[level-membership-for-critical-care-medicine-category]
CHAPTER 95 THE IMMUNOLOGY OF TRAUMA
Following trauma, the immune system is called into action by signals from injured tissues. Injuries, hypoxia, and hypotension, as well as secondary insults such as ischemia/reperfusion injuries, compartment syndromes, operative interventions, and infections induce a host response that is characterized by local and systemic release of proinflammatory cytokines, arachidonic acid metabolites, and activation of complement factors, kinins, and coagulation as well as hormonal mediators. Clinically, this is the systemic inflammatory response syndrome (SIRS). Paralleling SIRS is an anti-inflammatory response referred to as the compensatory anti-inflammatory response syndrome (CARS). An imbalance between these responses appears to be responsible for increased susceptibility to infection and organ dysfunction. The aim of this chapter is to provide an overview of the immune response following trauma.
TWO-HIT MODEL
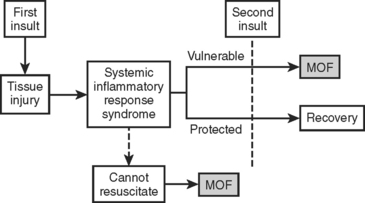
In the two-hit model, the inciting injury induces a systemic inflammatory response (Figure 1). This “first hit” primes the immune system for an exaggerated and potentially lethal inflammatory reaction to a secondary, otherwise nonlethal, stimulus (“second hit”). This secondary stimulus may be either endogenous or exogenous. Endogenous second hits include cardiovascular instability, respiratory distress, metabolic derangements, and ischemia/reperfusion injuries. In contrast, exogenous second hits include surgical interventions, blood product transfusions, and missed injuries. The two-hit model proposes that this second hit results in destructive inflammation leading to multiple organ failure (MOF) and potentially death. This model has been supported by the work of Moore and colleagues who linked postinjury opportunistic infections to SIRS and MOF.
SYSTEMIC INFLAMMATORY RESPONSE SYNDROME
In 1991, a consensus conference of the American College of Chest Physicians and the American Society of Critical Care Medicine (ACCP/SCCM) defined SIRS as a generalized inflammatory response triggered by a variety of infectious and noninfectious events. They arbitrarily established clinical parameters through a process of consensus. Table 1 summarizes the diagnostic criteria for SIRS. At least two of the four criteria must be present to fulfill the diagnosis of SIRS. Note that this definition emphasizes the inflammatory process regardless of the presence of infection. The term sepsis is reserved for SIRS when infection is suspected or proven. Subsequent studies have validated these criteria as predictive of increased intensive care unit (ICU) mortality, and that this risk increases concurrent with the number of criteria present.
Table 1 Clinical Parameters of Systemic Inflammatory Response Syndrome
PaCO2, Arterial CO2 partial pressure.
COMPENSATORY ANTI-INFLAMMATORY RESPONSE SYNDROME
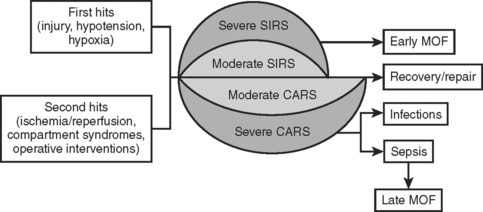
Trauma not only stimulates the release of proinflammatory mediators, but also the parallel release of anti-inflammatory mediators. This compensatory anti-inflammatory response is present concurrently with SIRS (Figure 2). When these two opposing responses are appropriately balanced, the traumatized individual is able to effectively heal the injury without incurring secondary injury from the autoimmune inflammatory response. However, overwhelming CARS appears responsible for post-traumatic immunosuppression, which leads to increased susceptibility to infections and sepsis. With time, SIRS ceases to exist and CARS is the predominant force.
CYTOKINE RESPONSE
Following trauma, IL-12 production is decreased, stimulating a shift in favor of TH2 cells and the subsequent production of antiinflammatory mediators IL-4, IL-10, IL-13, and transforming growth factor beta (TGF-β). This decrease in IL-12 and resultant increase in TH2 cells correlates with adverse outcomes. A listing of pro and anti-inflammatory mediators appears in Tables 2 and 3.
| Mediator | Action |
|---|---|
| IL-1 | IL-1 is pleiotropic. Locally, it stimulates cytokine and cytokine receptor production by T cells as well as stimulating B-cell proliferation. Systemically, IL-1 modulates endocrine responses and induces the acute phase response. |
| IL-6 | Il-6 induces acute-phase reactants in hepatocytes and plays an essential role in the final differentiation of B cells into Ig-secreting cells. Additionally, IL-6 has antiinflammatory properties. |
| IL-8 | IL-8 is a major mediator of the inflammatory response. It functions as a chemoattractant and is also a potent angiogenic factor. |
| IL-12 | IL-12 regulates the differentiation of naive T cells into TH1 cells. It stimulates the growth and function of T cells and alters the normal cycle of apoptotic cell death. |
| TNF-α | TNF-α is pleiotropic. TNF-α and IL-1 act alone or together to induce systemic inflammation as above. TNF-α is also chemotactic for neutrophils and monocytes, as well as increasing neutrophil activity. |
| MIF | MIF forms a crucial link between the immune and neuroendocrine systems. It acts systemically to enhance the secretion of IL-1 and TNF-α |
Ig, Immunoglobulin; IL, interleukin; MIF, migration inhibitory factor; TNF, tumor necrosis factor.
Table 3 Anti-Inflammatory Mediators
| Mediator | Action |
|---|---|
| IL-4 | IL-4, IL-3, IL-5, IL-13, and CSF2 form a cytokine gene cluster on chromosome 5q, with this gene particularly close to IL-13. |
| IL-10 | IL-10 has pleiotropic effects in immunoregulation and inflammation. It downregulates the expression of TH1 cytokines, MHC class II antigens, and costimulatory molecules on macrophages. It also enhances B-cell survival, proliferation, and antibody production. In addition, it can block NF-kappa B activity, and is involved in the regulation of the JAK-STAT signaling pathway. |
| IL-11 | IL-11 stimulates the T-cell–dependent development of immunoglobulin-producing B cells. It is also found to support the proliferation of hematopoietic stem cells and megakaryocyte progenitor cells. |
| IL-13 | IL-13 is involved in several stages of B-cell maturation and differentiation. It upregulates CD23 and MHC class II expression, and promotes IgE isotype switching of B cells. It downregulates macrophage activity, thereby inhibiting the production of proinflammatory cytokines and chemokines. |
| IFN-α | IFN-α enhances and modifies the immune response. |
| TGF-β | TGF-β regulates the proliferation and differentiation of cells, wound healing, and angiogenesis. |
| α-MSH | α-MSH modulates inflammation by way of three mechanisms: direct action on peripheral inflammatory cells, actions on brain inflammatory cells to modulate local reactions, and indirect activation of descending neural anti-inflammatory pathways that control peripheral tissue inflammation. |
CSF, Colony-stimulating factor; IFN, interferon; Ig, immunoglobulin; IL, interleukin; MHC, major histo-compatibility complex; MSH, melanocyte stimulating hormone; TGF, transforming growth factor; TH, T helper.
CELL-MEDIATED RESPONSE
Trauma alters the ability of splenic, peritoneal, and alveolar macrophages to release IL-1, IL-6, and TNF-α leading to decreased levels of these proinflammatory cytokines. Kupffer cells however, have an enhanced capacity for production of proinflammatory cytokines. Cell-mediated immunity not only requires functional macrophage and T cells but also intact macrophage–T-cell interaction. Following injury, human leukocyte antigen (HLA-DR) receptor expression is decreased leading to a loss of antigen-presenting capacity and decreased TNF-α production. Prostaglandin E2, IL-10, and TGF-β all contribute to this “immunoparalysis.”
ACUTE-PHASE REACTION
The acute-phase reaction describes the early systemic response following trauma and other insult states. During this phase, the biosynthetic profile of the liver is significantly altered. Under normal circumstances, the liver synthesizes a range of plasma proteins at steady state concentrations. However, during the acute phase reaction, hepatocytes increase the synthesis of positive acute-phase proteins (i.e., C-reactive protein [CRP], serum amyloid A [SAA], complement proteins, coagulation proteins, proteinase inhibitors, metal-binding proteins, and other proteins) essential to the inflammatory process at the expense of the negative acute-phase proteins. The list of acute-phase proteins is in Table 4.
| Group | Individual Proteins |
|---|---|
| Positive Acute-Phase Proteins | |
| Major acute phase proteins | C-reactive protein, serum amyloid A |
| Complement proteins | C2, C3, C4, C5, C9, B, C1 inhibitor, C4 binding protein |
| Coagulation proteins | Fibrinogen, prothrombin, von Willebrand factor |
| Proteinase proteins | α1-Pntitrypsin, α1-antichymotrypsin, α2-antiplasmin, heparin cofactor II, plasminogen activator inhibitor I |
| Metal-binding proteins | Haptoglobin, hemopexin, ceruloplasmin, manganese superoxide dismutase |
| Other proteins | α1-acid glycoprotein, heme oxygenase, mannose-binding protein, leukocyte protein I, lipoprotein (a), lipopolysac-charide-binding protein |
| Negative Acute-Phase Proteins | Albumin, prealbumin, transferrin, apolipoprotein AI, apolipoprotein AII, α2-Heremans-Schmid glycoprotein, inter– α-trypsin inhibitor, histidine-rich glycoprotein, protein c, protein s, antithrombin III, high-density lipoprotein |
Note: Positive acute-phase proteins increase production during an acute-phase response. Negative acute-phase proteins have decreased production during an acute-phase response.
SUMMARY
Injury triggers a tremendously complex response involving a multitude of systems. The individual’s immune system must balance the proinflammatory response, which is necessary to clear injured tissue, yet not cause overwhelming endogenous injury with a necessary downregulation of the inflammatory process in order to provide an environment of minimal inflammation that can nurture the cell proliferation and tissue remodeling needed for healing. A loss of this balance can cause additional tissue injury from the immune response itself, or leave the individual susceptible to infection and sepsis. As our understanding of the immune response following injury grows, the more likely that we will be able to monitor and effectively manage the traumatized, critically ill patient.
Angele MK, Chaundry IH. Surgical trauma and immunosuppression: pathophysiology and potential immunomodulatory approaches. Arch Surg. 2005;390:333-341.
Ayala A, Chung C-S, Grutkoski PS, Song GY. Mechanisms of immune resolution. Crit Care Med. 2003;31:S558-S571.
Cook MC. Immunology of trauma. J Trauma. 2001;3:79-88.
DeLong WG, Born CT. Cytokines in patients with polytrauma. Clin Orthop Relat Res. 2004;422:57-65.
Faist E, Angele MK, Wichmann M. The Immune Response. In Trauma, 5th ed., New York: McGraw Hill; 2004:1383-1396.
Faist E, Schinkel C, Zimmer S. Update on the mechanisms of immune suppression of injury and immune modulation. World J Surg. 1996;20:454-459.
Fosse E, Pillgram-Larsen J, Svennevig JL, Nordby C, Skulberg A, Mollnes TE, Abdelnoor M. Complement activation in injured patients occurs immediately and is dependent on the severity of the trauma. Injury. 1998;29:509-514.
Goris RJA. MODS/SIRS: result of an overwhelming inflammatory response? World J Surg. 1996;20:418-421.
Gruys E, Toussaint MJM, Niewold TA, Koopmans SJ. Acute phase reaction and acute phase proteins. J Zhejiang Univ Sci 6B. 2005;11:1045-1056.
Harris BH, Gelfand JA. The immune response to trauma. Semin Pediatr Surg. 1995;4:77-82.
Keel M, Trentz O. Pathophysiology of trauma. Injury. 2005;36:691-709.
Kubes P, Ward PA. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. 2000;10:127-135.
Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301-305.
Menger MD, vollmar B. Surgical trauma: hyperinflammation versus immunosuppression? Arch Surg. 2004;389:475-484.
Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995;75:257-277.
Murphy TJ, Paterson HM, Kriynovich S, et al. Linking the “two-hit” response following injury to enhanced TLR4 reactivity. J Leukocyte Biol. 2005;77:16-23.
Nathens AB, Marshall JC. Sepsis, SIRS, and MODS: what’s in a name? World J Surg. 1996;20:386-391.
Sauaia A, Moore FA, Moore EE, Lezotte DC. Early risk factors for postinjury multiple organ failure. World J Surg. 1996;20:392-400.
Schinkel C, Wick M, Muhr G, Köller. Analysis of systemic interleukin-11 after major trauma. Shock. 2005;23:30-34.
Schlag G, Redl H. Mediators of injury and inflammation. World J Surg. 1996;20:406-410.
Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005;7:151-185.
Sugimoto K, Hirata M, Majima M, Katori M, Ohwada T. Evidence for a role of kallikrein-kinin system in patients with shock after blunt trauma. Am J Physiol Regul Integr Comp Physiol. 1998;274:R1556-R1560.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
CHAPTER 95 THE IMMUNOLOGY OF TRAUMA
Following trauma, the immune system is called into action by signals from injured tissues. Injuries, hypoxia, and hypotension, as well as secondary insults such as ischemia/reperfusion injuries, compartment syndromes, operative interventions, and infections induce a host response that is characterized by local and systemic release of proinflammatory cytokines, arachidonic acid metabolites, and activation of complement factors, kinins, and coagulation as well as hormonal mediators. Clinically, this is the systemic inflammatory response syndrome (SIRS). Paralleling SIRS is an anti-inflammatory response referred to as the compensatory anti-inflammatory response syndrome (CARS). An imbalance between these responses appears to be responsible for increased susceptibility to infection and organ dysfunction. The aim of this chapter is to provide an overview of the immune response following trauma.
TWO-HIT MODEL
In the two-hit model, the inciting injury induces a systemic inflammatory response (Figure 1). This “first hit” primes the immune system for an exaggerated and potentially lethal inflammatory reaction to a secondary, otherwise nonlethal, stimulus (“second hit”). This secondary stimulus may be either endogenous or exogenous. Endogenous second hits include cardiovascular instability, respiratory distress, metabolic derangements, and ischemia/reperfusion injuries. In contrast, exogenous second hits include surgical interventions, blood product transfusions, and missed injuries. The two-hit model proposes that this second hit results in destructive inflammation leading to multiple organ failure (MOF) and potentially death. This model has been supported by the work of Moore and colleagues who linked postinjury opportunistic infections to SIRS and MOF.
SYSTEMIC INFLAMMATORY RESPONSE SYNDROME
In 1991, a consensus conference of the American College of Chest Physicians and the American Society of Critical Care Medicine (ACCP/SCCM) defined SIRS as a generalized inflammatory response triggered by a variety of infectious and noninfectious events. They arbitrarily established clinical parameters through a process of consensus. Table 1 summarizes the diagnostic criteria for SIRS. At least two of the four criteria must be present to fulfill the diagnosis of SIRS. Note that this definition emphasizes the inflammatory process regardless of the presence of infection. The term sepsis is reserved for SIRS when infection is suspected or proven. Subsequent studies have validated these criteria as predictive of increased intensive care unit (ICU) mortality, and that this risk increases concurrent with the number of criteria present.
Table 1 Clinical Parameters of Systemic Inflammatory Response Syndrome
PaCO2, Arterial CO2 partial pressure.
COMPENSATORY ANTI-INFLAMMATORY RESPONSE SYNDROME
Trauma not only stimulates the release of proinflammatory mediators, but also the parallel release of anti-inflammatory mediators. This compensatory anti-inflammatory response is present concurrently with SIRS (Figure 2). When these two opposing responses are appropriately balanced, the traumatized individual is able to effectively heal the injury without incurring secondary injury from the autoimmune inflammatory response. However, overwhelming CARS appears responsible for post-traumatic immunosuppression, which leads to increased susceptibility to infections and sepsis. With time, SIRS ceases to exist and CARS is the predominant force.
CYTOKINE RESPONSE
Following trauma, IL-12 production is decreased, stimulating a shift in favor of TH2 cells and the subsequent production of antiinflammatory mediators IL-4, IL-10, IL-13, and transforming growth factor beta (TGF-β). This decrease in IL-12 and resultant increase in TH2 cells correlates with adverse outcomes. A listing of pro and anti-inflammatory mediators appears in Tables 2 and 3.
| Mediator | Action |
|---|---|
| IL-1 | IL-1 is pleiotropic. Locally, it stimulates cytokine and cytokine receptor production by T cells as well as stimulating B-cell proliferation. Systemically, IL-1 modulates endocrine responses and induces the acute phase response. |
| IL-6 | Il-6 induces acute-phase reactants in hepatocytes and plays an essential role in the final differentiation of B cells into Ig-secreting cells. Additionally, IL-6 has antiinflammatory properties. |
| IL-8 | IL-8 is a major mediator of the inflammatory response. It functions as a chemoattractant and is also a potent angiogenic factor. |
| IL-12 | IL-12 regulates the differentiation of naive T cells into TH1 cells. It stimulates the growth and function of T cells and alters the normal cycle of apoptotic cell death. |
| TNF-α | TNF-α is pleiotropic. TNF-α and IL-1 act alone or together to induce systemic inflammation as above. TNF-α is also chemotactic for neutrophils and monocytes, as well as increasing neutrophil activity. |
| MIF | MIF forms a crucial link between the immune and neuroendocrine systems. It acts systemically to enhance the secretion of IL-1 and TNF-α |
Ig, Immunoglobulin; IL, interleukin; MIF, migration inhibitory factor; TNF, tumor necrosis factor.
Table 3 Anti-Inflammatory Mediators
| Mediator | Action |
|---|---|
| IL-4 | IL-4, IL-3, IL-5, IL-13, and CSF2 form a cytokine gene cluster on chromosome 5q, with this gene particularly close to IL-13. |
| IL-10 | IL-10 has pleiotropic effects in immunoregulation and inflammation. It downregulates the expression of TH1 cytokines, MHC class II antigens, and costimulatory molecules on macrophages. It also enhances B-cell survival, proliferation, and antibody production. In addition, it can block NF-kappa B activity, and is involved in the regulation of the JAK-STAT signaling pathway. |
| IL-11 | IL-11 stimulates the T-cell–dependent development of immunoglobulin-producing B cells. It is also found to support the proliferation of hematopoietic stem cells and megakaryocyte progenitor cells. |
| IL-13 |
