[level-membership-for-basic-science-category]
CHAPTER 2 The Cellular and Molecular Basis of Inheritance
The hereditary material is present in the nucleus of the cell, whereas protein synthesis takes place in the cytoplasm. What is the chain of events that leads from the gene to the final product?
The Cell
Within each cell of the body, visible with the light microscope, is the cytoplasm and a darkly staining body, the nucleus, the latter containing the hereditary material in the form of chromosomes (Figure 2.1). The phospholipid bilayer of the plasma membrane protects the interior of the cell but remains selectively permeable and has integral proteins involved in recognition and signaling between cells. The nucleus has a darkly staining area, the nucleolus. The nucleus is surrounded by a membrane, the nuclear envelope, which separates it from the cytoplasm but still allows communication through nuclear pores.
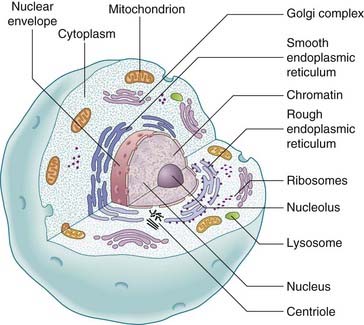
The cytoplasm contains the cytosol, which is semifluid in consistency, containing both soluble elements and cytoskeletal structural elements. In addition, in the cytoplasm there is a complex arrangement of very fine, highly convoluted, interconnecting channels, the endoplasmic reticulum. The endoplasmic reticulum, in association with the ribosomes, is involved in the biosynthesis of proteins and lipids. Also situated within the cytoplasm are other even more minute cellular organelles that can be visualized only with an electron microscope. These include the Golgi apparatus, which is responsible for the secretion of cellular products, the mitochondria, which are involved in energy production through the oxidative phosphorylation metabolic pathways, and the peroxisomes (p. 180) and lysosomes, both of which are involved in the degradation and disposal of cellular waste material and toxic molecules.
DNA: The Hereditary Material
Structure
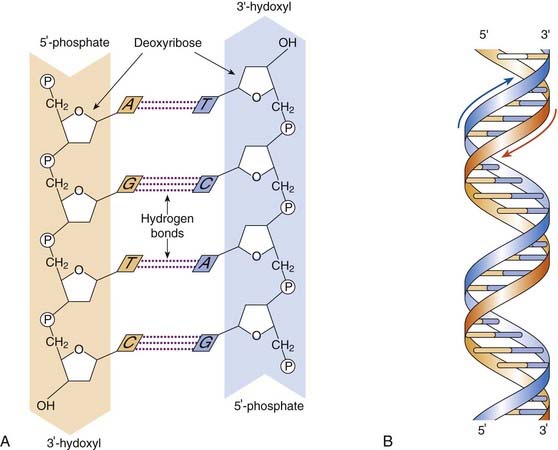
For genes to be composed of DNA, it is necessary that the latter should have a structure sufficiently versatile to account for the great variety of different genes and yet, at the same time, be able to reproduce itself in such a manner that an identical replica is formed at each cell division. In 1953, Watson and Crick, based on x-ray diffraction studies by themselves and others, proposed a structure for the DNA molecule that fulfilled all the essential requirements. They suggested that the DNA molecule is composed of two chains of nucleotides arranged in a double helix. The backbone of each chain is formed by phosphodiester bonds between the 3′ and 5′ carbons of adjacent sugars, the two chains being held together by hydrogen bonds between the nitrogenous bases, which point in toward the center of the helix. Each DNA chain has a polarity determined by the orientation of the sugar–phosphate backbone. The chain end terminated by the 5′ carbon atom of the sugar molecule is referred to as the 5′ end, and the end terminated by the 3′ carbon atom is called the 3′ end. In the DNA duplex, the 5′ end of one strand is opposite the 3′ end of the other, that is, they have opposite orientations and are said to be antiparallel.
The arrangement of the bases in the DNA molecule is not random. A purine in one chain always pairs with a pyrimidine in the other chain, with specific pairing of the base pairs: guanine in one chain always pairs with cytosine in the other chain, and adenine always pairs with thymine, so that this base pairing forms complementary strands (Figure 2.2). For their work Watson and Crick, along with Maurice Wilkins, were awarded the Nobel Prize for Medicine or Physiology in 1962 (p. 10).
Replication
DNA replication, through the action of the enzyme DNA polymerase, takes place at multiple points known as origins of replication, forming bifurcated Y-shaped structures known as replication forks. The synthesis of both complementary antiparallel DNA strands occurs in the 5′ to 3′ direction. One strand, known as the leading strand, is synthesized as a continuous process. The other strand, known as the lagging strand, is synthesized in pieces called Okazaki fragments, which are then joined together as a continuous strand by the enzyme DNA ligase (Figure 2.3A).
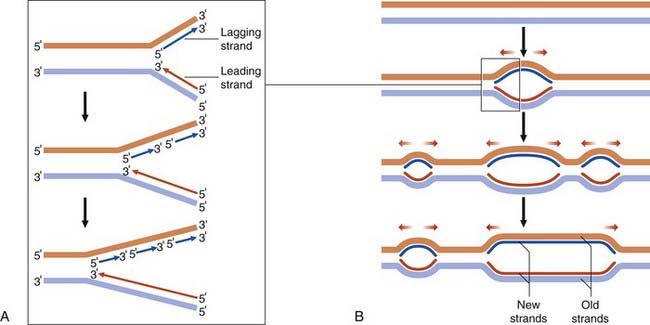
DNA replication progresses in both directions from these points of origin, forming bubble-shaped structures, or replication bubbles (Figure 2.3B). Neighboring replication origins are approximately 50 to 300 kilobases (kb) apart and occur in clusters or replication units of 20 to 80 origins of replication. DNA replication in individual replication units takes place at different times in the S phase of the cell cycle (p. 39), adjacent replication units fusing until all the DNA is copied, forming two complete identical daughter molecules.
Chromosome Structure
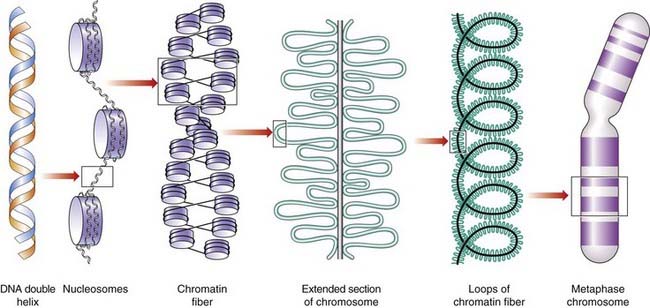
The packaging of DNA into chromosomes involves several orders of DNA coiling and folding. In addition to the primary coiling of the DNA double helix, there is secondary coiling around spherical histone ‘beads’, forming what are called nucleosomes. There is a tertiary coiling of the nucleosomes to form the chromatin fibers that form long loops on a scaffold of non-histone acidic proteins, which are further wound in a tight coil to make up the chromosome as visualized under the light microscope (Figure 2.4), the whole structure making up the so-called solenoid model of chromosome structure.
Types of DNA Sequence
DNA, if denatured, will reassociate as a duplex at a rate that is dependent on the proportion of unique and repeat sequences present, the latter occurring more rapidly. Analysis of the results of the kinetics of the reassociation of human DNA have shown that approximately 60% to 70% of the human genome consists of single- or low-copy number DNA sequences. The remainder of the genome, 30% to 40%, consists of either moderately or highly repetitive DNA sequences that are not transcribed. This latter portion consists of mainly satellite DNA and interspersed DNA sequences (Box 2.1).
Nuclear Genes
It is estimated that there are between 25,000 and 30,000 genes in the nuclear genome. The distribution of these genes varies greatly between chromosomal regions. For example, heterochromatic and centromeric (p. 32) regions are mostly non-coding, with the highest gene density observed in subtelomeric regions. Chromosomes 19 and 22 are gene rich, whereas 4 and 18 are relatively gene poor. The size of genes also shows great variability: from small genes with single exons to genes with up to 79 exons (e.g., dystrophin, which occupies 2.5 Mb of the genome).
Multigene Families
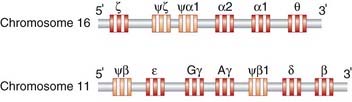
Many genes have similar functions, having arisen through gene duplication events with subsequent evolutionary divergence making up what are known as multigene families. Some are found physically close together in clusters; for example, the α- and β-globin gene clusters on chromosomes 16 and 11 (Figure 2.5), whereas others are widely dispersed throughout the genome occurring on different chromosomes, such as the HOX homeobox gene family (p. 87).
Classic Gene Families
Examples of classic gene families include the numerous copies of genes coding for the various ribosomal RNAs, which are clustered as tandem arrays at the nucleolar organizing regions on the short arms of the five acrocentric chromosomes (p. 32), and the different transfer RNA (p. 20) gene families, which are dispersed in numerous clusters throughout the human genome.
Gene Superfamilies
Examples of gene superfamilies include the HLA (human leukocyte antigen) genes on chromosome 6 (p. 200) and the T-cell receptor genes, which have structural homology with the immunoglobulin (Ig) genes (p. 200). It is thought that these are almost certainly derived from duplication of a precursor gene, with subsequent evolutionary divergence forming the Ig superfamily.
Gene Structure
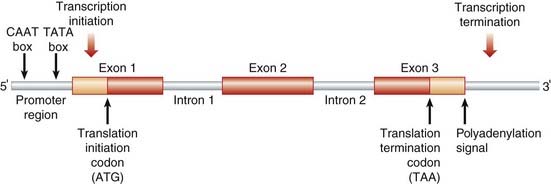
The original concept of a gene as a continuous sequence of DNA coding for a protein was turned on its head in the early 1980s by detailed analysis of the structure of the human β-globin gene. It was revealed that the gene was much longer than necessary to code for the β-globin protein, containing non-coding intervening sequences, or introns, that separate the coding sequences or exons (Figure 2.6). Most human genes contain introns, but the number and size of both introns and exons is extremely variable. Individual introns can be far larger than the coding sequences and some have been found to contain coding sequences for other genes (i.e., genes occurring within genes). Genes in humans do not usually overlap, being separated from each other by an average of 30 kb, although some of the genes in the HLA complex (p. 200) have been shown to be overlapping.
Extragenic DNA
Tandemly Repeated DNA Sequences
Minisatellite DNA
Telomeric DNA
The terminal portion of the telomeres of the chromosomes (p. 32) contains 10 to 15 kb of tandem repeats of a 6-base pair (bp) DNA sequence known as telomeric DNA. The telomeric repeat sequences are necessary for chromosomal integrity in replication and are added to the chromosome by an enzyme known as telomerase (p. 32).
Hypervariable minisatellite DNA
Hypervariable minisatellite DNA is made up of highly polymorphic DNA sequences consisting of short tandem repeats of a common core sequence. The highly variable number of repeat units in different hypervariable minisatellites forms the basis of the DNA fingerprinting technique developed by Professor Sir Alec Jeffreys in 1984 (p. 69).
Microsatellite DNA
Microsatellite DNA consists of tandem single, di-, tri-, and tetra-nucleotide repeat base-pair sequences located throughout the genome. Microsatellite repeats rarely occur within coding sequences but trinucleotide repeats in or near genes are associated with certain inherited disorders (p. 59).
This variation in repeat number is thought to arise by incorrect pairing of the tandem repeats of the two complementary DNA strands during DNA replication, or what is known as slipped strand mispairing. Duplications or deletions of longer sequences of tandemly repeated DNA are thought to arise through unequal crossover of non-allelic DNA sequences on chromatids of homologous chromosomes or sister chromatids (p. 32).
Nowadays DNA microsatellites are used for forensic and paternity tests (p. 69). They can also be helpful for gene tracking in families with a genetic disorder but no identified mutation (p. 70).
Highly Repeated Interspersed Repetitive DNA Sequences
Long Interspersed Nuclear Elements
The function of these interspersed repeat sequences is not clear. Members of the Alu repeat family are flanked by short direct repeat sequences and therefore resemble unstable DNA sequences called transposable elements or transposons. Transposons, originally identified in maize by Barbara McClintock (p. 10), move spontaneously throughout the genome from one chromosome location to another and appear to be ubiquitous in the plant and animal kingdoms. It is postulated that Alu repeats could promote unequal recombination, which could lead to pathogenic mutations (p. 22) or provide selective advantage in evolution by gene duplication. Both Alu and LINE-1 repeat elements have been implicated as a cause of mutation in inherited human disease.
Mitochondrial DNA
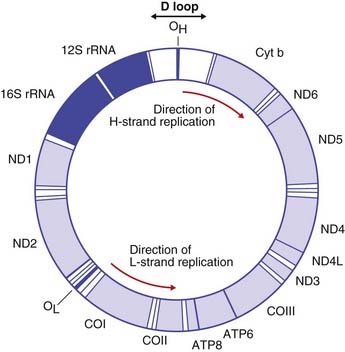
In addition to nuclear DNA, the several thousand mitochondria of each cell possess their own 16.6 kb circular double-stranded DNA, mitochondrial DNA (or mtDNA) (Figure 2.7). The mtDNA genome is very compact, containing little repetitive DNA, and codes for 37 genes, which include two types of ribosomal RNA, 22 transfer RNAs (p. 20) and 13 protein subunits for enzymes, such as cytochrome b and cytochrome oxidase, which are involved in the energy producing oxidative phosphorylation pathways. The genetic code of the mtDNA differs slightly from that of nuclear DNA.
The mitochondria of the fertilized zygote are inherited almost exclusively from the oocyte, leading to the maternal pattern of inheritance that characterizes many mitochondrial disorders (p. 181).
Transcription
In any particular gene, only one DNA strand of the double helix acts as the so-called template strand. The transcribed mRNA molecule is a copy of the complementary strand, or what is called the sense strand of the DNA double helix. The template strand is sometimes called the antisense strand. The particular strand of the DNA double helix used for RNA synthesis appears to differ throughout different regions of the genome.
RNA Processing
mRNA Splicing
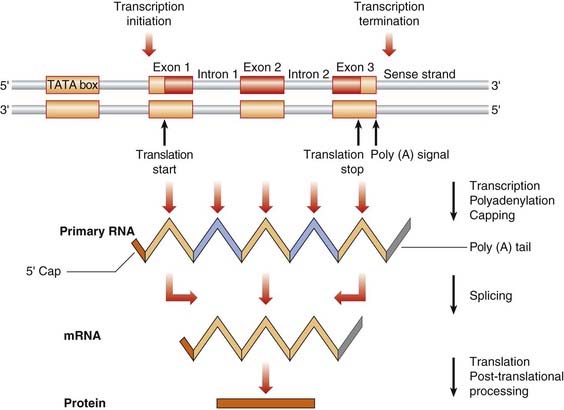
During and after transcription, the non-coding introns in the precursor (pre) mRNA are excised, and the non-contiguous coding exons are spliced together to form a shorter mature mRNA before its transportation to the ribosomes in the cytoplasm for translation. The process is known as mRNA splicing (Figure 2.8). The boundary between the introns and exons consists of a 5′ donor GT dinucleotide and a 3′ acceptor AG dinucleotide. These, along with surrounding short splicing consensus sequences, another intronic sequence known as the branch site, small nuclear RNA (snRNA) molecules and associated proteins, are necessary for the splicing process.
Translation
Transfer RNA
In the cytoplasm there is another form of RNA called transfer RNA, or tRNA. The incorporation of amino acids into a polypeptide chain requires the amino acids to be covalently bound by reacting with ATP to the specific tRNA molecule by the activity of the enzyme aminoacyl tRNA synthetase. The ribosome, with its associated rRNAs, moves along the mRNA, the amino acids linking up by the formation of peptide bonds through the action of the enzyme peptidyl transferase to form a polypeptide chain (Figure 2.9).
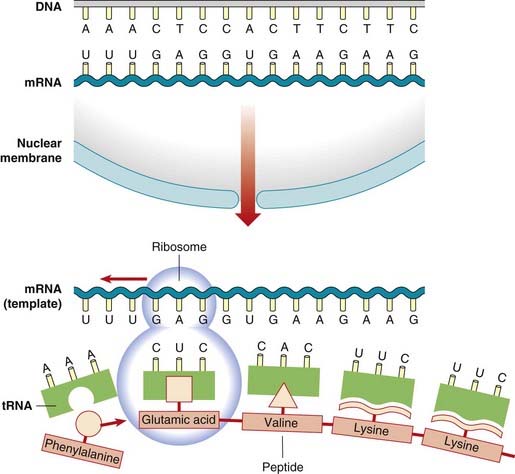
The Genetic Code
Triplet Codons
The triplet of nucleotide bases in the mRNA that codes for a particular amino acid is called a codon. Each triplet codon in sequence codes for a specific amino acid in sequence and so the genetic code is non-overlapping. The order of the triplet codons in a gene is known as the translational reading frame. However, some amino acids are coded for by more than one triplet, so the code is said to be degenerate (Table 2.1). Each tRNA species for a particular amino acid has a specific trinucleotide sequence called the anticodon, which is complementary to the codon of the mRNA. Although there are 64 codons, there are only 30 cytoplasmic tRNAs, the anticodons of a number of the tRNAs recognizing codons that differ at the position of the third base, with guanine being able to pair with uracil as well as cytosine. Termination of translation of the mRNA is signaled by the presence of one of the three stop or termination codons.

The genetic code of mtDNA differs from that of the nuclear genome. Eight of the 22 tRNAs are able to recognize codons that differ only at the third base of the codon, 14 can recognize pairs of codons that are identical at the first two bases, with either a purine or pyrimidine for the third base, the other four codons acting as stop codons (see Table 2.1).
Regulation of Gene Expression
Many cellular processes, and therefore the genes that are expressed, are common to all cells, for example ribosomal, chromosomal and cytoskeleton proteins, constituting what are called the housekeeping genes. Some cells express large quantities of a specific protein in certain tissues or at specific times in development, such as hemoglobin in red blood cells (p. 155). This differential control of gene expression can occur at a variety of stages.
Control of Transcription
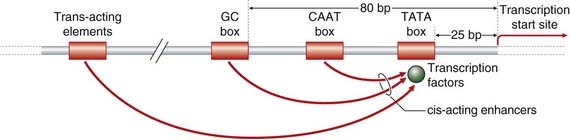
All of these mechanisms ultimately affect transcription through the binding of the general transcription factors to short specific DNA promoter elements located within 200 bp 5′ or upstream of most eukaryotic genes in the so-called core promoter region that leads to activation of RNA polymerase (Figure 2.10). Promoters can be broadly classed into two types, TATA box-containing and GC rich. The TATA box, which is about 25 bp upstream of the transcription start site, is involved in the initiation of transcription at a basal constitutive level and mutations in it can lead to alteration of the transcription start site. The GC box, which is about 80 bp upstream, increases the basal level of transcriptional activity of the TATA box.
RNA-Mediated Control of Gene Expression
RNA-mediated silencing was first described in the early 1990s, but it is only recently that its key role in controlling post-transcriptional gene expression has been both recognized and exploited (see Chapter 23). Small interfering RNAs (siRNAs) were discovered in 1998 and are the effector molecules of the RNA interference pathway (RNAi). These short double-stranded RNAs (21 to 23 nucleotides) bind to mRNAs in a sequence-specific manner and result in their degradation via a ribonuclease-containing RNA-induced silencing complex (RISC). MicroRNAs (miRNAs) also bind to mRNAs in a sequence-specific manner. They can either cause endonucleolytic cleavage of the mRNA or act by blocking translation.
RNA-directed DNA Synthesis
The process of the transfer of the genetic information from DNA to RNA to protein has been called the central dogma. It was initially believed that genetic information was transferred only from DNA to RNA and thence translated into protein. However, there is evidence from the study of certain types of virus—retroviruses—that genetic information can occasionally flow in the reverse direction, from RNA to DNA (p. 210). This is referred to as RNA-directed DNA synthesis. It has been suggested that regions of DNA in normal cells serve as templates for the synthesis of RNA, which in turn then acts as a template for the synthesis of DNA that later becomes integrated into the nuclear DNA of other cells. Homology between human and retroviral oncogene sequences could reflect this process (p. 211), which could be an important therapeutic approach for the treatment of inherited disease in humans.
Mutations
A mutation is defined as a heritable alteration or change in the genetic material. Mutations drive evolution but can also be pathogenic. Mutations can arise through exposure to mutagenic agents (p. 27), but the vast majority occur spontaneously through errors in DNA replication and repair. Sequence variants with no obvious effect upon phenotype may be termed polymorphisms.
There are also rare examples of ‘back mutation’ in patients with recessive disorders. For example, reversion of inherited deleterious mutations has been demonstrated in phenotypically normal cells present in a small number of patients with Fanconi anemia.
Types of Mutation
Mutations can range from single base substitutions, through insertions and deletions of single or multiple bases to loss or gain of entire chromosomes (Table 2.2). Base substitutions are most prevalent (Table 2.3) and missense mutations account for nearly half of all mutations. A standard nomenclature to describe mutations (Table 2.4) has been agreed on (see http://www.hgvs.org/mutnomen/). Examples of chromosome abnormalities are discussed in Chapter 3.
| Type of Mutation | Percentage of Total |
|---|---|
| Missense or nonsense | 56 |
| Splicing | 10 |
| Regulatory | 2 |
| Small deletions, insertions or indels* | 24 |
| Gross deletions or insertions | 7 |
| Other (complex rearrangements or repeat variations) | <1 |
* Indels are mutations that involve both an insertion and a deletion of nucleotides.
Data from http://www.hgmd.org

Substitutions
A substitution is the replacement of a single nucleotide by another. These are the most common type of mutation. If the substitution involves replacement by the same type of nucleotide—a pyrimidine for a pyrimidine (C for T or vice versa) or a purine for a purine (A for G or vice versa); this is termed a transition. Substitution of a pyrimidine by a purine or vice versa is termed a transversion. Transitions occur more frequently than transversions. This may be due to the relatively high frequency of C to T transitions, which is likely to be the result of the nucleotides cytosine and guanine occurring together, or what are known as CpG dinucleotides (p represents the phosphate) frequently being methylated in genomic DNA with spontaneous deamination of methylcytosine converting them to thymine. CpG dinucleotides have been termed ‘hotspots’ for mutation.
Deletions
A deletion involves the loss of one or more nucleotides. If this occurs in coding sequences and involves one, two, or more nucleotides that are not a multiple of three, the reading frame will be disrupted. Larger deletions may result in partial or whole gene deletions and may arise through unequal crossover between repeat sequences (e.g., hereditary neuropathy with liability to pressure palsies; see p. 296).
Insertions
An insertion involves the addition of one or more nucleotides into a gene. Again, if an insertion occurs in a coding sequence and involves one, two, or more nucleotides that are not a multiple of three, it will disrupt the reading frame. Large insertions can also result from unequal crossover (e.g., hereditary sensory and motor neuropathy type 1a; see p. 296) or the insertion of transposable elements (p. 18).
In 1991, expansion of trinucleotide repeat sequences was identified as a mutational mechanism. A number of single-gene disorders have subsequently been shown to be associated with triplet repeat expansions (Table 2.5). These are described as dynamic mutations because the repeat sequence becomes more unstable as it expands in size. The mechanism by which amplification or expansion of the triplet repeat sequence occurs is not clear at present. Triplet repeats below a certain length for each disorder are faithfully and stably transmitted in mitosis and meiosis. Above a certain repeat number for each disorder, they are more likely to be transmitted unstably, usually with an increase or decrease in repeat number. A variety of possible explanations has been offered as to how the increase in triplet repeat number occurs. These include unequal crossover or unequal sister chromatid exchange (see Chapter 18) in non-replicating DNA, and slipped-strand mispairing and polymerase slippage in replicating DNA.
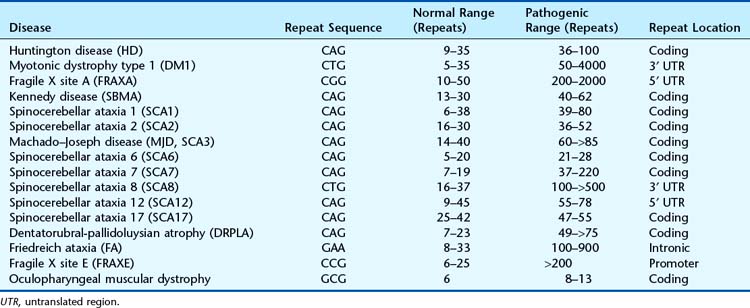
Triplet repeat expansions usually take place over a number of generations within a family, providing an explanation for some unusual aspects of patterns of inheritance as well as possibly being the basis of the previously unexplained phenomenon of anticipation (p. 120).
The exact mechanisms by which repeat expansions cause disease are not known. Unstable trinucleotide repeats may be within coding or non-coding regions of genes and hence vary in their pathogenic mechanisms. Expansion of the CAG repeat in the coding region of the HD gene and some SCA genes results in a protein with an elongated polyglutamine tract that forms toxic aggregates within certain cells. In fragile X the CGG repeat expansion in the 5′ untranslated region (UTR) results in methylation of promoter sequences and lack of expression of the FMR1 protein. In myotonic dystrophy (MD) it is thought that a gain-of-function RNA mechanism results from both the CTG expansion in the 3′ UTR of the DMPK (type 1 MD) and the CCTG expansion within intron 1 of the ZNF9 gene. The expanded transcripts bind splice regulatory proteins to form RNA-protein complexes that accumulate in the nuclei of cells. The disruption of these splice regulators causes abnormal developmental processing where embryonic isoforms of the resulting proteins are expressed in adult myotonic dystrophy tissues. The immature proteins then appear to cause the clinical features common to both diseases (p. 295).
The spectrum of repeat expansion mutations also includes a dodecamer repeat expansion upstream from the cystatin B gene that causes progressive myoclonus epilepsy (EPM1) and a pentanucleotide repeat expansion in intron 9 of the ATXN10 gene shown in families with spinocerebellar ataxia type 10. Spinocerebellar ataxia is an extremely heterogeneous disorder and, in addition to the dynamic mutations shown in Table 2.5, non-repeat expansion mutations have been reported in four additional genes.
Structural Effects of Mutations on the Protein
Non-Synonymous Mutations
Non-synonymous mutations can occur in one of three main ways.
Missense
A single base-pair substitution can result in coding for a different amino acid and the synthesis of an altered protein, a so-called missense mutation. If the mutation codes for an amino acid that is chemically dissimilar, for example has a different charge, the structure of the protein will be altered. This is termed a non-conservative substitution and can lead to a gross reduction, or even a complete loss, of biological activity. Single base-pair mutations can lead to qualitative rather than quantitative changes in the function of a protein, such that it retains its normal biological activity (e.g., enzyme activity) but differs in characteristics such as its mobility on electrophoresis, its pH optimum, or its stability so that it is more rapidly broken down in vivo. Many of the abnormal hemoglobins (p. 157) are the result of missense mutations.
Nonsense
A substitution that leads to the generation of one of the stop codons (see Table 2.1) will result in premature termination of translation of a peptide chain, or what is termed a nonsense mutation. In most cases the shortened chain is unlikely to retain normal biological activity, particularly if the termination codon results in the loss of an important functional domain(s) of the protein. mRNA transcripts containing premature termination codons are frequently degraded by a process known as nonsense-mediated decay. This is a form of RNA surveillance that is believed to have evolved to protect the body from the possible consequences of truncated proteins interfering with normal function.
Mutations in Non-Coding DNA
Splicing Mutations
Mutations of the highly conserved splice donor (GT) and splice acceptor (AG) sites (p. 19) usually result in aberrant splicing. This can result in the loss of coding sequence (exon skipping) or retention of intronic sequence, and may lead to frameshift mutations. Cryptic splice sites, which resemble the sequence of an authentic splice site, may be activated when the conserved splice sites are mutated. In addition, base substitutions resulting in apparent silent, missense and nonsense mutations can cause aberrant splicing through mutation of exon splicing enhancer sequences. These purine-rich sequences are required for the correct splicing of exons with weak splice-site consensus sequences.
Functional Effects of Mutations on the Protein
Mutations exert their phenotypic effect in one of two ways, through either loss or gain of function.
Loss-of-Function Mutations
Haplo-insufficiency
Loss-of-function mutations in the heterozygous state in which half normal levels of the gene product result in phenotypic effects are termed haplo-insufficiency mutations. The phenotypic manifestations sensitive to gene dosage are a result of mutations occurring in genes that code for either receptors, or more rarely enzymes, the functions of which are rate limiting; for example, familial hypercholesterolemia (p. 175) and acute intermittent porphyria (p. 179).
In a number of autosomal dominant disorders, the mutational basis of the functional abnormality is the result of haplo-insufficiency in which, not surprisingly, homozygous mutations result in more severe phenotypic effects; examples are angioneurotic edema and familial hypercholesterolemia (p. 175).
Gain-of-Function Mutations
Gain-of-function mutations, as the name suggests, result in either increased levels of gene expression or the development of a new function(s) of the gene product. Increased expression levels from activating point mutations or increased gene dosage are responsible for one type of Charcot-Marie-Tooth disease, hereditary motor, and sensory neuropathy type I (p. 296). The expanded triplet repeat mutations in the Huntington gene cause qualitative changes in the gene product that result in its aggregation in the central nervous system leading to the classic clinical features of the disorder (p. 293).
Mutations that alter the timing or tissue specificity of the expression of a gene can also be considered to be gain-of-function mutations. Examples include the chromosomal rearrangements that result in the combination of sequences from two different genes seen with specific tumors (p. 212). The novel function of the resulting chimeric gene causes the neoplastic process.
Gain-of-function mutations are dominantly inherited and the rare instances of gain-of-function mutations occurring in the homozygous state are often associated with a much more severe phenotype, which is often a prenatally lethal disorder, for example homozygous achondroplasia (p. 93) or Waardenburg syndrome type I (p. 91).
Genotype-Phenotype Correlation
Many genetic disorders are well recognized as being very variable in severity, or in the particular features manifested by a person with the disorder (p. 112). Developments in molecular genetics increasingly allow identification of the mutational basis of the specific features that occur in a person with a particular inherited disease, or what is known as the phenotype. This has resulted in attempts to correlate the presence of a particular mutation, which is often called the genotype, with the specific features seen in a person with an inherited disorder, this being referred to as genotype-phenotype correlation. This can be important in the management of a patient. One example includes the association of mutations in the BRCA1 gene with the risk of developing ovarian cancer as well as breast cancer (p. 224). Particularly striking examples are mutations in the receptor tyrosine kinase gene RET which, depending on their location, can lead to four different syndromes that differ in the functional mechanism and clinical phenotype. Loss-of-function nonsense mutations lead to lack of migration of neural crest–derived cells to form the ganglia of the myenteric plexus of the large bowel, leading to Hirschsprung disease, whereas gain-of-function missense mutations result in familial medullary thyroid carcinoma or one of the two types of multiple endocrine neoplasia type 2 (p. 100). Mutations in the LMNA gene are associated with an even broader spectrum of disease (p. 112).
Mutations and Mutagenesis
Radiation
Dosimetry is the measurement of radiation. The dose of radiation is expressed in relation to the amount received by the gonads because it is the effects of radiation on germ cells rather than somatic cells that are important as far as transmission of mutations to future progeny is concerned. The gonad dose of radiation is often expressed as the amount received in 30 years. This period has been chosen because it corresponds roughly to the generation time in humans.
The various sources and average annual doses of the different types of natural and artificial ionizing radiation are listed in Table 2.6. Natural sources of radiation include cosmic rays, external radiation from radioactive materials in certain rocks, and internal radiation from radioactive materials in tissues. Artificial sources include diagnostic and therapeutic radiology, occupational exposure and fallout from nuclear explosions.
Table 2.6 Approximate Average Doses of Ionizing Radiation from Various Sources to the Gonads of the General Population
| Source of Radiation | Average Dose per Year (mSv) | Average Dose per 30 Years (mSv) |
|---|---|---|
| Natural | ||
| Cosmic radiation | 0.25 | 7.5 |
| External γ radiation* | 1.50 | 45.0 |
| Internal γ radiation | 0.30 | 9.0 |
| Artificial | ||
| Medical radiology | 0.30 | 9.0 |
| Radioactive fallout | 0.01 | 0.3 |
| Occupational and miscellaneous | 0.04 | 1.2 |
| Total | 2.40 | 72.0 |
* Including radon in dwelling.
Data from Clarke RH, Southwood TRE 1989 Risks from ionizing radiation. Nature 338:197–198
DNA Repair
The occurrence of mutations in DNA, if left unrepaired, would have serious consequences for both the individual and subsequent generations. The stability of DNA is dependent upon continuous DNA repair by a number of different mechanisms (Table 2.7). Some types of DNA damage can be repaired directly. Examples include the dealkylation of O6-alkyl guanine or the removal of thymine dimers by photoreactivation in bacteria. The majority of DNA repair mechanisms involve cleavage of the DNA strand by an endonuclease, removal of the damaged region by an exonuclease, insertion of new bases by the enzyme DNA polymerase, and sealing of the break by DNA ligase.

Nucleotide excision repair removes thymine dimers and large chemical adducts. It is a complex process involving more than 30 proteins that remove fragments of approximately 30 nucleotides. Mutations in at least eight of the genes encoding these proteins can cause xeroderma pigmentosum (p. 289), characterized by extreme sensitivity to ultraviolet light and a high frequency of skin cancer. A different set of repair enzymes is used to excise single abnormal bases (base excision repair), with mutations in the gene encoding the DNA glycosylase MYH having recently been shown to cause an autosomal recessive form of colorectal cancer (p. 223).
Naturally occurring reactive oxygen species and ionizing radiation induce breakage of DNA strands. Double-strand breaks result in chromosome breaks that can be lethal if not repaired. Post-replication repair is required to correct double-strand breaks and usually involves homologous recombination with a sister DNA molecule. Human genes involved in this pathway include NBS, BLM, and BRCA1/2, mutated in Nijmegen breakage syndrome, Bloom syndrome (p. 288), and hereditary breast cancer (p. 224), respectively. Alternatively, the broken ends may be rejoined by non-homologous end-joining, which is an error-prone pathway.
Mismatch repair (MMR) corrects mismatched bases introduced during DNA replication. Cells defective in MMR have very high mutation rates (up to 1000 times higher than normal). Mutations in at least six different MMR genes cause hereditary non-polyposis colorectal cancer (hereditary non-polyposis colorectal cancer; see p. 222).
Although DNA repair pathways have evolved to correct DNA damage and hence protect the cell from the deleterious consequences of mutations, some mutations arise from the cell’s attempts to tolerate damage. One example is translesion DNA synthesis, in which the DNA replication machinery bypasses sites of DNA damage, allowing normal DNA replication and gene expression to proceed downstream. Human disease may also be caused by defective cellular responses to DNA damage. Cells have complex signaling pathways that allow cell-cycle arrest to provide increased time for DNA repair. If the DNA damage is irreparable, the cell may initiate programmed cell death (apoptosis). The ATM protein is involved in sensing DNA damage and has been described as the ‘guardian of the genome’. Mutations in the ATM gene cause ataxia telangiectasia (see p. 204), characterized by hypersensitivity to radiation and a high risk of cancer.
Alberts B, Johnson A, Lewis J, et al. Molecular biology of the cell, 5th ed. London: Garland; 2007.
Dawkins R. The selfish gene, 3rd ed. Oxford: Oxford University Press; 1989.
An interesting, controversial concept.
Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806-811.
Landmark paper describing the discovery of RNAi.
Lewin B 2011 Genes X, 10th ed. Oxford: Oxford University Press.
Mettler Mettler FA Upton AC 2008 medical effects of ionising radiation, 3rd ed. Philadelphia: Saunders.
Good overview of all aspects of the medical consequences of ionizing radiation.
Schull WJ, Neel JV. Radiation and the sex ratio in man. Sex ratio among children of survivors of atomic bombings suggests induced sex-linked lethal mutations. Science. 1958;228:434-438.
The original report of possible evidence of the effects of atomic radiation.
Strachan T, Read AP. Human molecular genetics, 4th ed. London: Garland Science; 2011.
Turner JE. Atoms, radiation and radiation protection. Chichester, UK: John Wiley; 1995.
Basis of the physics of radiation, applications, and harmful effects.
Watson JD, Crick FHC. Molecular structure of nucleic acids—a structure for deoxyribose nucleic acid. Nature. 1953;171:737-738.
Elements
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 2 The Cellular and Molecular Basis of Inheritance
The hereditary material is present in the nucleus of the cell, whereas protein synthesis takes place in the cytoplasm. What is the chain of events that leads from the gene to the final product?
The Cell
Within each cell of the body, visible with the light microscope, is the cytoplasm and a darkly staining body, the nucleus, the latter containing the hereditary material in the form of chromosomes (Figure 2.1). The phospholipid bilayer of the plasma membrane protects the interior of the cell but remains selectively permeable and has integral proteins involved in recognition and signaling between cells. The nucleus has a darkly staining area, the nucleolus. The nucleus is surrounded by a membrane, the nuclear envelope, which separates it from the cytoplasm but still allows communication through nuclear pores.
The cytoplasm contains the cytosol, which is semifluid in consistency, containing both soluble elements and cytoskeletal structural elements. In addition, in the cytoplasm there is a complex arrangement of very fine, highly convoluted, interconnecting channels, the endoplasmic reticulum. The endoplasmic reticulum, in association with the ribosomes, is involved in the biosynthesis of proteins and lipids. Also situated within the cytoplasm are other even more minute cellular organelles that can be visualized only with an electron microscope. These include the Golgi apparatus, which is responsible for the secretion of cellular products, the mitochondria, which are involved in energy production through the oxidative phosphorylation metabolic pathways, and the peroxisomes (p. 180) and lysosomes, both of which are involved in the degradation and disposal of cellular waste material and toxic molecules.
DNA: The Hereditary Material
Structure
For genes to be composed of DNA, it is necessary that the latter should have a structure sufficiently versatile to account for the great variety of different genes and yet, at the same time, be able to reproduce itself in such a manner that an identical replica is formed at each cell division. In 1953, Watson and Crick, based on x-ray diffraction studies by themselves and others, proposed a structure for the DNA molecule that fulfilled all the essential requirements. They suggested that the DNA molecule is composed of two chains of nucleotides arranged in a double helix. The backbone of each chain is formed by phosphodiester bonds between the 3′ and 5′ carbons of adjacent sugars, the two chains being held together by hydrogen bonds between the nitrogenous bases, which point in toward the center of the helix. Each DNA chain has a polarity determined by the orientation of the sugar–phosphate backbone. The chain end terminated by the 5′ carbon atom of the sugar molecule is referred to as the 5′ end, and the end terminated by the 3′ carbon atom is called the 3′ end. In the DNA duplex, the 5′ end of one strand is opposite the 3′ end of the other, that is, they have opposite orientations and are said to be antiparallel.
The arrangement of the bases in the DNA molecule is not random. A purine in one chain always pairs with a pyrimidine in the other chain, with specific pairing of the base pairs: guanine in one chain always pairs with cytosine in the other chain, and adenine always pairs with thymine, so that this base pairing forms complementary strands (Figure 2.2). For their work Watson and Crick, along with Maurice Wilkins, were awarded the Nobel Prize for Medicine or Physiology in 1962 (p. 10).
Replication
DNA replication, through the action of the enzyme DNA polymerase, takes place at multiple points known as origins of replication, forming bifurcated Y-shaped structures known as replication forks. The synthesis of both complementary antiparallel DNA strands occurs in the 5′ to 3′ direction. One strand, known as the leading strand, is synthesized as a continuous process. The other strand, known as the lagging strand, is synthesized in pieces called Okazaki fragments, which are then joined together as a continuous strand by the enzyme DNA ligase (Figure 2.3A).
DNA replication progresses in both directions from these points of origin, forming bubble-shaped structures, or replication bubbles (Figure 2.3B). Neighboring replication origins are approximately 50 to 300 kilobases (kb) apart and occur in clusters or replication units of 20 to 80 origins of replication. DNA replication in individual replication units takes place at different times in the S phase of the cell cycle (p. 39), adjacent replication units fusing until all the DNA is copied, forming two complete identical daughter molecules.
Chromosome Structure
The packaging of DNA into chromosomes involves several orders of DNA coiling and folding. In addition to the primary coiling of the DNA double helix, there is secondary coiling around spherical histone ‘beads’, forming what are called nucleosomes. There is a tertiary coiling of the nucleosomes to form the chromatin fibers that form long loops on a scaffold of non-histone acidic proteins, which are further wound in a tight coil to make up the chromosome as visualized under the light microscope (Figure 2.4), the whole structure making up the so-called solenoid model of chromosome structure.
Types of DNA Sequence
DNA, if denatured, will reassociate as a duplex at a rate that is dependent on the proportion of unique and repeat sequences present, the latter occurring more rapidly. Analysis of the results of the kinetics of the reassociation of human DNA have shown that approximately 60% to 70% of the human genome consists of single- or low-copy number DNA sequences. The remainder of the genome, 30% to 40%, consists of either moderately or highly repetitive DNA sequences that are not transcribed. This latter portion consists of mainly satellite DNA and interspersed DNA sequences (Box 2.1).
Nuclear Genes
It is estimated that there are between 25,000 and 30,000 genes in the nuclear genome. The distribution of these genes varies greatly between chromosomal regions. For example, heterochromatic and centromeric (p. 32) regions are mostly non-coding, with the highest gene density observed in subtelomeric regions. Chromosomes 19 and 22 are gene rich, whereas 4 and 18 are relatively gene poor. The size of genes also shows great variability: from small genes with single exons to genes with up to 79 exons (e.g., dystrophin, which occupies 2.5 Mb of the genome).
Multigene Families
Many genes have similar functions, having arisen through gene duplication events with subsequent evolutionary divergence making up what are known as multigene families. Some are found physically close together in clusters; for example, the α- and β-globin gene clusters on chromosomes 16 and 11 (Figure 2.5), whereas others are widely dispersed throughout the genome occurring on different chromosomes, such as the HOX homeobox gene family (p. 87).
Classic Gene Families
Examples of classic gene families include the numerous copies of genes coding for the various ribosomal RNAs, which are clustered as tandem arrays at the nucleolar organizing regions on the short arms of the five acrocentric chromosomes (p. 32), and the different transfer RNA (p. 20) gene families, which are dispersed in numerous clusters throughout the human genome.
Gene Superfamilies
Examples of gene superfamilies include the HLA (human leukocyte antigen) genes on chromosome 6 (p. 200) and the T-cell receptor genes, which have structural homology with the immunoglobulin (Ig) genes (p. 200). It is thought that these are almost certainly derived from duplication of a precursor gene, with subsequent evolutionary divergence forming the Ig superfamily.
Gene Structure
The original concept of a gene as a continuous sequence of DNA coding for a protein was turned on its head in the early 1980s by detailed analysis of the structure of the human β-globin gene. It was revealed that the gene was much longer than necessary to code for the β-globin protein, containing non-coding intervening sequences, or introns, that separate the coding sequences or exons (Figure 2.6). Most human genes contain introns, but the number and size of both introns and exons is extremely variable. Individual introns can be far larger than the coding sequences and some have been found to contain coding sequences for other genes (i.e., genes occurring within genes). Genes in humans do not usually overlap, being separated from each other by an average of 30 kb, although some of the genes in the HLA complex (p. 200) have been shown to be overlapping.
Extragenic DNA
Tandemly Repeated DNA Sequences
Minisatellite DNA
Telomeric DNA
The terminal portion of the telomeres of the chromosomes (p. 32) contains 10 to 15 kb of tandem repeats of a 6-base pair (bp) DNA sequence known as telomeric DNA. The telomeric repeat sequences are necessary for chromosomal integrity in replication and are added to the chromosome by an enzyme known as telomerase (p. 32).
Hypervariable minisatellite DNA
Hypervariable minisatellite DNA is made up of highly polymorphic DNA sequences consisting of short tandem repeats of a common core sequence. The highly variable number of repeat units in different hypervariable minisatellites forms the basis of the DNA fingerprinting technique developed by Professor Sir Alec Jeffreys in 1984 (p. 69).
Microsatellite DNA
Microsatellite DNA consists of tandem single, di-, tri-, and tetra-nucleotide repeat base-pair sequences located throughout the genome. Microsatellite repeats rarely occur within coding sequences but trinucleotide repeats in or near genes are associated with certain inherited disorders (p. 59).
This variation in repeat number is thought to arise by incorrect pairing of the tandem repeats of the two complementary DNA strands during DNA replication, or what is known as slipped strand mispairing. Duplications or deletions of longer sequences of tandemly repeated DNA are thought to arise through unequal crossover of non-allelic DNA sequences on chromatids of homologous chromosomes or sister chromatids (p. 32).
Nowadays DNA microsatellites are used for forensic and paternity tests (p. 69). They can also be helpful for gene tracking in families with a genetic disorder but no identified mutation (p. 70).
Highly Repeated Interspersed Repetitive DNA Sequences
Long Interspersed Nuclear Elements
The function of these interspersed repeat sequences is not clear. Members of the Alu repeat family are flanked by short direct repeat sequences and therefore resemble unstable DNA sequences called transposable elements or transposons. Transposons, originally identified in maize by Barbara McClintock (p. 10), move spontaneously throughout the genome from one chromosome location to another and appear to be ubiquitous in the plant and animal kingdoms. It is postulated that Alu repeats could promote unequal recombination, which could lead to pathogenic mutations (p. 22) or provide selective advantage in evolution by gene duplication. Both Alu and LINE-1 repeat elements have been implicated as a cause of mutation in inherited human disease.
Mitochondrial DNA
In addition to nuclear DNA, the several thousand mitochondria of each cell possess their own 16.6 kb circular double-stranded DNA, mitochondrial DNA (or mtDNA) (Figure 2.7). The mtDNA genome is very compact, containing little repetitive DNA, and codes for 37 genes, which include two types of ribosomal RNA, 22 transfer RNAs (p. 20) and 13 protein subunits for enzymes, such as cytochrome b and cytochrome oxidase, which are involved in the energy producing oxidative phosphorylation pathways. The genetic code of the mtDNA differs slightly from that of nuclear DNA.
[/not-level-membership-for-basic-science-category]
