7 Tetralogy of Fallot
I. CASE
A. Fetal echocardiography findings
1. The fetal echo reveals situs solitus of the atria, levocardia, right aortic arch, and heart rate 122 bpm.
2. The four-chamber view is normal, with equal sized ventricles.
3. The cardiac axis is abnormal (more toward the left, 90 degrees).
4. Position and size are abnormal (cardiothoracic ratio = 0.32).
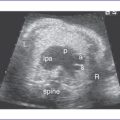
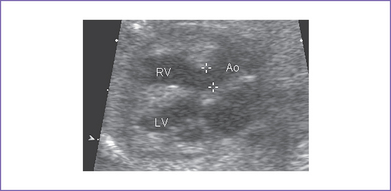
5. There is a moderate-size perimembranous subaortic ventricular septal defect (VSD), large overriding aorta, and anterior malalignment of the conal septum with subpulmonary narrowing (Fig. 7-1).
6. There is a small main pulmonary artery with confluent branches.
7. The right ventricular outflow tract (RVOT) velocity is 1.4 m/s.
8. The pulmonary artery annulus is 2 mm; the aortic artery valve annulus is 3 mm.
9. Ductal flow is normal (right to left).
10. The foramen ovale is patent, with unrestricted right-to-left shunt.
11. There is a left aortic arch and ductal arch, which are of similar size. Both arches have antegrade flow.
12. The pulmonary venous flow pattern is normal.
13. Tei index (myocardial performance index) in both ventricles is normal.
D. Fetal management and counseling
1. Management: Diagnosis of tetralogy of Fallot (TOF) should prompt referral for the following:
a. Thorough anatomic examination by ultrasound.
a. Serial antenatal studies were made at 6-week intervals.
b. If a significant gradient develops through either the VSD or the RVOT with high blood velocities, restriction of the VSD should be excluded.
c. Development of hydrops fetalis is uncommon in fetal TOF unless there is a chromosomal abnormality (not related to structural heart defect) or restriction of the VSD.
F. Neonatal management
b. Classic TOF with pulmonary stenosis.
a. Hypoxic spells should be recognized and treated appropriately.
b. Oral propranolol 2 to 4 mg/kg per day may be used to prevent hypoxic spells and delay corrective surgery.
a. Palliation: In infants with severe RV outflow obstruction and cyanosis or uncontrollable hypoxic spells in whom corrective surgery cannot be performed, palliation in the form of a modified Blalock–Taussig shunt may be created using a Gortex tube to anastomose the subclavian artery or brachiocephalic artery and the ipsilateral pulmonary artery.
G. Follow-up
1. The baby should be monitored routinely with echocardiography for the development of RV dysfunction, significant pulmonary insufficiency, and pulmonary outflow tract obstruction.
2. Varying levels of activity limitation may be indicated.
3. Prophylaxis for subacute bacterial endocarditis (SBE) should be used as indicated.
4. Arrhythmias, particularly ventricular tachycardia, can develop later and can cause sudden death. Holter monitoring, antiarrhythmic agents, and pacemaker therapy may be indicated.
5. Pulmonary valve replacement is needed in up to 20% of patients. The timing of and criteria for this intervention are currently being developed.
6. RV–to–pulmonary artery conduit follow-up.
a. Subsequent interventional catheterization(s) with balloon dilation and stent placement in the conduit may be needed to relieve significant conduit stenosis.
b. Surgical replacement of the conduit should be anticipated.
7. Branch pulmonary artery stenosis (diagnosed by cardiac magnetic resonance imaging [MRI] and lung isotope scan) can require postoperative balloon angioplasty with stent placement or surgical repair. The aim is to normalize blood flow to both lungs, thus reducing the RV systolic pressure. These additional procedures carry a risk of morbidity and mortality.
H. Risk of recurrence
1. The fetal karyotype done on amniocytes showed a male fetus with trisomy 21 (47,XY,+21).
2. The estimated recurrence risk of TOF in a fetus with normal karyotype, including FISH for 22q11.2 deletion, is 1.5% to 2%.
3. When the baby has trisomy 21 or other underlying condition, the recurrence risk is determined by the underlying etiology.
I. Outcome of this case
1. The baby was born vaginally at term with a good Apgar score and good weight.
2. Mild cyanosis was noted at birth; pulse oximeter reading was 90% in room air. There was no need for the hyperoxia test because cyanotic heart disease was diagnosed during gestation.
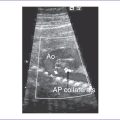
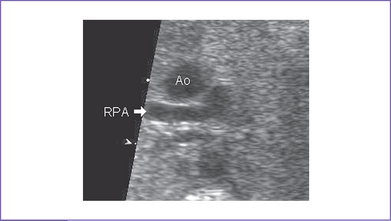
3. Postnatal echocardiography confirmed the diagnosis of TOF with large subpulmonary VSD with posterior deviation of the outlet septum causing valvar and subvalvar pulmonary stenosis. The pulmonary arteries were of good size, with a transpulmonary Doppler velocity of 4.5 m/s (Fig. 7-3).
4. The foramen ovale and ductus arteriosus were patent and of a good size.
5. In view of the baby’s anatomy and the current degree of cyanosis, prostaglandin E1 (PGE1) infusion was not started. It was decided to let the ductus arteriosus start to close and then decide based on the degree of cyanosis.
6. The baby maintained a pulse oximeter saturation of 85% to 90% and was feeding well, so he was discharged home and followed monthly at the cardiology outpatient clinic. The mother was made aware of the possibility of cyanotic spells.
7. The baby had successful surgical repair (patch closure of the VSD, widening of the RVOT, and pulmonary valvuloplasty) at 4 months of life.
II. YOUR HANDY REFERENCE
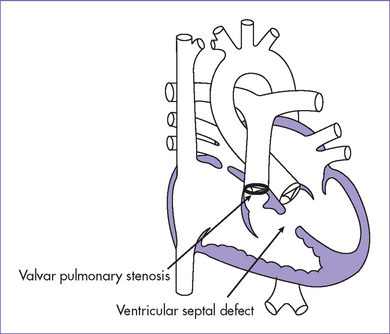
A. Tetralogy of Fallot (Figs. 7-4 and 7-5)
a. TOF accounts for 10% of all congenital heart disease. It is the most common form of cyanotic heart disease beyond infancy.
b. TOF is one of the more commonly encountered forms of heart disease in the fetus, and in one series it is the third most commonly identified form of structural heart disease. This could be due to the very common occurrence of aneuploidy and extracardiac structural pathology rather than recognition of the pathology at routine obstetrics assessment.
c. Classic TOF can be missed if echocardiographic examination of the fetal heart is confined to the four-chamber view, because an abnormal four-chamber view is rarely observed in this condition. One might see an abnormal four-chamber view in tetralogy with absent pulmonary valve, with mitral valve obstruction, or with restrictive VSD, all of which are less common than the classic form of TOF.
d. Allan and Sharland (1992) studied a total of 125 cases of TOF diagnosed prenatally. They found:
a. Two studies (Berning and colleagues [1996] and Hornberger and colleagues [1999]) have shown that perinatal mortality may be as high as 35% to 75%.
b. The perinatal outcome of fetal TOF is worse than that observed for postnatally identified TOF. The possible explanation is the relatively high incidence of aneuploidy and extracardiac anomalies.
3. Associated syndromes and extracardiac anomalies.
a. Extracardiac abnormalities in TOF occur in as many as 30% of affected infants and children.
b. In fetal TOF, the incidence of extracardiac lesions may be 50% to 60%.
c. Chromosomal abnormalities occur in 25% of fetuses with TOF and extracardiac lesions, including:
d. Down syndrome is present in approximately 75% to 80% of those with the combination of AVSD and TOF.
e. Midline defects (e.g., pentalogy of Cantrell, omphalocele) and renal, skeletal, gastrointestinal, and central nervous system abnormalities may be found in fetal TOF.
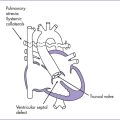
4. Clues to fetal sonographic diagnosis (Fig. 7-6).
a. Normal four-chamber view is typical, but often with a more leftward axis than normal.
b. Usually a subaortic VSD is seen in the long-axis view of the left ventricle (also known as a five-chamber view) with sweeps to the outflow tracts.
c. Anterior displacement of the ascending aorta results in the aorta overriding the VSD.
d. The ratio of the ascending aorta diameter to the pulmonary artery diameter is increased, with forward flow in the pulmonary artery. The pulmonary artery diameter might be within the normal range for gestation.
e. The ascending aorta tends to be larger than normal for gestation, particularly later in gestation.
f. Blood flow is seen into the ascending aorta from both ventricles on color flow mapping.
g. A gradient across the RVOT is usually not present as a result of the VSD and fetal physiology. The absence of a flow gradient does not rule out pulmonary stenosis.
h. Infundibular or subpulmonary narrowing is seen and is caused by anterior ventricular septal deviation (this tends to be more appreciated later in gestation).
5. Cardiovascular profile score.
a. Usually, fetuses with TOF do not develop CHF unless there are extracardiac causes such as chromosomal abnormality.
b. The score might help in the presence of restrictive VSD leading to RV hypertension and potentially the development of RV dysfunction and tricuspid regurgitation.
a. Right aortic arch (about 25%).
b. Peripheral pulmonary stenosis.
c. Discontinuous pulmonary arteries (anomalous origin of branch pulmonary arteries).
h. Aberrant left subclavian artery.
i. Anomalous origin of coronary arteries.
7. Immediate postnatal management for patients without prenatal diagnosis of TOF.
a. Check pulse oximeter; acceptable reading is above 92%. If the reading is lower, do a hyperoxia test. If the hyperoxia test is positive, start IV prostaglandin infusion.
b. Check four-limb blood pressure.
c. Transfer the patient to the neonatal or pediatric (or cardiac, if available) intensive care unit (ICU) for further assessment and management.
d. If TOF is diagnosed postnatally, acceptable pulse oximeter reading is greater than 75% to 80%. The systemic saturation usually indicates the degree of pulmonary outflow tract obstruction.
a. The two most important features of TOF are a VSD large enough to equalize pressures in both ventricles and an RVOT obstruction.
b. Because of the nonrestrictive VSD, systolic pressures in the RV and the LV are identical.
9. Risk of recurrence: For trisomy 21 (Down syndrome) see Chapter 6, Complete Atrioventricular Septal Defect.
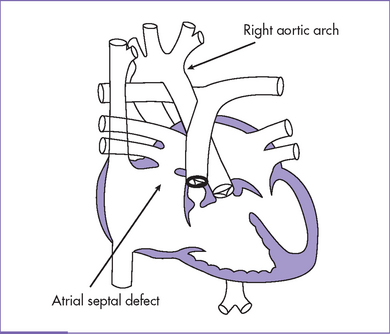
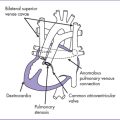
Fig. 7-4 Tetralogy of Fallot with secundum atrial septal defect and right aortic arch.
(Modified from Mullins CE, Mayer DC: Congenital Heart Disease: A Diagrammatic Atlas. New York, Liss, 1988.)
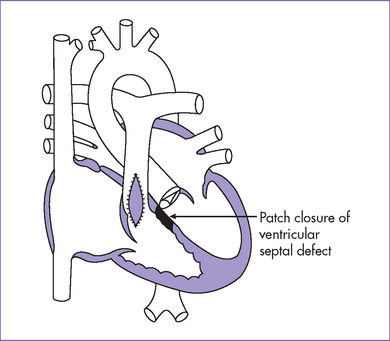
Fig. 7-5 Postoperative anatomy for tetralogy of Fallot.
(Modified from Mullins CE, Mayer DC: Congenital Heart Disease: A Diagrammatic Atlas. New York, Liss, 1988.)
![]() B.Congenital diaphragmatic hernia
B.Congenital diaphragmatic hernia
1. Prevalence: Congenital diaphragmatic hernia (CDH) occurs sporadically, affecting one per 2000 to 5000 live births.
a. In CDH, most (84%) lesions are left-sided, 13% are right-sided, and 2% bilateral.
b. In just over 50% of cases, the condition is isolated; the rest may have associated chromosomal, syndromal, or structural anomalies, which are independent determinants of survival.
a. The reported mortality for prenatally detected CDH is high. In spite of significant therapeutic progress, the prognosis of CDH remains poor in forms in which the liver is herniated into the chest.
b. The clinical outcome is largely determined by the degree of associated pulmonary hypoplasia. Pulmonary hypoplasia is probably at least in part due to lung compression by the herniated viscera in the thoracic cavity, particularly the liver.
c. Outcomes for patients with CDH vary widely depending on when the disease is found. Fetuses with CDH have lower survival rates than live-born infants and infants presenting to a neonatal surgical center.
d. In 2000, Cohen and colleagues from the Center for Fetal Diagnosis and Treatment at the Children’s Hospital of Philadelphia reported 58% survival for a large group of live-born babies with isolated CDH treated at that center. In that study, 174 patients with CDH, most of whom had prenatal diagnosis, were treated between 1996 and 2000.
e. New postnatal therapies have led to improvements in survival.
f. Despite application of even the most sophisticated and controlled treatment protocols, at least 25% of fetuses with severe isolated CDH die.
3. Associated syndromes and extracardiac anomalies.
a. Congenital heart defects are associated with diaphragmatic hernia in about 10% to 20% of cases.
b. Examples of such heart defects include VSD, VSD with pulmonary stenosis, TOF, AVSD, and hypoplastic left heart syndrome.
4. Clues to fetal sonographic diagnosis.
a. Abnormal cardiac position and/or axis, depending upon the location of the defect in the diaphragm and the amount and type of herniated viscera.
b. Abnormal four-chamber view with signs of displacement.
c. Direct imaging of the diaphragmatic defect is the hallmark of diagnosis of CHD, with the absence of the stomach from the abdomen and its presence in the chest.
d. If there is liver in the hernia, then hepatic tissue extends across the diaphragm into the chest.
e. Diaphragmatic hernia must be distinguished from:
a. The etiology of CDH remains largely unknown.
b. Any space-occupying lesion within the confines of the thorax can displace the heart or the lung tissue from their normal position. The site and the size of the diaphragmatic defect influence the volume of the abdominal contents that herniate into the chest.
c. The defect is due to failure of the posthepatic mesenchymal plate in closing the pleuroperitoneal canals, which are part of the early development before 8 weeks of gestation.
d. The defect is most common in the posterolateral aspect of the left diaphragm, although anterior and right-sided defects can occur.
e. The lungs of infants with CDH show delayed maturation, with fewer alveoli, thickened alveolar walls, increased interstitial tissue, and markedly diminished alveolar air space and gas-exchange surface area.
f. CDH is also associated with abnormal development of the pulmonary vasculature, consisting of a reduced total vascular bed, decreased number of vessels per volume of lung, medial hyperplasia, and peripheral extension of the muscle layer into the smaller intra-acinary arterioles, as well as adventitial thickening.
g. These morphological alterations probably result in pulmonary hypertension.
6. Fetal management and counseling.
b. Concept of antenatal intervention.
a. It has been shown that lethal pulmonary hypoplasia may be predicted on the basis of indirect assessment of lung development. Several methods to quantitate this have been investigated, including high-resolution two- and three-dimensional ultrasound and fetal MRI, as well as direct or indirect assessment of resistance within the pulmonary circulation.
b. Polyhydramnios and manifestation in early pregnancy have been suggested as high-risk factors adversely affecting outcome.
c. In one study, evidence of cardiac ventricular disproportion before 24 weeks’ gestation in isolated CDH was associated with 100% mortality. Development of ventricular disproportion during the third trimester was associated with a survival rate of 75%.
d. For left-sided CDH, herniation of the left liver lobe into the thorax and a low lung-to-head ratio during midgestation have been suggested as the best available methods for predicting neonatal mortality and morbidity as well as the need for ECMO.
e. Right-sided lesions are more uncommon, and typically a poorer prognosis is quoted. The lung-to-head ratio and liver herniation for right-sided lesions as criteria for lethal pulmonary hypoplasia have not been well validated, but massive liver herniation and small lung volumes have been used as selection criteria of severity and hence for fetal therapy.
f. In recent reports, the branch pulmonary artery diameters have been shown to reflect lung mass and to potentially predict postnatal respiratory morbidity.
a. Ideally, patients should be referred in utero to tertiary centers for timed term delivery in the hospital where they will be resuscitated.
b. When they are stable enough, they can undergo neonatal repair of the defect.
III. TAKE-HOME MESSAGE
A. Tetralogy of Fallot
a. TOF is a spectrum of disease that includes a large subaortic VSD and subpulmonary obstruction. The subpulmonary obstruction, in addition to the degree of hypoplasia of the pulmonary valve and branch pulmonary arteries, is a critical factor in determining the management and counseling.
b. The postnatal spectrum of pulmonary artery size in TOF can be attributed to variable patterns of growth in utero.
c. The pulmonary artery can have a diameter within the normal range for gestation; however, the aortic–to–pulmonary artery ratio is typically the reverse of normal.
d. In the classic TOF, it is unusual to have any significant gradient through the VSD or RVOT. If a significant gradient develops through either with high blood velocities, the restriction of the VSD should be excluded. This finding may be associated with a worse perinatal and postnatal outcome.
e. In fetal life, absence of a significant gradient across the pulmonary valve does not rule out pulmonary stenosis.
f. Chromosome 22q11 microdeletion has been associated with TOF (found in about 20%). Chromosome and FISH studies of the parents should be recommended when a fetus is found to have the deletion 22q11.2.
g. The association between increased nuchal translucency thickness and aneuploidy plus chromosome 22q11 microdeletion should be considered in diagnostic procedures.
h. The sonographic diagnosis of increased nuchal translucency thickness and intracardiac echogenic foci requires detailed ultrasonographic and echocardiographic examination.
a. Main pulmonary artery size, main pulmonary artery–to–aorta diameter ratio, and pattern of pulmonary artery growth can predict the severity of postnatal pulmonary outflow obstruction.
b. Documentation of antegrade flow through the pulmonary artery throughout gestation is essential. Absence of antegrade flow in the pulmonary artery or reverse ductal flow during fetal life is an indication of more severe pulmonary outflow obstruction—even pulmonary atresia—and denotes a spectrum of diseases with a worse prognosis than TOF.
B. Congenital diaphramatic hernia
a. In CDH, the heart is displaced by the physical presence of the herniated abdominal contents.
b. In left-sided CDH, lung hypoplasia with reduced pulmonary flow returning to the left atrium and altered left atrial hemodynamics can result in obvious left versus right heart disproportion, with a smaller left heart.
a. Despite significant advances in neonatal surgical care, the mortality and morbidity associated with this condition remain high, particularly among those with a prenatal diagnosis and with more severe disease.
b. CDH with congenital heart disease indicates a poor prognosis. Cardiac evaluation is vital in evaluating a fetus with CDH.
c. CDH has high mortality due to lung hypoplasia and pulmonary hypertension. Efforts to improve survival and outcome have included fetal intervention, delivery at specialist centers, elective operation after stabilization of labile physiology, and minimizing barotraumas.
d. Fetal pulmonary artery diameter measurements correlate with respiratory morbidity in postnatal CDH and can assist with prediction of outcome. Survival has improved, possibly because of improved postnatal management of CDH, limiting this measurement in assessing survival.
Allan LD, Sharland GK. Prognosis in fetal tetralogy of Fallot. Pediatr Cardiol. 1992;13(1):1-4.
Berning RA, Silverman NH, Villegas M, et al. Reversed shunting across the ductus arteriosus or atrial septum in utero heralds severe congenital heart disease. J Am Coll Cardiol. 1996;27(2):481-486.
DeVore GR, Siassi B, Platt LD. Fetal echocardiography. VIII. Aortic root dilatation—a marker for tetralogy of Fallot. Am J Obstet Gynecol. 1988;159(1):129-136.
Flanagan MF, Foran RB, Van Praagh R, et al. Tetralogy of Fallot with obstruction of the ventricular septal defect: Spectrum of echocardiographic findings. J Am Coll Cardiol. 1988;11(2):386-395.
Hornberger LK, Sanders SP, Sahn DJ, et al. In utero pulmonary artery and aortic growth and potential for progression of pulmonary outflow tract obstruction in tetralogy of Fallot. J Am Coll Cardiol. 1999;25(3):739-745.
Maeda J, Yamagishi H, Matsuoka R, et al. Frequent association of 22q11.2 deletion with tetralogy of Fallot. Am J Med Genet. 2000;92(4):269-272. Erratum in Am J Med Genet 94(1):following 84, 2000.
Congenital diaphragmatic hernia
Bargy F, Be audoin S, Barbet P. Fetal lung growth in congenital diaphragmatic hernia. Fetal Diagn Ther. 2006;21(1):39-44.
Baumgart S, Paul JJ, Huhta JC, et al. Cardiac malposition, redistribution of fetal cardiac output, and left heart hypoplasia reduce survival in neonates with congenital diaphragmatic hernia requiring extracorporeal membrane oxygenation. J Pediatr. 1998;133(1):57-62.
Cass DL. Fetal surgery for congenital diaphragmatic hernia: The North American experience. Semin Perinatol. 2004;29(2):104-111.
Chervenak FA, McCullough LB. A comprehensive ethical framework for fetal research and its application to fetal surgery for spina bifida. Am J Obstet Gynecol. 2002;187(1):10-14.
Cohen MS, Rychik J, Bush DM, et al. Influence of congenital heart disease on survival in children with congenital diaphragmatic hernia. J Pediatr. 2002;141(1):25-30.
Dillon E, Renwick M, Wright C. Congenital diaphragmatic herniation: Antenatal detection and outcome. Br J Radiol. 2000;73(868):360-365.
Harrison MR, Adzick NS, Bullard KM, et al. Correction of congenital diaphragmatic hernia in utero VII: A prospective trial. J Pediatr Surg. 1997;32(11):1637-1642.
Lipshutz GS, Albanese CT, Feldstein VA, et al. Prospective analysis of lung-to-head ratio predicts survival for patients with prenatally diagnosed congenital diaphragmatic hernia. J Pediatr Surg. 1997;32(11):1634-1636.
Sharland GK, Lockhart SM, Heward AJ, Allan LD. Prognosis in fetal diaphragmatic hernia. Am J Obstet Gynecol. 1992;166(1 Pt 1):9-13.
Skari H, Bjornland K, Haugen G, et al. Congenital diaphragmatic hernia: A meta-analysis of mortality factors. J Pediatr Surg. 2000;35(8):1187-1197.
Sokol J, Shimizu N, Bohn D, et al. Fetal pulmonary artery diameter measurements as a predictor of morbidity in antenatally diagnosed congenital diaphragmatic hernia: A prospective study. Am J Obstet Gynecol. 2006;195(2):470-477.
Wilcox DT, Irish MS, Holm BA, Glick PL. Prenatal diagnosis of congenital diaphragmatic hernia with predictors of mortality. Clin Perinatol. 1996;23(4):701-709.
Witters I, Legius E, Moerman P, et al. Associated malformations and chromosomal anomalies in 42 cases of prenatally diagnosed diaphragmatic hernia. Am J Med Genet. 2001;103(4):278-282.