed in glial cells in the murine brain. Glia. 2003;44:283-295.
304 Neely JD, Amiry-Moghaddam M, Ottersen OP, et al. Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci U S A. 2001;98:14108-14113.
305 Verloes A, Maquet P, Sadzot B, et al. Nasu-Hakola syndrome: polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy and presenile dementia. J Med Genet. 1997;34:753-757.
306 Malandrini A, Scarpini C, Palmeri S, et al. Palatal myoclonus and unusual MRI findings in a patient with membranous lipodystrophy. Brain Dev. 1996;18:59-63.
307 Haruta K, Matsunaga S, Ito H, et al. Membranous lipodystrophy (Nasu-Hakola disease) presenting an unusually benign clinical course. Oncol Rep. 2003;10:1007-1010.
308 Nasu T, Tsukahara Y, Terayama K. A lipid metabolic disease—“membranous lipodystrophy”—an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol Jpn. 1973;23:539-558.
309 Paloneva J, Autti T, Raininko R, et al. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. 2001;56:1552-1558.
310 Iivanainen M, Hakola P, Erkinjuntti T, et al. Cerebral MR and CT imaging in polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. J Comput Assist Tomogr. 1984;8:940-943.
311 Paloneva J, Kestila M, Wu J, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25:357-361.
312 Colonna M. Immunology. Unmasking the killer’s accomplice. Nature. 1998;391:642-643.
313 Bakker AB, Wu J, Phillips JH, et al. NK cell activation: distinct stimulatory pathways counterbalancing inhibitory signals. Hum Immunol. 2000;61:18-27.
314 Verhoeven NM, Huck JH, Roos B, et al. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am J Hum Genet. 2001;68:1086-1092.
315 van der Knaap MS, Wevers RA, Struys EA, et al. Leukoencephalopathy associated with a disturbance in the metabolism of polyols. Ann Neurol. 1999;46:925-928.
316 Huck JH, Verhoeven NM, Struys EA, et al. Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy. Am J Hum Genet. 2004;74:745-751.
317 van der Knaap MS, Naidu S, Pouwels PJ, et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol. 2002;23:1466-1474.
318 van der Knaap MS, van der Voorn P, Barkhof F, et al. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann Neurol. 2003;53:252-258.
319 Henneke M, Preuss N, Engelbrecht V, et al. Cystic leukoencephalopathy without megalencephaly: a distinct disease entity in 15 children. Neurology. 2005;64:1411-1416.
320 Nagae-Poetscher LM, Bibat G, Philippart M, et al. Leukoencephalopathy, cerebral calcifications, and cysts: new observations. Neurology. 2004;62:1206-1209.
321 Chretien F, Servan J, Elghozi D, et al. [Familial orthochromatic leukodystrophy: clinicopathological study of two cases]. Rev Neurol (Paris). 2001;157:178-186.
322 Shannon P, Wherrett JR, Nag S. A rare form of adult onset leukodystrophy: orthochromatic leukodystrophy with pigmented glia. Can J Neurol Sci. 1997;24:146-150.
323 Axelsson R, Roytta M, Sourander P, et al. Hereditary diffuse leukoencephalopathy with spheroids. Acta Psychiatr Scand. 1984;314:7-65.
324 van der Knaap MS, Naidu S, Kleinschmidt-Demasters BK, et al. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology. 2000;54:463-468.
325 Harbord MG, Finn JP, Hall-Craggs MA, et al. Early onset leukodystrophy with distinct facial features in 2 siblings. Neuropediatrics. 1989;20:154-157.
326 Leuzzi V, Rinna A, Gallucci M, et al. Ataxia, deafness, leukodystrophy: inherited disorder of the white matter in three related patients. Neurology. 2000;54:2325-2328.
327 Eldridge R, Anayiotos CP, Schlesinger S, et al. Hereditary adultonset leukodystrophy simulating chronic progressive multiple sclerosis. N Engl J Med. 1984;311:948-953.
328 Abe K, Ikeda M, Watase K, et al. A kindred of hereditary adultonset leukodystrophy with sparing of the optic radiations. Neuroradiology. 1993;35:281-283.
329 Hagen K, Boman H, Mellgren SI, et al. Progressive central and peripheral demyelinating disease of adult onset in a Nor-wegian family. Arch Neurol. 1998;55:1467-1472.
330 Tagawa A, Ono S, Inoue K, et al. A new familial adultonset leukodystrophy manifesting as cerebellar ataxia and dementia. J Neurol Sci. 2001;183:47-55.
331 Autti T, Muttilainen M, Raininko R, et al. Extensive cerebral white matter abnormality without clinical symptoms: a new hereditary condition? Ann Neurol. 1999;45:801-805.
332 Nomoto N, Iwasaki Y, Arasaki K, et al. Autosomal dominant childhood onset slowly progressive leukodystrophy—a Japanese family with spastic paraparesis, ataxia, mental deterioration, and skeletal abnormality. J Neurol Sci. 2004;221:35-39.
333 Lossos A, Cooperman H, Soffer D, et al. Hereditary leukoencephalopathy and palmoplantar keratoderma: a new disorder with increased skin collagen content. Neurology. 1995;45:331-337.
334 Norton KK, Carey JC, Gutmann DH. Oculodentodigital dysplasia with cerebral white matter abnormalities in a two-generation family. Am J Med Genet. 1995;57:458-461.
335 Dichgans M, Mayer M, Uttner I, et al. The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol. 1998;44:731-739.
336 Markus HS, Martin RJ, Simpson MA, et al. Diagnostic strategies in CADASIL. Neurology. 2002;59:1134-1138.
337 Pantoni L, Garcia JH. The significance of cerebral white matter abnormalities 100 years after Binswanger’s report. A review. Stroke. 1995;26:1293-1301.
338 Kang PB, Hunter JV, Melvin JJ, et al. Infantile leukoencephalopathy owing to mitochondrial enzyme dysfunction. J Child Neurol. 2002;17:421-428.
339 Harpey JP, Heron D, Prudent M, et al. Diffuse leukodystrophy in an infant with cytochrome-c oxidase deficiency. J Inherit Metab Dis. 1998;21:748-752.
340 Rahman S, Brown RM, Chong WK, et al. A SURF1 gene mutation presenting as isolated leukodystrophy. Ann Neurol. 2001;49:797-800.
341 Barone R, Parano E, Trifiletti RR, et al. White matter changes mimicking a leukodystrophy in a patient with mucopolysac-charidosis: characterization by MRI. J Neurol Sci. 2002;195:171-175.
342 Bischof F, Nagele T, Wanders RJ, et al. 3-Hydroxy-3-methyl-glutaryl–CoA lyase deficiency in an adult with leukoencephalopathy. Ann Neurol. 2004;56:727-730.
343 Surtees R. Demyelination and inborn errors of the single carbon transfer pathway. Eur J Pediatr. 1998;157(Suppl 2):S118-S121.
344 Tsao CY, Mendell JR, Rusin J, et al. Congenital muscular dystrophy with complete laminin-α2-deficiency, cortical dysplasia, and cerebral white-matter changes in children. J Child Neurol. 1998;13:253-256.
345 van Engelen BG, Leyten QH, Bernsen PL, et al. Familial adultonset muscular dystrophy with leukoencephalopathy. Ann Neurol. 1992;32:577-580.
346 Kato T, Funahashi M, Matsui A, et al. MRI of disseminated developmental dysmyelination in Fukuyama type of CMD. Pediatr Neurol. 2000;23:385-388.
347 Eichler FS, Wang P, Wityk RJ, et al. Diffuse metabolic abnormalities in reversible posterior leukoencephalopathy syndrome. AJNR Am J Neuroradiol. 2002;23:833-837.






