[level-membership-for-cardiovascular-category]
Chapter 2 The Endothelium
In 1839, the German physiologist Theodor Schwann became the first to describe a “thin, but distinctly perceptible membrane” that he observed as part of the capillary vessel wall that separated circulating blood from tissue.1,2 The cellular monolayer that formed this membrane would later be named the endothelium; however, the term endothelium did not appear until 1865 when it was introduced by the Swiss anatomist Wilhelm His in his essay, “Die Häute und Höhlen des Körpers (The Membranes and Cavities of the Body).”2,3 Owing to its anatomical location, the endothelium was believed initially to be a passive receptacle for circulating blood, cells, and macromolecules. It is now known that the endothelium is a dynamic cellular structure, and its biological and functional properties extend beyond that of a physical anatomical boundary. In its totality, the endothelium comprises approximately 10 trillion (1013) cells with a surface area of 7 m2, weighs 1.0 to 1.8 kilograms, and contributes 1.4% to total body mass.4,5 Endothelium exists as a monolayer of cells that is present in all arteries, veins, capillaries, and the lymphatic system, and lies at the interface of the bloodstream or lymph and the vessel wall.
Homeostatic Functions of the Endothelium
The endothelium exhibits considerable regional heterogeneity that reflects its arterial or venous location in the vascular tree, as well as the specialized metabolic and functional demands of the underlying tissues.5–7 Despite this heterogeneity, there are basal homeostatic properties that are common to all endothelial cell (EC) populations, although some of these functions may achieve greater importance in selected vascular beds7 (Box 2-1).
 Box 2-1
Box 2-1Maintenance of a Thromboresistant Surface and Regulation of Hemostasis
The endothelium was first recognized as a cellular structure that compartmentalizes circulating blood.4 As such, the endothelial luminal surface is exposed to cells and proteins in the bloodstream that possess prothrombotic and procoagulant activity and, when necessary, support hemostasis. Normal endothelium preserves blood fluidity by synthesizing and secreting factors that limit activation of the clotting cascade, inhibit platelet aggregation, and promote fibrinolysis.8 These include the cell surface–associated anticoagulant factors thrombomodulin, protein C, tissue factor pathway inhibitor (TFPI), and heparan sulfate proteoglycans (HSPG) that act in concert to limit coagulation at the luminal surface of the endothelium.8–10 For instance, thrombin-mediated activation of protein C is accelerated 104-fold by binding to thrombomodulin, Ca2 +, and the endothelial protein C receptor. Activated protein C (APC) engages circulating protein S, which is also synthesized and released by the endothelium, to inactivate factors Va and VIIIa proteolytically.8,11 Tissue factor pathway inhibitor is a Kunitz-type protease inhibitor that binds to and inhibits factor VIIa; about 80% of TFPI is bound to the endothelium via a glycosylphosphatidylinositol anchor and forms a quaternary complex with tissue factor – factor VIIa to diminish its procoagulant activity.12,13 Proteoglycan heparan sulfates that are present in the EC glycocalyx attain anticoagulant properties by catalyzing the association of the circulating serine protease inhibitor antithrombin III to factors Xa, IXa, and thrombin.8 Thus, these anticoagulant factors serve to limit activation and propagation of the clotting cascade at the endothelial luminal surface and thereby maintain vascular patency.
The endothelium also synthesizes and secretes tissue plasminogen activator (tPA) and the ecto-adenosine diphosphatase (ecto-ADPase) CD39 to promote fibrinolysis and inhibit platelet activation, respectively. Tissue plasminogen activator is produced and released into the bloodstream continuously, but unless tPA binds fibrin, it is cleared from the plasma within 15 minutes by the liver.8 Fibrin binding accelerates tPA amidolytic activity by increasing the catalytic efficiency for plasminogen activation and plasmin generation. Platelet activation at the endothelial luminal surface is inhibited by the actions of the ectonucleotidase CD39/NTPDase1 that hydrolyzes adenosine diphosphate (ADP), prostacyclin (PGI2), and nitric oxide (NO).8,14,15 Together these agents maintain an environment on the endothelial surface that is profibrinolytic and antithrombotic.
By contrast, in the setting of an acute vascular injury or trauma, the endothelium initiates a rapid and measured hemostatic response through regulated synthesis and release of tissue factor and von Willebrand factor (vWF). Tissue factor is a multidomain transmembrane glycoprotein (GP) that forms a complex with circulating factor VIIa to activate the coagulation cascade and generate thrombin.16 Tissue factor is expressed by vascular smooth muscle cells (VSMCs) and fibroblasts and by ECs only after activation. Tissue factor acquires its biological activity by phosphatidylserine exposure, dedimerization, decreased exposure to TFPI, or posttranslational modification(s) including disulfide bond formation between Cys186 and Cys209.17–19 This disulfide bond is important for tissue factor coagulation activity and may be reduced by protein disulfide isomerase, which is located on the EC surface.
The endothelium also synthesizes and stores vWF, a large polymeric GP that is expressed rapidly in response to injury. Propeptides and multimers of vWF are packaged in Weibel-Palade bodies that are unique to the endothelium. Once released, vWF multimers form elongated strings that retain platelets at sites of endothelial injury. Weibel-Palade bodies also contain P-selectin, angiopoietin-2, osteoprotegerin, the tetraspanin CD63/Lamp3, as well as cytokines, which are believed to be present as a result of incidental packaging.20 The stored pool of vWF may be mobilized quickly to the endothelial surface, where it binds to exposed collagen and participates in formation of a primary platelet hemostatic plug. The endothelium modulates this response further by regulating vWF size, and thereby its activity, through the action of the EC product ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type I motif, number 13).21 This protease cleaves released vWF at Tyr1605-Met1606 to generate smaller-sized polymers and decrease the propensity for platelet thrombus formation.21 Thus, the endothelium uses geographical separation of factors that regulate its anti- and prothrombotic functions to maintain blood fluidity yet allow for a hemostatic response to vascular injury.
Semipermeable Barrier and Transendothelial Transport Pathways
The endothelial monolayer serves as a size-selective semipermeable barrier that restricts the free bidirectional transit of water, macromolecules, and circulating or resident cells between the bloodstream and underlying vessel wall or tissues. Permeability function is determined in part by the architectural arrangement of the endothelial monolayer, as well as the activation of pathways that facilitate the transendothelial transport of fluids, molecules, and cells. This transport occurs via either transcellular pathways that involve vesicle formation, trafficking, and transcytosis, or by the loosening of interendothelial junctions and paracellular pathways22 (Fig. 2-1). Molecules that traverse the endothelium by paracellular pathways are size restricted to a radius of 3 nm or less, whereas those of larger diameter may be actively transported across the cell in vesicles.23 Although the diffusive flux of water occurs in ECs through aquaporin transmembrane water channels, the contribution of these channels to hydraulic conductivity and cellular permeability is limited.24
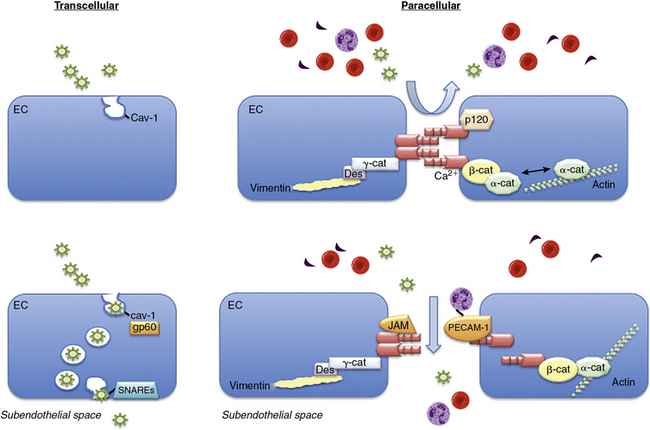
Figure 2-1 Transendothelial transport mechanisms.
(Adapted from Komarova Y, Malik AB: Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72:463–493, 2010.)
There is significant macrostructural heterogeneity of the endothelial monolayer that reflects the functional and metabolic requirements of the underlying tissue and has consequences for its permeability function. Endothelium may be arranged in either a continuous or discontinuous manner: continuous endothelium is either nonfenestrated or fenestrated.4–6
Continuous nonfenestrated endothelium forms a highly exclusive barrier and is found in the arterial and venous blood vessels of the heart, lung, skin, connective tissue, muscle, retina, spinal cord, brain, and mesentery.4–6 By contrast, continuous fenestrated endothelium is located in vessels that supply organs involved in filtration or with a high demand for transendothelial transport, including renal glomeruli, the ascending vasa recta and peritubular capillaries of the kidney, endocrine, and exocrine glands, intestinal villi, and the choroid plexus of the brain.4–6 These ECs are characterized by fenestrae, or transcellular pores, with a diameter of 50 to 80 nm that, in the majority of cells, has a 5- to 6-mm nonmembranous diaphragm across the pore opening.4–6,22 The distribution of these fenestrae may be polarized within the EC and allow for enhanced barrier size selectivity owing to the diaphragm.4–6
Discontinuous endothelium is found in the bone marrow, spleen, and liver sinusoids. This type of endothelial monolayer is notable for its large-diameter fenestrae (100-200 nm) with absent diaphragms and gaps, and a poorly organized underlying basement membrane that is permissive for transcellular flow of water and solutes as well as cellular trafficking.4–6
Transcellular and paracellular pathways are two distinct routes by which plasma proteins, solutes, and fluids traverse the endothelial monolayer. The transcellular pathway provides a receptor-mediated mechanism to transport albumin, lipids, and hormones across the endothelium.22,25,26 The paracellular pathway is dependent upon the structural integrity of adherens, tight, and gap junctions and allows fluids and solutes to permeate between ECs but restricts passage of large molecules.22,25,26 Although these pathways were believed to function independently, it is now recognized that they are interrelated and together modulate permeability under basal conditions.
The transcellular transport of albumin and albumin-bound macromolecules is initiated by albumin binding to gp60, or albondin, a 60-kDa albumin-binding protein located in flask-shaped caveolae that reside at the cell surface.27,28 These caveolae are cholesterol- and sphingolipid-rich structures that contain caveolin-1. Once activated, gp60 interacts with caveolin-1, followed by constriction of the caveolae neck and fission from the cell surface.29,30 These actions lead to formation of vesicles with a diameter of about 70 nm and vesicle transcytosis. Caveolae may contain as much as 15% to 20% of the cell volume, so they are capable of moving significant amounts of fluid across the cell through this mechanism.29,30 Once vesicles have detached from the membrane, they undergo vectorial transit to the abluminal membrane, where they dock and fuse with the plasma membrane by interacting with vesicle-associated and membrane-associated target soluble N-ethylmaleimide-sensitive factor attachment receptors (SNAREs).31 Once docked, the vesicles release their cargo to the interstitial space. Vesicles may traverse the cell as individual structures or cluster to form channel-like structures with a diameter of 80 to 200 nm that span the cell.5,6 Although transcellular vesicle trafficking is the predominant mechanism by which cells transport albumin, it is now appreciated that this pathway is not absolutely necessary for permeability function, owing to the compensatory capabilities of the paracellular pathway.
The junctions between ECs include the adherens, tight, and gap junctions; only the former two modulate permeability and comprise the paracellular pathway.32 Adherens junctions are normally impermeant to albumin and other large molecules and are the major determinant of endothelial barrier function and permeability. The expression of tight junctions, by contrast, is limited to the blood-brain or blood-retinal barriers where they restrict or prevent passage of small molecules (< 1 kDa) and some inorganic ions.22 Gap junctions are composed of connexins that form a channel between adjacent cells to enhance cell-cell communication and facilitate the transit of water, small molecules, and ions.22
Adherens junctions are critical for maintaining endothelial barrier functional integrity and are composed of complexes of vascular endothelial (VE)-cadherin and catenins. Vascular endothelial cadherin is a transmembrane GP with five extracellular repeats, a transmembrane segment, and a cytoplasmic tail. The external domains mediate the calcium-dependent hemophilic adhesion between VE-cadherin molecules expressed in adjacent cells.25,26,33 The cytoplasmic tail interacts with β-catenin, plakoglobin (γ-catenin), and p120 catenin to control the organization of VE-cadherin and the actin cytoskeleton at adherens junctions. The actin binding proteins α-actinin, annexin 2, formin-1, and eplin may further stabilize this interaction. Other proteins located in adherens junctions thought to provide stability include junctional adhesion molecules (JAMs) and platelet–EC adhesion molecule 1 (PECAM-1).22
Endothelial permeability may be increased or decreased through mechanisms that involve adherens junction remodeling or through interactions with the actin cytoskeleton.25,26,34 These events may occur rapidly, be transient or sustained, and are reversible. Most commonly, mediators that increase endothelial permeability either destabilize adherens junctions through phosphorylation, and thereby internalization, of VE-cadherin or by RhoA activation and actin cytoskeletal rearrangement to physically pull apart VE-cadherin molecules and adherens junctions, resulting in intercellular gaps.22 To counteract these effects, other mediators that attenuate permeability are present in the plasma or interstitial space. Fibroblast growth factor (FGF) stabilizes VE-cadherin by stabilizing VE-cadherin-gp120-catenin interaction. Sphingosine-1-phosphate, generated by breakdown of the membrane phospholipid sphingomyelin or released from activated platelets, also stabilizes adherens junctions. This effect occurs through activation of Rac1/Rap1/Cdc42 signaling and reorganization of the actin cytoskeleton, recycling of VE-cadherin to the cell surface, and (re)assembly of adherens junctions. The cytokine angiopoietin-1 stabilizes adherens junctions by inhibiting endocytosis of VE-cadherin.22,25,26,35,36
Endothelial tight junctions predominate in specialized vascular beds that require an impermeable barrier. These tight junctions are composed of the specific tight junction proteins occludin, claudins (3/5), and JAM-A.22,33,36,37 Occludin and claudins are membrane proteins that contain four transmembrane and two extracellular loop domains. The extracellular loop domains of these proteins bind similar domains on neighboring cells to seal the intercellular cleft and prevent permeability. Occludin, claudins, and JAM-A are also tethered to the actin cytoskeleton by α-catenin and zona occludens proteins (ZO-1, ZO-2).22 The ZO proteins also function as guanylyl kinases or scaffolding proteins and use PDZ and Sc homology 3 (SH3)-binding domains to recruit other signaling molecules. Connections between tight junctions and the actin cytoskeleton are stabilized further via the actin cross-linking proteins spectrin or filamen or by the accessory proteins cingulin and AF-6.22,36 In this manner, the junctions remain stabilized and sealed to limit or prevent transendothelial transport of fluids and molecules.
Regulation of Vascular Tone
Since the early seminal studies of Furchgott and Zawadski, it has been increasingly recognized that the endothelium regulates vascular tone via endothelium-derived factors that maintain a balance between vasoconstriction and vasodilation38,39 (Fig. 2-2). The endothelium produces both gaseous and peptide vasodilators, including NO, hydrogen sulfide, PGI2, and endothelium-derived hyperpolarizing factor (EDHF). The effects of these substances on vascular tone are counterbalanced by vasoconstrictors that are either synthesized or processed by the endothelium, such as thromboxane A2 TxA2, a product of arachidonic acid metabolism, and the peptides endothelin-1 (ET-1) and angiotensin II (Ang-II). The relative importance of these vasodilator or vasoconstrictor substances for maintaining vascular tone differs between vascular beds, with NO serving as the primary vasodilator in large conduit elastic vessels and non-NO mechanisms playing a greater role in the microcirculation.
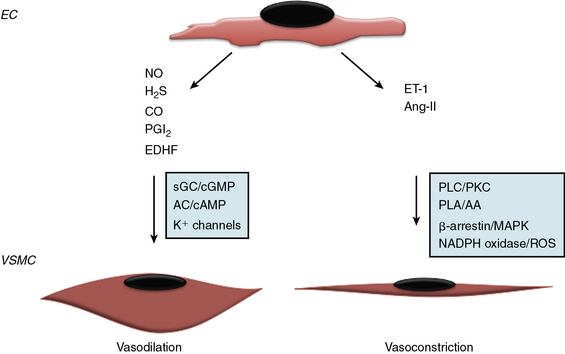
Nitric oxide is synthesized by three structurally similar NO synthase (NOS) isoenzymes: the constitutive enzyme identified in the endothelium (eNOS or NOS3) and neuronal cells (nNOS or NOS1) or the inducible enzyme (iNOS or NOS2) found in smooth muscle cells (SMCs), neutrophils, and macrophages following exposure to endotoxin or inflammatory cytokines.40–42 Nitric oxide is generated via a five-electron oxidation reaction of L-arginine to form L-citrulline and stoichiometric amounts of NO, and requires molecular oxygen and NADPH as co-substrates and flavin adenine dinucleotide, flavin mononucleotide, heme, and tetrahydrobiopterin as cofactors.43–45 In the endothelium, eNOS expression is up-regulated by a diverse array of stimuli including transforming growth factor (TGF)-β1, lysophosphatidylcholine, hydrogen peroxide, tumor necrosis factor (TNF)-α, oxidized low-density lipoprotein (LDL) cholesterol, laminar shear stress, and hypoxia, and is subject to both posttranscriptional and posttranslational modifications that influence activity, including phosphorylation, acetylation, palmitoylation and myristolation, as well as localization to caveolae.45 Once generated, NO diffuses into SMCs and reacts with the heme iron of guanylyl cyclase to increase cyclic guanosine monophosphate (cGMP) levels and promote vasodilation.42 Nitric oxide can also react with SH-containing molecules and proteins (e.g., peroxynitrite, N2O2) to generate S-nitrosothiols, a stable reservoir of bioavailable NO with recognized antiplatelet and vasodilator effects.46–48 In the presence of oxygen, NO can be oxidized to nitrite and nitrate, which are stable end-products of NO metabolism; nitrite serves as a vasodilator, predominantly in the pulmonary and cerebral circulations.48,49 In addition to vasodilator and antiplatelet effects, NO has other paracrine effects that include regulation of VSMC proliferation and migration, and leukocyte adhesion and activation.15
Hydrogen sulfide gas generated by the endothelium also possesses vasodilator properties. Hydrogen sulfide is membrane permeable and released as a byproduct of cysteine or homocysteine metabolism via the transulfuration/cystathionine-β-synthase and cystathionine-γ-lyase pathway or by the catabolism of cysteine via cysteine aminotransferase and 3-mercaptopyruvate sulfur transferase. Hydrogen sulfide–mediated vasodilation results from activation of KATP and transient receptor membrane channel currents.50–52
Prostacyclin is an eicosanoid generated by cyclooxygenase (COX) and arachidonic acid metabolism in the endothelium. It promotes vasodilation via adenylyl cyclase/cyclic adenosine monophosphate (cAMP) signal transduction pathways. Prostacyclin also induces smooth muscle relaxation by reducing cytoplasmic Ca2 + availability; decreases VSMC proliferation through a cAMP–peroxisome proliferator-activated receptor (PPAR)-γ-mediated mechanism, and limits inflammation by decreasing interleukin (IL)-1 and IL-6.53 Importantly, PGI2 has significant antiplatelet effects and by decreasing TxA2 levels, limits platelet aggregation. Because both COX-1 (constitutively expressed) and COX-2 (induced) contribute to basal PGI2 production, selective pharmacological inhibition of either isoform may result in diminished PGI2 levels, increased platelet aggregation, and impaired vasodilation.54
No single molecule has been identified as the vasodilator referred to as endothelium-derived hyperpolarizing factor, and the effects attributed to Endothelium-derived hyperpolarizing factor likely represent the composite actions of several agents that share a common mechanism. Endothelium-derived hyperpolarizing factor is an important vasodilator in the microcirculation and acts by opening K+ channels to allow for K+ efflux, hyperpolarization, and vascular smooth muscle relaxation. Candidate EDHFs include the 11, 12-epoxyeicosatrienoic acids and hydrogen peroxide.39,55–58
To counterbalance the effects of endothelium-derived vasodilators, the endothelium also synthesizes the vasoconstrictor ET-1 and metabolizes Ang I to Ang II. Endothelin-1, a 21-amino-acid peptide, is synthesized initially as inactive pre-proET-1 that is processed by endothelin-converting enzymes to its active form.59,60 Endothelin-1 binds to the G protein–coupled receptors (GPCRs) ETA and ETB: ECs express ETB, whereas SMCs express both receptors. Although activation of endothelial ETB increases NO production, concomitant activation of SMC ETA and ETB results in prolonged and long-lasting vasoconstriction that predominates.61
There is no evidence that ET-1 is stored for immediate early release in the endothelium, indicating that acute stimuli such as hypoxia, TGF-β, and shear stress that increase ET-1 production do so via a transcriptional mechanism; however, ET-1 and endothelin-converting enzyme are packaged in Weibel-Palade bodies.62 Endothelium also expresses angiotensin-converting enzyme (ACE) and, as such, modulates processing of Ang-I to the vasoconstrictor peptide Ang-II.63 Ang-II–stimulated activation of the Ang-I receptor results in vasoconstriction and SMC hypertrophy and proliferation, in part, by activating NADPH oxidase to increase reactive oxygen species (ROS) production.64–66 Vascular tone, therefore, is determined by the balance of vasodilator and vasoconstrictor substances synthesized or processed by the endothelium in response to stimuli: each vasoactive mediator may attain individual importance in a different vascular bed.
Regulating Response to Inflammatory and Immune Stimuli
The endothelium monitors circulating blood for foreign pathogens and participates in immunosurveillance by expressing Toll-like receptors (TLRs) 2, 3, and 4.67–69 These TLRs identify pathogen-associated molecular patterns that are common to bacterial cell wall proteins or viral deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) in the bloodstream. Once activated, TLRs elicit an inflammatory response through activation of nuclear factor (NF)-κB and generation of chemokines that promote transendothelial migration of leukocytes, have chemoattractant and mitogenic effects, and increase endothelial oxidant stress and apoptosis.67,68
The quiescent endothelium maintains its antiinflammatory phenotype through expression of cytokines with antiinflammatory properties and cytoprotective antioxidant enzymes that limit oxidant stress. The endothelium synthesizes TGF-β1, which inhibits synthesis of the proinflammatory cytokines monocyte chemotactic protein-1 (MCP-1) and IL-8; expression of the TNF-α receptor; NF-κB-mediated proinflammatory signaling; and leukocyte adherence to the luminal surface of the endothelium.70,71 Endothelium also expresses a wide array of antioxidant enzymes, including catalase, the superoxide dismutases, glutathione peroxidase-1, peroxiredoxins, and glucose-6-phosphate dehydrogenase.48 Through the actions of these antioxidant enzymes, ROS are reduced, and the redox environment remains stable. This homeostatic redox modulation also limits activation of ROS-stimulated transcription factors such as NF-κB, activator protein-1, specificity protein-1, and PPARs.48 The inflammatory phenotype of the endothelium is also influenced by other circulating or paracrine factors that have antioxidant or antiinflammatory properties, such as high-density lipoprotein (HDL) cholesterol, IL-4, IL-10, IL-13, and IL-1 receptor antagonist.5,6,72,73
The endothelium is capable of mounting a rapid inflammatory response that involves the actions of chemoattractant cytokines, or chemokines, and their associated receptors to facilitate interactions between leukocytes and the endothelium. Endothelial cells express the chemokine receptors CXCR4, CCR2, and CCR8 on the luminal or abluminal surface of cells.74 These receptors bind and transport chemokines to the opposite side of the cell to generate a chemoattractant gradient for inflammatory cell homing. Heparan sulfate (HS), which is present in the endothelial glycocalyx, may serve as a chemokine presenter and is necessary for the action of some chemokines such as CXCL8, CCL2, CCL4, and CCL5.75,76
Endothelial cells also express the Duffy antigen receptor for chemokines (DARC) that participates in chemokine transcytosis across cells. Duffy antigen receptor for chemokines is a member of the silent chemokine receptor family that has high homology to GPCRs and can bind a broad spectrum of inflammatory CC and CXC chemokines, including MCP-1, IL-8, and CCL5 or Regulated upon Activation, Normal T-cell Expressed, and Secreted (RANTES), but does not activate G-protein signaling.77–79 Exposure to chemokines, in turn, activates cellular signaling pathways that promote EC–leukocyte interactions; however, homing of leukocytes to tissues is mediated directly by cell surface adhesion molecules.
Endothelium expresses selectins and immunoglobulin (Ig)-like cell surface adhesion molecules that regulate endothelial-leukocyte interactions. P-selectin and E-selectin are lectin-like transmembrane GPs. These selectins mediate leukocyte adhesion through Ca2 +-dependent binding of their N-terminal C-type lectin-like domain with a sialyl-Lewis X capping structure ligand present on leukocytes.80–82 P-selectin is stored in Weibel-Palade bodies where it can be mobilized rapidly to the cell surface in response to thrombin, histamine, complement activation, ROS, and inflammatory cytokines. Cell surface expression of P-selectin is limited to minutes.80,82 By contrast, E-selectin requires de novo protein synthesis for its expression. E-selectin is expressed on the cell surface, but it may also be found in its biologically active form in serum as a result of proteolytic cleavage from the cell surface.5,81,82 These selectins bind the leukocyte ligands P-selectin glycoprotein ligand-1 (PSGL-1), E-selectin-ligand-1, and CD44, each of which appears to have a distinct function: PSGL1 is implicated in the initial tethering of leukocytes to the endothelium, E-selectin-ligand-1 converts transient initial tethers to slower and more stable rolling, and CD44 controls the speed of rolling.81,82
The Ig-like cell surface adhesion molecules expressed by the endothelium are intercellular adhesion molecule (ICAM)-1,ICAM-2, vascular cell adhesion molecule (VCAM)-1, and PECAM-1. Intercellular adhesion molecule-1 is expressed at low levels in the endothelium, but its expression is up-regulated several-fold by TNF-α or IL-1. Intercelluar adhesion molecule-1 is active when it exists as a dimer and is able to bind macrophage adhesion ligand-1 or lymphocyte function–associated antigen-1 on leukocytes to facilitate transendothelial migration.82,83 Clustering of ICAM-1 stimulates endothelial cytoskeletal rearrangements to form cuplike structures on the endothelial surface and remodel adherens junction complexes to enhance leukocyte transendothelial migration.82,84,85 Intercellular adhesion molecule-2, by contrast, is constitutively expressed at high levels by the endothelium, but its expression is down-regulated by inflammatory cytokines; however, ICAM-2 is believed to play a role in cytokine-stimulated migration of eosinophils and dendritic cells.86,87 Vascular cell adhesion molecule-1 is also up-regulated by inflammatory cytokines, binds to very late antigen-4 on leukocytes, and activates Rac-1 to increase NADPH oxidase activity and ROS production.82 PECAM-1 is expressed abundantly in adherens junctions and is involved in homophilic interaction between endothelial and leukocyte PECAM-1. This interaction stimulates targeted trafficking of segments of EC membrane to surround a leukocyte in preparation for transendothelial migration and typically occurs within 1 or 2 μm of an intact endothelial junction.82 The determination as to whether a leukocyte migrates paracellularly or transcellularly, therefore, appears to be dependent upon the relative tightness of endothelial junctions.
Vascular Repair and Remodeling
The vessel wall undergoes little proliferation or remodeling under ambient conditions, with the exception of repair or remodeling associated with physiological processes such as wound healing or menses. When the endothelial monolayer sustains a biochemical or biomechanical injury resulting in EC death and denudation, loss of contact inhibition stimulates the normally quiescent adjacent ECs to proliferate. If the injury is limited, locally proliferating ECs will cover the injured site. However, if the area of injury is larger, circulating blood cells are recruited to aide proliferating resident ECs and reestablish vascular integrity.88
A subset of circulating blood cells that participate in vascular repair expresses cell surface proteins that were thought to be endothelial-specific and subsequently referred to as endothelial progenitor cells (EPCs). These cells could be expanded in vitro to phenotypically resemble mature ECs, and when given in vivo could promote vascular repair and regeneration at sites of ischemia. It is now recognized that these putative EPCs are likely not true progenitor cells for the endothelium, but represent a mixed population of cells that include proangiogenic hematopoietic cells (myeloid or monocyte lineage), circulating ECs that that are viable but nonproliferative, and endothelial colony-forming cells that are viable, proliferative, and emerge at day 14 when cultured in vitro.88–90 These cells reside in the bone marrow as well as in specific niches in postnatal organs and vessel wall. Within blood vessels, it is believed that they are located in niches in the subendothelial matrix or in the vasculogenic zone in the adventitia.91
Putative EPCs were initially thought to promote vascular repair by incorporating into and contributing structurally to the vessel wall, but more recent evidence supports a paracrine role. Once these cells are recruited to sites of injury, they secrete growth and angiogenic factors that promote and support endothelial proliferation. In fact, these cells are known to secrete high levels of vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor.88,89 These cells also provide transient residence as immediate placeholders at the site of endothelial injury and may reside there until proliferation of the endothelial monolayer is complete.89
Mechanotransduction of Hemodynamic Forces
The endothelium is subjected to the effects of hemodynamic forces such as hydrostatic pressure, cyclic stretch, and fluid shear stress, which occur as a consequence of blood pressure and pulsatile blood flow in the vasculature (Fig. 2-3). In the vascular tree, there is a gradient of pulsatile pressure that is proportional to vessel diameter, ranges from around 120 to 100 mmHg in the aorta to about 0 to 30 mmHg in the microcirculation, and modulates other hemodynamic forces.92 Endothelial cells mechanotransduce these forces into cellular responses via ion channels, integrins, and GPCRs, as well as cytoskeletal deformations or displacements.92,93
The endothelial monolayer is exposed to variable levels of shear stress in the vascular tree that are inversely proportional to the radius of the vessel and range from 1 to 6 dyn/cm2 in veins and from 10 to 70 dyn/cm2 in arteries.93 Physiological shear stress promotes a quiescent endothelial phenotype with cells that are aligned morphologically in the direction of flow, owing to the influence of laminar flow and shear on NO release. Increases in shear stress stimulate compensatory EC and SMC hypertrophy to expand the vessel and thereby return shear forces to basal levels. Conversely, a decrease in shear can narrow the lumen of the vessel in an endothelium-dependent manner.93 Flow in tortuous vessels or at bifurcations is characterized by flow reversals, low flow velocities, and flow separation that cause shear stress gradients. Here, ECs acquire a polygonal shape with diminished cell and cytoskeletal alignment with flow.5–7 This disturbed flow profile contributes to development of endothelial dysfunction at these susceptible locations.6,7,93
Cyclic strain is circumferential deformation of the blood vessel wall associated with distension and relaxation with each cardiac cycle.92 Under ambient conditions, cyclic strain averages roughly 2% at 1 Hz in the aorta, but may increase to over 30% when hypertension is present.94,95 In the endothelial monolayer, individual cells are typically arranged so they are oriented perpendicular to the stretch axis. However, when strain levels are increased to pathophysiological levels, this orientation is lost, and stress fibers parallel the direction of stretch.96,97 Elevated levels of cyclic strain increase endothelial matrix metalloproteinases (MMPs) and induce remodeling of the extracellular matrix (ECM) as well as VE-cadherin and adherens junctions.98
In addition to physical forces imposed upon them, ECs are capable of generating traction stress and exerting force against the extracellular environment. These traction forces are mediated by stress fibers, actin-myosin interactions, and other proteins that anchor cells to focal adhesions. These self-generated forces are important for cell shape stability, regulate endothelial permeability and connectivity by applying force to cell junctions, and promote endothelial network formation by creating tension-based guidance pathways by which ECs sense each other at a distance.92,99–102
Endothelial Heterogeneity
Within the vascular tree, there is significant regional heterogeneity of the endothelium that occurs as a result of differences in developmental assignment, cellular structure, and surrounding environmental factors.5,6,103 This heterogeneity exists to support the specialized functions of the underlying vascular beds and tissues. As a result of these differences, the normal adult endothelium also exhibits functional heterogeneity in the homeostatic properties common to all ECs (Fig. 2-4). For instance, the endothelium functions as a semipermeable membrane that regulates transport of fluid, proteins, and macromolecules. Under basal conditions, this takes place primarily across capillaries, albeit at differing rates throughout the vascular beds. However, when stimulated with histamine, serotonin, bradykinin, or VEGF, the endothelium in postcapillary venules responds by increasing permeability either through retraction of adherens junctions and formation of interendothelial gaps, or via increased transendothelial transcytosis. This phenomenon is supported by increased expression of receptors for these agonists in the postcapillary venules.5–7,104,105
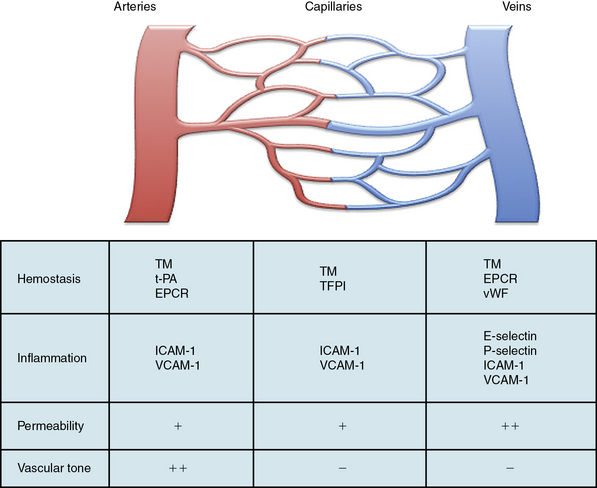
Transendothelial migration of leukocytes occurs as postcapillary venules in the skin, mesentery, and muscle, whereas in the lung and liver, this function takes place mostly at the level of the capillaries. In lymph nodes, this function occurs at the high endothelial venules.106 Activated ECs that are largely restricted to postcapillary venules and express E-selectin mediate this function.107 P-selectin, which is stored in Weibel-Palade bodies, is also preferentially expressed by endothelium in postcapillary venules, with levels of highest expression in the lung and mesentery.108 By contrast, ICAM-1 and VCAM-1 may be expressed throughout the vasculature and respond rapidly to induction by lipopolysaccharide or cytokines. Although interactions between leukocytes and the endothelium occur typically in postcapillary venules, they can also occur in arterioles, capillaries, and large veins.5–7
The endothelium regulates hemostatic functions largely through expression of both anticoagulant and antiplatelet factors that are unevenly distributed throughout the vasculature. For instance, endothelium in the arterial system expresses thrombomodulin, tPA, and the endothelial protein C receptor; capillaries express thrombomodulin and TFPI; and thrombomodulin, the endothelial protein C receptor, and vWF are typically expressed in veins.5–7,109 Endothelium also regulates vascular tone and does so at the level of the resistance arterioles through release of site-specific vasodilator and vasoconstrictor molecules. The endothelium is the predominant source of NO generated by eNOS, and expression of eNOS is greater in the arterial than the venous system.7 Thus, many of these functional heterogeneities allow the endothelium to respond to (patho)physiological stimuli and adapt to a changing environment.
Endothelial Dysfunction and Vascular Disease
Thrombosis
In conjunction with exposure to these pathophysiological stimuli, the activated endothelium is faced with loss of its anticoagulant cell surface–associated molecules, lower levels of antithrombotic NO, and expression of the prothrombotic factors tissue factor and vWF, as well as platelets that are recruited to the site of injury.40,42,110–113 Thrombosis is augmented further by increases in endothelial ROS and oxidant stress, inhibition of tPA activity by plasminogen activator inhibitor-1 (PAI-1) generated by activated ECs, and alterations in shear and other mechanical forces as blood fluidity is diminished.8,81,93
Vasculitis
The primary systemic vasculitides differentially affect vessels based on size and, as such, are grouped accordingly. Takayasu’s arteritis is a large-vessel type that affects the aorta and its major branches, whereas granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis) affects mostly small vessels and occurs as a vasculitis that primarily affects the kidneys and lungs.114,115 Although these vasculitides represent heterogeneous disease processes, they share the endothelium as the common target and propagator of an immuno-inflammatory reaction that occurs in the vessel wall. This immuno-inflammatory reaction may be so profound, as is seen in systemic lupus erythematosus (SLE), that antiendothelial antibodies are generated. These processes result in vascular immune-complex deposition, complement activation, and neutrophil-induced injury to the endothelial monolayer that results in EC activation, apoptosis, and in some areas, denudation.116,117 Other resident activated ECs synthesize and secrete cytokines, growth factors, and chemokines that include IL-1, IL-6, IL-8, and MCP-1.110 Repeated injury to the endothelium from prolonged attack by immune and inflammatory cells can stimulate a prothrombotic and profibrotic response that ultimately leads to vessel occlusion and abnormal vascular remodeling.
Atherosclerosis
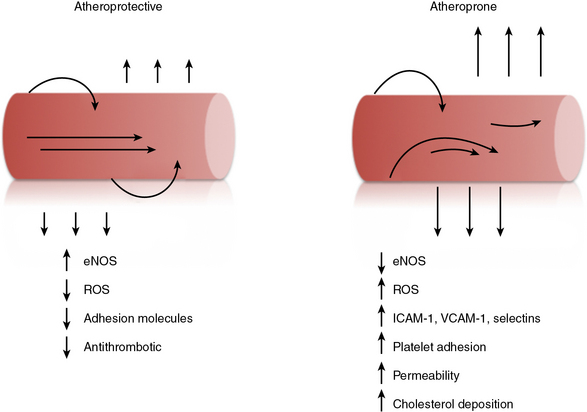
Atherosclerosis is a progressive disease of blood vessels that is initiated by endothelial dysfunction and is now recognized as a chronic inflammatory and immune process. Atherosclerosis is characterized by the accumulation of lipid, thrombus, and inflammatory cells within the vessel wall.48,118–120 This process may acutely occlude the vessel lumen, as occurs with plaque rupture and thrombosis, or result in a more chronic but stable process that eventually encroaches on the vessel lumen. In either event, atherosclerosis can lead to end-organ ischemia and ensuing infarction of the heart, brain, vital organs, or extremities. Early endothelial dysfunction associated with atherosclerosis is evidenced by the presence of a subendothelial accumulation of lipids and infiltration of monocyte-derived macrophages and other immune cells to form the fatty streak. Among the risk factors associated with development of atherosclerosis, diabetes mellitus, tobacco use, hyperlipidemia, and hypertension are all known to induce endothelial dysfunction.121 Within the vasculature, however, the branch points and bifurcations tend to be the most atherosclerosis-prone segments, indicating that hemodynamic profiles and complex non-uniform flow is also of importance for endothelial dysfunction.93,122 Once atherosclerosis is established, the endothelium continues to modify the progression of disease by recruiting inflammatory and immune cells and platelets; diminished NO production, enhanced permeability, and the production of prothrombotic species are believed to contribute to plaque progression.48,118–120,123
Functional Assessment of the Endothelium
Nitric Oxide–Mediated Vasodilation
Owing to the importance of endothelial function for vascular health, assessments of endothelial-dependent vasodilator responses, which reflect endothelial NO generation and NO bioavailability, have been advanced as predictors of adverse cardiovascular events. These studies are based on the principle that a healthy endothelium, when challenged with a physiological stress such as shear stress or an endothelium-dependent vasodilator such as acetylcholine, will release NO, leading to a measurable vasodilatory response. In contrast, when the endothelium is dysfunctional or diseased, these stimuli will elicit a vasoconstrictor or significantly diminished vasodilator response. In humans, this phenomenon, which recapitulates the preclinical studies of Furchgott and Zawadski, was first demonstrated following the intracoronary administration of acetylcholine to patients with angiographically diseased or normal epicardial coronary arteries. Here, the patients with prevalent atherosclerosis demonstrated paradoxical vasoconstriction when infused with acetylcholine, but normal vasodilator responses when challenged with the NO donor nitroglycerin. Patients with normal vessels dilated appropriately to both agents.124
Subsequently, a close correlation between coronary artery vasodilation in response to acetylcholine and noninvasive measurements of flow-mediated dilation of the brachial artery was demonstrated. Imaging of the brachial artery with high-resolution vascular ultrasound to detect flow-mediated dilation or the use of strain-gauge forearm plethysmography to assess forearm blood flow in response to pharmacological stimuli that release NO are both accepted methodologies for evaluating endothelial function.125–127 To date, these methods have been used to demonstrate impaired endothelium-dependent vascular reactivity in adults with risk factors for atherosclerosis in the absence of overt atherothrombotic cardiovascular disease; in children with diabetes mellitus, hypercholesterolemia, and congenital heart disease; and to demonstrate improved function in patients treated with 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins) or ACE inhibitors.128–133
Measurement of peripheral arterial tonometry is emerging as a newer methodology to examine endothelial function. This device utilizes finger-mounted probes with an inflatable membrane that record a pulse wave in the presence and absence of flow-mediated dilation. This method has been shown to correlate well with endothelial dysfunction assessed by brachial artery flow-mediated dilation.134
ADMA as a Biochemical Marker of Nitric Oxide Bioavailability
The endogenous competitive NOS inhibitor asymmetrical dimethylarginine (ADMA) has been suggested as a biomarker for decreased NO bioavailability and endothelial function. Asymmetrical dimethylarginine generated by the hydrolysis of methylated arginine residues is subject to intracellular degradation by dimethylarginine dimethylaminohydrolase (DDAH), but the activity of this enzyme is decreased significantly by oxidant stress.135–138 This in turn leads to increases in plasma ADMA levels, a finding that has been demonstrated in patients with risk factors for atherosclerosis or established coronary artery disease (CAD).139–142
With respect to endothelial function, a cross-sectional study of individuals enrolled in the Cardiovascular Risk in Young Finns Study confirmed a significant, albeit modest, inverse relationship between ADMA levels and endothelial function assessed by flow-mediated vasodilation.143 Despite these findings, in a community-based sample, ADMA levels were not associated with cardiovascular disease incidence or all-cause mortality in diabetic patients.144 Based on these observations, in certain populations, ADMA levels alone may not provide a full assessment of endothelial function; direct measurements of endothelial vasodilator capacity may be required.
Endothelial Microparticles
Endothelial microparticles are emerging as a surrogate biomarker for endothelial dysfunction.145 Endothelial cells can release membrane vesicles with a diameter of approximately 0.1 to 1.0 μm that include microparticles, exosomes, and apoptotic bodies. These microparticles are formed from plasma membrane blebbing and package endothelial proteins that include VE-cadherin, PECAM-1, ICAM-1, E-selectin, endoglin, VEGF receptor-2, S-endo, αv integrin, and eNOS.145,146 Although many of these proteins are expressed by microparticles derived from other cell types, the presence of VE-cadherin and E-selectin indicates EC origin. Endothelial microparticle formation is stimulated by TNF-α, ROS, inflammatory cytokines, lipopolysaccharides, thrombin, and low shear stress.146 They have procoagulant properties as a result of exposed phosphatidylserines and tissue factor that is present in the microparticle, as well as proinflammatory properties.
Techniques to measure circulating endothelial microparticles rely on differential centrifugation in platelet-free plasma and on the identification of cell-surface CD antigens.145,146 Thus, they may not be as convenient a measure of endothelial function as currently available noninvasive imaging techniques. Nonetheless, circulating endothelial microparticles have been measured and found to be elevated in a number of patient populations with risk factors or diseases associated with endothelial dysfunction.146 Increased levels of endothelial microparticles have been demonstrated and shown to correlate with flow-mediated dilation in individuals with end-stage renal disease, acute coronary syndromes (ACS), metabolic syndrome, diabetes, and systemic and pulmonary hypertension.147–152
1 Schwann T. Microscopical researches into the accordance in the structure and growth of animals and plants. London: Syndenham Society; 1847.
2 Hwa C., Aird W.C. The history of the capillary wall: doctors, discoveries, and debates. Am J Physiol Heart Circ Physiol. 2007;293:H2667–H2679.
3 His W. Die häute und höhlen des körpers. Basel: Schwighauser; 1865.
4 Gimbrone M. Vascular endothelium: Nature’s blood container. Vascular endothelium in hemostasis and thrombosis. New York: Churchill Livingstone; 1986. pp 1–13
5 Aird W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173.
6 Aird W.C. Phenotypic heterogeneity of the endothelium: II. Representative vascular beds. Circ Res. 2007;100:174–190.
7 dela Paz N.G., D’Amore P.A. Arterial versus venous endothelial cells. Cell Tissue Res. 2009;335:5–16.
8 van Hinsbergh V.W. Endothelium-role in regulation of coagulation and inflammation. Semin Immunopathol. 2011.
9 Navarro S., Bonet E., Estelles A., et al. The endothelial cell protein C receptor: Its role in thrombosis. Thromb Res. 2011;128:410–416.
10 Rezaie A.R. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr Med Chem. 2010;17:2059–2069.
11 Conway E.M. Thrombomodulin and its role in inflammation. Semin Immunopathol. 2011.
12 Kasthuri R.S., Glover S.L., Boles J., et al. Tissue factor and tissue factor pathway inhibitor as key regulators of global hemostasis: Measurement of their levels in coagulation assays. Semin Thromb Hemost. 2010;36:764–771.
13 Zhang J., Piro O., Lu L., et al. Glycosyl phosphatidylinositol anchorage of tissue factor pathway inhibitor. Circulation. 2003;108:623–627.
14 Atkinson B., Dwyer K., Enjyoji K., et al. Ecto-nucleotidases of the cd39/ntpdase family modulate platelet activation and thrombus formation: potential as therapeutic targets. Blood Cells Mol Dis. 2006;36:217–222.
15 Welch G., Loscalzo J. Nitric oxide and the cardiovascular system. J Card Surg. 1994;9:361–371.
16 Bazan J.F. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci U S A. 1990;87:6934–6938.
17 Jasuja R., Furie B., Furie B.C. Endothelium-derived but not platelet-derived protein disulfide isomerase is required for thrombus formation in vivo. Blood. 2010;116:4665–4674.
18 Breitenstein A., Tanner F.C., Luscher T.F. Tissue factor and cardiovascular disease: quo vadis? Circ J. 2010;74:3–12.
19 Bach R.R. Tissue factor encryption. Arterioscler Thromb Vasc Biol. 2006;26:456–461.
20 Valentijn K.M., Sadler J.E., Valentijn J.A., et al. Functional architecture of Weibel-Palade bodies. Blood. 2011;117:5033–5043.
21 Lowenberg E.C., Meijers J.C., Levi M. Platelet-vessel wall interaction in health and disease. Neth J Med. 2010;68:242–251.
22 Komarova Y., Malik A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72:463–493.
23 Pappenheimer J.R., Renkin E.M., Borrero L.M. Filtration, diffusion and molecular sieving through peripheral capillary membranes; a contribution to the pore theory of capillary permeability. Am J Physiol. 1951;167:13–46.
24 Fischbarg J. Fluid transport across leaky epithelia: central role of the tight junction and supporting role of aquaporins. Physiol Rev. 2010;90:1271–1290.
25 Dejana E., Orsenigo F., Molendini C., et al. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009;335:17–25.
26 Dejana E., Tournier-Lasserve E., Weinstein B.M. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009;16:209–221.
27 Tiruppathi C., Finnegan A., Malik A.B. Isolation and characterization of a cell surface albumin-binding protein from vascular endothelial cells. Proc Natl Acad Sci U S A. 1996;93:250–254.
28 Tiruppathi C., Song W., Bergenfeldt M., et al. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway. J Biol Chem. 1997;272:25968–25975.
29 Predescu D., Palade G.E. Plasmalemmal vesicles represent the large pore system of continuous microvascular endothelium. Am J Physiol. 1993;265:H725–H733.
30 Predescu S.A., Predescu D.N., Palade G.E. Endothelial transcytotic machinery involves supramolecular protein-lipid complexes. Mol Biol Cell. 2001;12:1019–1033.
31 Hu C., Ahmed M., Melia T.J., et al. Fusion of cells by flipped SNAREs. Science. 2003;300:1745–1749.
32 Mehta D., Malik A.B. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367.
33 Weber C., Fraemohs L., Dejana E. The role of junctional adhesion molecules in vascular inflammation. Nat Rev Immunol. 2007;7:467–477.
34 Spindler V., Schlegel N., Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253.
35 Mochizuki N. Vascular integrity mediated by vascular endothelial cadherin and regulated by sphingosine 1-phosphate and angiopoietin-1. Circ J. 2009;73:2183–2191.
36 Curry F.R., Adamson R.H. Vascular permeability modulation at the cell, microvessel, or whole organ level: towards closing gaps in our knowledge. Cardiovasc Res. 2010;87:218–229.
37 Taddei A., Giampietro C., Conti A., et al. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008;10:923–934.
38 Furchgott R.F., Zawadzki J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376.
39 Triggle C.R., Ding H. The endothelium in compliance and resistance vessels. Front Biosci (Schol Ed). 2011;3:730–744.
40 Michel T., Vanhoutte P.M. Cellular signaling and no production. Pflugers Arch. 2010;459:807–816.
41 Searles C.D., Miwa Y., Harrison D.G., et al. Posttranscriptional regulation of endothelial nitric oxide synthase during cell growth. Circ Res. 1999;85:588–595.
42 Walford G., Loscalzo J. Nitric oxide in vascular biology. J Thromb Haemost. 2003;1:2112–2118.
43 Bredt D.S., Hwang P.M., Glatt C.E., et al. Cloned and expressed nitric oxide synthase structurally resembles cytochrome p-450 reductase. Nature. 1991;351:714–718.
44 Bredt D.S., Hwang P.M., Snyder S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770.
45 Dudzinski D.M., Michel T. Life history of eNOS: Partners and pathways. Cardiovasc Res. 2007;75:247–260.
46 Handy D.E., Loscalzo J. Nitric oxide and posttranslational modification of the vascular proteome: S-nitrosylation of reactive thiols. Arterioscler Thromb Vasc Biol. 2006;26:1207–1214.
47 Upchurch G.R., Welch G.N., Loscalzo J. The vascular biology of S-nitrosothiols, nitrosated derivatives of thiols. Vasc Med. 1996;1:25–33.
48 Leopold J.A., Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free Radic Biol Med. 2009;47:1673–1706.
49 Stamler J.S., Singel D.J., Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902.
50 Bhatia M. Hydrogen sulfide as a vasodilator. IUBMB Life. 2005;57:603–606.
51 Wang R. Hydrogen sulfide: A new EDRF. Kidney Int. 2009;76:700–704.
52 Li L., Rose P., Moore P.K. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187.
53 Parkington H.C., Coleman H.A., Tare M. Prostacyclin and endothelium-dependent hyperpolarization. Pharmacol Res. 2004;49:509–514.
54 Feletou M., Huang Y., Vanhoutte P.M. Endothelium-mediated control of vascular tone: COX-1 and COX-2 products. Br J Pharmacol. 2011;164:894–912.
55 Matoba T., Shimokawa H., Nakashima M., et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530.
56 Edwards G., Dora K.A., Gardener M.J., et al. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272.
57 Campbell W.B., Fleming I. Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch. 2010;459:881–895.
58 Shimokawa H. Hydrogen peroxide as an endothelium-derived hyperpolarizing factor. Pflugers Arch. 2010;459:915–922.
59 Kohan D.E., Rossi N.F., Inscho E.W., et al. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77.
60 Yanagisawa M., Kurihara H., Kimura S., et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415.
61 Watts S.W. Endothelin receptors: what’s new and what do we need to know? Am J Physiol Regul Integr Comp Physiol. 2010;298:R254–R260.
62 Rondaij M.G., Bierings R., Kragt A., et al. Dynamics and plasticity of Weibel-Palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:1002–1007.
63 Danser A.H., Saris J.J., Schuijt M.P., et al. Is there a local renin-angiotensin system in the heart? Cardiovasc Res. 1999;44:252–265.
64 Griendling K.K., Minieri C.A., Ollerenshaw J.D., et al. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148.
65 Zafari A.M., Ushio-Fukai M., Akers M., et al. Role of NADH/NADPH oxidase-derived h2o2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495.
66 Garrido A.M., Griendling K.K. NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol. 2009;302:148–158.
67 Tobias P.S. TLRS in disease. Semin Immunopathol. 2008;30:1–2.
68 Tobias P.S., Curtiss L.K. Toll-like receptors in atherosclerosis. Biochem Soc Trans. 2007;35:1453–1455.
69 Zimmer S., Steinmetz M., Asdonk T., et al. Activation of endothelial Toll-like receptor 3 impairs endothelial function. Circ Res. 2011;108:1358–1366.
70 Feinberg M.W., Jain M.K. Role of transforming growth factor-beta1/smads in regulating vascular inflammation and atherogenesis. Panminerva Med. 2005;47:169–186.
71 Kofler S., Nickel T., Weis M. Role of cytokines in cardiovascular diseases: a focus on endothelial responses to inflammation. Clin Sci (Lond). 2005;108:205–213.
72 Haas M.J., Mooradian A.D. Inflammation, high-density lipoprotein and cardiovascular dysfunction. Curr Opin Infect Dis. 2011;24:265–272.
73 de Vries J.E. The role of IL-13 and its receptor in allergy and inflammatory responses. J Allergy Clin Immunol. 1998;102:165–169.
74 Speyer C.L., Ward P.A. Role of endothelial chemokines and their receptors during inflammation. J Invest Surg. 2011;24:18–27.
75 Lortat-Jacob H. The molecular basis and functional implications of chemokine interactions with heparan sulphate. Curr Opin Struct Biol. 2009;19:543–548.
76 Celie J.W., Beelen R.H., van den Born J. Heparan sulfate proteoglycans in extravasation: assisting leukocyte guidance. Front Biosci. 2009;14:4932–4949.
77 Peiper S.C., Wang Z.X., Neote K., et al. The Duffy antigen/receptor for chemokines (DARC) is expressed in endothelial cells of Duffy-negative individuals who lack the erythrocyte receptor. J Exp Med. 1995;181:1311–1317.
78 Schnabel R.B., Baumert J., Barbalic M., et al. Duffy antigen receptor for chemokines (DARC) polymorphism regulates circulating concentrations of monocyte chemoattractant protein-1 and other inflammatory mediators. Blood. 2010;115:5289–5299.
79 Horne K., Woolley I.J. Shedding light on DARC: the role of the Duffy antigen/receptor for chemokines in inflammation, infection and malignancy. Inflamm Res. 2009;58:431–435.
80 Huo Y., Xia L. P-selectin glycoprotein ligand-1 plays a crucial role in the selective recruitment of leukocytes into the atherosclerotic arterial wall. Trends Cardiovasc Med. 2009;19:140–145.
81 Langer H.F., Chavakis T. Leukocyte-endothelial interactions in inflammation. J Cell Mol Med. 2009;13:1211–1220.
82 Muller W.A. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2010;6:323–344.
83 Miller J., Knorr R., Ferrone M., et al. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. J Exp Med. 1995;182:1231–1241.
84 Shaw S.K., Ma S., Kim M.B., et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration. J Exp Med. 2004;200:1571–1580.
85 Yang L., Froio R.M., Sciuto T.E., et al. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106:584–592.
86 Woodfin A., Voisin M.B., Imhof B.A., et al. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood. 2009;113:6246–6257.
87 Huang M.T., Larbi K.Y., Scheiermann C., et al. ICAM-2 mediates neutrophil transmigration in vivo: evidence for stimulus specificity and a role in PECAM-1-independent transmigration. Blood. 2006;107:4721–4727.
88 Becher M.U., Nickenig G., Werner N. Regeneration of the vascular compartment. Herz. 2010;35:342–351.
89 Richardson M.R., Yoder M.C. Endothelial progenitor cells: quo vadis? J Mol Cell Cardiol. 2011;50:266–272.
90 Torsney E., Xu Q. Resident vascular progenitor cells. J Mol Cell Cardiol. 2011;50:304–311.
91 Watt S.M., Athanassopoulos A., Harris A.L., et al. Human endothelial stem/progenitor cells, angiogenic factors and vascular repair. J R Soc Interface. 2010;7(Suppl 6):S731–S751.
92 Califano J.P., Reinhart-King C.A. Exogenous and endogenous force regulation of endothelial cell behavior. J Biomech. 2010;43:79–86.
93 Chiu J.J., Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327–387.
94 Lee T., Sumpio B.E. Cell signalling in vascular cells exposed to cyclic strain: the emerging role of protein phosphatases. Biotechnol Appl Biochem. 2004;39:129–139.
95 Wedding K.L., Draney M.T., Herfkens R.J., et al. Measurement of vessel wall strain using cine phase contrast MRI. J Magn Reson Imaging. 2002;15:418–428.
96 Iba T., Sumpio B.E. Morphological response of human endothelial cells subjected to cyclic strain in vitro. Microvasc Res. 1991;42:245–254.
97 Kaunas R., Nguyen P., Usami S., et al. Cooperative effects of Rho and mechanical stretch on stress fiber organization. Proc Natl Acad Sci U S A. 2005;102:15895–15900.
98 Cummins P.M., von Offenberg Sweeney N., Killeen M.T., et al. Cyclic strain-mediated matrix metalloproteinase regulation within the vascular endothelium: a force to be reckoned with. Am J Physiol Heart Circ Physiol. 2007;292:H28–H42.
99 Lu L., Feng Y., Hucker W.J., et al. Actin stress fiber pre-extension in human aortic endothelial cells. Cell Motil Cytoskeleton. 2008;65:281–294.
100 Lu L., Oswald S.J., Ngu H., et al. Mechanical properties of actin stress fibers in living cells. Biophys J. 2008;95:6060–6071.
101 Costa K.D., Sim A.J., Yin F.C. Non-hertzian approach to analyzing mechanical properties of endothelial cells probed by atomic force microscopy. J Biomech Eng. 2006;128:176–184.
102 Kniazeva E., Putnam A.J. Endothelial cell traction and ECM density influence both capillary morphogenesis and maintenance in 3-D. Am J Physiol Cell Physiol. 2009;297:C179–C187.
103 Atkins G.B., Jain M.K., Hamik A. Endothelial differentiation: molecular mechanisms of specification and heterogeneity. Arterioscler Thromb Vasc Biol. 2011;31:1476–1484.
104 McDonald D.M., Thurston G., Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation. 1999;6:7–22.
105 Feng D., Nagy J.A., Hipp J., et al. Reinterpretation of endothelial cell gaps induced by vasoactive mediators in guinea-pig, mouse and rat: many are transcellular pores. J Physiol. 1997;504(Pt 3):747–761.
106 Miyasaka M., Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol. 2004;4:360–370.
107 Petzelbauer P., Bender J.R., Wilson J., et al. Heterogeneity of dermal microvascular endothelial cell antigen expression and cytokine responsiveness in situ and in cell culture. J Immunol. 1993;151:5062–5072.
108 McEver R.P., Beckstead J.H., Moore K.L., et al. Gmp-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest. 1989;84:92–99.
109 Laszik Z., Mitro A., Taylor F.B.Jr, et al. Human protein C receptor is present primarily on endothelium of large blood vessels: implications for the control of the protein C pathway. Circulation. 1997;96:3633–3640.
110 Levi M. The coagulant response in sepsis and inflammation. Hamostaseologie. 2010;30(10–12):14–16.
111 Granger D.N., Rodrigues S.F., Yildirim A., et al. Microvascular responses to cardiovascular risk factors. Microcirculation. 2010;17:192–205.
112 Antoniades C., Bakogiannis C., Tousoulis D., et al. Platelet activation in atherogenesis associated with low-grade inflammation. Inflamm Allergy Drug Targets. 2010;9:334–345.
113 Freedman J.E., Loscalzo J., Barnard M.R., et al. Nitric oxide released from activated platelets inhibits platelet recruitment. J Clin Invest. 1997;100:350–356.
114 Arnaud L., Haroche J., Mathian A., et al. Pathogenesis of Takayasu’s arteritis: a 2011 update. Autoimmun Rev. 2011;11:61–67.
115 Jennette J.C. Nomenclature and classification of vasculitis: lessons learned from granulomatosis with polyangiitis (Wegener’s granulomatosis). Clin Exp Immunol. 2011;164(Suppl 1):7–10.
116 Duval A., Helley D., Capron L., et al. Endothelial dysfunction in systemic lupus patients with low disease activity: evaluation by quantification and characterization of circulating endothelial microparticles, role of anti-endothelial cell antibodies. Rheumatology (Oxford). 2010;49:1049–1055.
117 Savage C.O. Vascular biology and vasculitis. APMIS Suppl. 2009:37–40.
118 Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J Lipid Res. 2009;50(Suppl):S352–S357.
119 Libby P., Ridker P.M., Hansson G.K. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138.
120 Libby P., Ridker P.M., Hansson G.K. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325.
121 Reriani M.K., Lerman L.O., Lerman A. Endothelial function as a functional expression of cardiovascular risk factors. Biomark Med. 2010;4:351–360.
122 Kwon G.P., Schroeder J.L., Amar M.J., et al. Contribution of macromolecular structure to the retention of low-density lipoprotein at arterial branch points. Circulation. 2008;117:2919–2927.
123 Borissoff J.I., Spronk H.M., ten Cate H. The hemostatic system as a modulator of atherosclerosis. N Engl J Med. 2011;364:1746–1760.
124 Ludmer P.L., Selwyn A.P., Shook T.L., et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046–1051.
125 Charakida M., Masi S., Luscher T.F., et al. Assessment of atherosclerosis: the role of flow-mediated dilatation. Eur Heart J. 2010;31:2854–2861.
126 Corretti M.C., Anderson T.J., Benjamin E.J., et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the international brachial artery reactivity task force. J Am Coll Cardiol. 2002;39:257–265.
127 Joyner M.J., Dietz N.M., Shepherd J.T. From Belfast to Mayo and beyond: the use and future of plethysmography to study blood flow in human limbs. J Appl Physiol. 2001;91:2431–2441.
128 Jarvisalo M.J., Lehtimaki T., Raitakari O.T. Determinants of arterial nitrate-mediated dilatation in children: role of oxidized low-density lipoprotein, endothelial function, and carotid intima-media thickness. Circulation. 2004;109:2885–2889.
129 Jarvisalo M.J., Raitakari M., Toikka J.O., et al. Endothelial dysfunction and increased arterial intima-media thickness in children with type 1 diabetes. Circulation. 2004;109:1750–1755.
130 Pasquali S.K., Marino B.S., Powell D.J., et al. Following the arterial switch operation, obese children have risk factors for early cardiovascular disease. Congenit Heart Dis. 2010;5:16–24.
131 Wallace S.M., Maki-Petaja K.M., Cheriyan J., et al. Simvastatin prevents inflammation-induced aortic stiffening and endothelial dysfunction. Br J Clin Pharmacol. 2010;70:799–806.
132 Ostad M.A., Eggeling S., Tschentscher P., et al. Flow-mediated dilation in patients with coronary artery disease is enhanced by high dose atorvastatin compared to combined low dose atorvastatin and ezetimibe: results of the CEZAR study. Atherosclerosis. 2009;205:227–232.
133 Shahin Y., Khan J.A., Samuel N., et al. Angiotensin converting enzyme inhibitors effect on endothelial dysfunction: a meta-analysis of randomised controlled trials. Atherosclerosis. 2011;216:7–16.
134 Lekakis J., Abraham P., Balbarini A., et al. Methods for evaluating endothelial function: a position statement from the European Society of Cardiology Working Group on Peripheral Circulation. Eur J Cardiovasc Prev Rehabil. 2011.
135 Cooke J.P. Asymmetrical dimethylarginine: the uber marker? Circulation. 2004;109:1813–1818.
136 Cooke J.P., Ghebremariam Y.T. DDAH says NO to ADMA. Arterioscler Thromb Vasc Biol. 2011;31:1462–1464.
137 Teerlink T. ADMA metabolism and clearance. Vasc Med. 2005;10(Suppl 1):S73–S81.
138 Ito A., Tsao P.S., Adimoolam S., et al. Novel mechanism for endothelial dysfunction: dysregulation of dimethylarginine dimethylaminohydrolase. Circulation. 1999;99:3092–3095.
139 Sibal L., Agarwal S.C., Home P.D., et al. The role of asymmetric dimethylarginine (ADMA) in endothelial dysfunction and cardiovascular disease. Curr Cardiol Rev. 2011;6:82–90.
140 Abbasi F., Asagmi T., Cooke J.P., et al. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am J Cardiol. 2001;88:1201–1203.
141 Kielstein J.T., Donnerstag F., Gasper S., et al. ADMA increases arterial stiffness and decreases cerebral blood flow in humans. Stroke. 2006;37:2024–2029.
142 Kielstein J.T., Impraim B., Simmel S., et al. Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation. 2004;109:172–177.
143 Juonala M., Viikari J.S., Alfthan G., et al. Brachial artery flow-mediated dilation and asymmetrical dimethylarginine in the cardiovascular risk in Young Finns study. Circulation. 2007;116:1367–1373.
144 Boger R.H., Sullivan L.M., Schwedhelm E., et al. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation. 2009;119:1592–1600.
145 Dignat-George F., Boulanger C.M. The many faces of endothelial microparticles. Arterioscler Thromb Vasc Biol. 2010;31:27–33.
146 Chironi G.N., Boulanger C.M., Simon A., et al. Endothelial microparticles in diseases. Cell Tissue Res. 2009;335:143–151.
147 Amabile N., Guerin A.P., Leroyer A., et al. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol. 2005;16:3381–3388.
148 Mallat Z., Benamer H., Hugel B., et al. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. 2000;101:841–843.
149 Arteaga R.B., Chirinos J.A., Soriano A.O., et al. Endothelial microparticles and platelet and leukocyte activation in patients with the metabolic syndrome. Am J Cardiol. 2006;98:70–74.
150 Sabatier F., Darmon P., Hugel B., et al. Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes. 2002;51:2840–2845.
151 Preston R.A., Jy W., Jimenez J.J., et al. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension. 2003;41:211–217.
152 Bakouboula B., Morel O., Faure A., et al. Procoagulant membrane microparticles correlate with the severity of pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;177:536–543.
[/level-membership-for-cardiovascular-category][not-level-membership-for-cardiovascular-category]
Chapter 2 The Endothelium
In 1839, the German physiologist Theodor Schwann became the first to describe a “thin, but distinctly perceptible membrane” that he observed as part of the capillary vessel wall that separated circulating blood from tissue.1,2 The cellular monolayer that formed this membrane would later be named the endothelium; however, the term endothelium did not appear until 1865 when it was introduced by the Swiss anatomist Wilhelm His in his essay, “Die Häute und Höhlen des Körpers (The Membranes and Cavities of the Body).”2,3 Owing to its anatomical location, the endothelium was believed initially to be a passive receptacle for circulating blood, cells, and macromolecules. It is now known that the endothelium is a dynamic cellular structure, and its biological and functional properties extend beyond that of a physical anatomical boundary. In its totality, the endothelium comprises approximately 10 trillion (1013) cells with a surface area of 7 m2, weighs 1.0 to 1.8 kilograms, and contributes 1.4% to total body mass.4,5 Endothelium exists as a monolayer of cells that is present in all arteries, veins, capillaries, and the lymphatic system, and lies at the interface of the bloodstream or lymph and the vessel wall.
Homeostatic Functions of the Endothelium
The endothelium exhibits considerable regional heterogeneity that reflects its arterial or venous location in the vascular tree, as well as the specialized metabolic and functional demands of the underlying tissues.5–7 Despite this heterogeneity, there are basal homeostatic properties that are common to all endothelial cell (EC) populations, although some of these functions may achieve greater importance in selected vascular beds7 (Box 2-1).
Maintenance of a Thromboresistant Surface and Regulation of Hemostasis
The endothelium was first recognized as a cellular structure that compartmentalizes circulating blood.4 As such, the endothelial luminal surface is exposed to cells and proteins in the bloodstream that possess prothrombotic and procoagulant activity and, when necessary, support hemostasis. Normal endothelium preserves blood fluidity by synthesizing and secreting factors that limit activation of the clotting cascade, inhibit platelet aggregation, and promote fibrinolysis.8 These include the cell surface–associated anticoagulant factors thrombomodulin, protein C, tissue factor pathway inhibitor (TFPI), and heparan sulfate proteoglycans (HSPG) that act in concert to limit coagulation at the luminal surface of the endothelium.8–10 For instance, thrombin-mediated activation of protein C is accelerated 104-fold by binding to thrombomodulin, Ca2 +, and the endothelial protein C receptor. Activated protein C (APC) engages circulating protein S, which is also synthesized and released by the endothelium, to inactivate factors Va and VIIIa proteolytically.8,11 Tissue factor pathway inhibitor is a Kunitz-type protease inhibitor that binds to and inhibits factor VIIa; about 80% of TFPI is bound to the endothelium via a glycosylphosphatidylinositol anchor and forms a quaternary complex with tissue factor – factor VIIa to diminish its procoagulant activity.12,13 Proteoglycan heparan sulfates that are present in the EC glycocalyx attain anticoagulant properties by catalyzing the association of the circulating serine protease inhibitor antithrombin III to factors Xa, IXa, and thrombin.8 Thus, these anticoagulant factors serve to limit activation and propagation of the clotting cascade at the endothelial luminal surface and thereby maintain vascular patency.
The endothelium also synthesizes and secretes tissue plasminogen activator (tPA) and the ecto-adenosine diphosphatase (ecto-ADPase) CD39 to promote fibrinolysis and inhibit platelet activation, respectively. Tissue plasminogen activator is produced and released into the bloodstream continuously, but unless tPA binds fibrin, it is cleared from the plasma within 15 minutes by the liver.8 Fibrin binding accelerates tPA amidolytic activity by increasing the catalytic efficiency for plasminogen activation and plasmin generation. Platelet activation at the endothelial luminal surface is inhibited by the actions of the ectonucleotidase CD39/NTPDase1 that hydrolyzes adenosine diphosphate (ADP), prostacyclin (PGI2), and nitric oxide (NO).8,14,15 Together these agents maintain an environment on the endothelial surface that is profibrinolytic and antithrombotic.
By contrast, in the setting of an acute vascular injury or trauma, the endothelium initiates a rapid and measured hemostatic response through regulated synthesis and release of tissue factor and von Willebrand factor (vWF). Tissue factor is a multidomain transmembrane glycoprotein (GP) that forms a complex with circulating factor VIIa to activate the coagulation cascade and generate thrombin.16 Tissue factor is expressed by vascular smooth muscle cells (VSMCs) and fibroblasts and by ECs only after activation. Tissue factor acquires its biological activity by phosphatidylserine exposure, dedimerization, decreased exposure to TFPI, or posttranslational modification(s) including disulfide bond formation between Cys186 and Cys209.17–19 This disulfide bond is important for tissue factor coagulation activity and may be reduced by protein disulfide isomerase, which is located on the EC surface.
The endothelium also synthesizes and stores vWF, a large polymeric GP that is expressed rapidly in response to injury. Propeptides and multimers of vWF are packaged in Weibel-Palade bodies that are unique to the endothelium. Once released, vWF multimers form elongated strings that retain platelets at sites of endothelial injury. Weibel-Palade bodies also contain P-selectin, angiopoietin-2, osteoprotegerin, the tetraspanin CD63/Lamp3, as well as cytokines, which are believed to be present as a result of incidental packaging.20 The stored pool of vWF may be mobilized quickly to the endothelial surface, where it binds to exposed collagen and participates in formation of a primary platelet hemostatic plug. The endothelium modulates this response further by regulating vWF size, and thereby its activity, through the action of the EC product ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type I motif, number 13).21 This protease cleaves released vWF at Tyr1605-Met1606 to generate smaller-sized polymers and decrease the propensity for platelet thrombus formation.21 Thus, the endothelium uses geographical separation of factors that regulate its anti- and prothrombotic functions to maintain blood fluidity yet allow for a hemostatic response to vascular injury.
Semipermeable Barrier and Transendothelial Transport Pathways
The endothelial monolayer serves as a size-selective semipermeable barrier that restricts the free bidirectional transit of water, macromolecules, and circulating or resident cells between the bloodstream and underlying vessel wall or tissues. Permeability function is determined in part by the architectural arrangement of the endothelial monolayer, as well as the activation of pathways that facilitate the transendothelial transport of fluids, molecules, and cells. This transport occurs via either transcellular pathways that involve vesicle formation, trafficking, and transcytosis, or by the loosening of interendothelial junctions and paracellular pathways22 (Fig. 2-1). Molecules that traverse the endothelium by paracellular pathways are size restricted to a radius of 3 nm or less, whereas those of larger diameter may be actively transported across the cell in vesicles.23 Although the diffusive flux of water occurs in ECs through aquaporin transmembrane water channels, the contribution of these channels to hydraulic conductivity and cellular permeability is limited.24
Figure 2-1 Transendothelial transport mechanisms.
(Adapted from Komarova Y, Malik AB: Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol 72:463–493, 2010.)
There is significant macrostructural heterogeneity of the endothelial monolayer that reflects the functional and metabolic requirements of the underlying tissue and has consequences for its permeability function. Endothelium may be arranged in either a continuous or discontinuous manner: continuous endothelium is either nonfenestrated or fenestrated.4–6
Continuous nonfenestrated endothelium forms a highly exclusive barrier and is found in the arterial and venous blood vessels of the heart, lung, skin, connective tissue, muscle, retina, spinal cord, brain, and mesentery.4–6 By contrast, continuous fenestrated endothelium is located in vessels that supply organs involved in filtration or with a high demand for transendothelial transport, including renal glomeruli, the ascending vasa recta and peritubular capillaries of the kidney, endocrine, and exocrine glands, intestinal villi, and the choroid plexus of the brain.4–6 These ECs are characterized by fenestrae, or transcellular pores, with a diameter of 50 to 80 nm that, in the majority of cells, has a 5- to 6-mm nonmembranous diaphragm across the pore opening.4–6,22 The distribution of these fenestrae may be polarized within the EC and allow for enhanced barrier size selectivity owing to the diaphragm.4–6
Discontinuous endothelium is found in the bone marrow, spleen, and liver sinusoids. This type of endothelial monolayer is notable for its large-diameter fenestrae (100-200 nm) with absent diaphragms and gaps, and a poorly organized underlying basement membrane that is permissive for transcellular flow of water and solutes as well as cellular trafficking.4–6
Transcellular and paracellular pathways are two distinct routes by which plasma proteins, solutes, and fluids traverse the endothelial monolayer. The transcellular pathway provides a receptor-mediated mechanism to transport albumin, lipids, and hormones across the endothelium.22,25,26 The paracellular pathway is dependent upon the structural integrity of adherens, tight, and gap junctions and allows fluids and solutes to permeate between ECs but restricts passage of large molecules.22,25,26 Although these pathways were believed to function independently, it is now recognized that they are interrelated and together modulate permeability under basal conditions.
The transcellular transport of albumin and albumin-bound macromolecules is initiated by albumin binding to gp60, or albondin, a 60-kDa albumin-binding protein located in flask-shaped caveolae that reside at the cell surface.27,28 These caveolae are cholesterol- and sphingolipid-rich structures that contain caveolin-1. Once activated, gp60 interacts with caveolin-1, followed by constriction of the caveolae neck and fission from the cell surface.29,30 These actions lead to formation of vesicles with a diameter of about 70 nm and vesicle transcytosis. Caveolae may contain as much as 15% to 20% of the cell volume, so they are capable of moving significant amounts of fluid across the cell through this mechanism.29,30 Once vesicles have detached from the membrane, they undergo vectorial transit to the abluminal membrane, where they dock and fuse with the plasma membrane by interacting with vesicle-associated and membrane-associated target soluble N-ethylmaleimide-sensitive factor attachment receptors (SNAREs).31 Once docked, the vesicles release their cargo to the interstitial space. Vesicles may traverse the cell as individual structures or cluster to form channel-like structures with a diameter of 80 to 200 nm that span the cell.5,6 Although transcellular vesicle trafficking is the predominant mechanism by which cells transport albumin, it is now appreciated that this pathway is not absolutely necessary for permeability function, owing to the compensatory capabilities of the paracellular pathway.
The junctions between ECs include the adherens, tight, and gap junctions; only the former two modulate permeability and comprise the paracellular pathway.32 Adherens junctions are normally impermeant to albumin and other large molecules and are the major determinant of endothelial barrier function and permeability. The expression of tight junctions, by contrast, is limited to the blood-brain or blood-retinal barriers where they restrict or prevent passage of small molecules (< 1 kDa) and some inorganic ions.22 Gap junctions are composed of connexins that form a channel between adjacent cells to enhance cell-cell communication and facilitate the transit of water, small molecules, and ions.22
Adherens junctions are critical for maintaining endothelial barrier functional integrity and are composed of complexes of vascular endothelial (VE)-cadherin and catenins. Vascular endothelial cadherin is a transmembrane GP with five extracellular repeats, a transmembrane segment, and a cytoplasmic tail. The external domains mediate the calcium-dependent hemophilic adhesion between VE-cadherin molecules expressed in adjacent cells.25,26,33 The cytoplasmic tail interacts with β-catenin, plakoglobin (γ-catenin), and p120 catenin to control the organization of VE-cadherin and the actin cytoskeleton at adherens junctions. The actin binding proteins α-actinin, annexin 2, formin-1, and eplin may further stabilize this interaction. Other proteins located in adherens junctions thought to provide stability include junctional adhesion molecules (JAMs) and platelet–EC adhesion molecule 1 (PECAM-1).22
Endothelial permeability may be increased or decreased through mechanisms that involve adherens junction remodeling or through interactions with the actin cytoskeleton.25,26,34 These events may occur rapidly, be transient or sustained, and are reversible. Most commonly, mediators that increase endothelial permeability either destabilize adherens junctions through phosphorylation, and thereby internalization, of VE-cadherin or by RhoA activation and actin cytoskeletal rearrangement to physically pull apart VE-cadherin molecules and adherens junctions, resulting in intercellular gaps.22 To counteract these effects, other mediators that attenuate permeability are present in the plasma or interstitial space. Fibroblast growth factor (FGF) stabilizes VE-cadherin by stabilizing VE-cadherin-gp120-catenin interaction. Sphingosine-1-phosphate, generated by breakdown of the membrane phospholipid sphingomyelin or released from activated platelets, also stabilizes adherens junctions. This effect occurs through activation of Rac1/Rap1/Cdc42 signaling and reorganization of the actin cytoskeleton, recycling of VE-cadherin to the cell surface, and (re)assembly of adherens junctions. The cytokine angiopoietin-1 stabilizes adherens junctions by inhibiting endocytosis of VE-cadherin.22,25,26,35,36
Endothelial tight junctions predominate in specialized vascular beds that require an impermeable barrier. These tight junctions are composed of the specific tight junction proteins occludin, claudins (3/5), and JAM-A.22,33,36,37 Occludin and claudins are membrane proteins that contain four transmembrane and two extracellular loop domains. The extracellular loop domains of these proteins bind similar domains on neighboring cells to seal the intercellular cleft and prevent permeability. Occludin, claudins, and JAM-A are also tethered to the actin cytoskeleton by α-catenin and zona occludens proteins (ZO-1, ZO-2).22 The ZO proteins also function as guanylyl kinases or scaffolding proteins and use PDZ and Sc homology 3 (SH3)-binding domains to recruit other signaling molecules. Connections between tight junctions and the actin cytoskeleton are stabilized further via the actin cross-linking proteins spectrin or filamen or by the accessory proteins cingulin and AF-6.22,36 In this manner, the junctions remain stabilized and sealed to limit or prevent transendothelial transport of fluids and molecules.
Regulation of Vascular Tone
Since the early seminal studies of Furchgott and Zawadski, it has been increasingly recognized that the endothelium regulates vascular tone via endothelium-derived factors that maintain a balance between vasoconstriction and vasodilation38,39 (Fig. 2-2). The endothelium produces both gaseous and peptide vasodilators, including NO, hydrogen sulfide, PGI2, and endothelium-derived hyperpolarizing factor (EDHF). The effects of these substances on vascular tone are counterbalanced by vasoconstrictors that are either synthesized or processed by the endothelium, such as thromboxane A2 TxA2, a product of arachidonic acid metabolism, and the peptides endothelin-1 (ET-1) and angiotensin II (Ang-II). The relative importance of these vasodilator or vasoconstrictor substances for maintaining vascular tone differs between vascular beds, with NO serving as the primary vasodilator in large conduit elastic vessels and non-NO mechanisms playing a greater role in the microcirculation.
Nitric oxide is synthesized by three structurally similar NO synthase (NOS) isoenzymes: the constitutive enzyme identified in the endothelium (eNOS or NOS3) and neuronal cells (nNOS or NOS1) or the inducible enzyme (iNOS or NOS2) found in smooth muscle cells (SMCs), neutrophils, and macrophages following exposure to endotoxin or inflammatory cytokines.40–42 Nitric oxide is generated via a five-electron oxidation reaction of L-arginine to form L-citrulline and stoichiometric amounts of NO, and requires molecular oxygen and NADPH as co-substrates and flavin adenine dinucleotide, flavin mononucleotide, heme, and tetrahydrobiopterin as cofactors.43–45 In the endothelium, eNOS expression is up-regulated by a diverse array of stimuli including transforming growth factor (TGF)-β1, lysophosphatidylcholine, hydrogen peroxide, tumor necrosis factor (TNF)-α, oxidized low-density lipoprotein (LDL) cholesterol, laminar shear stress, and hypoxia, and is subject to both posttranscriptional and posttranslational modifications that influence activity, including phosphorylation, acetylation, palmitoylation and myristolation, as well as localization to caveolae.45 Once generated, NO diffuses into SMCs and reacts with the heme iron of guanylyl cyclase to increase cyclic guanosine monophosphate (cGMP) levels and promote vasodilation.42 Nitric oxide can also react with SH-containing molecules and proteins (e.g., peroxynitrite, N2O2) to generate S-nitrosothiols, a stable reservoir of bioavailable NO with recognized antiplatelet and vasodilator effects.46–48 In the presence of oxygen, NO can be oxidized to nitrite and nitrate, which are stable end-products of NO metabolism; nitrite serves as a vasodilator, predominantly in the pulmonary and cerebral circulations.48,49 In addition to vasodilator and antiplatelet effects, NO has other paracrine effects that include regulation of VSMC proliferation and migration, and leukocyte adhesion and activation.15
Hydrogen sulfide gas generated by the endothelium also possesses vasodilator properties. Hydrogen sulfide is membrane permeable and released as a byproduct of cysteine or homocysteine metabolism via the transulfuration/cystathionine-β-synthase and cystathionine-γ-lyase pathway or by the catabolism of cysteine via cysteine aminotransferase and 3-mercaptopyruvate sulfur transferase. Hydrogen sulfide–mediated vasodilation results from activation of KATP and transient receptor membrane channel currents.50–52
Prostacyclin is an eicosanoid generated by cyclooxygenase (COX) and arachidonic acid metabolism in the endothelium. It promotes vasodilation via adenylyl cyclase/cyclic adenosine monophosphate (cAMP) signal transduction pathways. Prostacyclin also induces smooth muscle relaxation by reducing cytoplasmic Ca2 + availability; decreases VSMC proliferation through a cAMP–peroxisome proliferator-activated receptor (PPAR)-γ-mediated mechanism, and limits inflammation by decreasing interleukin (IL)-1 and IL-6.53 Importantly, PGI2 has significant antiplatelet effects and by decreasing TxA2 levels, limits platelet aggregation. Because both COX-1 (constitutively expressed) and COX-2 (induced) contribute to basal PGI2 production, selective pharmacological inhibition of either isoform may result in diminished PGI2 levels, increased platelet aggregation, and impaired vasodilation.54
No single molecule has been identified as the vasodilator referred to as endothelium-derived hyperpolarizing factor, and the effects attributed to Endothelium-derived hyperpolarizing factor likely represent the composite actions of several agents that share a common mechanism. Endothelium-derived hyperpolarizing factor is an important vasodilator in the microcirculation and acts by opening K+ channels to allow for K+ efflux, hyperpolarization, and vascular smooth muscle relaxation. Candidate EDHFs include the 11, 12-epoxyeicosatrienoic acids and hydrogen peroxide.39,55–58
To counterbalance the effects of endothelium-derived vasodilators, the endothelium also synthesizes the vasoconstrictor ET-1 and metabolizes Ang I to Ang II. Endothelin-1, a 21-amino-acid peptide, is synthesized initially as inactive pre-proET-1 that is processed by endothelin-converting enzymes to its active form.59,60 Endothelin-1 binds to the G protein–coupled receptors (GPCRs) ETA and ETB: ECs express ETB, whereas SMCs express both receptors. Although activation of endothelial ETB increases NO production, concomitant activation of SMC ETA and ETB results in prolonged and long-lasting vasoconstriction that predominates.61
There is no evidence that ET-1 is stored for immediate early release in the endothelium, indicating that acute stimuli such as hypoxia, TGF-β, and shear stress that increase ET-1 production do so via a transcriptional mechanism; however, ET-1 and endothelin-converting enzyme are packaged in Weibel-Palade bodies.62 Endothelium also expresses angiotensin-converting enzyme (ACE) and, as such, modulates processing of Ang-I to the vasoconstrictor peptide Ang-II.63 Ang-II–stimulated activation of the Ang-I receptor results in vasoconstriction and SMC hypertrophy and proliferation, in part, by activating NADPH oxidase to increase reactive oxygen species (ROS) production.64–66 Vascular tone, therefore, is determined by the balance of vasodilator and vasoconstrictor substances synthesized or processed by the endothelium in response to stimuli: each vasoactive mediator may attain individual importance in a different vascular bed.
Regulating Response to Inflammatory and Immune Stimuli
The endothelium monitors circulating blood for foreign pathogens and participates in immunosurveillance by expressing Toll-like receptors (TLRs) 2, 3, and 4.67–69 These TLRs identify pathogen-associated molecular patterns that are common to bacterial cell wall proteins or viral deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) in the bloodstream. Once activated, TLRs elicit an inflammatory response through activation of nuclear factor (NF)-κB and generation of chemokines that promote transendothelial migration of leukocytes, have chemoattractant and mitogenic effects, and increase endothelial oxidant stress and apoptosis.67,68
[/not-level-membership-for-cardiovascular-category]
