[level-membership-for-obstetrics-gynecology-category]
CHAPTER 35 The genetics and molecular biology of gynaecological cancer
Introduction
Gynaecological cancers provide examples of a number of contrasting mechanisms of carcinogenesis (Figure 35.1). For example, ovarian and endometrial cancer can occur as components of familial cancer syndromes (familial breast/ovarian cancer and Lynch II syndrome) due to germline inheritance of predisposing genetic abnormalities. However, most ovarian cancers are not thought to be due to inherited genetic alterations, but result from somatic mutations occurring in ovarian cells with an initially normal genome. These somatic mutations are thought to be secondary to environmental factors that act to increase the opportunity for spontaneous mutation in a number of critical genes. One of the genetic alterations that lead to cervical cancer is caused by an environmental factor, human papilloma virus (HPV), and in at least some endometrial cancers, unopposed oestrogen exposure or hyperinsulinism leads to carcinogenesis. High-risk subtypes of HPV are much less common in vulval cancer. This suggests that vulval and cervical carcinomas are not identical aetiologically, and that factors other than HPV are more important in vulval carcinogenesis. There is recent evidence that different genetic events occur between HPV-positive and -negative vulval cancers, with a greater number of molecular alterations in HPV-negative vulval cancers compared with HPV-positive tumours (Flowers et al 1999). HPV-positive vulval cancers are more frequently found in younger patients. Vulval cancers in older patients are more often HPV negative, and more frequently show allelic loss of the TP53 gene.
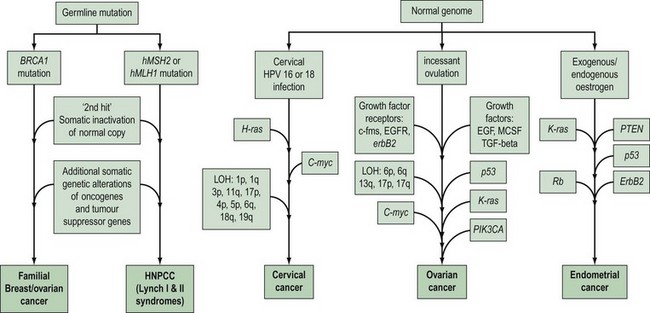
The histological features, and biological and clinical behaviour of fallopian tube cancers are similar to those of ovarian cancer. In addition, there is evidence that similar genetic alterations occur with the same frequency in fallopian tube and ovarian cancers of the same histological subtype. This suggests a common molecular pathogenesis between these cancer types (Levanon et al 2008).
To date, little is known about the molecular genetics of vaginal cancer.
Molecular Basis of Carcinogenesis
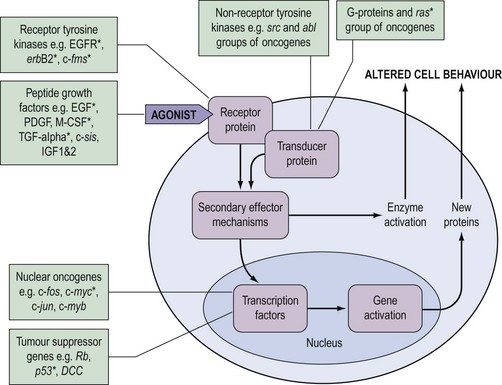
During the last decade, it has become clear that the genetic changes in cancer involve genes which control normal cell growth and proliferation. Cells communicate via a complex interacting set of signalling pathways, the principles of which, unlike the details, are well established (Figure 35.2). The best understood mechanism of communication between cells is via the release of molecules which interact as agonists with receptors either within the cell or, more frequently, in the cell membrane. This interaction results in a further signal within the cell which, via transducer proteins and secondary mechanisms, results in modified cell behaviour either by activating already synthesized target enzymes directly or by initiating transcription of genes and synthesis of their protein product. The genetic abnormalities observed in cancer are known to affect genes coding for all of the groups of molecules in Figure 35.2.
Oncogenes
The identification of oncogenes resulted from in-vitro and animal work with virus-induced cancers. As the viral genome is relatively small, it was possible to identify the specific genes (viral oncogenes) involved in the transforming activity of retroviruses, such as the Rous sarcoma virus. It was possible to identify homologous genes in normal cells, and a number of viral oncogenes were found to have normal cellular homologues known as ‘proto-oncogenes’. Other workers identified oncogenes by transfecting cell lines with DNA from human cancer cell lines, and found that some of the transforming oncogenes identified were mutated versions of the proto-oncogene identified by the retroviral approach. Subsequently, it was demonstrated that chromosome translocations such as the Philadelphia chromosome in chronic myeloid leukaemia involved known oncogenes (de Klein et al 1982).
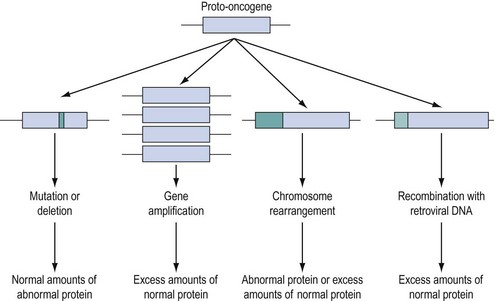
Approximately 100 proto-oncogenes have been identified, and many can be assigned to the groups of functions summarized in Figure 35.2. The mutation resulting in conversion of a proto-oncogene into an oncogene may occur in several different ways (Figure 35.3), and may result in an abnormal protein produced in a normal quantity, a normal protein produced in an excessive amount, or a normal protein produced inappropriately due to loss or alteration of control regions of the gene.
Tumour suppressor genes
The existence of tumour suppressor genes was suggested by cell fusion studies of transformed cells and non-transformed cells which resulted in a non-transformed hybrid cell. The subsequent identification of the first tumour suppressor gene followed from the study of retinoblastoma, a rare childhood cancer which may occur in a hereditary and sporadic form. On the basis of epidemiological and statistical analysis, Knudson suggested that development of the cancer required two events. He postulated that in the hereditary form, the first event was a germline mutation present in every cell of the individual, and that a second event in any of the many million retinoblasts could result in tumour formation. The rarity, later onset and unilateral nature of the sporadic form could be attributed to the need for two events in a single retinal cell in the absence of a germline mutation. When the retinoblastoma (Rb) gene was mapped to chromosome 13q and cloned, it was confirmed that in both familial and sporadic retinoblastoma, the two copies of the gene were inactivated in tumour cells, consistent with Knudson’s ‘two-hit’ hypothesis (Cavenee et al 1983).
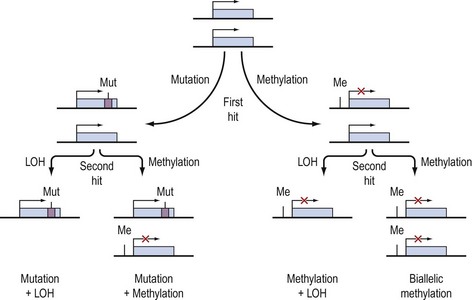
Knudson’s hypothesis that two hits are required for the full inactivation of a tumour suppressor gene has been shown to be fundamentally correct in almost all cases of human cancer. However, the fact that methylation of CpG islands located in the promoters of genes can cause transcriptional silencing, coupled with the observation that DNA methylation patterns are perturbed in cancer cells, has led to the suggestion that abnormal methylation of the promoters of tumour suppressor genes might be implicated in carcinogenesis, and can serve as a third pathway to inactivation of a tumour suppressor gene (Figure 35.4) (Jones and Laird 1999).
Epigenetics and DNA methylation in cancer
As outlined above, aberrant gene function and altered patterns of gene expression are key features of cancer. Growing evidence shows that acquired epigenetic abnormalities participate with genetic alterations to cause this dysregulation. The modern definition of epigenetics is modifications of the DNA or associated proteins, other than DNA sequence variation, that carry information content during cell division. The best understood example of epigenetic modification is DNA methylation, a covalent addition of a methyl group derived from S-adenosyl-L-methionine to the fifth carbon of the cytosine ring to form the fifth base, 5-methylcytosine (5meC) (Laird 2003, Jones and Baylin 2007). The reaction is catalysed by DNA methyltransferases together with accessory proteins. Among all eukaryotic species, methylation occurs predominantly in cytosines located 5′ of guanines, and known as ‘CpG dinucleotides’ (CpGs). In the mammalian genome, the distribution of CpGs is far from random. CpGs are greatly under-represented in the genome through evolutionary loss of 5meC through deamination to thymine. However, clusters of CpGs known as ‘CpG islands’ (CGIs) are present in 1–2% of the genome. Typically, they range in length from 200 to 5000 base pairs (bp). Most are unmethylated under normal circumstances, with the exception of those associated with imprinted genes, genes subjected to X-chromosome inactivation and transposable elements. In this last respect, DNA methylation is thought to repress faulty expression of endogenous transposons that may disrupt the genome. It is also involved in the parental-specific silencing of one allele of imprinted genes. In addition, approximately 70% of CGIs are associated with DNA sequences 200–2000 bp long found in the promoter, the first and second exons, and the first intron regions of all genes (5′ CGIs). This suggests that CGIs are critical in gene regulation. There is, for instance, usually an inverse relationship between the degree of methylation of a regulatory CGI and the extent of gene transcription (Figure 35.5) (Laird 2003, Jones and Baylin 2007).
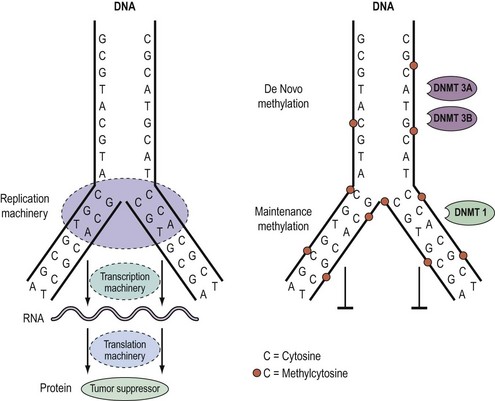
DNA methylation patterns are established during defined phases in embryonic development. With the exception of imprinted genes, gamete methylation patterns are erased by a genome-wide demethylation at around the eight-cell stage of blastocyst formation. During the implantation stage, methylation patterns are re-established via de-novo methylation. During adulthood, the degree and patterns of methylation are both tissue- and cell-type specific. Disruption of these preset patterns of DNA methylation in adult life has been linked to ageing and to disease. Furthermore, dysregulation of developmental programming by maternal factors or environmental mimics is thought to induce abnormal DNA methylation of specific genes and hence their faulty expression, leading to disease (Laird 2003, Jones and Baylin 2007).
Over the last decade, the importance of epigenetic gene regulation has become increasingly evident as studies have shown that each epithelial tumour only contains approximately 10–15 tumour suppressor genes silenced through mutations, but several hundred silenced through DNA methylation (Jones and Baylin 2007, Thomas et al 2007). Numerous genes involved in central pathways can be silenced by means of DNA methylation (Figure 35.3).
Germline and somatic genetic alterations
A common feature of tumours is that they accumulate somatic genetic changes during the development of the cancer. A proportion of cancers may also develop as a result of germline mutations in cancer predisposing genes. An important distinction must be made between germline and somatic mutations. Somatic mutations are found in the tumour cells but not the normal cells of an individual with cancer, and cannot therefore be passed on to descendants through the germline. Germline mutations, however, are present in all the cells of the individual and can initiate tumour development. They can be passed on to descendants of the individual, and can be identified in DNA obtained from all diploid cell populations (e.g. peripheral blood samples) and not just the tissues that are susceptible to disease. It is worth pointing out that the same genes mutated in the germline are also frequently mutated in the somatic cells of a cancer, which suggests a functional significance for specific genes in cancer development. For example, mutations of the p53 gene are one of the most common genetic changes found in human cancer. In most cancers, p53 mutations occur as somatic events, but germline p53 mutations are often found in patients from families with the rare Li-Fraumeni syndrome, which is characterized by sarcomas, breast cancer and other malignancies occurring at a young age. Another example is the colorectal cancer predisposition syndrome, familial adenomatous polyposis (FAP), caused by germline mutations in the APC gene (Kinzler et al 1991). The FAP syndrome is responsible for approximately 1% of all colorectal cancers, but somatic mutations of APC have been detected in over 80% of sporadic colorectal cancers and appear to be one of the earliest events in colorectal carcinogenesis.
Most of the familial cancer syndromes identified to date are rare, single gene disorders and have an autosomal-dominant pattern of inheritance. Individuals inheriting the germline abnormality are generally at high risk of developing malignancy because the penetrance of most of these genes is of the order of 80%. Another class of cancer predisposition genes that confer more modest penetrance has been identified recently. Although these genes are associated with much lower penetrance, they are also much more common in the population than the rare high-risk genes, and so are probably responsible for a greater proportion of cancer overall (reviewed in Easton and Eeles 2008). However, the identification of individuals in the population that are likely to carry these low penetrance genes is not easy because they are not often associated with a significant family history of disease.
Genetics of Familial Gynaecological Cancer
Although there are anecdotal reports of more than one case of vulval cancer occurring in close relatives, there is no good evidence of inherited predisposition to this disease. For cervical cancer, the absence of any obvious clustering of multiple cases within families suggests that no highly penetrant susceptibility loci for the disease await identification. However, population-based epidemiological studies suggest that shared genetic factors do play a role in the development of cervical cancer, and possibly account for approximately 27% of cases. This perhaps suggests the presence of several common genetic variants that confer low penetrance disease risk. Ovarian and endometrial cancers occur as part of well-defined cancer family syndromes with an autosomal-dominant inheritance pattern. Pedigree studies of cancer within families have revealed three main syndromes associated with ovarian cancer. The most common hereditary form of ovarian cancer occurs in association with breast cancer. A large number of hereditary breast/ovarian cancer families have been described in which there is a high frequency of both cancers and an association with an early age of onset. Hereditary ovarian cancer may also occur as a site-specific disease, although genetic studies suggest that a large proportion of site-specific ovarian and breast/ovarian cancer families are part of the same disease spectrum. Nevertheless, there remains evidence that there is a rare genetic component that confers high penetrance susceptibility specifically to ovarian cancer (reviewed in Ramus and Gayther 2009). Less frequently, ovarian cancer occurs in hereditary non-polyposis colorectal cancer (HNPCC) families, and germline genetic mutations in highly penetrant genes that cause HNPCC have been found in ovarian cancer cases from these families. Women from HNPCC families most frequently develop colorectal or endometrial cancer, and this is the syndrome most commonly associated with genetic predisposition to endometrial cancer. Some families have recently been described with an apparently high risk of site-specific endometrial cancer. It is important to recognize that when all of these familial syndromes are combined, they are probably responsible for less than 5% of ovarian cancer cases and a smaller proportion of endometrial cancer cases.
The BRCA1 and BRCA2 genes
The identification and detailed pedigree-based descriptions of families at high risk of breast and ovarian cancer, together with the development of polymorphic DNA markers, made it possible to perform detailed linkage analysis with the aim of locating genes involved in familial cancer. A major step forward was the report of linkage of early-onset breast cancer to the polymorphic marker CMM86 located on chromosome 17q (Hall et al 1990). Subsequently, linkage to 17q in 214 families was confirmed by a consortium of 13 research groups from Europe and the USA (Easton et al 1993). Almost all breast/ovarian cancer families and 40% of families with breast cancer alone were found to have a linkage with the BRCA1 (Breast Cancer 1) locus on chromosome 17q. Subsequently, the consortium localized the gene to a 2cM region on 17q, and in 1994, a group in Utah described a large gene on 17q within which they had identified mutations in five affected families (Miki et al 1994). Subsequent work by other research groups has confirmed that this gene is the BRCA1 gene, and over 12,000 carriers of a BRCA1 mutation have now been described in breast/ovarian cancer families and cancer cases unselected for a family history [described on the Breast Cancer Information Core (BIC) database: http://research.nhgri.nih.gov/bic/].
BRCA1 is a large gene with 22 exons encoding a protein of 1863 amino acids. There are no specific hotspots for mutation in the gene, and only a small proportion of mutations are recurrent. Overall, the penetrance of a BRCA1 mutation for ovarian cancer and breast cancer appears to be 40–50% and 80%, respectively Ford et al 1994, 1998). However, there is evidence that penetrance may be modified by both environmental factors and other genetic influences. Furthermore, the location of a mutation within the BRCA1 gene may influence both the overall risk of cancer and the type of cancer. Gayther et al (1995) reported a correlation between mutations toward the 3′ end of the gene and a greater risk of ovarian cancer than breast cancer. Well-defined founder mutations have been described in BRCA1. Mutations at 185delAG and 5382insC have been documented in approximately 1% of Ashkenazi Jewish cancers, and up to 40% of Ashkenazi Jewish women with ovarian cancer or early-onset breast cancer. Identification of BRCA2 closely followed the cloning of BRCA1. Wooster et al (1995) identified the gene following studies involving high-risk families in which the disease pattern was not linked to BRCA1.
BRCA2, located on chromosome 13q, is also a large gene with 26 coding exons encoding a protein with 3418 amino acids. More than 11,000 mutation carriers have been described to date (the BIC database). As for BRCA1, there are no hotspots for mutation, although there are founder mutations in some ethnic groups such as Ashkenazi Jews (Levy-Lahad et al 1997, Thorlacius et al 1997). Although the penetrance for breast cancer is approximately 80%, the penetrance for ovarian cancer seems to be lower than that for BRCA1 mutations at approximately 25% (Ford et al 1998). There is a suggestion that mutations in exon 11 confer a higher risk of ovarian cancer than mutations in other areas (Gayther et al 1997). This has been confirmed in a more recent meta-analysis (reviewed in Ramus and Gayther 2009). Overall, BRCA1 and BRCA2 are believed to account for over 95% of hereditary cases of ovarian cancer and over 80% of hereditary cases of breast cancers.
The functions of BRCA1 and BRCA2 are still unclear. Most of the mutations of BRCA1 are small insertions or deletions, which result in loss of protein synthesis or production of a truncated protein. This observation supports the concept that BRCA1 functions as a tumour suppressor gene. In this ‘two-hit’ model, although one copy of the gene is inherited as an inactive mutant form, the development of cancer requires somatic mutation of the other wild-type (‘normal’) gene copy. Analysis of tumours from familial breast/ovarian cancer families has demonstrated that loss of heterozygosity (LOH) in these tumours involves the wild-type gene, thus providing further evidence that the BRCA1 gene is a tumour suppressor (Smith et al 1992). Both BRCA1 and BRCA2 are expressed in the largest amounts in the testis and thymus, and at lower levels in the breast and ovary. Although the genes have limited sequence homology, they do have similarities including being A-T rich, having a large exon 11 with the start of translation at codon 2. The presence of a zinc finger motif in BRCA1 has suggested a role as a transcription factor, whilst BRCA2 has homology with known transcription factors. However, the most compelling functional evidence indicates that BRCA1 and BRCA2 both play a major role in homologous and non-homologous DNA double-strand break (DSB) repair through their interaction with the RAD51 DSB protein. It is still not known why abrogation of such a ubiquitous cellular function should predispose individuals specifically to breast and ovarian cancer.
HNPCC-associated gynaecological cancer and DNA repair genes
HNPCC is associated with autosomal-dominant inheritance of colorectal cancer in those who do not have the multiple adenomas which occur in FAP. The identification of the genetic basis of Lynch II syndrome is a notable example of the power of molecular technology. HNPCC was classified by Henry Lynch into site-specific hereditary colon cancer (Lynch I syndrome), and families with a predisposition to non-polyposis colorectal cancer in association with other cancers including stomach, small bowel, ureter, renal pelvis and brain, as well as ovarian and endometrial cancer (Lynch II syndrome) (Lynch 1985). The identification of the genes responsible for HNPCC was a result of several developments which showed that HNPCC is associated with hereditary defects in one of five DNA mismatch repair genes [mutS homolog 2 (MSH2), mutL homolog 1 (MLH1), postmeiotic segregation increased 1 (PMS1), postmeiotic segregation increased 2 (PMS2) and mutS homolog 6/G/T mismatch-binding protein (MSH6/GTBP)]. Hundreds of mutations have been identified in these five genes in HNPCC families, making mutation testing a difficult challenge in these families (Lynch and de la Chapelle 1999). Available evidence suggests that these genes function as tumour suppressor genes in a manner consistent with Knudson’s ‘two-hit’ model. The inherited germline mutation inactivates one copy of the mismatch repair gene, but inactivation of the remaining wild-type copy by a second somatic mutation is required prior to tumour formation. The secondary, acquired mutations which result from loss of DNA repair gene function include defects of APC, K-ras, DPC4 and p53 (Huang et al 1996, Kinzler and Vogelstein 1996). The penetrance for colorectal cancer in mutation carriers is in excess of 80% in men and 30% in women, and HNPCC accounts for approximately 5% of colorectal cancers (Dunlop et al 1997, Aarnio et al 1999, Salovaara et al 2000). Endometrial cancer is the second most common cancer in HNPCC families, with female carriers having a 42% risk by 70 years of age (Dunlop et al 1997). The penetrance for ovarian cancer is less, with a reported lifetime risk of 9% (Marra and Roland 1995).
Management of Familial Gynaecological Cancer
Risk assessment
Prevention of familial cancer
Use of the oral contraceptive pill may be suggested for HNPCC and breast/ovarian cancer family members on the basis of case–control studies in the general population which indicate a protective effect against ovarian and endometrial cancer. While one study has suggested a protective effect of the oral contraceptive pill in BRCA1 and BRCA2 carriers, this was not confirmed in a second study and the situation remains unclear (Modan et al 2001). Furthermore, there is concern in breast/ovarian cancer families that a reduction in risk of ovarian cancer may be offset by an increased risk of breast cancer.
Prophylactic salpingo-oophorectomy and hysterectomy as a primary procedure is justifiable in women from breast/ovarian cancer and HNPCC families after completion of their family and after thorough counselling. There is convincing evidence that salpingo-oophorectomy is an effective method of prevention of cancer of the ovary, fallopian tube and breast. A recent meta-analysis (Rebbeck et al 2009) of all reports of preventative surgery in BRCA1 and BRCA2 mutation carriers from 1999 to 2007 revealed a reduction in risk of breast cancer (hazard ratio 0.49) and ovarian/fallopian tube cancer (hazard ratio 0.21). Women undergoing prophylactic surgery should be counselled that the procedure will prevent ovarian and tubal cancer, but that they may still be at risk of primary peritoneal cancer.
It should also be noted that cases of intra-abdominal carcinomatosis following oophorectomy have been reported in which subsequent review of the oophorectomy specimen revealed a small focus of ovarian cancer (Chen et al 1985). Surgery should therefore include a careful inspection of the abdomen and pelvis, and thorough histological examination of the ovary. Fallopian tube cancer does seem to be more common in BRCA1 and BRCA2 families, so prophylactic surgery should include salpingectomy. Hysterectomy should be advised for women from HNPCC families, but women from breast/ovarian cancer families do not appear to be at increased risk of endometrial cancer and the decision regarding hysterectomy will depend on other factors.
Screening for familial cancer
The efficacy of ovarian cancer screening is unproven to date, although a survival benefit was noted in a pilot randomized trial in the general population (Jacobs et al 1999). The preliminary results of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS), a larger randomized trial involving 202,000 postmenopausal women from the general population, are encouraging (Menon et al 2009) but there will not be definitive data on mortality impact until 2015. Although the value of ovarian cancer screening is therefore unclear, there is a consensus that it is reasonable to screen women from breast/ovarian cancer families with transvaginal ultrasonography and serum CA125 measurement in view of their level of risk. Screening is usually performed on an annual basis from the mid 30s or 5 years prior to the earliest age of onset of ovarian cancer in the family.
It should be noted that both CA125 and ultrasound are associated with a substantial risk of false-positive results, leading to unnecessary surgery (Campbell et al 1993, Jacobs et al 1993). It is essential to warn women clearly about this risk. The false-positive rate for both CA125 and ultrasonography is approximately 1–2% in postmenopausal women and higher in premenopausal women. It is therefore likely that a 30-year-old woman undergoing annual screening for up to 50 years will have a false-positive result at some stage, with consequent anxiety and the risk of unnecessary surgery.
Molecular Genetics of Sporadic Gynaecological Cancer
Cervical cancer
The histopathological progression of cervical neoplasia from mild to increasingly severe dysplasia and ultimately invasive cervical cancer is well described, as is the evidence for an association with HPV infection. HPV infection is a necessary but insufficient cause for cervical cancer. Infection with oncogenic types of HPV is very common, but most of these infections go unnoticed. Carcinogenesis is a very rare outcome of HPV infection and it clearly involves interactions of the host, the environment and the virus. During recent years, important progress has been made in understanding the mechanism of HPV-induced carcinogenesis and incorporating these findings with the principles of multistep carcinogenesis. HPVs form a group of more than 80 viruses, most of which give rise to benign tumours such as wart infections. The risk of cervical neoplasia is associated with particular HPV types (HPV 16 and 18) (zur Hausen 1987). The genomes of all HPVs have a similar organization which encodes eight major open reading frames encoding early (E1–7) and late (L1–2) viral proteins. It is the early proteins E6 and E7 which are responsible for the transforming activity of the virus (Whiteside et al 2008). The E6 and E7 proteins of high-risk HPV types (HPV 16 and 18), but not those of low-risk types, cause in-vitro changes which parallel the changes in cervical intraepithelial neoplasia (Whiteside et al 2008).
During a normal viral infection resulting in production and release of new virus, the viral genome exists episomally (closed circular DNA) without integration in the nucleus of the infected cell. In tumour cells, the viral DNA is integrated with the DNA of the host cell. Although the sites of integration of viral DNA are random in cancer cell lines and many of the viral genes are lost, the E6 and E7 viral open reading frames are always conserved and are the most abundant viral transcripts in such cell lines (Whiteside et al 2008). Furthermore, integration of E6 and E7 usually occurs in a manner which disrupts the normal viral regulation of expression of these proteins. These observations suggest that E6 and E7 have an important role in cervical carcinogenesis, and are supported by evidence that these proteins can interact with and inactivate several important cellular proteins. E7 binds to the product of the Rb tumour suppressor gene, interfering with normal protein complex formation and disrupting the normal function of Rb protein. E6 interacts with the protein product of the p53 tumour suppressor gene, causing rapid breakdown of the protein and loss of normal p53 function. The overall effect of E6 and E7 expression in the cell is therefore equivalent to loss of the Rb and p53 tumour suppressor genes. This is consistent with the observation that p53 mutation is uncommon in cervical cancer but is found in HPV-16- and HPV-18-negative cervical cancer cell lines. E6 and E7 also interact with several other important cellular proteins (involved in adhesion, apoptosis, cell cycle, DNA repair, signal transduction and transcription) affecting the integrity of key pathways, as demonstrated in several in-vitro studies (Whiteside et al 2008).
Clinical and experimental evidence suggests that in common with other mechanisms of carcinogenesis, HPV infection alone is not sufficient to cause cervical cancer, but requires other factors which may influence local immune responses or result in other genetic alterations. First, high-risk genital HPV infection is very common, and the majority of individuals clear their infection with time. However, a proportion of women, approximately 15%, cannot clear the virus effectively, and the persistence of a high-risk HPV is the major risk factor for the development of anogenital malignancies. Immunosuppressed women (e.g. human immunodeficiency virus infection, transplant recipients) have a substantially increased risk of developing cervical cancer. Second, epidemiological studies suggest that other factors such as smoking and herpes simplex infection may play a role. Third, a number of genetic abnormalities have been identified in cervical cancers. These include amplification or overexpression of c-myc, mutation of c-H-ras and LOH at a number of chromosomal loci. Recurrent LOH is identified in chromosome regions 3p14–22, 4p16, 5p15, 6q21–22, 11q23, 17p, 18q12–22 and 19q13. All these regions may harbour potential tumour suppressor genes. Several studies reported amplification of chromosome 3q24–28 in up to 90% of cervical cancers. Comparative genomic hybridization (CGH) is a powerful molecular tool which allows screening of the entire genome of a tumour for genetic alterations by highlighting regions of altered DNA sequence copy numbers. CGH from preinvasive (dysplastic) cervical cells reveals several chromosomal imbalances. Most frequent gains were found on chromosomes 1p, 2q, 4 and 5, whereas losses could be found on chromosome 13q (Aubele et al 1998). As outlined above, epigenetic alterations regulate gene expression without changing the DNA sequence. As well as methylation of cytosine in DNA, these alterations include acetylation of histone proteins. Epigenetic alterations are increasingly recognized as important oncogenic mechanisms, and HPV-associated oncogenesis is no exception (reviewed in Whiteside et al 2008). DNA methylation is also presumed to be a cellular defence to silence foreign DNA transcription. Methylation of HPV is known to occur. Recent studies of HPV 16 and 18 have shown that the extent of methylation varies by region of the genome, and changes in the methylation patterns may be associated with the grade of neoplasia. Epigenetic changes could also occur through direct interaction between HPV and the cellular proteins involved in DNA methylation and chromatin remodelling, including histone deacetylase, DNA methyltransferase and p300 (reviewed in Whiteside et al 2008). Numerous genes [e.g. cadherin 1 (CDH1), fragile histidine triad gene (FHIT), telomerase reverse transcriptase (TERT), cadherin 13 (CDH13), O-6-methylguanine–DNA methyltransferase (MGMT), tissue inhibitor of metallopeptidase 3 (TIMP3) and hypermethylated in cancer 1 (HIC1)] have been demonstrated to be methlyated in preneoplastic and neoplastic cervical tissue (reviewed in Wentzensen et al 2009). Currently, studies are underway to determine whether aberrant epigenetic changes are a prerequisite for HPV-mediated carcinogenesis.
Ovarian cancer
The majority of cases of ovarian cancer are not associated with familial predisposition. Although it is widely believed that ovarian epithelial tumours arise in the coelomic epithelium that covers the ovarian surface, there is now accumulating evidence suggesting that they could arise from tissues that are embryologically derived from the Müllerian ducts. The observation that women with a greater number of ovulatory cycles have an increased risk of ovarian cancer led to the incessant ovulation hypothesis by Fathalla in 1971. According to this hypothesis, as ovulation occurs, ovarian surface epithelial cells are internalized and damaged, and the subsequent repair mechanisms place the cells at an increased risk of developing mutations and subsequent malignancies. Consistent with this hypothesis, women with a history of multiple pregnancies, increased time of lactation and oral contraceptive use are at decreased risk. However, this theory is weakened by other observations. For example, endometriosis, the presence of endometrium outside the uterus, is associated with increased risk of ovarian cancer (Melin et al 2006). Likewise, hysterectomy (particularly at an early age) and tubal ligation, both of which result in either removal or alteration of Müllerian tissue, are associated with decreased risk of epithelial ovarian cancer (Irwin et al 1991, Hankinson et al 1993, Parazzini et al 1993, Narod et al 2001). The fact that a substantial residual risk for peritoneal cancer in BRCA1 and BRCA2 mutation carriers remains for a long time following prophylactic salpingo-oophorectomy (Finch et al 2006) supports the view that the cell of origin for ovarian cancer may originate from outside the ovary. Dubeau was the first to challenge the coelomic hypothesis (Dubeau 1999), as it is surprising that tumours currently regarded as being of primary ovarian origin would resemble tumours derived from various segments of the Müllerian tract (cervix, endometrium and fallopian tube), despite the fact that the ovary is not embryologically related to this tract. Proponents of the coelomic metaplasia hypothesis account for the Müllerian appearance of ovarian tumours by stipulating that the coelomic epithelium is not the direct precursor of ovarian tumours, but must first change into Müllerian-like epithelium by metaplasia. This implies that ovarian carcinomas are better differentiated than the cells from which they originate. This notion is at odds with our current understanding of cancer development (Dubeau 2008a).
Early events in ovarian carcinogenesis
Like most cancers, epithelial ovarian cancers are thought to arise from a single cell in 90% of cases. Evidence for the clonality of ovarian cancer lies in the similarity between primary and metastatic lesions during the examination of LOH, X-chromosome inactivation and specific gene mutations (reviewed in Landen et al 2008). In contrast to cervical cancer, where early changes can be studied due to the fact that the cervix is easily accessible, it is very difficult to identify early changes in ovarian carcinogenesis, even more so because the cell of origin is still under debate.
Studying genetic disorders can provide great insight into the aetiology and early events in carcinogenesis. As outlined above, hereditary genetic disorders account for approximately 10% of ovarian cancers, and 90% of these are either BRCA1 or BRCA2 mutations. In recent years, a major effort to prevent serous cancer in genetically susceptible women with mutations in BRCA1 or BRCA2 has spawned the practice of prophylactic salpingo-oophorectomy. Histological surveillance of prophylactic salpingo-oophorectomy specimens revealed that many early cancers in these women arise in the fallopian tube, and further studies have pinpointed the distal (fimbrial) portion as the most common site of origin. In addition, a high proportion of histologically normal fimbriae from BRCA mutation carriers stain positive for p53 (Crum et al 2007). Another convincing piece of molecular evidence favouring the idea that the cell of origin for ovarian cancer comes from the Müllerian duct is provided by recent expression profiling studies. Gene expression profiles of laser capture microdissected non-malignant distal fallopian tube epithelium from BRCA1 and BRCA2 mutation carriers and control women, as well as high-grade tubal and ovarian serous carcinoma, have been analysed (Tone et al 2008). Interestingly, fallopian tube samples from mutation carriers clustered closely with serous carcinomas rather than normal control fallopian tube epithelium. These findings support a common molecular pathway for adnexal serous cancers, and provide further evidence that cells from the Müllerian duct serve as the cells of origin for ovarian cancer. All this leaves us with the question of why BRCA mutation carriers do not carry an exaggerated high risk for endometrial cancer. The endometrium contains by far the highest number of epithelial cells originating from the Müllerian duct, and hence the statistical chances would be higher for a cancer in this organ rather than in the ovary, where cancers develop extremely frequently in BRCA mutation carriers. This paradox can be resolved by looking at the contribution of the stroma and the microenvironment to tumorigenesis. Dubeau’s group inactivated the BRCA1 gene in mouse granulosa cells (Chodankar et al 2005, Dubeau 2008b). Two-thirds of the mice developed epithelial cysts in their reproductive organs by the time they reached 12–18 months of age. Some of those cysts involved the ovary and were very similar to human ovarian cystadenomas. Although the tumours were benign, preliminary results suggested that crossing the mutant mice with mice carrying a homozygous knock-out of p53 increases the rate of malignant transformation. The fact that the cystic tumours showed no evidence of rearrangement of BRCA1, implying that they expressed a functional BRCA1 protein, strongly supports the view that the ovarian stroma — in particular, granulosa cells — influences the development of ovarian epithelial tumours (Chodankar et al 2005, Dubeau 2008b).
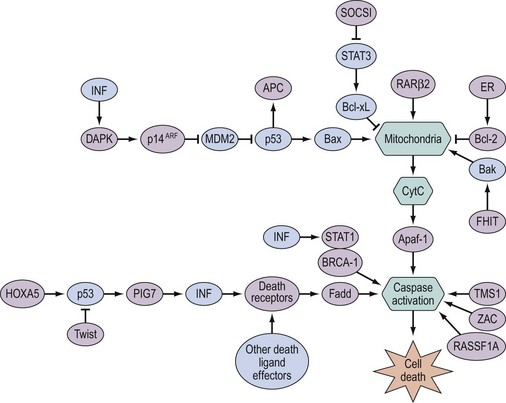
Most of the evidence on genetic and epigenetic alterations in ovarian cancer is based on studies of late-stage cancers. However, current understanding of these processes allows speculation that many alterations must occur early to achieve a clinically recognized tumour. The genetic and epigenetic abnormalities in ovarian cancer involve genes at each step in the complex pathway of cell regulation, which are summarized in Figure 35.1. These include involving growth factors [macrophage colony-stimulating factor, transforming growth factor (TGF)-β], growth factor receptors [fms, epidermal growth factor receptor (EGFR), Her-2/neu], genes involved in signal transduction (ras), genes involved in transcriptional regulation (myc, p53) and LOH at various loci, which occur in a proportion of epithelial ovarian cancers.
Oncogenes in sporadic ovarian cancer
Over 60 different oncogenes have been identified and they are categorized according to their cellular location and function. The subject is reviewed in detail by Landen et al (2008) and Barton et al (2008).
Growth signals
There is good evidence that the response of ovarian cancer cells to growth factors is altered. The proliferative response of ovarian cancer cells in culture to epidermal growth factor (EGF) and TGF-α is variable but usually less than normal ovarian epithelium (Berchuck et al 1990), whilst the inhibitory effect of TGF-β is less marked in ovarian cancer cell lines than in normal ovarian epithelium. TGF-β appears to function as an autocrine inhibitory factor in normal ovarian epithelial growth, and the loss of this regulatory loop may represent a step in the process of ovarian carcinogenesis. During early phases of carcinogenesis, antigrowth signals must be overcome. The restriction point after which a cell is committed to divide is controlled by cyclin D and E’s regulation of E2F release by Rb. Cyclin E is overexpressed in malignant tumours and is associated with poor prognosis in early-stage ovarian cancer (Marchini et al 2008).
The initiation of DNA replication represents a final and critical step in growth regulation, and lies downstream at the convergence point of growth regulatory pathways (Williams et al 1998). Minichromosome maintenance proteins (Mcm2–7) participate in the assembly of prereplicative complexes to establish competence for initiation of DNA synthesis (DNA replication licensing). All six Mcm proteins are essential for replication, are present in all phases of the proliferative cell cycle, but are tightly downregulated in the quiescent, terminally differentiated and senescent ‘out-of-cycle’ states. The presence of one protein reflects the presence of the other five, as all six are loaded together on to DNA as a heterohexamer on exit from metaphase. Mcm proteins have not been shown to be good prognostic markers in ovarian cancer (Gakiopoulou et al 2007, Marchini et al 2008).
Telomeres are tandem repeats of a short DNA sequence at the end of each chromosome. The length of telomeres decreases in somatic cells each time they divide. Most mammalian cells are therefore limited in terms of the number of times they can undergo cell division, because chromosomes cannot replicate unless their telomeres are longer than a minimal critical length. This mitotic clock does not operate in cancer cells because almost all cancers express telomerase, ensuring maintenance of the telomeres above the critical length necessary to support cell division, activating p53 and other policing proteins that propel a cell into an apoptotic pathway. Most ovarian cancer cells maintain telomere length by production of telomerase, a reverse transcriptase composed of an RNA component (hTR) and a catalytic subunit (hTERT). The hTR subunit is expressed by all cells, but hTERT expression increases with increasing tumorigenicity, which suggests that it is the rate-limiting step in telomerase activity (reviewed in Landen et al 2008).
Growth factor receptors
The growth factor receptors are a family of cell membrane tyrosine kinases which act as receptors for growth factors and are involved in signal transduction via autophosphorylation and phosphorylation of intracellular proteins. EGFR (also referred to as erbB) and Her-2/neu (also referred to as erbB2) are structurally similar, and both have been shown to be overexpressed in human cancers. EGFR is expressed by most advanced-stage ovarian cancers and there is some evidence that EGFR-positive tumours have a worse prognosis than tumours that do not express the receptor. Her-2/neu is overexpressed in approximately one-third of ovarian cancers (Slamon et al 1989), and overexpression is associated with gene amplification. Many proliferation pathways mediate signals through the RAS (rat sarcoma viral oncogene homolog) oncoprotein, a G-protein attached to the cell membrane and activated by many tyrosine kinase receptors, and BRAF (v-raf murine sarcoma viral oncogene homolog B1), which is downstream of RAS. RAS activates a cascade of serine/threonine and tyrosine non-receptor kinases, which leads to phosphorylation and activation of Erk1 and Erk2 transcription factors that make their way to the nucleus to initiate signals of growth and progression through the cell cycle. RASSF1A (Ras association domain family 1 isoform A) is a recently discovered tumour suppressor whose inactivation — mainly by DNA methylation (Barton et al 2008) — is implicated in the development of many human cancers. RASSF1A lacks apparent enzymatic activity but contains a Ras association domain and is potentially an effector of the Ras oncoprotein. RASSF1A modulates multiple apoptotic and cell cycle checkpoint pathways. Current evidence supports the hypothesis that it serves as a scaffold for the assembly of multiple tumour suppressor complexes, and may relay proapoptotic signalling by K-ras.
Oncogenes
Chromosome 3q26 is found to be increased in copy number in approximately 40% of ovarian cancers. This region contains a recently identified oncogene, PIK3CA, which encodes the p110-α subunit of phosphatidylinositol 3-kinase (PI 3-kinase). PI 3-kinase-mediated signalling is involved in a broad range of cancer-related functions, including glucose transport, catabolism, apoptosis, cell adhesion, RAS signalling and oncogenic transcription. Little is known about the role of most nuclear oncogenes in ovarian cancer. Amplification of c-myc and increased expression of the protein product has been described in approximately one-third of ovarian cancers. Overexpression of c-myc is found in 38% of ovarian tumours (Tashiro et al 1992).
Tumour suppressor genes in sporadic ovarian cancer
Mutations of the p53 gene are the most frequent genetic alterations in cancer, and occur most frequently in regions of the gene which show the greatest degree of conservation between p53 proteins of different species. Immunohistochemical studies have revealed overexpression of the p53 protein in 50% of advanced-stage ovarian cancers (Marks et al 1991). Sequencing of p53 from ovarian cancers overexpressing the p53 protein has, in most cases, confirmed point mutations in conserved regions of the gene (Marks et al 1991). In most of these tumours, LOH studies have revealed that the second copy of the gene is inactivated through deletion of the gene. p53 mutations are less common in stage I disease than advanced-stage disease, and clonal analysis suggests that the occurrence of p53 mutations precedes but is temporally associated with metastases (Jacobs et al 1992). No clear relationship between p53 mutation and histological grade or prognosis has been established. The location and nature of p53 mutations in ovarian cancer have now been reported in 149 tumours. Over 100 different mutations involving approximately 60 different codons have been identified, and most occur in the highly conserved exons 5–8 of the gene. The codons most frequently involved in p53 mutations are the same as those described in other cancers. The pattern of mutations suggests that most p53 alterations in ovarian cancer arise due to endogenous mutagenic processes rather than exposure to a carcinogen.
Several novel potential candidate tumour suppressor genes have recently been reported for ovarian cancer, and their role in ovarian carcinogenesis has yet to be elucidated. Analysis of LOH at 11q25 identified OPCML (opioid binding protein/cell adhesion molecule-like), a member of the IgLON family of immunoglobulin domain-containing glycosylphosphatidylinositol-anchored cell adhesion molecules, as a candidate tumour suppressor gene in ovarian cancer (Sellar et al 2003). OPCML is frequently somatically inactivated by allele loss, and up to 83% of ovarian cancers demonstrate CGI methylation of this gene.
Defects in programmed cell death can promote oncogenesis and resistance to chemotherapy. Apoptosis (type I programmed cell death) has been well studied as a caspase-regulated cellular response to environmental stress and to the activation of oncogenes. Numerous genes involved in apoptosis are methylated and silenced in ovarian cancer (Barton et al 2008, Figure 35.3). Autophagy (type II programmed cell death) is characterized by the accumulation of multilamellar vesicles that engulf cytoplasm and organelles, forming autophagosomes marked by microtubule-associated protein light chain 3. The maternally imprinted Ras-related tumour suppressor gene, aplasia Ras homolog member I (ARHI), is downregulated in more than 60% of ovarian cancers, and re-expression of ARHI in multiple human ovarian cancer cell lines induces autophagy (Lu et al 2008). In addition, death-associated protein kinase (DAPK) also contributes to the autophagic pathways, and loss of their function — a feature frequently observed in ovarian cancer — could inhibit the induction of autophagy and increase the incidence of cancer.
Loss of heterozygosity
Studies of LOH in ovarian cancer have revealed a number of regions with high frequencies of loss. The reported frequency of LOH for each chromosome arm is summarized in Figure 35.6. Losses have been observed on almost all chromosome arms, and the background ‘random’ rate of loss in ovarian cancer is high (range 15–25%). A high frequency of allelic deletion (>33%) based upon more than 50 tumours in at least three separate studies has been documented for seven chromosome arms: 6p, 6q, 13q, 17p (possibly associated with p53), 17q, 18q and Xp. A number of other chromosome arms have frequencies of loss in the range 25–33% which may be above background rates: 4p, 8p, 9p, 9q, 11p, 14q, 16q, 19p and 21q and 22q (McCluskey and Dubeau 1997). Some of these allelic deletions have been correlated with clinicopathological parameters. Recent studies have shown a distinct relationship between allelic imbalance at 8p12-p21 and 8p22-pter in ovarian cancer and tumour grade, stage and histological subtype. Poorly differentiated tumours, advanced-stage cancers and serous tumours were more prone to allelic imbalance at these regions (Pribill et al 2001).
Further localization of putative tumour suppressor genes in regions with a high rate of LOH requires detailed analysis with a panel of polymorphic markers for the relevant chromosome arm. Available data suggest that a number of as yet unidentified tumour suppressor genes are involved in ovarian carcinogenesis, and recent reports have defined deletion units on chromosome arms with the highest rates of LOH including 6p, 6q, 11p, 13q, 17q and Xp. The nature of specific chromosomes affected by LOH can influence tumour biological aggressiveness, as losses on certain chromosomes such as 13q and Xq are strongly associated with poorly differentiated tumours (Cheng et al 1996). LOH in chromosomes 6q or 17 is more frequently found in well-differentiated tumours.
Comparative genomic hybridization
The application of this technique has shown that 53–69% of human ovarian cancers demonstrate amplifications on chromosomes 1q, 3q, 8q, 13q, 19p and 20q (Kiechle et al 2001). Under-representations were found for chromosomes 4q, 13q and 18q in approximately 50% of ovarian cancers. Undifferentiated tumours were found to correlate significantly with under-representation of 11p and 13q, as well as with amplification of 7p and 8q.
cDNA microarrays
The development of cancer is the result of a series of molecular changes at DNA level, and these events lead to changes in gene expression (mRNA) levels of numerous genes resulting in different phenotypic characteristics of tumours. cDNA microarray analysis enables the identification of differences in gene expressions between normal and malignant tissues by analysing thousands of genes at the same time. A recent study comparing 5766 different gene expressions between normal and malignant ovarian tissue revealed that several genes were under- or overexpressed (Wang et al 1999). Several genes were highly overexpressed in ovarian cancer, such as epithelial glycoprotein (GA733, gene antigen 733), PUMP1 (Pump-1 protease) (MMP7, matrix metallopeptidase 7) and cytokeratin 8, and some of these have also been shown to be overexpressed in other cancers such as colorectal cancer. This illustrates that phenotypical similarity between different tumour types is also reflected at molecular level.
Another study, using a DNA microarray of 9121 genes, identified 55 genes commonly upregulated and 48 genes downregulated in ovarian cancer specimens compared with the corresponding normal ovarian tissues (Ono et al 2000). The 55 genes that were often upregulated in the nine adenocarcinomas analysed represent candidates for stimulating cell growth and preventing apoptosis. Serial analysis of gene expression has recently been introduced to generate global gene expression profiles from various ovarian cell lines and tissues including primary ovarian cancers, ovarian surface epithelial cells and cystadenoma cells. More than 56,000 gene expressions (10 different libraries) were generated (Hough et al 2000). Interestingly, ovarian cancer cell lines showed high levels of similarity to libraries from other cancer cell lines such as colorectal cancer, indicating that these cell lines had lost many of their tissue-specific expression patterns. Many of the genes upregulated in ovarian cancer represent surface or secreted proteins such as mucin-1, HE4 (human epididymal protein 4), epithelial cellular adhesion molecule, mesothelin and several keratins (e.g. keratins 18 and 19).
MicroRNAs
MicroRNAs (miRNAs) are endogenous non-coding small RNAs which negatively regulate gene expression. In human cancer, miRNAs might function as either oncogenes or tumour suppressor genes. Increasing evidence shows that expression of miRNAs is deregulated in human cancer. High-throughput miRNA quantification technologies have provided powerful tools to study global miRNA profiles. It has become progressively obvious that although the number of miRNAs (~600) is much smaller than that of the protein-coding genes (~22,000), miRNA expression signatures reflect the developmental lineage or tissue origin of human cancers more accurately. Recently, it has been demonstrated that genomic copy number loss and epigenetic silencing, respectively, may account for the downregulation of ~15% and at least ~36% of miRNAs in advanced ovarian tumours, and miRNA downregulation contributes to a genome-wide transcriptional deregulation. Eight miRNAs located in the chromosome 14 miRNA cluster (Dlk1–Gtl2 domain) were identified as potential tumour suppressor genes. Tumours with lower expression of these eight miRNAs were associated with a higher proliferation index and significantly shorter survival. Although the function of this miRNA cluster is largely unknown, it may play a critical role in embryonic development (Zhang et al 2008).
DNA methylation
As outlined above, genome-wide demethylation of normally methylated and silenced chromosomal regions, and hypermethylation and silencing of genes including tumour suppressors are among the most common features of cancer cells (reviewed in Barton et al 2008). Epigenetic alterations, including CGI DNA methylation, frequently occur in ovarian cancer, and the identification of specific genes that are altered by epigenetic events is currently an area of intense research. Aberrant DNA methylation in ovarian cancer is observed in early cancer development, can be detected in serum/plasma DNA and hence provides the promise of a non-invasive cancer detection test. In addition, identification of ovarian-cancer-specific epigenetic changes has promise in molecular classification and disease stratification.
Endometrial cancer
Endometrial cancers have long been classified into two major divisions (types I and II) based on light microscopic appearance, clinical behaviour and epidemiology. Type I (endometrioid histology) comprise 70–80% of newly diagnosed cases of endometrial cancer. They are associated with unopposed oestrogen exposure and are often preceded by premalignant disease (atypical complex hyperplasia). In contrast, type II endometrial cancers (usually papillary serous or clear cell histology) have not been identified in association with hormonal factors, and do not have a readily observed premalignant phase. In addition, they demonstrate an aggressive clinical course compared with the type I tumours. The morphologic and clinical differences are paralleled by genetic distinctions, in that type I and II cancers carry mutations of independent sets of genes (Hecht and Mutter 2006). While most type II cancers contain mutations of p53, type I adenocarcinomas demonstrate larger numbers of genetic changes in which the temporal sequence of mutation and the final combination of defects differ substantially between individual examples. Common genetic changes in endometrioid endometrial cancers (type I) include, but are not limited to, microsatellite instability or specific mutation of PTEN (phosphatase and tensin homolog), K-ras and β-catenin genes (Hecht et al 2006). With regard to epigenetics, promoter hypermethylation is common in type I but not type II tumours. Many of the tumour suppressor pathways that are mutated in type I endometrial cancer such as PTEN, hMLH1, MGMT and APC can also be inactivated by hypermethylation. In addition, PR-B (progesterone receptor B) promoter hypermethylation is established as the dominant mechanism of PR-B silencing (Zhou et al 2007).
Vulval cancer
CGH has recently been used to study alterations (losses or gains) on all chromosomes in vulval cancer (Jee et al 2001). Frequent chromosomal losses were found on 3p, 4p13-pter and 5q, and less frequent losses were found on 6q, 11q and 13q. Most frequent chromosomal gains were observed on 3q and 8p, and less frequent gains were found on 9p, 14, 17 and 20q. The pattern of chromosomal imbalance in vulval cancer detected by CGH was revealed to be very similar to that in cervical cancer. These results suggest that the molecular pathways in vulval and cervical carcinomas may be similar. However, it is not clear whether HPV-positive and -negative vulval cancers have similar or different molecular pathways.
Vulval squamous cell carcinomas (VSCCs) exhibit a broad range of allelic losses irrespective of HPV status, with high frequencies of LOH on certain chromosomal arms. High frequencies of LOH are found on 1q, 2q, 3p, 5q, 8p, 8q, 10p, 10q, 11p, 11q, 15q, 17p, 18q, 21q and 22q (Pinto et al 1999). This suggests that despite their differences in pathogenesis, both HPV-positive and -negative VSCCs share similarities in type and range of genetic losses during their evolution. Another study observed a greater number of molecular alterations in HPV-negative vulval cancers compared with HPV-positive tumours (Flowers et al 1999). Allelic losses at 3p are common early events in vulval carcinogenesis in HPV-negative cancers, and are detected at a high rate in the corresponding high-grade precursor lesions (VIN II/III). TP53 gene mutations with associated 17p13.1 LOH are also more common in HPV-negative cancers. Fractional regional loss index, an index of total allelic loss at chromosomal regions 3p, 13q14 and 17p13.1, is greater in HPV-negative vulval cancers than in HPV-positive tumours (Flowers et al 1999). Similar observations have been found in HPV-negative high-grade VINs compared with HPV-positive lesions. Overall, LOH at any 3p region is common (80%) in both groups of cancers and in their associated VIN lesions. Although TP53 gene mutations are present in a minority of vulval cancers (20%), allelic losses at the TP53 locus are frequently present, especially in HPV-negative vulval cancers, compared with HPV-positive tumours.
Fallopian tube cancer
The histological features, and biological and clinical behaviour of fallopian tube cancer are similar to those of ovarian cancer. In addition, there is recent evidence that similar genetic alterations occur with the same frequency between fallopian tube and ovarian cancer when considering the same histological subtype. This suggests a common molecular pathogenesis between these cancer types (Crum et al 2007). Recent molecular genetic analyses using CGH reveal that different histological subtypes differ in respect to their genomic alterations, but similar subtypes of different cancers have similar patterns of genomic abnormalities. For example, the frequency and pattern of chromosomal changes detected in serous tubal carcinomas (95% of all fallopian tube carcinomas) are strikingly similar to those observed in serous ovarian carcinomas, suggesting a common molecular pathogenesis. In fallopian tube carcinoma, frequent gains are found on chromosomes 3q and 8q and frequent losses are found on chromosomes 4q, 5q, 8q and 18q (Pere et al 1998). There is also evidence that fallopian tube cancer is similar to ovarian cancer with respect to the proportion of tumours with abnormal expression of Her-2/neu and p53. The prognostic significance and predictive drug response of these two genes needs to be explored in fallopian tube cancer.
Clinical Aspects of the Genetics of Gynaecological Cancer
Early detection of cancer
Knowledge of specific genetic and epigenetic alterations associated with cancer, along with the high sensitivity of molecular techniques such as the polymerase chain reaction, may provide new methods for detection of cancer. As direct sampling of the ovary requires an invasive procedure, any screening test for ovarian cancer based upon genetic markers will be directed towards identification of a gene product in peripheral blood. Tumour-specific hypermethylation of at least one of a panel of six tumour suppressor gene promoters, including RASSF1A (Ras association domain family member 1A), BRCA1 (breast cancer 1 early onset), APC (adenomatous polyposis coli), CDKN2A (cyclin-dependent kinase inbibitor 2A) and DAPK (death-associated protein kinase 1), could be detected in the serum or plasma of ovarian cancer patients with 100% specificity and 82% sensitivity, including 13/17 cases of stage I (confined to the ovary) disease. Methylation was only observed in one peritoneal fluid sample from 15 stage IA or B patients, but 11/15 paired sera were positive for methylation (Ibanez de Caceres et al 2004). In addition to proof of principle, these data indicate that circulating ovarian tumour DNA is more readily accessible in the bloodstream than in the peritoneum. In addition, serum DNA methylation of SFRP1 (secreted frizzled-related protein 1), SOX1 (SRY( sex determining region Y)-box 1) and LMX1A (LIM homeobox transcription factor 1 alpha) revealed sensitivity and specificity of 73% and 75%, respectively (Su et al 2009).
DNA is a very stable molecule compared with RNA or protein. It can easily be analysed in any body fluid, including vaginal secretions. In a proof of principle study, the authors aimed to define a new and simple strategy for detection of endometrial cancer using epigenetic markers. They investigated DNA isolated from vaginal secretions collected from tampons for aberrant methylation of five genes [(CDH13, HSPA2 (heat shock 70kDa protein 2), MLH1, RASSF1A, and SOCS2 (suppressor of cytokine signalling 2)] using MethyLight in 15 patients with endometrial cancer and 109 patients without endometrial cancer. All endometrial cancer patients revealed three or more methylated genes, whereas 91% (99 of 109) of the patients without endometrial cancer had fewer than three genes methylated in their vaginal secretion. The methods developed in this study provided the basis for a prospective clinical trial to screen asymptomatic women who are at high risk for endometrial cancer (Fiegl et al 2004).
Prognostic and predictive indicators
Numerous genetic and epigenetic prognostic and predictive markers have been discovered over the past few years (reviewed in Crijns et al 2006, Barton et al 2008), but none of these markers have yet been integrated into clinical practice. Silencing of hMLH1, a DNA mismatch repair gene, by hypermethylation of its promoter CGI has been linked with acquired resistance to platinum-based drugs in ovarian cell line models. Moreover, methylation of MLH1 is increased at relapse in epithelial ovarian cancer patients, with 25% (34/138) of plasma samples from relapsed patients showing methylation of MLHI which is not evident in matched prechemotherapy plasma samples (Barton et al 2008). The acquisition of MLH1 methylation at relapse predicts poor overall patient survival, and is associated with drug resistance (Gifford et al 2004, Barton et al 2008). The concept of predicting and monitoring response to systemic therapies by means of serum/plasma DNA analysis is currently under investigation.
Strategies for gene therapy
Promising preclinical and clinical data led to the initiation of an international randomized phase II/III trial of p53 gene therapy for first-line treatment of patients with ovarian cancer. In that trial, replication-deficient adenoviral vectors carrying wild-type p53 were given intraperitoneally in combination with standard chemotherapy to patients with ovarian cancers harbouring p53 mutations. The study was closed after the first interim analysis because an adequate therapeutic benefit was not shown (Zeimet and Marth 2003). There may be various reasons for the failure of this strategy; for example, the repair of single genes might not be a suitable strategy for the treatment of cancer, heterogeneity or lack of expression of coxsackie-adenovirus receptors and integrin coreceptors in ovarian tumours, and the presence of adenovirus-neutralizing antibodies in ovarian cancer-related ascites. Although the safety of many other treatment strategies has been demonstrated in early-phase clinical trials, efficacy has been mostly limited. Major challenges include improving the vectors used, with the aim of more effective and selective delivery. In addition, effective penetration into and spreading within advanced and complex tumour masses and metastases remains challenging (Kanerva et al 2007).
KEY POINTS
Aarnio M, Sankila R, Pukkala E, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. International Journal of Cancer. 1999;12:214-218.
Aubele M, Zitzelsberger H, Schenck U, Walch A, Hofler H, Werner M. Distinct cytogenetic alterations in squamous intraepithelial lesions of the cervix revealed by laser-assisted microdissection and comparative genomic hybridization. Cancer. 1998;84:375-379.
Barton CA, Hacker NF, Clark SJ, O’Brien PM. DNA methylation changes in ovarian cancer: implications for early diagnosis, prognosis and treatment. Gynecological Oncology. 2008;109:129-139.
Berchuck A, Rodriguez G, Kamel A, Soper JT, Clarke-Pearson DL, Bast RCJr. Expression of epidermal growth factor receptor and HER-2/neu in normal and neoplastic cervix, vulva, and vagina. Obstetrics and Gynecology. 1990;76:381-387.
Campbell S, Bourne T, Bradley E. Screening for ovarian cancer by transvaginal sonography and colour Doppler. European Journal of Obstetrics, Gynecology and Reproductive Biology. 1993;49:33.
Cavenee WK, Dryja TP, Phillips RA, et al. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305:779-784.
Chen KT, Schooley JL, Flam MS. Peritoneal carcinomatosis after prophylactic oophorectomy in familial ovarian cancer syndrome. Obstetrics and Gynecology. 1985;66:93.
Cheng PC, Gosewehr JA, Kim TM, et al. Potential role of the inactivated X chromosome in ovarian epithelial tumor development. Journal of the National Cancer Institute. 1996;88:510-518.
Chodankar R, Kwang S, Sangiorgi F, et al. Cell-nonautonomous induction of ovarian and uterine serous cystadenomas in mice lacking a functional Brca1 in ovarian granulosa cells. Current Biology. 2005;15:561-565.
Crijns AP, Duiker EW, de Jong S, Willemse PH, van der Zee AG, de Vries EG. Molecular prognostic markers in ovarian cancer: toward patient-tailored therapy. International Journal of Gynecological Cancer. 2006;16(Suppl 1):152-165.
Crum CP, Drapkin R, Kindelberger D, Medeiros F, Miron A, Lee Y. Lessons from BRCA: the tubal fimbria emerges as an origin for pelvic serous cancer. Clinical Medicine and Research. 2007;5:35-44.
de Klein A, van Kessel AG, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765-767.
Dubeau L. The cell of origin of ovarian epithelial tumors and the ovarian surface epithelium dogma: does the emperor have no clothes? Gynecological Oncology. 1999;72:437-442.
Dubeau L. The cell of origin of ovarian epithelial tumours. The Lancet Oncology. 2008;9:1191-1197.
Dubeau L. BRCA1-induced ovarian oncogenesis. Advances in Experimental Medicine and Biology. 2008;622:89-97.
Dunlop MG, Farrington SM, Carothers AD, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Human Molecular Genetics. 1997;6:105-110.
Easton DF, Eeles RA. Genome-wide association studies in cancer. Human Molecular Genetics. 2008;17(R2):R109-R115.
Easton DF, Bishop DT, Ford D, Crockford GP. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. The Breast Cancer Linkage Consortium. American Journal of Human Genetics. 1993;52:678.
Fathalla MF. Incessant ovulation: a factor in ovarian neoplasia? Lancet. 1971;2(7716):163.
Fiegl H, Gattringer C, Widschwendter A, et al. Methylated DNA collected by tampons — a new tool to detect endometrial cancer. Cancer Epidemiology, Biomarkers and Prevention. 2004;13:882-888.
Finch A, Beiner M, Lubinski J, et al. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 mutation. JAMA: the Journal of the American Medical Association. 2006;296:185-192.
Flowers LC, Wistuba II, Scurry J, et al. Genetic changes during the multistage pathogenesis of human papillomavirus positive and negative vulvar carcinomas. Journal of the Society for Gynecologic Investigation. 1999;6:213-221.
Ford D, Easton DF, Bishop DT, et al. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994;343:692-695.
Ford D, Easton DF, Stratton M, et al. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. American Journal of Human Genetics. 1998;62:676-689.
Gakiopoulou H, Korkolopoulou P, Levidou G, et al. Minichromosome maintenance proteins 2 and 5 in non-benign epithelial ovarian tumours: relationship with cell cycle regulators and prognostic implications. British Journal of Cancer. 2007;97:1124-1134.
Gayther SA, Warren W, Mazoyer S, et al. Germline mutations of the BRCAI gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nature Genetics. 1995;11:428-433.
Gayther SA, Mangion J, Russell P, et al. Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nature Genetics. 1997;15:103-105.
Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clinical Cancer Research. 2004;10:4420-4426.
Hall JM, Lee MK, Newman B, et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250:1684-1689.
Hankinson SE, Hunter DJ, Colditz GA, et al. Tubal ligation, hysterectomy, and risk of ovarian cancer. A prospective study. JAMA: the Journal of the American Medical Association. 1993;270:2813-2818.
Hecht JL, Mutter GL. Molecular and pathologic aspects of endometrial carcinogenesis. Journal of Clinical Oncology. 2006;24:4783-4791.
Hough CD, Sherman-Baust CA, Pizer ES, et al. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Cancer Research. 2000;60:6281-6287.
Huang J, Papadopoulus N, McKinley AJ, et al. APC mutations in colorectal tumours with mismatch repair deficiency. Proceeding of the National Academy of Sciences USA. 1996;93:9049-9054.
Ibanez de Caceres I, Battagli C, Esteller M, et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Research. 2004;64:6476-6481.
Irwin KL, Weiss NS, Lee NC, Peterson HB. Tubal sterilization, hysterectomy, and the subsequent occurrence of epithelial ovarian cancer. American Journal of Epidemiology. 1991;134:362-369.
Jacobs I, Davies AP, Bridges J, et al. Prevalence screening for ovarian cancer in postmenopausal women by CA 125 measurement and ultrasonography [see comments]. British Medical Journal. 1993;306:1030.
Jacobs IJ, Kohler MF, Wiseman RW, et al. Clonal origin of epithelial ovarian carcinoma: analysis by loss of heterozygosity, p53 mutation, and X-chromosome inactivation. Journal of the National Cancer Institute. 1992;84:1793-1798.
Jacobs IJ, Skates SJ, MacDonald N, et al. Screening for ovarian cancer: a pilot randomized controlled trial. Lancet. 1999;353(9160):1207-1210.
Jee KJ, Kim YT, Kim KR, Kim HS, Yan A, Knuutila S. Loss in 3p and 4p and gain of 3q are concomitant aberrations in squamous cell carcinoma of the vulva. Modern Pathology. 2001;14:377-381.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683-692.
Jones PA, Laird PW. Cancer epigenetics comes of age. Nature Genetics. 1999;21:163-167.
Kanerva A, Raki M, Hemminki A. Gene therapy of gynaecological diseases. Expert Opinion on Biological Therapy. 2007;7:1347-1361.
Kiechle M, Jacobsen A, Schwarz-Boeger U, Hedderich J, Pfisterer J, Arnold N. Comparative genomic hybridization detects genetic imbalances in primary ovarian carcinomas as correlated with grade of differentiation. Cancer. 2001;91:534-540.
Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-170.
Kinzler KW, Nilbert MC, Su L-K, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661-665.
Laird PW. The power and the promise of DNA methylation markers. Nature Reviews. Cancer. 2003;3:253-266.
Landen CNJr, Birrer MJ, Sood AK. Early events in the pathogenesis of epithelial ovarian cancer. Journal of Clinical Oncology. 2008;26:995-1005.
Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. Journal of Clinical Oncology. 2008;26:5284-5293.
Levy-Lahad E, Catane R, Eisenberg S, et al. Founder BRCA1 and BRCA2 mutations in Ashkenazi Jews in Israel: frequency and differential penetrance in ovarian cancer and in breast-ovarian cancer families. American Journal of Human Genetics. 1997;60:1059-1067.
Lu Z, Luo RZ, Lu Y, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. Journal of Clinical Investigation. 2008;118:3917-3929.
Lynch HT, de la Chapelle H. Genetic susceptibility to non-polyposis colorectal cancer. Journal of Medical Genetics. 1999;36:801-808.
Lynch HT, Kimberling W, Albano WA, et al. Hereditary nonpolyposis colorectal cancer (Lynch syndromes I and II). Cancer. 15, 1985. 938–938
Marchini S, Mariani P, Chiorino G, et al. Analysis of gene expression in early-stage ovarian cancer. Clinical Cancer Research. 2008;14:7850-7860.
Marks JR, Davidoff AM, Kerns BJ, et al. Overexpression and mutation of p53 in epithelial ovarian cancer. Cancer Research. 1991;51:2979-2984.
Marra G, Boland CR. Hereditary nonpolyposis colorectal cancer. Journal of the National Cancer Institute. 1995;87:1114-1125.
McCluskey LL, Dubeau L. Biology of ovarian cancer. Current Opinion in Oncology. 1997;9:465-470.
Melin A, Sparen P, Persson I, Bergqvist A. Endometriosis and the risk of cancer with special emphasis on ovarian cancer. Human Reproduction. 2006;21:1237-1242.
Menon U, Gentry-Maharaj A, Hallett R, et al. Sensitivity and specificity of multimodal and ultrasound screening for ovarian cancer, and stage distribution of detected cancers: results of the prevalence screen of the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS). The Lancet Oncology. 2009;10:327-340.
Miki Y, Swensen J, Schattuck-Eidens D, et al. Isolation of BRCA1, the 17q linked breast and ovarian cancer susceptibility gene. Science. 1994;266:66-71.
Modan B, Hartge P, Hirsh-Yechezkel G, et al. Parity, oral contraceptives, and the risk of ovarian cancer among carriers and noncarriers of a BRCA1 or BRCA2 mutation. for the National Israel Ovarian Cancer Study Group. New England Journal of Medicine, 2001;345(4); 235-240.
Narod SA, Sun P, Ghadirian P, et al. Tubal ligation and risk of ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case–control study. The Lancet. 2001;357:1467-1470.
Ono K, Tanaka T, Tsunoda T, et al. Identification by cDNA microarray of genes involved in ovarian carcinogenesis. Cancer Research. 2000;60:5007-5011.
Parazzini F, Negri E, La Vecchia C, Luchini L, Mezzopane R. Hysterectomy, oophorectomy, and subsequent ovarian cancer risk. Obstetrics and Gynecology. 1993;81:363-366.
Pere H, Tapper J, Seppala M, Knuutila S, Butzow R. Genomic alterations in fallopian tube carcinoma: comparison to serous uterine and ovarian carcinomas reveals similarity suggesting likeness in molecular pathogenesis. Cancer Research. 1998;58:4274-4276.
Pinto AP, Lin MC, Mutter GL, Sun D, Villa LL, Crum CP. Allelic loss in human papillomavirus-positive and -negative vulvar squamous cell carcinomas. American Journal of Pathology. 1999;154:1009-1015.
Pribill I, Speiser P, Leary J, et al. High frequency of allelic imbalance at regions of chromosome arm 8p in ovarian carcinoma. Cancer Genetics and Cytogenetics. 2001;129:23-29.
Ramus SJ, Gayther SA. The contribution of BRCA1 and BRCA2 to ovarian cancer. Molecular Oncology. 2009;3(2):138-150.
Rebbeck TR, Kauff ND, Domchek SM. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. Journal of the National Cancer Institute. 2009;101:80-87.
Salovaara R, Loukola A, Kristo P, et al. Population-based detection of hereditary nonpolyosis colorectal cancer. Journal of Clinical Oncology. 2000;18:2193-2200.
Sellar GC, Watt KP, Rabiasz GJ, et al. OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nature Genetics. 2003;34:337-343.
Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707-712.
Smith SA, Easton DF, Evans DG, Ponder BA. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nature Genetics. 1992;2:128.
Su HY, Lai HC, Lin YW, Chou YC, Liu CY, Yu MH. An epigenetic marker panel for screening and prognostic prediction of ovarian cancer. International Journal of Cancer. 2009;124:387-393.
Tashiro H, Miyazaki K, Okamura H, Iwai A, Fukumoto M. c-myc over-expression in human primary ovarian tumours: its relevance to tumour progression. International Journal of Cancer. 1992;50:828-833.
Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nature Genetics. 2007;39:347-351.
Thorlacius S, Sigurdsson S, Bjarnadottir H, et al. Study of a single BRCA2 mutation with high carrier frequency in a small population. American Journal of Human Genetics. 1997;60:1079-1084.
Tone AA, Begley H, Sharma M, et al. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clinical Cancer Research. 2008;14:4067-4078.
Wang K, Gan L, Jeffery E, et al. Monitoring gene expression profile changes in ovarian carcinomas using cDNA microarray. Gene. 1999;229:101-108.
Wentzensen N, Sherman ME, Schiffman M, Wang SS. Utility of methylation markers in cervical cancer early detection: appraisal of the state-of-the-science. Gynecological Oncology. 2009;112:293-299.
Whiteside MA, Siegel EM, Unger ER. Human papillomavirus and molecular considerations for cancer risk. Cancer. 2008;113(Suppl):2981-2994.
Williams GH, Romanowski P, Morris L, et al. Improved cervical smear assessment using antibodies against proteins that regulate DNA replication. Proceedings of the National Academy of Science USA. 1998;95:14932-14937.
Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789-792.
Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer? The Lancet Oncology. 2003;4:415-422.
Zhang L, Volinia S, Bonome T, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proceedings of the National Academy of Science USA. 2008;105:7004-7009.
Zhou XC, Dowdy SC, Podratz KC, Jiang SW. Epigenetic considerations for endometrial cancer prevention, diagnosis and treatment. Gynecological Oncology. 2007;107:143-153.
zur Hausen H. Papillomaviruses in human cancer. Applied Pathology. 1987;5:19-24.
[/level-membership-for-obstetrics-gynecology-category][not-level-membership-for-obstetrics-gynecology-category]
CHAPTER 35 The genetics and molecular biology of gynaecological cancer
Introduction
Gynaecological cancers provide examples of a number of contrasting mechanisms of carcinogenesis (Figure 35.1). For example, ovarian and endometrial cancer can occur as components of familial cancer syndromes (familial breast/ovarian cancer and Lynch II syndrome) due to germline inheritance of predisposing genetic abnormalities. However, most ovarian cancers are not thought to be due to inherited genetic alterations, but result from somatic mutations occurring in ovarian cells with an initially normal genome. These somatic mutations are thought to be secondary to environmental factors that act to increase the opportunity for spontaneous mutation in a number of critical genes. One of the genetic alterations that lead to cervical cancer is caused by an environmental factor, human papilloma virus (HPV), and in at least some endometrial cancers, unopposed oestrogen exposure or hyperinsulinism leads to carcinogenesis. High-risk subtypes of HPV are much less common in vulval cancer. This suggests that vulval and cervical carcinomas are not identical aetiologically, and that factors other than HPV are more important in vulval carcinogenesis. There is recent evidence that different genetic events occur between HPV-positive and -negative vulval cancers, with a greater number of molecular alterations in HPV-negative vulval cancers compared with HPV-positive tumours (Flowers et al 1999). HPV-positive vulval cancers are more frequently found in younger patients. Vulval cancers in older patients are more often HPV negative, and more frequently show allelic loss of the TP53 gene.
The histological features, and biological and clinical behaviour of fallopian tube cancers are similar to those of ovarian cancer. In addition, there is evidence that similar genetic alterations occur with the same frequency in fallopian tube and ovarian cancers of the same histological subtype. This suggests a common molecular pathogenesis between these cancer types (Levanon et al 2008).
To date, little is known about the molecular genetics of vaginal cancer.
Molecular Basis of Carcinogenesis
During the last decade, it has become clear that the genetic changes in cancer involve genes which control normal cell growth and proliferation. Cells communicate via a complex interacting set of signalling pathways, the principles of which, unlike the details, are well established (Figure 35.2). The best understood mechanism of communication between cells is via the release of molecules which interact as agonists with receptors either within the cell or, more frequently, in the cell membrane. This interaction results in a further signal within the cell which, via transducer proteins and secondary mechanisms, results in modified cell behaviour either by activating already synthesized target enzymes directly or by initiating transcription of genes and synthesis of their protein product. The genetic abnormalities observed in cancer are known to affect genes coding for all of the groups of molecules in Figure 35.2.
Oncogenes
The identification of oncogenes resulted from in-vitro and animal work with virus-induced cancers. As the viral genome is relatively small, it was possible to identify the specific genes (viral oncogenes) involved in the transforming activity of retroviruses, such as the Rous sarcoma virus. It was possible to identify homologous genes in normal cells, and a number of viral oncogenes were found to have normal cellular homologues known as ‘proto-oncogenes’. Other workers identified oncogenes by transfecting cell lines with DNA from human cancer cell lines, and found that some of the transforming oncogenes identified were mutated versions of the proto-oncogene identified by the retroviral approach. Subsequently, it was demonstrated that chromosome translocations such as the Philadelphia chromosome in chronic myeloid leukaemia involved known oncogenes (de Klein et al 1982).
Approximately 100 proto-oncogenes have been identified, and many can be assigned to the groups of functions summarized in Figure 35.2. The mutation resulting in conversion of a proto-oncogene into an oncogene may occur in several different ways (Figure 35.3), and may result in an abnormal protein produced in a normal quantity, a normal protein produced in an excessive amount, or a normal protein produced inappropriately due to loss or alteration of control regions of the gene.
Tumour suppressor genes
The existence of tumour suppressor genes was suggested by cell fusion studies of transformed cells and non-transformed cells which resulted in a non-transformed hybrid cell. The subsequent identification of the first tumour suppressor gene followed from the study of retinoblastoma, a rare childhood cancer which may occur in a hereditary and sporadic form. On the basis of epidemiological and statistical analysis, Knudson suggested that development of the cancer required two events. He postulated that in the hereditary form, the first event was a germline mutation present in every cell of the individual, and that a second event in any of the many million retinoblasts could result in tumour formation. The rarity, later onset and unilateral nature of the sporadic form could be attributed to the need for two events in a single retinal cell in the absence of a germline mutation. When the retinoblastoma (Rb) gene was mapped to chromosome 13q and cloned, it was confirmed that in both familial and sporadic retinoblastoma, the two copies of the gene were inactivated in tumour cells, consistent with Knudson’s ‘two-hit’ hypothesis (Cavenee et al 1983).
Knudson’s hypothesis that two hits are required for the full inactivation of a tumour suppressor gene has been shown to be fundamentally correct in almost all cases of human cancer. However, the fact that methylation of CpG islands located in the promoters of genes can cause transcriptional silencing, coupled with the observation that DNA methylation patterns are perturbed in cancer cells, has led to the suggestion that abnormal methylation of the promoters of tumour suppressor genes might be implicated in carcinogenesis, and can serve as a third pathway to inactivation of a tumour suppressor gene (Figure 35.4) (Jones and Laird 1999).
Epigenetics and DNA methylation in cancer
As outlined above, aberrant gene function and altered patterns of gene expression are key features of cancer. Growing evidence shows that acquired epigenetic abnormalities participate with genetic alterations to cause this dysregulation. The modern definition of epigenetics is modifications of the DNA or associated proteins, other than DNA sequence variation, that carry information content during cell division. The best understood example of epigenetic modification is DNA methylation, a covalent addition of a methyl group derived from S-adenosyl-L-methionine to the fifth carbon of the cytosine ring to form the fifth base, 5-methylcytosine (5meC) (Laird 2003, Jones and Baylin 2007). The reaction is catalysed by DNA methyltransferases together with accessory proteins. Among all eukaryotic species, methylation occurs predominantly in cytosines located 5′ of guanines, and known as ‘CpG dinucleotides’ (CpGs). In the mammalian genome, the distribution of CpGs is far from random. CpGs are greatly under-represented in the genome through evolutionary loss of 5meC through deamination to thymine. However, clusters of CpGs known as ‘CpG islands’ (CGIs) are present in 1–2% of the genome. Typically, they range in length from 200 to 5000 base pairs (bp). Most are unmethylated under normal circumstances, with the exception of those associated with imprinted genes, genes subjected to X-chromosome inactivation and transposable elements. In this last respect, DNA methylation is thought to repress faulty expression of endogenous transposons that may disrupt the genome. It is also involved in the parental-specific silencing of one allele of imprinted genes. In addition, approximately 70% of CGIs are associated with DNA sequences 200–2000 bp long found in the promoter, the first and second exons, and the first intron regions of all genes (5′ CGIs). This suggests that CGIs are critical in gene regulation. There is, for instance, usually an inverse relationship between the degree of methylation of a regulatory CGI and the extent of gene transcription (Figure 35.5) (Laird 2003, Jones and Baylin 2007).
DNA methylation patterns are established during defined phases in embryonic development. With the exception of imprinted genes, gamete methylation patterns are erased by a genome-wide demethylation at around the eight-cell stage of blastocyst formation. During the implantation stage, methylation patterns are re-established via de-novo methylation. During adulthood, the degree and patterns of methylation are both tissue- and cell-type specific. Disruption of these preset patterns of DNA methylation in adult life has been linked to ageing and to disease. Furthermore, dysregulation of developmental programming by maternal factors or environmental mimics is thought to induce abnormal DNA methylation of specific genes and hence their faulty expression, leading to disease (Laird 2003, Jones and Baylin 2007).
Over the last decade, the importance of epigenetic gene regulation has become increasingly evident as studies have shown that each epithelial tumour only contains approximately 10–15 tumour suppressor genes silenced through mutations, but several hundred silenced through DNA methylation (Jones and Baylin 2007, Thomas et al 2007). Numerous genes involved in central pathways can be silenced by means of DNA methylation (Figure 35.3).
Germline and somatic genetic alterations
A common feature of tumours is that they accumulate somatic genetic changes during the development of the cancer. A proportion of cancers may also develop as a result of germline mutations in cancer predisposing genes. An important distinction must be made between germline and somatic mutations. Somatic mutations are found in the tumour cells but not the normal cells of an individual with cancer, and cannot therefore be passed on to descendants through the germline. Germline mutations, however, are present in all the cells of the individual and can initiate tumour development. They can be passed on to descendants of the individual, and can be identified in DNA obtained from all diploid cell populations (e.g. peripheral blood samples) and not just the tissues that are susceptible to disease. It is worth pointing out that the same genes mutated in the germline are also frequently mutated in the somatic cells of a cancer, which suggests a functional significance for specific genes in cancer development. For example, mutations of the p53 gene are one of the most common genetic changes found in human cancer. In most cancers, p53 mutations occur as somatic events, but germline p53 mutations are often found in patients from families with the rare Li-Fraumeni syndrome, which is characterized by sarcomas, breast cancer and other malignancies occurring at a young age. Another example is the colorectal cancer predisposition syndrome, familial adenomatous polyposis (FAP), caused by germline mutations in the APC gene (Kinzler et al 1991). The FAP syndrome is responsible for approximately 1% of all colorectal cancers, but somatic mutations of APC have been detected in over 80% of sporadic colorectal cancers and appear to be one of the earliest events in colorectal carcinogenesis.
Most of the familial cancer syndromes identified to date are rare, single gene disorders and have an autosomal-dominant pattern of inheritance. Individuals inheriting the germline abnormality are generally at high risk of developing malignancy because the penetrance of most of these genes is of the order of 80%. Another class of cancer predisposition genes that confer more modest penetrance has been identified recently. Although these genes are associated with much lower penetrance, they are also much more common in the population than the rare high-risk genes, and so are probably responsible for a greater proportion of cancer overall (reviewed in Easton and Eeles 2008). However, the identification of individuals in the population that are likely to carry these low penetrance genes is not easy because they are not often associated with a significant family history of disease.
Genetics of Familial Gynaecological Cancer
Although there are anecdotal reports of more than one case of vulval cancer occurring in close relatives, there is no good evidence of inherited predisposition to this disease. For cervical cancer, the absence of any obvious clustering of multiple cases within families suggests that no highly penetrant susceptibility loci for the disease await identification. However, population-based epidemiological studies suggest that shared genetic factors do play a role in the development of cervical cancer, and possibly account for approximately 27% of cases. This perhaps suggests the presence of several common genetic variants that confer low penetrance disease risk. Ovarian and endometrial cancers occur as part of well-defined cancer family syndromes with an autosomal-dominant inheritance pattern. Pedigree studies of cancer within families have revealed three main syndromes associated with ovarian cancer. The most common hereditary form of ovarian cancer occurs in association with breast cancer. A large number of hereditary breast/ovarian cancer families have been described in which there is a high frequency of both cancers and an association with an early age of onset. Hereditary ovarian cancer may also occur as a site-specific disease, although genetic studies suggest that a large proportion of site-specific ovarian and breast/ovarian cancer families are part of the same disease spectrum. Nevertheless, there remains evidence that there is a rare genetic component that confers high penetrance susceptibility specifically to ovarian cancer (reviewed in Ramus and Gayther 2009). Less frequently, ovarian cancer occurs in hereditary non-polyposis colorectal cancer (HNPCC) families, and germline genetic mutations in highly penetrant genes that cause HNPCC have been found in ovarian cancer cases from these families. Women from HNPCC families most frequently develop colorectal or endometrial cancer, and this is the syndrome most commonly associated with genetic predisposition to endometrial cancer. Some families have recently been described with an apparently high risk of site-specific endometrial cancer. It is important to recognize that when all of these familial syndromes are combined, they are probably responsible for less than 5% of ovarian cancer cases and a smaller proportion of endometrial cancer cases.
The BRCA1 and BRCA2 genes
The identification and detailed pedigree-based descriptions of families at high risk of breast and ovarian cancer, together with the development of polymorphic DNA markers, made it possible to perform detailed linkage analysis with the aim of locating genes involved in familial cancer. A major step forward was the report of linkage of early-onset breast cancer to the polymorphic marker CMM86 located on chromosome 17q (Hall et al 1990). Subsequently, linkage to 17q in 214 families was confirmed by a consortium of 13 research groups from Europe and the USA (Easton et al 1993). Almost all breast/ovarian cancer families and 40% of families with breast cancer alone were found to have a linkage with the BRCA1 (Breast Cancer 1) locus on chromosome 17q. Subsequently, the consortium localized the gene to a 2cM region on 17q, and in 1994, a group in Utah described a large gene on 17q within which they had identified mutations in five affected families (Miki et al 1994). Subsequent work by other research groups has confirmed that this gene is the BRCA1 gene, and over 12,000 carriers of a BRCA1 mutation have now been described in breast/ovarian cancer families and cancer cases unselected for a family history [described on the Breast Cancer Information Core (BIC) database: http://research.nhgri.nih.gov/bic/].
BRCA1 is a large gene with 22 exons encoding a protein of 1863 amino acids. There are no specific hotspots for mutation in the gene, and only a small proportion of mutations are recurrent. Overall, the penetrance of a BRCA1 mutation for ovarian cancer and breast cancer appears to be 40–50% and 80%, respectively Ford et al 1994, 1998). However, there is evidence that penetrance may be modified by both environmental factors and other genetic influences. Furthermore, the location of a mutation within the BRCA1 gene may influence both the overall risk of cancer and the type of cancer. Gayther et al (1995) reported a correlation between mutations toward the 3′ end of the gene and a greater risk of ovarian cancer than breast cancer. Well-defined founder mutations have been described in BRCA1. Mutations at 185delAG and 5382insC have been documented in approximately 1% of Ashkenazi Jewish cancers, and up to 40% of Ashkenazi Jewish women with ovarian cancer or early-onset breast cancer. Identification of BRCA2 closely followed the cloning of BRCA1. Wooster et al (1995) identified the gene following studies involving high-risk families in which the disease pattern was not linked to BRCA1.
BRCA2, located on chromosome 13q, is also a large gene with 26 coding exons encoding a protein with 3418 amino acids. More than 11,000 mutation carriers have been described to date (the BIC database). As for BRCA1, there are no hotspots for mutation, although there are founder mutations in some ethnic groups such as Ashkenazi Jews (Levy-Lahad et al 1997, Thorlacius et al 1997). Although the penetrance for breast cancer is approximately 80%, the penetrance for ovarian cancer seems to be lower than that for BRCA1 mutations at approximately 25% (Ford et al 1998). There is a suggestion that mutations in exon 11 confer a higher risk of ovarian cancer than mutations in other areas (Gayther et al 1997). This has been confirmed in a more recent meta-analysis (reviewed in Ramus and Gayther 2009). Overall, BRCA1 and BRCA2 are believed to account for over 95% of hereditary cases of ovarian cancer and over 80% of hereditary cases of breast cancers.
The functions of BRCA1 and BRCA2 are still unclear. Most of the mutations of BRCA1 are small insertions or deletions, which result in loss of protein synthesis or production of a truncated protein. This observation supports the concept that BRCA1 functions as a tumour suppressor gene. In this ‘two-hit’ model, although one copy of the gene is inherited as an inactive mutant form, the development of cancer requires somatic mutation of the other wild-type (‘normal’) gene copy. Analysis of tumours from familial breast/ovarian cancer families has demonstrated that loss of heterozygosity (LOH) in these tumours involves the wild-type gene, thus providing further evidence that the BRCA1 gene is a tumour suppressor (Smith et al 1992). Both BRCA1 and BRCA2 are expressed in the largest amounts in the testis and thymus, and at lower levels in the breast and ovary. Although the genes have limited sequence homology, they do have similarities including being A-T rich, having a large exon 11 with the start of translation at codon 2. The presence of a zinc finger motif in BRCA1 has suggested a role as a transcription factor, whilst BRCA2 has homology with known transcription factors. However, the most compelling functional evidence indicates that BRCA1 and BRCA2 both play a major role in homologous and non-homologous DNA double-strand break (DSB) repair through their interaction with the RAD51 DSB protein. It is still not known why abrogation of such a ubiquitous cellular function should predispose individuals specifically to breast and ovarian cancer.
HNPCC-associated gynaecological cancer and DNA repair genes
HNPCC is associated with autosomal-dominant inheritance of colorectal cancer in those who do not have the multiple adenomas which occur in FAP. The identification of the genetic basis of Lynch II syndrome is a notable example of the power of molecular technology. HNPCC was classified by Henry Lynch into site-specific hereditary colon cancer (Lynch I syndrome), and families with a predisposition to non-polyposis colorectal cancer in association with other cancers including stomach, small bowel, ureter, renal pelvis and brain, as well as ovarian and endometrial cancer (Lynch II syndrome) (Lynch 1985). The identification of the genes responsible for HNPCC was a result of several developments which showed that HNPCC is associated with hereditary defects in one of five DNA mismatch repair genes [mutS homolog 2 (MSH2), mutL homolog 1 (MLH1), postmeiotic segregation increased 1 (PMS1), postmeiotic segregation increased 2 (PMS2) and mutS homolog 6/G/T mismatch-binding protein (MSH6/GTBP)]. Hundreds of mutations have been identified in these five genes in HNPCC families, making mutation testing a difficult challenge in these families (Lynch and de la Chapelle 1999). Available evidence suggests that these genes function as tumour suppressor genes in a manner consistent with Knudson’s ‘two-hit’ model. The inherited germline mutation inactivates one copy of the mismatch repair gene, but inactivation of the remaining wild-type copy by a second somatic mutation is required prior to tumour formation. The secondary, acquired mutations which result from loss of DNA repair gene function include defects of APC, K-ras, DPC4 and p53 (Huang et al 1996, Kinzler and Vogelstein 1996). The penetrance for colorectal cancer in mutation carriers is in excess of 80% in men and 30% in women, and HNPCC accounts for approximately 5% of colorectal cancers (Dunlop et al 1997, Aarnio et al 1999, Salovaara et al 2000). Endometrial cancer is the second most common cancer in HNPCC families, with female carriers having a 42% risk by 70 years of age (Dunlop et al 1997). The penetrance for ovarian cancer is less, with a reported lifetime risk of 9% (Marra and Roland 1995).
Management of Familial Gynaecological Cancer
Risk assessment
Prevention of familial cancer
Use of the oral contraceptive pill may be suggested for HNPCC and breast/ovarian cancer family members on the basis of case–control studies in the general population which indicate a protective effect against ovarian and endometrial cancer. While one study has suggested a protective effect of the oral contraceptive pill in BRCA1 and BRCA2 carriers, this was not confirmed in a second study and the situation remains unclear (Modan et al 2001). Furthermore, there is concern in breast/ovarian cancer families that a reduction in risk of ovarian cancer may be offset by an increased risk of breast cancer.
Prophylactic salpingo-oophorectomy and hysterectomy as a primary procedure is justifiable in women from breast/ovarian cancer and HNPCC families after completion of their family and after thorough counselling. There is convincing evidence that salpingo-oophorectomy is an effective method of prevention of cancer of the ovary, fallopian tube and breast. A recent meta-analysis (Rebbeck et al 2009) of all reports of preventative surgery in BRCA1 and BRCA2 mutation carriers from 1999 to 2007 revealed a reduction in risk of breast cancer (hazard ratio 0.49) and ovarian/fallopian tube cancer (hazard ratio 0.21). Women undergoing prophylactic surgery should be counselled that the procedure will prevent ovarian and tubal cancer, but that they may still be at risk of primary peritoneal cancer.
It should also be noted that cases of intra-abdominal carcinomatosis following oophorectomy have been reported in which subsequent review of the oophorectomy specimen revealed a small focus of ovarian cancer (Chen et al 1985
[/not-level-membership-for-obstetrics-gynecology-category]
