11 The basal ganglia
 Clinical cases for thought
Clinical cases for thoughtCase 11.2
Questions
• 11.2.1 What are the cardinal classic symptoms of idiopathic Parkinson’s disease? Which of these does this man exhibit?
• 11.2.2 Describe the neuronal circuits of the basal ganglia thought to be responsible for hypokinetic dyskinesias.
• 11.2.3 What treatment options are available for this patient?
Case 11.3
Questions
• 11.3.1 Describe the motion of a patient with the following movement disorders: chorea, athetosis, and ballismus.
• 11.3.2 Describe the neuronal circuits of the basal ganglia thought to be responsible for hyperkinetic dyskinesias.
• 11.3.3 Describe the differences between Huntington’s disease and Sydenham’s chorea.
Introduction
Under normal conditions the inhibition and excitation of the thalamus from the basal ganglia occurs at the appropriate time and in the appropriate amounts to support the activities of the cortex. However, in certain circumstances dysfunction of the basal ganglionic circuits can result in a number of conditions that affect movement and thought processes: idiopathic Parkinson’s disease (PD), Huntington’s disease (HD), Sydenham’s chorea (SC), Tourette’s syndrome (TS), ballismus, dystonias, obsessive-compulsive disorder (OCD), attention deficit hyperactivity disorder (ADHD), schizophrenia, depression, substance abuse disorders, and temporal lobe epilepsy (Marsden 1984; Javoy-Agid et al. 1984; Swerdlow & Koob 1987; Reiner et al. 1988; Modell et al. 1990; Swerdlow 1996; Castellanos 1997; Van Paesschen et al. 1997; Leckman et al. 1998).
In this chapter we will consider the neurocircuitry of the cortico-thalamo-thalamic-cortico system and the disorders of movement that can arise from dysfunctions of this system. Other non-motor dysfunctions are discussed in Chapter 16.
Anatomy of the basal ganglia
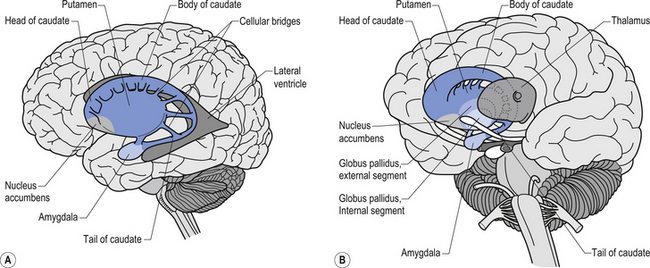
The caudate nucleus and the putamen are embryological homologues that have maintained similar morphological structure and function as they matured. For this reason these two nuclei, and the nuclei formed by the merger of these structures, the ventral striatum, are grouped into a single functional structure called the neostriatum (Kandel et al. 2000) (Fig. 11.1).
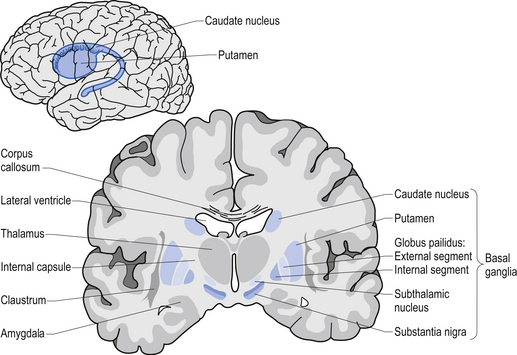
The neostriatum receives projection axons from virtually all areas of cortex and acts as the gatekeeper for all input to the basal ganglia. The caudate nucleus is a large C-shaped structure composed of a head, body, and tail that maintains a constant relationship with the lateral ventricle of the brain. Except for an area located anteriorly and ventrally where these two nuclei merge as the ventral striatum, the caudate nucleus and the putamen are separated by the fibre tracts of axons of the internal capsule. Most of the area composing the ventral striatum, which receives projections from areas of the limbic system, is taken up by the nucleus accumbens (Blumenfeld 2002) (Fig. 11.2).
The substantia nigra is a broad layer of pigmented grey substance separating the ventral portion of the mesencephalon from the tectum and extending from the upper surface of the pons to the hypothalamus. The substantia nigra can be separated into two areas which have different cell types. The most ventral area is referred to as the substantia nigra pars reticulata (SNr) and the more dorsal portion the substantia nigra pars compacta (SNc). The SNc contains a large population of dopaminergic neurons that contain a darkly pigmented grey substance, neuromelanin, which accumulates with age in dopaminergic neurons. The neuromelanin is thought to be composed of oxidised polymers of dopamine that accumulate in lysosomal storage granules in the neurons (Kandel et al. 2000).
The neostriatum is the input nucleus of the basal ganglia
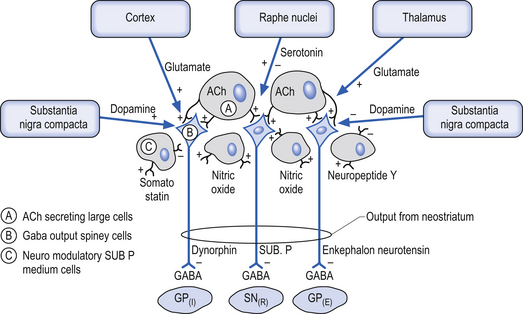
The neostriatum, which is composed of the caudate nucleus and the putamen, is the major input nucleus of the basal ganglia. The neostriatum has been estimated to contain some 110 million neurons per hemisphere (Alexander & DeLong 1992) compared to the 12 million neurons receiving cortical projections in each half of the basis pontis (Tomasch 1969). The striatum receives excitatory glutaminergic topographic projections from all areas of cortex and the intralaminar (centromedian and parafascicular) nuclei of the thalamus (Kunzle 1975, 1977; Selemon & Goldman-Rakic 1985). Dopaminergic input projections are also received from the SNc via the nigrostriatal pathway. The influence of this pathway on the neostriatal neurons involves complex interactions with various classes of dopamine receptors that result in excitation in some neurons and inhibition in others. Serotonergic axons from the raphe nuclei also project to the neostriatum.
The output neurons of the neostriatum produce spontaneous discharges in the default or resting state at about 20 spikes per second. The neurons of the globus pallidus, on the other hand, maintain a much higher spontaneous discharge rate approaching 200 spikes per second. This results in a spontaneous inhibition of the thalamus in the default or resting state. This inhibition can be modulated by the activity of the neostriatal inhibitory neurons (Kropotov 2009).
The small cell group of interneurons releases a variety of inhibitory neuroactive substances such as somatostatin, neuropeptide Y, and nitric oxide synthase (Fig. 11.3).
Direct and indirect pathways from the neostriatum to the GPi and SNr can modulate inhibition of the thalamus and pontomedullary reticular formation
There are two predominant pathways from the neostriatum to the output nuclei of the basal ganglia—the globus pallidus pars internus and the substantia nigra pars reticulata. Understanding the inhibition and excitation circuits involved in these two pathways will help one understand the spectrum of functional disorders ranging from hyperkinetic to hypokinetic, involving movement and thought processes caused by basal ganglia disorders.
The output neurons in the GPi and SNr are inhibitory in nature and release the neurotransmitter GABA (Fig. 11.4).
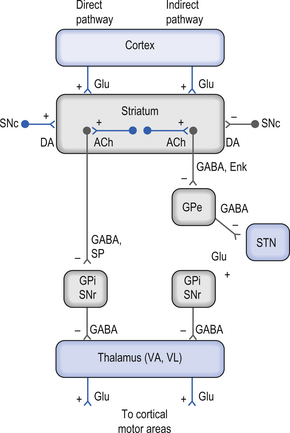
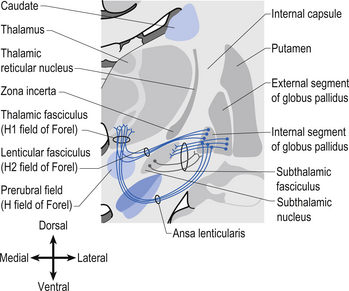
The neurons in GPi project axons via the anterior thalamic fasciculus to the ventral lateral and ventral anterior nuclei of the thalamus. These projections are mainly associated with motor control functions of the body below the head and neck. GPi neurons also project to the intralaminar nuclei (centromedian and parafascicular) and the mediodorsal nuclei of the thalamus. These projections are largely associated with limbic activities (Fig. 11.5). Output projections of the GPi reach the thalamic fasciculus via two different pathways. The first pathway, called the ansa lenticularis, loops ventrally and passes beneath the internal capsule before swinging dorsally to join the thalamic fasciculus and reach the thalamus. The second pathway, called the lenticular fasciculus, passes straight through the internal capsule to join the ansa lenticularis to form the thalamic fasciculus and enter the thalamus (Chusid 1982). The point at which the two pathways combine to form the thalamic fasciculus is sometimes referred to as the H fields of Forel. The H1 field of Forel refers to the thalamic fasciculus, the H2 field of Forel refers to the lenticular fasciculus, and the H or prerubral field of Forel refers to the area where the ansa lenticularis joins the thalamic fasciculus (Fig. 11.6). Finally, the GPi neurons also project to the complex reticular neurons in the pons and medulla known as the pontomedullary reticular formation (PMRF). These projections are involved in the modulation of the reticulospinal tracts (Afifi 1994).
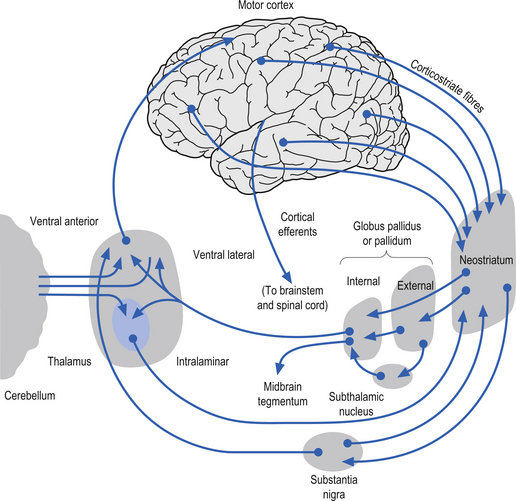
The neurons in the SNr also project to the ventral anterior and ventrolateral nuclei of the thalamus. These projections are associated with motor control of the head and neck. The SNr neurons also project to the superior colliculus where they modulate actions of the tectospinal pathways. Finally, the SNr neurons project to the PMRF, where they also modulate the output of the reticulospinal tract neurons (Fig. 11.5).
The neuron in the substantia nigra pars compacta release dopamine as their neuromodulators. These neurons project to the neostriatum where they have complex modulatory effects on the output neurons of the neostriatum. The net effect of the SNc release of dopamine in the neostriatum is an excitation of the output neurons of the direct pathway and an inhibition of the output neurons of the indirect pathway (Parent & Cicchetti 1998).
Functional modulatory outputs of the direct and indirect pathways may result in movement and cognitive dysfunctions
The activation pathways of the cortico-neostriatal-thalamo-cortical system are thought to operate through parallel segregated circuits that maintain their segregation throughout the neostriatal-thalamo-cortical projections. Several loops including a motor loop, a limbic loop, and a frontal cortical loop function to modulate motor, limbic, and frontal cortical activities, respectively (Fig. 11.7).
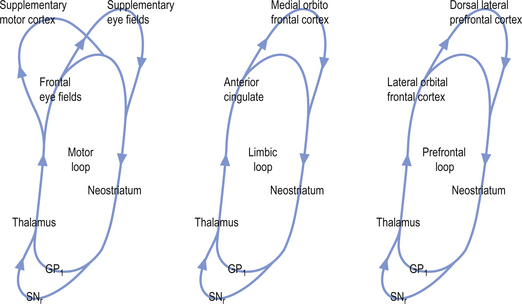
Cortical activation of the direct basal ganglionic pathway results in a net excitation of the thalamus via disinhibition of the basal ganglionic projections and subsequently excitation of the cortical areas that receive thalamic projections. Cortical activation of the indirect basal ganglionic pathway results in inhibition of the thalamus and subsequently inhibition of the cortical areas receiving thalamic projections (see Fig. 11.4). The large ACh-releasing neurons in the neostriatum tend to preferentially form excitatory synapses on the output neurons of the indirect pathway; thus, excitation of these neurons would result in an increased activation of the indirect pathway or an inhibition of movement and thought processes.
 Quick facts 11.1
Quick facts 11.1Summary of outputs from basal ganglionic structures
| Neostriatum (caudate, putamen) | All inhibitory |
| Globus pallidus pars internus | All inhibitory |
| Globus pallidus pars externus | All inhibitory |
| Subthalamic nucleus | All excitatory |
| Substantia nigra pars reticulata | All inhibitory |
| Substantia nigra pars compacta | Both excitatory and inhibitory |
 Quick facts 11.2
Quick facts 11.2Under normal conditions the inhibition and excitation of the thalamus from the basal ganglia occurs at the appropriate time and in the appropriate amounts to support the activities of the cortex. However, in certain circumstances dysfunction of the basal ganglionic circuits can result in a number of conditions that affect movement and thought processes. These include idiopathic Parkinson’s disease (PD), Huntington’s disease (HD), Sydenham’s chorea (SC), Tourette’s syndrome (TS), ballismus, dystonias, obsessive-compulsive disorder (OCD), attention deficit hyperactivity disorders (ADHD), schizophrenia, depression, substance abuse disorders, and temporal lobe epilepsy (Marsden 1984; Javoy-Agid et al. 1984; Swerdlow & Koob 1987; Reiner et al. 1988; Modell et al. 1990; Baxter et al. 1992; Swerdlow 1996; Castellanos 1997; Van Paesschen et al. 1997; Leckman et al. 1998).
Idiopathic Parkinson’s disease
Idiopathic Parkinson’s disease (PD) is associated with the degeneration of the dopaminergic neurons of the SNc which, as stated previously, have an excitatory effect on the direct pathway and an inhibitory effect on the indirect pathway. A loss of dopaminergic stimulation in the neostriatum would result in a net inhibition of movement through both direct and indirect pathways (Fig. 11.8). The onset of PD is gradual in nature, but slowly and progressively continues until eventual severe disability. The cardinal signs and symptoms include tremor at rest, bradykinesia (slowness of movement), muscular rigidity, akinesia (impairment in initiation and poverty of movement), and loss of postural reflexes (Wichmann & DeLong 2002). The clinical diagnosis of PD can only be tentative because the major symptoms described above are not specific for PD. Some degree of certainty of the diagnosis can be achieved if the patient responds favourably to levodopa, a precursor in the formation of dopamine synthesis.
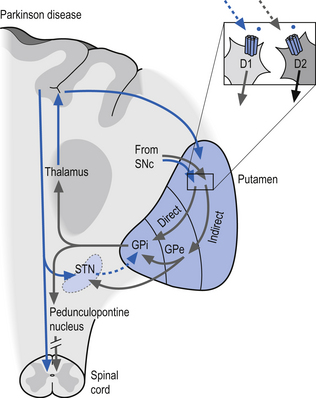
Figure 11.8 Pathological function of the dopaminergic neurons in the development of idiopathic Parkinson’s disease (PD).
 Quick facts 11.3
Quick facts 11.3The effects of excitation and inhibition on the direct and indirect basal ganglionic pathways
| Excitation of direct pathway | Excitation of thalamus | Movement, thought, limbic excitement |
| Excitation of indirect pathway | Inhibition of thalamus | Inhibition of movement, thought, limbic activity |
| Inhibition of direct pathway | Inhibition of thalamus | Inhibition of movement, thought, limbic activity |
| Inhibition of indirect pathway | Excitation of thalamus | Movement, thought, limbic excitement |
 Quick facts 11.4
Quick facts 11.4What do they mean?
• Dyskinesia—abnormal movements
• Bradykinesia—slowing of movements
• Hypokinesia—reduced amounts of movements
• Akinesia—absence of movement
• Rigidity—increased resistance to passive movement of a limb or joint
• Paratonia—active resistance against movement of limbs
• Dystonia—prolonged muscle spasms resulting in distorted positions or postures
• Athetosis—slow, twisting movements of the face, limbs, or trunk
• Chorea—dance-like movements that have a fluid, jerky, constant quality
• Ballismus—flinging, ballistic movements of the limbs
• Tremor—slow or fast rhythmic or semirhythmic oscillating movements
The pathological hallmark of PD is intracellular inclusions called Lewy bodies. These occur inside the dopamine-producing neurons in the substantia nigra pars compacta. These inclusions probably accumulate in neurons as breakdown products of dopamine and probably increase in concentration in neurons undergoing degeneration. During the past decade it has become clear that Lewy bodies are not limited to the substantia nigra in PD, but may occur in a widespread distribution in the cortex. Diffuse Lewy body disease is a pathological entity whose clinical correlates have not yet been defined. Patients commonly have cognitive decline and Parkinsonian features, and either one may dominate the picture (Korczyn 2000).
The number of dopamine (DA)-producing neurons progressively diminishes in PD over time. It is important to note that only DA neurons in the substantia nigra whose axons are destined to go to the putamen (less so to the caudate) in the nigrostriatal tract are affected. Chemical analysis shows progressive loss of DA in the striatum, with the clinical symptoms first becoming apparent when DA content in the striatum is reduced by about 70%. This process may take as long as 20 years before symptoms become apparent (Hornykiewicz 1988; Scherman et al. 1989).
The aetiology of PD is uncertain but is most probably multifactorial in nature with both genetic and environmental factors contributing to the development of the disease. One theory that has gained some popularity recently is that excessive concentrations of excitatory amino acids, particularly glutamate, may be involved in causing irreversible neuronal damage (Sonsalla et al. 1989). This is particularly relevant for PD because of the massive cortical, glutaminergic innervation received by the corpus striatum. The neurotoxicity is thought to be produced by over- or sustained activation of N-methyl- D-aspartate (NMDA) receptors on the neuron membranes. One environmental hypothesis suggests that a selective increase in lipid peroxidation in the substantia nigra neurons may occur in PD. This process may lead to excessive production of free radicals, which may in turn result in cellular damage and death (Ben Shachar et al. 1991). A particularly relevant fact concerning this theory is that DA degradation may involve the sequestration of iron in free radical formation in the process of lipid peroxidation. Both the substantia nigra and globus pallidum are rich in iron, and the iron concentration increases with age, particularly in PD.
The gold standard treatment of PD is replacement of DA using levodopa. Levodopa is absorbed from the gastrointestinal tract and converted to DA in both the brain and the periphery by the enzyme 1-amino acid decarboxylase (1-AAD) (Clough 1991). The peripheral conversion of levodopa to DA can be inhibited by the actions of benserazide and carbidopa. Most patients today are treated by a combination of levodopa and benserazide or carbidopa. The aim of using this combination is to prevent the peripheral conversion of levodopa to DA, because DA may act in the periphery to produce undesirable side effects such as orthostatic hypotension and nausea (Cederbaum et al. 1991).
Surgical interventions of PD include ablative and transplanting approaches. Targets for functional stereotactic neurosurgical lesions, which reduce tremor, are the ventrolateral thalamus and the posteroventral pallidum. There has been extensive interest in transplanting DA tissue removed from aborted fetal midbrains into the caudate or putamen in PD; however, the results remained confusing because of small cohort sizes and disease severity issues of the participants (Widner & Rehncrona 1993).
Depression is also rather common in PD. Because depression is potentially treatable, every patient with PD must be assessed for possible depressive symptomatology (Cummings 1992). Several tests are available for diagnosing depression. These include neuropsychological evaluations, self-reports, and projection tests. However, while all these tests have important roles in research, none is superior to the clinical assessment by a competent clinician. The clinical evaluation of the affective state of PD patients may be difficult because the motionless face, the slowness of movement, and the bradyphrenia may create an erroneous impression of depression. The distinction from depressive motor retardation is obviously very important.
 Quick facts 11.5
Quick facts 11.5Huntington’s disease
In Huntington’s disease (HD) the neurons in the neostriatum degenerate. The degeneration appears to be more pronounced in the output neostriatal neurons of the indirect pathway (Albin et al. 1992). This results in the disinhibition of the GPe, which in turn results in an overinhibition of the subthalamic nucleus. The functional overinhibition of the subthalamic nucleus results in a situation that resembles an ablative lesion to the subthalamic nucleus and results in a hyperkinetic movement disorder (Fig. 11.9). In the latter stages of HD neostriatal degeneration spreads to include the output neurons of both the direct and indirect pathways, resulting in hypokinetic Parkinson-like activities (Young et al. 1986).
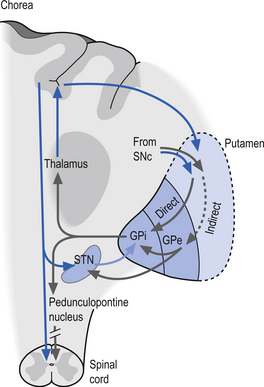
Figure 11.9 The pathology observed in Huntington’s disease (HD) in which the neurons in the neostriatum degenerate.
Although a juvenile form of HD does occur and the onset of HD can range from as young as 2 years to as old as 80 years, disease onset typically occurs in adults in their mid-thirties to mid-forties. The disease affects men and women in equal frequencies, ranging from 5 to 10 per 100 000 (Kandel et al. 2000). The disorder is characterised by insidious onset of both neurological and psychiatric symptoms. Initial symptoms include personality change and the gradual appearance of small involuntary movements; as the disease progresses, chorea becomes more obvious and incapacitating (Harper 1996). Over time, motor symptoms worsen such that walking, speaking, and eating becomes more difficult, and weight loss is common because of the extra energy required for movement and an increase in their basal metabolic rate. A large percentage of HD patients eventually succumb to aspiration pneumonia, resulting from the inability to coordinate pharyngeal muscles and vocal cords, which results in swallowing difficulties. It has a large genetic component.
The juvenile form of HD, which is also referred to as the Westphal variant form of HD, occurs in about 10% of reported cases. The initial presentation is more Parkinsonian in nature with bradykinesia, rigidity, and tremor rather than chorea as the prominent symptoms. Juvenile-onset HD is usually the result from paternal transmission and in individuals who develop symptoms before age 10; more than 90% have an affected father (Folstein 1989). There is a unique tendency for juvenile HD to have a younger age of onset in successive generations, which is referred to as anticipation. Anticipation in juvenile HD is especially pronounced in cases of paternal transmission.
Within the striatum, HD differentially affects subpopulations of neurons, with projection neurons rather than interneurons preferentially being lost (DiFiglia 1990). Consistent with the finding of loss of projection neurons is the fact that GABA levels are markedly reduced in the caudate–putamen of HD patients. Of the two populations of striatal projection neurons, the neurons of the indirect pathway are affected first; thus, the indirect pathway is predominantly disrupted. With interruption of the indirect pathway, the current models of basal ganglionic circuitry predict an overall increase in movement, manifested as chorea and ballism. The functional result of degeneration of both the direct and indirect pathways is a rigid bradykinetic state, which occurs in the later stages of adult HD. In the case of juvenile HD where the symptoms resemble Parkinson’s disease early in the presentation, degeneration of both direct and indirect pathway striatal neurons occurs from the onset (Albin et al. 1989).
The mode by which neurons die in HD is still unclear although the process of apoptosis or preprogrammed cell death may be the final common pathway through which neurons are terminated. Prior to the discovery of the HD gene, the leading hypotheses concerning the pathogenesis of HD implicated either excitotoxicity or metabolic dysfunction. The protein huntingtin, which is coded for by the huntingtin gene, has no clear relationship to excitatory amino acid neurotransmission, or to mitochondrial energetics. The normal function of huntingtin is not completely known, although it has been implicated in membrane recycling (DiFiglia et al. 1995). Thus, the influence of the huntingtin protein remains unclear as it relates to these hypotheses.
Excitotoxicity is the process in which neuronal cells die as a result of excessive excitatory amino acid neurotransmission. This process has been well documented with respect to overstimulation or excessive stimulation of glutaminergic NMDA receptors. This overstimulation can result in excessive amounts of Ca++ ions entering neurons and triggering preprogrammed genetic termination pathways in the neuron. Glutamate has been postulated to trigger neuron death in a number of neurological disorders, including hypoxia-ischaemia, head trauma, epilepsy, schizophrenia, and neurodegenerative disorders such as Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis (Fagg et al. 1986; Choi & Rothman 1990; Kornbuber & Wiltfang 1998).
Mitochondrial dysfunction has also been implicated as a pathologic mechanism in HD, potentially rendering cells vulnerable to normal ambient levels of extracellular glutamate (Albin & Greenamyre 1992). Positron emission tomography and MRI spectroscopy studies have demonstrated abnormalities in glucose metabolism in HD patients (Mazziotta et al. 1987). The impact or contribution of mitochondrial dysfunction in the development of HD remains under investigation; however, at least one study has demonstrated that administration of coenzyme Q10, an essential cofactor of the mitochondrial electron transport chain, lowers elevated cortical lactate levels in HD patients back to levels seen in normal controls (Koroshetz et al. 1997). This suggests that mitochondrial energetic processes are in some way contributing to the development of HD.
The discovery of the huntingtin gene was unusual in that the usual genetic mutations observed in human diseases include point mutations, deletions, duplications, or missense mutations. The mutation in the huntingtin gene (IT-15 gene), however, resides in an unstable region of the gene, where mutation can result in an expansion of a normally appearing trinucleotide repeat motif present in the alleles of HD patients. CAG is the codon for glutamine, and the trinucleotide repeat of this motif gives rise to a polyglutamine moiety within the huntingtin protein. Normal huntingtin alleles contain from 6 to 35 CAG repeats, giving rise to 6 to 35 glutamines in the mature protein. Patients with Huntington’s disease invariably have alleles with greater than 35 repeats. While repeats greater than 40 invariably give rise to Huntington’s disease, there is a ‘grey area’, between 35 and 39 repeats, where there is some uncertainty whether the disease will develop (Cha & Young 2000).
There are currently no effective therapies for preventing the onset or slowing the progression of HD. Current therapies are symptomatic, and include the use of neuroleptics to decrease chorea, and the use of psychotropic medications to address depression, obsessive-compulsive symptoms, or psychosis. In addition, speech therapy and physiotherapy are useful in addressing the swallowing and walking difficulties that many HD patients experience (Ranen et al. 1993).
Ballismus
Ballismus or ballism includes a group of conditions characterised by flinging, large-amplitude, rotary movements, usually involving the proximal limb muscles. The most common form of this condition occurs unilaterally and is referred to as hemiballism. The cause is classically a basal ganglionic lesion in the contralateral subthalamic nucleus, but contralateral lesions in the neostriatum can also result in ballismic dyskinesia (Provenzale & Schwarzschild 1994) (Fig. 11.10).
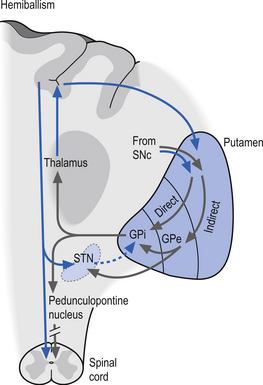
Tourette’s syndrome (TS)
The diagnosis of TS is based solely on the patient history presented. The DSM-IV diagnostic criteria for TS is the frequent occurrence of multiple motor tics and one or more types of vocal tic present and occurring over a continuous interval for most of 1 year. Usually, the onset of symptoms must have occurred early in life, before the age of 21, to be considered as TS (American Psychiatric Association 1994).
Tics are sudden, rapid, recurrent, nonrhythmic, stereotyped movements or vocalisations. Simple tics are brief circumscribed movements or sounds that resemble ‘chunks’ of movement or sounds rather than meaningful or recognisable actions. These may include facial grimaces, mouth movements, head jerking, and shoulder, arm, and leg jerks. Complex tics are more sustained and elaborate movements or more recognisable words or sounds that can give the perception of being intentionally produced (Swerdlow & Leckman 2002). In a small percentage (10%) of those with TS vocal tics can involve vulgar or obscene expletives. This form of expression is referred to as coprolalia. Tics can be voluntarily suppressed but, like obsessive-compulsive tendencies, the suppression builds up anxiety and results in a more forceful expression when the tic is eventually expressed. Many children express tics as a normal activity as they pass through various phases of development. These normal or developmental tics have usually completely disappeared by 18 years of age (Shapiro et al. 1978).
 Quick facts 11.6
Quick facts 11.6The usual presentation of TS typically begins between the ages of 3 and 8 years old, with periods of worsening and remission of the tics throughout childhood. The period of 8–12 years of age seems to be the period of greatest severity in most children, with a steady decline to the age of 18 years where as many as 50% of the children will present as tic free (Leckman et al. 1998). For those who maintain their tics into adulthood a more predictable pattern usually emerges with the frequency and intensity of the tics increasing during periods of increased stress or emotional excitement and generally over time.
1. Intrinsic neostriatal neuron abnormalities such as increased packing density of the neurons in the neostriatum (Balthasar 1957);
2. A decrease in the activity of the output neurons of the direct pathway (Haber & Wolfer 1992);
3. Increased dopaminergic innervation of the neostriatum, with increased density of dopamine transporter sites (Singer et al. 1992); and
4. Reduction in the glutaminergic output of the subthalamic nucleus (Anderson et al. 1992).
Treatment of TS is aimed at developing flexible, integrated biosocial and biopsychological strategies to allow the build-up of anxiety to be dissipated in a controlled fashion, and control the excitement in emotional situations.
Dystonia
The characteristic features of dystonia are the distorted postures and movements caused by spasmodic muscular activity in people with this condition. If the spasmodic muscular activity is maintained for long periods it is referred to as dystonic posturing. If the spasmodic muscular activity results in slowly changing repetitive activity then it is referred to as dystonic movements (Rothwell et al. 1983).
The hallmarks of dystonia include:
1. Excessive contraction of antagonistic muscles during voluntary movement;
2. Overflow of contraction to remote muscles not usually involved in the attempted movement; and
3. Development of spontaneous spasms on contraction of muscles.
The excessive involvement of antagonistic muscles and overflow of contraction suggest that the normal spinal inhibitory feedback mechanisms are dysfunctional in this condition. However, this does not appear to be the case. The classic spinal disynaptic pathway involving 1a reciprocal inhibition of antagonist muscles remains intact in dystonic patients (Nakashima et al. 1989). However, the presynaptic or supraspinal inhibition of these reflexes is dysfunctional. The cause of the dysfunctional descending inhibition is thought to be due to altered function of the basal ganglionic circuits that relay back to the cortex via the thalamus.
 Clinical case answers
Clinical case answersCase 11.1
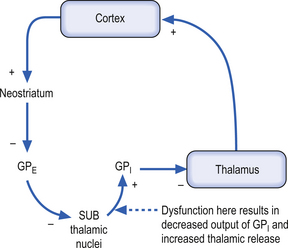
Case 11.2
11.2.1
Idiopathic Parkinson’s disease (PD) is associated with the degeneration of the dopaminergic neurons of the substantia nigra pars compacta (SNc) which, as stated previously, have an excitatory effect on the direct pathway and an inhibitory effect on the indirect pathway. A loss of dopaminergic stimulation in the neostriatum would result in a net inhibition of movement through both direct and indirect pathways. The onset of PD is gradual in nature, but slowly and progressively continues until eventual severe disability. The cardinal signs and symptoms include tremor at rest, bradykinesia (slowness of movement), muscular rigidity, akinesia (impairment in initiation and poverty of movement), and loss of postural reflexes. This gentleman is exhibiting tremor at rest, and muscular rigidity.
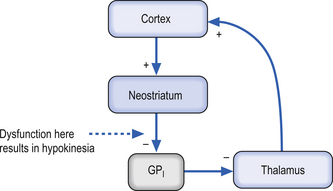
Case 11.3
11.3.3
Huntington’s disease is characterised by insidious onset of both neurological and psychiatric symptoms. Initial symptoms include personality change and the gradual appearance of small involuntary movements; as the disease progresses, chorea becomes more obvious and incapacitating (Harper 1996). Over time, motor symptoms worsen such that walking, speaking, and eating becomes more difficult, and weight loss is common because of the extra energy required for movement and an increase in their basal metabolic rate. A large percentage of HD patients eventually succumb to aspiration pneumonia, resulting from the inability to coordinate pharyngeal muscles and vocal cords which results in swallowing difficulties. It has a large genetic component.
References
Albin R.L., Greenamyre J.T. Alternative excitotoxic hypotheses. Neurology. 1992;42:733-738.
Clough C.G. Parkinson’s disease: management. Lancet. 1991;337:1324-1327.
Cummings J.L. Depression and Parkinson’s disease: review. Am. J. Psychiatry. 1992;149:443-454.
Kropotov J. Quantitative and Event Related Potentials in Neurotherapy. London: Elsevier, 2009.
Marsden C.D. Motor disorders in basal ganglia disease. Hum. Neurobiol.. 1984;2:245-255.
Provenzale J.M., Schwarzschild M.A. Hemiballism. Am. J. Neuroradiol.. 1994;15(7):1377-1382.

