[level-membership-for-pediatrics-category]
Teratomas, Dermoids, and Other Soft Tissue Tumors
Teratomas
Embryology and Pathology
Teratoma, from the Greek teratos (‘of the monster’) and onkoma (‘swelling’), is a term first applied by Virchow in 1869 to ‘sacrococcygeal growths’.1 Teratomas are composed of multiple tissues foreign to the organ or site from which they arise.2 Although teratomas are sometimes defined as having three embryonic layers (endoderm, mesoderm, and ectoderm), recent classifications also include monodermal types.2,3
Teratomas are thought by some to arise from totipotent primordial germ cells.3 These cells develop among the endodermal cells of the yolk sac near the origin of the allantois and migrate to the gonadal ridges during weeks 4 and 5 of gestation (Fig. 68-1).4 Some cells may miss their target destination and give rise to a teratoma anywhere from the brain to the coccygeal area, usually in the midline. Another theory has teratomas arising from remnants of the primitive streak or primitive node.4–6 During week 3 of development, midline cells at the caudal end of the embryo divide rapidly and, in a process called gastrulation, give rise to all three cell layers of the embryo (Fig. 68-2).4 By the end of week 3, the primitive streak shortens and disappears. This theory explains the more common occurrence of teratomas in the sacrococcygeal region. With either theory, the totipotent cells could give rise to monoclonal neoplasms. Evidence shows that, whereas immature teratomas may be monoclonal, mature teratomas can be polyclonal, more like a hamartoma than a neoplasm.7 This finding is compatible with the third theory that teratomas are a form of incomplete twinning.2,3
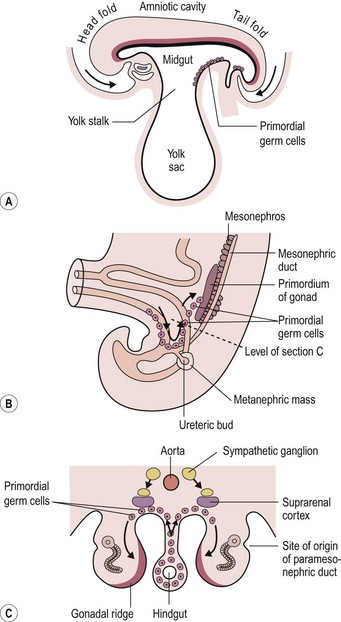
FIGURE 68-1 Commonly cited theory on the origin of teratomas. (A) Drawing of embryo during week 4 (longitudinal section), showing primordial germ cells at the base of the yolk sac. (B, C) During week 5, these cells migrate toward the gonadal ridges. According to this theory, some cells could miss their intended destination. (Modified from Moore KL, Persaud TVN. The Developing Human. Philadelphia: WB Saunders; 1993. p. 71, 181.)
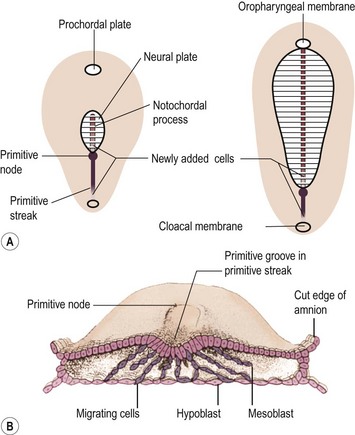
FIGURE 68-2 Alternate theory on embryogenesis of teratomas. (A) Sketches of dorsal views of the embryonic disk on days 17 and 18, showing the primitive streak and primitive node. (B) Drawing of a transverse cut of the embryonic disk during week 3. This shows that cells from the primitive streak migrate to form the mesoblast (the origin of all mesenchymal tissues) and also displace the hypoblast to form the endoderm. Hence, remnants of these pluripotent primitive streak cells could give rise to teratomas and could account for the more frequent sacrococcygeal location. (From Moore KL, Persaud TVN. The Developing Human. Philadelphia: WB Saunders; 1993. p. 55–6.)
The primordial germ cell is the principal but probably not the exclusive progenitor of a teratoma. The recent trend is to include teratomas under the classification of germ cell tumors.2,3,5 This histologic classification also includes germinomas (formerly dysgerminomas), embryonal carcinomas, yolk sac tumors (YST), choriocarcinomas, gonadoblastomas, and mixed germ cell tumors. Gonadal and extragonadal teratomas may have a different origin, explaining the different behavior according to tumor site.
Teratomas are fascinating tumors owing to the diversity of tissues they may contain and the varying degree of organization of these tissues. Many tumors contain skin elements, neural tissue, teeth, fat, cartilage, and intestinal mucosa, often with normal ganglion cells. These tissues are usually present as disorganized islands of cells with cystic spaces. The tumor sometimes consists of more organized tissue, such as small bowel, limbs, and even a beating heart. These have been called fetiform teratomas (Fig. 68-3).2,3,5,8,9 When the mass includes vertebrae or notochord and a high degree of structural organization, the term fetus in fetu is used. This is viewed by some as a variant of conjoined twinning but is classified as a teratoma by others, owing to the absence of a recognizable umbilical cord in its vascular pedicle.3,10 Whether teratomas are at one end of a spectrum that includes fetus in fetu, parasitic twins, conjoined twins, and normal twins is the subject of controversy. One certainly cannot dismiss the many reports of teratomas associated with fetus in fetu in the same patient and with a twin pregnancy.2,3,11–13
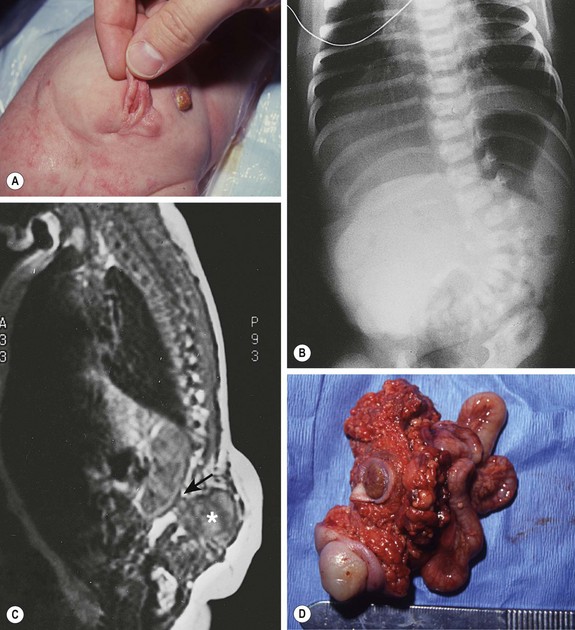
FIGURE 68-3 (A) This child had a large fluctuant lumbar mass at birth. The patient is prone with the head at the top of the photograph. A family history of myelomeningocele existed in a great aunt. She also had an atrophic right leg with neurologic impairment below the L3 root and clubbing of the right foot. Note the ulcerated, arachnoid-looking area cranially and the pedunculated skin caudally, which had the appearance of a vulva and was oozing serous fluid. (B) Plain radiograph shows a severe lumbosacral scoliosis with vertebral anomalies. CT confirmed the vertebral anomalies with spina bifida and demonstrated a pattern of intestine with inspissated or calcified meconium in the teratoma. (C) MR image reveals that the mass (asterisk) extended into the retroperitoneum, where it was contiguous with the lower pole of the right kidney (arrow). (D) At operation, normal-looking blind bowel loops were found deep to the vulva-like structure. Part of the mass extended along the spinal cord, which required dissection and untethering by a neurosurgeon. The pathologic diagnosis was a mature fetiform teratoma that contained, among many other things, two adrenals, two ovaries, renal tissue with some glomeruli and tubules, bone with bone marrow, and portions of stomach and small and large bowel. The child recovered well neurologically but required spinal instrumentation owing to progressive scoliosis at age 2 years.
The overall tissue architecture is variable in teratomas. Moreover, a spectrum of cellular differentiation exists. Most benign teratomas are composed of mature cells, but 20–25% also contain immature elements, most often neuroepithelium. However, the degree of histologic immaturity is of proven prognostic significance only in ovarian teratomas.3,14 Even this concept is being questioned, since one large cooperative study demonstrated that overlooked microscopic foci of YST, rather than the grade of immaturity, was more predictive of recurrence.15 In neonatal teratoma, immature tissue is considered normal and without any influence on prognosis.2,5,16 Spontaneous maturation of malignant tumors has been reported after partial excision of giant SCTs in two fetuses at 23 and 27 weeks of gestation.17
Teratomas may also contain or develop foci of malignancy. A malignant germ cell tumor may be found in sites typical for teratomas, such as the mediastinum or sacrococcygeal area. Whether the lesion was malignant from the onset or the malignant cells destroyed and replaced the benign teratoma component is often difficult to ascertain. The most common malignant component within a teratoma is a YST (formerly also called ‘endodermal sinus tumor’). Other malignant germ cell tumors can occur, and, rarely, malignancy in other tissues composing the teratoma, such as neuroblastoma,18,19 squamous cell carcinoma,20 carcinoid,21 and others, can develop. Malignancy at birth is uncommon but increases with age and with incomplete resection. An apparently mature teratoma may recur several months or years after resection as a malignant YST, illustrating the difficulties in histologic sampling of large tumors and the need for close follow-up.2,3
Most YSTs and some embryonal carcinomas secrete α-fetoprotein (AFP), which can be measured in the serum and demonstrated in the cells by immunohistochemistry.22 This marker is particularly useful for assessing the presence of residual or recurrent disease. AFP levels are normally very high in neonates and decrease with time.22,23 The postoperative half-life is about six days. Persistently high levels may be an indication for the need for further surgical procedures or chemotherapy. Other markers that may be elevated are β-human chorionic gonadotropin (β-hCG), produced by choriocarcinomas, and, rarely, carcinoembryonic antigen. CA 125 was also found helpful in the follow-up of patients with SCTs, but not CA 19–9.24 Secretion of β-hCG by the tumor may be sufficient to cause precocious puberty.25
The genetic basis of teratomas is not yet understood. Most germ cell tumors appear to have an amplification, or isochromosome, in a region of the short arm of chromosome 12, designated i(12p).3,26 This has been well described in adults but was not confirmed in one pediatric series in which deletions on chromosomes 1 and 6 were found instead.27 Similarly, oncogenes and tumor suppressor genes did not appear to correlate with prognosis in one study.28 MYCN gene amplification was present in immature teratomas, but absent in mature teratomas in another report.29 BAX mutation and overexpression correlated with survival in another study of childhood germ cell tumors.30 The clinical usefulness of these findings remains unclear.
Associated Anomalies
Teratomas are usually isolated lesions. A well-recognized association is the Currarino triad of anorectal malformation, sacral anomaly, and a presacral mass.31,32 The presacral mass is usually a teratoma or an anterior meningocele. However, hamartomas, duplication cysts, and dermoid cysts have been described, as have combinations of these lesions.
An extensive review of the English and German literature found 51 cases of infants with the Currarino triad and highlighted several important facts.33 Twenty per cent of patients were older than 12 years at the time of diagnosis, yet no reports of malignancy were found. This contrasts to a 75% malignancy rate in patients older than 1 year who had the usual SCT.34 However, a subsequent case report described a malignant recurrence that resulted in the patient’s demise at 4 years of age despite chemotherapy.35 Since then, more reports of malignant transformation of a presacral teratoma in the context of the Currarino triad have appeared, and the risk of malignant transformation has been estimated at 1%.36,37 This can occur well into adulthood.38 Hence, one should not dismiss the presacral mass as always being benign in these patients. The female preponderance for patients with this triad is 1.5 : 1, which is less than the 3 : 1 ratio noted in isolated SCTs. A familial predisposition is noted in 57% of cases and has an autosomal dominant inheritance pattern.39 Although all variants of anorectal malformations have been described, by far the most common is anal or anorectal stenosis. In one report, this triad was present in 38% of all patients with anorectal stenosis and in 1.6% of patients with low imperforate anus.40
Anal anomalies also have been reported in conjunction with a presacral mass, but in the absence of sacral defects. Hirschsprung disease has been incorrectly diagnosed in some cases,41,42 indicating the need to eliminate the presence of a presacral mass by digital rectal examination, by a metal sound when the anus is too tight, or by imaging studies. In the screening of family members, normal plain radiographs of the sacrum are not adequate, because a presacral mass may exist in the absence of a bony defect.43 The low incidence of malignancy has led one author to conclude that the presacral lesion in this context is a hamartoma rather than a teratoma.44 This is supported by the demonstration of deletions or mutations of the homeobox gene HLXB9, located at 7q36, in several affected families.45 In two families where one member had died from a malignant neuroendocrine tumor originating from a presacral teratoma, the diagnosis of Currarino syndrome was made through identification of a HLXB9 mutation.36,38
The mutated gene has recently been renamed ‘motor neuron and pancreas homeobox 1 (MNX1).’45a Urogenital anomalies, such as hypospadias, vesicoureteral reflux, vaginal or uterine duplications, and other anomalies are associated with teratomas.32,34,46,47 Congenital dislocation of the hip has been found in 7% of patients with SCTs which can lead to late orthopedic sequelae.48 Vertebral anomalies can also be found (see Fig 68-3). Central nervous system lesions, such as anencephaly, trigonocephaly, Dandy–Walker malformations, spina bifida, and myelomeningocele can occur as well.2,3,49,50 Another peculiar association with SCTs is a family history of twins in as many as 10% of the patients.39,51,52 Although not confirmed in all series, this finding, combined with reports of simultaneous twin pregnancy or sequential familial occurrences of fetus in fetu and teratoma, supports the theory that teratomas may be just one end of the spectrum of conjoint twinning.2,3,9
Klinefelter syndrome is strongly associated with mediastinal teratoma50,53 and has been reported in patients with intracranial54 and retroperitoneal tumors.55 It is estimated that 8% of male patients with primary mediastinal germ cell tumors have Klinefelter syndrome, which is 50 times the expected frequency.53 These tumors are often malignant, are of the choriocarcinoma type, secrete β-hCG, and produce precocious puberty. Histiocytosis and leukemia are also associated with mediastinal teratoma, both with and without Klinefelter syndrome.56–58 Other hematologic malignancies, such Hodgkin disease, occur rarely.59
The following rare associations have been reported, most often with nonsacrococcygeal lesions: trisomy 13, trisomy 21, Morgagni hernia, congenital heart defects, Beckwith–Wiedemann syndrome, pterygium, cleft lip and palate, and rare syndromes, such as Proteus and Schinzel-Giedion syndromes.49,50,60–64
Diagnosis and Management by Tumor Site
Sacrococcygeal Teratoma
SCTs account for 35–60% of teratomas (gonadal included) in large series (Table 68-1).65–67 This is the most common tumor in the newborn, even when stillbirths are considered.49 The estimated incidence is 1 per 35,000 to 40,000 live births.3,16
TABLE 68-1
Relative Frequency of Teratomas by Site

Data are from five series of teratomas in children.
Modified from Dehner LP. Gonadal and extragonadal germ cell neoplasms: Teratomas in childhood. In: Finegold M, editor. Pathology of Neoplasia in Children and Adolescents. Philadelphia: WB Saunders; 1986. p. 282–312.
Diagnosis.
In countries where prenatal ultrasonography (US) is not readily available, most SCTs are seen as a visible mass at birth, making the diagnosis obvious (Fig. 68-4). Prenatal diagnosis has important implications and will be discussed further.
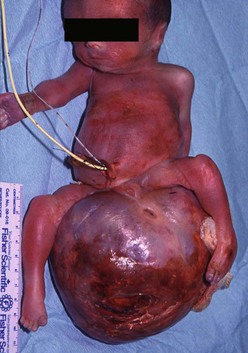
FIGURE 68-4 This infant was diagnosed with a large sacrococcygeal teratoma in utero. Within days, premature labor occurred, prompting cesarian delivery at 25 weeks gestation. The baby died despite resuscitation attempts.
There is an unexplained female preponderance of 3 : 1.34,65,67 The main differential diagnosis is meningocele. Typically, meningoceles occur cephalad to the sacrum and are covered by dura, but sometimes they are covered by skin. Examination of the child reveals bulging of the fontanelle with gentle pressure on a sacral meningocele, helping to establish the diagnosis before plain radiography, ultrasound, and magnetic resonance imaging (MRI) confirms it. The co-existence of meningocele with teratoma is well recognized in the familial form, but these are usually presacral. Rarely, a typical exophytic teratoma may have an intradural extension.68,69 Other lesions in the differential diagnosis of neonatal sacrococcygeal masses include lymphangiomas, lipomas, tail-like remnants (Fig. 68-5), meconium pseudocysts, rectal duplications, and several other rare conditions.34,70–73
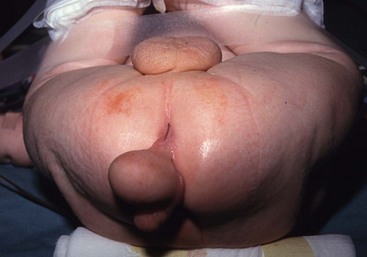
FIGURE 68-5 This patient had a scrotum-like perianal mass with anal stenosis at birth. An anoplasty was done with removal of the mass, which was not attached to the coccyx. Pathology showed only fibroadipose tissue with smooth muscle, vascular structures, and cartilage, consistent with a hamartomatous process or caudal vestige (also called a tail remnant).
Although many neonates with SCTs do not have symptoms, some require intensive care because of prematurity, high-output cardiac failure, disseminated intravascular coagulation, and tumor rupture or bleeding within the tumor.74–78 Lethal hyperkalemia from tumor necrosis has also been described.79 Lesions with a large intrapelvic component may cause urinary obstruction.78,80 Besides looking for signs of a myelomeningocele, the physical examination should always include a rectal examination to evaluate for an intrapelvic component. The most helpful imaging studies consist of plain anteroposterior and lateral radiographs of the pelvis and spine, looking for calcifications in the tumor and for spinal defects, and ultrasound of the abdomen, pelvis, and spine. Further preoperative studies are unnecessary in most newborns.
The diagnosis of purely intrapelvic teratomas is often delayed.34 Children develop constipation, urinary retention, an abdominal mass, or symptoms of malignancy, such as failure to thrive. Age is a predictor of malignancy in patients with testicular, mediastinal, and SCTs.2 The risk of malignancy is less than 10% at birth, but rises to more than 75% after age 1 year for SCTs, with the exception of familial presacral teratomas. The risk of malignancy also is high for incompletely excised lesions. Complete excision of the tumor should be carried out as soon as the neonate is stable enough to undergo the operation. Serum markers should be determined before the operation for later comparison.
In recent decades, the diagnosis has often been made on prenatal ultrasound, especially when this examination is routinely performed in the second trimester. The site of the lesion, its complex appearance, and intrapelvic extension, with or without urinary tract obstruction, are easily recognized. Although most small teratomas do not adversely affect the fetus, the presence of a large solid vascular tumor is associated with a significant mortality rate, both in utero and perinatally.75,76,78,81–83 Perinatal mortality is usually related to prematurity or tumor rupture with exsanguination (or both). Premature delivery may occur spontaneously from polyhydramnios or may be induced urgently because of fetal distress or maternal pre-eclampsia.
Repeated ultrasound assessment of tumor size is important because the fetus should be delivered by cesarean section if the tumor is larger than 5 cm or larger than the fetal biparietal diameter.84 Dystocia during vaginal delivery is associated with tumor rupture and hemorrhage, and is an avoidable obstetric complication. The options in managing unexpected cases with dystocia include emergency cesarean completion of the partially delivered fetus, who has been intubated and ventilated after vaginal presentation of the head.84
Polyhydramnios with larger tumors may lead to premature labor; amnioreduction is often required to decrease uterine irritability.78,83 Tumors that are larger than the fetal biparietal diameter at diagnosis, or that grow faster than the fetus or grow faster than 150 mL/week, are associated with a poor prognosis.82,85 As the tumor enlarges, the fetus may develop placentomegaly or hydrops, caused by high-output cardiac failure from vascular shunting within the tumor, with fetal anemia from intratumoral bleeding also playing a role. This is a harbinger of impending fetal death and should lead to urgent cesarean delivery, especially when the maternal mirror syndrome has developed.86 Open fetal surgical excision/debulking is one option in fetuses considered too premature to deliver. It has been performed with success in three of eight cases in one center and in three of four cases in another.76,87 Interestingly, there were no cases of fetal resection in the latter center in a more recent cohort of 23 fetuses with SCT, emphasizing that the indications for such a procedure are limited.82
As a result of the maternal and fetal risks, less invasive therapeutic options have been sought. Successful intrauterine endoscopic laser ablation of the large feeding arteries has been described.88 There have been several other reports, a few with good outcomes, but some with poor outcomes often related to severe fetal anemia and cardiac failure.78,89,90 Alcohol injection has also been used.78 Attempts at interrupting the high vascular flow have also been described by using radiofrequency ablation, with two survivors in the first four attempts.91 One survivor had significant perineal damage.92 As the technology has improved, there seems to be a renewed interest in this approach.93–95 However, it remains unclear when early delivery is preferable to intervention in utero in fetuses with early hydrops and placentomegaly. Some advocate a fetal operation before 30 weeks of gestation,82 with the same group recently suggesting early delivery in selected fetuses after 27 weeks of gestation.96 Others have reported survival after emergency delivery as early as 26 weeks of gestation.97 The EXIT procedure (ex utero intrapartum treatment) has been used as an adjunct to allow safer resection of a ruptured teratoma in a 27 week gestation fetus, but long-term neurological sequelae were noted.96
Purely cystic teratomas occur in 10–15% of cases. Prenatal diagnosis allows percutaneous aspiration to facilitate delivery (Fig. 68-6), to decrease uterine irritability, or to prevent tumor rupture at delivery.76,78,81,98,99 In another report, prenatal decompression using a cyst-amniotic shunt was successfully performed to relieve the obstructive uropathy caused by the cystic teratoma.100 Fetal MRI is a useful adjunct to the prenatal evaluation, providing additional information that aids in counseling and preoperative planning.101 It also helps in the differential diagnosis when the teratoma is entirely presacral.102
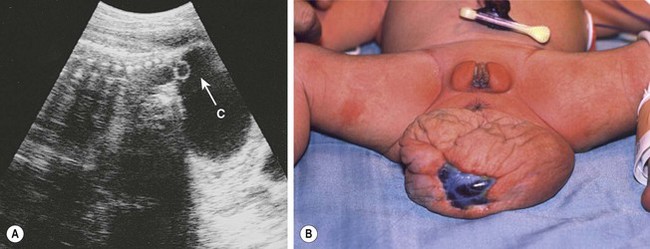
FIGURE 68-6 (A) Ultrasound image of a female fetus at 38 weeks of gestation, showing a large cystic mass (C) attached to the coccyx, with tiny cysts anterior to the sacrum (arrow). An ultrasound evaluation at 18 weeks was normal. The cyst was gradually enlarging from an initial diameter of 9.5 cm at 31 weeks of gestation. The cyst was aspirated for 650 mL of fluid, permitting external rotation from breech to the vertex position. Two days later, when labor was induced, another 200 mL of fluid was removed to permit an uncomplicated vaginal delivery. (B) Twenty-four hours postnatally, the lesion remained floppy with an area of skin ulceration, likely a consequence of excessive in utero distention. A mature cystic teratoma was confirmed histologically.
Operative Approach.
For most tumors, the major component is extrapelvic and the patient is placed in the prone position. If a significant intrapelvic or intra-abdominal component is present, or if the tumor is highly vascular and bleeding within the tumor is suspected, it may be wise to begin with a laparotomy or laparoscopy (see above). Generally, most resections can be achieved completely in the prone position, especially if the internal portion is cystic (Fig. 68-7).78 When in doubt, a safe approach is to prepare the skin from the lower chest to the toes, allowing the infant to be turned to the supine position without having to re-drape. Vaseline packing in the rectum facilitates its identification throughout the procedure. En bloc excision, including the coccyx, is preferable. Failure to remove the coccyx is associated with a high recurrence rate.2,3,103 An acceptable gluteal crease and perineum is formed by the appropriate positioning of the perianal musculature. The use of plastic surgical principles to close the skin improves the cosmetic appearance of the scar (see Fig. 68-7, inset).104
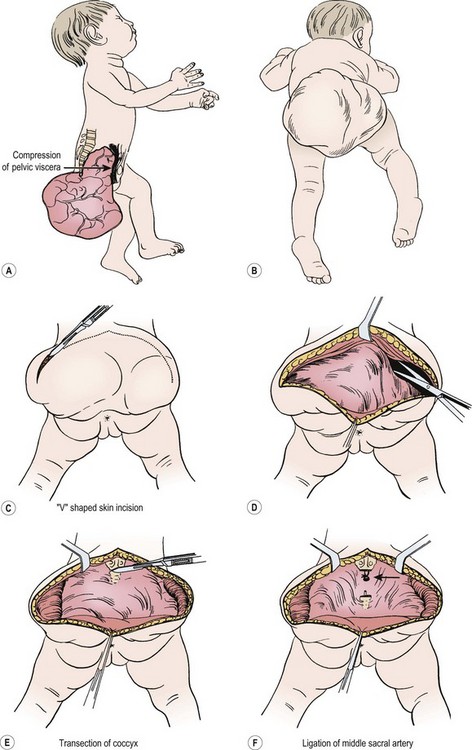
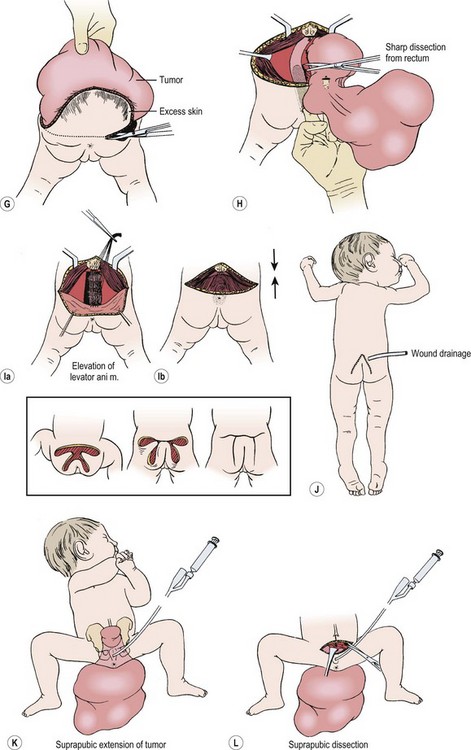
FIGURE 68-7 (A) The teratomatous attachment may compress the rectum, vagina, and bladder anteriorly. (B) The patient is placed on the operating table in a prone jackknife position, with general endotracheal anesthesia. An appropriate intravenous cannula should be placed in an arm vein. (C) The incision is an inverted V-shape to allow excision of the tumor and to facilitate an eventually satisfactory cosmetic closure. The amount of skin excised is dependent on the size and shape of the tumor. (D) The tumor is dissected from the gluteus maximus muscle. (E) The coccyx is transected and removed in continuity with the tumor. (F) The middle sacral artery is the major blood supply to the tumor and is ligated (arrow) after transection of the coccyx. (G) Excess skin is excised to facilitate closure. (H) As the tumor is adherent to the rectum, sharp dissection can be directed by placing a finger in the rectum. (I) Placement of sutures between the anal sphincter and the presacral fascia (a). When the sutures are tied, the anal sphincter is pulled upward to the sacrum to form a gluteal crease (b). (J) A drain is left in the surgical site for drainage of postoperative serosanguineous fluid. Inset, Recently described technique for closure after excision of large teratomas. Using plastic surgery principles, this avoids ‘dog ears’ and places the scars along natural skin lines for a much-improved long-term cosmetic result. (K) If the tumor extends through the bony pelvis into the retroperitoneum, a urinary bladder catheter is inserted to facilitate suprapubic dissection. (L) Lower abdominal transverse incision allows interruption of the middle sacral artery and dissection of the tumor from the sacrum and pelvis, which is eventually removed from the perineum. (Inset redrawn from Fishman SJ, Jennings RW, Johnson SM, et al. Contouring buttock reconstruction after sacrococcygeal teratoma resection. J Pediatr Surg 2004;39:439–41; with permission.)
Although the chevron incision has been used by most surgeons, a vertical incision is sometimes advantageous. It is preferred for smaller teratomas because it leaves a nearly normal-looking median raphe (Fig. 68-8). Resection of the excess skin at the closure gives an optimal cosmetic result. Others have reported using this approach for large tumors as well.105
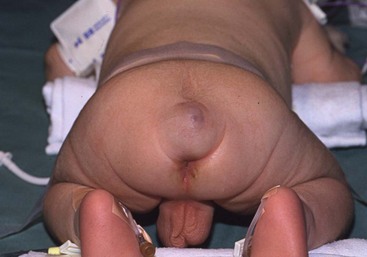
FIGURE 68-8 Smaller cystic teratoma at age 1 month, initially mistaken for a hemangioma owing to its soft compressible nature and bluish skin discoloration. This tumor lends itself well to a longitudinal elliptical excision with midline closure, as in a posterior sagittal anorectoplasty.
Several techniques have been described to help in the management of giant SCTs. These include intraoperative snaring of the aorta,77 laparoscopic division of the median sacral artery,106,107 the use of extracorporeal membrane oxygenation and hypothermic perfusion,108 devascularization and staged resection,97,109 and preoperative embolization with or without radiofrequency ablation.110,111 Autologous cord blood transfusion is another useful adjunct.112
Prognosis.
Fetuses with an SCT diagnosed in utero have a survival rate in excess of 90% if the tumor is small and discovered by routine prenatal ultrasound. If a complicated pregnancy is the indication for ultrasound evaluation, the mortality increases to 60%. Nearly 100% of patients die when hydrops or placentomegaly develop.76,78,81,113 Dystocia or tumor rupture during delivery are likely underreported as a cause of mortality.83,114 In one series, 10% of babies died during transfer, all before the widespread use of antenatal ultrasound.115 In a report describing 24 patients with a SCT diagnosed on routine obstetric ultrasound, 3 were aborted electively, four died in utero at 20 to 27 weeks of gestation, and three died of tumor rupture during delivery at 29 to 35 weeks of gestation (one after vaginal and two after cesarean delivery).75 The incidence of placentomegaly (none), hydrops (5%), and polyhydramnios (19%) was lower than in series in which ultrasound was performed for an obstetric reason.76,84,116 Tumor size, vascularity, and content were used to develop a prognostic classification from a cohort of 44 fetal SCTs.117 Fifty per cent of infants with tumors 10 cm or greater that were highly vascular or fast growing died, whereas none died if these features were absent or if the tumor was predominantly cystic. Recently, a growth rate >150 mL/week was found to be associated with an increased risk of perinatal mortality.82 A vena cava diameter ≥6 mm and cardiac output ≥600 mL/kg/min have also been used to detect high-output cardiac failure in the early stages.82 Another prognostic classification involves the ratio of solid tumor volume over the head volume.118 In a series of 28 cases, none of the fetuses with STV/HV ratio <1 died, while 61% with a ratio >1 died. The same center has also reported risk stratification based on fetal echocardiographic findings.119 Finally, the ratio of tumor volume over fetal weight was found to be an early prognostic marker.120
In the absence of severe prematurity and perinatal complications, the prognosis is dependent on the presence of malignancy and is therefore related to age at operation and completeness of resection.121,122 When the tumor is benign and completely excised, recurrence is low, unless the tumor is large and mostly solid.123 The recurrent tumor may be benign or malignant, and benign metastatic tissue may become evident in lymph nodes.124 Although immature or fetal elements in gonadal teratomas are associated with a higher risk of aggressive behavior, this is not considered true for SCTs.2,5,16 However, a multicenter study identified immature histology, in addition to malignancy and incomplete resection, as risk factors for recurrence.122 Although malignant recurrence of a ‘benign’ teratoma may be as high as 10–15%,6,122 the original benign diagnosis may have been due to sampling error,15 an undetected residual microscopic focus of malignant tumor,125 or secondary to incomplete coccygectomy at the initial operation.35 Patients whose tumors are resected after the newborn period have a higher risk of malignant recurrence, especially when an elevated AFP level is present at diagnosis. The elevated AFP likely signifies the presence of malignancy in the original tumor.15,126 It is important to monitor all patients by physical examination, including rectal examination and serum markers (AFP and CA 125), every two or three months for at least 3 years because most recurrences occur within three years of operation.24,127
Recurrent disease is usually local, but metastases to inguinal nodes, lung, liver, brain, and peritoneum can occur, including pseudomyxoma peritonei.128 The prognosis of a malignant tumor or a malignant recurrence was dismal until the advent of platinum-based chemotherapy.125 Survival rates of 80–90% are now achieved, even in the presence of metastatic disease, but the risk of late recurrences or second malignancies persists.129–131 Older patients presenting with large malignant tumors usually undergo biopsy followed by chemotherapy before resection is attempted to avoid sacrificing vital structures.16,130,132
A Children’s Cancer Group (CCG) review illustrates the shift in mortality causes, from late diagnosis/malignancy to perinatal events.129 The mortality was 10% in 126 patients treated in 15 institutions from 1972 to 1994. Three patients died of severe associated anomalies. Two died of hemorrhagic shock postoperatively and six due to combinations of severe prematurity, birth asphyxia due to failed vaginal delivery, or preoperative tumor rupture. Death from a metastatic YST occurred in one patient. A second patient with metastatic disease was lost to follow-up and is presumed dead. Thus, only two deaths occurred from malignancy, despite a total of 20 YSTs (13 malignant at initial operation, seven malignant recurrences after resection of ‘benign’ teratomas). Owing to the effectiveness of current chemotherapy in treating recurrent disease, as well as its toxicity in young infants, it appears that a completely excised malignant YST does not require adjuvant therapy. These patients should be closely monitored clinically and with serial AFP measurements.133 Similar encouraging results have been found in the German Cooperative Studies.134
In the current era of the rather routine use of ultrasound in pregnancy, the prognosis for patients with an SCT is not dependent on Altman’s classification34 but rather on tumor size, physiologic consequences, histology, and associated anomalies. The prognosis of malignant tumors depends on tumor type, stage, location, and patient age.129 Functional results in survivors have been reported as excellent in most series.78,135,136 However, several reports draw attention to fecal and urinary continence problems, as well as lower limb weakness.121,137–141 Some of these problems are clearly related to associated anomalies,115 the need for reoperation,142 or to the presence of large presacral or intra-abdominal tumors,80,135 but they can occur after excision of purely extrapelvic benign tumors. A good outcome requires meticulous dissection along the tumor capsule, preservation and reconstruction of muscular structures, and long-term follow-up. One group advocates earlier cesarean delivery to minimize urologic sequelae in patients with large tumors causing urinary tract dilatation.139 Others have placed vesico-amniotic shunts in such cases with good outcomes.78,100 Urodynamic studies and surveillance ultrasound are now being performed more regularly in some centers.143 Patients with Currarino syndrome, who often have a tethered cord in addition to the presacral tumor, appear to have an increased risk of bladder and bowel dysfunction.144,145 A poor cosmetic result was noted in more than half of the patients in another review.146 These authors advocate early assessment of bladder, anorectal, and sexual function along with cosmetic results within a structured oncology follow-up program. The technique described by the Boston Children’s group is a major step to improve cosmesis (see Fig. 68-7),104 while others favor a sagittal incision.105
Thoracic Teratomas
The anterior mediastinum is the most common site of thoracic teratomas, which account for 7–10% of all teratomas (see Table 68-1).2,3
Mediastinal Teratomas.
Mediastinal teratomas are diagnosed from the fetal period to adolescence and even adulthood.2,147–149 Most are located in the anterior mediastinum, but a few have been described in the posterior mediastinum, some with epidural extension.150–152 In infants, respiratory distress is a common presenting manifestation,153 but in older children the teratoma is often an incidental finding on chest radiograph (Fig. 68-9).2 Any patient with a mediastinal mass that presents with orthopnea or a reduction in the tracheal cross-sectional diameter of greater than 50% on axial imaging is at a significant risk for airway collapse during general anesthesia.154 In these cases, procedures using local or regional anesthesia may be required to obtain a confirmatory diagnosis. Mediastinal teratomas may be first seen as a chest wall tumor and may even erode through the skin. They also can erode into a bronchus, with hemoptysis as the initial manifestation,155 or rupture into the pleural cavity.156 Secondary pericardial effusion and tamponade have also been described.157 A strong association has been found with Klinefelter syndrome. In these cases, choriocarcinoma in the teratoma often leads to precocious puberty (see under Associated Anomalies).2,25,58 Mediastinal germ cell tumors have also been observed concurrently or after treatment for hematologic malignancies such as Langerhan’s cell histiocytosis and hemophagocytic syndrome.57,158,159 Histologically, the presence of immature tissue does not affect the prognosis in children younger than 15 years. After age 15, mediastinal teratomas have a high incidence of malignant behavior, which is usually indicated by elevated levels of AFP or β-hCG, or both.149 For those with malignancy, YST is most prevalent in girls and young boys while mixed histology prevails in over 50% of older adolescent males.160
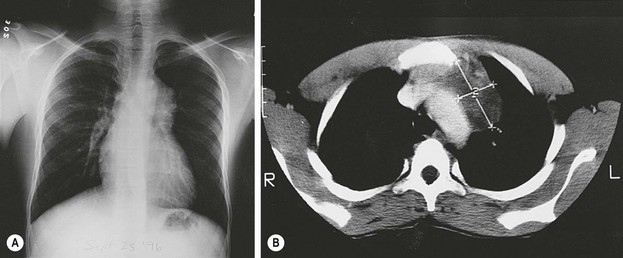
FIGURE 68-9 (A) A 13-year-old African boy has an asymptomatic anterior mediastinal mass that was discovered on routine immigration chest radiograph. (B) The CT scan shows a heterogeneous mass adjacent to the aorta, suggestive of a neoplasm (thymoma or lymphoma). During consideration of a fine-needle aspiration biopsy, ultrasonography was done and suggested the presence of cysts with debris (not shown). MRI (not shown) confirmed the presence of the cystic components as well as fat. A mature teratoma was excised through a small left anterior mediastinotomy, removing the left second costal cartilage.
Tumors should be excised through either a median sternotomy or a thoracotomy.16,161 Smaller tumors may be approached through an anterior mediastinotomy (see Fig. 68-9) or by thoracoscopy,162 although tumor seeding is a concern with the latter. Complete resection is the goal, but often these masses are too large at presentation and require initial biopsy followed by chemotherapy.16,163,164 During chemotherapy, attention should be paid to the ‘growing teratoma syndrome’ that occurs when the benign elements within a germ cell tumor continue to grow.165 A Pediatric Oncology Group (POG) and Children Cancer Group (CCG) intergroup study involving 38 patients demonstrated that primary resection could be achieved in 14 patients.160 In another 18, chemotherapy reduced tumor size in 57% while the remainder were stable or increased in size. All patients with residual disease underwent successful post-chemotherapy resection. The overall survival for malignant mediastinal germ cell tumors was 71% which is less than for other extragonadal sites. The prognosis is best for young children with YSTs and worst in older adolescents with mixed germ cell histology.149,160 In a recent series from Memorial Sloan-Kettering Cancer Center, normalization or reduction in preoperative tumor markers was the strongest predictor of increased survival.161
Intrapericardial Teratomas.
Intrapericardial teratomas are most commonly seen in the newborn period or in utero, with evidence of cardiorespiratory distress secondary to pericardial effusions or nonimmune fetal hydrops resulting from cardiac compression.2,3,166 While a fetal diagnosis allows for early postnatal treatment in most newborns, it may also offer an opportunity for antenatal interventions such as fetal pericardiocentesis.167 Early delivery for emergency surgical excision should be considered if the baby develops signs of cardiac tamponade.168 Intrapericardial teratomas are also the leading cause of massive pericardial effusion in the neonate and any delays in diagnosis could be fatal.3,169 In older infants, it may present with respiratory distress or poor feeding. Ultrasound imaging usually demonstrates a cystic or solid teratoma located anterior to the right atrium and ventricle with attachments to the great vessels (see Fig. 25-4).170 The tumor may also be found incidentally on chest radiographs performed for other reasons. The only treatment option for this lesion is excision. On histologic examination, these teratomas are usually composed of mature tissue with or without neuroglial elements.2,3
Abdominal Teratomas
Retroperitoneal Teratomas.
Retroperitoneal teratomas occur outside the pelvis, often in a suprarenal location. They represent about 4% of all childhood teratomas, and 75% occur in children younger than 5 years.3,173 They occur twice as frequently in females and may have an association with Klinefelter syndrome.16,55 While more than 90% are benign, up to one-quarter may be malignant when diagnosed in the first month of life.174 Usually the tumor is discovered as an abdominal mass that compresses the gastrointestinal tract, causing symptoms such as vomiting, difficulty feeding and constipation.163 Presentation with an acute abdomen from infection has also been described.175 Abdominal radiographs may show calcifications or bony structures within the tumor (Fig. 68-10). ultrasound, CT scan, or MRI, and the assessment of serum markers are the essential aspects of preoperative evaluation. Operative excision is usually straightforward,176,177 but occasionally the tumor may encase major vessels, making resection difficult.178 Malignant lesions present as bulky masses that are not easily resectable.163 In a POG/CCG intergroup study involving 25 patients, only five could be resected primarily.179 The remainder underwent initial biopsy and platinum-based chemotherapy, of which four did not require further surgery. The remaining patients in this series underwent total or partial resection with a 6-year event-free survival of over 80% and an overall survival close to 90%. The retroperitoneum is also the most common site for the fetus in fetu malformations and intermediate fetiform teratomas.2,3,180,181
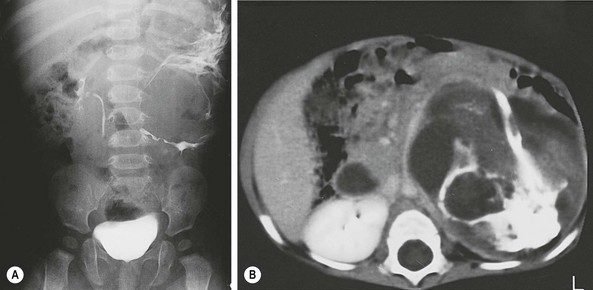
FIGURE 68-10 This 7-month-old girl was found to have an abdominal mass on physical examination. (A) Plain radiograph films showed a large calcified left upper quadrant mass, which can be seen to displace the kidney inferiorly after injection of intravenous contrast. Ultrasonography (not shown) revealed multiple cystic areas. (B) This was confirmed by CT, which also revealed areas of fat density, making teratoma much more likely than neuroblastoma. The mature teratoma contained all types of cerebral and cerebellar tissues; respiratory, transitional, and squamous epithelium; sebaceous and salivary glands; smooth muscle; cartilage; and fat. Serum markers were normal.
Gastric Teratomas.
Gastric teratomas are rare lesions seen most commonly in infant boys.2,182 They account for 1% of all teratomas. Clinically, the tumors present with hematemesis or vomiting due to gastric-outlet obstruction in the postnatal period, but rapid growth in utero has also been described leading to fetal distress and emergency cesarean delivery.183 A palpable mass is common. The tumor is usually an exophytic mass in the lesser curvature or posterior wall of the stomach, but the whole stomach may be involved. Most gastric teratomas are benign with mature and immature elements, most frequently containing neuroglial tissue. Benign peritoneal gliomatosis has been found incidentally in hernia sacs after resection of a gastric teratoma as an infant, illustrating the unusual behavior of some of these tumors.184 Excision is curative. Recurrence and malignancy are rare, despite local infiltration or nodal metastasis.182,185 Periodic follow-up including AFP measurements is nevertheless important.186
Head and Neck Teratomas
More than 10% of teratomas in children originate from the neck, head, and central nervous system.2,3,193,194 Many of these tumors are now diagnosed with prenatal ultrasound (Fig. 68-11). They are associated with an increased incidence of stillbirth, especially in the absence of prenatal diagnosis.174,195
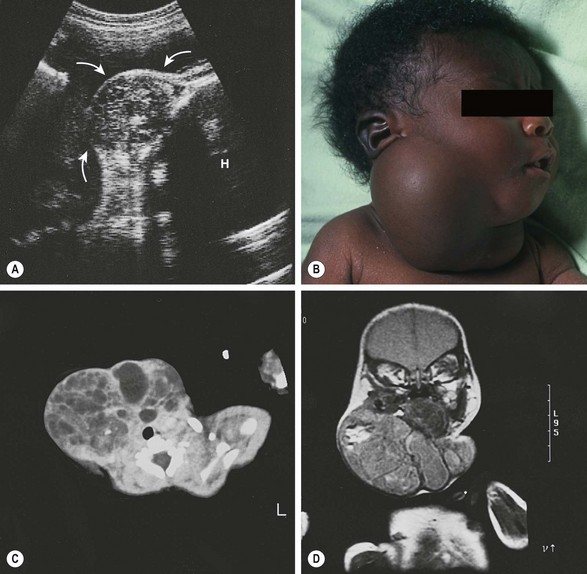
FIGURE 68-11 (A) Fetal ultrasound image at 34 weeks of gestation shows a 7 cm cervical mass of mixed echogenicity, containing blood flow by Doppler (arrows point to the mass; H, head). (B) After birth by cesarean section, the neonate had only mild tachypnea despite the large right cervical mass extending to the left side. Parts of the mass transilluminated, suggesting the diagnosis of a lymphangioma. (C) CT scan of the lower cervical area shows an intact trachea and multiple cysts in the mass. (D) MRI confirmed the presence of fat, which appears bright on T1-weighted images as well as on the proton density–weighted imaging sequence shown here. At operation, the mass appeared to originate from the right lobe of the thyroid gland. It contained epithelium-lined cysts, cartilage, bone, glandular tissue, and complex papillary structures. A predominance of neuroepithelial tissue was found with a few small areas of immature, neuroblastoma-like tissue. Preoperative vanillylmandelic acid levels were normal. The patient required postoperative thyroid hormone supplementation because of subclinical hypothyroidism (elevated thyroid-stimulating hormone with normal thyroxine and triiodothyronine levels).
Cervical Teratomas.
Cervical teratomas represent up to 8% of all teratomas.18,195 These tumors are initially seen as a partly or completely cystic neck mass. Large teratomas often lead to severe polyhydramnios, presumably because of esophageal compression; in turn, this may lead to premature labor. Serial amnioreduction may be required to prevent this complication (see Fig. 68-12). Tracheal deviation and/or obstruction may also be identified on prenatal imaging studies such as fetal MRI,196 thus necessitating a strategy to secure the airway immediately after birth. Indeed, death may result in the inability to intubate a severely compressed or deviated trachea.18,197 Predictors of airway obstruction based on prenatal ultrasound have also been identified, including the presence of polyhydramnios, a presumptive diagnosis of teratoma as well as a tracheoesophageal displacement index greater than 12.198 Prenatal diagnosis permits cesarean section and the establishment of an airway by the surgical team before the cord is clamped.197 Refinements of this technique (EXIT procedure) have been well documented in a series of 87 infants from Children’s Hospital of Philadelphia, of whom 17 had giant cervical teratomas.199 Extension of tumor to the mediastinum or displacement of the trachea and carina may cause pulmonary hypoplasia, which increases respiratory morbidity and mortality.200 The tumor is usually well defined and may contain calcifications. The differential diagnosis includes cervical lymphangioma, congenital goiter, foregut duplication cyst, branchial cleft cyst, or rarely cystic neuroblastoma (Fig. 68-12).166,169 Postnatal investigations should include plain radiographs, ultrasound, and a measurement of AFP and β-hCG, as well as urinary catecholamine metabolites. CT and MRI may be useful adjuncts to establish the diagnosis and to define anatomic relationships.
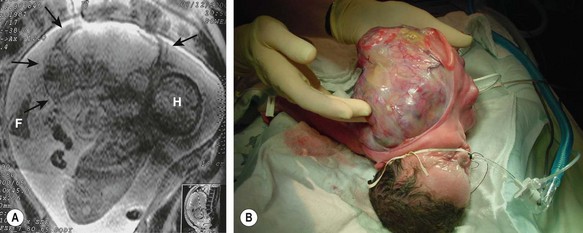
FIGURE 68-12 (A) A fetus has a giant cervical teratoma which is seen on magnetic resonance imaging at 30 weeks of gestation. The lesion was first diagnosed on routine ultrasonography at 18 weeks and grew much faster than the fetus. Several amnioreductions were required because of polyhydramnios, and an EXIT procedure was performed at 34 weeks of gestation (H, head; F, foot; arrows outline the tumor). (B) After securing the airway during the EXIT procedure and stabilizing the neonate, resection was completed successfully. During resection, it was found that the left carotid artery and jugular vein entered the tumor. The left vagus nerve (hence the left recurrent nerve) was never seen, and the left glossopharyngeal nerve also was sacrificed. The tumor originated from the left pharyngeal wall. Including the fluid within the cystic parts, the teratoma weighed 1.4 kg, and the neonate weighed 1.6 kg postoperatively.
Complete excision is accomplished through a wide collar incision. The tumor is usually not difficult to separate from the strap muscles and the fascial planes, but the pretracheal fascia is sometimes very adherent. Often, the site of origin cannot be identified, yet in many instances, the tumor is firmly attached to and appears to originate from the thyroid gland. A thyroid lobectomy should be performed in these cases. In other instances, the tumor is adherent to the pharynx. In these cases, meticulous dissection and pharyngotomy, if necessary, are important to prevent tumor recurrence. Giant teratomas may distort the anatomy, leading to permanent sequelae. Generally, the tumor is composed of both mature and immature neuroglial tissue, but cartilage and bronchial epithelium are not uncommon.18,201 In 35% of cases, the tumor contains thyroid tissue, and hypothyroidism is a well-known postoperative complication.202 Cervical teratomas are usually benign, but malignancy has been reported, even in infants. A report from the CCG showed that 20% of tumors clearly contained malignant elements, most often neuroblastoma, but also teratocarcinoma, neuroblastoma-like tumor, and neuroectodermal tumor.18 Complete excision in the newborn period results in a survival rate of 80–90%. As for teratomas in other sites, one newborn was reported with a benign thyroid teratoma accompanied by neuroglial tissue deposits in cervical nodes.203 A prognostic classification for cervical teratomas takes into account birth status, age at diagnosis, and presence of respiratory distress.201 In neonates without respiratory distress, the mortality rate was 2.7%, compared with 43.4% in those with respiratory compromise. Prenatal diagnosis and delivery using the EXIT procedure can undoubtedly increase survival.199
Thyroid teratomas may be present in older children and adults, and are often malignant in the latter.204,205 Spindle epithelial tumor with thymus-like elements (SETTLE) is a malignant thyroid neoplasm that can sometimes be confused with thyroid teratomas due to their spindle cell and epithelial components.206 These unique neoplasms have a propensity for delayed metastases, particularly to the lungs, kidney, mediastinum, lymph nodes, liver and vertebrae. Adjuvant chemotherapy and radiotherapy do not seem to have a significant impact on overall outcome for metastatic disease. In these cases, survival is poor.206
Craniofacial Teratomas.
Craniofacial teratomas include a spectrum of lesions that may be life-threatening.
Epignathus.
Epignathus is a term used to describe teratomas protruding from the mouth (Fig. 68-13). These tumors arise from the soft or hard palate in the region of Rathke’s pouch.207,208 They generally fill the oral cavity and extend out through the mouth. They can prevent fetal swallowing, which leads to polyhydramnios. An EXIT procedure may be required.209 Surgical excision is mandatory. These lesions are usually benign, and recurrence is rare. A high degree of organization often gives them the appearance of a parasitic fetus. Prenatally, the differential diagnosis includes a large epulis, a benign lesion which will be discussed further in this chapter.210
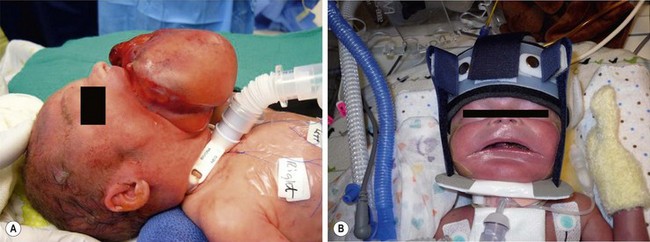
FIGURE 68-13 On prenatal ultrasound, this baby was found to have a large epignathus and was born via an EXIT approach. (A) A tracheostomy was performed during the EXIT procedure. (B) The epignathus has been excised and the baby is in a mandibular molding device to help re-shape the mandible which was splayed out due to the large intraoral mass.
Nasopharyngeal Teratomas.
Nasopharyngeal teratomas arise from the posterior aspect of the nasopharynx.211 Large tumors can interfere with fetal swallowing and produce polyhydramnios, cause severe respiratory distress at birth, and lead to stillbirth.2 Most pharyngeal teratomas are benign, and the treatment is excision.
Oropharyngeal Teratomas.
Oropharyngeal teratomas represent 2% of all teratomas.212 These tumors can originate from the tongue, sinuses, mandible, and tonsils. Airway compromise requires immediate care at the time of delivery. For oro- and nasopharyngeal teratomas, the EXIT procedure may be indicated when a large tumor detected prenatally appears to obstruct the airway.213 Most tumors are benign, and recurrence is uncommon after complete resection. Separate tumors may occur in the same infant, and intracranial extension has been described.214
Other Sites.
Craniofacial teratomas can be found in the orbit, the middle ear, and skull.215,216 Intracranial teratomas account for only 2–4% of all teratomas, but represent nearly 50% of brain tumors in the first two months of life.2,3 Most are benign in neonates but can be malignant in older children and young adults, and are often unresectable.217,218 These teratomas can appear in utero and cause massive hydrocephalus. Perinatal mortality is high, with only a 6% survival when diagnosed in the fetus or newborn.3,219 The pineal gland is the most common site of origin, but intracranial teratomas can be seen in different areas, such as the hypothalamus, ventricles, cavernous sinus, cerebellum, and suprasellar region.218,220 Pineal gland teratomas can secrete enough β-hCG to cause precocious puberty.2,219
Miscellaneous Sites
Teratomas have been reported in other sites, such as skin, parotid, vulva, perianal region well away from the coccyx, spinal canal, umbilical cord (possibly associated with omphalocele), and placenta.2,3,221–224 Some of the spinal teratomas are now considered to be enterogenous cysts.225
Dermoid, Epidermoid, and Related Cysts
Dermoid Cysts
Dermoid cysts are congenital cysts that are lined by skin with fully mature pilosebaceous structures.226 They are the result of sequestration of skin along lines of embryonic closure. Typical locations are under the lateral part of the eyebrow, the scalp, the glabella, the tip of the nose, and the orbit. The head and neck are the sites of predilection, but these lesions have been described in other midline sites, including the sacral area, perineal raphe, scrotum, and presternal area. The so-called dermoids of the ovary are, in fact, cystic teratomas and are discussed in Chapter 74.
Dermoids are usually round, soft, and often fixed to deep tissues or to bone. They usually present as a painless mass of 1–2 cm in diameter but can grow up to 4 cm or more if untreated. Some are associated with a sinus tract, especially those on the nose. This site is also typical for intracranial extension and a familial occurrence.227 Dermoids in the head area are usually deep to muscles and often cause an indentation in the outer table of the skull. They can even erode through both bony tables and extend intracranially. A skull radiograph may show the defect, but it may be normal if the cyst is situated over a fontanelle or an unfused suture. Further imaging is essential in those cases, and neurosurgical consultation is advisable.
Dermoids deep to the lateral part of the eyebrow are usually approached through an incision just above the eyebrow because an incision within the eyebrow leaves a more visible scar. Very good cosmetic results are achieved long-term.228 We have been impressed with an alternative incision through the palpebral crease.229,230 This requires a slightly longer incision and more retraction but leaves an invisible scar. This approach also has the advantage of allowing access to the orbit for the rare cases in which the cyst penetrates through the orbital bone in a dumbbell fashion (Fig. 68-14).229 Another alternative is an endoscopic approach through a scalp incision.231 This approach requires special instruments, is more indirect, and the scar posterior to the hairline may become visible later in life in balding males.
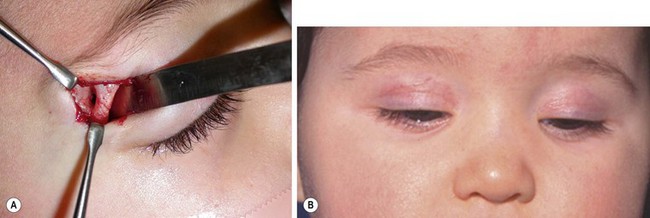
FIGURE 68-14 Two different infants with right supraorbital dermoid cysts, for which a palpebral skin incision was used for excision. This approach requires a slightly longer incision and retraction, but the scar is almost invisible. (A) In this patient the approach was ideal since the cyst had eroded into the orbit (courtesy of Patricia Bortoluzzi, MD). (B) A different patient shown at one month follow-up; the scar is invisible when the eyes are open.
Dermoids in the cervical area are usually midline, mostly suprahyoid or submental. Because they are deep within muscles, they tend to move with swallowing, just as thyroglossal duct cysts do. On ultrasound, they usually appear echogenic and are often misinterpreted as being solid rather than cystic. They can be differentiated intraoperatively by their yellowish appearance and their soft, buttery content with sebaceous material and hair (Fig. 68-15). This appearance alleviates the need for a hyoidectomy.
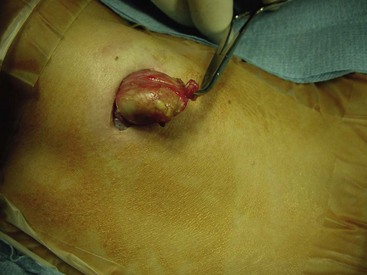
FIGURE 68-15 Dermoid cysts in the cervical region are found in the midline. They are usually suprahyoid or submental. This operative photograph shows a midline cervical dermoid cyst with its characteristic yellowish appearance. The head is to the left. The cyst consists of a soft, buttery, cheesy content with sebaceous material. Preoperatively, these lesions are usually confused with a thyroglossal duct cyst. However, removal of the hyoid bone is not needed if a dermoid cyst is found.
Epidermal Cysts
Epidermal or epidermoid cysts have a wall composed of true epidermis, as seen on the skin surface and in the infundibulum of hair follicles (hence, they also are called infundibular cysts).226 They do not contain pilosebaceous units or hair. Some have a congenital origin like dermoid cysts, whereas others are acquired, either spontaneously arising from hair follicles or secondary to trauma with implantation of epidermis into the dermis or subcutaneous tissue.
Epidermal cysts are slow growing and are formed by the desquamation of epithelial cells. They are round, intradermal, or subcutaneous lesions that stop growing after having reached 1–5 cm in diameter. They occur most commonly on the face, scalp, neck, and trunk (i.e., hair-bearing areas). They may be associated with a small sinus tract or dimpling of the skin. In the neck and infraclavicular area, they may be confused with branchial cleft remnants. Preauricular sinuses and cysts are often considered epidermal cysts. Epidermoid cysts of the spleen are discussed in Chapter 47.
Some patients may have more than one cyst, but the presence of multiple cysts, especially on the scalp and face, should raise the possibility of Gardner syndrome.226,232,233
Treatment is excision, which often can be achieved under local anesthesia, even in young children. Preauricular cysts are better excised under general anesthesia, owing to their deep attachment to the helix cartilage.234 Spontaneous rupture of any epidermal cyst leads to an intense foreign-body reaction, and the child presents with an abscess-like mass. This may require incision and drainage but often can be treated with antibiotics and local warm compresses. This mode of presentation increases the risk of cyst recurrence after excision, and often results in a wider scar than would have occurred with earlier excision. Infection of the cyst also may be caused by bacteria tracking along the small sinus tract that is sometimes present. These lesions rarely degenerate to epidermoid or basal cell carcinomas.233 The treatment of small preauricular sinuses is controversial as they may remain asymptomatic, but generally excision is recommended with a palpable cyst or discharge of material from the sinus tract.
Trichilemmal Cysts
Trichilemmal cysts, also called pilar or sebaceous cysts, are thought to arise from hair follicles.226 Most are acquired and appear in adulthood. They often show an autosomal dominant inheritance pattern and are solitary in only 30% of the cases. Some authors classify these as epidermal cysts that occur on the scalp.233
Soft Tissue and Nerve Tumors
Numerous soft tissue tumors have been described in children and are of mainly ectodermal and mesodermal origin. Some of these neoplasms are classified in Table 68-2. Only those soft tissue tumors likely to be encountered by pediatric surgeons are discussed here. More extensive discussions of soft tissue tumors are available.235 Many benign soft tissue tumors are cutaneous or subcutaneous and are amenable to excision under local anesthesia.

Epidermal and Adnexal Tumors
Pyogenic Granulomas
Pyogenic granulomas (also known as lobular capillary hemangiomas) are solitary polypoid capillary lesions often associated with trauma or local irritation. They are commonly found on the skin as red, raised, occasionally bleeding lesions or in the mouth in association with pregnancy.236 They are usually treated with topical silver nitrate or liquid nitrogen or ligature of the polyp neck. Rarely, excision or electrocautery is needed.
Warts
Warts are uncommon before age 4 years but are a common pediatric complaint. Various subtypes of human papillomavirus affect different body areas. Verrucae spread through families, sports teams, and schoolmates, and are most common on the hands and feet. Topical treatment includes salicylic or trichloroacetic acid, liquid nitrogen, or fine-tip electrocautery. A Cochrane review on topical treatment suggested cryotherapy may be only equivalent to older remedies.237 Excision is occasionally required.
Condylomata acuminata occur in the perineal skin and suggest, but do not prove, child sexual abuse. The virus may be transmitted by hand contact during diaper changes in infants or acquired at birth during vaginal delivery, but the lesions may take months to develop. One study suggested that sexual abuse is an unlikely source of transmission in children younger than 3 years if no other signs of abuse are found.238 These lesions have a core of connective tissue covered in epithelium, occurring as solitary or cauliflower-like lesions. Spontaneous regression is known, but topical podophyllin may be required. Some cases may need cauterization under general anesthesia as a thorough rectoscopic and vaginoscopic assessment can also be performed.
Calcifying Epithelioma of Malherbe
The calcifying epithelioma of Malherbe or pilomatrixoma is a solitary benign calcifying tumor of hair follicles. This is one of the most common acquired soft tissue lesions in children. Clinically, a circumscribed, firm, intracutaneous or subcutaneous nodule is palpable, with occasional yellowish or bluish coloration (Fig. 68-16).227 These lesions are most common before age 20 years, and 60– 70% are found in the head and neck region.236 Extremities are also common locations, followed by the trunk. Most are smaller than 1 cm, although lesions up to 4 cm have been reported.239 Only 2–3% are multiple, and in such cases there may be a familial predisposition and an association with Gardner syndrome.227 They are more common than sebaceous cysts in younger patients. Local excision is curative.
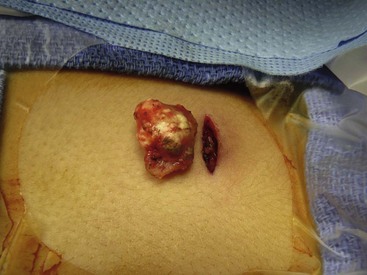
FIGURE 68-16 This operative photograph shows a pilomatrixoma (calcifying epithelioma of Malherbe). These lesions are circumscribed, are firm, and have a distinctive ‘knobby’ appearance. Once removed, they are seen to have a yellowish content. Most of these lesions are found in the head and neck region.
Malignant Epithelial Tumors
Malignant epithelial tumors are rare in children. General treatment principles include wide local excision and radiation therapy for certain high-risk tumors.240 The basal cell nevus syndrome, also known as Gorlin syndrome, is an autosomal dominant disease with basal cell lesions of the eyes, nose, and cheeks in association with anomalies of the mouth, skin, skeleton, central nervous system, eyes, and genitals.241 These patients may have concomitant xeroderma pigmentosum. Epidermoid cancers in pediatric transplant recipients also have been reported.242
Tumors of Nerve Tissue
Neurofibromas
A neurofibroma is a benign neoplasm of Schwann cells, usually of peripheral nerves. When multiple, or associated with multiple café-au-lait skin lesions, it suggests the presence of neurofibromatosis type 1 (NF-1) or von Recklinghausen disease, an autosomal dominant disorder due to a mutation of the NF-1 gene on chromosome 17.243 Diagnosis may be delayed but affected probands can be diagnosed early by genetic testing or in utero by ultrasound.244 Mucosal neurofibromas are associated with MEN 2B.245 The NF-2 genetic anomaly occurs on chromosome 22 and is associated with bilateral acoustic neuromas. NF-1 has several clinical forms summarized in Table 68-3. NF-1 and NF-2 are compared in Table 68-4.243 About half of the cases of NF-1 represent de novo mutations.246
TABLE 68-3
Clinical Patterns in Neurofibromatosis
| Clinical Pattern | Description |
| Fibroma molluscum | Hundreds or thousands of pedunculated nodules; number makes resection impractical |
| Plexiform neurofibroma | Occur usually in the face and scalp, causing bony deformity by pressure erosion; resections for cosmesis may be repeated because curative resection is rare |
| Elephantiasis nervorum | With neurofibromas of the extremities, these cause greatly thickened skin simulating limb hypertrophy; resection is done to manage disfigurement |
| Thoracic neurofibroma | May have intraspinal extension (dumbbell tumor); has a high incidence of malignancy |
| Visceral neurofibroma | May affect intestine, kidney, and bladder because of the presence of associated nerves; when large, neurofibrosarcoma incidence increases |
| Skeletal syndromes | Include kyphoscoliosis, pseudarthrosis of tibia and ulna |
| Cranial syndromes | Meningiomas, gliomas, and optic gliomas have been reported |
| Endocrine syndromes | Sexual precocity, medullary thyroid carcinoma, and pheochromocytoma have been reported |
| Cardiovascular syndromes | Heart is rarely involved, but coarctation of the aorta and renal artery lesions have been reported |
TABLE 68-4
Comparison of Neurofibromatosis Types 1 and 2

Modified from Curatolo P, Riva D. Neurocutaneous syndromes in children. Montrouge. France: John Libbey Eurotext; 2006. p. 170.
Operative management is not curative of this genetic disorder and these children optimally have multidisciplinary approach to their care. NF 1 patients may have additional functional and cognitive disabilities requiring screening247 and benefit from parent support groups (such as http://www.nfmidatlantic.org).
Highly aggressive sarcomas may develop and new symptoms require appropriate imaging using ultrasound, CT, or MRI. NF-1 patients have a 7–12% lifetime incidence of malignant peripheral nerve sheath tumors (MPNST), often arising from plexiform neurofibromas.248 Malignant transformation may be heralded by pain, rapid growth, and new neurologic deficits in a previously stable lesion.249 MRI criteria for malignancy include large size (median 14.5 cm), central necrosis, peripheral enhancement, and perilesional edema-like zone.250 Other reports also suggest of a positive role for PET scans in identifying malignancy.246,251
Where symptoms warrant, excision or debulking remain the mainstay for treatment of benign disease (Figs 68-17 and 68-18) while novel therapies are being explored, including rapamycin and antiangiogenic drugs.246 MPNSTs are highly malignant, especially in the context of NF-1; standard of care consists of wide surgical excision followed by radiotherapy. The tumors are usually chemoresistant, metastases are common, and five-year survival has been reported at 28–33%.252,253 The Children’s Tumor Foundation (www.ctf.org) is currently sponsoring drug trials and encouraging networking between NF clinics.
FIGURE 68-17 (A) This 8-year-old child with NF-1 has developed abdominal pain and a large neurofibroma (asterisk) is seen in the left abdomen on the CT scan. (B) At laparoscopy, the mass (asterisk) is also visualized and involves the transverse mesocolon. (C) The transverse colon and mass were mobilized and were then exteriorized by enlarging the umbilical incision. The mass was very adherent to the transverse colon and colon resection was also required. (D) The port sites used for the laparoscopic-assisted operation are seen. Note the café-au-lait spots on the abdominal wall.
FIGURE 68-18 This 15-year-old has been followed for several years and has previously required excision of a large lumbar neurofibroma which was causing discomfort. (A) This neurofibroma located between the ninth and tenth ribs (arrow) was causing significant pain. (B) Note the café-au-lait spot on the skin on the left abdominal wall. The lesion is seen through the left transverse chest wall incision. (D) The lesion is visualized following its complete excision.
Xanthomas
A xanthoma is a tumor of lipid-laden histiocytes or foam cells forming yellowish skin nodules.254 It may be due to uncontrolled diabetes mellitus or biliary tract obstruction, which unbalances triglyceride and cholesterol metabolism, leading to lipid accumulation in histiocytes. Xanthomas are typical features of Alagille syndrome (syndromic paucity of bile ducts) and familial hypercholesterolemia (Fig. 68-19). Correcting the underlying disorder reverses these lesions, but excisional biopsy may be indicated for bothersome lesions or for diagnostic purposes.
Tumors of Mesoderm
Myxomas
A myxoma is a benign primitive connective tissue cell and stroma tumor resembling mesenchyme. It occurs mainly in the heart, producing symptoms by obstructing normal blood flow, and is removed using cardiopulmonary bypass.255 Cutaneous and other sites are uncommon in children. They may be associated with the Carney complex.235,255
Fibrous Tissue Tumors
Nodular Fasciitis
Nodular fasciitis is the most common fibrous tissue tumor in all age groups. It is a self-limiting reactive process. These tumors may be subcutaneous, intramuscular, or fascial in location and are commonly found in the head and neck of children.256 Half of cases are associated with discomfort, and 40% of lesions occur in patients younger than 20 years.257 Excisional biopsy is necessary to differentiate these rapidly growing lesions from malignancy. Recurrence is rare.
Myofibromas/myofibromatosis
This category represents the most common fibrous tumor of childhood. Most present as a solitary skin nodule involving the head and neck, trunk or extremity, but they can involve bone or internal organs.257,258 Spontaneous regression is possible. Infantile myofibromatosis (formerly called congenital fibromatosis) usually is first seen in infancy with multiple firm rubbery masses in the soft tissues, generally in the lower extremities and head and neck. Multifocal superficial lesions have a good prognosis, but a generalized form with visceral lesions (1–2% of cases) is often lethal due to pulmonary lesions.257
Fibrosarcomas
Fibrosarcomas include several specific entities, among which congenital infantile fibrosarcoma, presenting in the first two years of life as a large superficial or visceral mass (Fig. 68-20), and adult-type fibrosarcoma, which has a less favorable prognosis.258 The infantile type is treated with complete excision, preceded by low-dose chemotherapy if necessary to avoid mutilating surgery. Cytogenetic studies are often required to separate the two entities and differentiate the congenital type from infantile myofibromatosis.257,259
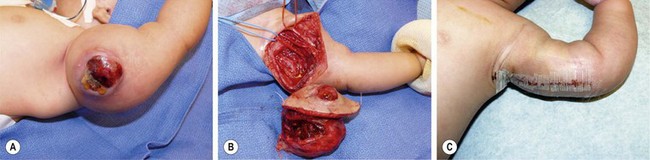
FIGURE 68-20 (A) This 4-week-old baby was initially seen with a large mass in his left arm which had become ulcerated. (B) The baby underwent excision of the lesion with sparing of the underlying neurovascular structures. (C) The postoperative appearance. This patient has recovered uneventfully and remains disease free after 4 years. The lesion was an infantile fibrosarcoma.
Congenital Epulis
Congenital epulis is a benign granular cell tumor occurring almost exclusively in girls. It can be detected on prenatal ultrasound.210 It originates from the gingival mucosa and averages 1–2 cm in diameter. Larger masses could be confused with epignathus (see Fig. 68-13). Its exact nature is not clear.260 Some classify it under tumors of peripheral nerves, whereas others consider it a fibrous tumor, hence the synonym granular cell fibroblastoma. Spontaneous regression is possible,261 and simple excision is curative.
Keloid
A keloid is a sharply elevated, irregularly shaped, progressively enlarging scar caused by excessive collagen in the dermis during connective tissue re-modeling. Unlike the hypertrophic scar (which does not progress), a keloid extends beyond the wound and may recur after excision. Treatment with intradermal injection of corticosteroids, silicone pressure garments (as used for burn patients), pulsed dye laser, and cryosurgery may be attempted. Rarely, judicious excision with radiation therapy may be used.233 Keloids developing after minor procedures are common. Their incidence is higher in blacks, but they can occur in any racial group. Patients prone to keloids are advised to avoid body piercing and tattooing.
Desmoid Tumors
Desmoid tumors are part of a broad group of benign fibroblastic proliferations called fibromatoses, which include palmar and plantar fibromatoses as well as fibromatosis colli (congenital torticollis). Desmoid tumors can be found in extra-abdominal, intra-abdominal, and retroperitoneal locations.235,257 They are not encapsulated and are locally invasive, although they do not metastasize. An association with Gardner syndrome or familial adenomatous polyposis syndrome occurs in 10–15% and similar mutations of the APC gene can be demonstrated.262
First line therapy is complete surgical excision, but when they arise from the mesentery or retroperitoneum, complete resection may not be possible without risking damage to splanchnic vessels. High-dose radiation therapy or interstitial brachytherapy may be considered for residual tumor.263 Antihormonal therapy (tamoxifen), nonsteroidal anti-inflammatory drugs (indomethacin and sulindac), and various chemotherapy protocols have been used in recurrent and inoperable tumors with positive, but variable results.262 Because of the unpredictable behavior of desmoid tumors, some advocate a conservative approach in selected patients.264 Nonsteroidal anti-inflammatory medications are often used as first line therapy for unresectable asymptomatic disease, and chemotherapy is indicated for rapidly growing or life-threatening tumors.262
Adipose Tissue Tumors
A lipoma is a benign, soft, rubbery, encapsulated tumor of adipose tissue composed of mature fat cells occurring on the trunk, neck, and forearms. In one review of adipose tumors, lipomas represented 94%; lipoblastoma, 4.7%; and liposarcoma, 1.3%.257 All are slow-growing tumors. Diagnosed often before age 3 years, lipoblastoma may be superficial and well encapsulated, or it may be deep and infiltrative. Definitive treatment of adipose tumors is complete resection. Chemotherapy may play a role in treating residual liposarcoma. Local recurrence rates for lipoblastoma and liposarcoma are 10% to 20%. Characterized by a myxoid stroma, embryonal lipoblasts, and mature fat cells, the myxoid variant of liposarcoma is similar histologically to lipoblastoma. Tumor karyotyping may be useful in differentiating these adipose tumors.265
Tumors of Synovial Tissue
Synovial Cysts
Pathology texts separate synovial cysts that have a true synovial lining (e.g., Baker’s cysts) from ganglia, which are thought to be degenerative and are without a synovial lining. Symptoms of pain and weakness can occur, but most children present with an asymptomatic mass. Spontaneous resolution of all types of synovial cysts and ganglia is common in children. Surgical treatment is reserved for patients with persistently symptomatic lesions.266 Traumatic disruption (‘strike it with the family Bible’) or corticosteroid injection should be discouraged because of the high recurrence rate and associated pain. The use of nonsteroidal anti-inflammatory agents, coupled with rest or wrist splinting, is usually sufficient if the cyst causes transient discomfort. Ultrasound examination may useful in the evaluation of Baker’s cysts, especially in the context of juvenile arthritis.267
References
1. Virchow, R. Ueber Die Sakralgeschwulst Des Schliewener Kindes. Klin Wschr. 1869; 46:132.
2. Dehner, LP. Gonadal and extragonadal germ cell neoplasms: Teratomas in childhood. In: Finegold M, ed. Pathology of Neoplasia in Children and Adolescents. Philadelphia: W B Saunders; 1986:282–312.
3. Isaacs, H. Germ cell tumors. In: Isaacs H, ed. Tumors of the Fetus and Newborn. Philadelphia: W B Saunders; 1997:15–38.
4. Moore, KL, Persaud, TVN. The Developing Human: Clinically Oriented Embryology, 8th ed. Philadelphia: WB Saunders Elsevier; 2008.
5. Isaacs, H. Tumors. In: Gilbert-Barness A, ed. Potter’s Pathology of the Fetus and Infant. Philadelphia: Mosby Elsevier; 2007:1677–1709.
6. Bale, PM. Sacrococcygeal developmental abnormalities and tumors in children. Perspect Pediatr Pathol. 1984; 8:9–56.
7. Sinnock, KL, Perez-Atayde, AR, Boynton, KA, et al. Clonal analysis of sacrococcygeal ‘teratomas’. Pediatr Pathol Lab Med. 1996; 16:865–875.
8. Chadha, R, Bagga, D, Malhotra, CJ, et al. Accessory limb attached to the back. J Pediatr Surg. 1993; 28:1615–1617.
9. de Lagausie, P, de Napoli Cocci, S, Stempfle, N, et al. Highly differentiated teratoma and fetus-in-fetu: A single pathology? J Pediatr Surg. 1997; 32:115–116.
10. Heifetz, SA, Alrabeeah, A, Brown, BS, et al. Fetus in fetu: A fetiform teratoma. Pediatr Pathol. 1988; 8:215–226.
11. Hanquinet, S, Damry, N, Heimann, P, et al. Association of a fetus in fetu and two teratomas: Ultrasound and MRI. Pediatr Radiol. 1997; 27:336–338.
12. Drut, RM, Drut, R, Fontana, A, et al. Mature presacral sacrococcygeal teratoma associated with a sacral ‘epignathus’. Pediatr Pathol. 1992; 12:99–103.
13. Parizek, J, Nemecek, S, Pospisilova, B, et al. Mature sacrococcygeal teratoma containing the lower half of a human body. Childs Nerv Syst. 1992; 8:108–110.
14. Kooijman, CD. Immature teratomas in children. Histopathology. 1988; 12:491–502.
15. Heifetz, SA, Cushing, B, Giller, R, et al. Immature teratomas in children: Pathologic considerations: A report from the combined Pediatric Oncology Group/Children’s Cancer Group. Am J Surg Pathol. 1998; 22:1115–1124.
16. Rescorla, FJ. Pediatric germ cell tumors. Semin Pediatr Surg. 2012; 21:51–60.
17. Graf, JL, Housely, HT, Albanese, CT, et al. A surprising histological evolution of preterm sacrococcygeal teratoma. J Pediatr Surg. 1998; 33:177–179.
18. Azizkhan, RG, Haase, GM, Applebaum, H, et al. Diagnosis, management, and outcome of cervicofacial teratomas in neonates: A Childrens Cancer Group study. J Pediatr Surg. 1995; 30:312–316.
19. Unal, E, Koksal, Y, Toy, H, et al. Neuroblastoma arising from an unresected sacrococcygeal teratoma in a child. J Pediatr Hematol Oncol. 2010; 32:233.
20. Hijiya, N, Horikawa, R, Matsushita, T, et al. Malignant mediastinal germ-cell tumors in childhood: A report of two cases achieving long-term disease-free survival. Am J Pediatr Hematol Oncol. 1989; 11:437–440.
21. Stringer, DA, Sprigg, A, Kerrigan, D, et al. Malignant carcinoid within a recurrent sacrococcygeal teratoma in childhood. Can Assoc Radiol J. 1990; 41:105–107.
22. Tsuchida, Y, Endo, Y, Saito, S, et al. Evaluation of alpha-fetoprotein in early infancy. J Pediatr Surg. 1978; 13:155–162.
23. Wu, JT, Book, L, Sudar, K. Serum alpha fetoprotein (AFP) levels in normal infants. Pediatr Res. 1981; 15:50–52.
24. Pauniaho, SL, Tatti, O, Lahdenne, P, et al. Tumor markers AFP, CA 125, and CA 19-9 in the long-term follow-up of sacrococcygeal teratomas in infancy and childhood. Tumour Biol. 2010; 31:261–265.
25. Volkl, TM, Langer, T, Aigner, T, et al. Klinefelter syndrome and mediastinal germ cell tumors. Am J Med Genet A. 2006; 140:471–481.
26. Rodriguez, E, Reuter, VE, Mies, C, et al. Abnormalities of 2q: A common genetic link between rhabdomyosarcoma and hepatoblastoma? Genes Chromosomes Cancer. 1991; 3:122–127.
27. Perlman, EJ, Cushing, B, Hawkins, E, et al. Cytogenetic analysis of childhood endodermal sinus tumors: A Pediatric Oncology Group study. Pediatr Pathol. 1994; 14:695–708.
28. Kruslin, B, Hrascan, R, Manojlovic, S, et al. Oncoproteins and tumor suppressor proteins in congenital sacrococcygeal teratomas. Pediatr Pathol Lab Med. 1997; 17:43–52.
29. Ishiwata, I, Ishiwata, C, Soma, M, et al. N-myc gene amplification and neuron specific enolase production in immature teratomas. Virchows Arch A Pathol Anat Histopathol. 1991; 418:333–338.
30. Addeo, R, Crisci, S, D’Angelo, V, et al. Bax mutation and overexpression inversely correlate with immature phenotype and prognosis of childhood germ cell tumors. Oncol Rep. 2007; 17:1155–1161.
31. Currarino, G, Coln, D, Votteler, T. Triad of anorectal, sacral, and presacral anomalies. AJR Am J Roentgenol. 1981; 137:395–398.
32. Ng, WT, Ng, TK, Cheng, PW. Sacrococcygeal teratoma and anorectal malformation. Aust N Z J Surg. 1997; 67:218–220.
33. Tsuchida, Y, Watanasupt, W, Nakajo, T. Anorectal malformations associated with a presacral tumor and sacral defect. Pediatr Surg Int. 1989; 4:398–402.
34. Altman, RP, Randolph, JG, Lilly, JR. Sacrococcygeal teratoma: American Academy of Pediatrics Surgical Section Survey-1973. J Pediatr Surg. 1974; 9:389–398.
35. Tander, B, Baskin, D, Bulut, M. A case of incomplete Currarino triad with malignant transformation. Pediatr Surg Int. 1999; 15:409–410.
36. Urioste, M, Garcia-Andrade, MdC, Valle, L, et al. Malignant degeneration of presacral teratoma in the Currarino anomaly. Am J Med Genet A. 2004; 128A:299–304.
37. Sen, G, Sebire, NJ, Olsen, O, et al. Familial Currarino syndrome presenting with peripheral primitive neuroectodermal tumour arising with a sacral teratoma. Pediatr Blood Cancer. 2008; 50:172–175.
38. Ciotti, P, Mandich, P, Bellone, E, et al. Currarino syndrome with pelvic neuroendocrine tumor diagnosed by post-mortem genetic analysis of tissue specimens. Am J Med Genet A. 2011; 155A:2750–2753.
39. Ashcraft, KW, Holder, TM. Hereditary presacral teratoma. J Pediatr Surg. 1974; 9:691–697.
40. Lee, SC, Chun, YS, Jung, SE, et al. Currarino triad: Anorectal malformation, sacral bony abnormality, and presacral mass–a review of 11 cases. J Pediatr Surg. 1997; 32:58–61.
41. Shija, JK. Anorectal malformation presenting as Hirschsprung’s disease: A case report. East Afr Med J. 1995; 72:130–131.
42. Sonnino, RE, Chou, S, Guttman, FM. Hereditary sacrococcygeal teratomas. J Pediatr Surg. 1989; 24:1074–1075.
43. Singh, SJ, Rao, P, Stockton, V, et al. Familial presacral masses: Screening pitfalls. J Pediatr Surg. 2001; 36:1841–1844.
44. Weinberg, AG. ‘Teratomas’ in the Currarino triad: A misnomer. Pediatr Dev Pathol. 2000; 3:110–111.
45. Ross, AJ, Ruiz-Perez, V, Wang, Y, et al. A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nat Genet. 1998; 20:358–361.
45a. Zu, S, Winberg, J, Arnberg, F, et al. Mutation analysis of the motor neuron and pancreas homeobox 1 (MNX1, former HLXB9) gene in Swedish patients with Currarino syndrome. J Pediatr Surg. 2011; 46:1390–1395.
46. Subbarao, P, Bhatnagar, V, Mitra, DK. The association of sacrococcygeal teratoma with high anorectal and genital malformations. Aust N Z J Surg. 1994; 64:214–215.
47. Shalaby, MS, O’Toole, S, Driver, C, et al. Urogenital anomalies in girls with sacrococcygeal teratoma: A commonly missed association. J Pediatr Surg. 2012; 47:371–374.
48. Lahdenne, P, Heikinheimo, M, Jaaskelainen, J, et al. Vertebral abnormalities associated with congenital sacrococcygeal teratomas. J Pediatr Orthop. 1991; 11:603–607.
49. Werb, P, Scurry, J, Ostor, A, et al. Survey of congenital tumors in perinatal necropsies. Pathology. 1992; 24:247–253.
50. Lakhoo, K. Neonatal teratomas. Early Hum Dev. 2010; 86:643–647.
51. Rowe, MI, O’Neill, JA, Grosfeld, JL, et al. Teratomas and germ cell tumors. In: Rowe MI, O’Neill JA, Grosfeld JL, et al, eds. Essentials of Pediatric Surgery. St-Louis: CV Mosby; 1995:296–305.
52. Gross, RW, Clatworthy, HW, Jr., Meeker, IA, Jr. Sacrococcygeal teratomas in infants and children; A report of 40 cases. Surg Gynecol Obstet. 1951; 92:341–354.
53. Hasle, H, Jacobsen, BB, Asschenfeldt, P, et al. Mediastinal germ cell tumour associated with Klinefelter syndrome. A report of case and review of the literature. Eur J Pediatr. 1992; 151:735–739.
54. Casalone, R, Righi, R, Granata, P, et al. Cerebral germ cell tumor and XXY karyotype. Cancer Genet Cytogenet. 1994; 74:25–29.
55. Czauderna, P, Stoba, C, Wysocka, B, et al. Association of Klinefelter syndrome and abdominal teratoma: A case report. J Pediatr Surg. 1998; 33:774–775.
56. Zon, R, Orazi, A, Neiman, RS, et al. Benign hematologic neoplasm associated with mediastinal mature teratoma in a patient with Klinefelter’s syndrome: A case report. Med Pediatr Oncol. 1994; 23:376–379.
57. Sasou, S, Nakamura, SI, Habano, W, et al. True malignant histiocytosis developed during chemotherapy for mediastinal immature teratoma. Hum Pathol. 1996; 27:1099–1103.
58. Hiramatsu, H, Morishima, T, Nakanishi, H, et al. Successful treatment of a patient with Klinefelter’s syndrome complicated by mediastinal germ cell tumor and AML (M7). Bone Marrow Transplant. 2008; 41:907–908.
59. Zambudio, AR, Lanzas, JT, Calvo, MJ, et al. Mediastinal cystic teratoma associated with a Hodgkin’s lymphoma. Eur J Cardiothorac Surg. 2001; 20:650–651.
60. Dische, MR, Gardner, HA. Mixed teratoid tumors of the liver and neck in trisomy 13. Am J Clin Pathol. 1978; 69:631–637.
61. Quah, BS, Menon, BS. Down syndrome associated with a retroperitoneal teratoma and Morgagni hernia. Clin Genet. 1996; 50:232–234.
62. Falik-Borenstein, TC, Korenberg, JR, Davos, I, et al. Congenital gastric teratoma in Wiedemann-Beckwith syndrome. Am J Med Genet. 1991; 38:52–57.
63. Zachariou, Z, Krug, M, Benz, G, et al. Proteus syndrome associated with a sacrococcygeal teratoma; A rare combination. Eur J Pediatr Surg. 1996; 6:249–251.
64. Robin, NH, Grace, K, DeSouza, TG, et al. New finding of Schinzel-Giedion syndrome: A case with a malignant sacrococcygeal teratoma. Am J Med Genet. 1993; 47:852–856.
65. Billmire, DF, Grosfeld, JL. Teratomas in childhood: Analysis of 142 cases. J Pediatr Surg. 1986; 21:548–551.
66. Schropp, KP, Lobe, TE, Rao, B, et al. Sacrococcygeal teratoma: The experience of four decades. J Pediatr Surg. 1992; 27:1075–1078.
67. Isaacs, H, Jr. Perinatal (fetal and neonatal) germ cell tumors. J Pediatr Surg. 2004; 39:1003–1013.
68. Powell, RW, Weber, ED, Manci, EA. Intradural extension of a sacrococcygeal teratoma. J Pediatr Surg. 1993; 28:770–772.
69. Kunisaki, SM, Maher, CO, Powelson, I, et al. Benign sacrococcygeal teratoma with spinal canal invasion and paraplegia. J Pediatr Surg. 2011; 46:e1–e4.
70. West, DK, Touloukian, RJ. Meconium pseudocyst presenting as a buttock mass. J Pediatr Surg. 1988; 23:864–865.
71. York, DG, Wolfe, H, von Allmen, D. Fetal abdomino-perineal lymphangioma: Differential diagnosis and management. Prenat Diagn. 2006; 26:692–695.
72. Lu, T, Zhao, GP, Wang, K, et al. An unusual case of tubular rectal duplication mimicking teratoma recurrence and review of the literature. European Journal of Radiology Extra. 2010; 73:e17–e19.
73. Al-Salem, AH. Congenital-infantile fibrosarcoma masquerading as sacrococcygeal teratoma. J Pediatr Surg. 2011; 46:2177–2180.
74. Murphy, JJ, Blair, GK, Fraser, GC. Coagulopathy associated with large sacrococcygeal teratomas. J Pediatr Surg. 1992; 27:1308–1310.
75. Holterman, AX, Filiatrault, D, Lallier, M, et al. The natural history of sacrococcygeal teratomas diagnosed through routine obstetric sonogram: A single institution experience. J Pediatr Surg. 1998; 33:899–903.
76. Hedrick, HL, Flake, AW, Crombleholme, TM, et al. Sacrococcygeal teratoma: Prenatal assessment, fetal intervention, and outcome. J Pediatr Surg. 2004; 39:430–438.
77. Angel, CA, Murillo, C, Mayhew, J. Experience with vascular control before excision of giant, highly vascular sacrococcygeal teratomas in neonates. J Pediatr Surg. 1998; 33:1840–1842.
78. Makin, EC, Hyett, J, Ade-Ajayi, N, et al. Outcome of antenatally diagnosed sacrococcygeal teratomas: Single-center experience (1993-2004). J Pediatr Surg. 2006; 41:388–393.
79. Jona, JZ. Progressive tumor necrosis and lethal hyperkalemia in a neonate with sacrococcygeal teratoma (SCT). J Perinatol. 1999; 19:538–540.
80. Le, LD, Alam, S, Lim, FY, et al. Prenatal and postnatal urologic complications of sacrococcygeal teratomas. J Pediatr Surg. 2011; 46:1186–1190.
81. Brace, V, Grant, SR, Brackley, KJ, et al. Prenatal diagnosis and outcome in sacrococcygeal teratomas: A review of cases between 1992 and 1998. Prenat Diagn. 2000; 20:51–55.
82. Wilson, RD, Hedrick, H, Flake, AW, et al. Sacrococcygeal teratomas: Prenatal surveillance, growth and pregnancy outcome. Fetal Diagn Ther. 2009; 25:15–20.
83. Usui, N, Kitano, Y, Sago, H, et al. Outcomes of prenatally diagnosed sacrococcygeal teratomas: The results of a Japanese nationwide survey. J Pediatr Surg. 2012; 47:441–447.
84. Flake, AW. Fetal sacrococcygeal teratoma. Semin Pediatr Surg. 1993; 2:113–120.
85. Veschambre, P, Wartanian, R, Lebouvier, B, et al. Facteurs pronostiques anténatals des tératomes sacro-coccygiens. Rev Fr Gynecol Obstet. 1993; 88:325–330.
86. Finamore, PS, Kontopoulos, E, Price, M, et al. Mirror syndrome associated with sacrococcygeal teratoma: A case report. J Reprod Med. 2007; 52:225–227.
87. Heerema-McKenney, A, Harrison, MR, Bratton, B, et al. Congenital teratoma: A clinicopathologic study of 22 fetal and neonatal tumors. Am J Surg Pathol. 2005; 29:29.
88. Hecher, K, Hackeloer, BJ. Intrauterine endoscopic laser surgery for fetal sacrococcygeal teratoma. Lancet. 1996; 347:470.
89. Ding, J, Chen, Q, Stone, P. Percutaneous laser photocoagulation of tumour vessels for the treatment of a rapidly growing sacrococcygeal teratoma in an extremely premature fetus. J Matern Fetal Neonatal Med. 2010; 23:1516.
90. Ruano, R, Duarte, S, Zugaib, M. Percutaneous laser ablation of sacrococcygeal teratoma in a hydropic fetus with severe heart failure–Too late for a surgical procedure? Fetal Diagn Ther. 2009; 25:26–30.
91. Paek, BW, Jennings, RW, Harrison, MR, et al. Radiofrequency ablation of human fetal sacrococcygeal teratoma. Am J Obstet Gynecol. 2001; 184:503–507.
92. Ibrahim, D, Ho, E, Scherl, SA, et al. Newborn with an open posterior hip dislocation and sciatic nerve injury after intrauterine radiofrequency ablation of a sacrococcygeal teratoma. J Pediatr Surg. 2003; 38:248–250.
93. Ryan, G, Beecroft, R, Finan, E, et al. P27. 10: Successful fetal therapy for massive fetal sacrococcygeal teratoma (SCT) using radio frequency ablation (RFA). Ultrasound Obstet Gynecol. 2010; 36:275.
94. Kim, J, Won, H, Shim, J, et al. P27. 09: Prenatal diagnosis of type II sacrococcygeal teratoma and its management in-utero using percutaneous radiofrequency ablation (RFA): Case report. Ultrasound Obstet Gynecol. 2010; 36:275.
95. Lee, MY, Won, HS, Hyun, MK, et al. Perinatal outcome of sacrococcygeal teratoma. Prenat Diagn. 2011; 31:1217–1221.
96. Roybal, JL, Moldenhauer, JS, Khalek, N, et al. Early delivery as an alternative management strategy for selected high-risk fetal sacrococcygeal teratomas. J Pediatr Surg. 2011; 46:1325–1332.
97. Robertson, FM, Crombleholme, TM, Frantz, ID, 3rd., et al. Devascularization and staged resection of giant sacrococcygeal teratoma in the premature infant. J Pediatr Surg. 1995; 30:309–311.
98. Kay, S, Khalife, S, Laberge, JM, et al. Prenatal percutaneous needle drainage of cystic sacrococcygeal teratomas. J Pediatr Surg. 1999; 34:1148–1151.
99. Stefanovic, V, Halmesmäki, E. Peripartum ultrasound-guided drainage of cystic fetal sacrococcygeal teratoma for the prevention of the labor dystocia: A report of two cases. Am J Perinatol Rep. 2011; 1:87–90.
100. Jouannic, JM, Dommergues, M, Auber, F, et al. Successful intrauterine shunting of a sacrococcygeal teratoma (SCT) causing fetal bladder obstruction. Prenat Diagn. 2001; 21:824–826.
101. Danzer, E, Hubbard, AM, Hedrick, HL, et al. Diagnosis and characterization of fetal sacrococcygeal teratoma with prenatal MRI. AJR Am J Roentgenol. 2006; 187:W350–W356.
102. Garel, C, Mizouni, L, Menez, F, et al. Prenatal diagnosis of a cystic type IV sacrococcygeal teratoma mimicking a cloacal anomaly: Contribution of MR. Prenat Diagn. 2005; 25:216–219.
103. De Backer, A, Madern, GC, Hakvoort-Cammel, FGAJ, et al. Study of the factors associated with recurrence in children with sacrococcygeal teratoma. J Pediatr Surg. 2006; 41:173–181.
104. Fishman, SJ, Jennings, RW, Johnson, SM, et al. Contouring buttock reconstruction after sacrococcygeal teratoma resection. J Pediatr Surg. 2004; 39:439–441.
105. Jan, IA, Khan, EA, Yasmeen, N, et al. Posterior sagittal approach for resection of sacrococcygeal teratomas. Pediatr Surg Int. 2011; 27:545–548.
106. Bax, N, van der Zee, DC. The laparoscopic approach to sacrococcygeal teratomas. Surg Endosc. 2004; 18:128–130.
107. Solari, V, Jawaid, W, Jesudason, EC. Enhancing safety of laparoscopic vascular control for neonatal sacrococcygeal teratoma. J Pediatr Surg. 2011; 46:e5.
108. Lund, DP, Soriano, SG, Fauza, D, et al. Resection of a massive sacrococcygeal teratoma using hypothermic hypoperfusion: A novel use of extracorporeal membrane oxygenation. J Pediatr Surg. 1995; 30:1557–1559.
109. Smithers, CJ, Javid, PJ, Turner, CG, et al. Damage control operation for massive sacrococcygeal teratoma. J Pediatr Surg. 2011; 46:566–569.
110. Cowles, RA, Stolar, CJ, Kandel, JJ, et al. Preoperative angiography with embolization and radiofrequency ablation as novel adjuncts to safe surgical resection of a large, vascular sacrococcygeal teratoma. Pediatr Surg Int. 2006; 22:554–556.
111. Lahdes-Vasama, TT, Korhonen, PH, Seppanen, JM, et al. Preoperative embolization of giant sacrococcygeal teratoma in a premature newborn. J Pediatr Surg. 2011; 46:e5–e8.
112. Hosono, S, Mugishima, H, Nakano, Y, et al. Autologous cord blood transfusion in an infant with a huge sacrococcygeal teratoma. J Neonatal Perinatal Med. 2004; 32:187–189.
113. Gucciardo, L, Uyttebroek, A, De Wever, I, et al. Prenatal assessment and management of sacrococcygeal teratoma. Prenat Diagn. 2011; 31:678–688.
114. Hoehn, T, Krause, MF, Wilhelm, C, et al. Fatal rupture of a sacrococcygeal teratoma during delivery. J Perinatol. 1999; 19:596–598.
115. Shanbhogue, LKR, Bianchi, A, Doig, CM, et al. Management of benign sacrococcygeal teratoma: Reducing mortality and morbidity. Pediatr Surg Int. 1990; 5:41–44.
116. Chisholm, CA, Heider, AL, Kuller, JA, et al. Prenatal diagnosis and perinatal management of fetal sacrococcygeal teratoma. Am J Perinatol. 1999; 16(1):47–50.
117. Benachi, A, Durin, L, Maurer, SV, et al. Prenatally diagnosed sacrococcygeal teratoma: A prognostic classification. J Pediatr Surg. 2006; 41:1517–1521.
118. Sy, ED, Filly, RA, Cheong, ML, et al. Prognostic role of tumor-head volume ratio in fetal sacrococcygeal teratoma. Fetal Diagn Ther. 2009; 26:75–80.
119. Byrne, FA, Lee, H, Kipps, AK, et al. Echocardiographic risk stratification of fetuses with sacrococcygeal teratoma and twin-reversed arterial perfusion. Fetal Diagn Ther. 2011; 30:280–286.
120. Rodriguez, MA, Cass, DL, Lazar, DA, et al. Tumor volume to fetal weight ratio as an early prognostic classification for fetal sacrococcygeal teratoma. J Pediatr Surg. 2011; 46:1182–1185.
121. Schmidt, B, Haberlik, A, Uray, E, et al. Sacrococcygeal teratoma: Clinical course and prognosis with a special view to long-term functional results. Pediatr Surg Int. 1999; 15:573–576.
122. Derikx, JP, De Backer, A, van de Schoot, L, et al. Factors associated with recurrence and metastasis in sacrococcygeal teratoma. Br J Surg. 2006; 93:1543–1548.
123. Bilik, R, Shandling, B, Pope, M, et al. Malignant benign neonatal sacrococcygeal teratoma. J Pediatr Surg. 1993; 28:1158–1160.
124. Ouimet, A, Russo, P. Fetus in fetu or not? J Pediatr Surg. 1989; 24:926–927.
125. Gilcrease, MZ, Brandt, ML, Hawkins, EP. Yolk sac tumor identified at autopsy after surgical excision of immature sacrococcygeal teratoma. J Pediatr Surg. 1995; 30:875–877.
126. Malogolowkin, MH, Ortega, JA, Krailo, M, et al. Immature teratomas: Identification of patients at risk for malignant recurrence. J Natl Cancer Inst. 1989; 81:870–874.
127. Hawkins, E, Issacs, H, Cushing, B, et al. Occult malignancy in neonatal sacrococcygeal teratomas. A report from a Combined Pediatric Oncology Group and Children’s Cancer Group study. Am J Pediatr Hematol Oncol. 1993; 15:406–409.
128. McKenney, JK, Longacre, TA. Low-grade mucinous epithelial neoplasm (intestinal type) arising in a mature sacrococcygeal teratoma with late recurrence as pseudomyxoma peritonei. Hum Pathol. 2008; 39:629–632.
129. Rescorla, FJ, Sawin, RS, Coran, AG, et al. Long-term outcome for infants and children with sacrococcygeal teratoma: A report from the Childrens Cancer Group. J Pediatr Surg. 1998; 33:171–176.
130. Gobel, U, Schneider, DT, Calaminus, G, et al. Multimodal treatment of malignant sacrococcygeal germ cell tumors: A prospective analysis of 66 patients of the German cooperative protocols MAKEI 83/86 and 89. J Clin Oncol. 2001; 19:1943–1950.
131. Rescorla, F, Billmire, D, Stolar, C, et al. The effect of cisplatin dose and surgical resection in children with malignant germ cell tumors at the sacrococcygeal region: A pediatric intergroup trial (POG 9049/CCG 8882). J Pediatr Surg. 2001; 36:12–17.
132. Khalil, BA, Aziz, A, Kapur, P, et al. Long-term outcomes of surgery for malignant sacrococcygeal teratoma: 20-year experience of a regional UK centre. Pediatr Surg Int. 2009; 25:247–250.
133. Marina, NM, Cushing, B, Giller, R, et al. Complete surgical excision is effective treatment for children with immature teratomas with or without malignant elements: A Pediatric Oncology Group/Children’s Cancer Group Intergroup Study. J Clin Oncol. 1999; 17:2137–2143.
134. Gobel, U, Calaminus, G, Schneider, DT, et al. The malignant potential of teratomas in infancy and childhood: The MAKEI experiences in non-testicular teratoma and implications for a new protocol. Klinische Padiatrie. 2006; 218:309–314.
135. Draper, H, Chitayat, D, Ein, SH, et al. Long-term functional results following resection of neonatal sacrococcygeal teratoma. Pediatr Surg Int. 2009; 25:243–246.
136. Cozzi, F, Schiavetti, A, Zani, A, et al. The functional sequelae of sacrococcygeal teratoma: A longitudinal and cross-sectional follow-up study. J Pediatr Surg. 2008; 43:658–661.
137. Malone, PS, Spitz, L, Kiely, EM, et al. The functional sequelae of sacrococcygeal teratoma. J Pediatr Surg. 1990; 25:679–680.
138. Havranek, P, Hedlund, H, Rubenson, A, et al. Sacrococcygeal teratoma in Sweden between 1978 and 1989: Long-term functional results. J Pediatr Surg. 1992; 27:916–918.
139. Uchiyama, M, Iwafuchi, M, Naitoh, M, et al. Sacrococcygeal teratoma: A series of 19 cases with long-term follow-up. Eur J Pediatr Surg. 1999; 9:158–162. [[et al] = Zeitschrift fur Kinderchirurgie].
140. Derikx, JP, De Backer, A, van de Schoot, L, et al. Long-term functional sequelae of sacrococcygeal teratoma: A national study in The Netherlands. J Pediatr Surg. 2007; 42:1122–1126.
141. Gabra, HO, Jesudason, EC, McDowell, HP, et al. Sacrococcygeal teratoma–a 25-year experience in a UK regional center. J Pediatr Surg. 2006; 41:1513–1516.
142. Berger, M, Heinrich, M, Lacher, M, et al. Postoperative bladder and rectal function in children with sacrococcygeal teratoma. Pediatr Blood Cancer. 2011; 56:397–402.
143. Tailor, J, Roy, PG, Hitchcock, R, et al. Long-term functional outcome of sacrococcygeal teratoma in a UK regional center (1993 to 2006). J Pediatr Hematol Oncol. 2009; 31:183.
144. Lee, NG, Gana, R, Borer, JG, et al. Urodynamic findings in patients with Currarino syndrome. J Urol. 2012; 187:2195–2200.
145. Yoshida, A, Maoate, K, Blakelock, R, et al. Long-term functional outcomes in children with Currarino syndrome. Pediatr Surg Int. 2010; 26:677–681.
146. Bittmann, S, Bittmann, V. Surgical experience and cosmetic outcomes in children with sacrococcygeal teratoma. Curr Surg. 2006; 63:51–54.
147. Avci, A, Eren, S. Life-threatening giant mediastinal cystic teratoma in a 4-month-old male baby. Gen Thorac Cardiovasc Surg. 2009; 57:389–391.
148. Takayasu, H, Kitano, Y, Kuroda, T, et al. Successful management of a large fetal mediastinal teratoma complicated by hydrops fetalis. J Pediatr Surg. 2010; 45:e21–e24.
149. Schneider, DT, Calaminus, G, Reinhard, H, et al. Primary mediastinal germ cell tumors in children and adolescents: Results of the German cooperative protocols MAKEI 83/86, 89, and 96. Clin Oncol. 2000; 18:832–839.
150. Magu, S, Rattan, KN, Mishra, DS. Posterior mediastinal teratomas. Indian J Pediatr. 2000; 67:236–240.
151. Kaneko, M, Ohkawa, H, Iwakawa, M, et al. Extensive epidural teratoma in early infancy treated by multi-stage surgery. Pediatr Surg Int. 1999; 15:280–283.
152. Hari, S, Kumar, J, Kumar, A, et al. Posterior mediastinal teratoma. Intern Med J. 2008; 38:448–449.
153. Kuroiwa, M, Suzuki, N, Takahashi, A, et al. Life-threatening mediastinal teratoma in a neonate. Pediatr Surg Int. 2001; 17:235–238.
154. Perger, L, Lee, EY, Shamberger, RC. Management of children and adolescents with a critical airway due to compression by an anterior mediastinal mass. J Pediatr Surg. 2008; 43:1990–1997.
155. Nenna, R, Papoff, P, Moretti, C, et al. What could hemoptysis hide in an otherwise healthy child? Pediatr Pulmonol. 2011; 46:1146–1148.
156. Miyazawa, M, Yoshida, K, Komatsu, K, et al. Mediastinal mature teratoma with rupture into pleural cavity due to blunt trauma. Ann Thorac Surg. 2012; 93:990–992.
157. Revere, DJ, Makaryus, AN, Bonaros, EP, Jr., et al. Chylopericardium presenting as cardiac tamponade secondary to an anterior mediastinal cystic teratoma. Tex Heart Inst J. 2007; 34:379–382.
158. Chaudary, IU, Bojal, SA, Attia, A, et al. Mediastinal endodermal sinus tumor associated with fatal hemophagocytic syndrome. Hematol Oncol Stem Cell Ther. 2011; 4:138–141.
159. Urban, C, Lackner, H, Schwinger, W, et al. Fatal hemophagocytic syndrome as initial manifestation of a mediastinal germ cell tumor. Med Pediatr Oncol. 2003; 40:247–249.
160. Billmire, D, Vinocur, C, Rescorla, F, et al. Malignant mediastinal germ cell tumors: An intergroup study. J Pediatr Surg. 2001; 36:18–24.
161. Sarkaria, IS, Bains, MS, Sood, S, et al. Resection of primary mediastinal non-seminomatous germ cell tumors: A 28-year experience at Memorial Sloan-Kettering Cancer Center. J Thorac Oncol. 2011; 6:1236–1241.
162. Nakajima, K, Fukuzawa, M, Minami, M, et al. Videothoracoscopic resection of anterior mediastinal teratoma in a child. Report of a case. Surg Endosc. 1998; 12:54–56.
163. Billmire, DF. Malignant germ cell tumors in childhood. Semin Pediatr Surg. 2006; 15:30–36.
164. Zamboni, M, Lannes, DC, Cordeiro Pde, B, et al. Transthoracic biopsy with core cutting needle (Trucut) for the diagnosis of mediastinal tumors. Rev Port Pneumol. 2009; 15:589–595.
165. Logothetis, CJ, Samuels, ML, Trindade, A, et al. The growing teratoma syndrome. Cancer. 1982; 50:1629–1635.
166. Liddle, AD, Anderson, DR, Mishra, PK. Intrapericardial teratoma presenting in fetal life: Intrauterine diagnosis and neonatal management. Congenit Heart Dis. 2008; 3(6):449–451.
167. Goldberg, SP, Boston, US, Turpin, DA, et al. Surgical management of intrapericardial teratoma in the fetus. J Pediatr. 2010; 156:848–849.
168. Tollens, T, Casselman, F, Devlieger, H, et al. Fetal cardiac tamponade due to an intrapericardial teratoma. Ann Thorac Surg. 1998; 66:559–560.
169. Pratt, JW, Cohen, DM, Mutabagani, KH, et al. Neonatal intrapericardial teratomas: Clinical and surgical considerations. Cardiol Young. 2000; 10:27–31.
170. Sumner, TE, Crowe, JE, Klein, A, et al. Intrapericardial teratoma in infancy. Pediatr Radiol. 1980; 10(1):51–53.
171. Gunther, T, Schreiber, C, Noebauer, C, et al. Treatment strategies for pediatric patients with primary cardiac and pericardial tumors: A 30-year review. Pediatr Cardiol. 2008; 29:1071–1076.
172. Rana, SS, Swami, N, Mehta, S, et al. Intrapulmonary teratoma: An exceptional disease. Ann Thorac Surg. 2007; 83:1194–1196.
173. Horton, Z, Schlatter, M, Schultz, S. Pediatric germ cell tumors. Surg Oncol. 2007; 16:205–213.
174. Barksdale, EM, Jr., Obokhare, I. Teratomas in infants and children. Curr Opin Pediatr. 2009; 21:344.
175. Nguyen, CT, Kratovil, T, Edwards, MJ. Retroperitoneal teratoma presenting as an abscess in childhood. J Pediatr Surg. 2007; 42:E21–E23.
176. Luo, CC, Huang, CS, Chu, SM, et al. Retroperitoneal teratomas in infancy and childhood. Pediatr Surg Int. 2005; 21:536–540.
177. Chaudhary, A, Misra, S, Wakhlu, A, et al. Retroperitoneal teratomas in children. Indian J Pediatr. 2006; 73:221–223.
178. Jones, NM, Kiely, EM. Retroperitoneal teratomas-potential for surgical misadventure. J Pediatr Surg. 2008; 43:184–187.
179. Billmire, D, Vinocur, C, Rescorla, F, et al. Malignant retroperitoneal and abdominal germ cell tumors: An intergroup study. J Pediatr Surg. 2003; 38:315–318.
180. Federici, S, Prestipino, M, Domenichelli, V, et al. Fetus in fetu: Report of an additional, well-developed case. Pediatr Surg Int. 2001; 17:483–485.
181. Khadaroo, RG, Evans, MG, Honore, LH, et al. Fetus-in-fetu presenting as cystic meconium peritonitis: Diagnosis, pathology, and surgical management. J Pediatr Surg. 2000; 35:721–723.
182. Gupta, DK, Srinivas, M, Dave, S, et al. Gastric teratoma in children. Pediatr Surg Int. 2000; 16:329–332.
183. Jeong, HC, Cha, SJ, Kim, GJ. Rapidly grown congenital fetal immature gastric teratoma causing severe neonatal respiratory distress. J Obstet Gynaecol Res. 2012; 38:449–451.
184. Yeo, DM, Lim, GY, Lee, YS, et al. Gliomatosis peritonei of the scrotal sac associated with an immature gastric teratoma. Pediatr Radiol. 2010; 40:1288–1292.
185. Saha, M. Malignant gastric teratoma: Report of two cases from a single center. Pediatr Surg Int. 2010; 26:931–934.
186. Ukiyama, E, Endo, M, Yoshida, F, et al. Recurrent yolk sac tumor following resection of a neonatal immature gastric teratoma. Pediatr Surg Int. 2005; 21:585–588.
187. Shah, RS, Kaddu, SJ, Kirtane, JM. Benign mature teratoma of the large bowel: A case report. J Pediatr Surg. 1996; 31:701–702.
188. Cakmak, O, Senel, E, Erdogan, D, et al. Ileal teratoma with multiple congenital anomalies. J Pediatr Surg. 2000; 35:1370–1371.
189. Torikai, M, Tahara, H, Kaji, T, et al. Immature teratoma of gallbladder associated with gliomatosis peritonei, a case report. J Pediatr Surg. 2007; 42:E25–E27.
190. Rivkine, E, Goasguen, N, Chelbi, E, et al. [Cystic teratoma of the pancreas]. Gastroenterol Clin Biol. 2007; 31:1016–1019.
191. Yoshida, A, Murabayashi, N, Shiozaki, T, et al. Case of mature cystic teratoma of the greater omentum misdiagnosed as ovarian cyst. J Obstet Gynaecol Res. 2005; 31:399–403.
192. Ariizumi, T, Ogose, A, Hotta, T, et al. Cystic teratoma of the diaphragm which mimicked soft tissue lipoma. Skeletal Radiol. 2007; 36:991–994.
193. Filston, HC. Hemangiomas, cystic hygromas, and teratomas of the head and neck. Semin Pediatr Surg. 1994; 3:147–159.
194. Kountakis, SE, Minotti, AM, Maillard, A, et al. Teratomas of the head and neck. Am J Otolaryngol. 1994; 15:292–296.
195. Garmel, SH, Crombleholme, TM, Semple, JP, et al. Prenatal diagnosis and management of fetal tumors. Semin Perinatol. 1994; 18:350–365.
196. Figueiredo, G, Pinto, PS, Graham, EM, et al. Congenital giant cervical teratoma: Pre- and postnatal imaging. Fetal Diagn Ther. 2010; 27:231–232.
197. Langer, JC, Tabb, T, Thompson, P, et al. Management of prenatally diagnosed tracheal obstruction: Access to the airway in-utero prior to delivery. Fetal Diagn Ther. 1992; 7:12–16.
198. Lazar, DA, Cassady, CI, Olutoye, OO, et al. Tracheoesophageal displacement index and predictors of airway obstruction for fetuses with neck masses. J Pediatr Surg. 2012; 47:46–50.
199. Laje, P, Johnson, MP, Howell, LJ, et al. Ex utero intrapartum treatment in the management of giant cervical teratomas. J Pediatr Surg. 2012; 47:1208–1216.
200. Liechty, KW, Hedrick, HL, Hubbard, AM, et al. Severe pulmonary hypoplasia associated with giant cervical teratomas. J Pediatr Surg. 2006; 41:230–233.
201. Jordan, RB, Gauderer, MW. Cervical teratomas: An analysis. Literature review and proposed classification. J Pediatr Surg. 1988; 23:583–591.
202. Martino, F, Avila, LF, Encinas, JL, et al. Teratomas of the neck and mediastinum in children. Pediatr Surg Int. 2006; 22:627–634.
203. Keen, CE, Said, AJ, Agrawal, M, et al. Congenital thyroid teratoma: A case with persistent neuroglial involvement of cervical lymph nodes. Pediatr Dev Pathol. 1998; 1:322–327.
204. Craver, RD, Lipscomb, JT, Suskind, D, et al. Malignant teratoma of the thyroid with primitive neuroepithelial and mesenchymal sarcomatous components. Ann Diagn Pathol. 2001; 5:285–292.
205. Thompson, LD, Rosai, J, Heffess, CS. Primary thyroid teratomas: A clinicopathologic study of 30 cases. Cancer. 2000; 88:1149–1158.
206. Grushka, JR, Ryckman, J, Mueller, C, et al. Spindle epithelial tumor with thymus-like elements of the thyroid: A multi-institutional case series and review of the literature. J Pediatr Surg. 2009; 44:944–948.
207. Oliveira-Filho, AG, Carvalho, MH, Bustorff-Silva, JM, et al. Epignathus: Report of a case with successful outcome. J Pediatr Surg. 1998; 33:520–521.
208. Wilson, JW, Gehweiler, JA. Teratoma of the face associated with a patent canal extending into the cranial cavity (Rathke’s pouch) in a three-week-old child. J Pediatr Surg. 1970; 5:349–359.
209. Chung, JH, Farinelli, CK, Porto, M, et al. Fetal epignathus: The case of an early EXIT (ex utero intrapartum treatment). Obstet Gynecol. 2012; 119:466–470.
210. Kovacs, L, Volpe, C, Laberge, JM, et al. Gingival mass in a newborn infant diagnosed in-utero. J Pediatr. 2002; 141:837.
211. Maartens, IA, Wassenberg, T, Halbertsma, FJ, et al. Neonatal airway obstruction caused by rapidly growing nasopharyngeal teratoma. Acta Paediatr (Oslo, Norway: 1992). 2009; 98:1852–1854.
212. Sauter, ER, Diaz, JH, Arensman, RM, et al. The perioperative management of neonates with congenital oropharyngeal teratomas. J Pediatr Surg. 1990; 25:925–928.
213. Morof, D, Levine, D, Grable, I, et al. Oropharyngeal teratoma: Prenatal diagnosis and assessment using sonography, MRI, and CT with management by ex utero intrapartum treatment procedure. AJR Am J Roentgenol. 2004; 183:493–496.
214. Jarrahy, R, Cha, ST, Mathiasen, RA, et al. Congenital teratoma of the oropharyngeal cavity with intracranial extension: Case report and literature review. J Craniofac Surg. 2000; 11:106–112.
215. Lee, GA, Sullivan, TJ, Tsikleas, GP, et al. Congenital orbital teratoma. Aust N Z J Ophthalmol. 1997; 25:63–66.
216. Roncaroli, F, Scheithauer, BW, Pires, MM, et al. Mature teratoma of the middle ear. Otol Neurotol. 2001; 22:76–78.
217. Washburne, JF, Magann, EF, Chauhan, SP, et al. Massive congenital intracranial teratoma with skull rupture at delivery. Am J Obstet Gynecol. 1995; 173:226–228.
218. Hunt, SJ, Johnson, PC, Coons, SW, et al. Neonatal intracranial teratomas. Surg Neurol. 1990; 34:336–342.
219. Chien, YH, Tsao, PN, Lee, WT, et al. Congenital intracranial teratoma. Pediatr Neurol. 2000; 22:72–74.
220. Sawamura, Y, Kato, T, Ikeda, J, et al. Teratomas of the central nervous system: Treatment considerations based on 34 cases. J Neurosurg. 1998; 89:728–737.
221. Jona, JZ. Congenital anorectal teratoma: Report of a case. J Pediatr Surg. 1996; 31:709–710.
222. Kreczy, A, Alge, A, Menardi, G, et al. Teratoma of the umbilical cord. Case report with review of the literature. Arch Pathol Lab Med. 1994; 118:934–937.
223. Pirodda, A, Ferri, GG, Truzzi, M, et al. Benign cystic teratoma of the parotid gland. Otolaryngol Head Neck Surg. 2001; 125:429–430.
224. Cakmak, M, Savas, C, Ozbasar, D, et al. Congenital vulvar teratoma in a newborn. J Pediatr Surg. 2001; 36:620–621.
225. Daszkiewicz, P, Roszkowski, M, Przasnek, S, et al. Teratoma or enterogenous cyst? The histopathological and clinical dilemma in co-existing occult neural tube dysraphism. Folia Neuropathol. 2006; 44:24–33.
226. Kirkham, N. Tumors and cysts of the epidermis. In Elder DE, Elenitsas R, Johnson BL, et al, eds.: Lever’s Histopathology of the Skin, 10th ed, Philadelphia: Lippincott Williams&Wilkins, 2011.
227. Li, WY, Reinisch, JF. Cysts, pits, and tumors. Plast Reconstr Surg. 2009; 124:106e.
228. Gur, E, Drielsma, R, Thomson, HG. Angular dermoid cysts in the endoscopic era: Retrospective analysis of aesthetic results using the direct, classic method. Plast Reconstr Surg. 2004; 113:1324.
229. Ruszkowski, A, Caouette-Laberge, L, Bortoluzzi, P, et al. Superior eyelid incision: An alternative approach for frontozygomatic dermoid cyst excision. Ann Plast Surg. 2000; 44:591.
230. Cozzi, DA, Mele, E, d’Ambrosio, G, et al. The eyelid crease approach to angular dermoid cysts in pediatric general surgery. J Pediatr Surg. 2008; 43:1502–1506.
231. Dutta, S, Lorenz, HP, Albanese, CT. Endoscopic excision of benign forehead masses: A novel approach for pediatric general surgeons. J Pediatr Surg. 2006; 41:1874–1878.
232. Vijay, K, Choudhary, AK. Multiple scalp epidermoid cysts in a child with Gardner syndrome. Pediatr Radiol. 2010; 40:S172.
233. Paller, ASMAJ. Cutaneous tumors and tumor syndromes. In: Paller ASMAJ, ed. Hurwitz Clinical Pediatric Dermatology. 3rd ed. Philadelphia: Elsevier Saunders; 2006:205–255.
234. Ban, R, Shinohara, H, Matsuo, K, et al. Limited distribution of gravitation abscess caused by infected preauricular sinus depends on anatomical structure. Eur J Plast Surg. 2008; 31:59–63.
235. Weiss, SWGJR. Enzinger and Weiss’s Soft Tissue Tumors, 5th ed. Philadelphia: Mosby Elsevier; 2008.
236. Reddy, VB, Husain, AN. The skin. In: Stocker JT, Dehner LP, Husain AN, eds. Pediatric Pathology. 3rd ed. Philadelphia: Wolters Kluwer Lippincott Williams&Wilkins; 2011:1105–1146.
237. Gibbs, S, Harvey, I. Topical treatments for cutaneous warts. Cochrane Database Syst Rev. 2006.
238. Cohen, BA, Honig, P, Androphy, E. Anogenital warts in children. Clinical and virologic evaluation for sexual abuse. Arch Dermatol. 1990; 126:1575–1580.
239. Schlechter, R, Hartsough, NA, Guttman, FM. Multiple pilomatricomas (calcifying epitheliomas of Malherbe). Pediatr Dermatol. 1984; 2:23–25.
240. Lee, EH, Levine, VJ, Nehal, KS. Cutaneous Squamous Cell Carcinoma. In: Alam M, ed. Evidence-Based Procedural Dermatology. New York NY: Springer; 2012:57–74.
241. Tom, WL, Hurley, MY, Oliver, DS, et al. Features of basal cell carcinomas in basal cell nevus syndrome. Am J Med Genet A. 2011; 155A:2098–2104.
242. Chow, CW, Tabrizi, SN, Tiedemann, K, et al. Squamous cell carcinomas in children and young adults: A new wave of a very rare tumor? J Pediatr Surg. 2007; 42:2035–2039.
243. Curatolo, P, Riva, D. Neurocutaneous syndromes in children. Montrouge, France: John Libbey Eurotext; 2006.
244. Friedman, JM, Birch, PH. Type 1 neurofibromatosis: A descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997; 70:138–143.
245. Pujol, RM, Matias-Guiu, X, Miralles, J, et al. Multiple idiopathic mucosal neuromas: A minor form of multiple endocrine neoplasia type 2B or a new entity? J Am Acad Dermatol. 1997; 37:349–352.
246. Williams, VC, Lucas, J, Babcock, MA, et al. Neurofibromatosis type 1 revisited. Pediatrics. 2009; 123:124–133.
247. Gilboa, Y, Rosenblum, S, Fattal-Valevski, A, et al. Application of the International Classification of Functioning, Disability and Health in children with neurofibromatosis type 1: A review. Dev Med Child Neurol. 2010; 52:612–619.
248. Ferner, RE. Neurofibromatosis 1. Eur J Hum Genet. 2007; 15:131–138.
249. Ferner, RE, Huson, SM, Thomas, N, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007; 44:81–88.
250. Wasa, J, Nishida, Y, Tsukushi, S, et al. MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 2010; 194:1568–1574.
251. Bredella, MA, Torriani, M, Hornicek, F, et al. Value of PET in the assessment of patients with neurofibromatosis type 1. AJR Am J Roentgenol. 2007; 189:928–935.
252. Ferrari, A, Bisogno, G, Macaluso, A, et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer. 2007; 109:1406–1412.
253. Porter, D, Prasad, V, Foster, L, et al. Survival in malignant peripheral nerve sheath tumours: A comparison between sporadic and neurofibromatosis type 1-associated tumours. Sarcoma. 2009; 2009:756395.
254. Weiss, SWGJR. Benign fibrohistiocytic tumors. In: Weiss SWGJR, ed. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia: Mosby Elsevier; 2008:331–370.
255. Bielefeld, KJ, Moller, JH. Cardiac tumors in infants and children: study of 120 operated patients. Pediatr Cardiol. 2012; 1–4.
256. Weiss, SWGJR. Benign fibroblastic/myofibroblastic proliferations. In: Weiss SWGJR, ed. Enzinger and Weiss’s Soft Tissue Tumors. 5th ed. Philadelphia: Mosby Elsevier; 2008:175–225.
257. Dehner, LP. Soft tissue. In: Stocker JT, Dehner LP, Husain AN, eds. Pediatric Pathology. 3rd ed. Philadelphia: Wolters Kluwer/ Lippincott Williams&Wilkins; 2011:1040–1104.
258. Weiss, SW. Fibrous tumors of infancy and childhood. In: Enzinger FM, Weiss SW, eds. Soft Tissue Tumors. 5th ed. Philadelphia: Mosby Elsevier; 2008:257–302.
259. Triche, TJHJ, Sorensen, PHB. Diagnostic Pathology of Pediatric Malignancies. In Pizzo PAPDG, ed.: Principles and Practice of Pediatric Oncology, 6th ed, Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2011.
260. Childers, ELB, Fanburg-Smith, JC. Congenital epulis of the newborn: 10 new cases of a rare oral tumor. Ann Diagn Pathol. 2011; 15:157–161.
261. Sakai, VT, Oliveira, TM, Silva, TC, et al. Complete spontaneous regression of congenital epulis in a baby by 8 months of age. Int J Paediatr Dent. 2007; 17:309–312.
262. Kasper, B, Ströbel, P, Hohenberger, P. Desmoid tumors: Clinical features and treatment options for advanced disease. Oncologist. 2011; 16:682–693.
263. Jabbari, S, Andolino, D, Weinberg, V, et al. Successful treatment of high risk and recurrent pediatric desmoids using radiation as a component of multimodality therapy. Int J Radiat Oncol Biol Phys. 2009; 75:177–182.
264. Stoeckle, E, Coindre, J, Longy, M, et al. A critical analysis of treatment strategies in desmoid tumours: A review of a series of 106 cases. Eur J Surg Oncol. 2009; 35:129–134.
265. Miller, GG, Yanchar, NL, Magee, JF, et al. Lipoblastoma and liposarcoma in children: An analysis of 9 cases and a review of the literature. Can J Surg. 1998; 41:455–458.
266. Cypel, TKS, Mrad, A, Somers, G, et al. Ganglion cyst in children: Reviewing treatment and recurrence rates. Can J Plast Surg. 2011; 19:53–55.
267. Alessi, S, Depaoli, R, Canepari, M, et al. Baker’s cyst in pediatric patients: Ultrasonographic characteristics. J Ultrasound. 2012; 15:76–81.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Teratomas, Dermoids, and Other Soft Tissue Tumors
Teratomas
Embryology and Pathology
Teratoma, from the Greek teratos (‘of the monster’) and onkoma (‘swelling’), is a term first applied by Virchow in 1869 to ‘sacrococcygeal growths’.1 Teratomas are composed of multiple tissues foreign to the organ or site from which they arise.2 Although teratomas are sometimes defined as having three embryonic layers (endoderm, mesoderm, and ectoderm), recent classifications also include monodermal types.2,3
Teratomas are thought by some to arise from totipotent primordial germ cells.3 These cells develop among the endodermal cells of the yolk sac near the origin of the allantois and migrate to the gonadal ridges during weeks 4 and 5 of gestation (Fig. 68-1).4 Some cells may miss their target destination and give rise to a teratoma anywhere from the brain to the coccygeal area, usually in the midline. Another theory has teratomas arising from remnants of the primitive streak or primitive node.4–6 During week 3 of development, midline cells at the caudal end of the embryo divide rapidly and, in a process called gastrulation, give rise to all three cell layers of the embryo (Fig. 68-2).4 By the end of week 3, the primitive streak shortens and disappears. This theory explains the more common occurrence of teratomas in the sacrococcygeal region. With either theory, the totipotent cells could give rise to monoclonal neoplasms. Evidence shows that, whereas immature teratomas may be monoclonal, mature teratomas can be polyclonal, more like a hamartoma than a neoplasm.7 This finding is compatible with the third theory that teratomas are a form of incomplete twinning.2,3
FIGURE 68-1 Commonly cited theory on the origin of teratomas. (A) Drawing of embryo during week 4 (longitudinal section), showing primordial germ cells at the base of the yolk sac. (B, C) During week 5, these cells migrate toward the gonadal ridges. According to this theory, some cells could miss their intended destination. (Modified from Moore KL, Persaud TVN. The Developing Human. Philadelphia: WB Saunders; 1993. p. 71, 181.)
FIGURE 68-2 Alternate theory on embryogenesis of teratomas. (A) Sketches of dorsal views of the embryonic disk on days 17 and 18, showing the primitive streak and primitive node. (B) Drawing of a transverse cut of the embryonic disk during week 3. This shows that cells from the primitive streak migrate to form the mesoblast (the origin of all mesenchymal tissues) and also displace the hypoblast to form the endoderm. Hence, remnants of these pluripotent primitive streak cells could give rise to teratomas and could account for the more frequent sacrococcygeal location. (From Moore KL, Persaud TVN. The Developing Human. Philadelphia: WB Saunders; 1993. p. 55–6.)
The primordial germ cell is the principal but probably not the exclusive progenitor of a teratoma. The recent trend is to include teratomas under the classification of germ cell tumors.2,3,5 This histologic classification also includes germinomas (formerly dysgerminomas), embryonal carcinomas, yolk sac tumors (YST), choriocarcinomas, gonadoblastomas, and mixed germ cell tumors. Gonadal and extragonadal teratomas may have a different origin, explaining the different behavior according to tumor site.
Teratomas are fascinating tumors owing to the diversity of tissues they may contain and the varying degree of organization of these tissues. Many tumors contain skin elements, neural tissue, teeth, fat, cartilage, and intestinal mucosa, often with normal ganglion cells. These tissues are usually present as disorganized islands of cells with cystic spaces. The tumor sometimes consists of more organized tissue, such as small bowel, limbs, and even a beating heart. These have been called fetiform teratomas (Fig. 68-3).2,3,5,8,9 When the mass includes vertebrae or notochord and a high degree of structural organization, the term fetus in fetu is used. This is viewed by some as a variant of conjoined twinning but is classified as a teratoma by others, owing to the absence of a recognizable umbilical cord in its vascular pedicle.3,10 Whether teratomas are at one end of a spectrum that includes fetus in fetu, parasitic twins, conjoined twins, and normal twins is the subject of controversy. One certainly cannot dismiss the many reports of teratomas associated with fetus in fetu in the same patient and with a twin pregnancy.2,3,11–13
FIGURE 68-3 (A) This child had a large fluctuant lumbar mass at birth. The patient is prone with the head at the top of the photograph. A family history of myelomeningocele existed in a great aunt. She also had an atrophic right leg with neurologic impairment below the L3 root and clubbing of the right foot. Note the ulcerated, arachnoid-looking area cranially and the pedunculated skin caudally, which had the appearance of a vulva and was oozing serous fluid. (B) Plain radiograph shows a severe lumbosacral scoliosis with vertebral anomalies. CT confirmed the vertebral anomalies with spina bifida and demonstrated a pattern of intestine with inspissated or calcified meconium in the teratoma. (C) MR image reveals that the mass (asterisk) extended into the retroperitoneum, where it was contiguous with the lower pole of the right kidney (arrow). (D) At operation, normal-looking blind bowel loops were found deep to the vulva-like structure. Part of the mass extended along the spinal cord, which required dissection and untethering by a neurosurgeon. The pathologic diagnosis was a mature fetiform teratoma that contained, among many other things, two adrenals, two ovaries, renal tissue with some glomeruli and tubules, bone with bone marrow, and portions of stomach and small and large bowel. The child recovered well neurologically but required spinal instrumentation owing to progressive scoliosis at age 2 years.
The overall tissue architecture is variable in teratomas. Moreover, a spectrum of cellular differentiation exists. Most benign teratomas are composed of mature cells, but 20–25% also contain immature elements, most often neuroepithelium. However, the degree of histologic immaturity is of proven prognostic significance only in ovarian teratomas.3,14 Even this concept is being questioned, since one large cooperative study demonstrated that overlooked microscopic foci of YST, rather than the grade of immaturity, was more predictive of recurrence.15 In neonatal teratoma, immature tissue is considered normal and without any influence on prognosis.2,5,16 Spontaneous maturation of malignant tumors has been reported after partial excision of giant SCTs in two fetuses at 23 and 27 weeks of gestation.17
Teratomas may also contain or develop foci of malignancy. A malignant germ cell tumor may be found in sites typical for teratomas, such as the mediastinum or sacrococcygeal area. Whether the lesion was malignant from the onset or the malignant cells destroyed and replaced the benign teratoma component is often difficult to ascertain. The most common malignant component within a teratoma is a YST (formerly also called ‘endodermal sinus tumor’). Other malignant germ cell tumors can occur, and, rarely, malignancy in other tissues composing the teratoma, such as neuroblastoma,18,19 squamous cell carcinoma,20 carcinoid,21 and others, can develop. Malignancy at birth is uncommon but increases with age and with incomplete resection. An apparently mature teratoma may recur several months or years after resection as a malignant YST, illustrating the difficulties in histologic sampling of large tumors and the need for close follow-up.2,3
Most YSTs and some embryonal carcinomas secrete α-fetoprotein (AFP), which can be measured in the serum and demonstrated in the cells by immunohistochemistry.22 This marker is particularly useful for assessing the presence of residual or recurrent disease. AFP levels are normally very high in neonates and decrease with time.22,23 The postoperative half-life is about six days. Persistently high levels may be an indication for the need for further surgical procedures or chemotherapy. Other markers that may be elevated are β-human chorionic gonadotropin (β-hCG), produced by choriocarcinomas, and, rarely, carcinoembryonic antigen. CA 125 was also found helpful in the follow-up of patients with SCTs, but not CA 19–9.24 Secretion of β-hCG by the tumor may be sufficient to cause precocious puberty.25
The genetic basis of teratomas is not yet understood. Most germ cell tumors appear to have an amplification, or isochromosome, in a region of the short arm of chromosome 12, designated i(12p).3,26 This has been well described in adults but was not confirmed in one pediatric series in which deletions on chromosomes 1 and 6 were found instead.27 Similarly, oncogenes and tumor suppressor genes did not appear to correlate with prognosis in one study.28 MYCN gene amplification was present in immature teratomas, but absent in mature teratomas in another report.29 BAX mutation and overexpression correlated with survival in another study of childhood germ cell tumors.30 The clinical usefulness of these findings remains unclear.
Associated Anomalies
Teratomas are usually isolated lesions. A well-recognized association is the Currarino triad of anorectal malformation, sacral anomaly, and a presacral mass.31,32 The presacral mass is usually a teratoma or an anterior meningocele. However, hamartomas, duplication cysts, and dermoid cysts have been described, as have combinations of these lesions.
An extensive review of the English and German literature found 51 cases of infants with the Currarino triad and highlighted several important facts.33 Twenty per cent of patients were older than 12 years at the time of diagnosis, yet no reports of malignancy were found. This contrasts to a 75% malignancy rate in patients older than 1 year who had the usual SCT.34 However, a subsequent case report described a malignant recurrence that resulted in the patient’s demise at 4 years of age despite chemotherapy.35 Since then, more reports of malignant transformation of a presacral teratoma in the context of the Currarino triad have appeared, and the risk of malignant transformation has been estimated at 1%.36,37 This can occur well into adulthood.38 Hence, one should not dismiss the presacral mass as always being benign in these patients. The female preponderance for patients with this triad is 1.5 : 1, which is less than the 3 : 1 ratio noted in isolated SCTs. A familial predisposition is noted in 57% of cases and has an autosomal dominant inheritance pattern.39 Although all variants of anorectal malformations have been described, by far the most common is anal or anorectal stenosis. In one report, this triad was present in 38% of all patients with anorectal stenosis and in 1.6% of patients with low imperforate anus.40
Anal anomalies also have been reported in conjunction with a presacral mass, but in the absence of sacral defects. Hirschsprung disease has been incorrectly diagnosed in some cases,41,42 indicating the need to eliminate the presence of a presacral mass by digital rectal examination, by a metal sound when the anus is too tight, or by imaging studies. In the screening of family members, normal plain radiographs of the sacrum are not adequate, because a presacral mass may exist in the absence of a bony defect.43 The low incidence of malignancy has led one author to conclude that the presacral lesion in this context is a hamartoma rather than a teratoma.44 This is supported by the demonstration of deletions or mutations of the homeobox gene HLXB9, located at 7q36, in several affected families.45 In two families where one member had died from a malignant neuroendocrine tumor originating from a presacral teratoma, the diagnosis of Currarino syndrome was made through identification of a HLXB9 mutation.36,38
The mutated gene has recently been renamed ‘motor neuron and pancreas homeobox 1 (MNX1).’45a Urogenital anomalies, such as hypospadias, vesicoureteral reflux, vaginal or uterine duplications, and other anomalies are associated with teratomas.32,34,46,47 Congenital dislocation of the hip has been found in 7% of patients with SCTs which can lead to late orthopedic sequelae.48 Vertebral anomalies can also be found (see Fig 68-3). Central nervous system lesions, such as anencephaly, trigonocephaly, Dandy–Walker malformations, spina bifida, and myelomeningocele can occur as well.2,3,49,50 Another peculiar association with SCTs is a family history of twins in as many as 10% of the patients.39,51,52 Although not confirmed in all series, this finding, combined with reports of simultaneous twin pregnancy or sequential familial occurrences of fetus in fetu and teratoma, supports the theory that teratomas may be just one end of the spectrum of conjoint twinning.2,3,9
Klinefelter syndrome is strongly associated with mediastinal teratoma50,53 and has been reported in patients with intracranial54 and retroperitoneal tumors.55 It is estimated that 8% of male patients with primary mediastinal germ cell tumors have Klinefelter syndrome, which is 50 times the expected frequency.53 These tumors are often malignant, are of the choriocarcinoma type, secrete β-hCG, and produce precocious puberty. Histiocytosis and leukemia are also associated with mediastinal teratoma, both with and without Klinefelter syndrome.56–58 Other hematologic malignancies, such Hodgkin disease, occur rarely.59
The following rare associations have been reported, most often with nonsacrococcygeal lesions: trisomy 13, trisomy 21, Morgagni hernia, congenital heart defects, Beckwith–Wiedemann syndrome, pterygium, cleft lip and palate, and rare syndromes, such as Proteus and Schinzel-Giedion syndromes.49,50,60–64
Diagnosis and Management by Tumor Site
Sacrococcygeal Teratoma
SCTs account for 35–60% of teratomas (gonadal included) in large series (Table 68-1).65–67 This is the most common tumor in the newborn, even when stillbirths are considered.49 The estimated incidence is 1 per 35,000 to 40,000 live births.3,16
TABLE 68-1
Relative Frequency of Teratomas by Site
Data are from five series of teratomas in children.
Modified from Dehner LP. Gonadal and extragonadal germ cell neoplasms: Teratomas in childhood. In: Finegold M, editor. Pathology of Neoplasia in Children and Adolescents. Philadelphia: WB Saunders; 1986. p. 282–312.
Diagnosis.
In countries where prenatal ultrasonography (US) is not readily available, most SCTs are seen as a visible mass at birth, making the diagnosis obvious (Fig. 68-4). Prenatal diagnosis has important implications and will be discussed further.
FIGURE 68-4 This infant was diagnosed with a large sacrococcygeal teratoma in utero. Within days, premature labor occurred, prompting cesarian delivery at 25 weeks gestation. The baby died despite resuscitation attempts.
There is an unexplained female preponderance of 3 : 1.34,65,67 The main differential diagnosis is meningocele. Typically, meningoceles occur cephalad to the sacrum and are covered by dura, but sometimes they are covered by skin. Examination of the child reveals bulging of the fontanelle with gentle pressure on a sacral meningocele, helping to establish the diagnosis before plain radiography, ultrasound, and magnetic resonance imaging (MRI) confirms it. The co-existence of meningocele with teratoma is well recognized in the familial form, but these are usually presacral. Rarely, a typical exophytic teratoma may have an intradural extension.68,69 Other lesions in the differential diagnosis of neonatal sacrococcygeal masses include lymphangiomas, lipomas, tail-like remnants (Fig. 68-5), meconium pseudocysts, rectal duplications, and several other rare conditions.34,70–73
FIGURE 68-5 This patient had a scrotum-like perianal mass with anal stenosis at birth. An anoplasty was done with removal of the mass, which was not attached to the coccyx. Pathology showed only fibroadipose tissue with smooth muscle, vascular structures, and cartilage, consistent with a hamartomatous process or caudal vestige (also called a tail remnant).
Although many neonates with SCTs do not have symptoms, some require intensive care because of prematurity, high-output cardiac failure, disseminated intravascular coagulation, and tumor rupture or bleeding within the tumor.74–78 Lethal hyperkalemia from tumor necrosis has also been described.79 Lesions with a large intrapelvic component may cause urinary obstruction.78,80 Besides looking for signs of a myelomeningocele, the physical examination should always include a rectal examination to evaluate for an intrapelvic component. The most helpful imaging studies consist of plain anteroposterior and lateral radiographs of the pelvis and spine, looking for calcifications in the tumor and for spinal defects, and ultrasound of the abdomen, pelvis, and spine. Further preoperative studies are unnecessary in most newborns.
The diagnosis of purely intrapelvic teratomas is often delayed.34 Children develop constipation, urinary retention, an abdominal mass, or symptoms of malignancy, such as failure to thrive. Age is a predictor of malignancy in patients with testicular, mediastinal, and SCTs.2 The risk of malignancy is less than 10% at birth, but rises to more than 75% after age 1 year for SCTs, with the exception of familial presacral teratomas. The risk of malignancy also is high for incompletely excised lesions. Complete excision of the tumor should be carried out as soon as the neonate is stable enough to undergo the operation. Serum markers should be determined before the operation for later comparison.
In recent decades, the diagnosis has often been made on prenatal ultrasound, especially when this examination is routinely performed in the second trimester. The site of the lesion, its complex appearance, and intrapelvic extension, with or without urinary tract obstruction, are easily recognized. Although most small teratomas do not adversely affect the fetus, the presence of a large solid vascular tumor is associated with a significant mortality rate, both in utero and perinatally.75,76,78,81–83 Perinatal mortality is usually related to prematurity or tumor rupture with exsanguination (or both). Premature delivery may occur spontaneously from polyhydramnios or may be induced urgently because of fetal distress or maternal pre-eclampsia.
Repeated ultrasound assessment of tumor size is important because the fetus should be delivered by cesarean section if the tumor is larger than 5 cm or larger than the fetal biparietal diameter.84 Dystocia during vaginal delivery is associated with tumor rupture and hemorrhage, and is an avoidable obstetric complication. The options in managing unexpected cases with dystocia include emergency cesarean completion of the partially delivered fetus, who has been intubated and ventilated after vaginal presentation of the head.84
Polyhydramnios with larger tumors may lead to premature labor; amnioreduction is often required to decrease uterine irritability.78,83 Tumors that are larger than the fetal biparietal diameter at diagnosis, or that grow faster than the fetus or grow faster than 150 mL/week, are associated with a poor prognosis.82,85 As the tumor enlarges, the fetus may develop placentomegaly or hydrops, caused by high-output cardiac failure from vascular shunting within the tumor, with fetal anemia from intratumoral bleeding also playing a role. This is a harbinger of impending fetal death and should lead to urgent cesarean delivery, especially when the maternal mirror syndrome has developed.86 Open fetal surgical excision/debulking is one option in fetuses considered too premature to deliver. It has been performed with success in three of eight cases in one center and in three of four cases in another.76,87 Interestingly, there were no cases of fetal resection in the latter center in a more recent cohort of 23 fetuses with SCT, emphasizing that the indications for such a procedure are limited.82
As a result of the maternal and fetal risks, less invasive therapeutic options have been sought. Successful intrauterine endoscopic laser ablation of the large feeding arteries has been described.88 There have been several other reports, a few with good outcomes, but some with poor outcomes often related to severe fetal anemia and cardiac failure.78,89,90 Alcohol injection has also been used.78 Attempts at interrupting the high vascular flow have also been described by using radiofrequency ablation, with two survivors in the first four attempts.91 One survivor had significant perineal damage.92 As the technology has improved, there seems to be a renewed interest in this approach.93–95 However, it remains unclear when early delivery is preferable to intervention in utero in fetuses with early hydrops and placentomegaly. Some advocate a fetal operation before 30 weeks of gestation,82 with the same group recently suggesting early delivery in selected fetuses after 27 weeks of gestation.96 Others have reported survival after emergency delivery as early as 26 weeks of gestation.97 The EXIT procedure (ex utero intrapartum treatment) has been used as an adjunct to allow safer resection of a ruptured teratoma in a 27 week gestation fetus, but long-term neurological sequelae were noted.96
Purely cystic teratomas occur in 10–15% of cases. Prenatal diagnosis allows percutaneous aspiration to facilitate delivery (Fig. 68-6), to decrease uterine irritability, or to prevent tumor rupture at delivery.76,78,81,98,99 In another report, prenatal decompression using a cyst-amniotic shunt was successfully performed to relieve the obstructive uropathy caused by the cystic teratoma.100 Fetal MRI is a useful adjunct to the prenatal evaluation, providing additional information that aids in counseling and preoperative planning.101 It also helps in the differential diagnosis when the teratoma is entirely presacral.102
FIGURE 68-6 (A) Ultrasound image of a female fetus at 38 weeks of gestation, showing a large cystic mass (C) attached to the coccyx, with tiny cysts anterior to the sacrum (arrow). An ultrasound evaluation at 18 weeks was normal. The cyst was gradually enlarging from an initial diameter of 9.5 cm at 31 weeks of gestation. The cyst was aspirated for 650 mL of fluid, permitting external rotation from breech to the vertex position. Two days later, when labor was induced, another 200 mL of fluid was removed to permit an uncomplicated vaginal delivery. (B) Twenty-four hours postnatally, the lesion remained floppy with an area of skin ulceration, likely a consequence of excessive in utero distention. A mature cystic teratoma was confirmed histologically.
Operative Approach.
For most tumors, the major component is extrapelvic and the patient is placed in the prone position. If a significant intrapelvic or intra-abdominal component is present, or if the tumor is highly vascular and bleeding within the tumor is suspected, it may be wise to begin with a laparotomy or laparoscopy (see above). Generally, most resections can be achieved completely in the prone position, especially if the internal portion is cystic (Fig. 68-7).78 When in doubt, a safe approach is to prepare the skin from the lower chest to the toes, allowing the infant to be turned to the supine position without having to re-drape. Vaseline packing in the rectum facilitates its identification throughout the procedure. En bloc excision, including the coccyx, is preferable. Failure to remove the coccyx is associated with a high recurrence rate.2,3,103 An acceptable gluteal crease and perineum is formed by the appropriate positioning of the perianal musculature. The use of plastic surgical principles to close the skin improves the cosmetic appearance of the scar (see Fig. 68-7, inset).104
FIGURE 68-7 (A) The teratomatous attachment may compress the rectum, vagina, and bladder anteriorly. (B) The patient is placed on the operating table in a prone jackknife position, with general endotracheal anesthesia. An appropriate intravenous cannula should be placed in an arm vein. (C) The incision is an inverted V-shape to allow excision of the tumor and to facilitate an eventually satisfactory cosmetic closure. The amount of skin excised is dependent on the size and shape of the tumor. (D) The tumor is dissected from the gluteus maximus muscle. (E) The coccyx is transected and removed in continuity with the tumor. (F) The middle sacral artery is the major blood supply to the tumor and is ligated (arrow) after transection of the coccyx. (G) Excess skin is excised to facilitate closure. (H) As the tumor is adherent to the rectum, sharp dissection can be directed by placing a finger in the rectum. (I) Placement of sutures between the anal sphincter and the presacral fascia (a). When the sutures are tied, the anal sphincter is pulled upward to the sacrum to form a gluteal crease (b). (J) A drain is left in the surgical site for drainage of postoperative serosanguineous fluid. Inset, Recently described technique for closure after excision of large teratomas. Using plastic surgery principles, this avoids ‘dog ears’ and places the scars along natural skin lines for a much-improved long-term cosmetic result. (K) If the tumor extends through the bony pelvis into the retroperitoneum, a urinary bladder catheter is inserted to facilitate suprapubic dissection. (L) Lower abdominal transverse incision allows interruption of the middle sacral artery and dissection of the tumor from the sacrum and pelvis, which is eventually removed from the perineum. (Inset redrawn from Fishman SJ, Jennings RW, Johnson SM, et al. Contouring buttock reconstruction after sacrococcygeal teratoma resection. J Pediatr Surg 2004;39:439–41; with permission.)
Although the chevron incision has been used by most surgeons, a vertical incision is sometimes advantageous. It is preferred for smaller teratomas because it leaves a nearly normal-looking median raphe (Fig. 68-8). Resection of the excess skin at the closure gives an optimal cosmetic result. Others have reported using this approach for large tumors as well.105
FIGURE 68-8 Smaller cystic teratoma at age 1 month, initially mistaken for a hemangioma owing to its soft compressible nature and bluish skin discoloration. This tumor lends itself well to a longitudinal elliptical excision with midline closure, as in a posterior sagittal anorectoplasty.
Several techniques have been described to help in the management of giant SCTs. These include intraoperative snaring of the aorta,77 laparoscopic division of the median sacral artery,106,107 the use of extracorporeal membrane oxygenation and hypothermic perfusion,108 devascularization and staged resection,97,109 and preoperative embolization with or without radiofrequency ablation.110,111 Autologous cord blood transfusion is another useful adjunct.112
Prognosis.
Fetuses with an SCT diagnosed in utero have a survival rate in excess of 90% if the tumor is small and discovered by routine prenatal ultrasound. If a complicated pregnancy is the indication for ultrasound evaluation, the mortality increases to 60%. Nearly 100% of patients die when hydrops or placentomegaly develop.76,78,81,113 Dystocia or tumor rupture during delivery are likely underreported as a cause of mortality.83,114 In one series, 10% of babies died during transfer, all before the widespread use of antenatal ultrasound.115 In a report describing 24 patients with a SCT diagnosed on routine obstetric ultrasound, 3 were aborted electively, four died in utero at 20 to 27 weeks of gestation, and three died of tumor rupture during delivery at 29 to 35 weeks of gestation (one after vaginal and two after cesarean delivery).75 The incidence of placentomegaly (none), hydrops (5%), and polyhydramnios (19%) was lower than in series in which ultrasound was performed for an obstetric reason.76,84,116 Tumor size, vascularity, and content were used to develop a prognostic classification from a cohort of 44 fetal SCTs.117 Fifty per cent of infants with tumors 10 cm or greater that were highly vascular or fast growing died, whereas none died if these features were absent or if the tumor was predominantly cystic. Recently, a growth rate >150 mL/week was found to be associated with an increased risk of perinatal mortality.82 A vena cava diameter ≥6 mm and cardiac output ≥600 mL/kg/min have also been used to detect high-output cardiac failure in the early stages.82 Another prognostic classification involves the ratio of solid tumor volume over the head volume.118 In a series of 28 cases, none of the fetuses with STV/HV ratio <1 died, while 61% with a ratio >1 died. The same center has also reported risk stratification based on fetal echocardiographic findings.119 Finally, the ratio of tumor volume over fetal weight was found to be an early prognostic marker.120
In the absence of severe prematurity and perinatal complications, the prognosis is dependent on the presence of malignancy and is therefore related to age at operation and completeness of resection.121,122 When the tumor is benign and completely excised, recurrence is low, unless the tumor is large and mostly solid.123 The recurrent tumor may be benign or malignant, and benign metastatic tissue may become evident in lymph nodes.124 Although immature or fetal elements in gonadal teratomas are associated with a higher risk of aggressive behavior, this is not considered true for SCTs.2,5,16 However, a multicenter study identified immature histology, in addition to malignancy and incomplete resection, as risk factors for recurrence.122 Although malignant recurrence of a ‘benign’ teratoma may be as high as 10–15%,6,122 the original benign diagnosis may have been due to sampling error,15 an undetected residual microscopic focus of malignant tumor,125 or secondary to incomplete coccygectomy at the initial operation.35 Patients whose tumors are resected after the newborn period have a higher risk of malignant recurrence, especially when an elevated AFP level is present at diagnosis. The elevated AFP likely signifies the presence of malignancy in the original tumor.15,126 It is important to monitor all patients by physical examination, including rectal examination and serum markers (AFP and CA 125), every two or three months for at least 3 years because most recurrences occur within three years of operation.24,127
Recurrent disease is usually local, but metastases to inguinal nodes, lung, liver, brain, and peritoneum can occur, including pseudomyxoma peritonei.128 The prognosis of a malignant tumor or a malignant recurrence was dismal until the advent of platinum-based chemotherapy.125 Survival rates of 80–90% are now achieved, even in the presence of metastatic disease, but the risk of late recurrences or second malignancies persists.129–131 Older patients presenting with large malignant tumors usually undergo biopsy followed by chemotherapy before resection is attempted to avoid sacrificing vital structures.16,130,132
A Children’s Cancer Group (CCG) review illustrates the shift in mortality causes, from late diagnosis/malignancy to perinatal events.129 The mortality was 10% in 126 patients treated in 15 institutions from 1972 to 1994. Three patients died of severe associated anomalies. Two died of hemorrhagic shock postoperatively and six due to combinations of severe prematurity, birth asphyxia due to failed vaginal delivery, or preoperative tumor rupture. Death from a metastatic YST occurred in one patient. A second patient with metastatic disease was lost to follow-up and is presumed dead. Thus, only two deaths occurred from malignancy, despite a total of 20 YSTs (13 malignant at initial operation, seven malignant recurrences after resection of ‘benign’ teratomas). Owing to the effectiveness of current chemotherapy in treating recurrent disease, as well as its toxicity in young infants, it appears that a completely excised malignant YST does not require adjuvant therapy. These patients should be closely monitored clinically and with serial AFP measurements.133 Similar encouraging results have been found in the German Cooperative Studies.134
In the current era of the rather routine use of ultrasound in pregnancy, the prognosis for patients with an SCT is not dependent on Altman’s classification34 but rather on tumor size, physiologic consequences, histology, and associated anomalies. The prognosis of malignant tumors depends on tumor type, stage, location, and patient age.129 Functional results in survivors have been reported as excellent in most series.78,135,136 However, several reports draw attention to fecal and urinary continence problems, as well as lower limb weakness.121,137–141 Some of these problems are clearly related to associated anomalies,115 the need for reoperation,142 or to the presence of large presacral or intra-abdominal tumors,80,135 but they can occur after excision of purely extrapelvic benign tumors. A good outcome requires meticulous dissection along the tumor capsule, preservation and reconstruction of muscular structures, and long-term follow-up. One group advocates earlier cesarean delivery to minimize urologic sequelae in patients with large tumors causing urinary tract dilatation.139 Others have placed vesico-amniotic shunts in such cases with good outcomes.78,100 Urodynamic studies and surveillance ultrasound are now being performed more regularly in some centers.143 Patients with Currarino syndrome, who often have a tethered cord in addition to the presacral tumor, appear to have an increased risk of bladder and bowel dysfunction.144,145 A poor cosmetic result was noted in more than half of the patients in another review.146 These authors advocate early assessment of bladder, anorectal, and sexual function along with cosmetic results within a structured oncology follow-up program. The technique described by the Boston Children’s group is a major step to improve cosmesis (see Fig. 68-7),104 while others favor a sagittal incision.105
Thoracic Teratomas
The anterior mediastinum is the most common site of thoracic teratomas, which account for 7–10% of all teratomas (see Table 68-1).2,3
Mediastinal Teratomas.
Mediastinal teratomas are diagnosed from the fetal period to adolescence and even adulthood.2,147–149 Most are located in the anterior mediastinum, but a few have been described in the posterior mediastinum, some with epidural extension.150–152 In infants, respiratory distress is a common presenting manifestation,153 but in older children the teratoma is often an incidental finding on chest radiograph (Fig. 68-9).2 Any patient with a mediastinal mass that presents with orthopnea or a reduction in the tracheal cross-sectional diameter of greater than 50% on axial imaging is at a significant risk for airway collapse during general anesthesia.154 In these cases, procedures using local or regional anesthesia may be required to obtain a confirmatory diagnosis. Mediastinal teratomas may be first seen as a chest wall tumor and may even erode through the skin. They also can erode into a bronchus, with hemoptysis as the initial manifestation,155 or rupture into the pleural cavity.156 Secondary pericardial effusion and tamponade have also been described.157 A strong association has been found with Klinefelter syndrome. In these cases, choriocarcinoma in the teratoma often leads to precocious puberty (see under Associated Anomalies).2,25,58 Mediastinal germ cell tumors have also been observed concurrently or after treatment for hematologic malignancies such as Langerhan’s cell histiocytosis and hemophagocytic syndrome.57,158,159 Histologically, the presence of immature tissue does not affect the prognosis in children younger than 15 years. After age 15, mediastinal teratomas have a high incidence of malignant behavior, which is usually indicated by elevated levels of AFP or β-hCG, or both.149 For those with malignancy, YST is most prevalent in girls and young boys while mixed histology prevails in over 50% of older adolescent males.160
FIGURE 68-9 (A) A 13-year-old African boy has an asymptomatic anterior mediastinal mass that was discovered on routine immigration chest radiograph. (B) The CT scan shows a heterogeneous mass adjacent to the aorta, suggestive of a neoplasm (thymoma or lymphoma). During consideration of a fine-needle aspiration biopsy, ultrasonography was done and suggested the presence of cysts with debris (not shown). MRI (not shown) confirmed the presence of the cystic components as well as fat. A mature teratoma was excised through a small left anterior mediastinotomy, removing the left second costal cartilage.
Tumors should be excised through either a median sternotomy or a thoracotomy.16,161 Smaller tumors may be approached through an anterior mediastinotomy (see Fig. 68-9) or by thoracoscopy,162 although tumor seeding is a concern with the latter. Complete resection is the goal, but often these masses are too large at presentation and require initial biopsy followed by chemotherapy.16,163,164 During chemotherapy, attention should be paid to the ‘growing teratoma syndrome’ that occurs when the benign elements within a germ cell tumor continue to grow.165 A Pediatric Oncology Group (POG) and Children Cancer Group (CCG) intergroup study involving 38 patients demonstrated that primary resection could be achieved in 14 patients.160 In another 18, chemotherapy reduced tumor size in 57% while the remainder were stable or increased in size. All patients with residual disease underwent successful post-chemotherapy resection. The overall survival for malignant mediastinal germ cell tumors was 71% which is less than for other extragonadal sites. The prognosis is best for young children with YSTs and worst in older adolescents with mixed germ cell histology.149,160 In a recent series from Memorial Sloan-Kettering Cancer Center, normalization or reduction in preoperative tumor markers was the strongest predictor of increased survival.161
Intrapericardial Teratomas.
Intrapericardial teratomas are most commonly seen in the newborn period or in utero, with evidence of cardiorespiratory distress secondary to pericardial effusions or nonimmune fetal hydrops resulting from cardiac compression.2,3,166 While a fetal diagnosis allows for early postnatal treatment in most newborns, it may also offer an opportunity for antenatal interventions such as fetal pericardiocentesis.167 Early delivery for emergency surgical excision should be considered if the baby develops signs of cardiac tamponade.168 Intrapericardial teratomas are also the leading cause of massive pericardial effusion in the neonate and any delays in diagnosis could be fatal.3,169 In older infants, it may present with respiratory distress or poor feeding. Ultrasound imaging usually demonstrates a cystic or solid teratoma located anterior to the right atrium and ventricle with attachments to the great vessels (see Fig. 25-4).170 The tumor may also be found incidentally on chest radiographs performed for other reasons. The only treatment option for this lesion is excision. On histologic examination, these teratomas are usually composed of mature tissue with or without neuroglial elements.2,3
Abdominal Teratomas
Retroperitoneal Teratomas.
Retroperitoneal teratomas occur outside the pelvis, often in a suprarenal location. They represent about 4% of all childhood teratomas, and 75% occur in children younger than 5 years.3,173 They occur twice as frequently in females and may have an association with Klinefelter syndrome.16,55
[/not-level-membership-for-pediatrics-category]
