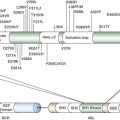
Molecular Biology of the Most Common Primary Central Nervous System Tumors
Astrocytic Tumors
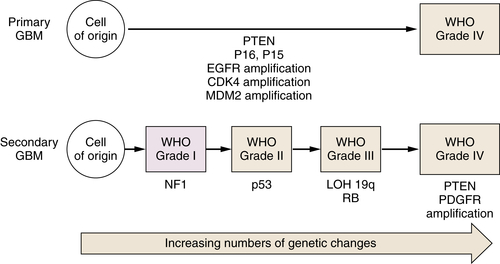
Table 40-2
Tumor Suppressor Genes and Proto-oncogenes Implicated in Brain Tumor Development
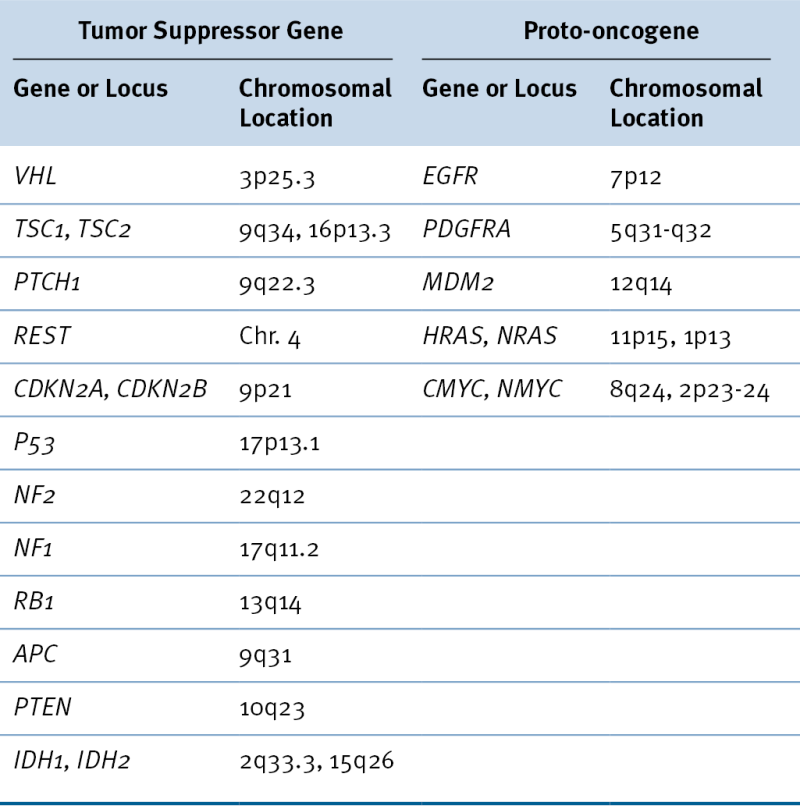
Table 40-3
Glioblastoma Multiforme Subtypes as Suggested by Verhaak et al.
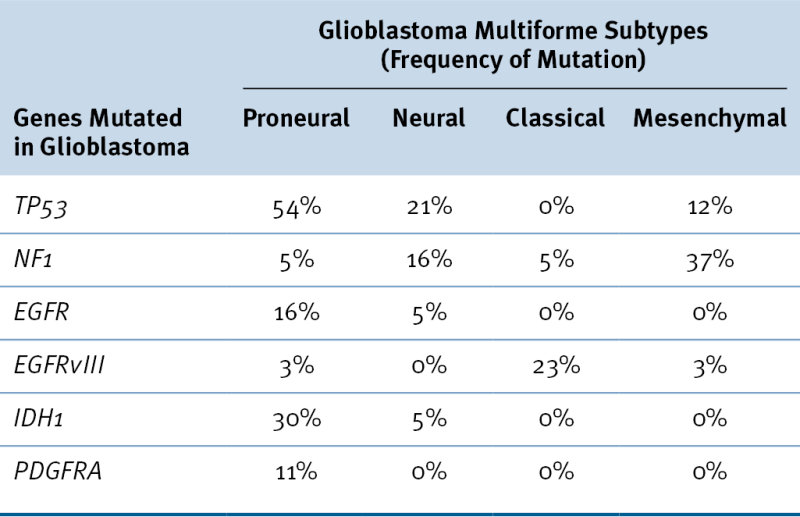
Data from Verhaak RG et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17:98-110.
Oligodendroglioma
Meningioma
Medulloblastoma
Molecular Pathophysiology of Primary Brain Tumors
Therapeutic Resistance of Primary Central Nervous System Tumors
Future Directions/Perspective
1. SEER Cancer Statistics Review, 1975-2009 . Bethesda, Md : National Cancer Institute ; 2012 : Based on November 2011 SEER data submission: http://seer.cancer.gov/csr/1975_2009_pops09/ .
2. Surveillance, Epidemiology, and End Results (SEER) Program. Surveillance Research Program, Surveillance Systems Branch, April 2012, based on November 2011 submission (www.seer.cancer.gov)
3. Characterizing mutational heterogeneity in a glioblastoma patient with double recurrence . PLoS One . 2012 ; 7 : e35262 .
4. Intracranial gliomas in neurofibromatosis type 1 . Am J Med Genet . 1999 ; 89 : 38 – 44 .
5. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis) . N Engl J Med . 1988 ; 318 : 684 – 688 .
6. Germ-line mutations of TP53 in Li-Fraumeni families: an extended study of 39 families . Cancer Res . 1997 ; 57 : 3245 – 3252 .
7. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma . Cancer Cell . 2005 ; 8 : 119 – 130 .
8. Mosaic analysis with double markers reveals tumor cell of origin in glioma . Cell . 2011 ; 146 : 209 – 221 .
9. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis . Cancer Cell . 2002 ; 1 : 269 – 277 .
10. Identification of human brain tumour initiating cells . Nature . 2004 ; 432 : 396 – 401 .
11. The 2007 WHO classification of tumours of the central nervous system . Acta Neuropathol . 2007 ; 114 : 97 – 109 .
12. A genetic model for colorectal tumorigenesis . Cell . 1990 ; 61 : 759 – 767 .
13. Prognostic impact of TP53 mutation status for adult patients with supratentorial World Health Organization Grade II astrocytoma or oligoastrocytoma: a long-term analysis . Cancer . 2004 ; 101 : 1028 – 1035 .
14. An integrated genomic analysis of human glioblastoma multiforme . Science . 2008 ; 321 : 1807 – 1812 .
15. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas . Cancer Res . 2000 ; 60 : 1383 – 1387 .
16. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance . J Neuropathol Exp Neurol . 2004 ; 63 : 700 – 707 .
17. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas . Lab Invest . 2004 ; 84 : 941 – 951 .
18. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1 . Cancer Cell . 2010 ; 17 : 98 – 110 .
19. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme . Proc Natl Acad Sci U S A . 2005 ; 102 : 5814 – 5819 .
20. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis . Cancer Cell . 2006 ; 9 : 157 – 173 .
21. Non-stem cell origin for oligodendroglioma . Cancer Cell . 2010 ; 18 : 669 – 682 .
22. Gene expression profiling with oligonucleotide microarrays distinguishes World Health Organization grade of oligodendrogliomas . Cancer Res . 2001 ; 61 : 1825 – 1829 .
23. Mutations in CIC and FUBP1 contribute to human oligodendroglioma . Science . 2011 ; 333 : 1453 – 1455 .
24. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p . Am J Pathol . 1994 ; 145 : 1175 – 1190 .
25. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas . J Natl Cancer Inst . 1998 ; 90 : 1473 – 1479 .
26. NF2 gene expression in sporadic meningiomas: relation to grades or histotypes real time-pCR study . Neuropathology . 2007 ; 27 : 36 – 42 .
27. Quantitative analysis of neurofibromatosis type 2 gene transcripts in meningiomas supports the concept of distinct molecular variants . Lab Invest . 1997 ; 77 : 601 – 606 .
28. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum . Genes Dev . 2000 ; 14 : 994 – 1004 .
29. Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy . J Neurosurg . 1994 ; 81 : 690 – 698 .
30. Incidence and trends in pediatric malignancies medulloblastoma/primitive neuroectodermal tumor: a SEER update. Surveillance Epidemiology and End Results . Med Pediatr Oncol . 2002 ; 39 : 190 – 194 .
31. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system . Cancer Res . 1998 ; 58 : 1798 – 1803 .
32. TrkC expression predicts good clinical outcome in primitive neuroectodermal brain tumors . J Clin Oncol . 2000 ; 18 : 1027 – 1035 .
33. The neuronal repressor REST/NRSF is an essential regulator in medulloblastoma cells . Nat Med . 2000 ; 6 : 826 – 831 .
34. Nestin expression in embryonic human neuroepithelium and in human neuroepithelial tumor cells . Lab Invest . 1992 ; 66 : 303 – 313 .
35. Molecular mechanisms of glioma cell migration and invasion . J Neurooncol . 2004 ; 70 : 217 – 228 .
36. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets . Neoplasia . 2005 ; 7 : 7 – 16 .
37. Biological mechanisms of glioma invasion and potential therapeutic targets . J Neurooncol . 2001 ; 53 : 129 – 147 .
38. Integrins: molecular determinants of glioma invasion . J Clin Neurosci . 2007 ; 14 : 1041 – 1048 .
39. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo . Cancer Res . 1994 ; 54 : 3656 – 3661 .
40. Gelatinase-A (MMP-2), gelatinase-B (MMP-9) and membrane type matrix metalloproteinase-1 (MT1-MMP) are involved in different aspects of the pathophysiology of malignant gliomas . Br J Cancer . 1999 ; 79 : 1828 – 1835 .
41. Tumor angiogensis: role in regulation of tumor growth . Symp Soc Dev Biol . 1974 ; 30 : 43 – 52 .
42. Vascular microenvironment in gliomas . J Neurooncol . 2000 ; 50 : 99 – 108 .
43. Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms . Int J Cancer . 1994 ; 59 : 520 – 529 .
44. Expression of vascular endothelial growth factor and its receptors in the anaplastic progression of astrocytoma, oligodendroglioma, and ependymoma . Am J Surg Pathol . 1998 ; 22 : 816 – 826 .
45. Bevacizumab for the treatment of recurrent glioblastoma . Clin Med Insights Oncol . 2011 ; 5 : 117 – 129 .
46. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis . J Clin Oncol . 2005 ; 23 : 2411 – 2422 .
47. Chemotherapy for brain tumors of astrocytic and oligodendroglial lineage: the past decade and where we are heading . Neuro Oncol . 1999 ; 1 : 69 – 80 .
48. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma . N Engl J Med . 2005 ; 352 : 987 – 996 .
49. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance . Curr Cancer Drug Targets . 2012 ; 12 : 197 – 209 .
50. Chemoresistance of glioblastoma cancer stem cells—much more complex than expected . Mol Cancer . 2011 ; 10 : 128 .