[level-membership-for-neurology-category]
CHAPTER 86 THE CONGENITAL MYOPATHIES
The congenital myopathies are a clinically and genetically heterogeneous group of congenital muscle disorders with characteristic structural abnormalities evident on muscle biopsy, visible after preparation with specific histochemical stains and/or on electron microscopy. Central core disease (CCD),1,2 nemaline myopathy,3 myotubular (centronuclear) myopathy,4 and minicore myopathy (or multiminicore disease [MmD])5 are the major disease entities. Other conditions with more unusual structural abnormalities are very rare, and it is not clear whether all are genetic entities.6
Autosomal dominant, autosomal recessive, and X-linked inheritance are all recognized in this group of disorders, and some conditions such as nemaline myopathy, CCD, and myotubular myopathy may have more than one mode of inheritance. Genetic advances have implicated several genes encoding sarcomeric and sarcotubular proteins (Table 86-1). The boundaries between these conditions, originally defined according to histopathological and clinical criteria, are often indistinct and do not necessarily reflect underlying molecular mechanisms: Mutations in the same gene can indeed give rise to diverse clinical and histopathological phenotypes, and, conversely, a similar histopathological and clinical phenotype may arise from mutations in a variety of genes. Although clinical management is currently the main form of treatment of the congenital myopathies, further advances in the understanding of the precise molecular mechanisms underlying each disorder may result in more rational therapeutic options in the future.
EPIDEMIOLOGY
Epidemiological data on the congenital myopathies are few, and larger geographical surveys are limited. The overall incidence of the congenital myopathies is estimated at 6 per 100,000 live births, representing approximately 10% of all neuromuscular disorders.7 Studies in northern Ireland8 and western Sweden9 suggest that the prevalence of the congenital myopathies in a pediatric population is between 3.5 and 5.0 per 100,000. The relative frequency of individual conditions is unknown, but CCD and conditions associated with mutations in the skeletal muscle ryanodine receptor gene (RYR1) appear to be more common in the patient population (see later discussion) than are nemaline myopathy and the much rarer centronuclear myopathies. Also, the prevalence of specific congenital myopathies may have been previously underestimated, inasmuch as not all muscle biopsy specimens from individuals carrying disease-causing mutations exhibit the characteristic structural abnormalities.10
CLINICAL FEATURES
Most of the clinical features are nonspecific, despite some variations in overall severity, distribution of weakness, and associated features. The diagnosis of a specific congenital myopathy can therefore be made only tentatively on clinical grounds alone, and muscle biopsy is required for a precise diagnosis in individual cases.
Presentation of the patient is either as a “floppy infant” with generalized hypotonia at birth followed by developmental delay or with weakness of variable severity and distribution later in childhood. Presentation in adulthood has also been described, but other disease processes mimicking the appearance of a congenital myopathy should be considered.11 Presentation at birth is often associated with “myopathic” facial features, feeding difficulties, and respiratory difficulties; thinning of the ribs and chest deformities may indicate antenatal onset. Arthrogryposis may be present in the most severe cases of nemaline myopathy12 and CCD13 but is not a common feature. Most cases manifest later in infancy or childhood with motor developmental delay or predominantly proximal weakness that mimics a limb girdle muscular dystrophy or a mild form of spinal muscular atrophy. In other patients, there is marked weakness of the axial muscles and/or the face; a minority have prominent distal involvement. Extraocular involvement is common in centronuclear myopathy and specific subgroups of minicore myopathy, but not in CCD and nemaline myopathy.
Spinal deformities include exaggerated lordosis, scoliosis, and spinal rigidity.14 Scoliosis may be present at birth but typically manifests in a progressive manner around the time of the pubertal growth spurt. Ligamentous laxity is common. Joint contractures, mainly of the Achilles tendon, tend to progress over time. Tendon reflexes are weak or absent. Congenital dislocation of the hips is a common feature in CCD15 but can occur in several other neuromuscular disorders.
Respiratory involvement secondary to diaphragmatic and/or intercostal muscle weakness is the main prognostic factor.16,17 Respiratory impairment is common in centronuclear myopathy, nemaline myopathy, and subgroups of minicore myopathy; it may occur only rarely in CCD,18 with the exception of the severe congenital variant, in which it is common.13 Susceptibility to respiratory infections is frequent, particularly early in life, but may improve with time.13,19 Cardiac involvement other than cor pulmonale secondary to respiratory impairment is not usually a feature, although structural abnormalities such as mitral valve prolapse have been occasionally documented. However, a few patients have had congenital myopathy with rods and/or cores and cardiac involvement in association with mutations in the skeletal muscle αactin gene (ACTA1).20
There are no associated structural central nervous system or peripheral nerve abnormalities, and intelligence is usually normal. In the few cases in which an autopsy was performed, no central or peripheral nerve abnormalities could be identified.21–23
Progression of muscle weakness in the absence of substantial respiratory impairment does not usually occur, but deterioration is occasionally associated with growth spurts or marked weight gain. The most severely affected neonates may die from respiratory failure, but long-term survival has been reported even in infants with severe hypotonia and marked respiratory impairment.13,19,24,25
INVESTIGATIONS
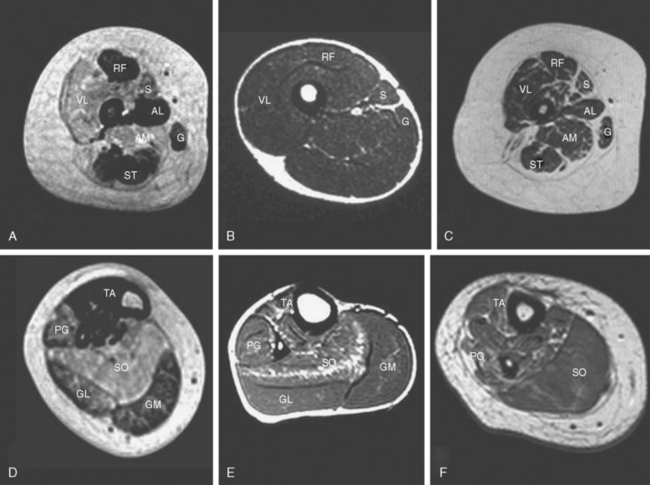
Other investigations may support the suspicion of an underlying myopathic process, but, with the possible exception of muscle imaging, they rarely aid in the diagnosis. Diseased muscle exhibits characteristic changes in echogenicity and signal intensity on ultrasonography, computed tomography, and magnetic resonance imaging (MRI), which reflect an increase in adipose and connective tissue.7,25–27 Muscle MRI in particular may reveal a characteristic pattern of selective involvement in conditions such as CCD and nemaline myopathy, thereby guiding the molecular diagnosis in clinically and histopathologically equivocal cases.28,29
The serum creatine kinase level is usually normal or only mildly elevated. The electromyogram may appear normal in the young patients or in mild cases but usually reveals nonspecific myopathic features, comprising small-amplitude polyphasic potentials.30,31 Additional “neurogenic” changes may be present in distal muscles, particularly of patients with nemaline myopathy.31–33 Spontaneous activity resembling findings in spinal muscular atrophy have been reported in the most severely affected neonates.34
MANAGEMENT
Respiratory impairment is the most important complication and should be anticipated by regular monitoring of respiratory function: regular measurement of forced vital capacity, in sitting and lying positions in order to evaluate diaphragmatic function, and annual overnight oxygen saturation monitoring if the forced vital capacity falls under 60% or, if clinically indicated by symptoms of nocturnal hypoventilation such as early morning headaches, loss of appetite, and daytime tiredness.35 Respiratory impairment may be disproportionately severe, and respiratory failure may occur even in ambulant patients.16,17,19,35–37 Nighttime noninvasive ventilation offers an effective means of improving quality of life and overall prognosis in otherwise only moderately disabled patients. Respiratory infections should be treated aggressively. A cardiac ultrasound study should be performed to document baseline cardiac function and occasionally associated structural cardiac abnormalities. Regular cardiac ultrasonography should be performed in patients with respiratory impairment, although appropriate respiratory management should prevent cor pulmonale.16 Primary cardiomyopathies are rare in the congenital myopathies but have been reported in individual cases with mutations in the ACTA1 gene20 and in a few patients with minicores evident in muscle biopsy specimens (see later discussion).
Spinal posture should be monitored closely and surgical procedures for scoliosis planned carefully to ensure that respiratory function is still sufficient at the time of operative intervention.38 Regular physiotherapy is aimed at preservation of muscle power and prevention of contractures. When orthopedic intervention is planned, the potential benefit has to be weighed against adverse effects of prolonged immobilization.
Patients with CCD and other congenital myopathies associated with mutations in the RYR1 gene are at increased risk of malignant hyperthermia susceptibility,39 and potentially triggering anesthetic agents should be rigorously avoided in these cases. Although a higher anesthetic risk is not clearly documented in other congenital myopathies, similar precautions should be taken in the anesthetic preparation of other patients,35 particularly in those in whom the genetic background is unclear.
Pharmacological interventions have not been widely used in the treatment of the congenital myopathies. A small pilot study has demonstrated some beneficial effect of salbutamol in the treatment of patients with CCD and minicore myopathy,40 but this has yet to be confirmed in a larger controlled trial.
CENTRAL CORE DISEASE
Histopathology
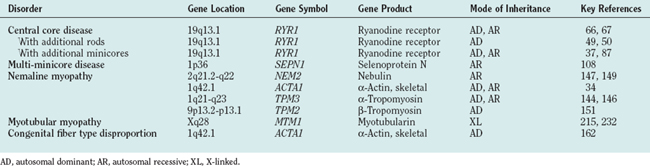
The term central core disease (Mendelian Inheritance in Man [MIM] number 117000)41 was introduced in 195842 after the original description of a family with five affected individuals in three generations who showed amorphous central areas in muscle fibers with the modified Gomori trichrome stain1; absence of oxidative enzyme activity within the core area was subsequently identified as the characteristic histopathological feature (Fig. 86-1A).27 The cores have a predilection for type 1 fibers and extend along a significant length of the longitudinal muscle fiber axis43; localization is characteristically single and central, but multiple, peripheral, and eccentric cores may occur within the same sample.10 In some cases, large typical cores may not be apparent, but only unevenness of oxidative enzyme stain or small core areas are visible, producing a confusing pathological overlap with minicore disease. Pronounced type 1 predominance or uniformity is common and may be the only feature at presentation, particular in very young patients.10,43 The degree of associated increases in fat and connective tissue depends both on the age of the patient and on the sampling site, because the degree of selective muscle involvement may be considerable29; centrally placed nuclei may also be present but are not a consistent feature. Necrosis and regeneration are not usually seen in CCD.
Ultrastructurally, typical cores show a reduction or absence of mitochondria, reflected in the absence of oxidative enzyme staining, accompanied by varying degrees of sarcomeric disruption and accumulation of Z line material.43 In most cases, cores are “structured” with hypercontracted myofibrils that retain adenosine triphosphatase activity but are depleted in mitochondria and sarcoplasmic reticulum; “unstructured” cores, on the contrary, show an absence of adenosine triphosphatase activity, marked disruption of myofibrils, and accumulation of Z line material.
Immunohistochemical studies have demonstrated accumulation of the intermediate filament protein desmin within cores and at their perimeter.10,44,45 Accumulation of other proteins also occurs, including the actin cross-linking protein γ-filamin, αβ-crystallin, small heat shock protein 27, and myotilin46; however, these are nonspecific findings and may be observed in core formation of different etiology.
Core formation is a nonspecific secondary phenomenon and is not correlated with the degree of the muscle weakness. Core formation can be observed after tenotomy,47 in the muscle of patients with long-standing neurogenic atrophy (“target fibers”), and in association with several other gene defects. Central corelike structures may be found in association with dilated cardiomyopathy in patients with mutations in ACTA120 or in the β-myosin heavy-chain gene (MYH7)48; however, in the latter group, there is usually no or little associated weakness. Central core and nemaline rods are occasionally observed in the same biopsy specimen in patients with mutations in the RYR1 gene.49,50
Clinical Features
“Classical” CCD is usually inherited as a dominant trait with a fairly consistent clinical phenotype; sporadic cases with similar clinical features are increasingly recognized. Presentation is in infancy with hypotonia or in early childhood with motor developmental delay or proximal weakness17; however, more severe presentations with features of the fetal akinesia sequence have been reported.13,51 Most of these infants required ventilation from birth, and their course was severe and eventually fatal; however, one infant was eventually weaned off the ventilator and became independently mobile.13
Weakness in most familial cases is pronounced in the hip girdle and in the axial muscle groups17; in rare cases, there is associated muscle wasting.52 Facial involvement is usually mild, and lack of complete eye closure may be the only finding. Bulbar and extraocular involvement is usually absent. Some patients may have prominent exercise-induced myalgia.53
Orthopedic complications are common and comprise congenital dislocation of the hips, foot deformities, and scoliosis.15,18,54 Contractures other than Achilles tendon tightness are rare, and many affected individuals have marked ligamentous laxity, occasionally in association with patellar instability.54
Cardiac abnormalities other than occasional mitral valve prolapse have rarely been reported.55 Respiratory involvement is typically milder than in other congenital myopathies but may be severe in some sporadic and recessive cases.51
Malignant hyperthermia susceptibility (MHS) is a frequent complication39,55,56 and should be anticipated in the anesthetic preparation of patients with CCD. Some patients with MHS may show consistent dysmorphic features (King-Denborough syndrome), including ptosis, down-slanting palpebral fissures, neck webbing, scoliosis, pectus deformity, short stature, and cryptorchism57,58; additional findings in other families may include vertebral fusion, eventration of the diaphragm, and spinal cord tethering.58,59
Except for patients with the most severe neonatal cases and some patients with congenital dislocation of the hips,37,60 most patients achieve the ability to walk independently. The course of CCD is static or only slowly progressive, even over prolonged periods of follow-up.61
The serum creatine kinase level is usually normal or only mildly elevated.62,63 Muscle ultrasonography often shows a striking increase in echogenicity and differential muscle involvement, even in paucisymptomatic individuals.64 Muscle MRI reveals a characteristic pattern of selective muscle involvement in patients with typical CCD29 (Fig. 86-2A and D) and may complement clinical assessment.
Genetics of Central Core Disease and Malignant Hyperthermia Susceptibility
MHS was recognized in 1960 as a familial, autosomal dominant trait by M. A. Denborough and R. R. H. Lovell in Australia and has been linked to several loci. Both CCD and MHS show considerable clinical overlap and have been associated with mutations in the RYR1 gene at chromosome 19q13.1.65–67 CCD is usually inherited as a dominant trait; however, sporadic cases either caused by de novo dominant mutations or associated with recessive inheritance are increasingly recognized.
Although it is now apparent that mutations in the RYR1 gene (MHS1) account for the majority of familial MHS cases,68 locus heterogeneity has been suggested by variable degrees of linkage evidence for several loci (MHS2 to MHS6) and cosegregation of MHS susceptibility with mutations in candidate genes.69–74 There is also the possibility that the MHS phenotype may reflect the compound influence of several genes rather than a major gene defect.75 Genetic homogeneity in CCD has been suggested by RYR1 linkage in most families without confirmed mutation. Although dominant mutations in the ACTA1 gene20 and the MYH7 genes48 may mimic the histopathological appearance, clinical features such as an associated cardiomyopathy are divergent from those in classic CCD.
More than 50 RYR1 mutations have been reported to date in association with CCD, MmD (described later), and MHS with or without cores evident on muscle biopsy specimens.76–80 Most RYR1 mutations are missense mutations; a few small deletions78,79,81 and a cryptic splicing site mutation51 have also been documented. The first reported RYR1 mutations gave rise predominantly to the MHS phenotype and affected mainly two regions of the protein: the cytoplasmic N-terminal domain (MHS/CCD region 1, amino acids 35 to 614) and the cytoplasmic central domain (MHS/CCD region 2, amino acids 2163 to 2458). Although mutations associated with a congenital myopathy phenotype have been identified in these regions,66,67,82,83 there has been increasing evidence that mutations affecting the C-terminal portion of the receptor molecule (MHS/CCD region 3, amino acids 4550 to 4940) are common in patients with CCD.49,78–80,84 Although the mutations are occasionally reported in the C-terminal portion of the protein,85 a series of 124 unrelated North American patients with MHS confirmed the N-terminal and central portion of the protein as the predominant sites of MHS-related RYR1 mutations.86
Although the majority of RYR1 mutations associated with MHS or CCD described to date are dominant missense mutations, recessive inheritance of RYR1 mutations has been reported in mild cases with histopathological features of MmD and CCD37,87 and in a severe form of CCD manifesting with a fetal akinesia syndrome.13 These findings suggest that clinically silent MHS mutations may give rise to a severe phenotype in the compound heterozygous or homozygous state, caused by a combined deleterious effect on the tetrameric RyR1 protein.
The effects of specific RYR1 mutations on excitability and calcium homeostasis have been studied in various homologous and heterologous expression systems. Two models for mutation-induced receptor malfunction have been proposed: depletion of sarcoplasmic reticulum calcium stores with resulting increases in cytosolic calcium levels (the “leaky channel” hypothesis)88 and a specific disturbance of excitation-contraction coupling (the “E-C uncoupling” hypothesis).89 The “ectopic” RYR1 expression in B lymphocytes has been recognized; this offers an easily accessible model for studying the effects of RYR1 mutations in vitro. B lymphocytes harboring CCD-related RYR1 mutations exhibit unprompted calcium release with resulting depletion of sarcoplasmic reticulum stores78,90; increased release of inflammatory cytokines in the same study91 may also indicate a role of RYR1 in immunomodulation.
MULTIMINICORE DISEASE
Multicore disease (MIM numbers 255320 and 157550)41 was originally described in two unrelated children with hypotonia and muscle weakness dating from infancy and a characteristic histopathological appearance of multiple, small, well-circumscribed foci of myofibrillar degeneration with reduction of oxidative enzyme staining.5 Since the original description, at least 100 other cases with similar histopathological features and a wide range of clinical phenotypes have been reported, under different names such as minicore myopathy, myopathy with multiple minicore, pleocore disease, or myopathy with focal loss of cross-striations. The designation multiminicore disease92 used in this chapter reflects the larger number and smaller size of characteristic lesions compared with classic CCD.
Histopathology
Minicores are areas of focal myofibrillar disruption with a variable degree of Z line disorganization and may occur in both type 1 and type 2 fibers (see Fig. 86-1B). They extend only a short distance along the longitudinal axis of the muscle fiber, affecting a variable number of sarcomeres.5,43 In rare cases, the entire fiber diameter may be involved (focal loss of cross-striations).93,94 Pathological distinction between CCD and MmD is occasionally difficult, because in some biopsy specimens from patients with RYR1 mutations, only unevenness of oxidative enzyme stain or minicores are seen, and minicores may evolve into central cores with time.10,87
Commonly associated features are predominance or uniformity of type 1 fibers, often hypotrophic, with or without type 2 hypertrophy.95 This is particularly true for cases secondary to RYR1 but not selenoprotein N, 1 (SEPN1) mutations (see later discussion). Although central nuclei may be a feature in cases caused by RYR1 mutations, they are not usually associated with SEPN1 mutations. The presence of whorled fibers and increases in fat and connective tissue in some affected families43 are suggestive of a potential overlap with the milder congenital muscular dystrophies.
On electron microscopy, minicores are characterized by focal areas of myofibrillar disruption with paucity of mitochondria.43 The sizes of minicores and the degree of myofibrillar disruption are variable, ranging from mild Z line streaming to areas with complete loss of sarcomeric organization. These may be observed in the same biopsy speciment.5 In some areas, ultrastructural abnormalities may consist only of subtle misalignment of myofibrils accompanied by an absence of mitochondria.
In immunohistochemical studies, authors have reported abnormalities on staining with desmin45 and γ-filamin antibodies,46 similar to those observed in CCD.
Minicores are nonspecific; they may be found in various disorders such as muscular dystrophies; denervation; and inflammatory, endocrine, and metabolic myopathies. Also, minicore-like structures may coexist with central cores, central nuclei, and nemaline rods in the same biopsy specimen10,20,96–102 and in association with various gene defects.
Clinical Features
MmD usually manifests in infancy or childhood with hypotonia or proximal weakness95,103; cases with antenatal onset or presentation in adulthood104–107 have been reported, but the molecular defect in these is not known. Clinical features associated with the histopathological appearance of MmD are markedly heterogeneous, and at least four different subgroups have been recognized and are gradually being resolved molecularly (see later discussion).
The most instantly recognizable classic phenotype of MmD95,103,108 usually manifests in infancy with hypotonia and is characterized by spinal rigidity, scoliosis, and early respiratory impairment. There is predominant truncal and proximal weakness, often in association with wasting pronounced in the shoulder girdle and the hip adductors (“bracket-like thighs”). There may be mild facial weakness and a high-pitched nasal voice. Severe scoliosis and respiratory failure have usually evolved by the early teens.95,103,109 Respiratory impairment is often disproportionate to the overall degree of muscle weakness and has to be anticipated even in ambulant patients. Most of these cases are secondary to mutations in the SEPN1 gene (discussed later).
In a subset of patients with a similar phenotype, extraocular muscle involvement pronounced on abduction and upward gaze may evolve over time.94,95,110 With the exception of the most severely affected neonatal cases,51 respiratory impairment is usually milder than in the classic form and may improve with time. Mutations in the RYR1 gene have been identified in these patients (discussed later).
Another group has a milder phenotype similar to CCD and characterized by predominant hip girdle weakness with relative sparing of respiratory and bulbar muscles.37 A common complaint is exercise-induced myalgia. In male patients, cryptorchism may be an additional feature (personal observation). Patients in this group may also demonstrate muscle imaging findings similar to those observed in classic CCD caused by mutations in the RYR1 C-terminal portion.37,87,110 In some patients, there is additional marked distal weakness and wasting, predominantly affecting the hands.37,87 The observation of extraocular muscle involvement evolving over time in this group is suggestive of a clinical continuum between the latter groups rather than distinct clinical entities.110 Mutations in the RYR1 gene also account for this group of disorders.
A severe form with antenatal onset, generalized arthrogryposis, dysmorphic features, and moderate respiratory impairment has been described in only a few patients.103
Congenital cardiac defects—namely, mitral valve prolapse—have been occasionally reported.5,37 Secondary right ventricular impairment is common in the classic phenotype with untreated respiratory impairment but not in other clinical subgroups.37 A primary cardiomyopathy has been reported in some cases with minicores on muscle biopsy specimens,55,106,111,112 but these were genetically unresolved, and additional desmin accumulation in muscle and dominant inheritance were suggestive of a pathogenic mechanism distinct from that in other families (see later discussion).
Malignant hyperthermia has been reported occasionally113,114 and has to be anticipated in the anesthetic management of these patients. Minicores have been noted in muscle biopsy specimens from a few families with mutations in the RYR1 gene and MHS but no other clinical features of a congenital myopathy.115,116
Minicores have been reported in the multiple pterygium syndrome117 and in two siblings with mental retardation and dysmorphic features similar to the King-Denborough syndrome118; the molecular basis of these associations is currently unclear.
The clinical course is usually static or only slowly progressive37,119 but depends largely on the degree of cardiorespiratory impairment.
Genetics
Investigation of SEPN1 on chromosome 1p36 as a candidate for causing MmD was prompted by the considerable clinical and histopathological overlap between the classic phenotype of MmD and congenital muscular dystrophy with rigidity of the spine, previously attributed to SEPN1 mutations.120 SEPN1 involvement in the classic phenotype of MmD was suggested both by linkage data and by direct mutational analysis in approximately 50% of patients with these clinical features.108 More than 30, mainly truncating SEPN1 mutations associated with a congenital myopathy phenotype have been identified to date. Mutations in the SEPN1 gene were also identified in cases with unusual pathological structures resembling Mallory bodies (Mallory body myopathy), further expanding the pathological spectrum of the condition.121 Homozygous mutations are unexpectedly common even in nonconsanguineous families, which reflects the presence of a few founder mutations in different European populations. The precise function of selenoprotein N, a glycoprotein localized in the endoplasmic reticulum, is unclear, but a structural motif similar to those observed in calcium-binding proteins120 is suggestive of possible involvement in calcium homeostasis in muscle. Although selenoprotein N is expressed in all adult tissues, prominent expression in fetal muscle precursor cells is suggestive of a potential role in myogenesis.
Involvement of the RYR1 gene as a cause of multi-minicores was to some extent unexpected, in view of differences in core structure and mode of inheritance between recessively inherited MmD and CCD caused by dominant mutations in the same gene. Attempting to classify individual cases as MmD or CCD may be difficult because there is a histopathological and clinical continuum between the two conditions. Although the histopathologic appearance of MmD appears to be more closely associated with recessively inherited RYR1 mutations, dominant RYR1 mutations occasionally give rise to minicores on muscle biopsy specimens10 and may have accounted for a proportion of MmD pedigrees with autosomal dominant inheritance reported before the molecular area. Also, the histopathological appearance of MmD caused by recessive RYR1 mutations may evolve into the classic picture of CCD over long periods of follow-up.87 A particular feature of RYR1-related recessive cases with minicores is extraocular involvement, not present in dominant CCD. MmD with external ophthalmoplegia in an isolated case from a consanguineous Tunisian family was attributed to a homozygous RYR1 mutation, introducing a cryptic splice site in intron 101,51 carried by asymptomatic parents. RYR1 involvement was also suggested by linkage evidence in four additional families with a similar phenotype,110 including the original family reported by Swash and Schwartz.94 RYR1 involvement in the moderate form of MmD with hand involvement was suggested by considerable clinical overlap with CCD and an identical pattern of selective involvement on muscle imaging.37,87 In a consanguineous Algerian family and a consanguineous British family, homozygous RYR1 mutations were subsequently identified, and RYR1 involvement has been suggested by linkage evidence in other families. The RYR1 gene is also a likely candidate for causing the severe form of MmD with neonatal onset and arthrogryposis, in view of phenotypic overlap with the form of CCD with fetal akinesia sequence.13
There is evidence for further genetic heterogeneity in MmD, because only one half of all cases with the classic phenotype have linkage or mutational evidence of SEPN1 involvement.108 Also, although a proportion of MmD in pedigrees with autosomal dominant inheritance are likely to be caused by dominant RYR1 mutations, unusual clinical features such as a primary cardiomyopathy55,111 are suggestive of a different genetic background in other families with this unusual mode of transmission. Specimens from patients with dominantly inherited desmin myopathy may demonstrate minicores in addition to inclusions on muscle biopsy. This observation and the desmin accumulation reported in some patients with MmD and cardiomyopathy111 suggest that the desmin gene is a possible candidate for causing this subgroup disorder. Also, a cardiomyopathy associated with cores on muscle biopsy has been described as part of the expanding clinical spectrum associated with dominant mutations in the ACTA1 gene.20
NEMALINE MYOPATHY
Nemaline myopathy was first described in 1963.3 The name nemaline myopathy is derived from the abundance of threadlike or rodlike structures (Greek nema means “thread”) in muscle that turn red with the modified Gomori trichrome technique (see Fig. 86-1C).
Histopathology
Nemaline rods are often in subsarcolemmal clusters (see Fig. 86-1C) but may also be observed within the fiber and/or intranuclearly, particularly in severe cases122 caused by mutations in the ACTA1 gene; these may also feature additional accumulation of actin filaments.34,123 The number of rods varies between fibers and between muscles and is not correlated with clinical severity of the disease. Associated features include type 1 predominance or uniformity with or without type 1 hypotrophy43 and deficiency of type 2B muscle fibers in patients and heterozygous persons in the recessive form.124 Necrosis, regeneration, fibrosis, and internal nuclei are not usually seen in nemaline myopathy. Because the presence of nemaline rods is highly nonspecific and has been described in a number of different clinical contexts43 and in association with histopathological features of other congenital myopathies,96 a diagnosis of nemaline myopathy can be made only in the context of specific clinical features.
On electron microscopy, nemaline rods are often observed in continuity with the Z lines and have a similar lattice structure.43 They are often, but not consistently, orientated parallel to the long axis of the fiber.
The close structural relationship between nemaline rods and the Z line has been further supported by immunohistochemical studies demonstrating the Z line component α-actinin as the main component of nemaline bodies.125 Nemaline bodies are associated with actin and tropomyosin and are also labeled with antibodies against myotilin and telethonin.126,127 In immunohistochemical studies, researchers have also investigated the effect of specific genetic defects on the expression of affected proteins,128,129 and these investigations may in a few selected cases be helpful in directing molecular genetic studies.23,130 Research studies with noncommercial antibodies to nebulin suggested that absence of antibody binding to the C-terminal SH3 domain occurs in some cases with mutations in the nebulin gene (NEB)129; however, secondary alterations in nebulin may also occur in cases with an ACTA1 mutation and in Duchenne muscular dystrophy.
Clinical Features
The clinical spectrum of nemaline myopathy is wide. A classification suggested by the European Neuromuscular Consortium on nemaline myopathy131 distinguishes six different clinical phenotypes; the “typical” form is a reference point and other forms are defined on the basis of differences in onset or severity and the presence of unusual features: The “typical” congenital form of nemaline myopathy is characterized by onset in infancy or early childhood with pronounced facial and bulbar involvement.131 Achievement of motor milestones is usually delayed, and in the absence of substantial respiratory problems, the course is slowly progressive or static. Feeding difficulties may be disproportionately severe. The severe congenital form is characterized by absence of both spontaneous movements and respiration at birth, occasionally preceded by signs of antenatal onset within the spectrum of the fetal akinesia sequence.12,132 Other forms of nemaline myopathy include the intermediate congenital form, in which patients move and breathe at birth but are later unable to achieve ambulation or respiratory independence; mild nemaline myopathy, with childhood onset; adult-onset nemaline myopathy, which may or may not be genetic in origin; and other manifestations with unusual associated features such as cardiomyopathy, ophthalmoplegia, or an uncommon distribution of weakness.19,23,130,133
Patients with the typical congenital form commonly have marked axial weakness and facial and bulbar involvement.134,135 Distal involvement may evolve later and mimic the appearance of a peripheral neuropathy or a distal myopathy. Skeletal involvement consisting of facial dysmorphism, scoliosis, spinal rigidity,136 and foot deformities is frequent both in the typical and intermediate forms. Although there is considerable phenotypic overlap between genetically distinct forms of nemaline myopathy, one study of 60 patients with mutations in the nebulin or actin genes revealed clinically distinct selective involvement of specific muscles, depending on the underlying gene defect.130 These observations have been corroborated by comparisons of muscle MRI results in molecularly characterized patients with nemaline myopathy (see Fig. 86-2).28
Respiratory involvement is frequent in all forms of nemaline myopathy, and respiratory failure may occur at any age. Nocturnal hypoventilation may be the presenting symptom in adulthood101 and may be frequently underestimated in otherwise paucisymptomatic patients. Patients with nemaline myopathy must be closely monitored for signs and symptoms of nocturnal hypoventilation, and noninvasive nighttime ventilation ought to be commenced early when indicated.35,137 Respiratory infections are frequent and must be treated vigorously.
Cardiac evaluation of a large series of patients with the typical congenital form of nemaline myopathy revealed normal cardiac function.138 Although cardiac involvement has been suggested in some genetically unresolved cases with nemaline rods in skeletal or cardiac muscles, those were only rarely associated with a congenital myopathy phenotype.139,140 Furthermore, nemaline bodies may develop in evolving cardiac hypertrophy in the absence of a clinically overt cardiomyopathy or muscle weakness.141
Although to the authors’ knowledge there have been no reports of clear malignant hyperthermia reactions in nemaline myopathy, a respiratory decompensation with anesthesia needs to be anticipated.142 Also, RYR1 mutations may give rise to both cores and rods on muscle biopsy,49,79 and MHS thus must be considered in genetically unresolved cases with unusual histopathological features.
The course of the “typical” form of congenital nemaline myopathy is slowly progressive or nonprogressive, but the severe neonatal form is often fatal. Neonatal respiratory failure and arthrogryposis are indicators of a poor outcome, whereas achievement of motor milestones within the first year of life is correlated with a good prognosis.142 The degree of cardiorespiratory involvement is the main prognostic factor in all age groups. There is a potential for deterioration in affected women during pregnancy.143
Genetics
Nemaline myopathy is inherited in both an autosomal recessive and, less frequently, an autosomal dominant manner. To date, five genes have been implicated in nemaline rod formation: the gene for slow α-tropomyosin (TPM3)144–146 (Wallgren-Pettersson, 2003), NEB,135,147–149 ACTA13234 (Sparrow et al, 2003), the muscle troponin T gene (TNNT1),150 and the β-tropomyosin gene (TPM2).151 Recessive mutations in NEB and dominant mutations in ACTA1 are the most common genetic causes. The typical form of nemaline myopathy131,134 is usually caused by mutations in the NEB147,149,152,153 (Wallgren-Pettersson et al, 2003) but sometimes by de novo dominant mutations in the actin gene.130,154,155 In sporadic cases, mutations in the relatively small ACTA1 ought to be ruled out first before screening of the giant NEB is considered.156 Further genetic heterogeneity in nemaline myopathy is expected, because some families show no linkage to any of the known loci.135,142,151
NEB on chromosome 2q22 is large, with 249 kilobases of genomic sequence and 183 exons.156,157 NEB encodes nebulin, a giant protein that both stabilizes the length of actin filaments and forms a composite Ca2+-linked regulatory complex on the thin filament.157,158 NEB mutations reported to date have been exclusively recessive and are most commonly involved in the typical congenital form of nemaline myopathy149,152,153; a more severe phenotype with neonatal presentation159 and milder cases occasionally occur. A particular NEB deletion has been described in the Ashkenazi Jewish population with a carrier frequency of approximately 1:100.153
ACTA1 on chromosome 1q42 contains only six exons.34 A study of Australian patients with nemaline myopathy revealed a heterozygosity frequency of 15% for ACTA1 missense mutations,154 which suggests that changes in this gene are the second most common cause of nemaline myopathy. ACTA1 mutations are predominantly autosomal dominant and only rarely autosomal recessive missense mutations.19,34,130,133,154,155 There is a high rate of de novo mutations, accounting for many sporadic cases, often with a severe congenital phenotype34,123,133,154,155,160,161; muscle biopsy specimens from those patients may feature intranuclear rods with or without cytoplasmic rods. Parental somatic mosaicism has been reported in two families.155 Less frequently, dominant ACTA1 mutations give rise to much milder cases with significant clinical overlap with the form caused by mutations in NEB.34,101,154 Mutations in the actin gene can also cause accumulation of thin filaments with or without nemaline bodies,34 congenital fiber type disproportion,162 or cores.20 There is an overall correlation between genotype and phenotype; the most severely affected patients carry nonsense mutations. Interestingly, cardiac actin can functionally compensate for the defective skeletal actin, and in a few patients, this has led to improved muscle function (F. Muntoni, N. G. Laing, and C. A. Sewry, personal observation, July 2005). A mouse model is currently being generated to investigate whether the upregulation of cardiac actin may alleviate the myopathy caused by ACTA1 mutations.163
The tropomyosin genes are rarely involved in nemaline myopathy. A dominant mutation in the first exon of TPM3 at chromosome 1q22-q23 has been identified in a large Australian pedigree with a mild form of the condition and predominant lower limb weakness144,145; in addition, another dominant de novo164 and a few recessive mutations146,165 have also been reported. Dominant mutations in TPM2 have so far been identified in only two families with a mild form of nemaline myopathy.151 TPM2 mutations may also give rise to autosomal dominant distal arthrogryposis without features of nemaline myopathy.166 Nemaline myopathy caused by mutations in TNNT1 appears to be limited to the Old Order Amish community,150 and symptoms may include tremor and progressive contractures.
The concurrence of rods and cores has been associated with ACTA1 mutations,101 mutations in RYR1,49,50 and mutations in an as yet unidentified gene on chromosome 15.167 Muscle MRI may help in the selection of genetic tests in cases with equivocal histopathological features (see Fig. 86-2B, C, E, and F).28,110
Nemaline myopathy exemplifies how individual structural congenital myopathies may represent syndromes with different genetic etiologies but similar histopathological appearances, caused by the involvement of defective gene products in a common pathway. All genes identified in nemaline myopathy to date code for thin filament associated proteins, several of which are also associated with Z line proteins, which suggests that disturbed assembly or interplay of myofibrillar structure is a pivotal mechanism in the evolution of rod pathology. Other genes encoding proteins involved in thin filament assembly or function are likely candidates for as yet genetically unresolved forms of nemaline myopathy.162
The pathogenetic effects of specific mutations associated with nemaline myopathy have been studied in transgenic mice,168–170 in cultured cardiomyocytes,171–173 in cultured myocytes transfected with mutant proteins,174 and through functional studies of RNA and proteins.175
Transgenic mice harboring mutations in TPM3 and ACTA1 show nemaline bodies163,168–170 whose number appears to be correlated with the degree of muscle weakness. In the mouse with TPM3 mutation, muscle weakness is correlated with the degree of type 1 hypotrophy and appears to be delayed by compensatory type 2 hypertrophy,168 and muscle regeneration may be abnormal.169 When expressed in rat adult cardiomyocytes, TPM3 mutations associated with skeletal muscle phenotypes produce hyposensitivity of Ca2+-activated force production that may underlie the muscle weakness observed in nemaline myopathy.171–173
CENTRONUCLEAR (MYOTUBULAR) MYOPATHY
Myotubular myopathy was initially described by Spiro and associates4 and is characterized by centrally located nuclei, often large, surrounded by a zone devoid of myofibrils and containing mitochondrial aggregates. The term myotubular myopathy was derived from the resemblance to fetal myotubes with central nuclei, and the presence in these fibers of high levels of desmin and vimentin was considered an indicator of maturational arrest; the descriptive term centronuclear myopathy avoids the ongoing controversy over the occurrence of maturational arrest.45,176–180 The term myotubular myopathy, however, continues to be widely used for the X-linked form, whereas the designation centronuclear myopathy is usually applied to autosomal forms.
Histopathology
In the X-linked form of centronuclear myopathy (myotubular myopathy), type 1 predominance and hypotrophy are common; these changes may progress over time with marked increases in fat and connective tissue (see Fig. 86-1D).102,181 Despite a superficial resemblance to fetal myotubes, lack of fetal myosin in fibers with central nuclei is suggestive of normal maturation in terms of myosin transition. Histopathological findings in carriers of the X-linked form range from absent to the full-fledged centronuclear myopathy pathology seen in affected male patients.102,182
In presumably autosomal forms, oxidative stains may reveal radial distribution of sarcoplasmic strands in fibers with central nuclei183; similar radial strands may be seen on periodic acid–Schiff stains. It is not clear whether this is simply an age-related effect, inasmuch as biopsy specimens from patients with autosomal cases are often taken at later ages than those from patients with the X-linked form. Corelike structures are occasionally present.99,184
Central internal nuclei are not specific, and other neuromuscular disorders with an associated increase in internal nuclei must be ruled out in the differential diagnosis of myotubular myopathy. In particular, patients with congenital myotonic dystrophy may exhibit histopathological findings identical to those in X-linked myotubular myopathy.185
Clinical Features
The X-linked form of centronuclear myopathy (myotubular myopathy) usually causes a severe phenotype in male patients, who present at birth with marked hypotonia, external ophthalmoplegia, and respiratory failure.143,180,186,187 The family history often includes miscarriages and male neonatal deaths in the mother’s family. Signs of antenatal onset include reduced fetal movements, polyhydramnios, and thinning of the ribs evident on chest radiographs,188 features rarely seen in other congenital myopathies. Birth asphyxia may be the presenting feature.182,189 Affected infants are often long and have increased head circumference, which may serve as a diagnostic clue.190 Many affected infants lack spontaneous antigravity movements, and some fail to establish spontaneous respiration at birth.187 Contractures of the hips and knees are commonly present. The course is usually fatal over days or weeks, but a proportion of affected boys may survive into their teens or beyond, sometimes with little residual disability and usually with normal intellect.191 Other patients may be mildly affected from the neonatal period onward.24,143,187,192–194 A range of medical complications in long-term survivors includes pyloric stenosis and cavernous liver hemangiomas191 and indicates widespread expression of the defective protein. Genital abnormalities have been described in affected boys with a contiguous gene syndrome.195 Most carriers of the X-linked form are either normal or show mild weakness196,197; the manifestation may be more severe if there are additional genetic abnormalities such as skewed X-inactivation102,196,198–201 or X chromosome abnormalities.202 Urinary incontinence may indicate smooth muscle involvement.102,197 Carrier status is best determined by molecular genetic methods. Manifesting heterozygosity should therefore always be considered in a female patient with histopathological features of myotubular myopathy.
Autosomal recessive and autosomal dominant forms differ from the X-linked form in age at onset, severity, clinical characteristics, and prognosis and are likely to be genetically heterogeneous.203,204
The autosomal recessive form of centronuclear myopathy is characterized by facial weakness, including extraocular muscle involvement182,203,205; Jeannet and coworkers reported two families with manifestations compatible with recessive inheritance and seven sporadic cases, and they distinguished between an early-onset form with or without ophthalmoplegia and a late-onset form without ophthalmoplegia.183 Weakness and wasting may be pronounced distally in the lower limbs. Skeletal deformities, including high arched palate, foot deformities, and scoliosis, are common.206 Respiratory involvement may be severe and cardiomyopathy has been associated in few probably recessive cases207–209 and in two sporadic cases.210,211 In the absence of severe cardiorespiratory involvement, the prognosis is favorable.
Jeannet and coworkers183 suggested autosomal dominant inheritance in two forms of centronuclear myopathy, both with onset occurring from infancy to adulthood and with only a slowly progressive course, one of which included diffuse muscle hypertrophy. Autosomal dominant centronuclear myopathy has to be differentiated from myotonic dystrophy and other autosomal dominant disorders with increased central nuclei evident on muscle biopsy specimens. Clinical findings such as cataracts or electrical myotonia98,212 strongly suggest that some of the families reported before the molecular era had, in fact, myotonic dystrophy rather than autosomal dominant centronuclear myopathy. Facioscapulohumeral distribution of weakness was reported in other families,213 which indicates that facioscapulohumeral muscular dystrophy should be considered in the differential diagnosis. Clinical findings in families without features suggestive of other neuromuscular disorders are usually milder than those of the recessive form.
The creatine kinase level is normal or only slightly elevated in all forms of centronuclear myopathy. Malignant hyperthermia has been reported in a patient with adult-onset centronuclear myopathy.214
Genetics
Initial linkage studies assigned a locus on chromosome Xq28 to the severe X-linked form of myotubular myopathy and recessive mutations in the myotubularin gene (MTM1) at this locus have subsequently been identified.215–219 In addition, a contiguous gene syndrome characterized by clinical and histopathological features of myotubular myopathy and additional abnormal genital development resulting in intersexual genitalia or severe hypospadias has been described in male patients harboring large deletions on Xq28.195,216,219 The initial suggestion of genetic heterogeneity of X-linked myotubular myopathy220 was later refuted.221
MTM1, a small gene with 15 exons,222 encodes myotubularin, a lipid phosphatase acting on phosphatidylinositol-3-monophosphate (PI3P). Myotubularin is involved in regulating intracellular and endosomal trafficking and vesicular transport processes thought to be required for myogenesis.223–230 There are homologues of myotubularin in many species, including mice and yeast, as well as many human homologues.215,231–234 Myotubularin shows structural homology to the hMTMR5/SBF1 protein, involved in the differentiation of myoblasts231; to the hMTMR2 protein, mutated in autosomal recessive Charcot-Marie-Tooth disease type 4B1234; and to the SBF2 protein, mutated in autosomal recessive Charcot-Marie-Tooth disease type 4B2.235,236 Mutations in MTM1 that are associated with the human disease affect residues preserved in drosophila homologues231 and markedly compromise the ability of the phosphatase to regulate PI3P levels; this illustrates the importance of this function for normal muscle metabolism. Observations in myotubularin knockout mice supports the previous suggestion of a structural maintenance defect in myotubularin-deficient muscle rather than a degenerative process.
To date, more than 200 mutations affecting all four active sites of the MTM1 gene have been identified in families with X-linked myotubular myopathy.215,218,232,237,238 Exons 12, 8, and 14 are more commonly affected, harboring about 50% of mutations, and seven mutations account for about 25% of cases.184,217–219,232,238–241 Most point mutations are truncating, but almost one third are missense mutations affecting conserved residues.184,232 Disease-causing mutations in the MTM1 gene have not been identified in all patients with typical features of X-linked myotubular myopathy, which suggests that the remainder may harbor mutations in a functionally related gene or in noncoding regions of the MTM1 gene. The latter hypothesis is supported by the finding of abnormal myotubularin expression in a number of patients without confirmed mutation in MTM1 coding regions.228 Intronic variants causing partial exon skipping238 and splice site mutations242 have been reported. De novo mutations are rare,232 and most mothers of affected patients are carriers184; somatic mosaicism has been occasionally documented.232,243,244
Genotypephenotype correlation studies are complicated by the fact that many of the mutations identified to date are unique.238 Moreover, marked intrafamilial phenotypical variability24 indicates that mutations in MTM1 may not be the only determinants of phenotype. A recent genotypephenotype correlation study187 suggested that some nontruncating mutations are associated with a milder clinical course,24,193,194 whereas other nontruncating and all known truncating mutations are associated with the common very severe neonatal form.
The genetic defects underlying the autosomal recessive and the autosomal dominant forms of centronuclear myopathy have not yet been identified. On Western blot, specimens from presumed autosomal recessive cases exhibit normal myotubularin expression.228 Several human myotubularin-related genes have been mapped to autosomal chromosomes233 and are candidates for causing autosomal dominant and autosomal recessive forms of myotubular myopathy. An autosomal recessive canine disorder resembling myotubular myopathy has been identified,245 but no obvious candidate gene in the orthologous human genomic region has yet been identified.
OTHER CONGENITAL MYOPATHIES WITH STRUCTURAL DEFECTS
In addition to the more common congenital myopathies previously described, a number of unusual structural defects have characterized rarer myopathies. Some are familial and may have a genetic basis; others are sporadic and may not be distinct genetic entities. Some are early-onset disorders with hypotonia and can be considered congenital myopathies; others have later onset and are better considered myopathies with structural defects. These unusual disorders include cap disease, reducing body myopathy, fingerprint body myopathy, tubular aggregate myopathy, sarcotubular myopathy, zebra body myopathy, broad A band disease, and cylindrical spiral myopathy, lamellar body myopathy.6,246 The molecular basis of some cases of hyaline body myopathy has been attributed to a mutation in the gene encoding slow/MYH7247 and can therefore be considered a myofibrillar/myosin storage myopathy.
Bertini E, Biancalana V, Bolino A, et al. 11th ENMC International Workshop on Advances in Myotbular Myopathy. 26–28 September 2003, Naarden, The Netherlands (5th Workshop of the International Consortium on Myotubular Myopathy). Neuromuscul Disord. 2004;14:387-396.
Jungbluth H, Beggs A, Bonnemann C, et al. 111th ENMC International Workshop on Multiminicore Disease. 2nd International MmD Workshop, 9–11 November 2002, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:754-766.
Sparrow JC, Nowak KJ, Durling HJ, et al. Muscle disease caused by mutation in the skeletal muscle αactin gene (ACTAQ1). Neuromuscul Disord. 2003;13:519-531.
Treves S, Anderson AA, Ducreux S, et al. Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul Disord. 2005;15:577-587.
Wallgren-Pettersson C, Laing NG. 109th ENMC International Workship: 5th workshop on nemaline myopathy, 11th-13th October 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:501-507.
1 Shy GM, Magee KR. A new congenital nonprogressive myopathy. Brain. 1956;79:610-621.
2 Treves S, Anderson AA, Ducreux S, et al: Ryanodine receptor 1 mutations, dysregulation of calcium homeostasis and neuromuscular disorders. Neuromuscul Disord 15:577–587.
3 Shy GM, Engel WK, Somers JE, et al. Nemaline myopathy. A new congenital myopathy. Brain. 1963;86:793-810.
4 Spiro AJ, Shy GM, Gonatas NK. Myotubular myopathy. Persistence of fetal muscle in an adolescent boy. Arch Neurol. 1966;14:1-14.
5 Engel AG, Gomez MR, Groover RV. Multicore disease. Mayo Clin Proc. 1971;10:666-681.
6 Goebel HH, Anderson JR. Structural congenital myopathies (excluding nemaline myopathy, myotubular myopathy and desminopathies): 56th European Neuromuscular Centre (ENMC) sponsored International Workshop. December 12–14, 1997, Naarden, The Netherlands. Neuromuscul Disord. 1999;9:50-57.
7 Wallgren-Pettersson C, Kivisaari L, Jääskeläinen J, et al. Ultrasonography, CT and MRI of muscles in congenital nemaline myopathy. Pediatr Neurol. 1990;6:20-28.
8 Hughes MI, Hicks EM, Nevin NC, et al. The prevalence of inherited neuromuscular disease in Northern Ireland. Neuromuscul Disord. 1996;6:69-73.
9 Darin N, Tulinius M. Neuromuscular disorders in childhood: a descriptive epidemiological study from western Sweden. Neuromuscular Disorders. 2000;10:1-9.
10 Sewry CA, Muller C, Davis M, et al. The spectrum of pathology in central core disease. Neuromuscul Disord. 2002;12:930-938.
11 Naumann M, Reiners K, Gold R, et al. Mitochondrial dysfunction in adult-onset myopathies with structural abnormalities. Acta Neuropathol. 1995;89:152-157.
12 Lammens M, Moerman P, Fryns JP, et al. Fetal akinesia sequence caused by nemaline myopathy. Neuropediatrics. 1997;28:116-119.
13 Romero NB, Monnier N, Viollet L, et al. Dominant and recessive central core disease associated with RYR1 mutations and fetal akinesia. Brain. 2003;126:2341-2349.
14 Merlini L, Granata C, Ballestrazi P, et al. Rigid spine in myopathies. J Neurol Sci. 1990;98:435.
15 Ramsey PL, Hensinger RN. Congenital dislocation of the hip associated with central core disease. J Bone Joint Surg Am. 1975;57:648-651.
16 Howard RS, Wiles CM, Hirsch NP, et al. Respiratory involvement in primary muscle disorders: assessment and management. Q J Med. 1993;86:175-189.
17 Dubowitz V. Muscle Disorders in Childhood, 2nd ed. London: WB Saunders, 1995.
18 Quinlivan RM, Muller CR, Davis M, et al. Central core disease: clinical, pathological, and genetic features. Arch Dis Child. 2003;88:1051-1055.
19 Ryan MM, Schnell C, Strickland CD, et al. Nemaline myopathy: a clinical study of 143 cases. Ann Neurol. 2001;50:312-320.
20 Kaindl AM, Ruschendorf F, Krause S, et al. Missense mutations of ACTA1 cause dominant congenital myopathy with cores. J Med Genet. 2004;41:842-848.
21 McComb RD, Markesbery WR, O’Connor WN. Fatal neonatal nemaline myopathy with multiple congenital anomalies. J Pediatr. 1979;94:47-51.
22 McMenamin JB, Curry B, Taylor GP, et al. Fatal nemaline myopathy in infancy. Can J Neurol Sci. 1984;11:305-309.
23 North KN, Laing NG, Wallgren-Pettersson C, et al. Nemaline myopathy: current concepts. J Med Genet. 1997;34:705-713.
24 Barth PG, Dubowitz V. X-linked myotubular myopathy—a long-term follow up study. Eur J Pediatr Neurol. 1998;1:49-56.
25 Tsuji M, Higuchi Y, Shiraishi K, et al. Congenital fiber type disproportion: severe form with marked improvement. Pediatr Neurol. 1999;21:658-660.
26 Heckmatt JZ, Leeman S, Dubowitz V. Ultrasound imaging in the diagnosis of muscle disease. J Pediatr. 1982;5:656-660.
27 Dubowitz V, Pearse AGE. Oxidative enzymes and phosphorylase in central core disease of muscle. Lancet. 1960;2:23-24.
28 Jungbluth H, Sewry CA, Councell S, et al. Magnetic resonance imaging of muscle in nemaline myopathy. Neuromuscul Disord. 2004;14:779-784.
29 Jungbluth H, Davis MR, Muller C, et al. Magnetic resonance imaging of muscle in congenital myopathies associated with RYR1 mutations. Neuromuscul Disord. 2004;14:785-790.
30 Dietzen CJ, D’Auria R, Fesenmeier J, et al. Electromyography in benign congenital myopathies. Muscle Nerve. 1993;16:328.
31 Bertorini TE, Staålberg E, Yuson CP, et al. Single fiber electromyography in neuromuscular disorders: correlation of muscle histochemistry, single fiber electromyography, and clinical findings. Muscle Nerve. 1994;17:345-353.
32 Coers C, Telerman Toppet N, Gerard JM, et al. Changes in motor innervation and histochemical pattern of muscle fibers in some congenital myopathies. Neurology. 1976;26:1046-1053.
33 Cruz Martinez A, Ferrer MT, Lopez Terradas JM, et al. Single fiber electromyography in central core disease. J Neurol Neurosurg Psychiatry. 1979;42:662-667.
34 Nowak KJ, Wattanasirichaigoon D, Goebel HH, et al. Mutations in the skeletal muscle αactin gene in patients with actin myopathy and nemaline myopathy. Nat Genet. 1999;23:208-212.
35 Wallgren-Pettersson C, Bushby K, Mellies U, et al. 117th ENMC workshop: Ventilatory Support in Congenital Neuromuscular Disorders: Congenital Myopathies, Congenital Muscular Dystrophies, Congenital Myotonic Dystrophy and SMA (II). April 4–6th 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:56-69.
36 Heckmatt JZ, Loh L, Dubowitz V. Night time nasal ventilation in neuromuscular disease. Lancet. 1990;335:579-582.
37 Jungbluth H, Müller CR, Halliger-Keller B, et al. Autosomal recessive inheritance of RYR1 mutations in a congenital myopathy with cores. Neurology. 2002;59:284-287.
38 Drennan JC. Surgical management of neuromuscular scoliosis. In: Serratrice G, Cros D, Desnuelle C, et al, editors. Neuromuscular Diseases. New York: Raven Press; 1984:551.
39 Denborough MA, Dennett X, Anderson R. Central core disease and malignant hyperpyrexia. BMJ. 1973;1:272-273.
40 Messina S, Hartley L, Main M, et al. Pilot trial of salbutamol in central core and multiminicore diseases. Neuropediatrics. 2004;35:262-266.
41 McKusick VA. Mendelian Inheritance in Man: A Catalog of Human Genes and Genetic Disorders, 12th ed. Baltimore: Johns Hopkins University Press, 1998.
42 Greenfield JG, Cornman T, Shy GM. The prognostic value of the muscle biopsy in the “floppy infant.”. Brain. 1958;81:461-484.
43 Dubowitz V. Muscle Biopsy: A Practical Approach, 2nd ed. London: Baillière Tindall, 1985.
44 Vita C, Migliorato A, Baradello A, et al. Expression of cytoskeleton proteins in central core disease. J Neurol Sci. 1994;124:71-76.
45 Sewry CA. The role of immunocytochemistry in congenital myopathies. Neuromuscul Disord. 1998;8:394-400.
46 Bonnemann CG, Thompson TG, van der Ven PF, et al. Filamin C accumulation is a strong but nonspecific immunohistochemical marker of core formation in muscle. J Neurol Sci. 2003;206:71-78.
47 Shafig SA, Gorycki MA, Asiedu SA, et al. Tenotomy. Effects on the fine structure of the soleus of the rat. Arch Neurol. 1969;20:625-633.
48 Fananapazir L, Dalakas MC, Cyran F, et al. Missense mutations in the β myosin heavy chain gene cause central core disease in hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 1993;90:3993-3997.
49 Scacheri PC, Hoffman EP, Fratkin JD, et al. A novel ryanodine receptor gene mutation causing both cores and rods in congenital myopathy. Neurology. 2000;55:1689-1696.
50 Monnier N, Romero NB, Lerale J, et al. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum Mol Genet. 2000;9:2599-2608.
51 Monnier N, Ferreiro A, Marty I, et al. A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multiminicore disease congenital myopathy with ophthalmoplegia. Hum Mol Genet. 2003;12:1171-1178.
52 Dubowitz V, Platts M. Central core disease of muscle with focal wasting. J Neurol Neurosurg Psychiatry. 1965;28:432-437.
53 Bethlem J, van Gool J, Haålsmann WC, et al. Familial non progressive myopathy with muscle cramps after exercise. A new disease associated with cores in the muscle fibers. Brain. 1966;89:569-588.
54 Gamble JG, Rinsky LA, Lee JH. Orthopaedic aspects of central core disease. J Bone Joint Surg Am. 1988;70:1061-1066.
55 Shuaib A, Paasuke RT, Brownell AKW. Central core disease. Clinical features in 13 patients. Medicine (Baltimore). 1987;66:389-396.
56 Robinson RL, Brooks C, Brown SL, et al. RYR1 mutations causing central core disease are associated with more severe malignant hyperthermia in vitro contracture test phenotypes. Hum Mutat. 2002;20:88-97.
57 King JO, Denborough MA. Anaesthetic-induced malignant hyperpyrexia in children. J Pediatr. 1973;83:37-40.
58 Graham GE, Silver K, Arlet V, et al. King syndrome: further clinical variability and review of the literature. Am J Med Genet. 1998;78:254-259.
59 Rueffert H, Olthoff D, Deutrich C, et al. A new mutation in the skeletal ryanodine receptor gene (RYR1) is potentially causative of malignant hyperthermia, central core disease, and severe skeletal malformation. Am J Med Genet. 2004;124:248-254.
60 Manzur AY, Sewry CA, Ziprin J, et al. A severe clinical and pathological variant of central core disease with possible autosomal recessive inheritance. Neuromuscul Disord. 1998;8:467-473.
61 Lamont PJ, Dubowitz V, Landon DN, et al. Fifty year follow-up of a patient with central core disease shows slow but definite progression. Neuromuscul Disord. 1998;8:385-391.
62 Isaacs H, Barlow MB. Central core disease associated with elevated creatine phosphokinase levels. Two members of a family known to be susceptible to malignant hyperpyrexia. S Afr Med J. 1974;48:640-642.
63 Patterson VH, Hill TR, Fletcher PJH, et al. Central core disease. Clinical and pathological evidence of progression within a family. Brain. 1979;102:581-594.
64 Heckmatt JZ, Dubowitz V. Ultrasound imaging and directed needle biopsy in the diagnosis of selective involvement in muscle disease. J Child Neurol. 1987;2:205-213.
65 Gillard EF, Otsu K, Fujii J, et al. A substitution of cysteine for arginine 614 in the ryanodine receptor is potentially causative of human malignant hyperthermia. Genomics. 1991;11:751-755.
66 Zhang Y, Chen HS, Khanna VK, et al. A mutation in the human ryanodine receptor gene associated with central core disease. Nat Genet. 1993;5:46-49.
67 Quane KA, Healy JMS, Keating KE, et al. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat Genet. 1993;5:51-55.
68 Robinson RL, Anetseder MJ, Brancadoro V, et al. Recent advances in the diagnosis of malignant hyperthermia susceptibility: how confident can we be of genetic testing? Eur J Hum Genet. 2003;11:342-348.
69 Levitt RC, Olckers A, Meyers S, et al. Evidence for the localization of a malignant hyperthermia susceptibility locus (MHS2) to human chromosome 17q. Genomics. 1992;14:562-566.
70 Moslehi R, Langlois S, Yam I, et al. Linkage of malignant hyperthermia and hyperkalemic periodic paralysis to the adult skeletal muscle sodium channel (SCN4A) gene in a large pedigree. Am J Med Genet. 1998;76:21-27.
71 Sudbrak R, Procaccio V, Klausnitzer M, et al. Mapping of a further malignant hyperthermia susceptibility locus to chromosome 3q13.1. Am J Hum Genet. 1995;56:684-691.
72 Monnier N, Procaccio V, Stieglitz P, et al. Malignant-hyperthermia susceptibility is associated with a mutation of the α1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. 1997;60:1316-1325.
73 Robinson RL, Monnier N, Wolz W, et al. A genome wide search for susceptibility loci in three European malignant hyperthermia pedigrees. Hum Mol Genet. 1997;6:953-961.
74 Stewart SL, Hogan K, Rosenberg H, et al. Identification of the Arg1086His mutation in the α subunit of the voltage-dependent calcium channel (CACNA1S) in a North American family with malignant hyperthermia. Clin Genet. 2001;59:178-184.
75 Robinson RL, Curran JL, Ellis FR, et al. Multiple interacting gene products may influence susceptibility to malignant hyperthermia. Ann Hum Genet. 2000;64:307-320.
76 McCarthy TV, Quane KA, Lynch PJ. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum Mutat. 2000;15:410-417.
77 Jurkat-Rott K, McCarthy T, Lehmann-Horn F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve. 2000;23:4-17.
78 Tilgen N, Zorzato F, Halliger-Keller B, et al. Identification of four novel mutations in the C-terminal membrane spanning domain of the ryanodine receptor 1: association with central core disease and alteration of calcium homeostasis. Hum Mol Genet. 2001;10:2879-2887.
79 Monnier N, Romero NB, Lerale J, et al. Familial and sporadic forms of central core disease are associated with mutations in the C-terminal domain of the skeletal muscle ryanodine receptor. Hum Mol Genet. 2001;10:2581-2592.
80 Davis MR, Haan E, Jungbluth H, et al. Principal mutation hotspot for central core disease and related myopathies in the C-terminal transmembrane region of the RYR1 gene. Neuromuscul Disord. 2003;13:151-157.
81 Sambuughin N, McWilliams S, de Bantel A, et al. Single–amino-acid deletion in the RYR1 gene, associated with malignant hyperthermia susceptibility and unusual contraction phenotype. Am J Hum Genet. 2001;69:204-208.
82 Quane KA, Keating KE, Manning BM, et al. Detection of a novel common mutation in the ryanodine receptor gene in malignant hyperthermia: implications for diagnosis and heterogeneity studies. Hum Mol Genet. 1994;3:471-476.
83 Shepherd S, Ellis F, Halsall J, et al. RYR1 mutations in UK central core disease patients: more than just the C-terminal transmembrane region of the RYR1 gene. J Med Genet. 2004;41(3):e33.
84 Lynch PJ, Tong J, Lehane M, et al. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc Natl Acad Sci U S A. 1999;96:4164-4169.
85 Galli L, Orrico A, Cozzolino S, et al. Mutations in the RYR1 gene in Italian patients at risk for malignant hyperthermia: evidence for a cluster of novel mutations in the C-terminal region. Cell Calcium. 2002;32:143-151.
86 Sei Y, Sambuughin NN, Davis EJ, et al. Malignant hyperthermia in North America: genetic screening of the three hot spots in the type I ryanodine receptor gene. Anesthesiology. 2004;101:824-830.
87 Ferreiro A, Monnier N, Romero NB, et al. A recessive form of central core disease, transiently presenting as multiminicore disease, is associated with a homozygous mutation in the ryanodine receptor type 1 gene. Ann Neurol. 2002;51:750-759.
88 Tong J, McCarthy TV, MacLennan DH. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J Biol Chem. 1999;274:693-702.
89 Dirksen RT, Avila G. Altered ryanodine receptor function in central core disease: leaky or uncoupled Ca(2+) release channels? Trends Cardiovasc Med. 2002;12:189-197.
90 Zorzato F, Yamaguchi N, Xu L, et al. Clinical and functional effects of a deletion in a COOH-terminal luminal loop of the skeletal muscle ryanodine receptor. Hum Mol Genet. 2003;12:379-388.
91 Ducreux S, Zorzato F, Muller C, et al. Effect of ryanodine receptor mutations on interleukin-6 release and intracellular calcium homeostasis in human myotubes from malignant hyperthermia-susceptible individuals and patients affected by central core disease. J Biol Chem. 2004;279:43838-43846.
92 Ferreiro A, Fardeau M. 80th ENMC International Workshop on Multi-Minicore Disease: 1st International MmD Workshop. 12–13th May, 2000, Soestduinen, The Netherlands. Neuromuscul Disord. 2002;12:60-68.
93 van Wijngaarden GK, Bethlem J, Dingemans KP, et al. Familial focal loss of cross striations. J Neurol. 1977;216:163-172.
94 Swash M, Schwartz MS. Familial multicore disease with focal loss of cross-striations and ophthalmoplegia. J Neurol Sci. 1981;52:1-10.
95 Jungbluth H, Sewry C, Brown SC, et al. Minicore myopathy in children—a clinical and histopathological study of 19 cases. Neuromuscul Disord. 2000;10:264-273.
96 Bethlem J, Arts WF, Dingemans KP. Common origin of rods, cores, miniature cores, and focal loss of cross striations. Arch Neurol. 1978;35:555-566.
97 Lee YS, Yip WCL. A fatal congenital myopathy with severe type I fiber atrophy, central nuclei and multicores. J Neurol Sci. 1981;50:277-290.
98 Hulsmann N, Gullotta F, Okur H. Cytopathology of an unusual case of centronuclear myopathy. J Neurol Sci. 1981;50:311-333.
99 Fitzsimons RB, McLeod JG. Myopathy with pathological features of both centronuclear myopathy and multicore disease. J Neurol Sci. 1982;57:395-405.
100 Vallat JM, de Lumley L, Loubet A, et al. Coexistence of minicores, cores, and rods in the same muscle biopsy. A new example of mixed congenital myopathy. Acta Neuropathol. 1982;58:229-232.
101 Jungbluth H, Sewry CA, Brown SC, et al. Mild phenotype of nemaline myopathy with sleep hypoventilation due to a mutation in the skeletal muscle αactin (ACTA1) gene. Neuromuscul Disord. 2001;11:35-40.
102 Jungbluth H, Sewry CA, Buj-Bello A, et al. Early and severe presentation of X-linked myotubular myopathy in a girl with skewed X-inactivation. Neuromuscul Disord. 2003;13:55-59.
103 Ferreiro A, Estournet B, Chateau D, et al. Multiminicore disease—searching for boundaries: phenotype analysis of 38 cases. Ann Neurol. 2000;48:745-757.
104 Bonnette H, Roelofs R, Olson WH. Multicore disease: report of a case with onset in middle age. Neurology. 1974;24:1039-1044.
105 Shuaib A, Martin JME, Mitchell LB, et al. Multicore myopathy: not always a benign entity. Can J Neurol Sci. 1988;15:10-14.
106 Magliocco AM, Mitchell LB, Brownell AK, et al. Dilated cardiomyopathy in multicore myopathy. Am J Cardiol. 1989;63:150-151.
107 Zeman AZJ, Dick DJ, Anderson JR, et al. Multicore myopathy presenting in adulthood with respiratory failure. Muscle Nerve. 1997;20:367-369.
108 Ferreiro A, Quijano-Roy S, Pichereau C, et al. Mutations of the selenoprotein N gene, which is implicated in rigid spine muscular dystrophy, cause the classical phenotype of multiminicore disease: reassessing the nosology of early-onset myopathies. Am J Hum Genet. 2002;71:739-749.
109 Rowe PW, Eagle M, Pollitt C, et al. Multicore myopathy: respiratory failure and paraspinal muscle contractures are important complications. Dev Med Child Neurol. 2000;42:340-343.
110 Jungbluth H, Beggs A, Bonnemann C, et al. 111th ENMC International Workshop on Multiminicore Disease. 2nd International MmD Workshop, 9–11 November 2002, Naarden, The Netherlands. Neuromuscul Disord. 2004;14:754-766.
111 Bertini E, Bosman C, Bevilacqua M, et al. Cardiomyopathy and multicore myopathy with accumulation of intermediate filaments. Eur J Pediatr. 1990;149:856-858.
112 Willemsen MAAP, van Oort AM, ter Laak HJ, et al. Multicore myopathy with restrictive cardiomyopathy. Acta Paediatr. 1997;86:1271-1274.
113 Koch BM, Bertorini TE, Eng GD, et al. Severe multicore disease associated with reaction to anesthesia. Arch Neurol. 1985;42:1204-1206.
114 Osada H, Masuda K, Seki K, et al. Multiminicore disease with susceptibility to malignant hyperthermia in pregnancy. Gynecol Obstet Invest. 2004;58:32-35.
115 Barone V, Massa O, Intravaia E, et al. Mutation screening of the RYR1 gene and identification of two novel mutations in Italian malignant hyperthermia families. J Med Genet. 1999;36:115-118.
116 Guis S, Figarella-Branger D, Monnier N, et al. Multiminicore disease in a family susceptible to malignant hyperthermia: histology, in vitro contracture tests, and genetic characterization. Arch Neurol. 2004;61:106-113.
117 Ohkubo M, Ino T, Shimazaki S, et al. Multicore myopathy associated with multiple pterygium syndrome and hypertrophic cardiomyopathy. Pediatr Cardiol. 1996;17:53-56.
118 Chudley AE, Rozdilsky B, Houston CS, et al. Multicore disease in sibs with severe mental retardation, short stature, facial anomalies, hypoplasia of the pituitary fossa, and hypogonadotrophic hypogonadism. Am J Med Genet. 1985;20:145-158.
119 Penegyres PK, Kakulas BA. The natural history of minicoremulticore myopathy. Muscle Nerve. 1991;14:411-415.
120 Moghadaszadeh B, Petit N, Jaillard C, et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat Genet. 2001;29:17-18.
121 Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desminrelated myopathy with Mallory body–like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol. 2004;55:676-686.
122 Barohn RJ, Jackson CE, Kagan Hallet KS. Neonatal nemaline myopathy with abundant intranuclear rods. Neuromuscul Disord. 1994;4:513-520.
123 Schröder JM, Durling H, Laing N. Actin myopathy with nemaline bodies, intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn). Acta Neuropathol. 2004;108:250-256.
124 Wallgren-Pettersson C, Kääriäinen H, Rapola J, et al. Genetics of congenital nemaline myopathy: a study of ten families. J Med Genet. 1990;27:480-487.
125 Jockusch BM, Veldman H, Griffiths G, et al. Immunofluorescence microscopy of a myopathy. α Actinin is a major constituent of nemaline rods. Exp Cell Res. 1980;127:409-420.
126 Vainzof M, Moreira ES, Suzuki OT, et al. Telethonin protein expression in neuromuscular disorders. Biochim Biophys Acta. 2002;1588:33-40.
127 Schröder R, Reimann J, Salmikangas P, et al. Beyond LGMD1A: myotilin is a component of central core lesions and nemaline rods. Neuromuscul Disord. 2003;13:451-455.
128 Gurgel-Giannetti J, Reed U, Bang ML, et al. Nebulin expression in patients with nemaline myopathy. Neuromuscul Disord. 2001;11:154-162.
129 Sewry CA, Brown SC, Pelin K, et al. Abnormalities in the expression of nebulin in chromosome-2 linked nemaline myopathy. Neuromuscul Disord. 2001;11:146-153.
130 Wallgren-Pettersson C, Pelin K, Nowak KJ, et al. Genotypephenotype correlations in nemaline myopathy caused by mutations in the genes for nebulin and skeletal muscle αactin. Neuromuscul Disord. 2004;14:461-470.
131 Wallgren-Pettersson C, Laing NG. Report of the 70th ENMC International Workshop: nemaline myopathy. Neuromuscul Disord. 2000;10:299-306.
132 Lacson AG, Donaldson G, Barness EG, et al. Infant with higharched palate, bell-shaped chest, joint contractures and intrauterine fractures. Pediatr Pathol Mol Med. 2002;21:569-584.
133 Agrawal PB, Strickland CD, Midgett C, et al. Heterogeneity of nemaline myopathy cases with skeletal muscle αactin gene mutations. Ann Neurol. 2004;56:86-96.
134 Wallgren-Pettersson C. Congenital nemaline myopathy: a clinical follow up study of twelve patients. J Neurol Sci. 1989;89:1-14.
135 Wallgren-Pettersson C, Pelin K, Hilpela P, et al. Clinical and genetic heterogeneity in autosomal recessive nemaline myopathy. Neuromuscul Disord. 1999;9:564-572.
136 Topaloglu H, Gögüs S, Yalaz K, et al. Two siblings with nemaline myopathy presenting with rigid spine syndrome. Neuromuscul Disord. 1994;4:263-267.
137 Iannaccone S, Guilfoile T. Long term mechanical ventilation in infants with neuromuscular disease. J Child Neurol. 1988;3:30-32.
138 Airenne AL, Wallgren-Pettersson C. Normal cardiac contractility in patients with congenital nemaline myopathy. Neuropediatrics. 1988;19:115-117.
139 Ishibashi Ueda H, Imakita M, Yutani C, et al. Congenital nemaline myopathy with dilated cardiomyopathy: an autopsy study. Hum Pathol. 1990;21:77-82.
140 Müller-Höcker J, Schafer S, Mendel B, et al. Nemaline cardiomyopathy in a young adult: an ultraimmunohistochemical study and review of the literature. Ultrastruct Pathol. 2000;24:407-416.
141 Davies MJ, Ballantine S, Darracott S, et al. Nemaline bodies in the heart: anomalous Z bands with a periodic structure suggesting tropomyosin in human cardiac muscle biopsies. Cardiovasc Res. 1973;7:408-411.
142 Wallgren-Pettersson C, Laing NG. 83rd ENMC International Workshop: 4th Workshop on nemaline myopathy. 22–24 September, 2000, Naarden, The Netherlands. Neuromuscul Disord. 2001;11:589-595.
143 Wallgren-Pettersson C, Hiilesmaa VK, Paatero H. Pregnancy and delivery in congenital nemaline myopathy. Acta Obstet Gynecol Scand. 1995;74:659-661.
144 Laing NG, Majda BT, Akkari PA, et al. Assignment of a gene (NEM1) for autosomal dominant nemaline myopathy to chromosome 1. Am J Hum Genet. 1992;50:576-583.
145 Laing NG, Wilton SD, Akkari PA, et al. A mutation in the α tropomyosin gene TPM3 associated with autosomal dominant nemaline myopathy NEM1.. Nat Genet. 1995;9:75-79.
146 Tan P, Briner J, Boltshauser E, et al. Homozygosity for a nonsense mutation in the α-tropomyosin gene TPM3 in a patient with severe congenital nemaline myopathy. Neuromuscul Disord. 1999;9:573-579.
147 Wallgren-Pettersson C, Avela K, Marchand S, et al. A gene for autosomal recessive nemaline myopathy assigned to chromosome 2q by linkage analysis. Neuromuscul Disord. 1995;5:441-443.
148 Pelin K, Ridanpaa M, Donner K, et al. Refined localisation of the genes for nebulin and titin on chromosome 2q allows the assignment of nebulin as a candidate gene for autosomalrecessive nemaline myopathy. Eur J Hum Genet. 1997;5:229-234.
149 Pelin K, Hilpela P, Donner K, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci U S A. 1999;96:2305-2310.
150 Johnston JJ, Kelley RI, Crawford TO, et al. A novel nemaline myopathy in the Amish caused by mutation in troponin T1. Am J Hum Genet. 2000;67:814-821.
151 Donner K, Ollikainen M, Ridanpaa M, et al. Mutations in the β-tropomyosin (TPM2) gene—a rare cause of nemaline myopathy. Neuromuscul Disord. 2002;12:151-158.
152 Pelin K, Donner K, Holmberg M, et al. Nebulin mutations in autosomal recessive nemaline myopathy: an update. Neuromuscul Disord. 2002;12:680-686.
153 Anderson SL, Ekstein J, Donnelly MC, et al. Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet. 2004;115:185-190.
154 Ilkovski B, Cooper ST, Nowak K, et al. Nemaline myopathy caused by mutations in the muscle α-skeletal-actin gene. Am J Hum Genet. 2001;68:1333-1343.
155 Sparrow JC, Nowak KJ, Durling HJ, et al. Muscle disease caused by mutations in the skeletal muscle αactin gene (ACTA1).. Neuromuscul Disord. 2003;13:519-531.
156 Donner K, Sandbacka M, Lehtokari VL, et al. Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet. 2004;12:744-751.
157 Labeit S, Kolmerer B. The complete primary structure of human nebulin and its correlation to muscle structure. J Mol Biol. 1995;248:308-315.
158 Wang K, Knipfer M, Huang QQ, et al. Human skeletal muscle nebulin sequence encodes a blueprint for thin filament architecture. Sequence motifs and affinity profiles of tandem repeats and terminal SH3. J Biol Chem. 1996;271:4304-4314.
159 Wallgren-Pettersson C, Donner K, Sewry C, et al. Mutations in the nebulin gene can cause severe congenital nemaline myopathy. Neuromuscul Disord. 2002;12:674-679.
160 Buxmann H, Schlösser R, Schlote W, et al. Congenital nemaline myopathy due to ACTA1-gene mutation and carnitine insufficiency: a case report. Neuropediatrics. 2001;32:267-270.
161 Ohlsson M, Tajsharghi H, Darin N, et al. Follow-up of nemaline myopathy in two patients with novel mutations in the skeletal muscle αactin gene (ACTA1). Neuromuscul Disord. 2004;14:471-475.
162 Laing NG, Clarke NF, Dye DE, et al. Actin mutations are one cause of congenital fibre type disproportion. Ann Neurol. 2004;56:689-694.
163 Wallgren-Pettersson C, Laing NG. 109th ENMC International Workshop: 5th workshop on nemaline myopathy, 11th-13th October 2002, Naarden, The Netherlands. Neuromuscul Disord. 2003;13:501-507.
164 Durling HJ, Reilich P, Müller-Höcker J, et al. De novo missense mutation in a constitutively expressed exon of the slow α-tropomyosin gene TPM3 associated with an atypical, sporadic case of nemaline myopathy. Neuromuscul Disord. 2002;12:947-951.
165 Wattanasirichaigoon D, Swoboda KJ, Takada F, et al. Mutations of the slow muscle α-tropomyosin gene, TPM3, are a rare cause of nemaline myopathy. Neurology. 2002;59:613-617.
166 Sung SS, Brassington A-M, Grannatt K, et al. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet. 2003;72:681-690.
167 Gommans IM, Davis M, Saar K, et al. A locus on chromosome 15q for a dominantly inherited nemaline myopathy with corelike lesions. Brain. 2003;126:1545-1551.
168 Corbett MA, Robinson CS, Dunglison GF, et al. A mutation in α-tropomyosin(slow) affects muscle strength, maturation and hypertrophy in a mouse model for nemaline myopathy. Hum Mol Genet. 2001;10:317-328.
169 Joya JE, Kee AJ, Nair-Shalliker V, et al. Muscle weakness in a mouse model of nemaline myopathy can be reversed with exercise and reveals a novel myofiber repair mechanism. Hum Mol Genet. 2004;13:2633-2645.
170 Nair-Shalliker V, Kee AJ, Joya JE, et al. Myofiber adaptational response to exercise in a mouse model of nemaline myopathy. Muscle Nerve. 2004;30:470-480.
171 Michele DE, Albayya FP, Metzger JM. A nemaline myopathy mutation in α-tropomyosin causes defective regulation of striated muscle force production. J Clin Invest. 1999;104:1575-1581.
172 Michele DE, Metzger JM. Physiological consequences of tropomyosin mutations associated with cardiac and skeletal myopathies. J Mol Med. 2000;78:543-553.
173 Michele DE, Coutu P, Metzger JM. Divergent abnormal muscle relaxation by hypertrophic cardiomyopathy and nemaline myopathy mutant tropomyosins. Physiol Genomics. 2002;9:103-111.
174 Ilkovski B, Nowak KJ, Domazetovska A, et al. Evidence for a dominant-negative effect in ACTA1 nemaline myopathy caused by abnormal folding, aggregation and altered polymerization of mutant actin isoforms. Hum Mol Genet. 2004;13:1727-1743.
175 Marston S, Mirza M, Abdulrazzak H, et al. Functional characterisation of a mutant actin (Met132Val) from a patient with nemaline myopathy. Neuromuscul Disord. 2004;14:167-174.
176 Sarnat HB. Myotubular myopathy: arrest of morphogenesis of myofibers associated with persistence of fetal vimentin and desmin. Four cases compared with fetal and neonatal muscle. Can J Neurol Sci. 1990;17:109-123.
177 Sarnat HB. Vimentin and desmin in maturing skeletal muscle and developmental myopathies. Neurology. 1992;42:1616-1624.
178 Mora M, Morandi L, Merlini L, et al. Fetus-like dystrophin expression and other cytoskeletal protein abnormalities in centronuclear myopathies. Muscle Nerve. 1994;17:1176-1184.
179 Helliwell TR, Ellis IH, Appleton RE. Myotubular myopathy: morphological, immunohistochemical and clinical variation. Neuromuscul Disord. 1998;8:152-161.
180 Wallgren-Pettersson C. 58th ENMC workshop: myotubular myopathy 20–22 March, 1998, Naarden, The Netherlands. Neuromuscul Disord. 1998;8:521-525.
181 van der Ven PFM, Jap PHK, Wetzels RHW, et al. Postnatal centralization of muscle fiber nuclei in centronuclear myopathy. Neuromuscul Disord. 1991;1:211-220.
182 Heckmatt JZ, Sewry CA, Hodes D, et al. Congenital centronuclear (myotubular) myopathy: a clinical, pathological and genetic study in eight children. Brain. 1985;108:941-964.
183 Jeannet PY, Bassez G, Eymard B, et al. Clinical and histologic findings in autosomal centronuclear myopathy. Neurology. 2004;62:1484-1490.
184 Bertini E, Biancalana V, Bolino A, et al. 118th ENMC International Workshop on Advances in Myotubular Myopathy. 26–28 September 2003, Naarden, The Netherlands. (5th Workshop of the International Consortium on Myotubular Myopathy). Neuromuscul Disord. 2004;14:387-396.
185 Dubowitz V. Lesson for the month: genetic counselling. Neuromuscul Disord. 1992;2:85-86.
186 Wallgren-Pettersson C, Thomas N. Report on the 20th ENMC sponsored international workshop: myotubular/centronuclear myopathy. Neuromuscul Disord. 1994;4:71-74.
187 McEntagart M, Parsons G, Buj-Bello A, et al. Genotypephenotype correlations in X-linked myotubular myopathy. Neuromuscul Disord. 2002;12:939-946.
188 Osborne JP, Murphy EG, Hill A. Thin ribs on chest X-ray: a useful sign in the differential diagnosis of the floppy newborn. Dev Med Child Neurol. 1983;25:343-345.
189 Barth PG, van Wijngaarden GK, Bethlem J. X linked myotubular myopathy with fatal neonatal asphyxia. Neurology. 1975;25:531-536.
190 LeGuennec JC, Bernier JP, Lamarche J. High stature in neonatal myotubular myopathy. Acta Paediatr Scand. 1988;77:610-611.
191 Herman GE, Finegold M, Zhao W, et al. Medical complications in long-term survivors with X-linked myotubular myopathy. J Pediatr. 1999;134:206-214.
192 Chanzy S, Routon MC, Moretti S, et al. Unusual good prognosis for X-linked myotubular myopathy. Arch Pediatr. 2003;10:707-709.
193 Biancalana V, Caron O, Gallati S, et al. Characterisation of mutations in 77 patients with X-linked myotubular myopathy, including a family with a very mild phenotype. Hum Genet. 2003;112:135-142.
194 Yu S, Manson J, White S, et al. X-linked myotubular myopathy in a family with three adult survivors. Clin Genet. 2003;64:148-152.
195 Hu LJ, Laporte J, Kress W, et al. Deletions in Xq28 in two boys with myotubular myopathy and abnormal genital development define a new contiguous gene syndrome in a 430 kb region. Hum Mol Genet. 1996;5:139-143.
196 Wallgren-Pettersson C. Report of the 72nd ENMC International Workshop on Myotubular Myopathy, Hilversum, The Netherlands, 1–3 October 1999. Neuromuscul Disord. 2000;10:521-525.
197 Hammans SR, Robinson DO, Moutou C, et al. A clinical and genetic study of a manifesting heterozygote with X-linked myotubular myopathy. Neuromuscul Disord. 2000;10:133-137.
198 Tanner SM, Orstavik KH, Kristiansen M, et al. Skewed X-inactivation in a manifesting carrier of X-linked myotubular myopathy and in her non-manifesting carrier mother. Hum Genet. 1999;104:249-253.
199 Sutton IJ, Winer JB, Norman AN, et al. Limb girdle and facial weakness in female carriers of X-linked myotubular myopathy mutations. Neurology. 2001;57:900-902.
200 Schara U, Kress W, Tucke J, et al. X-linked myotubular myopathy in a female infant caused by a new MTM1 gene mutation. Neurology. 2003;60:1363-1365.
201 Kristiansen M, Knudsen GP, Tanner SM, et al. X-inactivation patterns in carriers of X-linked myotubular myopathy. Neuromuscul Disord. 2003;13:468-471.
202 Dahl N, Hu LJ, Chery M, et al. Myotubular myopathy in a girl with a deletion at Xq27-q28 and unbalanced X inactivation assigns the MTM1 gene to a 600 kb region. Am J Hum Genet. 1995;56:1108-1115.
203 De Angelis MS, Palmucci L, Leone M, et al. Centronuclear myopathy: clinical, morphological and genetic characters. A review of 288 cases. J Neurol Sci. 1991;103:2-9.
204 Wallgren-Pettersson C, Clarke A, Samson F, et al. The myotubular myopathies: differential diagnosis of the X linked recessive, autosomal dominant and autosomal recessive forms and present state of DNA studies. J Med Genet. 1995;32:673-679.
205 Zanoteli E, Oliveira AS, Kiyomoto BH, et al. Centronuclear myopathy. Histopathological aspects in ten patients with childhood onset. Arq Neuropsiquiatr. 1998;56:1-8.
206 Pages M, Cesari JB, Pages AM. Centronuclear myopathy. Complete review of the literature apropos of a case. Ann Pathol. 1982;2:301-310.
207 Verhiest W, Brucher JM, Goddeeris P, et al. Familial centronuclear myopathy associated with “cardiomyopathy.”. Br Heart J. 1976;38:504-509.
208 Gospe SMJr, Armstrong DL, Gresik MV, et al. Life-threatening congestive heart failure as the presentation of centronuclear myopathy. Pediatr Neurol. 1987;3:117-120.
209 Bataille J, Guillon F, Urtizberea A, et al. [Pathological anatomy of the heart in myopathies and infantile muscular atrophies]. Ann Med Interne (Paris). 1991;142:5-8.
210 Bethlem J, van Wijngaarden GK, Meijer AEFH, et al. Neuromuscular disease with type 1 fiber atrophy, central nuclei, and myotube like structures. Neurology. 1969;19:705-710.
211 Al-Ruwaishid A, Vajsar J, Tein I, et al. Centronuclear myopathy and cardiomyopathy requiring heart transplant. Brain Dev. 2003;25:62-66.
212 Hawkes CH, Absolon MJ. Myotubular myopathy associated with cataract and electrical myotonia. J Neurol Neurosurg Psychiatry. 1975;38:761-764.
213 Felice KJ, Grunnet ML. Autosomal dominant centronuclear myopathy: report of a new family with clinical features simulating facioscapulohumeral syndrome. Muscle Nerve. 1997;20:1194-1196.
214 Quinn RD, Pae WE, McGary SA, et al. Development of malignant hyperthermia during mitral valve replacement. Ann Thorac Surg. 1992;53:1114-1116.
215 Laporte J, Hu LJ, Kretz C, et al. A gene mutated in X-linked myotubular myopathy defines a new putative tyrosine phosphatase family conserved in yeast. Nat Genet. 1996;13:175-182.
216 Laporte J, Kioschis P, Hu LJ, et al. Cloning and characterization of an alternatively spliced gene in proximal Xq28 deleted in two patients with intersexual genitalia and myotubular myopathy. Genomics. 1997;41:458-462.
217 de Gouyon BM, Zhao W, Laporte J, et al. Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum Mol Genet. 1997;6:1499-1504.
218 Buj-Bello A, Biancalana V, Moutou C, et al. Identification of novel mutations in the MTM1 gene causing severe and mild forms of X-linked myotubular myopathy. Hum Mutat. 1999;14:320-325.
219 Herman GE, Kopacz K, Zhao W, et al. Characterization of mutations in fifty North American patients with X-linked myotubular myopathy. Hum Mutat. 2002;19:114-121.
220 Samson F, Mesnard L, Heimburger M, et al. Genetic linkage heterogeneity in myotubular myopathies. Am J Hum Genet. 1995;57:120-126.
221 Guiraud-Chaumeil C, Vincent MC, Laporte J, et al. A mutation in the MTM1 gene invalidates a previous suggestion of nonallelic heterogeneity in X-linked myotubular myopathy. Am J Hum Genet. 1997;60:1542-1544.
222 Laporte J, Guiraud-Chaumeil C, Tanner SM, et al. Genomic organization of the MTM1 gene implicated in X-linked myotubular myopathy. Eur J Hum Genet. 1998;6:325-330.
223 Cui X, De IV, Slany R, et al. Association of SET domain and myotubularin-related proteins modulates growth control. Nat Genet. 1998;18:331-337.
224 Laporte J, Blondeau F, Buj-Bello A, et al. Characterization of the myotubularin dual specificity phosphatase gene family from yeast to human. Hum Mol Genet. 1998;7:1703-1712.
225 Blondeau F, Laporte J, Bodin S, et al. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. 2000;15:2223-2229.
226 Taylor GS, Maehama T, Dixon JE. Myotubularin, a protein tyrosine phosphatase mutated in myotubular myopathy, dephosphorylates the lipid second messenger, phosphatidylinositol 3-phosphate. Proc Natl Acad Sci U S A. 2000;97:8910-8915.
227 Laporte J, Blondeau F, Buj-Bello A, et al. The myotubularin family: from genetic disease to phosphoinositide metabolism. Trends Genet. 2001;17:221-228.
228 Laporte J, Kress W, Mandel JL. Diagnosis of X-linked myotubular myopathy by detection of myotubularin. Ann Neurol. 2001;50:42-46.
229 Chaussade C, Pirola L, Bonnafous S, et al. Expression of myotubularin by an adenoviral vector demonstrates its function as a phosphatidylinositol 3-phosphate [PtdIns(3)P] phosphatase in muscle cell lines: involvement of PtdIns(3)P in insulin-stimulated glucose transport. Mol Endocrinol. 2003;17:2448-2460.
230 Tsujita K, Itoh T, Ijuin T, et al. Myotubularin regulates the function of the late endosome through the gram domainphosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279:13817-13824.
231 Kioschis P, Wiemann S, Heiss NS, et al. Genomic organization of a 225-kb region in Xq28 containing the gene for X-linked myotubular myopathy (MTM1) and a related gene (MTMR1). Genomics. 1998;54:256-266.
232 Laporte J, Biancalana V, Tanner SM, et al. MTM1 mutations in X-linked myotubular myopathy. Hum Mutat. 2000;15:393-409.
233 Appel S, Reichwald K, Zimmermann W, et al. Identification and localization of a new myotubularin-related protein gene, MTMR8, on 8p22-p23. Genomics. 2001;75:6-8.
234 Bolino A, Muglia M, Conforti FL, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein–2. Nat Genet. 2000;25:17-19.
235 Azzedine H, Bolino A, Taieb T, et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72:1141-1153.
236 Senderek J, Bergmann C, Weber S, et al. Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot-Marie-Tooth neuropathy type 4B2/11p15. Hum Mol Genet. 2003;12:349-356.
237 Laporte J, Guiraud-Chaumeil C, Vincent MC, et al. Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. ENMC International Consortium on Myotubular Myopathy. European Neuro-Muscular Center. Hum Mol Genet. 1997;6:1505-1511.
238 Tanner SM, Schneider V, Thomas NS, et al. Characterization of 34 novel and six known MTM1 gene mutations in 47 unrelated X-linked myotubular myopathy patients. Neuromuscul Disord. 1999;9:41-49.
239 Nishino I, Minami N, Kobayashi O, et al. MTM1 gene mutations in Japanese patients with the severe infantile form of myotubular myopathy. Neuromuscul Disord. 1998;8:453-458.
240 Donnelly A, Haan E, Manson J, et al. A novel mutation in exon b (R259C) of the MTM1 gene is associated with a mild myotubular myopathy. Mutations in brief no. 125. Online. Hum Mutat. 1998;11:334.
241 De Luca A, Torrente I, Mangino M, et al. A novel mutation (R271X) in the myotubularin gene causes a severe miotubular myopathy. Hum Hered. 1999;49:59-60.
242 Watanabe T, Watanabe M, Saito T, et al. X-linked recessive myotubular myopathy with a splice-site mutation in the myotubularin gene. No To Hattatsu. 1998;30:523-527.
243 Vincent MC, Guiraud-Chaumeil C, Laporte J, et al. Extensive germinal mosaicism in a family with X linked myotubular myopathy simulates genetic heterogeneity. J Med Genet. 1998;35:241-243.
244 Hane BG, Rogers RC, Schwartz CE. Germline mosaicism in X-linked myotubular myopathy. Clin Genet. 1999;56:77-81.
245 Tiret L, Blot S, Kessler JL, et al. The cnm locus, a canine homologue of human autosomal forms of centronuclear myopathy, maps to chromosome 2. Hum Genet. 2003;113:297-306.
246 North K. Congenital myopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology. 3rd ed. New York: McGraw-Hill; 2004:1473-1533.
247 Tajsharghi H, Thornell LE, Lindberg C, et al. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7.. Ann Neurol. 2003;54:494-500.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
CHAPTER 86 THE CONGENITAL MYOPATHIES
The congenital myopathies are a clinically and genetically heterogeneous group of congenital muscle disorders with characteristic structural abnormalities evident on muscle biopsy, visible after preparation with specific histochemical stains and/or on electron microscopy. Central core disease (CCD),1,2 nemaline myopathy,3 myotubular (centronuclear) myopathy,4 and minicore myopathy (or multiminicore disease [MmD])5 are the major disease entities. Other conditions with more unusual structural abnormalities are very rare, and it is not clear whether all are genetic entities.6
Autosomal dominant, autosomal recessive, and X-linked inheritance are all recognized in this group of disorders, and some conditions such as nemaline myopathy, CCD, and myotubular myopathy may have more than one mode of inheritance. Genetic advances have implicated several genes encoding sarcomeric and sarcotubular proteins (Table 86-1). The boundaries between these conditions, originally defined according to histopathological and clinical criteria, are often indistinct and do not necessarily reflect underlying molecular mechanisms: Mutations in the same gene can indeed give rise to diverse clinical and histopathological phenotypes, and, conversely, a similar histopathological and clinical phenotype may arise from mutations in a variety of genes. Although clinical management is currently the main form of treatment of the congenital myopathies, further advances in the understanding of the precise molecular mechanisms underlying each disorder may result in more rational therapeutic options in the future.
EPIDEMIOLOGY
Epidemiological data on the congenital myopathies are few, and larger geographical surveys are limited. The overall incidence of the congenital myopathies is estimated at 6 per 100,000 live births, representing approximately 10% of all neuromuscular disorders.7 Studies in northern Ireland8 and western Sweden9 suggest that the prevalence of the congenital myopathies in a pediatric population is between 3.5 and 5.0 per 100,000. The relative frequency of individual conditions is unknown, but CCD and conditions associated with mutations in the skeletal muscle ryanodine receptor gene (RYR1) appear to be more common in the patient population (see later discussion) than are nemaline myopathy and the much rarer centronuclear myopathies. Also, the prevalence of specific congenital myopathies may have been previously underestimated, inasmuch as not all muscle biopsy specimens from individuals carrying disease-causing mutations exhibit the characteristic structural abnormalities.10
CLINICAL FEATURES
Most of the clinical features are nonspecific, despite some variations in overall severity, distribution of weakness, and associated features. The diagnosis of a specific congenital myopathy can therefore be made only tentatively on clinical grounds alone, and muscle biopsy is required for a precise diagnosis in individual cases.
Presentation of the patient is either as a “floppy infant” with generalized hypotonia at birth followed by developmental delay or with weakness of variable severity and distribution later in childhood. Presentation in adulthood has also been described, but other disease processes mimicking the appearance of a congenital myopathy should be considered.11 Presentation at birth is often associated with “myopathic” facial features, feeding difficulties, and respiratory difficulties; thinning of the ribs and chest deformities may indicate antenatal onset. Arthrogryposis may be present in the most severe cases of nemaline myopathy12 and CCD13 but is not a common feature. Most cases manifest later in infancy or childhood with motor developmental delay or predominantly proximal weakness that mimics a limb girdle muscular dystrophy or a mild form of spinal muscular atrophy. In other patients, there is marked weakness of the axial muscles and/or the face; a minority have prominent distal involvement. Extraocular involvement is common in centronuclear myopathy and specific subgroups of minicore myopathy, but not in CCD and nemaline myopathy.
Spinal deformities include exaggerated lordosis, scoliosis, and spinal rigidity.14 Scoliosis may be present at birth but typically manifests in a progressive manner around the time of the pubertal growth spurt. Ligamentous laxity is common. Joint contractures, mainly of the Achilles tendon, tend to progress over time. Tendon reflexes are weak or absent. Congenital dislocation of the hips is a common feature in CCD15 but can occur in several other neuromuscular disorders.
Respiratory involvement secondary to diaphragmatic and/or intercostal muscle weakness is the main prognostic factor.16,17 Respiratory impairment is common in centronuclear myopathy, nemaline myopathy, and subgroups of minicore myopathy; it may occur only rarely in CCD,18 with the exception of the severe congenital variant, in which it is common.13 Susceptibility to respiratory infections is frequent, particularly early in life, but may improve with time.13,19 Cardiac involvement other than cor pulmonale secondary to respiratory impairment is not usually a feature, although structural abnormalities such as mitral valve prolapse have been occasionally documented. However, a few patients have had congenital myopathy with rods and/or cores and cardiac involvement in association with mutations in the skeletal muscle αactin gene (ACTA1).20
There are no associated structural central nervous system or peripheral nerve abnormalities, and intelligence is usually normal. In the few cases in which an autopsy was performed, no central or peripheral nerve abnormalities could be identified.21–23
Progression of muscle weakness in the absence of substantial respiratory impairment does not usually occur, but deterioration is occasionally associated with growth spurts or marked weight gain. The most severely affected neonates may die from respiratory failure, but long-term survival has been reported even in infants with severe hypotonia and marked respiratory impairment.13,19,24,25
INVESTIGATIONS
Other investigations may support the suspicion of an underlying myopathic process, but, with the possible exception of muscle imaging, they rarely aid in the diagnosis. Diseased muscle exhibits characteristic changes in echogenicity and signal intensity on ultrasonography, computed tomography, and magnetic resonance imaging (MRI), which reflect an increase in adipose and connective tissue.7,25–27 Muscle MRI in particular may reveal a characteristic pattern of selective involvement in conditions such as CCD and nemaline myopathy, thereby guiding the molecular diagnosis in clinically and histopathologically equivocal cases.28,29
The serum creatine kinase level is usually normal or only mildly elevated. The electromyogram may appear normal in the young patients or in mild cases but usually reveals nonspecific myopathic features, comprising small-amplitude polyphasic potentials.30,31 Additional “neurogenic” changes may be present in distal muscles, particularly of patients with nemaline myopathy.31–33 Spontaneous activity resembling findings in spinal muscular atrophy have been reported in the most severely affected neonates.34
MANAGEMENT
Respiratory impairment is the most important complication and should be anticipated by regular monitoring of respiratory function: regular measurement of forced vital capacity, in sitting and lying positions in order to evaluate diaphragmatic function, and annual overnight oxygen saturation monitoring if the forced vital capacity falls under 60% or, if clinically indicated by symptoms of nocturnal hypoventilation such as early morning headaches, loss of appetite, and daytime tiredness.35 Respiratory impairment may be disproportionately severe, and respiratory failure may occur even in ambulant patients.16,17,19,35–37 Nighttime noninvasive ventilation offers an effective means of improving quality of life and overall prognosis in otherwise only moderately disabled patients. Respiratory infections should be treated aggressively. A cardiac ultrasound study should be performed to document baseline cardiac function and occasionally associated structural cardiac abnormalities. Regular cardiac ultrasonography should be performed in patients with respiratory impairment, although appropriate respiratory management should prevent cor pulmonale.16 Primary cardiomyopathies are rare in the congenital myopathies but have been reported in individual cases with mutations in the ACTA1 gene20 and in a few patients with minicores evident in muscle biopsy specimens (see later discussion).
Spinal posture should be monitored closely and surgical procedures for scoliosis planned carefully to ensure that respiratory function is still sufficient at the time of operative intervention.38 Regular physiotherapy is aimed at preservation of muscle power and prevention of contractures. When orthopedic intervention is planned, the potential benefit has to be weighed against adverse effects of prolonged immobilization.
Patients with CCD and other congenital myopathies associated with mutations in the RYR1 gene are at increased risk of malignant hyperthermia susceptibility,39 and potentially triggering anesthetic agents should be rigorously avoided in these cases. Although a higher anesthetic risk is not clearly documented in other congenital myopathies, similar precautions should be taken in the anesthetic preparation of other patients,35 particularly in those in whom the genetic background is unclear.
Pharmacological interventions have not been widely used in the treatment of the congenital myopathies. A small pilot study has demonstrated some beneficial effect of salbutamol in the treatment of patients with CCD and minicore myopathy,40 but this has yet to be confirmed in a larger controlled trial.
CENTRAL CORE DISEASE
Histopathology
The term central core disease (Mendelian Inheritance in Man [MIM] number 117000)41 was introduced in 195842 after the original description of a family with five affected individuals in three generations who showed amorphous central areas in muscle fibers with the modified Gomori trichrome stain1; absence of oxidative enzyme activity within the core area was subsequently identified as the characteristic histopathological feature (Fig. 86-1A).27 The cores have a predilection for type 1 fibers and extend along a significant length of the longitudinal muscle fiber axis43; localization is characteristically single and central, but multiple, peripheral, and eccentric cores may occur within the same sample.10 In some cases, large typical cores may not be apparent, but only unevenness of oxidative enzyme stain or small core areas are visible, producing a confusing pathological overlap with minicore disease. Pronounced type 1 predominance or uniformity is common and may be the only feature at presentation, particular in very young patients.10,43 The degree of associated increases in fat and connective tissue depends both on the age of the patient and on the sampling site, because the degree of selective muscle involvement may be considerable29; centrally placed nuclei may also be present but are not a consistent feature. Necrosis and regeneration are not usually seen in CCD.
Ultrastructurally, typical cores show a reduction or absence of mitochondria, reflected in the absence of oxidative enzyme staining, accompanied by varying degrees of sarcomeric disruption and accumulation of Z line material.43 In most cases, cores are “structured” with hypercontracted myofibrils that retain adenosine triphosphatase activity but are depleted in mitochondria and sarcoplasmic reticulum; “unstructured” cores, on the contrary, show an absence of adenosine triphosphatase activity, marked disruption of myofibrils, and accumulation of Z line material.
Immunohistochemical studies have demonstrated accumulation of the intermediate filament protein desmin within cores and at their perimeter.10,44,45 Accumulation of other proteins also occurs, including the actin cross-linking protein γ-filamin, αβ-crystallin, small heat shock protein 27, and myotilin46; however, these are nonspecific findings and may be observed in core formation of different etiology.
Core formation is a nonspecific secondary phenomenon and is not correlated with the degree of the muscle weakness. Core formation can be observed after tenotomy,47 in the muscle of patients with long-standing neurogenic atrophy (“target fibers”), and in association with several other gene defects. Central corelike structures may be found in association with dilated cardiomyopathy in patients with mutations in ACTA120 or in the β-myosin heavy-chain gene (MYH7)48; however, in the latter group, there is usually no or little associated weakness. Central core and nemaline rods are occasionally observed in the same biopsy specimen in patients with mutations in the RYR1 gene.49,50
Clinical Features
“Classical” CCD is usually inherited as a dominant trait with a fairly consistent clinical phenotype; sporadic cases with similar clinical features are increasingly recognized. Presentation is in infancy with hypotonia or in early childhood with motor developmental delay or proximal weakness17; however, more severe presentations with features of the fetal akinesia sequence have been reported.13,51 Most of these infants required ventilation from birth, and their course was severe and eventually fatal; however, one infant was eventually weaned off the ventilator and became independently mobile.13
Weakness in most familial cases is pronounced in the hip girdle and in the axial muscle groups17; in rare cases, there is associated muscle wasting.52 Facial involvement is usually mild, and lack of complete eye closure may be the only finding. Bulbar and extraocular involvement is usually absent. Some patients may have prominent exercise-induced myalgia.53
Orthopedic complications are common and comprise congenital dislocation of the hips, foot deformities, and scoliosis.15,18,54 Contractures other than Achilles tendon tightness are rare, and many affected individuals have marked ligamentous laxity, occasionally in association with patellar instability.54
Cardiac abnormalities other than occasional mitral valve prolapse have rarely been reported.55 Respiratory involvement is typically milder than in other congenital myopathies but may be severe in some sporadic and recessive cases.51
Malignant hyperthermia susceptibility (MHS) is a frequent complication39,55,56 and should be anticipated in the anesthetic preparation of patients with CCD. Some patients with MHS may show consistent dysmorphic features (King-Denborough syndrome), including ptosis, down-slanting palpebral fissures, neck webbing, scoliosis, pectus deformity, short stature, and cryptorchism57,58; additional findings in other families may include vertebral fusion, eventration of the diaphragm, and spinal cord tethering.58,59
Except for patients with the most severe neonatal cases and some patients with congenital dislocation of the hips,37,60 most patients achieve the ability to walk independently. The course of CCD is static or only slowly progressive, even over prolonged periods of follow-up.61
The serum creatine kinase level is usually normal or only mildly elevated.62,63 Muscle ultrasonography often shows a striking increase in echogenicity and differential muscle involvement, even in paucisymptomatic individuals.64 Muscle MRI reveals a characteristic pattern of selective muscle involvement in patients with typical CCD29 (Fig. 86-2A and D) and may complement clinical assessment.
Genetics of Central Core Disease and Malignant Hyperthermia Susceptibility
MHS was recognized in 1960 as a familial, autosomal dominant trait by M. A. Denborough and R. R. H. Lovell in Australia and has been linked to several loci. Both CCD and MHS show considerable clinical overlap and have been associated with mutations in the RYR1 gene at chromosome 19q13.1.65–67 CCD is usually inherited as a dominant trait; however, sporadic cases either caused by de novo dominant mutations or associated with recessive inheritance are increasingly recognized.
Although it is now apparent that mutations in the RYR1 gene (MHS1) account for the majority of familial MHS cases,68 locus heterogeneity has been suggested by variable degrees of linkage evidence for several loci (MHS2 to MHS6) and cosegregation of MHS susceptibility with mutations in candidate genes.69–74 There is also the possibility that the MHS phenotype may reflect the compound influence of several genes rather than a major gene defect.75 Genetic homogeneity in CCD has been suggested by RYR1 linkage in most families without confirmed mutation. Although dominant mutations in the ACTA1 gene20 and the MYH7 genes48 may mimic the histopathological appearance, clinical features such as an associated cardiomyopathy are divergent from those in classic CCD.
More than 50 RYR1 mutations have been reported to date in association with CCD, MmD (described later), and MHS with or without cores evident on muscle biopsy specimens.76–80 Most RYR1 mutations are missense mutations; a few small deletions78,79,81 and a cryptic splicing site mutation51 have also been documented. The first reported RYR1 mutations gave rise predominantly to the MHS phenotype and affected mainly two regions of the protein: the cytoplasmic N-terminal domain (MHS/CCD region 1, amino acids 35 to 614) and the cytoplasmic central domain (MHS/CCD region 2, amino acids 2163 to 2458). Although mutations associated with a congenital myopathy phenotype have been identified in these regions,66,67,82,83 there has been increasing evidence that mutations affecting the C-terminal portion of the receptor molecule (MHS/CCD region 3, amino acids 4550 to 4940) are common in patients with CCD.49,78–80,84 Although the mutations are occasionally reported in the C-terminal portion of the protein,85 a series of 124 unrelated North American patients with MHS confirmed the N-terminal and central portion of the protein as the predominant sites of MHS-related RYR1 mutations.86
Although the majority of RYR1 mutations associated with MHS or CCD described to date are dominant missense mutations, recessive inheritance of RYR1 mutations has been reported in mild cases with histopathological features of MmD and CCD37,87 and in a severe form of CCD manifesting with a fetal akinesia syndrome.13 These findings suggest that clinically silent MHS mutations may give rise to a severe phenotype in the compound heterozygous or homozygous state, caused by a combined deleterious effect on the tetrameric RyR1 protein.
The effects of specific RYR1 mutations on excitability and calcium homeostasis have been studied in various homologous and heterologous expression systems. Two models for mutation-induced receptor malfunction have been proposed: depletion of sarcoplasmic reticulum calcium stores with resulting increases in cytosolic calcium levels (the “leaky channel” hypothesis)88 and a specific disturbance of excitation-contraction coupling (the “E-C uncoupling” hypothesis).89 The “ectopic” RYR1 expression in B lymphocytes has been recognized; this offers an easily accessible model for studying the effects of RYR1 mutations in vitro. B lymphocytes harboring CCD-related RYR1 mutations exhibit unprompted calcium release with resulting depletion of sarcoplasmic reticulum stores78,90; increased release of inflammatory cytokines in the same study91 may also indicate a role of RYR1 in immunomodulation.
MULTIMINICORE DISEASE
Multicore disease (MIM numbers 255320 and 157550)41 was originally described in two unrelated children with hypotonia and muscle weakness dating from infancy and a characteristic histopathological appearance of multiple, small, well-circumscribed foci of myofibrillar degeneration with reduction of oxidative enzyme staining.5 Since the original description, at least 100 other cases with similar histopathological features and a wide range of clinical phenotypes have been reported, under different names such as minicore myopathy, myopathy with multiple minicore, pleocore disease, or myopathy with focal loss of cross-striations. The designation multiminicore disease92 used in this chapter reflects the larger number and smaller size of characteristic lesions compared with classic CCD.
Histopathology
Minicores are areas of focal myofibrillar disruption with a variable degree of Z line disorganization and may occur in both type 1 and type 2 fibers (see Fig. 86-1B). They extend only a short distance along the longitudinal axis of the muscle fiber, affecting a variable number of sarcomeres.5,43 In rare cases, the entire fiber diameter may be involved (focal loss of cross-striations).93,94 Pathological distinction between CCD and MmD is occasionally difficult, because in some biopsy specimens from patients with RYR1 mutations, only unevenness of oxidative enzyme stain or minicores are seen, and minicores may evolve into central cores with time.10,87
Commonly associated features are predominance or uniformity of type 1 fibers, often hypotrophic, with or without type 2 hypertrophy.95 This is particularly true for cases secondary to RYR1 but not selenoprotein N, 1 (SEPN1) mutations (see later discussion). Although central nuclei may be a feature in cases caused by RYR1 mutations, they are not usually associated with SEPN1 mutations. The presence of whorled fibers and increases in fat and connective tissue in some affected families43 are suggestive of a potential overlap with the milder congenital muscular dystrophies.
On electron microscopy, minicores are characterized by focal areas of myofibrillar disruption with paucity of mitochondria.43 The sizes of minicores and the degree of myofibrillar disruption are variable, ranging from mild Z line streaming to areas with complete loss of sarcomeric organization. These may be observed in the same biopsy speciment.5 In some areas, ultrastructural abnormalities may consist only of subtle misalignment of myofibrils accompanied by an absence of mitochondria.
In immunohistochemical studies, authors have reported abnormalities on staining with desmin45 and γ-filamin antibodies,46 similar to those observed in CCD.
Minicores are nonspecific; they may be found in various disorders such as muscular dystrophies; denervation; and inflammatory, endocrine, and metabolic myopathies. Also, minicore-like structures may coexist with central cores, central nuclei, and nemaline rods in the same biopsy specimen10,20,96–102 and in association with various gene defects.
Clinical Features
MmD usually manifests in infancy or childhood with hypotonia or proximal weakness95,103; cases with antenatal onset or presentation in adulthood104–107 have been reported, but the molecular defect in these is not known. Clinical features associated with the histopathological appearance of MmD are markedly heterogeneous, and at least four different subgroups have been recognized and are gradually being resolved molecularly (see later discussion).
The most instantly recognizable classic phenotype of MmD95,103,108 usually manifests in infancy with hypotonia and is characterized by spinal rigidity, scoliosis, and early respiratory impairment. There is predominant truncal and proximal weakness, often in association with wasting pronounced in the shoulder girdle and the hip adductors (“bracket-like thighs”). There may be mild facial weakness and a high-pitched nasal voice. Severe scoliosis and respiratory failure have usually evolved by the early teens.95,103,109 Respiratory impairment is often disproportionate to the overall degree of muscle weakness and has to be anticipated even in ambulant patients. Most of these cases are secondary to mutations in the SEPN1 gene (discussed later).
In a subset of patients with a similar phenotype, extraocular muscle involvement pronounced on abduction and upward gaze may evolve over time.94,95,110 With the exception of the most severely affected neonatal cases,51 respiratory impairment is usually milder than in the classic form and may improve with time. Mutations in the RYR1 gene have been identified in these patients (discussed later).
Another group has a milder phenotype similar to CCD and characterized by predominant hip girdle weakness with relative sparing of respiratory and bulbar muscles.37 A common complaint is exercise-induced myalgia. In male patients, cryptorchism may be an additional feature (personal observation). Patients in this group may also demonstrate muscle imaging findings similar to those observed in classic CCD caused by mutations in the RYR1 C-terminal portion.37,87,110 In some patients, there is additional marked distal weakness and wasting, predominantly affecting the hands.37,87 The observation of extraocular muscle involvement evolving over time in this group is suggestive of a clinical continuum between the latter groups rather than distinct clinical entities.110 Mutations in the RYR1 gene also account for this group of disorders.
A severe form with antenatal onset, generalized arthrogryposis, dysmorphic features, and moderate respiratory impairment has been described in only a few patients.103
Congenital cardiac defects—namely, mitral valve prolapse—have been occasionally reported.5,37 Secondary right ventricular impairment is common in the classic phenotype with untreated respiratory impairment but not in other clinical subgroups.37 A primary cardiomyopathy has been reported in some cases with minicores on muscle biopsy specimens,55,106,111,112 but these were genetically unresolved, and additional desmin accumulation in muscle and dominant inheritance were suggestive of a pathogenic mechanism distinct from that in other families (see later discussion).
Malignant hyperthermia has been reported occasionally113,114 and has to be anticipated in the anesthetic management of these patients. Minicores have been noted in muscle biopsy specimens from a few families with mutations in the RYR1 gene and MHS but no other clinical features of a congenital myopathy.115,116
Minicores have been reported in the multiple pterygium syndrome117 and in two siblings with mental retardation and dysmorphic features similar to the King-Denborough syndrome118; the molecular basis of these associations is currently unclear.
The clinical course is usually static or only slowly progressive37,119 but depends largely on the degree of cardiorespiratory impairment.
Genetics
Investigation of SEPN1 on chromosome 1p36 as a candidate for causing MmD was prompted by the considerable clinical and histopathological overlap between the classic phenotype of MmD and congenital muscular dystrophy with rigidity of the spine, previously attributed to SEPN1 mutations.120 SEPN1 involvement in the classic phenotype of MmD was suggested both by linkage data and by direct mutational analysis in approximately 50% of patients with these clinical features.108 More than 30, mainly truncating SEPN1 mutations associated with a congenital myopathy phenotype have been identified to date. Mutations in the SEPN1 gene were also identified in cases with unusual pathological structures resembling Mallory bodies (Mallory body myopathy), further expanding the pathological spectrum of the condition.121 Homozygous mutations are unexpectedly common even in nonconsanguineous families, which reflects the presence of a few founder mutations in different European populations. The precise function of selenoprotein N, a glycoprotein localized in the endoplasmic reticulum, is unclear, but a structural motif similar to those observed in calcium-binding proteins120 is suggestive of possible involvement in calcium homeostasis in muscle. Although selenoprotein N is expressed in all adult tissues, prominent expression in fetal muscle precursor cells is suggestive of a potential role in myogenesis.
Involvement of the RYR1 gene as a cause of multi-minicores was to some extent unexpected, in view of differences in core structure and mode of inheritance between recessively inherited MmD and CCD caused by dominant mutations in the same gene. Attempting to classify individual cases as MmD or CCD may be difficult because there is a histopathological and clinical continuum between the two conditions. Although the histopathologic appearance of MmD appears to be more closely associated with recessively inherited RYR1 mutations, dominant RYR1 mutations occasionally give rise to minicores on muscle biopsy specimens10 and may have accounted for a proportion of MmD pedigrees with autosomal dominant inheritance reported before the molecular area. Also, the histopathological appearance of MmD caused by recessive RYR1 mutations may evolve into the classic picture of CCD over long periods of follow-up.87 A particular feature of RYR1-related recessive cases with minicores is extraocular involvement, not present in dominant CCD. MmD with external ophthalmoplegia in an isolated case from a consanguineous Tunisian family was attributed to a homozygous RYR1 mutation, introducing a cryptic splice site in intron 101,51 carried by asymptomatic parents. RYR1 involvement was also suggested by linkage evidence in four additional families with a similar phenotype,110 including the original family reported by Swash and Schwartz.94 RYR1 involvement in the moderate form of MmD with hand involvement was suggested by considerable clinical overlap with CCD and an identical pattern of selective involvement on muscle imaging.37,87 In a consanguineous Algerian family and a consanguineous British family, homozygous RYR1 mutations were subsequently identified, and RYR1 involvement has been suggested by linkage evidence in other families. The RYR1 gene is also a likely candidate for causing the severe form of MmD with neonatal onset and arthrogryposis, in view of phenotypic overlap with the form of CCD with fetal akinesia sequence.13
There is evidence for further genetic heterogeneity in MmD, because only one half of all cases with the classic phenotype have linkage or mutational evidence of SEPN1 involvement.108 Also, although a proportion of MmD in pedigrees with autosomal dominant inheritance are likely to be caused by dominant RYR1 mutations, unusual clinical features such as a primary cardiomyopathy55,111
[/not-level-membership-for-neurology-category]
