8 Tetralogy of Fallot with Pulmonary Atresia
I. CASE
A. Fetal echocardiography findings
1. The fetal echo reveals situs solitus of the atria, levocardia, right aortic arch, heart rate 149 bpm.
2. Cardiac axis, position, and size are normal (cardiothoracic ratio = 0.29).
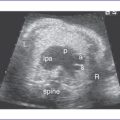
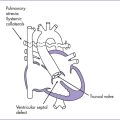
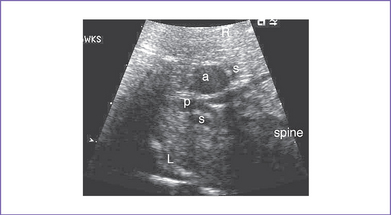
3. The four-chamber view is normal, with an axis that is approximately 90 degrees (Fig. 8-1). However, more superior scanning shows it to be abnormal, with a large outlet perimembranous subaortic ventricular septal defect (VSD) and a large overriding aorta.
4. The outflow assessment reveals normally related great arteries with significant asymmetry (aorta–to–pulmonary artery diameter ratio = 5.5:1.5).
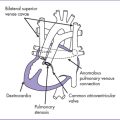
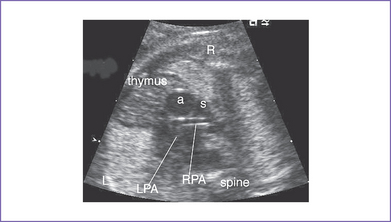
5. There is a blind outlet of the right ventricle (RV) connected to a small main pulmonary artery with hypoplastic and confluent branches (Fig. 8-2).
6. The foramen ovale is patent, with unrestricted right-to-left shunt.
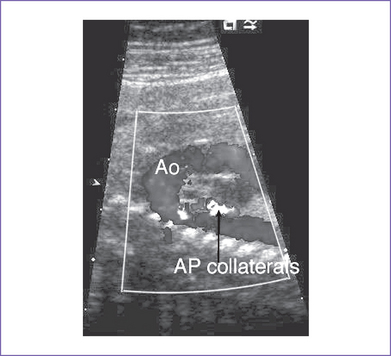
7. There is reversed ductal flow (aorta to pulmonary), and multiple small aortopulmonary collaterals come from the thoracic aorta.
8. The pulmonary venous flow pattern is normal.
9. Tei index (myocardial performance index).
a. Left ventricular (LV) Tei index is normal.
b. RV Tei index could not be calculated because of the atretic pulmonary valve.
D. Fetal management and counseling
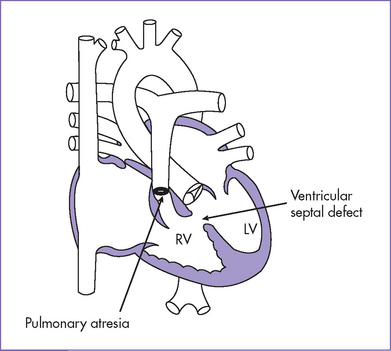
1. Diagnosis of tetralogy of Fallot (TOF) with pulmonary atresia (or pulmonary atresia and VSD) should prompt referral (Fig. 8-3).
a. Thorough anatomic examination by ultrasound.
2. Follow-up includes serial antenatal studies at 4- to 6-week intervals.
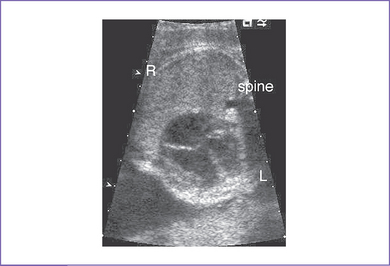
a. The branch pulmonary arteries may be hypoplastic or discontinuous. This will affect the surgical management of the baby postnatally (Fig. 8-4).
b. Absence of antegrade flow should be documented across the main pulmonary artery. In pulmonary atresia, there is no patent connection between the right ventricle and the pulmonary arteries.
c. Reversal of ductal flow suggests that pulmonary artery blood supply is from the arterial duct (pulmonary atresia with duct-dependant circulation).
d. Descending aorta to pulmonary artery collaterals should be identified. The presence of these collaterals can increase the likelihood of 22q11.2 deletion, based on postnatal experience.
e. Development of hydrops fetalis is uncommon in fetal TOF and pulmonary atresia, unless there is chromosomal abnormality not related to structural heart defect or inadequate cardiac output.
F. Neonatal management
a. Prostaglandin E1 (PGE1) infusion should be started to keep the ductus open to increase pulmonary blood flow and raise arterial oxygen saturation.
b. If there is no ductus arteriosus, or if it is uncertain whether the branch pulmonary arteries themselves are stenotic or if the pulmonary arteries are discontinuous with uncertain source of pulmonary blood flow, the infant requires cardiac catheterization.
c. If pulmonary blood flow is via aortopulmonary collaterals and the infant has sufficient oxygen saturations, some centers do not perform cardiac catheterization in the neonatal period, but rather closer to the time of repair with unifocalization of the collaterals, perhaps at 2 to 3 months.
d. Administration of oxygen can increase oxygen saturation by decreasing pulmonary vascular resistance and increasing blood flow.
e. At times, volume and inotropic support are indicated to improve ventricular function and increase the pulmonary blood flow through raising the systemic vascular resistance.
b. Staged repair consists of the initial systemic pulmonary shunt to encourage growth of the central pulmonary artery before primary repair.
c. When the central pulmonary arteries are nonconfluent, with multiple collaterals supplying different segments of the lungs, unifocalization of these collaterals may be necessary at the time of complete repair or with staged reconstruction.
d. Occlusion of the systemic collateral arteries can be done by coil embolization preoperatively (transcutaneous during cardiac catheterization or intraoperatively while off cardiac bypass) and would be considered if there is an antegrade source of pulmonary blood flow through the native branch pulmonary arteries.
G. Follow-up
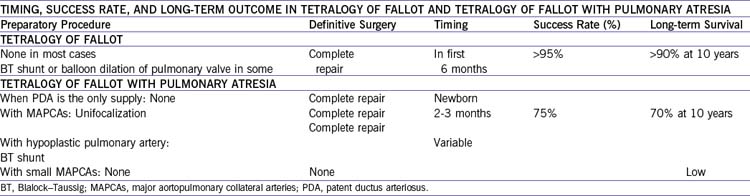
1. Usually the long-term prognosis in TOF with pulmonary atresia is worse than for the less severe spectrum of TOF.
2. Frequent follow-up is needed to assess the palliative surgery and to decide on the appropriate time to replace the conduit.
3. Repeated interventional catheterization procedures may be necessary to balloon dilate hypoplastic and stenotic branch pulmonary arteries and to coil occlude additional collaterals.
4. Refer to follow-up in the discussion of tetralogy of Fallot (Chapter 7).
H. Risk of recurrence
1. The patient had velocardiofacial syndrome with absent thymus and parathyroids, cleft palate, and facial anomalies.
2. Although TOF with pulmonary atresia occurs de novo in most cases, parental screening is necessary to identify familial cases for proper counseling.
3. If one of the parents is affected, then their risk of having another affected baby is 50% (autosomal dominant inheritance). As in almost all autosomal dominant conditions, the severity of the condition in an offspring cannot be predicted. Thus, genetic testing of the parents is important to provide accurate counseling regarding future pregnancies.
I. Outcome of this case
1. Amniocentesis showed normal karyotype, and FISH analysis confirmed 22q11.2 microdeletion in both the fetus and mother. The baby was delivered at term with good Apgar scores. Weight was 7 kg.
2. Pulse oximetry reading was 79% in room air, but there was no metabolic acidosis. PGE1 infusion was started at 0.05 μg /kg per minute because a small patent ductus arteriosus (PDA) was noted on echocardiography, supplying the confluent pulmonary arteries.
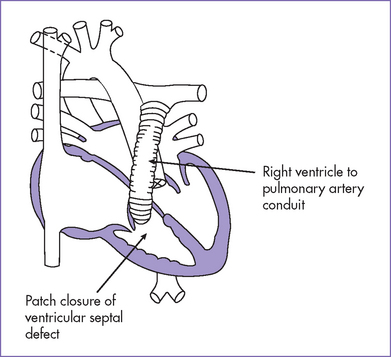
3. The baby had a palliative aortic–pulmonary shunt at 1 week of age and right ventricular outflow tract (RVOT) valved conduit and VSD closure at 5 months, with coil embolization of the systemic collaterals intraoperatively at the corrective surgery.
II. YOUR HANDY REFERENCE
A. Prevalence
1. TOF is found in about 10% of children with congenital heart disease. TOF with pulmonary atresia is one of the forms of TOF.
2. TOF with pulmonary atresia occurs in about 10% of patients with TOF.
3. TOF with pulmonary atresia may be missed if echocardiographic examination of the fetal heart is confined to the four-chamber view.
a. An abnormal four-chamber view is rare in TOF.
b. An abnormally leftward axis may be a clue to its diagnosis in the four-chamber view.
4. The fetal series of Allan and colleagues (1986) studied a total group of 2136 cases of congenital heart diseases diagnosed prenatally.
a. There were 125 cases of TOF (5.8%).
b. Out of the 125 cases, 32 (25.6%) were with TOF with pulmonary atresia.
B. Outcome (Table 8-1)
1. Reddy and colleagues (2000) studied 85 patients with pulmonary atresia with VSD and major aortopulmonary collateral arteries (MAPCAs). The study looked at the early and intermediate outcomes after repair of the anomalies. They found that:
a. Early one-stage complete unifocalization yields good functional results in more than 90% of patients with pulmonary atresia and MAPCAs, even those with absent true pulmonary arteries.
b. Complete repair during the same operation is achieved in two thirds of patients.
c. There remains room for improvement.
2. The results of this study must be appreciated within the context of the natural history of this lesion.
a. About 65% of patients survive to 1 year of age without surgery.
b. Slightly more than 50% survive to 2 years even with surgical intervention.
C. Associated syndromes and extracardiac anomalies
1. Extracardiac abnormalities in TOF occur in as many as 30% of affected infants and children.
2. In fetal TOF, the incidence of extracardiac lesions may be 50% to 60%, and 25% have chromosomal abnormalities.
3. TOF in combination with a cleft palate suggests velocardiofacial syndrome, which may have additional abnormalities including T-cell subset abnormalities, abnormal parathyroid function, and long-term neurodevelopmental delay.
4. TOF with or without pulmonary atresia can also occur in trisomies 21, 18, and 13.
D. Differential diagnosis
1. The truncal valve in a common arterial trunk is more often thickened and dysplastic in appearance, it may be either tricuspid or quadricuspid, and it may be stenotic or regurgitant.
2. The aortic valve in TOF with pulmonary atresia is typically tricuspid and thin, without regurgitation or stenosis.
3. The VSD in TOF with pulmonary atresia is usually perimembranous (in continuity with the tricuspid valve); it is the outlet muscular type in common arterial trunk.
4. Identifying the origin of the branch pulmonary arteries by two-dimensional imaging and color flow mapping is important for diagnosing a common arterial trunk.
5. The branch pulmonary arteries in common arterial trunk are usually of normal caliber or larger, whereas they are usually hypoplastic in TOF with pulmonary atresia.
6. An arterial duct is usually not present in most types of common arterial trunk. In TOF with pulmonary atresia, the pulmonary blood flow direction is retrograde in the arterial duct.
E. Clues to fetal sonographic diagnosis
1. The axis of the heart is leftward on four-chamber view assessment.
2. There is only a single large outlet, that of the aorta with a semilunar valve that overrides a large outlet VSD.
3. The pulmonary artery is hypoplastic or not visible.
4. The flow in the ductus arteriosus is reversed (aorta to pulmonary). The arterial duct in pulmonary atresia may be tortuous and commonly arises from the transverse rather than the distal aortic arch. It can also arise from the base of the brachiocephalic artery (left in a right arch and right in a left arch).
5. The collateral supply is directly from the aorta to the lungs (continuous low-velocity flow) (Fig. 8.6).
6. Branch pulmonary arteries are small (often) and might have a seagull wings appearance or might be impossible to identify. The seagull sign is often used to describe the angiographic appearance of the branch pulmonary arteries.
H. Immediate postnatal management
1. When there is a confluence of central pulmonary arteries, one of the following three approaches has been used initially:
a. An aortic–pulmonary shunt (such as modified Blalock–Taussig shunt)
b. RVOT reconstruction with patch.
c. For very small but confluent central pulmonary arteries, a central but end-to-side shunt has been proposed (Mee procedure).
2. When central pulmonary arteries are nonconfluent, with multiple large collaterals supplying different segments of the lungs, unifocalization of pulmonary blood flow surgery is performed.
a. This is a surgical connection between or among the isolated regions of the lungs so that the lung can be perfused by a single source.
b. Surgical mortality is about 5% to 15%.
c. Later, a right ventricle–to–pulmonary artery conduit can be placed and the VSD closed or fenestrated.
d. At long-term follow-up, if collateral artery stenosis is found, the intervention must be repeated.
I. Pathophysiology
1. In this extreme form of TOF, the intracardiac pathology resembles that of TOF in all respects except for the pulmonary atresia.
2. It has been documented that TOF with a patent pulmonary outflow tract can progress to pulmonary atresia in fetal life.
3. Pulmonary atresia can be either at the infundibular or the valvar level.
4. The main pulmonary artery might not be identifiable or may be very hypoplastic.
5. The branch pulmonary arteries might not be confluent and might have abnormal arborization (distribution).
6. The source of blood supply to the lungs is either through the ductus arteriosus or the major bronchopulmonary collateral arteries.
a. In two thirds of cases the pulmonary blood flow is mediated through the PDA, which courses downward from the transverse aortic arch (vertical ductus).
b. In one third, the pulmonary blood flow is through multiple systemic collaterals arising from the aorta. Collateral arteries arise most commonly from the descending thoracic aorta, less often from the brachiocephalic branches (innominate), and rarely from the coronary arteries.
J. Risk of recurrence
1. The risk of recurrence for TOF when only one first-degree relative is affected is about 2.5%; if father is affected, the estimated recurrence risk is 1.5%.
2. Chromosome 22q11 microdeletion is common in fetuses with TOF.
a. With this combination there is a higher incidence of pulmonary atresia, aortic arch anomalies, or absent pulmonary valve syndrome.
b. This specific chromosomal abnormality occurs in a syndromic pattern such as velocardiofacial (or Shprintzen’s) syndrome and DiGeorge’s syndrome.
c. Velocardiofacial syndrome is one of the most common genetic disorders in humans. It is characterized by cleft palate, heart abnormalities, learning disabilities, and more than 180 other clinical findings.
d. Clinical and laboratory features are described with the acronym CATCH 22: cardiac defect, thymic hypoplasia, cleft palate, hypocalcemia, and deletion of the long arm of chromosome 22. Although this syndrome occurs de novo in most cases, parental screening is necessary to identify familial cases for proper counseling.
III. TAKE-HOME MESSAGE
A. Diagnosis
1. Pulmonary atresia may be at the valvar or infundibular level. Atresia of longer segments can involve absence of the main pulmonary artery–to–RV outflow connection with continuous or discontinuous branch pulmonary arteries. The source of blood flow to the lungs may be either the ductus arteriosus or the major aortopulmonary collateral arteries.
2. The size discrepancy of the arterial trunks might not be apparent in early gestation but becomes evident with advancing gestation.
3. Pulmonary atresia with VSD is an extreme form of TOF. TOF with a patent pulmonary outflow can progress to pulmonary atresia in fetal life.
C. Prognosis
1. TOF with pulmonary atresia usually has a worse long-term prognosis and outcome than the less-severe forms of TOF.
2. Pulmonary artery hypoplasia at the initial echocardiographic fetal examination, little or no growth of the main and branch pulmonary arteries on follow-up, and retrograde ductus arteriosus flow are among the highly predictive echocardiographic findings of severe postnatal disease.
3. Neonatal cyanosis correlates with the severity of pulmonary outflow obstruction and the resulting right-to-left shunt across the VSD.
4. An estimated overall perinatal mortality for the TOF spectrum may be as high as 35% to 75% (including long-segment pulmonary atresia and absent pulmonary valve syndrome).
Allan LD, Crawford DC, Tynan MJ. Pulmonary atresia in prenatal life. J Am Coll Cardiol. 1986;8(5):1131-1136.
Berning RA, Silverman NH, Villegas M, et al. Reversed shunting across the ductus arteriosus or atrial septum in utero heralds severe congenital heart disease. J Am Coll Cardiol. 1996;27(2):481-486.
Boudjemline Y, Fermont L, Le Bidois J, et al. Can we predict 22q11 status of fetuses with tetralogy of Fallot? Prenat Diagn. 2002;22(3):231-234.
Chaoui R, Kalache KD, Heling KS, et al. Absent or hypoplastic thymus on ultrasound: A marker for deletion 22q11.2 in fetal cardiac defects. Ultrasound Obstet Gynecol. 2002;20(6):546-552.
Hornberger LK, Sanders SP, Sahn DJ, et al. In utero pulmonary artery and aortic growth and potential for progression of pulmonary outflow tract obstruction in tetralogy of Fallot. J Am Coll Cardiol. 1995;25(3):739-745.
Lee W, Smith RS, Comstock CH, et al. Tetralogy of Fallot: Prenatal diagnosis and postnatal survival. Obstet Gynecol. 1995;86(4 Pt 1):583-588.
Meijboom F, Szatmari A, Deckers JW, et al. Cardiac status and health-related quality of life in the long term after surgical repair of tetralogy of Fallot in infancy and childhood. J Thorac Cardiovasc Surg. 1995;110(4 Pt 1):883-891.
Miyashita S, Chiba Y. Prenatal demonstration of major aortopulmonary collateral arteries with tetralogy of Fallot and pulmonary atresia. Fetal Diagn Ther. 2004;19(1):100-105.
Pearl JM, Laks H, Drinkwater DCJr, et al. Repair of conotruncal abnormalities with the use of the valved conduit: Improved early and midterm results with the cryopreserved homograft. J Am Coll Cardiol. 1992;20(1):191-196.
Reddy VM, McElhinney DB, Amin Z, et al. Pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries: Experience with 85 patients. Circulation. 2000;101(15):1826-1832.
Yoo SJ, Lee YH, Kim ES, et al. Tetralogy of Fallot in the fetus: Findings at targeted sonography. Ultrasound Obstet Gynecol. 1999;14(1):29-37.