Chapter 415 The Fetal to Neonatal Circulatory Transition
415.1 The Fetal Circulation
The human fetal circulation and its adjustments after birth are similar to those of other large mammals, although rates of maturation differ. In the fetal circulation, the right and left ventricles exist in a parallel circuit, as opposed to the series circuit of a newborn or adult (see  Fig. 415-1A on the Nelson Textbook of Pediatrics website at www.expertconsult.com). In the fetus, the placenta provides for gas and metabolite exchange. Since the lungs do not provide gas exchange, the pulmonary vessels are vasoconstricted, diverting blood away from the pulmonary circulation. Three cardiovascular structures unique to the fetus are important for maintaining this parallel circulation: the ductus venosus, foramen ovale, and ductus arteriosus.
Fig. 415-1A on the Nelson Textbook of Pediatrics website at www.expertconsult.com). In the fetus, the placenta provides for gas and metabolite exchange. Since the lungs do not provide gas exchange, the pulmonary vessels are vasoconstricted, diverting blood away from the pulmonary circulation. Three cardiovascular structures unique to the fetus are important for maintaining this parallel circulation: the ductus venosus, foramen ovale, and ductus arteriosus.
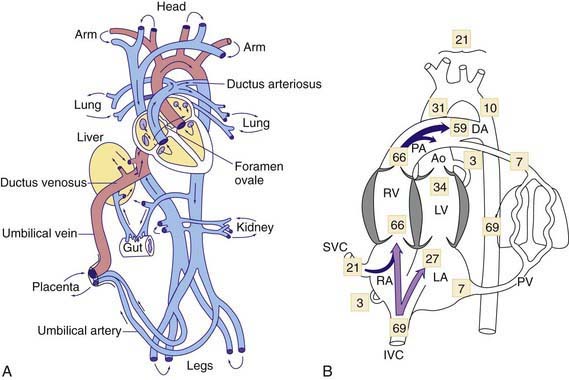
The placenta is not as efficient an oxygen exchange organ as the lungs, so that umbilical venous PO2 (the highest level of oxygen provided to the fetus) is only about 30-35 mm Hg. Approximately 50% of the umbilical venous blood enters the hepatic circulation, whereas the rest bypasses the liver and joins the inferior vena cava via the ductus venosus, where it partially mixes with poorly oxygenated inferior vena cava blood derived from the lower part of the fetal body. This combined lower body plus umbilical venous blood flow (PO2 of ≈26-28 mm Hg) enters the right atrium and is preferentially directed by a flap of tissue at the right atrial–inferior vena caval junction, the eustachian valve, across the foramen ovale to the left atrium (see Fig. 415-1B). This is the major source of left ventricular blood flow, since pulmonary venous return is minimal. Left ventricular blood is then ejected into the ascending aorta where it supplies predominantly the fetal upper body and brain.
It has been postulated that blood flow is an important determinant of growth of fetal cardiac chambers, valves, and blood vessels. Thus, in the presence of a narrowing (stenosis) of an upstream structure such as the mitral valve, flow downstream into the left ventricle is limited and left ventricular growth may be compromised, leading to hypoplastic left heart syndrome (Chapter 425.10). Similarly, stenosis of a downstream structure such as the aortic valve can similarly disrupt flow into the left ventricle and lead to hypoplastic left heart syndrome. Fetal cardiac interventional treatments, currently experimental, are aimed at opening stenotic aortic valves in mid-gestation fetuses, and allowing more normal left ventricular growth.
415.3 The Neonatal Circulation
At birth, the fetal circulation must immediately adapt to extrauterine life as gas exchange is transferred from the placenta to the lungs (Chapter 95.1). Some of these changes are virtually instantaneous with the 1st breath, whereas others develop over a period of hours or weeks. With the onset of ventilation, pulmonary vascular resistance is markedly decreased as a consequence of both active (PO2-related) and passive (mechanical related) pulmonary vasodilation. In a normal neonate, closure of the ductus arteriosus and the fall in pulmonary vascular resistance decreases pulmonary arterial and right ventricular pressures. The largest decline in pulmonary resistance from the high fetal levels to the low “adult” levels in the human infant at sea level usually occurs within the 1st 2-3 days but may be prolonged for 7 days or more. Over the next several weeks of life, pulmonary vascular resistance decreases even further, secondary to a remodeling of the pulmonary vasculature, including thinning of the vascular smooth muscle and recruitment of new vessels. This decrease in pulmonary vascular resistance significantly influences the timing of the clinical appearance of many congenital heart lesions that are dependent on the relative levels of systemic and pulmonary vascular resistances. The left-to-right shunt through an large ventricular septal defect may be minimal in the 1st wk after birth when pulmonary vascular resistance is still high. As pulmonary resistance decreases in the next week or two, the volume of the left-to-right shunt through the ventricular septal defect increases and eventually leads to symptoms of heart failure within the 1st month or two of life.
Significant differences between the neonatal circulation and that of older infants include: (1) right-to-left or left-to-right shunting may persist across the patent foramen ovale; (2) in the presence of cardiopulmonary disease, continued patency of the ductus arteriosus may allow left-to-right, right-to-left, or bidirectional shunting; (3) the neonatal pulmonary vasculature constricts more vigorously in response to hypoxemia, hypercapnia, and acidosis; (4) the wall thickness and muscle mass of the neonatal left and right ventricles are almost equal; and (5) newborn infants at rest have relatively high oxygen consumption, which is associated with relatively high cardiac output. The newborn cardiac output (about 350 mL/kg/min) falls in the 1st 2 mo of life to about 150 mL/kg/min and then more gradually to the normal adult cardiac output of about 75 mL/kg/min. Although fetal hemoglobin is beneficial to delivery of oxygen in the low PO2 fetal circulation, the high percentage of fetal hemoglobin present in the newborn may actually interfere with delivery of oxygen to tissues in the high systemic PO2 neonatal circulation (Chapter 95.1).
The foramen ovale is usually functionally closed by the 3rd mo of life, although it is possible to pass a probe through the overlapping flaps in a large percentage of children and in 15-25% of adults. Functional closure of the ductus arteriosus is usually complete by 10-15 hr in a normal neonate, although the ductus may remain patent much longer in the presence of congenital heart disease, especially when associated with cyanosis. In premature newborn infants, an evanescent systolic murmur with late accentuation or a continuous murmur may be audible, and in the context of respiratory distress syndrome, the presence of a patent ductus arteriosus should be suspected (Chapter 95.4).
Abman SH. Recent advances in the pathogenesis and treatment of persistent pulmonary hypertension of the newborn. Neonatology. 2007;91:283-290.
Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412-417.
Friedberg MK, Silverman NH, Moon-Grady AJ, et al. Prenatal detection of congenital heart disease. J Pediatr. 2009;155(1):26-31.
Gluckman PD, Hanson MA, Pinal C. The developmental origins of adult disease. Matern Child Nutr. 2005;1:130-141.
Ionescu-Ittu R, Marelli AJ, Mackie AS, et al. Prevalence of severe congenital heart disease after folic acid fortification of grain products: time trend analysis in Quebec, Canada. BMJ. 2009;338:b1673.
McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol Rev. 2005;85:571-633.
Ozanne SE, Fernandez-Twinn D, Hales CN. Fetal growth and adult diseases. Semin Perinatol. 2004;28:81-87.
Peterson AL, Deatsman S, Frommelt MA, et al. Correlation of echocardiographic markers and therapy in persistent pulmonary hypertension of the newborn. Pediatr Cardiol. 2009;30:160-165.
Rudolph AM. Aortopulmonary transposition in the fetus: speculation on pathophysiology and therapy. Pediatr Res. 2007;61:375-380.
Rudolph AM. Congenital diseases of the heart: clinical-physiological considerations, ed 2. New York: Futura; 2001.
Trines J, Hornberger LK. Evolution of heart disease in utero. Pediatr Cardiol. 2004;25:287-298.



