Renal Tumors
Renal tumors are the second most common abdominal tumor seen in infants and children behind neuroblastoma. They represent a wide spectrum from benign to extremely malignant tumors (Box 65-1). These tumors include: Wilms tumor (WT) (also referred to as nephroblastoma or renal embryoma), renal cell carcinoma (RCC), clear cell sarcoma of the kidney (CCSK), rhabdoid tumor of the kidney (RTK), congenital mesoblastic nephroma (CMN), cystic renal tumor, and angiomyolipoma. Advances in the management of these tumors have been significant over the past six decades since Sidney Farber first administered dactinomycin (Actinomycin D) for advanced-stage WTs.1 Multidisciplinary and multi-institutional prospective randomized cooperative group trials in both North America and Europe by the Children’s Oncology Group (COG-formerly the National Wilms Tumor Study Group [NWTSG]) and the Société Internationale d’Oncologie Pédiatrique (SIOP) have produced a large body of evidence-based knowledge to establish the optimal risk-based treatments (Table 65-1). The goal of this chapter is to describe the early history of WT, followed by a discussion of the etiologic factors in renal tumor formation, molecular genetics, pathologic subtypes and premalignant syndromes, and current treatment algorithms for children.

Wilms Tumor
History
The first descriptions of WT have been variably attributed to either Rance in 1814 or Wilms in 1899.2–4 The first known specimen of this tumor was preserved by the British surgeon John Hunter between 1763 and 1793.5 This specimen of a bilateral tumor in a young infant remains in the Hunterian Museum of the Royal College of Surgeons in London. Carl Max Wilhelm Wilms was a German pathologist and surgeon. Wilms’ name became indelibly linked to this tumor in children after publication of his comprehensive monograph in 1899 titled ‘Die Mischgeschwülste der Niere’ which described seven children with nephroblastoma as part of a monograph on ‘mixed tumors of the kidney’.3,4 In it he proposed that tumor cells originate during the development of the embryo. The first successful nephrectomy was probably performed by Thomas Jessop at the General Infirmary in Leeds, England on the 7th of June, 1877 on a 2-year-old child with hematuria and a tumor of the kidney.6,7
Resection was recognized early as an effective treatment for WT and remains as the cornerstone of all therapies. However, at the beginning of the 20th century, survival was approximately 5% and operative mortality was high. In 1916 radiation therapy was added as an adjuvant therapy by Friedlander.8 In the 1930s, Ladd and Gross described the principles of operative therapy for WT, including transperitoneal exposure and early ligation of the renal pedicle.7,9 They stressed the need to remove the perirenal fat to resect lymphatic extensions and to avoid rupture of the renal capsule, principles we continue to follow today. From 1931 to 1939, survival from resection alone, involving ligation of the renal pedicle before removal, was 32% at the Children’s Hospital in Boston.1,10 Gross and Neuhauser later proposed the routine addition of abdominal radiation to WT therapy and reported an estimated 47% cure.11 Thus in 1940,1 most patients began to receive postoperative irradiation to the renal fossa. This decreased the local recurrence rate, but did not significantly affect the incidence of pulmonary metastases or improve the long-term survival. Under Gross’s tutelage, pediatric surgeons in North America generally performed primary resection of WT, while in Europe, the Paris school led by Schweisguth and Bamberger reported early success with preoperative irradiation, establishing a precedent for initial adjuvant therapy.12
WT was the first malignancy in which the importance of adjuvant treatment was recognized. This principle was espoused by Sidney Farber decades before it would be applied to other pediatric and adult solid tumors.1 The concept of adjuvant therapy ‘was based upon the supposition that in the children with WT who died, the tumor must have metastasized already at the time of discovery of the primary tumor’, although no evidence of spread was available. Dactinomycin was the first active agent identified for the treatment of WT. Of the 53 patients who had no demonstrable metastases on admission treated with combined regimen of operation, local radiation, and dactinomycin from 1957 to 1964, an 89% 2-year disease-free survival was reported,1 a very reasonable rate of survival even today. In patients with metastases identified at presentation, 18 of 31 (58%) were alive and free of disease more than 2 years later. In the early 1960s, vincristine sulfate was identified as an active agent against WT and was added to the standard therapy.13
Epidemiology
WT is the most frequent tumor of the kidney in infants and children. Its incidence is 7.6 cases for every million children younger than 15 years, or one case per 10,000 infants.14 This translates into 600 to 650 cases a year in North America. It is less common in East Asian populations than in white children, but is more frequent in black children.15,16 The mean age at diagnosis is 36 months, with most children presenting between the ages of 12 and 48 months. Tumors tend to occur earlier in boys than girls. WT incidence decreases over the age of 10 and is less common under 6 months of age. However, it still constitutes 20% of all renal tumors in children less than 6 months. Bilateral Wilms tumors (BWT) occur in 4–13% of patients.15,17
WT occurs in several well-described syndromes. These comprise about 10% of all WT cases, and include sporadic aniridia, isolated hemihypertrophy, the Denys–Drash syndrome (nephropathy, renal failure, male pseudohermaphroditism, and WT), genital anomalies, Beckwith–Wiedemann syndrome (BWS; visceromegaly, macroglossia, omphalocele, and hyperinsulinemic hypoglycemia in infancy), and the WAGR complex (WT with aniridia, genitourinary malformations, and mental retardation). These congenital syndromes have helped identify the genetic and etiological mechanisms inherent in developing a WT. For example, children with WAGR syndrome, which is associated with a chromosomal defect in 11p13, are at a 30% higher risk of developing WT than a normal child. Aniridia is usually diagnosed at birth and helps identify these children.18 BWS is a second common syndrome associated with WT. BWS affects 1 in 14,000 children. These children are at an increased risk of several types of embryonal tumors, including WT. The most frequently observed tumors in BWS are WT and hepatoblastoma, which comprise 43% and 12% of reported associated cancers, respectively.19,20 The risk for malignant development is greatest in the first decade of life. Three large studies of children with BWS reported tumor frequencies of 7.1% (13/183), 7.5% (29/388), and 14% (22/159).19,21–23
Molecular Genetics of Wilms Tumor
WT is an embryonal tumor that was originally thought to fit the model proposed by Knudson’s ‘two-hit’ hypothesis for cancer development.24 However, the genesis of WT has been shown to be far more complex. Multiple mutated WT genes have been identified as well as areas of loss of genetic material and allelic uniqueness (loss of heterozygosity) that are important to tumor development. A few of these genetic anomalies have been evaluated in clinical trials to assess their prognostic significance and are now being used in conjunction with traditional factors, such as stage, to determine the intensity of therapy.19,25 A detailed discussion of all the genetic anomalies associated with WT is beyond the scope of this chapter; however, a summary of the key genes and genetic anomalies is presented in Table 65-2.
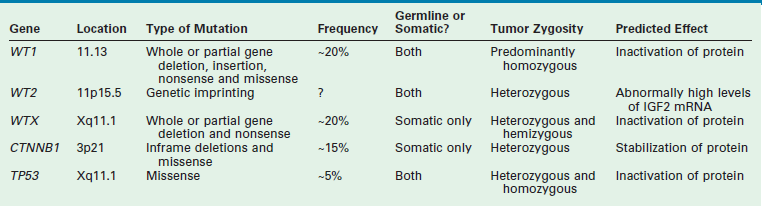
TP53
TP53 mutations in WT are almost exclusively found in tumors with anaplastic histology. Seventy-five per cent of anaplastic WT have p53 mutations.26,27 Interestingly, some tumors can contain both anaplastic and favorable histology. Studies in these tumors demonstrate that the p53 mutation is found only in the areas of anaplasia and not in the area of favorable histology.26 This implies that p53 mutations may be essential for anaplastic progression.
CTNNB1
CTNNB1 plays a central role in the Wnt signal transduction pathway. The Wnt signaling pathway describes a network of proteins known for their roles in embryogenesis and cancer. Deregulation of CTNNB1 has been linked to several malignancies. CTNNB1 mutations have been reported to occur in 15% of WT.28,29 The mutations resulted in areas of phosphorylation and degradation of beta catenin, which is highly correlated with WT1 mutation and the WTX gene.
WTX
The WTX gene (also known as AMER1 for adenomatous polyposis coli (APC) membrane recruitment 1) was found to be mutated in 15 of 51 (29%) WT tested, making it the most common known gene mutation in WT.30 Localization of WTX on the X-chromosome results in complete inactivation by a single mutational event in males, and in females if the active X-chromosome is affected. The exact clinical impact of WTX on WT development remains to be defined.
WT1 (11p13)
WT1 gene was the first gene to be linked with WT development. Evidence suggests that WT1 may act as a tumor suppressor gene or oncogene and is located at chromosome 11p13.31,32 Wild-type WT1 is important for normal cell development and survival. WT1 is involved in cell growth, differentiation, and apoptosis.33 Patients heterozygous for WT1 germline mutations are predisposed to WT, and WT1 is inactivated in tumors. This implies the loss of WT1 function is associated with enhanced cell viability and/or proliferation. Ablation of WT1 at the initial stages of kidney development results in apoptosis and renal agenesis, indicating that it has a crucial role in maintaining cell viability. In some leukemias, the increased expression of WT1 compared with normal bone marrow cells, along with some reports of WT1 expression being a marker of poor prognosis, suggest that WT1 functions as an oncogene.26 By contrast, observations of WT1 inactivating mutations in leukemias suggest it functions as a tumor suppressor gene. It should be emphasized that children with congenital syndromes and WT make up a very small proportion of the total WT population. However, loss of heterozygosity (LOH) is seen in 30–40% of WT patients in the region of WT1. Based on this observation, it would be expected that many patients with sporadic WT would have a mutation in the WT1 gene. Surprisingly, the incidence of mutations of WT1 associated with WT in the sporadic form of the disease is low (10–20%).34,35
Loss of Heterozygosity
LOH refers to loss of genetic material and allelic uniqueness. A major aim of the fifth National Wilms Tumor Study (NWTS-5) was to determine if tumor-specific LOH for chromosomes 11p, 1p, or 16q was associated with an adverse prognosis for children with favorable-histology (FH) WT, a finding suggested in earlier retrospective studies.25,36,37 The results of NWTS-5 demonstrated that outcomes for patients with LOH at 1p and 16q were at least 10% worse than those without LOH (Fig. 65-1). These findings were used as determinants for the intensity of therapy on the current COG renal tumor studies.
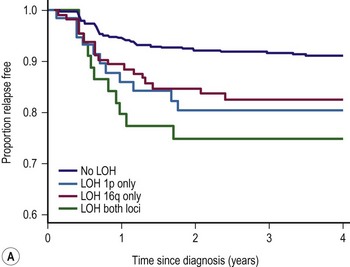
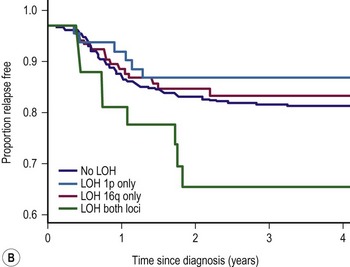
FIGURE 65-1 (A) Relapse-free survival with loss of heterozygosity (LOH) at chromosomes 1p and 16q for patients with stage I/II Wilms tumor of favorable histology. (B) Relapse-free survival for patients with stage III/IV Wilms tumor with favorable histology and LOH at chromosome 1p and 16q. (From Grundy PE, Breslow N, Li S. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms’ tumor: A report from the National Wilms’ Tumor Study Group. J Clin Oncol 2005;23:7312–21, with permission from the American Society of Clinical Oncology.)
Pathologic Precursors: Nephrogenic Rests, Nephroblastomatosis, and Multicystic Dysplastic Kidneys
Nephrogenesis in the normal kidney is usually complete by 34 to 36 weeks gestation. The presence of nephrogenic rests (NRs; persistent metanephric tissue in the kidney after the 36th week of gestation) has been associated with the occurrence of WT. NR are considered precursor lesions to WT. However, only a small number develop clonal transformation resulting in the development of WT. The presence of multiple or diffuse NRs is termed nephroblastomatosis.
These NRs may occur in a perilobar (PLNRs) or intralobar (ILNRs) location, and may be single or multiple (Fig. 65-2). In children with aniridia or the Denys–Drash syndrome, the lesions are primarily ILNRs, whereas children with hemihypertrophy or the BWS have predominantly PLNRs (Table 65-3).38 NRs may be further classified by their growth phase, which has been separated into three phases; (1) incipient or dormant nephrogenic rests which show few well-formed tubular structures, but no evidence of proliferation and no mitoses; (2) hyperplastic nephrogenic rests which are composed of epithelial elements with nodular expansive growth; and (3) sclerosing rests which consist of stromal and epithelial elements with few blastemal nephrogenic elements. Most NRs are dormant or in the sclerosing phase, and the majority will spontaneously resolve. Hyperplastic NR can produce masses as large as conventional WT and can present a diagnostic dilemma. NRs are rare in the general population. In an autopsy series of infants younger than 3 months, nine of 1,035 infants (0.87%) had PLNRs, while ILNRs occurred in only 2 of 2000 cases (0.1%).39
TABLE 65-3
Association of Nephrogenic Rests with Wilms Tumor Predisposition Syndromes and Congenital Anomalies
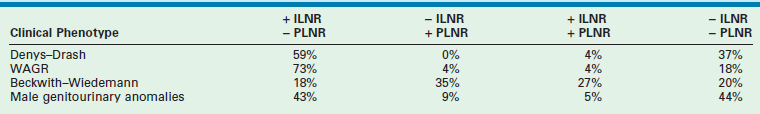
ILNR, intralobar nephrogenic rests; PLNR, perilobar nephrogenic rests; +, present; −, absent.
From Breslow NE, Beckwith JB, Perlman EJ, et al. Age distributions, birth weights, nephrogenic rests, and heterogeneity in the pathogenesis of Wilms tumor. Pediatr Blood Cancer 2006;47:260–7.
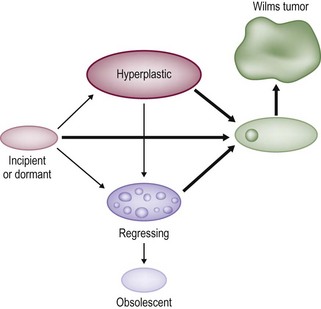
FIGURE 65-2 Diagrammatic depiction of nephrogenic rests and their classification. Thick arrows indicate tumor induction. (From Beckwith JB. Precursor lesions of Wilms’ tumor: Clinical and biological implications. Med Pediatr Oncol 1993;21:158–68.)
Pathologic distinction between NR and WT can be very difficult. In fact, it may be impossible to distinguish a hyperplastic NR from a WT based on an incisional or needle biopsy specimen that does not include the margin between the NR and the remaining kidney.38,40 Most hyperplastic nodules lack a pseudocapsule at their perpiphery, whereas most WTs will have one. Thus, if the biopsy specimen does not contain the lesion and its margin, it will be difficult to differentiate between these two lesions. In these situations, the current recommendation is to use the term ‘nephrogenic process, consistent with a WT or a nephrogenic rest’.40,41 This is one reason that it is recommended to avoid percutaneous biopsies in children with renal masses.
The incidence of NRs is about 100 times greater than that of WT (1/10,000 infants). In a review of cases of WT reported in the NWTS-4, 41% of the unilateral WTs were associated with NRs, whereas in children with synchronous bilateral WT, the incidence of NRs was 99%.38 These were primarily PLNRs. A child with a WT and NRs in the resected specimen is at increased risk of developing a metachronous tumor in the contralateral kidney.42 In a child under one year of age, this risk is very significant, and these children need to be followed very carefully with sequential ultrasound (US) examinations. It is very difficult to diagnose NRs on imaging. Although magnetic resonance imaging (MRI) may be helpful, there is no gold standard.
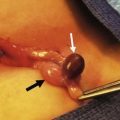
Diffuse hyperplastic perilobar nephroblastomatosis (DHPLN) is a unique category of nephroblastomatosis in which the rests form a thick rind around the periphery of the kidney. Infants with DHPLN may initially present with large unilateral or bilateral flank masses (Fig. 65-3). A characteristic radiographic finding is massively enlarged kidneys that maintain their normal configuration and lack evidence of necrosis. As with the isolated NRs, proliferation of the thin rind of NRs on the periphery of the kidney will preserve the normal configuration of the kidney, but result in marked enlargement of its size. This is in contrast to WT, in which the normal renal configuration and collecting system are generally distorted. The optimal diagnosis and management of a child with DHPLN is unclear. In a 2006 study of 52 children and 33 cases of DHPLN, histologic examination alone was unable to establish a diagnosis in 21 children that underwent biopsy at the time of initial presentation.41 In the same study, 24 children developed WT during long-term follow-up with eight being anaplastic. Thirteen had single tumors and 11 were multiple. Treatment was variable and included surgery, radiation and chemotherapy, or expectant observation. The three patients who received no therapy developed WT 10 months after diagnosis. Chemotherapy may decrease the proliferative element in NRs, but it is unclear whether it prevents the development of malignancy. A current COG study includes a treatment protocol to address some of the questions with respect to DHPLN and its optimal therapy.
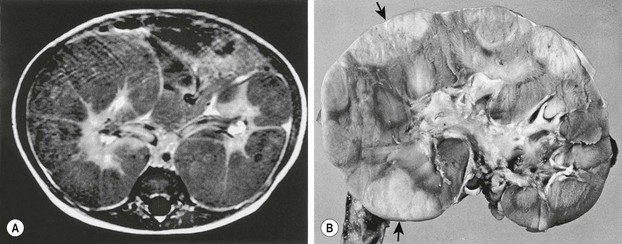
FIGURE 65-3 (A) MR image of a 10-month-old infant who presented with large bilateral flank masses demonstrates a picture characteristic of diffuse hyperplastic perilobar nephroblastomatosis (DHPLN) with extensive involvement of the entire cortex of the kidney with no evidence of necrosis and general preservation of the shape of the kidney. (B) A resected kidney with a similar pattern of DHPLN reveals extensive involvement of the periphery of the cortex by severely hypertrophied nephrogenic rests (arrows). Resection of such kidneys should be avoided because, in most cases, the hypertrophy will resolve and the kidney will have excellent preservation of its function. (B courtesy of J. B. Beckwith, by permission.)
An increased risk of WT arising in multicystic dysplastic kidneys has been suggested. This is primarily based on case reports and all children have been under the age of four. In contrast, a report describing 1041 infants and children with multicystic dysplastic kidneys found no case associated with a WT.43 The authors concluded that routine prophylactic nephrectomy was not needed in children with multicystic dysplastic kidneys. Furthermore, a review of the NWTS pathology files identified only three cases of dysplastic kidneys in more than 7,000 children with WT over a 26-year interval.44 Thus, prophylactic nephrectomy in children with multicystic dysplastic kidney for malignancy is not justified. However, screening ultrasound in these children until age 4 is suggested.43,44
Pathology
WTs are currently divided into those with ‘favorable’ histology and those with ‘unfavorable’ histology. Unfavorable histology proved to be the most important factor in patient outcome in NWTS-1, a finding that has remained consistent through the current trials.5,45 Fortunately, favorable histology comprises 90% of the tumors. These tumors are the ‘classic WT’ consisting of three elements: blastemal, stromal, and epithelial tubules (Fig. 65-4). Tumors contain various proportions of each of these elements. Triphasic are the most characteristic, but biphasic and monophasic lesions also occur.46 The proportion of these three elements in WTs have been studied, but have not been shown to predict outcomes. Under the current treatment protocols, they are not utilized to determine therapy.46,47 Abnormal mucinous or squamous epithelium, skeletal muscle, cartilage, osteoid, or fat are less frequent elements found in WT.5
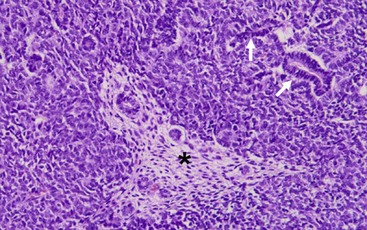
FIGURE 65-4 The classic ‘triphasic’ histologic pattern (blastemal, epithelial, and mesenchymal derivatives) of a Wilms tumor is seen on this H&E slide. There is a predominance of small undifferentiated blastemal cells in the image that surround a few neoplastic duct and tubular epithelial structures (solid arrows). In the center is an island (asterisk) of spindle-shaped fibroblastic mesenchymal cells that surround a few tubules. The neoplastic cells in the image lack the requisite features of an anaplastic variant (see Fig. 65-5).
Unfavorable tumors are those with focal or diffuse anaplasia.5,48,49 Anaplasia is defined by multipolar polyploid mitotic figures, marked nuclear enlargement (giant nuclei with diameters at least three times those of adjacent cells), and hyperchromasia (Fig. 65-5).50 Determining whether a tumor has diffuse or focal anaplasia is important for prognosis and therapy.48 Focal anaplasia is defined as the presence of one or a few sharply localized regions of anaplasia within a primary tumor, the majority of which contains no nuclear atypia. Tumor designated as diffuse anaplasia must have at least one of the following four criteria: anaplastic cells outside of the kidney, presence of anaplasia in a random kidney biopsy, anaplasia in more than one region of the kidney, or anaplasia in one region with extreme nuclear pleomorphism in another site. Anaplasia occurs primarily in children older than 2 years. In NWTS-1, 66.7% of patients with anaplasia experienced relapse and 58.3% died of their tumor.45 Of note, there was a higher frequency of relapse and death in the ‘diffuse’ subgroup. Additionally, anaplasia in extrarenal tumor sites and a predominantly blastemal tumor pattern were both adverse prognostic factors.50 Moreover, anaplasia is a marker of resistance to therapy, not of aggressive behavior.46,47,50 TP53 deletion on chromosome 17 has been associated with anaplasia. However, a recent study demonstrated recurrent anaplasia-specific genomic loss and under-expression in several additional regions, most strikingly 4q and 14q, suggesting anaplasia is not linked solely to deletions of chromosome 17, but is associated with loss at multiple loci.51
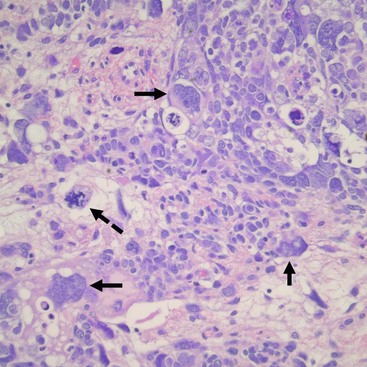
FIGURE 65-5 Salient features of the anaplastic Wilms’ tumor variant with H&E staining. Anaplasia is defined by three requisite criteria that are all depicted in the image: atypical mitotic figures (dotted arrow) (often tripolar or multipolar); nuclear enlargement (solid arrows) to greater than three times the size of resident cell nuclei of the same type; and nuclear hyperchromicity present in some of the cells. Enlargement and hyperchromicity reflect the fact that anaplastic Wilms tumors contain nearly twice the amount of DNA as normal cells with duplication of many whole chromosomes in each cell.
CCSK and malignant RTK were grouped in the initial NWTSG studies with the unfavorable histology WT. They are now considered distinct entities from WT, based on their pathologic appearance, biologic behavior, and response to different therapies.52,53 CCSK is a highly malignant tumor with an unusual proclivity for bony metastasis. It generally appears as a large unifocal and unilateral tumor with a homogeneous mucoid, tan, or gray/tan cut surface, often with foci of necrosis or prominent cysts.52 This tumor invades and surrounds the renal parenchyma rather than compresses the margin into a pseudocapsule as is seen in a WT. Its classic appearance is that of a deceptively bland tumor with uniform oval nuclei with a delicate chromatin pattern, a prominent nuclear membrane, and sparse, poorly stained vacuolated ‘water-clear’ cytoplasm with indistinct cell membranes. Although the cells generally appear in cords or nests divided by an arborizing network of vessels and supporting spindle cell septa, there are variations and nine major histologic patterns have been identified.52,53 Recently, a translocation t(10;17) and deletion (14q) have also been described in CCSK, suggesting that they may play a role in its pathogenesis.54 The cell of origin of this tumor is not known. In addition to osseous metastases, CCSKs also have a significant incidence of metastases to the brain. Long-term follow-up is important as one report found 30% of the relapses occur more than two years after diagnosis.55
Malignant RTK occur in young infants with a median age of 11 months.56 Most (85%) cases occur within the first two years of life. It is the most aggressive and lethal of all pediatric renal tumors, and fortunately only accounts for 2% of renal tumors. Grossly these tumors are unencapsulated and invasive with characteristic involvement of the perihilar renal parenchyma. Histologically, RTKs are characterized by monomorphous, discohesive, rounded to polygonal cells with acidophilic cytoplasm and eccentric nuclei containing prominent large ‘owl eye’ nucleoli reminiscent of skeletal muscle, but lacking its cytoplasmic striations, ultrastructural features, and immunochemical markers.57 A large periodic acid–Schiff (PAS)-positive hyaline cytoplasmic inclusion occurs in a variable population of tumor cells and is a hallmark of this tumor.58 Ultrastructural examination reveals parallel cytoplasmic filamentous inclusions packed in concentric whorled arrays, a distinctive feature of this tumor, which suggests a neuroectodermal origin. The tumor tends to infiltrate the adjacent renal parenchyma rather than to compress it. These tumors are notable for the occurrence of a second primary tumor in the midline of the brain, resembling medulloblastoma.59 A consistent deletion (22q11-12) has been described in both renal and extrarenal rhabdoid tumors.60,61 These deletions delineate an area of overlap at the site of the hSNF5/INI1 gene, and tumors have biallelic alterations or deletions of this gene.62–64 For all renal tumors except RTK, immunohistochemical staining for the wild type INI-1 protein shows nuclear positivity. In renal and extra renal rhabdoid tumors, this is absent.64
Staging
Two principal staging systems are used for children with WT (Table 65-4). The COG system is based on pretreatment findings prior to administration of chemotherapy or radiotherapy. Patients are given a local stage and a disease stage. The local stage defines the extent of abdominal disease, while the disease stage considers both the local extent of disease and distant metastasis. Both factors determine therapy with the use of local radiation therapy to the tumor bed based on the local stage, and the intensity of chemotherapy based on the disease stage.65

SIOP protocols generally recommend chemotherapy followed by nephrectomy, with surgicopathologic staging occurring at the time of nephrectomy. The SIOP classification was revised in 2010 based on a review of the histologic appearance of the tumors at resection (post neoadjuvant chemotherapy) and the corresponding outcomes.66,67 Tumors are now classified on SIOP protocols as completely necrotic (low-risk tumor), blastemal (high-risk tumor), and other histology (intermediate-risk tumors).
Prognostic Factors
The current prognostic factors used in COG trials are histology, stage, age, tumor weight, response to therapy and LOH at 1p and 16q. The two most important factors continue to be the histology and the stage of the tumor.25,68,69 The details and prognostic significance of tumor pathology has been previously discussed in the Pathology section. The tumor stage is determined by the results of the imaging studies, the findings at operation, and the histological findings.
Rapid response to chemotherapy is a prognostic category that is being evaluated in patients who present with stage IV disease due to lung metastasis. The goal in these patients is to avoid lung radiation. Response to therapy is also being assessed in BWTs. Also, LOH at both 1p and 16q is now being used for determination of therapy in the current COG renal tumor studies.25
Clinical Presentation and Diagnosis
The differential diagnosis for a malignant abdominal mass in a child includes WT, neuroblastoma, hepatoblastoma, rhabdomyosarcoma and lymphoma. WT is often noted during a bath or by the pediatrician at a routine visit. This is in marked contrast to neuroblastoma, which is seen in the same age group, but frequently presents with pain, often from osseous metastasis. WT also may be associated with hematuria (gross in 18.2% and microscopic in 24.5% of patients), but with a much lower frequency than is seen with RCC.16 Twenty to 25% of patients present with hypertension, and 10% with fever. Occasionally, a child may have abdominal trauma and demonstrate pain out of proportion to what is expected, and an abdominal mass is found that cannot be attributed to the trauma.
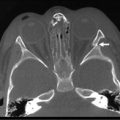
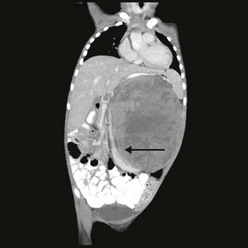
Radiographic imaging is performed to determine the anatomic location and extent of the mass. WT can extend into the renal vein (11% of cases) or the vena cava (4% of cases) (Fig. 65-6).70,71 Embolization of a caval thrombus to the pulmonary artery can be lethal, and the presence of a thrombus must be identified preoperatively to prevent this occurrence. Ultrasound has been the recommended screening study for an abdominal mass to determine its site of origin and to assess for possible intravascular or ureteral extension. However, a 2012 COG study demonstrated computed tomography (CT) can accurately identify cavo-atrial tumor thrombus as well.72 Routine Doppler ultrasound evaluation, after CT has already been performed, may not be needed. A CT/MRI scan of the abdomen will confirm a renal origin to the mass, and whether there are bilateral tumors. Historically, bilateral renal explorations were recommended by NWTSG protocols because early generations of CT scans missed 7–10% of bilateral lesions. However, with new helical CT scans only 0.25% of bilateral tumors are missed.73 Based on these results, bilateral exploration is not recommended on current protocols from the COG.
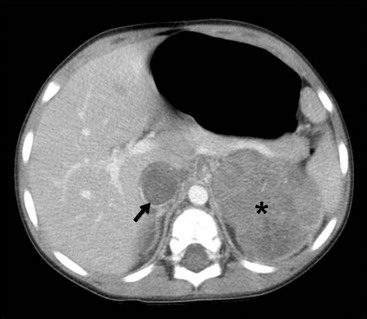
FIGURE 65-6 A transverse cut of a CT scan demonstrating a left Wilms tumor (asterisk) that extends into the inferior vena cava (arrow).
A neuroblastoma will generally indent the kidney whereas WTs arise from within the kidney and distort its internal configuration. Also, a thin lip of renal parenchyma can often be seen extending over the neoplasm in a WT, known as the claw sign (Fig. 65-7). Unfortunately, there are no characteristic radiographic findings of WTs that allow accurate diagnosis. Intra-abdominal staging has been difficult to assess radiographically unless extensive lymph node involvement or intrahepatic metastases are present. A recent study by Khanna and associates, using the COG database of 3,000 cases, found the preoperative imaging sensitivity and specificity of ultrasound in detecting WT rupture to be 53.7% and 88.4%, respectively, compared to the findings at operation.74 The common sites of metastatic spread are the lungs and the liver; hence CT scans of the chest should be included in the staging studies.
Surgery
Surgical Details
The essential tasks that are required of the surgeon, irrespective of when the WT resection occurs, are: (1) safe resection of the tumor; (2) accurate staging of the tumor; (3) avoidance of complications that ‘upstage’ the tumor (rupture or unnecessary biopsy); and (4) accurate documentation of the operative findings and details of the procedure in the operative notes. Tumor spill, failure to biopsy lymph nodes (for both unilateral and bilateral tumors), incomplete tumor removal, failure to assess for extrarenal tumor extension, and operative complications will adversely impact patient survival.75–77
Although WTs are usually initially seen as a large mass, most are resectable (Fig. 65-8). Renal tumors should be excised through an adequate subcostal or thoracoabdominal incision. Struggling through an inadequate incision will often result in tumor rupture, which increases both the stage of the tumor and the risk for intra-abdominal recurrence.77 A flank incision should not be used for resection of pediatric renal tumors because of the limited exposure it provides. Initial exploration of the abdomen includes inspection for hepatic metastases or intraperitoneal spread. The vena cava, if it is accessible, should be palpated to assess for intravascular extension of tumor. The colon is then mobilized off the anterior aspect of the kidney and the renal mass. Although early descriptions of the operative technique recommended initial control of the renal hilum, this is often not feasible with extremely large tumors and must await mobilization of the mass to allow safe exposure of the hilum. Premature attempts at vascular control, particularly for left-sided tumors, may result in ligation of the superior mesenteric artery.78 The renal mass should not be biopsied unless the decision is made to not proceed with a complete resection. Biopsy will produce contamination of the peritoneum and increase the tumor stage to stage III.
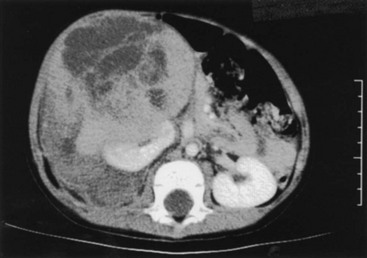
FIGURE 65-8 CT scan of a 3-year-old boy who presented with a history of abdominal pain, fever, and a right flank mass. The tumor is very large with extension outside the renal capsule into the retroperitoneal tissues posterior to the kidney. Despite its size, this lesion was able to be completely resected. Lymph nodes and specimen margins were negative for tumor. This tumor was therefore stage II, and the infant was able to avoid both anthracycline therapy and abdominal irradiation because of the complete resection.
Biopsy of lymph nodes in the renal hilum and along the vena cava and aorta is critical for adequate staging. Even in children with stage IV disease, local staging is vital because it will determine whether abdominal radiation therapy is used in children treated on COG protocols. Studies have demonstrated that the surgeon’s gross evaluation and assessment of lymph nodes does not reliably correspond with the histologic involvement of tumor in the nodes, with false-negative and false-positive rates of 31.3% and 18.1%, respectively.79 An increased incidence of local recurrence was seen in NWTS-4 children in whom biopsy of lymph nodes was not performed, particularly in stage I cases.77 This suggests that under treatment of local disease in these children, due to inadequate staging, resulted in an increased frequency of local recurrence. Although grossly involved lymph nodes are generally resected, an extensive retroperitoneal lymph node resection has not been demonstrated to improve local control.80 There has recently been increased interest in determining the extent of node dissection required for both WT and other renal tumors, although to date the data are inconclusive and retrospective.81,82 As the tumor is being mobilized, the ureter is palpated for tumor and is resected close to the bladder. Gross hematuria in children with WT is infrequent, but its occurrence suggests involvement of the renal pelvis with possible extension down into the ureter.83 Cystoscopy should be considered in these children to identify bladder involvement to avoid transection of the tumor thrombus during division of the ureter. Complete resection of the ureteral extension prevents this finding from increasing the local stage of the tumor. If the tumor involves the upper pole of the kidney, the adrenal gland is generally resected to provide adequate margins around the tumor. With lower-pole lesions, the adrenal gland may be preserved.
The factors associated with an increased risk of local recurrence include stage III disease, unfavorable histology, failure to biopsy the lymph nodes, and tumor rupture during operation.77 Tumor rupture and biopsy of the lymph nodes are the factors that the surgeon can impact. Multiple regression analysis adjusting for the combined effects of histology, lymph node involvement, and age reveal that tumor spillage remained significant and was greatest in children with stage II disease who received less intensive therapy. Most tumor ruptures occur during mobilization of the posterior aspects of the tumor where it is adherent to the diaphragm. This can be best prevented by the use of an adequate incision for exposure and resection of a segment of the adherent diaphragm if necessary. In a review of 2,000 cases of WT, size of tumor greater than 15 cm was the most significant risk for tumor rupture.84 Tumor rupture/spillage results in a stage III classification which results in additional chemotherapy and RT in a child who otherwise might have stage II disease.
‘Spill’ refers to a break in the tumor capsule during resection whether accidental, unavoidable, or by design. Spill is also considered to have occurred if the tumor is transected when the renal vein or ureter is divided. In COG protocols, a preoperative or intraoperative biopsy (needle or open) are stage III criteria. This is not true for patients on SIOP protocols where fine needle or Tru-cut biopsies are stage II criteria and incisional biopsies mean stage III. ‘Rupture’ refers to either the spontaneous or post-traumatic rupture of the tumor preoperatively with the result that tumor cells are disseminated throughout the peritoneal or retroperitoneal space.85 Blood in the peritoneal cavity may be a sign of rupture. When found, a thorough examination of the tumor surface is mandated. Rupture is also considered to have occurred if the tumor penetrates the renal capsule, with tumor extending into the peritoneal cavity. When encountered, all of these situations make the child stage III (COG and SIOP) and must be carefully documented in the operative note.
Resection of adjacent organs (liver, spleen, or pancreas) or resection of massive WT are discouraged because such extensive resections are associated with a significant increase in surgical complications.76,86 In this situation the surgeon should only sample the primary tumor.
Prior to operation, coagulation studies should be obtained because acquired von Willebrand disease (vWD) has been reported in WT and other malignancies.87,88 A single prospective study of 50 WT patients found the incidence of acquired vWD was 8%.88 However, the true incidence and prevalence is unknown. Historically, when coagulation abnormalities were identified, they were felt to be clinically insignificant.89 However, reports of profuse intraoperative bleeding (despite normalization of FVIII, vWF activity, and antigen level prior to operation) that only stopped after ligation of the renal vessels have contradicted this assumption.89,90 Once the renal vessels were ligated, all abnormal bleeding stopped. The mechanism of acquired vWD in WT is unknown.91,92 In all cases where bleeding occurred, the child had a prolonged PT and PTT. Correction of these abnormalities is important. The observation that bleeding stopped when the renal vessels were ligated suggests that preoperative embolization should be considered as a management strategy.
Surgical Complications
The complications detailed in the NWTS-4 study have also been assessed.76 In this study, surgeons were discouraged from performing extensive operations involving resection of adjacent organs or massive tumors. Complications occurred in 12.7% of a random sample of 534 of the 3335 patients treated in this study. Again, intestinal obstruction was the most frequent complication (5.1%), followed by extensive hemorrhage (1.9%), wound infection (1.9%), and vascular injury (1.5%). The factors associated with an increased risk of complications were assessed. Intravascular tumor extension into the inferior vena cava (IVC) or atrium, and nephrectomy performed through a flank or paramedian incision were both significant factors. Tumor diameter of 10 cm or larger also was associated with increased complications. Finally, the risk of complications was increased if the resection was performed by a general surgeon rather than a pediatric surgeon or pediatric urologist. In a study involving 598 patients registered on SIOP-9, a complication rate of 8% was reported. These patients were pretreated with vincristine, dactinomycin, and epirubicin or doxorubicin before nephrectomy.93 The most frequent events were small bowel obstruction (3.7%) and tumor rupture (2.8%).
Unresectable Tumors
The current guidelines for an unresectable tumor are: (1) when there is extension of tumor thrombus into the IVC above the level of the hepatic veins; (2) the tumor involves contiguous structures whereby the only means of removing the kidney tumor requires removal of the other structures (e.g., spleen, pancreas, colon, but excluding the adrenal gland and diaphragm); (3) bilateral tumors; (4) tumor in a solitary kidney; or (5) if there is pulmonary compromise due to extensive pulmonary metastases. SIOP studies have shown that pretreatment with chemotherapy almost always reduces the bulk of the tumor.93,94 This makes tumor removal easier, and may reduce the incidence of operative complications.
Intravascular Extension
Identification of vascular extension by preoperative radiographic studies (see Fig. 65-6) or during exploration is critical to avoid a tumor embolus during mobilization of the kidney. The presence of intravascular extension does not affect the prognosis of the tumor as long as it is successfully resected.95 Initially, intravascular extension was managed with removal of the tumor thrombus from the renal vein or IVC using cardiopulmonary bypass if necessary. However, this strategy has been found to have significant complications (70%).96,97 A review of all of the children treated on the NWTS-4 protocol described 165 of 2731 patients with intravascular extension into the IVC (134 patients) or atrium (31 patients).95 Sixty-nine of these patients received preoperative chemotherapy (55 with IVC extension and 14 with atrial extension). Five complications were encountered during preoperative chemotherapy, including tumor embolism and tumor progression in one patient each, and three patients developed adult respiratory distress syndrome, one of whom died. Intravascular extension of the tumor regressed in 39 of 49 children with comparable pre- and post-therapy radiographic studies, including regression in seven of 12 in whom the tumor regressed from an atrial location, avoiding the need for cardiopulmonary bypass. A high incidence of operative complications occurred in these patients, with 36.7% in the children with atrial extension and 17.2% in those with IVC involvement. The frequency of operative complications was 26% in the primary resection group versus 13.2% in children with preoperative therapy. When all the complications were considered, including those that occurred during preoperative chemotherapy (one of these five patients also had a surgical complication), the incidence of complications among those receiving preoperative therapy was not statistically different from the incidence among those who underwent initial resection. However, the most severe complications occurred in the primary resection group. Also, preoperative therapy was found to facilitate surgical resection by decreasing the extent of the tumor thrombus.
Management of Tumor Extension in the Ureter
Extension of WT into the ureter is a rare event which can be difficult to detect with preoperative imaging. In NWTS-5, the incidence of ureteral extension was 2%. However, preoperative imaging detected the ureteral involvement in only 30%.83 Clinical findings of ureteral involvement may include gross hematuria, passage of tissue per urethra, hydronephrosis, and a urethral mass. If these findings are encountered, ureteral involvement should be suspected. Cystoscopy with a retrograde ureterogram may aid in the preoperative evaluation. When encountered or suspected, the ureter with tumor extension should be resected with clear margins.
Horseshoe Kidney, Single Kidney and Nonfunctioning Kidney
Children with a tumor in a horseshoe kidney should be treated as having a unilateral tumor, not a bilateral tumor. The blood supply to horseshoe kidneys is quite variable as is the location of the ureters which should be delineated prior to surgery.98 The side of the kidney containing the tumor, the isthmus, and the ipsilateral ureter are resected and the lymph nodes are sampled for staging. Children with a single kidney, or a situation where a tumor occurs in one kidney but the second kidney is nonfunctioning, should be managed using a renal-sparing approach with preoperative chemotherapy to facilitate preservation of renal tissue.
Resection Alone for Very Low Risk Wilms Tumor (VLRWT)
The outcomes of children younger than 2 years with stage I tumor specimens weighing less than 550 g registered in the NWTSG studies were reviewed. Adjuvant therapy made little apparent impact on their survival which exceeded 90%.99 In a prospective pilot study at Children’s Hospital in Boston, eight children meeting these criteria were followed without adjuvant therapy.100 In seven of eight, there was no recurrence. In one child, a metachronous tumor was cured by resection and chemotherapy.
One treatment arm of NWTS-5 was a trial of operation only for children with these VLRWTs.101 Seventy-five infants were enrolled. In three infants, a metachronous, contralateral WT developed, and eight patients experienced relapse 0.3 to 1.05 years after diagnosis. The sites of relapse were pulmonary (five cases) and the operative bed (three cases). The 2-year disease-free survival, including both relapse and metachronous tumors, was 86.5%, and the two-year survival rate was 100%, with a median follow-up of 2.84 years. The 2-year disease-free survival, excluding metachronous contralateral WT, was 89.2%, and the 2-year cumulative risk of metachronous contralateral WT was 3.1%. The stopping rules for the study required closure after these 75 infants were enrolled. A recent long-term follow-up study was reported on this surgery-only cohort as well as the children entered on the protocol that eventually were treated with vincristine and actinomycin D (EE-4A).102 With a median follow-up of 8.2 years, the estimated 5-year event-free survival (EFS) for surgery-only was 84%; for the EE-4A patients it was 97% (P = 0.002). One death was observed in each group. The estimated 5-year overall survival (OS) was 98% for surgery-only and 99% for EE-4A (P = 0.70). The surgery-only EFS was lower than EE-4A, consistent with the earlier report. The salvage rate for the surgery-only cohort exceeded that seen with children who had received two-drug chemotherapy. Thus, 85% of the infants avoided any chemotherapy, while those who did receive it for relapse, were treated with three agents, vincristine, actinomycin D and doxorubicin (DD-4A), resulting in an OS equivalent to the group which received chemotherapy.
A recent biological study looking at the surgery-only arm in NWTS-5 found that a WT1 mutation and 11p15 LOH were associated with relapse in the patients with VLRWTs who did not receive chemotherapy.103 11p15 LOH was identified in 19 (41%) of 46 evaluable VLRWTs and was significantly associated with relapse (P < 0.001). A WT1 mutation was identified in nine (20%) of 45 evaluable VLRWTs and was significantly associated with relapse (P = 0.004); All nine cases also had 11p15 LOH. A current study in COG is assessing this cohort again and is evaluating biological markers for this very low-risk group.
Neonatal Wilms Tumor
Neonatal renal lesions are rare and include benign and malignant tumors.104,105 In the perinatal period, CMN accounts for greater than 50% of the renal tumors followed in rank by WT, RTK, and CCSK. Of note, after 3 months of age, CMN accounts for less than 10%. In a 2008 report, 210 renal tumors diagnosed prenatally (n = 47) and at birth (n = 163) were evaluated.105 The cohort consisted of four main tumors: CMN (139 cases or 66%), WT (41 or 20%), RTK (23 or 11%), and CCSK (7 or 3%). A recent international retrospective study of 750 neonatal renal tumors in children less than 7 months of age found that 63.4% were WT.104 Eighty-two per cent of these were stage I/II. In contrast, RTK presented with advanced disease (53% stage III/IV). Outcomes paralleled older children with excellent results for neonates with WT (5-year OS of 93.4%) and poor outcome for RTK (5-year OS of 16.4%).
Extrarenal Wilms Tumor
An extrarenal site of primary WT is uncommon. These extrarenal tumors behave identically to tumors arising within the kidney and should be treated both locally and systemically based on the same criteria.106,107 Common sites of occurrence of extrarenal WT include the retroperitoneum, inguinal canal, scrotum, and vagina. Less common sites are the uterus, cervix, ovary, and the presacral space.
Chemotherapy
WT was the first malignant pediatric solid tumor with a demonstrated response to dactinomycin.1 Many additional effective agents have been subsequently identified including vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide. Today the backbone of all WT chemotherapeutic regimens continues to be dactinomycin and vincristine, and they remain the sole chemotherapy for patients with stage I and II tumors.
NWTS-1 and 2 established the main chemotherapeutic regimen for stage I WT. NWTS-3 and NWTS-4 were the first studies to prospectively evaluate the benefit of additional/different chemotherapy for these tumors.108 Key results from NWTS-3 were: (1) children with stage I tumors, with an 11-week regimen composed of vincristine and dactinomycin only, had a 4-year relapse-free survival (RFS) and OS of 89.0% and 95.6%; (2) the addition of doxorubicin to the treatment of children with stage III tumors improved survival; (3) no benefit was found with the addition of doxorubicin or radiation therapy for children with stage II tumors; and (4) no benefit from the addition of cyclophosphamide to the treatment of children with stage IV tumors.109 A follow-up analysis of doxorubicin for stage III patients demonstrated an increase in the 8-year EFS and OS of randomized patients who received doxorubicin, dactinomycin, and vincristine (84% and 89%) when compared with those who received dactinomycin and vincristine alone (74% and 83%).110
NWTS-4 addressed dose intensification and also evaluated the use of two time intervals for the administration of chemotherapy: a short course (18 to 26 weeks, depending on the regimen and stage) versus a long course (54 to 66 weeks). Key findings were that the pulse-intensive regimens produced less hematologic toxicity than the standard regimens, allowing greater dose intensity with comparable outcomes.108,111 The second randomization demonstrated no benefit in any of the stages for the long interval of therapy compared with the short interval. In NWTS-4, the 4-year EFS and OS averaged 90% for patients with favorable histology.108,112
NWTS-5 focused on evaluating biological markers of prognosis, developing more effective therapy for recurrent disease, and reducing therapy in children with low-risk tumors. Patients with LOH at both 1p and 16q had EFS and OS which were 10% worse than those in patients without LOH (see Fig. 65-1). In the current COG protocols, children with LOH at these sites will receive more intensive therapy; children with stage I or II will receive three drug therapy with DD-4A and children with stage III and IV will receive regimen M (Table 65-5).
TABLE 65-5
COG Chemotherapy Regimens for Unilateral Wilms Tumor
| Regimen | Agents |
| EE-4A | Vincristine and dactinomycin |
| DD-4A | Vincristine, dactinomycin, doxorubicin, plus RT |
| Regimen I | Vincristine, dactinomycin, doxorubicin, cyclophosphamide, and etoposide, plus RT |
| Regimen M | Vincristine, dactinomycin, doxorubicin, cyclophosphamide, and etoposide, plus RT |
| Revised UH-1 | Vincristine, dactinomycin, doxorubicin, cyclophosphamide, carboplatin, etoposide, plus RT |
| Revised UH2 | Vincristine, dactinomycin, doxorubicin, cyclophosphamide, carboplatin, etoposide, irinotecan, plus RT |
| Vincristine/irinotecan Window therapy | Vincristine and irinotecan in conjunction with revised UH-1 or revised UH-2 depending on response |
NWTS-5 had specific relapse protocols for children with stage I and II, as well as stage III and IV disease. Fifty-eight children with initial stage I and II disease experienced relapse after treatment with EE-4A.113 Their relapse therapy included alternating courses of vincristine, doxorubicin, cyclophosphamide, and etoposide, along with surgery if feasible, and RT. After this specified relapse therapy, EFS at 4 years was 71.1% and OS was 81.8%. The lung was the only site of relapse in 31 children. Those with only pulmonary relapse had similar EFS (67.8%) and OS (81.0%). Sixty patients with stage III and IV disease relapsed after initial therapy with DD-4A and radiation.114 Their relapse therapy included alternating cycles of cyclophosphamide/etoposide and carboplatin/etoposide along with surgery and RT. After therapy, EFS in these patients at 4 years was 42.3% and OS was 48.0%. The lung was the sole site of relapse in 33 patients with an EFS of 48.9% and OS of 52.8%. At present, there is no open relapse study in SIOP or COG. The current COG chemotherapeutic regimens are shown in Table 65-5.
Treatment of children with diffuse anaplastic tumors remains a challenge. NWTS-3 and NWTS-4 studies demonstrated that children with focal anaplasia had an excellent outcome when treated with vincristine, doxorubicin, and dactinomycin.49 In NWTS-5, in order to reduce toxicity, patients with focal anaplasia or diffuse stage I were treated with EE-4A. Unfortunately, the four-year EFS and OS estimates for stage I (focal or diffuse) anaplastic WT were lower than in previous studies (EFS 69.5% and OS 82.6%). Thus, therapy with EE-4A above is inadequate. In NWTS-5, children with stage II to IV diffuse anaplastic WT were treated with alternating courses of vincristine, doxorubicin, and cyclophosphamide, and cyclophosphamide and etoposide (regimen I). The four-year EFS estimates for stages II-IV diffuse anaplastic WT in NWTS-5 were 82.6%, 64.7%, and 33.3%, respectively, with similar OS.47
The chemotherapy treatment for CCSK on NWTS-1 to NWTS-3 was the same as for WT and the outcomes were poor (OS 66.9% at 8 years).115 In NWTS-4, patients with CCSK were treated with vincristine, dactinomycin, and doxorubicin, and were randomized to standard (ST) versus pulse-intensive (PI) chemotherapy and short-duration versus long-duration chemotherapy. OS improved on NWTS-4 compared to NWTS-3 (OS 83% versus 66.9% at 8 years, P < 0.01).115 To further improve survival, patients on NWTS-5 with CCSK were treated using regimen I, because etoposide and cyclophosphamide were active against CCSK in preclinical models.116 Four-year OS for stage I patients was 100%. Stage II, III, and IV had 4-year OS of 88.9%, 94.8% and 41.7%, respectively. LOH was not found in patients with CCSK and is not predictive of outcomes. In the current COG study, children with stage I disease will continue to be treated with regimen I, but will not receive RT. In order to improve survival for children with higher stage disease, they will be treated with the more intensive revised UH-1 (see Table 65-5).
The rhabdoid tumors have remained the most resistant of all pediatric renal tumors. An analysis of 142 children treated on NWTS-1 to NWTS-5 showed an OS of 23.2% at four years.117 Survival was stage dependent and children with stage I/II disease had a 41.8% 4-year survival, while children with stage III, IV, or V tumors had a 15.9% four-year survival. Survival was also clearly related to the age at presentation. The 4-year survival was worst for those between birth and 5 months of age at diagnosis (8.8%) and best for those older than 2 years of age (41.1%). NWTS-5 used an intensive therapy with carboplatin, etoposide, and cyclophosphamide, and showed no significant improvement. Children with RTK on the current COG study are treated using revised UH-1 if they are stage I-IV after operation and have no measurable disease. If they have measureable disease (stage III, IV), they will receive a vincristine/irinotecan ‘window,’ followed by revised UH-2 if they have a clinical response. The rational for this treatment strategy was based on reviewing the outcomes from the IRS studies and several case reports that documented the successful treatment of advanced or metastatic RTK.118–120
Pretreatment Prior to Surgery
SIOP has promoted the use of preoperative treatment of children with WT with RT or chemotherapy since the early 1970s. The major driving force for the use of preoperative therapy by SIOP was the high rate of tumor rupture at operation that occurred in their early series during which patients did not receive preliminary treatment. The rupture rate decreased from 33% (20 of 60) to 4% (3 of 72) with preoperative abdominal irradiation (20 Gy) in the first randomized SIOP study of renal tumors (SIOP-1).121 It must be noted, however, that 33% is an extremely high rupture rate. In addition, survival was not improved by the decrease in operative rupture, and the incidence of local recurrence was not reported. It is difficult to compare rupture rates between SIOP and COG as the definition of what is a rupture differs between the two groups. SIOP reported an intraoperative rupture rate of less than 8%.122 The most recent COG data from 2012 reports an intraoperative rupture rate of 9.8%.84
Histologic confirmation of the diagnosis before therapy is not routinely performed in SIOP protocols. This approach has several risks. One is the potential for administration of chemotherapy for benign disease. Second, modification of tumor histology by the chemotherapy may occur. Third, staging information may be lost. Fourth, a malignant RTK or CCSK, if present, will not respond to standard therapies. NWTSG and SIOP studies have demonstrated a 7.6–9.9% rate of benign or altered malignant diagnosis in children with a prenephrectomy diagnosis of WT.123,124 The United Kingdom Children’s Cancer Study Group (UKCCSG) identified 12% of cases that were clinically and radiographically consistent with WT, but had other diagnoses established by biopsy.125 For SIOP-9 inappropriate preoperative therapy was given to 5.5% of the patients, including 1.6% with benign lesions or malignant lesions not expected to respond to the therapy, including neuroblastoma, lymphoma, RTK, and RCC.
In the SIOP-9 study, the most common subtype of tumors resected without neoadjuvant chemotherapy was triphasic mixed histology (45.1%), followed by blastemal (39.4%) and epithelial dominant (15.5%), whereas in tumors that received preoperative chemotherapy, the most common histology was regressive (37.6%), followed by mixed (29.4%), stromal (14%), blastemal (9.3%) and epithelial predominant (3.1%). 6.6% of tumors were completely necrotic.67,126
In the SIOP-9 study, a randomization was performed between 4 and 8 weeks of preoperative therapy with dactinomycin and vincristine to determine whether the additional 4 weeks of therapy produced a larger proportion of stage I tumors.93 No advantage was seen from the extended therapy in terms of staging at resection between the 4-week and 8-week courses (stage I,64% versus 62%) or the incidence of tumor rupture (1% vs 3%). For all cases, tumor size decreased by more than half in 52% of the cases. During the second 4 weeks of therapy, there was another 50% reduction in 33% of the cases.
Radiotherapy
The three principle fields for RT for renal tumors are whole abdominal, flank, and lung (metastatic lung disease). Favorable histology tumors are generally very radiosensitive. Current guidelines for radiation therapy in the North American protocols are based on the results of prior cooperative group studies. In 1950, Gross demonstrated that postoperative RT could improve survival to 47%.11 NWTS-1 concluded that RT provided no survival advantage in children younger than 24 months with stage I FH tumors who also received 15 months of dactinomycin. NWTS-1 also showed that stage III tumors with a confined spill did not require whole abdominal RT, but had comparable outcomes with flank radiation.123,127 Currently, whole abdominal radiation is only used in three instances: (1) peritoneal seeding; (2) preoperative tumor rupture;, and (3) an intraoperative spill that is widespread in the opinion of the operating surgeon.
NWTS-2 confirmed that RT could be avoided in all children with stage I WT if they received vincristine and dactinomycin.128 NWTS-2 also established that delaying RT longer than 10 to 14 days after resection was associated with a higher incidence of abdominal relapse, particularly among patients with unfavorable-histology tumors and a small radiation field.123,127,129 A recent review from NWTS-3 and -4 data confirmed this finding.130
NWTS-3 demonstrated that RT could be avoided in children with stage II tumors who received vincristine and dactinomycin, and showed that children with stage III favorable histology tumors who received 10.8 Gy radiotherapy, vincristine, dactinomycin, and doxorubicin had similar tumor control to those who received 20 Gy with vincristine and dactinomycin. This was an important finding, because it eliminated the need for an age-adjusted dose schedule, and significantly reduced the recommended dose of radiation and its subsequent early and late toxicity. Anaplastic tumors tend to be more resistant to chemotherapy, and are also resistant to RT. NWTS-5 did not give RT for stage I anaplastic histology(AH) tumors. However, patients with AH stage II and III tumors in NWTS-5 did receive 10 Gy in conjunction with nephrectomy and chemotherapy. The outcomes for both of these treatment strategies were suboptimal as described above. Fifty per cent of stage III recurrences were local, suggesting that the dose of 10 Gy was not adequate.
The management of pulmonary disease in children with WT has become more complicated. The use of CT scans instead of chest radiographs to establish the presence of pulmonary metastasis has resulted in more lesions being identified—not all of which may be cancer.131 Complicating the possibility of unnecessary radiation is the fact that radiation therapy is a major cause of long-term morbidity, particularly to the lung and heart, producing myocardial injury, pulmonary fibrosis and second malignancy.132–134 From NWTS-5, the 5-year EFS (95% CI) for stage IV category was: lung only 76% (72–80%); liver and lung 70% (57–80%).135
The current COG protocol has made significant changes with regard to patients with pulmonary disease. First, lesions seen by CT scan will be considered pulmonary metastasis unless biopsy proven not to be metastatic disease. Second, in an attempt to spare pulmonary radiation in those children who appear to have responsive disease, children will not receive radiation therapy if the pulmonary lesions radiographically resolve by CT scan after six weeks of chemotherapy. This second change was based on several observations. In SIOP-9, patients were spared whole lung radiation therapy if they rapidly responded to three drug chemotherapy by 6 weeks. The five-year RFS for stage IV patients receiving preoperative chemotherapy was 62.5%.136 A COG study of patients with pulmonary lesions detected by CT only (as opposed to CT and chest radiograph), and treated with only two chemotherapeutic agents, showed an inferior outcome compared to those treated with three drugs, irrespective of whether or not they received pulmonary RT.135 The results of these studies have been controversial. The UKCCSG-Wilms Tumor Study 1 followed a similar protocol, yet their six-year EFS was only 50%.137 In the second UKCCSG-Wilms tumor study (UKWT2), the majority of children with lung metastases received whole lung irradiation (WLI), and the 4-year survival rate improved to 75%.138
Bilateral Wilms Tumor
BWT occurs in 4–13% of patients.17,69,139,140 The EFS and OS in children with bilateral tumors have been lower than those with unilateral tumors. In NWTS-5, the four-year OS was 80.8% for a child with favorable histology and 43.8% for a child with anaplastic histology.47 SIOP-9 reported 28 children with BWT, all treated with individualized preoperative therapy.136 Overall survival at 5 years was 85.1% (95% CI: 71.6–98.6%), and RFS was 80.5% (95% CI: 65.2– 95.8%).136
COG initiated the first cooperative group trial of BWT in 2009. Reviews of patients treated prior to this time revealed that in addition to poor outcomes, children with BWT had significantly higher rates of renal failure and underwent prolonged chemotherapy regimens compared to those with unilateral tumors. BWT was the greatest risk factor for renal failure (16.4% for NWTS-1 and -2, 9.9% for NWTS-3, and 3.8% for NWTS-4).141 Other risk factors identified were: Denys–Drash syndrome, metachronous tumor, progressive disease requiring bilateral nephrectomies, and radiation nephritis. Breslow reported the 20-year end-stage renal disease (ESRD) outcomes in children treated for WT.142 The major risk factors he identified for renal failure were BWT, Denys–Drash and WAGR, and congenital genitourinary anomalies (hypospadias or cryptorchidism).
Two studies highlight the issue of prolonged chemotherapy in patients with BWT. One study found 38 of 188 patients with BWT had progressive or nonresponsive disease (PNRD).143 Patients with PNRD fell into two categories. The first category included patients with anaplastic tumors that were not sensitive to the administered therapy (four patients). Due to this lack of response to therapy once the diagnosis of anaplasia is made, a complete resection is needed.47,144–146 The second category included patients who had tumors with very mature rhabdomyomatous or differentiated stromal elements (14 patients) or with complete necrosis. These patients had good outcomes and are best served with resection. In the other 20 patients, 12 weeks of chemotherapy did not result in any tumor shrinkage. Another study evaluated the histological changes in BWT that did not respond to chemotherapy, and the relationship between these changes and prognosis.147 The results mirrored those of the NWTS study. The findings of these two reports emphasize that in cases of bilateral lesions that do not respond radiographically to therapy, it is critical to establish whether this is due to anaplasia or a mature histology.
How long to treat a child who has BWT with chemotherapy before intervening surgically remains an important consideration. In SIOP-9, patients with unilateral tumors were randomized to receive either 4 or 8 weeks of dactinomycin and vincristine preoperatively. On average, a 48% and 62% reduction in tumor volume was seen at four and eight weeks, respectively.93,148 A review by the German Pediatric Hematology Group (GPOH) looking at patients with BWT reported that maximum tumor shrinkage occurred in the first 12 weeks of chemotherapy.149
The two principal aims of the current COG study for BWT are to improve four-year EFS and prevent complete removal of at least one kidney in 50% of patients. This is a response-based protocol starting with chemotherapy followed by evaluation at 6 and 12 weeks with definitive operative therapy in all patients by 12 weeks. For most patients with BWT, the initial regimen is an intensive combination of three drugs (vincristine, actinomycin D and doxorubicin) based on regimens used with good results and minimal toxicities by both SIOP and the UKCCSG WT Group.150 This regimen was chosen to provide enhanced therapy for possible stage III disease. Additionally, it was elected to intensify the chemotherapy rather than administer radiotherapy, which would increase the development of radiation nephritis and loss of renal function in the remaining kidney. Finally, a more intensive therapy was selected to avoid the use of sequential regimens of increasing intensity, thereby delaying resection of tumors which was identified in the review of the prior cohort of NWTS-4 BWT patients.
Health Consequences for Survivors
WT treatment impacts general health, renal function, pregnancy, cardiac function, thoracic function, and the occurrence of second malignancies.134,151–153 Table 65-6 shows the causes of early and late mortality in a cohort of 10,000 children diagnosed with WT.151
TABLE 65-6
Causes of Early and Late Mortality in Children Diagnosed with Wilms’ Tumor (n = 11,157)
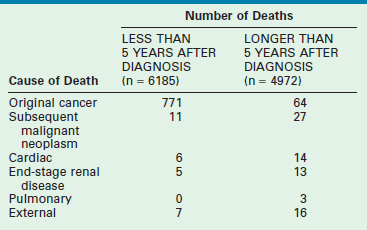
From Cotton CA, Peterson S, Norkool PA, et al. Early and late mortality after diagnosis of Wilms’ tumor. J Clin Oncol 2009;27:1304–9.
General Health
Chronic health conditions, health status, health care utilization, socioeconomic status, subsequent malignant neoplasms, and mortality of 1,256 WT survivors (1970–1986) were compared to the USA population, and a sibling cohort (n = 4023) in an effort to determine the long-term effects of WT on survivors (Fig. 65-9).154 The cumulative incidence of all and severe chronic health conditions was 65.4% and 24.2%, respectively, at 25 years. WT survivors reported a more adverse general health status than the sibling group. However, mental health status, socioeconomic outcome, and health care utilization were similar.
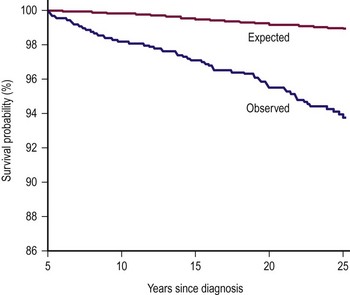
FIGURE 65-9 Graph of observed vs expected mortality in a 25 year follow-up of children with Wilms tumor. (Permission from Termuhlen AM, Tersak JM, Liu Q, et al. Twenty-five year follow-up of childhood Wilms tumor: A report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer 2011;57(7):1210–16.)
Renal Function
Factors that contribute to renal failure include intrinsic progressive renal disease, inadequate renal parenchyma after one or more tumor resections, nephrotoxic effects of chemotherapy and radiation, and the potential for hyperfiltration injury to the remaining renal parenchyma. Breslow reported the 20-year ESRD outcomes in children treated for WT in 2005.142 The major risk factors identified for renal failure were BWT and congenital syndromes. However, for a standard unilateral nonsyndromic WT patient, the risk of renal failure at 20 years was very low (0.6%).
Although transplantation is an option for children with renal failure, the current recommendations are to wait 2 years before considering a transplant.155
Pregnancy
The National Wilms Tumor Long-Term Follow-Up Study evaluated 700 maternal/offspring pairs.156 Hypertension complicating pregnancy (P < 0.001), or threatened labor (P = 0.002) and malposition of fetus (P = 0.04) were more frequent among irradiated women and were related to radiation therapy dose.
Secondary Malignancies
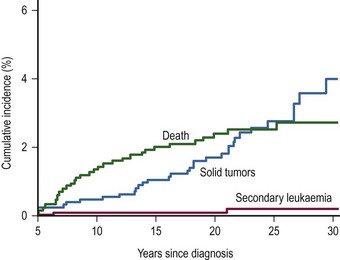
An international cohort of 13,351 children with WT diagnosed before 15 years of age from 1960 to 2004 was established to determine the risk of second malignant neoplasms.134 One hundred and seventy-four solid tumors and 28 leukemias were found in 195 people. The age-specific incidence of secondary solid tumors increased from approximately one case per 1,000 person years at age 15 to five cases per 1,000 person years at age 40. The cumulative incidence of solid tumors at age 40 was 6.7%. In those patients where WT was diagnosed after 1980, there was a lower age-specific incidence rate for second tumors compared to those treated before 1980. Paradoxically, the incidence of leukemia was higher in those diagnosed after 1990 (Fig. 65-10). This may be due to the decreasing use of radiation therapy and increasing the intensity of chemotherapy in modern protocols for treatment of WT.
Congestive Heart Failure
The relative risk of congestive heart failure was found to be increased in females (RR = 4.5; P = 0.004), and related to the cumulative doxorubicin dose (RR = 3.2/100 mg/m2; P < 0.001), lung irradiation (RR = 1.6 for every/10 Gy; P = 0.037), and left abdominal irradiation (RR = 1.8/10 Gy; P = 0.013).157 The cumulative risk of congestive heart failure was 4.4% 20 years after initial treatment with doxorubicin, and 17.4% 20 years after treatment with doxorubicin for a first or subsequent relapse. Preliminary results suggest that cardiotoxicity is lower with current radiation doses, but patients still have a substantial life-time risk of developing cardiac disease.151,158
Thoracic
The late effects of pulmonary radiation include pneumonitis, restrictive lung disease, scoliosis, kyphosis, reduced lung capacity, and secondary tumors. In girls, breast hypoplasia and cancer have also been described.132,133 Paulino and his associates reported on the late complications of pulmonary RT in 55 long-term survivors of WT.132 Two-thirds of the patients had at least one complication. Forty-three percent had scoliosis or kyphosis and 10% developed benign chest tumors (osteochondromas). Secondary tumors were noted in three patients within the lung field (two osteogenic sarcomas of the rib and one breast cancer), and all succumbed to these tumors. Pulmonary function was examined in another study.133 Subjectively, 63% percent of patients had mild to moderate exercise intolerance. Objective measurement of vital capacity and total lung capacity was decreased compared to age and height predicted values in patients. All females had breast hypoplasia.
Current Children’s Oncology Group Renal Tumor Studies
Renal Cell Carcinoma
Children with RCC are generally older than those with WT.159 RCC in children displays gross and histological features similar to those seen in adults. Clinical stage at the time of diagnosis is the most important prognostic factor, and the identification of renal vascular invasion does not appear to be an adverse predictor. Radical nephrectomy and regional lymphadenectomy have been the primary modality for cure, and children with distant spread have a grave prognosis. In a study of 22 children, the mean age at presentation with RCC was 14 years.159 Overall survival was much worse than for WT, with a five-year survival of only 30%. Analysis of multiple factors including age, tumor size, location, and histology failed to demonstrate predictors of survival. Only stage and complete tumor resection were meaningful prognostic factors. In another study, survival was 60% in children with complete resection of the primary tumor and zero in those with only partial resection.160 Survival was also stage dependent: 92.5% for stage I, 84.5% for stage II, 72.7% for stage III, and 12.7% for stage IV. It should be noted, however, that those with positive nodes but no distant metastasis had survival rates three times that of adult historical controls. RCC is remarkably resistant to chemotherapy, preventing cure in most children with metastatic disease.161 Ten to 20% of patients have nodal involvement identified at operation, but lack evidence of distant metastatic disease. No benefit has been found for adjuvant therapy in children.160
Nephron-sparing resection has been used in adults with small polar lesions in whom no evidence of a multicentric tumor is found. In these selected cases with tumors smaller than 4 cm and a normal contralateral kidney, the risk of local recurrence is reported to be 2% or less, which is comparable to the frequency of metachronous recurrence in the contralateral kidney after unilateral radical nephrectomy.161
The development of late recurrences long after nephrectomy, prolonged stability of disease in the absence of systemic therapy, and rare cases of spontaneous regression of tumors have led to an interest in immunotherapy comparable to that used for melanoma. Trials of immunomodulating therapy with interferon-alpha and interleukin-2 have demonstrated efficacy in some studies, but maintenance of a durable cure has been elusive.162 In contrast, a trial of 294 patients with advanced-stage RCC randomized to receive placebo or nine months of subcutaneous lymphoblastoid interferon demonstrated similar recurrence rates between the two groups and worse survival for the treatment group.163 With the significant toxicity involved with immunotherapy, demonstration of improved survival in randomized trials will be required before this can be adopted as standard therapy.
Mesoblastic Nephroma
Congenital mesoblastic nephroma, also referred to as fetal renal hamartoma or leiomyomatous hamartoma, is the most common renal tumor identified in the neonatal period. Although initially diagnosed and treated as a congenital Wilms tumor, mesoblastic nephroma was defined as a distinct entity in 1967.164 Mesoblastic nephromas are found most frequently in the neonatal period as a palpable flank mass which can be massive. Additional symptoms seen at presentation include hematuria, hypertension, vomiting, and jaundice.165
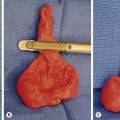
Mesoblastic nephroma accounted for 2.8% of 1,905 renal tumors submitted to the early NWTSG studies. Grossly, these tumors have a homogeneous rubbery appearance resembling a uterine fibroid in color and consistency (Fig. 65-11). Microscopically they are composed of sheets of fibrous or mesenchymal stroma, within which bizarre and dysplastic tubules and glomeruli are irregularly scattered.166 The tumor can invade intact renal parenchyma, and extrarenal infiltration into the perihilar connective tissues is common. The histologic subtypes of this tumor include classic type (24% of cases), cellular type (66%), and mixed type (10%). The pluripotency of these tumors is revealed by their differentiation into angiomatoid patterns, cartilaginous nests, and their elicitation of intratumoral hematopoiesis in addition to the tiny nephroblastic epithelial foci.

FIGURE 65-11 Cross section of a mesoblastic nephroma identified in an infant on a neonatal examination. Note the rubbery appearance of this tumor, which resembles a fibroid tumor of the uterus with a very thin margin of normal renal parenchyma around the periphery.
A characteristic chromosomal translocation, (t12;15)(p13;q25), has been recently found.167 It results in fusion of the ETV6 (also known as TEL) gene from 12p13 with the NRTK3 neurotrophin-3 receptor gene (also known as TRKC) from 15q25.27,29 This fusion results in a chimeric RNA, which is characteristic of both infantile fibrosarcoma and the cellular variant of CMN. This may be helpful in differentiating the cellular variant from other lesions that must be considered in the differential diagnosis, including CCSK and RTK. It also suggests a close relation between infantile fibrosarcoma and the cellular variant of mesoblastic nephroma.
This generally benign tumor usually can be cured with nephrectomy alone. This should include generous margins around the tumor to avoid local recurrence. Particular attention should be paid to the medial aspect of the kidney, including the hilum and great vessels, because of the tumor’s proclivity to have extensions into these perirenal soft tissues. Several children have been reported with local recurrence or metastases to the brain, bones, lungs, and heart.168–170 The histology in some cases has revealed an unusual degree of mesenchymal cell immaturity and hypercellularity, suggesting a more aggressive tumor. This also supports the concept that mesoblastic nephroma cannot be considered a simple hamartoma and that complete nephrectomy with negative margins is critical in all cases.166
In a series of 51 children with mesoblastic nephroma identified in the NWTSG series, eight had local extension and ten had tumor spillage during resection.165 The use of adjuvant therapy in these cases depended on the era in which the children were treated. Twenty-three infants treated after 1978 underwent resection alone. Prior to 1978, 24 underwent an operation and chemotherapy, and before 1976, four children also received irradiation. Survival was excellent in this entire group with only one child dying of sepsis during chemotherapy. One child’s tumor recurred at 6 months despite receiving dactinomycin and vincristine. The tumor was re-excised, and the child was treated with cyclophosphamide and doxorubicin and remained without disease 18 months later.
A SIOP report of 29 children with CMN confirmed the early age at which this tumor is seen.171 Only five infants were older than 4 months at presentation in the series. Interestingly, treatment of a neonate with an extensively infiltrating tumor with eight weekly courses of vincristine before resection has been reported.172 Shrinkage of the tumor occurred with this treatment, facilitating its eventual resection and cure.
Beckwith reported the largest cohort of children with recurrent or metastatic lesions from his large collected series.173 Twenty-four patients with an aggressive tumor were seen in a series of 330 mesoblastic nephromas. Of these 24 cases, eight had metastatic disease, 17 had relapse in the peritoneum or retroperitoneum, and six infants have died of persistent disease. Recurrences developed in children after initial chemotherapy or radiation, which suggests that conventional adjuvant therapy may not decrease the incidence of recurrence. Histologic criteria were not helpful in predicting outcome. Beckwith supports aggressive surgical attempts to remove all gross tumor.173 He also stresses the need for close monitoring for one year after resection because relapse in 23 of the 24 cases was apparent within 11 months of resection. Ultrasound of the local site is adequate, and scans for metastatic disease are unrewarding.
Cystic Nephroma
Cystic nephroma is indistinguishable grossly and radiographically from its malignant neoplastic ‘cousins,’ cystic partially differentiated nephroblastoma (CPDN) and cystic nephroblastoma. All lesions are composed of purely cystic masses characterized by multiple thin-walled septations (Fig. 65-12). In cystic nephroma, the septations are lined by flattened, cuboidal, or hobnail epithelium, and are composed entirely of differentiated tissues without blastemal or other embryonal elements that are the distinguishing characteristics of CPDN.174 Although the term multilocular cyst of the kidney has been used, cystic nephroma is preferred because the lesion appears to be neoplastic rather than congenital. In the cystic nephroblastoma or cystic WT, solid nodules on the septa of blastemal or embryonal elements are characteristic of WT. An unexplained synchronous occurrence has been reported between cystic nephroma and pleuropulmonary blastoma.175,176
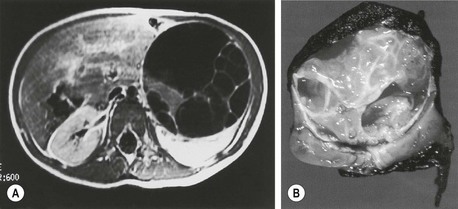
FIGURE 65-12 (A) MR image obtained in a 2-year-old infant with an asymptomatic left flank mass. Note the multilocular cystic mass extending out from the normal renal tissue posteriorly. (B) Cross section of the tumor and kidney reveals thin-walled septa within the mass. Histologic examination of this tumor revealed a cystic partially differentiated nephroblastoma.
These lesions should not be confused with cystic CCSK, cystic CMN, or multicystic dysplastic kidney.177 Cystic nephroma, CPDN, and cystic nephroblastoma can be distinguished from multicystic dysplastic kidney because the former are confined to only a portion of the kidney with normal renal parenchyma being identified, whereas the cystic changes of multicystic dysplastic kidney almost always involve the entire kidney due to the fact that it develops early in gestation from urinary traction obstruction. Contralateral renal anomalies are frequent in dysplastic kidneys, including ureteropelvic junction obstruction and reflux. A multicystic dysplastic kidney is often identified antenatally or in the neonatal period, whereas the other lesions occur later.
Generally, nephrectomy will be curative in both cystic nephroma and CPDN. Twenty-three children with these cystic lesions were identified in the NWTSG series: 5 with cystic nephroma and 18 with CPDN.178 Only one patient with CPDN had local recurrence, and no distant metastases were found. A more recent review of the GPOH and SIOP files of the CPDN cases has again confirmed that primary resection appears to be adequate for all lesions removed intact.179 In cases in which the lesion is isolated to one pole of the kidney, a partial nephrectomy can be considered. However, it must be remembered that these tumors can resemble cystic variants of CCSK, which carries an entirely different prognosis.180–182
Ossifying Renal Tumor of Infancy
Ossifying renal tumor of infancy is a relatively rare tumor occurring entirely in infancy. In many cases, children with this lesion initially have gross hematuria, although a palpable mass may be present.183 These lesions are attached to a renal papilla, but are seen primarily within the lumen of the calyx and may extend into the renal pelvis. They have been confused occasionally with staghorn calculi. Histologically, they contain osteoid, osteoblastic cells, and spindle cells. Their true histogenesis has not been established, although it has been suggested that they represent hyperplastic ILNRs. Metastasis or local spread of these tumors has not been reported. Renal-sparing procedures may be reasonable, although this may not result in significant ipsilateral renal function.184,185
References
1. Farber, S. Chemotherapy in the treatment of leukemia and Wilms’ tumor. JAMA. 1966; 198:826–836.
2. Rance, T. Case of fungus hematodes of the kidneys. Med Phys J. 1814; 32:19.
3. wilms, M. Diagnostischer und therapeutischer Werth der Lumbalpunction. Druckbestimmung mit Quicksilvermanometer. Munch Med Wochenschr. 1897; 3:53–57.
4. Wilms, M. Die Mischgeschwülste der Niere. Leipzig: Verlag von Arthur Georgi; 1899.
5. Beckwith, JB. Wilms’ tumor and other renal tumors of childhood: A selective review from the National Wilms’ Tumor Study Pathology Center. Hum Pathol. 1983; 14:481–492.
6. Willets, IE. Jessop and the Wilms Tumor. J Pediatr Surg. 2003; 38:1496–1498.
7. Gross, RE. Embryoma of the Kidney (Wilms Tumor). In: Gross RE, ed. The surgery of infancy and childhood. Philadelphia: WB Saunders; 1953:588–605.
8. Friedlander, A. Sarcoma of the kidney treated by roetgen ray. Am J Dis Child. 1916; 12:328–331.
9. Ladd, WE. Embryoma Of The Kidney (Wilms’ Tumor). Ann Surg. 1938; 108:885–902.
10. Mukherjee, S. The Emperor of All Maladies: A Biography of Cancer. New York: Scribner; 2010.
11. Gross, RE, Neuhauser, EB. Treatment of mixed tumors of the kidney in childhood. Pediatrics. 1950; 6:843–852.
12. Schweisguth, O, Bamberger, J. Le nephroblastome de l’enfant. Ann Chir Infant. 1963; 4:335–354.
13. Sutow, W. Vincristine (Leurocristine) sulfate in the treatment of children with metastatic Wilms’ tumor. Pediatrics. 1963; 32:880–887.
14. Bersntein, L, Linet, M, Smith, MA, et al. Renal Tumors. Bethesda: National Cancer Institute, SEER Program; 1999.
15. Breslow, N, Olshan, A, Beckwith, JB, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993; 21:172–181.
16. Green, DM. The diagnosis and management of Wilms’ tumor. Pediatr Clin North Am. 1985; 32:735–754.
17. Coppes, MJ, de Kraker, J, van Dijken, PJ. Bilateral Wilms’ tumor: Long-term survival and some epidemiological features. J Clin Oncol. 1989; 7:310–315.
18. Breslow, N, Norris, N, Norkool, P, et al. Characteristics and outcomes of children with the Wilms tumor-Aniridia syndrome: A report from the National Wilms Tumor Study Group. J Clin Oncol. 2003; 21:4579–4585.
19. Debaun, MR, Tucker, MA. Risk of cancer during the first four years of life in the children from the Beckwith-Wiedemann syndrome registry. J Pediatr. 1998; 132:398–400.
20. Rao, A, Rothman, J, Nichols, KE. Genetic testing and tumor surveillance for children with cancer predisposition syndromes. Curr Opin Pediatr. 2008; 20:1–7.
21. Tan, Ty, Armour, DJ. Tumour surveillance in Beckwith-Wiedemann syndrome and hemihyperplasia: A critical review of the evidence and suggested guidelines for local practice. J Paediatr Child Health. 2006; 42:486–490.
22. Wiedemann, HR, Wiedemann, H-R. Tumours and hemihypertrophy associated with Wiedemann-Beckwith syndrome [Letter]. Eur J Pediat. 1983; 141:129.
23. Goldman, M, Smith, A, Shuman, C, et al. Renal abnormalities in Beckwith-Wiedemann syndrome are associated with 11p15.5 uniparental disomy. J Am Soc Nephrol. 2002; 13:2077–2084.
24. Knudson, AG, Strong, LC. Mutation and Cancer: A model for Wilms’ tumor of the kidney. J Nat Cancer Inst. 1972; 48:313–324.
25. Grundy, PE, Breslow, N, Li, S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: A report from the National Wilms Tumor Study Group. J Clin Oncol. 2005; 23:7312–7321.
26. Huff, V. Wilms’ tumours: About tumour suppressor genes, an oncogene and a chameleon gene. Nat Rev Cancer. 2011; 11:111–121.
27. Malkin, D, Li, FP, Strong, LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990; 250:1233–1238.
28. Maiti, S, Alam, R, Amos, CI, et al. Frequent association of beta-catenin and WT1 mutations in Wilms tumors. Cancer Res. 2000; 60:6288–6292.
29. Koesters, R, Ridder, R, Kopp-Schneider, A, et al. Mutational activation of the beta-catenin proto-oncogene is a common event in the development of Wilms tumors. Cancer Res. 1999; 59:3880–3882.
30. Rivera, MN, Kim, WJ, Wells, J, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007; 315:642–645.
31. Call, KM, Glaser, T, Ito, CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms tumor locus. Cell. 1990; 60:509–520.
32. Yang, Y, Han, Y, Sauez Saiz, F, et al. A tumor suppressor and oncogene: The WT1 story. Leukemia. 2007; 21:868–876.
33. Rainer, S, Johnson, LA, Dobry, CJ, et al. Relaxation of imprinted genes in human cancer. Nature. 1993; 362:747–749.
34. Little, MH, Wells, C. A clinical overview of WT1 gene mutations. Hum Mutat. 1997; 9:209–225.
35. Fukuzawa, R, Heathcott, RW, More, HE, et al. Sequential WT1 and CTNNB1 mutations and alterations of beta-catenin localisation in intralobar nephrogenic rests and associated Wilms tumours: Two case studies. J Clin Pathol. 2007; 60:1013–1016.
36. Grundy, PE, Koufos, A, Morgan, K, et al. Familial predisposition to Wilms’ tumour does not map to the short arm of chromosome 11. Nature. 1988; 336:374–376.
37. Rahman, N, Arbour, L, Tonin, P, et al. Evidence for a familial Wilms’ tumour gene (FWT1) on chromosome 17q12-q21. Nat Genet. 1996; 13:461–463.
38. Beckwith, JB, Kiviai, NB, Bonadio, JF. Nephrogenic rests, nephroblastomatosis and the pathogenesis of Wilms tumor. Pediatr Pathol. 1990; 10:1–36.
39. Bennington, J, Beckwith, J. Tumor of the kidney, renal pelvis and ureter. In: Armed Forces Institute of Pathology; Atlas of Tumor Pathology. Washington DC: AFIP; 1975:31–91.
40. Perlman, EJ. Pediatric renal tumors: Practical updates for the pathologist. Pediatr Dev Pathol. 2005; 8:320–338.
41. Perlman, EJ, Faria, P, Soares, A, et al. Hyperplastic perilobar nephroblastomatosis: Long term survival in 52 patients. Pediatr Blood Cancer. 2006; 46:203–221.
42. Coppes, MJ, Beckwith, JB, Ritchey, ML, et al. Factors affecting the risk of contralateral Wilms tumor development (A report from the national Wilms tumor study group). Cancer. 1999; 85:1616–1625.
43. Cambio, A, Evans, C, Kurzrock, E. Non-surgical management of multicystic dysplastic kidney. BJU Int. 2008; 101:804–808.
44. Narchi, H, Narchi, H. Risk of Wilms’ tumour with multicystic kidney disease: A systematic review. Arch Dis Child. 2005; 90:147–149.
45. Beckwith, JB, Palmer, NF. Histopathology and prognosis of Wilms tumors: Results from the First National Wilms’ Tumor Study. Cancer Genet Cytogenet. 1978; 41:1937–1948.
46. Murphy, WM, Grignon, DJ, Perlman, EJ. Tumors of the kidney, bladder and related urinary structures. AFIP. 2004.
47. Dome, JS, Cotton, CA, Perlman, EJ. Treatment of anaplastic histology Wilms’ tumor: Results from the fifth National Wilms’ Tumor Study. J Clin Oncol. 2006; 24:2352–2358.
48. Faria, P, Beckwith, JB, Mishra, K, et al. Focal versus diffuse anaplasia in Wilms tumor – New definitions with prognostic significance. Am J Surg Pathol. 1996; 20:920.
49. Green, DM, Beckwith, JB, Breslow, N, et al. Treatment of children with stages II to IV anaplastic Wilms Tumor: A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994; 12:2126–2131.
50. Zuppan, CW, Beckwith, JB, Luckey, DW. Anaplasia in unilateral Wilms tumor. A report from the National Wilms Tumor Study Pathology Center. Hum Path. 2010; 19:1199–1209. [1988].
51. Williams, RD, Al-Saadi, R, Natrajan, R, et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer. 2011; 50:982–995.
52. Marsden, HB, Lawler, W, Kumar, PM, et al. Bone metastasizing renal tumor of childhood: Morphological and clinical features, and differences from Wilms’ tumor. Cancer. 1978; 42:1922–1928.
53. Argani, P, Perlman, EJ, Breslow, N, et al. Clear cell sarcoma of the kidney: A review of 351 cases from the National Wilms’ Tumour Study Pathology Center. Am J Surg Pathol. 2000; 24:4–18.
54. Brownlee, NA, Perkins, LA, Stewart, W. Recurring translocation (10;17) and deletion (14q) in clear cell sarcoma of the kidney. Arch Pathol Lab Med. 2007; 131:446–451.
55. Kusumakumary, P, Chellam, VG, Rojymon, J, et al. Late recurrence of clear cell sarcoma of the kidney. Med Pediatr Oncol. 1997; 28:355–357.
56. Weeks, DA, Beckwith, JB, Mierau, GW, et al. Rhabdoid tumor of kidney: A report of 111 cases from the National Wilms’ Tumor Study Pathology Center. Am J Surg Pathol. 1989; 13:439–458.
57. Argani, P, Hawkins, A, Griffin, CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol. 2001; 158:2089–2096.
58. Hass, JE, Palmer, NF, Weinberg, AG. Ultrastructure of malignant rhabdoid tumor of the kidney: A distinctive renal tumor of children. Hum Pathol. 1981; 12:646–657.
59. Bonnin, JM, Rubenstein, IJ, Palmer, NF, et al. The association of embryonal tumors originating in the kidney and the brain. Cancer. 1984; 54:2137–2146.
60. Perlman, EJ, Ali, SZ, Robinson, R, et al. Infantile extrarenal rhabdoid tumor. Pediatr Dev Pathol. 1998; 1:149–152.
61. Schofield, DE, Beckwith, JB, Sklar, J. Loss of heterozygosity at chromosome regions 22q11-12 and 11p15.5 in renal rhabdoid tumors. Genes Chromosomes Cancer. 1996; 15:10–17.
62. Versteege, I, Sevenet, N, Lange, J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998; 394:203–206.
63. Biegel, JA, Zhou, J, Rorke, L, et al. Germ-line and acquired mutations of INI1 in atypical teratoid and rhabdoid tumors. Cancer. 2012; 59:74–79.
64. Hoot, AC, Russo, P, Judkins, AR, et al. Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol. 2004; 28:1485–1491.
65. Grundy, PE, Dome, JD, Perlman, EJ, et al. Renal Tumors Classification, Biology, and Banking Study. https://members childrensoncologygroup org/Prot/AREN03B2/AREN03B2DOC pdf, 2006.
66. Vujanic, G, Sandstedt, B, Harms, D. Revised International Society of Paediatric Oncology (SIOP)Working Classification of Renal Tumors of Childhood. Med Pediatr Oncol. 2002; 38:79–82.
67. Vujanic, G, Sandstedt, B. The pathology of Wilms’ tumour (nephroblastoma): The International Society of Paediatric Oncology approach. J Clin Pathol. 2010; 63:102–109.
68. Grundy, PE, Perlman, EJ, Ehrlich, PF, et al. Current issues in Wilms tumor management. Cur Prob Cancer. 2005; 29:221–260.
69. Kalapurakal, JA, Dome, JS, Perlman, EJ, et al. Management of Wilms’ tumour: Current practice and future goals. Lancet Oncol. 2004; 5:37–46.
70. Breslow, N, Olshan, A, Beckwith, JB, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993; 21:172–181.
71. Dome, JD, Perlman, EJ, Ritchey, ML, et al. Renal tumours. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology. 5th ed. Philadelphia: Lippincott Williams and Wilkins; 2006:905–932.
72. Khanna, G, Rosen, N, Anderson, J, et al. Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2012; 58:551–555.
73. Ritchey, ML, Shamberger, RC, Hamilton, TE, et al. Fate of bilateral renal lesions missed on preoperative imaging: A report from the National Wilms Tumor Study Group. J Urol. 2005; 174:1519–1521.
74. Khanna, G, Naranjo, A, Hoffer, F, et al. Detection of preoperative Wilms tumor rupture with CT: a report from the Children’s Oncology Group. Radiology. 2013; 266(2):610–617.
75. Ehrlich, PF, Ritchey, ML, Hamilton, TE, et al. Quality assessment for Wilms’ tumor: A report from the National Wilms’ Tumor Study-5. J Pediatr Surg. 2005; 40:208–212.
76. Ritchey, ML, Shamberger, RC, Haase, G, et al. Surgical complications after primary nephrectomy for Wilms’ tumor: Report from the National Wilms’ Tumor Study Group. J Am Col Surg. 2001; 192:63–68.
77. Shamberger, RC, Guthrie, KA, Ritchey, ML, et al. Surgery related factors and local recurrence of Wilms tumor in the national Wilms tumor study 4. Ann Surg. 1999; 229:292–297.
78. Ritchey, ML, Lally, KP, Haase, GM, et al. Superior mesenteric artery injury during nephrectomy for Wilms’ tumor. J Pediatr Surg. 1992; 27:612–615.
79. Othersen, HB, Delorimier, A, Hrabovsky, E, et al. Surgical evaluation of lymph node metastases in Wilms tumor. J Pediatr Surg. 1990; 25:330–331.
80. D’Angio, GJ, Evans, AE, Breslow, N. The Treatment of Wilms’ Tumor: Results of the Second National Wilms’ Tumor Study. Am Cancer Soc. 1981; 4:2302–2310.
81. Kieran, K, Anderson, J, Dome, JS, et al. Lymph node involvement in Wilms tumor: Results from National Wilms Tumor Studies 4 and 5. J Pediatr Surg. 2012; 47:700–706.
82. Ehrlich, PF, Dome, JS, Shamberger, RC, et al. Clinico-pathologic Findings predictive of relapse in children with Stage III Favorable Histology Wilms Tumor: The importance of lymph nodes. Pediatr Blood Cancer. 2010; 55:794.
83. Ritchey, ML, Daley, S, Shamberger, RC, et al. Ureteral extension in Wilms’ tumor: A report from the National Wilms’ Tumor Study Group (NWTSG). J Pediatr Surg. 2008; 43:1625–1629.
84. Gow, K, Barnhart, DC, Hamilton, TE, et al. Primary nephrectomy and intraoperative tumor spill: Report from the Children’s Oncology Group (COG) Renal Tumors Committee. J Pediatr Surg. 2012.
85. Grundy, PE, Dome, JS, Ehrlich, PF. Renal tumors classification, biology and banking studies. https://members childrensoncologygroup org/Prot/AREN03B2/AREN03B2DOC pdf, 2007. [p. 18–33].
86. Ehrlich, PF, Ferrer, F, Ritchey, ML, et al. Hepatic metastasis at diagnosis in patients with Wilms tumor is not an independent adverse prognostic factor for stage IV Wilms tumor. A report from the childrens oncology group/national wilms tumor study group. Ann Surg. 2009; 250:642–648.
87. Elli, M, Pinarli, FG, Dagdemir, A. Acquired von Willebrand syndrome in a patient with Ewing sarcoma. Pediatr Hematol Oncol. 2006; 23:111–114.
88. Coppes, MJ, Zandvoort, SW, Sparling, CR. Acquired von Willebrand disease in Wilms’ tumor patients. J Clin Oncol. 1992; 10:422–427.
89. Scott, JP, Mongomery, RR, Tubergen, DG, et al. Acquired von Willebrand’s disease in association with Wilm’s tumor: Regression following treatment. Blood. 1981; 58:665–669.
90. Baxter, PA, Nutchtern, JG, Guillerman, RP, et al. Acquired von Willebrand Syndrome and Wilms Tumor: Not Always Benign. Pediatr Blood Cancer. 2009; 52:392–428.
91. Bracey, AW, Wu, AH, Aceves, J, et al. Platelet dysfunction associated with Wilms tumor and hyaluronic acid. Am J Hematol. 1987; 24:247–257.
92. Michiels, JJ, Budde, U, van der Planken, M, et al. Acquired von Willebrand syndromes: Clinical features, aetiology, pathophysiology, classification and management. Best Pract Res Clin Haematol. 2001; 14:401–436.
93. Tournade, MF, Com-Nougue, C, de Kraker, J, et al. Optimal duration of preoperative chemotherapy in unilateral non metastatic Wilms tumor in children older then six months. Results of the ninth International Society of Pediatric Oncology tumor trial. J Clin Oncol. 2001; 19:488–500.
94. Tournade, MF, Com-Nougue, C, Voute, PA, et al. Results of the sixth International Society of Pediatric Oncology Wilms’ tumor trial and study: A risk-adapted therapeutic approach in Wilms’ tumor. J Clin Oncol. 1993; 11:1014–1023.
95. Shamberger, RC, Ritchey, ML, Haase, G, et al. Intravascular extension of Wilms tumor. Ann Surg. 2001; 234:116–121.
96. Lall, A, Pritchard-Jones, K, Walker, J, et al. Wilms’ tumor with intracaval thrombus in the UK Children’s Cancer Study Group UKW3 trial. J Pediatr Surg. 2006; 41:382–387.
97. Nakayama, D.K, Norkool, P, Delorimier, A, et al. Intracardiac extension of Wilms’ tumor: A report of the National Wilms’ Tumor Study. Ann Surg. 1986; 204:693–697.
98. Ritchey, ML. Renal sparing surgery for Wilms tumor. J Urol. 2005; 174:1172–1173.
99. Green, DM, Breslow, N, Beckwith, JB, et al. Treatment outcomes in patients less than 2 years of age with small, stage I, favorable-histology Wilms’ tumors: A report from the National Wilms’ Tumor Study. J Clin Oncol. 1993; 11:91–95.
100. Larsen, E, Perez- Atayde, A, Green, DM, et al. Surgery only for the treatment of patients with stage I (Cassady) Wilms’ tumor. Cancer. 1990; 66:264–266.
101. Green, DM, Breslow, N, Beckwith, JB, et al. Treatment with nephrectomy only for small, stage I/favorable histology Wilms’ tumor: A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2001; 19:3719–3724.
102. Shamberger, RC, Anderson, J, Breslow, N, et al. Long-term outcomes for infants with very low risk Wilms tumor treated with surgery alone in National Wilms Tumor Study-5. Ann Surg. 2010; 251:555–558.
103. Sredni, ST, Gadd, S, Huang, CC. Subsets of very low risk Wilms tumor show distinctive gene expression, histologic, and clinical features. Clin Cancer Res. 2009; 15:6800–6809.
104. van den Heuvel-Eibrink, MM, Grundy, PE, Graf, N, et al. Characteristics and survival of 750 children diagnosed with a renal tumor in the first seven months of life: A collaborative study by the SIOP/GPOH/SFOP, NWTSG, and UKCCSG Wilms tumor study groups. Pediatr Blood Cancer. 2008; 50:1130–1134.
105. Isaacs, H. Fetal and neonatal renal tumors. J Pediatr Surg. 2008; 43:1587–1595.
106. Andrews, PE, Kelalis, P, Haase, G. Extrarenal Wilms’ tumor: Results of the National Wilms’ Tumor Study. J Pediatr Surg. 1992; 27:1181–1184.
107. Coppes, MJ, Wilson, PC, Weitzman, S. Extrarenal Wilms’ tumor: Staging, treatment, and prognosis. J Clin Oncol. 1991; 9:167–174.
108. Green, DM, Breslow, N, Beckwith, JB. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms’ tumor: A report from the National Wilms’ tumor study group. J Clin Oncol. 1998; 16:237–245.
109. D’Angio, GJ, Breslow, N, Beckwith, JB, et al. Treatment of Wilms’ Tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1989; 64:360.
110. Breslow, N, Ou, SS, Beckwith, JB, et al. Doxorubicin for favorable histology, Stage II-III Wilms tumor – Results from the National Wilms Tumor Studies. Cancer. 2004; 101:1072–1080.
111. Green, DM, Breslow, N, Evans, I, et al. The effect of chemotherapy dose intensity on the hematological toxicity of the treatment for Wilms’ tumor. A report of the National Wilms Tumor Study. Am J Pediatr Hematol Oncol. 1994; 16:207–212.
112. Green, DM, Breslow, N, Beckwith, JB, et al. Effect of duration of treatment on treatment outcome and cost of treatment for Wilms tumor: A report from the National Wilms Tumor Study Group. J Clin Oncol. 1998; 16:3744–3751.
113. Green, DM, Cotton, CA, Malogolowkin, M, et al. Treatment of Wilms tumor relapsing after initial treatment with vincristine snd actinomycin D: A report form the National Wilms Tumor Study. Pediatr Blood Cancer. 2007; 48:493–499.
114. Malogolowkin, M, Cotton, CA, Green, DM, et al. Treatment of Wilms tumor relapsing after initial treatment with vincristine, actinomycin D, and doxorubicin. A report from the National Wilms Tumor Study Group. Pediatr Blood Cancer. 2008; 50:236–241.
115. Seibel, NL, Breslow, N, Beckwith, JB, et al. Effect of duration of treatment on treatment outcome for patients with clear-cell sarcoma of the kidney: A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2004; 22:468–473.
116. Abu-Ghosh, AM. Ifosfamide, carboplatin and etoposide in children with poor risk relapsed Wilms’ tumor: A Children’s Cancer Group report. Ann Oncol. 2002; 13:460.
117. Tomlinson, GE, Breslow, NE, Dome, J, et al. Rhabdoid tumor of the kidney in the National Wilms’ Tumor Study: Age at diagnosis as a prognostic factor. J Clin Oncol. 2005; 23:7641–7645.
118. Waldron, PE, Rodgers, BM, Kelly, MD, et al. Successful treatment of a patient with stage IV rhabdoid tumor of the kidney: Case report and review. J Pediatr Hematol Oncol. 1999; 21:53–57.
119. Wagner, L, Hill, DA, Fuller, C, et al. Treatment of metastatic rhabdoid tumor of the kidney. J Pediatr Hematol Oncol. 2002; 24:385–388.
120. Gururangan, S, Bowman, LC, Parham, DM, et al. Primary extracranial rhabdoid tumors. Cancer Genet Cytogenet. 1993; 71:2653–2659.
121. Lemerle, J, Voute, PA, Tournade, MF, et al. Preoperative versus postoperative radiotherapy, single versus multiple courses of actinomycin D, in the treatment of Wilms’ tumor: Preliminary results of a controlled clinical trial conducted by the International Society of Paediatric Oncology. Cancer. 1976; 38:647–654.
122. Godzinski, J, Tournade, MF, deKraker, J, et al. Rarity of surgical complications after postchemotherapy nephrectomy for nephroblastoma. Experience of the International Society of Paediatric Oncology-Trial and Study ‘SIOP-9’. International Society of Paediatric Oncology Nephroblastoma Trial and Study Committee. Eur J Pediatr Surg. 1998; 8:83–86.
123. D’Angio, GJ, Evans, AE, Breslow, N, et al. The treatment of Wilms’ tumor: Results of the National Wilms’ Tumor Study. Cancer. 1976; 38:633–646.
124. Zoeller, G, Pekrun, A, Lakomek, M, et al. Wilms tumor: The problem of diagnostic accuracy in children undergoing preoperative chemotherapy without histological tumor verification. J Urol. 1994; 151:169–171.
125. Vujanic, G, Kelsey, A, Mitchell, C, et al. The role of biopsy in the diagnosis of renal tumors of childhood: Results of the UKCCSG Wilms’ tumor study 3. Med Pediatr Oncol. 2003; 40:18–22.
126. Weirich, A, Leuschner, I, Harms, D, et al. Clinical impact of histologic subtypes in localized non-anaplastic nephroblastomatreated according to the trial and study SIOP-9/GPOH. Ann Oncol. 2001; 12:311–319.
127. D’Angio, GJ, Tefft, M, Breslow, N, Meyer, JA. Radiation therapy of Wilms’ tumor: Results according to dose, field, post-operative timing and histology. Int J Radiat Oncol Biol Phys. 1978; 4:769–780.
128. Thomas, PRM, Tefft, M, Farewell, VT, et al. Abdominal relapses in irradiated second National Wilms’ Tumor Study patients. J Clin Oncol. 1984; 2:1098–1101.
129. Thomas, PRM, Tefft, M, Compaan, PJ, et al. Results of two radiotherapy randomizations in the Third National Wilms’ Tumor Study (NWTS-3). Cancer. 1991; 68:1703–1707.
130. Kalapurakal, JA, Li, SM, Breslow, N, et al. Influence of radiation therapy delay on abdominal tumor recurrence in patients with favorable histology Wilms’ tumor treated on NWTS-3 and NWTS-4: A report from the National Wilms’ Tumor Study Group. Int J Radiat Oncol Biol Phys. 2003; 57:495–499.
131. Green, DM. Use of chest computed tomography for staging and treatment of Wilms’ tumor in children. J Clin Oncol. 2002; 20:2763–2764.
132. Paulino, AC, Wen, BC, Brown, CK, et al. Late effects in children treated with radiation therapy for Wilms tumor. Int J Rad Oncol. 2000; 46:1239–1246.
133. Attard-Montalto, SP, Kingston, JE, Eden, OB, Plowman, PN. Late follow-up of lung function after whole lung irradiation for Wilms tumor. Br J Radiol. 1992; 65:1114–1118.
134. Breslow, N, Lange, JM, Friedman, DL, et al. Secondary malignant neoplasms after Wilms tumor: An international collaborative study. Int J Cancer. 2009; 127:657–666.
135. Grundy, PE, Green, DM, Dirks, A, et al. Clinical significance of pulmonary nodules detected by CT and Not CXR in patients treated for favorable histology Wilms tumor on national Wilms tumor studies-4 and -5: A report from the Children’s Oncology Group. Med Pediatr Oncol. 2003; 41:251.
136. Weirich, A, Ludwig, R, Graf, N. Survival in nephroblastoma treated according to the trial and study SIOP-9/GPOH with respect to relapse and morbidity. Ann Oncol. 2004; 15:808–820.
137. Prichard, J, Imeson, J, Barnes, J, et al. Results of the United Kingdoms children’s cancer study group first Wilms tumor study. J Clin Oncol. 1995; 13:124–133.
138. Mitchell, C, Jones, PM, Kelsey, A, et al. The treatment of Wilms’ tumour: Results of the United Kingdom Children’s Cancer Study Group (UKCCSG) second Wilms’ tumour study. Br J Cancer. 2000; 83:602–608.
139. Petruzzi, MJ, Green, DM. Wilms tumor. Pediatr Clin North Am. 1997; 44:939–952.
140. Ritchey, ML. Renal sparing surgery for children with bilateral Wilms tumor. Cancer. 2008; 112:1877–1878.
141. Ritchey, ML, Green, DM, Thomas, PRM. Renal failure in Wilms tumor patients: A report from the National Wilms Tumor Study Group. Med Pediatr Oncol. 1996; 26:75–80.
142. Breslow, N, Collins, AJ, Ritchey, ML, et al. End stage renal failure in patients with Wilms tumor: Results from the national Wilms tumor study group and the United States renal data system. J Urol. 2005; 174:1972–1975.
143. Shamberger, RC, Haase, GM, Argani, P. Bilateral Wilms’ tumors with progressive or nonresponsive disease. J Pediatr Surg. 2006; 41:642–657.
144. Green, DM, Beckwith, JB, Breslow, N, et al. Treatment of children with stages II to IV anaplastic Wilms Tumor: A report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994; 12:2126–2131.
145. Hamilton, TE, Green, DM, Perlman, EJ, et al. Bilateral Wilms’ tumor with anaplasia: Lessons from the National Wilms’ Tumor Study. J Pediatr Surg. 2006; 41:1641–1644.
146. Kumar, R, Fitzgerald, R, Breatnach, F. Conservative surgical managment of bilateral Wilms tumor: Results of the United Kingdom children’s cancer study group. J Urol. 1998; 160:1450–1453.
147. Anderson, J, Slater, O, McHugh, K, et al. Response without shrinkage in Bilateral Wilms Tumor: Significance of Rhabdomyomatous Histiology. J Pediatr Hem Oncol. 2002; 24:31–34.
148. Graf, N, Tournade, MF, de Kraker, J. The role of preoperative chemotherapy in the management of Wilms tumor. The SIOP studies. Urol Clin North Am. 2000; 27:443–454.
151. Cotton, CA, Peterson, SS, Norkool, PA. Early and late mortality after diagnosis of Wilms tumor. J Clin Oncol. 2009; 27:1304–1309.
152. Green, DM, Grigoriev, YA, Nan, B, et al. Congestive heart failure after treatment for Wilms’ tumor: A report from the National Wilms’ Tumor Study group. J Clin Oncol. 2001; 19:1926–1934.
153. van Dijk, IW, Oldenburger, F, Cardous-Ubbink, MC. Evaluation of late adverse events in long-term Wilms’ Tumor survivors. Int J Radiat Oncol Biol Phys. 2010; 78:370–378.
154. Termuhlen, AM, Tersak, JM, Liu, Q, et al. Twenty-five year follow-up of childhood Wilms tumor: A report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. 2011; 57:1210–1216.
155. Grigoriev, Y, Lange, J, Peterson, SM, et al. Treatments and outcomes for end-stage renal disease following Wilms tumor. Pediatr Nephrol. 2012; 27:1325–1333.
156. Green, DM, Lange, JM, Peabody, EM. Pregnancy Outcome after treatment for Wilms Tumor: A report from the National Wilms Tumor Long-Term Follow-Up Study. J Clin Oncol. 2010; 28:2824–2830.
157. Green, DM, Grigoriev, YA, Nan, B, et al. Correction to ‘Congestive heart failure after treatment for Wilms’ tumor’. J Clin Oncol. 2003; 21:2447–2448.
158. Pein, F, Sakiroglu, O, Dahan, M, et al. Cardiac abnormalities 15 years and more after adriamycin therapy in 229 childhood survivors of a solid tumour at the Institut Gustave Roussy. Br J Cancer. 2004; 91:37–44.
159. Aronson, DC, Medary, I, Findlay, JL. Renal cell carcinoma in childhood and adolescence: A retrospective survey for prognostic factors in 22 cases. J Pediatr Surg. 1996; 31:183–186.
160. Geller, JI, Dome, JS. Local lymph node involvement does not predict poor outcome in pediatric renal cell carcinoma. Cancer. 2004; 101:1575–1583.
161. Motzer, RJ, Bander, NH, Nanus, DM. Renal-cell carcinoma. N Engl J Med. 1996; 335:865–875.
162. Fyfe, G, Fisher, RI, Rosenberg, SA, et al. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995; 13:688–696.
163. Messing, EM, Manola, J, Wilding, G, et al. Phase III study of interferon alfa-NL as adjuvant treatment for resectable renal cell carcinoma: An Eastern Cooperative Oncology Group/Intergroup trial. J Clin Oncol. 2003; 21:1214–1222.
164. Bolande, RP, Brough, AJ, Izant, RJ. Congenital mesoblastic nephroma of infancy. A report of eight cases and the relationship to Wilms’ tumor. Pediatrics. 1967; 40:272–278.
165. Howell, CG, Othersen, HB, Kiviat, NE, et al. Therapy and outcome in 51 children with mesoblastic nephroma: A report of the National Wilms’ Tumor Study. J Pediatr Surg. 1982; 17:826–831.
166. Bolande, RP. Congenital and infantile neoplasia of the kidney. Lancet. 1974; 2:1497–1499.
167. Argani, P, Fritsch, M, Kadkol, SS, et al. Detection of the ETV6-NTRK3 chimeric RNA of infantile fibrosarcoma/cellular congenital mesoblastic nephroma in paraffin-embedded tissue: Application to challenging pediatric renal stromal. Mod Pathol. 2000; 13:29–36.
168. Beckwith, JB. Mesenchymal renal neoplasms of infancy revisited. J Pediatr Surg. 1974; 9:803–805.
169. Heidelberger, KP, Ritchey, ML, Dauser, RC, et al. Congenital mesoblastic nephroma metastatic to the brain. Cancer. 1993; 72:2499–2502.
170. Vujanic, G, Delemarre, JF, Moeslichan, S, et al. Mesoblastic nephroma metastatic to the lungs and heart–another face of this peculiar lesion: Case report and review of the literature. Pediatrc Pathol. 1993; 13:143–153.
171. Sandstedt, B, Delamarre, JFM, Krul, EJ, et al. Mesoblastic nephromas: A study of 29 tumours from the SIOP nephroblastoma file. Histopathology. 1985; 9:741–750.
172. Chan, KL, Chan, KW, Lee, CW. Preoperative chemotherapy for mesoblastic nephroma. Med Pediatr Oncol. 1995; 24:271–273.
173. Beckwith, JB. Reply. Pediatrc Pathol. 1993; 13:886–887.
174. Agrons, GA, Wagner, BJ, Davidson, AJ, et al. Multilocular cystic renal tumor in children: Radiologic-pathologic correlation. Radiographics. 1995; 15:653–669.
175. Delanhunt, B, Thomson, K, Ferguson, A. Familial cystic nephroma and pleuro-pulmonary blastoma. Cancer. 1993; 71:1338–1342.
176. Ishida, Y, Kato, K, Kigasawa, H, et al. Synchronous occurrence of pleuropulmonary blastoma and cystic nephroma: Possible genetic link in cystic lesions of the lung and the kidney. Med Pediatr Oncol. 2000; 35:85–87.
177. Joshi, VV, Beckwith, JB. Multilocular cyst of the kidney (cystic nephroma) and cystic, partially differentiated nephroblastoma. Terminology and criteria for diagnosis. Cancer. 1989; 64:466–479.
178. Blakely, ML, Shamberger, RC, Norkool, P, et al. Outcome of children with cystic partially differentiated nephroblastoma treated with or without chemotherapy. J Pediatr Surg. 2003; 38:897–900.
179. Luithle, T, Szavay, P, Furtwängler, R, et al. Treatment of cystic nephroma and cystic partially differentiated nephroblastoma–a report from the SIOP/GPOH study group. J Urol. 2007; 177:294–296.
180. Beckwith, JB. The John Lattimer lecture: Wilms’ tumor and other renal tumors of childhood: An update. J Urol. 1986; 136:320–324.
181. Steif, W, Gassner, I, Janetschek, G, et al. Partial nephrectomy in a cystic partially differentiated nephroblastoma. Med Pediatr Oncol. 1997; 28:416–419.
182. Sacher, P, Willi, U, Niggli, F, et al. Cystic nephroma: A rare benign renal tumor. Pediatr Surg Int. 1998; 13:197–199.
183. Mushtaq, I, Carachi, R, Roy, G, et al. Childhood renal tumours with intravascular extension. Br J Urol. 1996; 78:772–776.
184. Vazquez, JL, Barnewolt, CE, Shamberger, RC. Ossifying renal tumor of infancy presenting as a palpable abdominal mass. Pediatr Radiol. 1998; 28:454–457.
185. Steffens, J, Kraus, J, Misho, B, et al. Ossifying renal tumor of infancy. J Urol. 1993; 149:1080–1081.