[level-membership-for-emergency-medicine-category]
59 Tachydysrhythmias
 Key Points
Key Points• Tachydysrhythmia may be due to intrinsic cardiologic disease or external stimulation.
• Noncardiologic causes, such as hypoxia, inadequate perfusion, metabolic derangements, or toxicity from medications or other agents, should always be considered.
• The higher the tachycardic heart rate, the greater the likelihood of instability.
• Patients with ventricular tachycardia may not necessarily appear unstable.
• If there is uncertainty, wide QRS complex tachycardia should be assumed to be ventricular in origin.
• The most effective treatment of ventricular tachycardia is electrical cardioversion. However, antidysrhythmic medications may also be necessary to prevent recurrence.
• All ostensibly healthy young patients with syncope should be assessed for valvular disease, left ventricular hypertrophy, Wolff-Parkinson-White syndrome, abnormal QT interval or T-wave morphology, and Brugada syndrome.
Epidemiology
Heart rhythms with rapid rates, or tachydysrhythmias, have a range of causes and associated incidences, morbidities, and mortality. Atrial fibrillation (AF) occurs in up to 5% of people older than 65 years. Conversely, sustained ventricular tachycardia (VT) is rare and accounts for less than 0.1% of emergency department (ED) visits. Morbidity ranges from temporary lightheadedness to syncope and deterioration, ventricular fibrillation (VF), or embolization and subsequent permanent disability as a result of ischemic stroke. Mortality from atrial tachydysrhythmias is generally low. Mortality from VT is approximately 5%, and that from VF ranges from 80% to greater than 90%, depending on the clinical circumstances.1 A particular exception to the low mortality associated with atrial tachydysrhythmias is AF or flutter with rapid ventricular excitation via a bypass tract, such as in Wolff-Parkinson-White (WPW) syndrome.2
Pathophysiology and Mechanisms
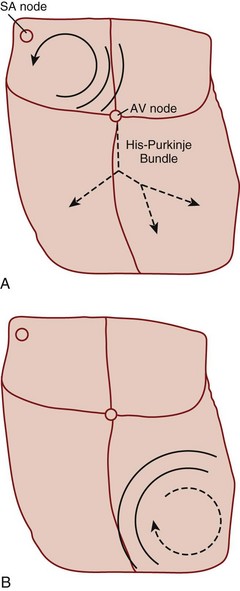
Reentry refers to a microscopic or macroscopic circular pattern of myocardial depolarization. A wave of excitation passes around the circuit and depolarizes the cells until it arrives at the beginning, and the process repeats itself. Reentry can occur if the circuit is sufficiently long or there is a slow-conducting portion that allows enough time for cells to have recovered from their refractory period before the wave of excitation returns (Fig. 59.1). The reentrant rhythm becomes the cardiac pacemaker if it is sufficiently fast to outpace the sinus node.
Presenting Signs and Symptoms
Treatment
The primary emergency therapies for tachydysrhythmias include medications to slow electrical conduction or to increase the cellular refractory period in the AV node or other groups of cardiac cells (Table 59.1). The goal of therapy is to terminate and suppress the abnormal rhythm or to slow the response in the remainder of the heart. Electrical therapy, including synchronized cardioversion and defibrillation, is used in potentially or frankly unstable patients. Electrical therapy has the advantages of high efficacy and safety, but shocks are painful and do not prevent recurrence of the tachydysrhythmia. Patients with higher heart rates, regardless of the underlying rhythm, have compromised diastolic filling and cardiac output. These patients should be treated more aggressively with early electrical therapy. The emergency physician (EP) should understand and use antidysrhythmic medications but should minimize the use of multiple agents in a single patient. Use of multiple drugs increases the likelihood of drug interactions and compromised cardiac output.
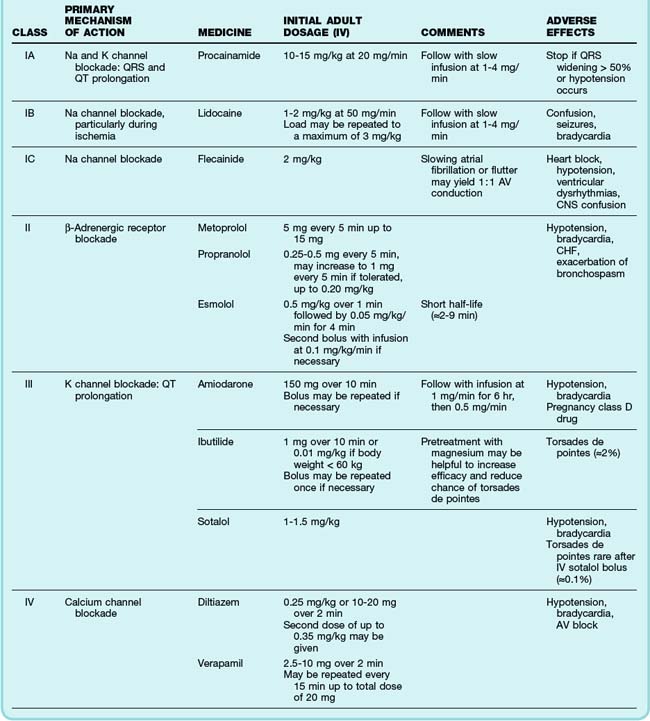
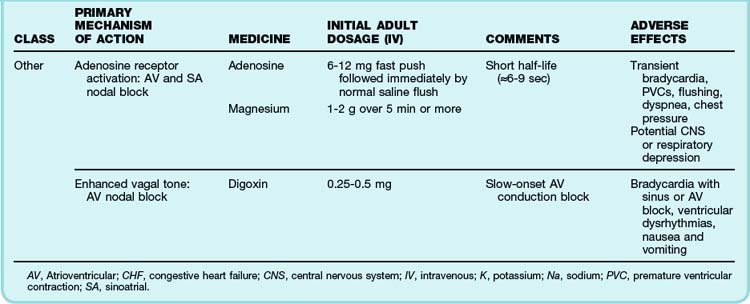
See Table 59.1, Select Tachydysrhythmia Medications, online at www.expertconsult.com
Specific Tachycardias
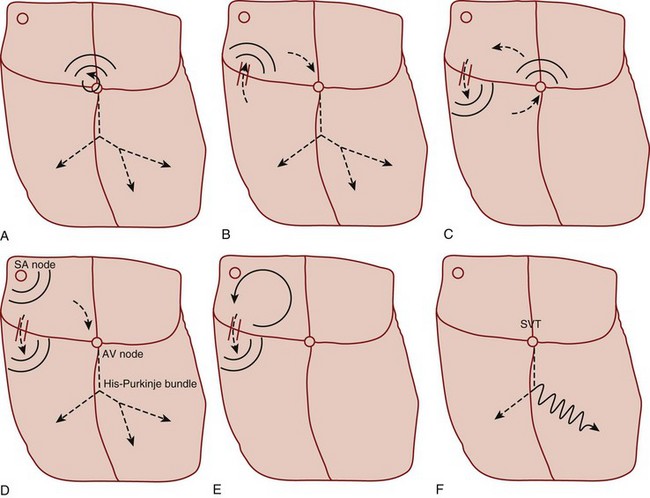
Primary tachycardiac conditions can be divided most simply into those of supraventricular and ventricular origin (Box 59.1; Fig. 59.2; see also Fig. 59.1). Supraventricular tachycardia (SVT) must involve tissue above the ventricles but may also involve ventricular tissue in a tachycardiac circuit, whereas VT involves only ventricular tissue below the AV node. The remainder of the chapter deals with the diagnosis and treatment of specific tachycardiac conditions.
Supraventricular Tachycardias
Irregular Supraventricular Tachycardias
Atrial Fibrillation
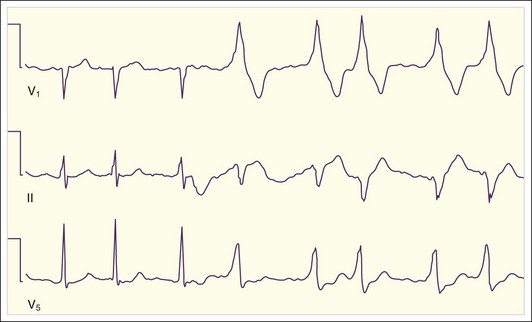
The clinician can usually diagnose AF at the bedside by feeling the pulse, listening to the heart sounds, and observing a single-lead cardiac monitor. The ECG may show fine or coarse irregular undulations of the baseline and an absence of regular P waves. The QRS complex is usually normal; however, if the ventricular response is rapid, the QRS complex may be transiently widened because of intermittent aberrant conduction through the His-Purkinje system (Fig. 59.3). This finding, termed the Ashman phenomenon, can occur when areas of the His-Purkinje tissue have a longer refractory period than the AV node does. Such bursts of wide QRS complex tachycardia can be confused with nonsustained VT, but on close inspection the rhythm remains irregularly irregular.
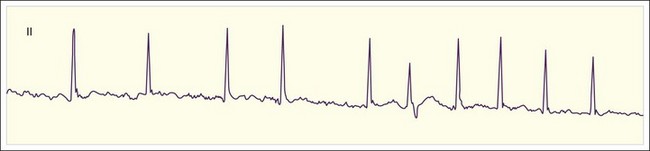
The most common treatment issue for patients with AF in the emergency setting is control of the ventricular rate. Depending on the function of the patient’s AV node, the ventricular rate may range up to approximately 150 beats/min in adults. Ventricular rates this fast allow insufficient time for diastolic filling of the heart. The rate can be slowed with a variety of agents that act to lengthen the refractory period of the AV nodal tissue, such as calcium channel blockers,3 beta-blockers,4 magnesium,5 digoxin, and amiodarone. Digoxin is less useful in the emergency setting because it takes a few hours to work, but the other agents must be used carefully because they can decrease blood pressure to varying degrees. The calcium channel blocker diltiazem usually provides excellent rate control with minimal loss of blood pressure. Beta-blockers may cause a greater loss of blood pressure and must be used with caution in patients with CHF or pulmonary disease. There is no indication for the administration of adenosine in patients with AF. The diagnosis should be made on the basis of findings from the history, physical examination, and ECG. Furthermore, adenosine, which only transiently blocks AV nodal conduction, would have no lasting therapeutic benefit.
Cardioversion can also be accomplished chemically when circumstances allow. Agents that slow conduction or increase the refractory period of atrial tissue are used to break the reentrant microcircuits within the atria. Vaughn-Williams class III and class I medications that can accomplish this goal include ibutilide, amiodarone, procainamide, propafenone, and flecainide.6,7 These agents can terminate AF alone and increase the likelihood of cardioversion of AF with subsequent DCCV. However, their use entails some risk. There is an approximately 2% to 5% chance of causing torsades de pointes (TdP) after the infusion of ibutilide, amiodarone, and procainamide because these agents variably increase the duration of the depolarized phase and the associated QT interval in the ventricles, as well as the atria. The newly developed agent vernakalant may be less likely to cause TdP because of its action in blocking the ultrarapid potassium channels located primarily in the atria. Hypokalemia and hypomagnesemia raise the risk for TdP. Pretreatment with magnesium may be protective in some circumstances, even in patients with normal serum electrolyte levels.8 Propafenone and flecainide can slow conduction, exacerbate heart failure, and cause ventricular dysrhythmias. Cardioversion also carries a risk for embolic stroke.
In the acute setting, clot may form within 48 hours after the onset of AF. Cardioversion of AF may cause a new clot to embolize into the systemic circulation. If the patient can clearly discern when he or she is in AF, symptoms have begun within the preceding 1 to 2 days, and the patient is not at high risk for stroke, it is generally safe to perform cardioversion without anticoagulation.9 However, if there is any uncertainty about the time of onset of the tachydysrhythmia or if it occurred more than 48 hours ago, the patient should not undergo immediate cardioversion. Instead, the cardiology service should be consulted and transesophageal echocardiography performed to search for atrial clot formation, or cardioversion should be deferred until adequate anticoagulation has been established.10 The only exception would be cardiovascular instability secondary to AF, which requires emergency DCCV. Continuation of anticoagulation should also be considered for 4 weeks after cardioversion because there may be some delay in return of normal atrial contraction.
The disposition of patients with AF depends on a number of factors, including age, comorbid illnesses and social and outpatient medical support, adequacy of control of the ventricular rate, plans for cardioversion, and the state of anticoagulation.11 If the patient requires ongoing adjustment of medications to control a rapid ventricular response, attempted cardioversion is planned, or new anticoagulation is initiated, hospitalization should be considered. The EP should make this decision in concert with the patient and the consulting cardiologist.
Multifocal Atrial Tachycardia
Multifocal AT (MFAT) is a relatively uncommon tachydysrhythmia that primarily affects older patients with chronic lung diseases such as chronic obstructive pulmonary disease, and it appears to be related to the administration of methylxanthine.12 Its mechanism is uncertain, but MFAT is thought to occur as a result of DADs in atrial tissue triggering ectopic beats. The diagnosis is made on the basis of the following ECG criteria: at least three consecutive P waves with different morphologies, variable P-P and PR intervals, and a heart rate greater than 100 beats/min. Treatment of MFAT is centered on treating the underlying pulmonary disease and possible hypoxia. Antidysrhythmic agents are poorly effective in slowing the atrial rate or the ventricular response or in terminating MFAT. Nevertheless, a carefully administered trial of a calcium channel blocker or amiodarone is reasonable. Generally, beta-blockers should be avoided in patients with any evidence of bronchospasm. Magnesium therapy, particularly in combination with potassium replacement, may slow the rate or terminate the tachydysrhythmia. Patients usually require admission to treat the pulmonary disease.
Regular Supraventricular Tachycardias
Atrial Flutter
Atrial flutter can occur as a result of a variety of cardiopulmonary and metabolic derangements, often in association with atrial dilation. Patients with atrial flutter also tend to experience AF, but atrial flutter is less common overall. The mechanism of atrial flutter is a macroreentrant circuit in the right atrium (see Fig. 59.1, A). The most common variant of atrial flutter is termed typical atrial flutter and involves a counterclockwise wave of roughly circular excitation when viewed from a position facing the front of the patient. The flutter pathway is constrained in the inferior portion of the circular loop by anatomic structures, and it travels between the tricuspid annulus anteriorly and the inferior vena cava and coronary sinus posteriorly. This segment of the pathway, the isthmus, contains the relatively slower-conducting tissue in the reentrant loop. Atypical atrial flutter most commonly travels over the same pathway, but in the opposite direction.
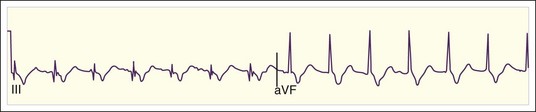
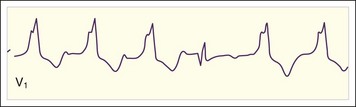
The rate of atrial flutter waves ranges from 250 to 350 beats/min. The diagnosis is made from the ECG tracing, which demonstrates typical sawtooth-shaped flutter waves at the appropriate rate, generally most prominent in the inferior limb leads (II, III, and aVF) and lead V1. The flutter waves tend to be identical and to recur regularly, and there is no isoelectric or flat ECG segment between the waves (Figs. 59.4 and 59.5). As in AF, the refractory period of the AV node usually controls the ventricular response to atrial flutter. The most common response is 2 : 1 AV conduction; however, higher, lower, or variable ratios of conduction are also possible. The ratio of conduction depends on the atrial flutter rate, the health of the AV node, and any modifying physiologic, metabolic, or pharmacologic factors. Because atrial flutter is most commonly manifested as flutter waves at 300 beats/min, 2 : 1 AV conduction, and a ventricular response rate of 150 beats/min, atrial flutter should be the first diagnosis considered in all patients with a regular tachycardia at 150 beats/min. It is important to remember, however, that pharmacologic agents can slow the rate of atrial flutter and decrease or increase AV nodal conduction, thereby altering the usual characteristics.
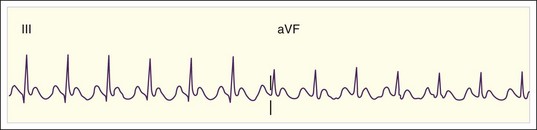
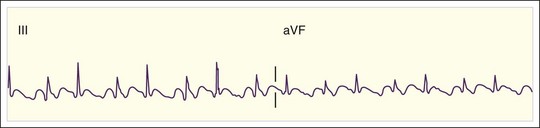
Cardioversion of atrial flutter is associated with thromboembolism, although the association is probably weaker than that with cardioversion of AF.10 For this reason, cardioversion of atrial flutter should probably not be undertaken in the ED unless the indication is an emergency because of cardiovascular instability, the combined duration of atrial flutter and AF is less than 48 hours, the patient has been adequately anticoagulated before arrival in the ED, or the patient is newly anticoagulated without evidence of atrial thrombi on transesophageal echocardiography. The decision to hospitalize patients with atrial flutter involves weighing issues similar to those described for AF.
Atrial Tachycardia
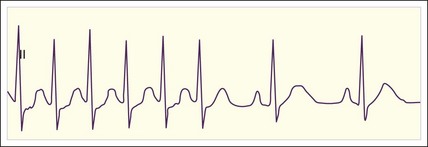
Regular AT is a relatively uncommon tachydysrhythmia. It may be due to cardiopulmonary disease with dilated atria and a reentrant mechanism; toxicity from digoxin, methylxanthines, or adrenergic agents with increased automaticity; or other causes. AT is distinguished from atrial flutter by separate identifiable P waves with an intervening isoelectric baseline and a rate lower than 200 beats/min (Fig. 59.6). Sometimes the P waves have an abnormal upward or rightward axis, or the onset of tachycardia is abrupt. These characteristics would distinguish the rhythm from sinus tachycardia. Finally, as discussed later, unlike reentrant atrial rhythms that involve the AV node, AT would not be expected to terminate with agents that slow AV nodal conduction. As with atrial flutter, decreasing AV nodal conduction dynamics might slow the ventricular response to the rhythm and facilitate the diagnosis. It would not generally terminate the rhythm because the origin of the tachycardia is contained entirely within the atria.
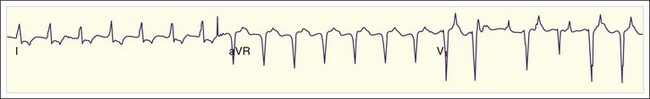
Reentrant Supraventricular Tachycardia
Mechanistically, the two major subtypes of reentrant SVT are distinguished by the route of the reentrant pathway. AVRT uses a macroreentrant circuit with atrial and ventricular segments and connecting segments that involve the AV node and an accessory conducting tract, or rarely, two accessory conducting tracts (see Fig. 59.2, B and C). The AV nodal segment usually serves as the necessary slow-conducting portion of the reentrant circuit.
Many patients have at least two discrete segments that can conduct the input wave of excitability from the right atrium into the AV node. The conducting properties of these inputs may differ, and frequently both a fast-conducting input or pathway and a relatively slow-conducting one are present. Usually, conduction to the AV node is via the fast pathway. AVNRT uses a small macroreentrant circuit consisting of the AV node, right atrial tissue, and connecting fast and slow pathway segments to form a full circle of conduction (see Fig. 59.2, A).
Within each subtype of reentrant SVT, the direction of the wave of excitation can vary. In AVRT, the wave of excitation most commonly proceeds down the AV node and normally through the His-Purkinje system, and it returns in a retrograde direction up the accessory tract to the atria to complete the circuit; this pathway is termed orthodromic conduction (see Fig. 59.2, B). Antidromic conduction occurs when the wave of excitation proceeds in the opposite direction, down the accessory pathway, and returns in retrograde fashion up the AV node (see Fig. 59.2, C). In this case, conduction down the His-Purkinje system and normal, nearly simultaneous excitation of the ventricles do not occur. Rather, the ventricles are activated from the terminus of the accessory tract, and the wave of excitation must propagate across the ventricular myocardium similar to excitation from a premature ventricular contraction (PVC). Consequently, it takes longer to excite the entire ventricular muscle mass, and the corresponding QRS wave is widened with antidromic conduction.
Reentrant SVTs are usually easily diagnosed from the ECG. The most common finding is a narrow QRS complex tachycardia at rate of 120 to 250 beats/min. The atria are activated in a retrograde direction from the AV node or an accessory tract. In AVRT, retrograde P waves that are inverted in V1 and the inferior leads may be observed just after the QRS complex (Fig. 59.7). In AVNRT, retrograde excitation of the atria is often simultaneous with ventricular excitation, and retrograde P waves are not observed because they are hidden or buried within the QRS complex (Fig. 59.8). In this case, P waves appear to be absent.
Reentrant SVT may be manifested as a wide QRS complex primarily for two reasons. Normal rapid conduction down the His-Purkinje system leads to nearly simultaneous excitation and depolarization of the ventricular myocardium and a narrow QRS complex on the ECG. If the left or right bundle of the Purkinje system fails to conduct, the QRS complex is widened and displays bundle branch block (BBB) morphology (see Fig. 59.2, F). This can occur as a result of intrinsic conduction system disease and chronic BBB, or the failure may be rate dependent and occur only during tachycardia. The other reason for a widened QRS complex is AVRT with antidromic conduction antegrade down the accessory pathway (see Fig. 59.2, C). This path does not use the His-Purkinje system, and a widened QRS complex without typical BBB morphology would be expected.
From an ED perspective, it is not usually important to distinguish AVRT from AVNRT. The acute treatment and prognosis for the two conditions are the same. Vagal maneuvers13 should be attempted or drugs administered to slow conduction and increase the refractory period through the AV nodal tissue so that the node is refractory to excitation and conduction when the reentrant wave approaches it. Agents such as adenosine and calcium channel blockers act directly on the nodal tissue, whereas other interventions, such as vagal maneuvers, digoxin, and beta-blockers, may alter vagal or adrenergic tone (Box 59.2). Adenosine may be the optimal agent because it is highly effective and safe with an ultrashort (9-second) half-life in serum. Rapid clearing, however, can be a disadvantage if the tachydysrhythmia is recurrent. In this case, the calcium channel blocker verapamil may be useful because it is equally effective for termination and may prevent recurrent episodes.14 Verapamil should not be used in infants, however, because of the risk for cardiovascular collapse.15
Preexcitation (Wolff-Parkinson-White) Syndrome
The diagnosis can be made while the patient is in normal sinus rhythm if the following characteristics are observed on the ECG: (1) shortened PR interval (<120 msec), (2) widened QRS complex beyond 120 msec, and (3) atypical initiation of the QRS complex with a delta-wave morphology (Fig. 59.9). Associated ST-T wave abnormalities are often present, with the T wave inverted with respect to the delta wave and QRS complex. These three diagnostic abnormalities are easily understood from an electrophysiologic perspective (see Fig. 59.2, D).
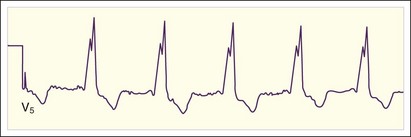
Patients with WPW syndrome are predisposed to AVRT, as well as to AF and atrial flutter, which often demonstrates a rapid ventricular response. The pathway of conduction of the AF wavelets from the atria to the ventricles depends on the relative refractory period of the accessory pathway with respect to the AV node. The slow-conducting cells of the AV node have a long refractory period, and the refractory period of the accessory conducting tissue is usually shorter. Consequently, the AV node remains refractory when the accessory pathway tissue is repolarized and ready for conduction. In WPW syndrome, AF usually conducts down the accessory pathway with a rapid ventricular response rate that is determined by the refractory period of the accessory conducting tissue (Fig. 59.10; also see Fig. 59.2, E). Similar to the situation with normal sinus rhythm, the ventricles are activated from the ectopic accessory pathway terminus, and an irregularly irregular tachycardia with a wide QRS complex results.
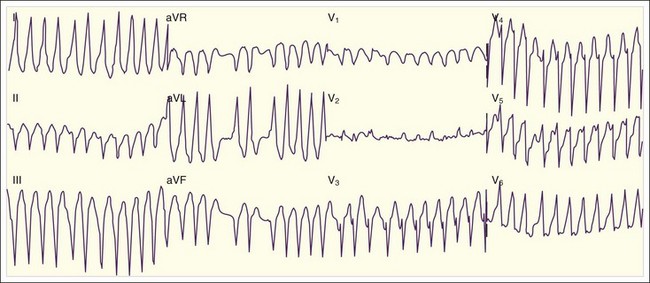
Perhaps the greatest concern regarding patients with WPW syndrome is their higher risk for sudden cardiac death. This risk is inversely associated with the minimum refractory period of the accessory conducting tissue.2 The mechanism of sudden cardiac death is thought to most commonly be AF with accessory tract conduction and a dangerously high ventricular response rate that causes ischemia and electrical instability and degenerates to VF.
It is important to remember that like the AV node and other cardiac tissue, accessory pathways have dynamic electrophysiologic properties that may vary with the hormonal, physiologic, or pharmacologic milieu. However, in this respect, accessory pathway tissue behaves more like myocardium and less like AV nodal tissue, which is highly sensitive to vagal stimulation. Depending on the current milieu, conduction in many patients with WPW syndrome may variably occur primarily down the accessory pathway or the AV node in normal sinus rhythm, and this variation even occasionally occurs in AF (Fig. 59.11).
The preferred pharmacologic agents for patients with WPW syndrome and AF with a rapid ventricular response are the class IA antidysrhythmic agent procainamide16 and, possibly, class III medications such as amiodarone and sotalol. These antidysrhythmics are used to decrease the ventricular response rate, and they may also terminate AF. Nevertheless, they have multiple potential dangers and must be used with caution. All these antidysrhythmic agents can decrease blood pressure. When administered intravenously, amiodarone blocks AV nodal conduction, so the concerns already described with other AV nodal blocking agents may be applicable.17 The safest and most effective treatment for patients with WPW syndrome and AF with a rapid ventricular response is synchronized DCCV. The EP should consider using DCCV with sedation before administering antidysrhythmics to patients with a ventricular response greater than 150 beats/min or borderline low blood pressure.
Ventricular Tachycardias
Premature Ventricular Contractions and Nonsustained Ventricular Tachycardia
• Cardiac ischemia, which may be due to local coronary artery disease or systemic hypoxia
Chronic ventricular ectopy does not generally require emergency treatment, and suppression of chronic ventricular ectopy after MI does not lower the mortality.18 Patients with chronic PVCs may be at variably higher risk for sudden death, depending on other clinical factors, but even so, it is often unclear whether the increased risk is directly attributable to the ectopy or to the underlying cardiac disease. EPs must commonly manage new ventricular ectopy in association with acute MI or other severe systemic disease. The greatest concern is that PVCs may induce sustained VT or VF. Theoretically, this could occur if the ectopic ventricular depolarization occurs during the vulnerable period or upstroke of the T wave of the preceding beat (R-on-T phenomenon).
The risk for VF and sudden death is elevated during acute MI, but this risk is not decreased with routine suppressive class I antidysrhythmic therapy.19 Conversely, beta-blockade with metoprolol may not affect the occurrence of PVCs or NSVT but does lower the rate of VF and death.20 Suppression of new ventricular ectopy should be considered if the PVCs or tachydysrhythmias are symptomatic and contributing to hemodynamic instability or the patient has already experienced sustained VT or VF. The first priority is to search for reversible causes of ventricular ectopy, such as hypoxia; potassium, magnesium, or other electrolyte imbalance; or drug toxicity. After reversible causes are addressed, the most commonly used agents include beta-blockers, amiodarone, lidocaine, and procainamide. The class I effect of myocardial sodium channel blockade may be particularly prominent for lidocaine in an ischemic and acidotic local cellular environment. Evidence suggests that amiodarone may be effective when lidocaine fails.21 The antidysrhythmics should be given as a loading dose followed by a sustained infusion, and the patient should be monitored closely for adverse effects and admitted to a cardiac care unit.
Monomorphic Ventricular Tachycardia
VT is considered sustained if it is continuous for at least 30 seconds. If the QRS complex has primarily a single morphology, the VT is monomorphic, whereas if the QRS complex varies, the VT is polymorphic. Monomorphic VT is an uncommon condition that underlies the chief complaint in approximately 1 in every 10,000 ED visits. The mechanism in the majority of cases is reentry within the ventricular myocardium, where the slow-conducting segment of the reentrant loop is associated with a scar from a previous MI (see Fig. 59.1, B). The scar tissue serves to enable and fix the location of the VT circuit within the myocardium. Other causes of monomorphic VT that are less commonly encountered in the ED are dilated cardiomyopathy, hypertrophic cardiomyopathy, previous surgical repair of congenital heart disease with a myocardial scar, arrhythmogenic right ventricular dysplasia, right ventricular outflow tract VT, and fascicular tachycardia.
Patients with stable VT may initially be treated with medical or electrical therapy. The primary principle of treatment with class I or III antidysrhythmic agents is to prolong the refractory period of the ventricular myocardium so that the cells remain refractory when the reentrant wave of excitation returns. Amiodarone is recommended by the American Heart Association, and procainamide and sotalol can be used as alternatives when cardiac output seems to be clinically preserved. Lidocaine is no longer recommended for this purpose. Unfortunately, neither amiodarone nor lidocaine immediately increases the refractory period of normal myocardium after intravenous administration. Both are unlikely to terminate VT within 20 minutes of treatment.22 However, amiodarone may become more effective in both terminating and preventing VT over the ensuing hours after administration.21 Procainamide and racemic sotalol do prolong the refractory period soon after administration and are more likely to terminate VT. However, procainamide blocks cardiac sodium channels, and the l isomer of sotalol is a beta-blocker; thus, both agents can cause a decrease in cardiac contractility and subsequent hemodynamic instability. Intravenous sotalol is not currently available in the United States. Other agents that have been used in this situation are pure beta-blockers and magnesium. Neither has been proved to be effective, however, and pure beta-blockers may cause hypotension.
Wide QRS Complex Tachycardia: Differentiating Supraventricular From Ventricular Tachycardia
Wide QRS complex tachycardia may be due to an SVT with aberrant His-Purkinje conduction or antegrade conduction down an accessory pathway or be due to VT. Diagnosis of the underlying mechanism is important for a number of reasons. The optimal short- and long-term treatment and prognosis may vary considerably, depending on the mechanism involved. The history, physical examination, and ECG findings can be used to make the diagnosis (Box 59.3). In the ED, the pretest probability favors VT by a ratio of 2 : 1 to 3 : 1.
Box 59.3 Diagnosing Regular Wide QRS Complex Tachycardia
Predictors Suggestive of Ventricular Tachycardia
Electrocardiographic Findings
Frontal axis −90 to −180 degrees
QRS interval > 140 msec (right bundle branch block) or >160 msec (left bundle branch block) or RS interval > 100 msec
Positive or negative concordance across the precordial leads
Wide QRS morphology grossly different from the QRS morphology in sinus rhythm
Atrioventricular dissociation (including fusion or capture beats)
No single historical factor has been found to be both sensitive and specific for diagnosing VT. Nevertheless, age older than 35 years and male sex are sensitive for VT, and a previous history of MI or coronary artery bypass graft surgery, recent angina, or CHF is a specific predictor of VT.23 The primary underlying theme for all these predictors is a greater likelihood of coronary artery disease and previous infarction with scar formation. In fact, the strongest single historical predictor of VT is a history of MI, which has a positive likelihood ratio of 13 : 1 to 20 : 1 and a negative likelihood ratio of 0.36.23,24
A number of approaches may be used to diagnose wide QRS complex tachycardia from the physical examination. Perhaps the first and most important principle to remember is that apparent hemodynamic stability does not rule out VT. AV dissociation is present about half the time in patients with VT.25 When it is present, the atria are contracting independently of the ventricles, so the atria may either contract during ventricular diastole and assist cardiac output or contract against closed valves during ventricular systole with a large retrograde venous pulsation. The EP can assess for beat-to-beat variation of the first heart sound or systolic blood pressure or for large cannon jugular venous waves. Unfortunately, even under controlled experimental conditions, none of these findings are both sensitive and specific.26 In clinical conditions in which about half the patients have some form of retrograde ventricular-atrial conduction, these tests for physical evidence of AV dissociation can approach a sensitivity for VT of only approximately 50% at best.
A similar principle of brief increased refractoriness of the AV node underlies the use of adenosine to diagnose and treat wide QRS complex tachycardia. The combined response to a 12-mg bolus of adenosine—either termination of the tachycardia or transient ventricular slowing—has a sensitivity of 90% and a specificity of 98% for an underlying SVT.27 Considered in reverse fashion, “nonresponse” to a 12-mg bolus of adenosine would have a positive likelihood ratio of about 9 and a negative likelihood ratio of 0.03 for VT. This test is useful for diagnosis, but is it safe?
The primary safety concern is the administration of adenosine to patients with atrial flutter and antegrade accessory pathway conduction, which might cause subsequent transient acceleration of this conduction and the ventricular response. This and other destabilizing scenarios have been reported after the administration of adenosine to patients with wide QRS complex tachycardia secondary to SVT and VT. Adenosine should never be given to patients with a rapid, irregularly irregular wide QRS complex tachycardia because it may actually be AF with accessory pathway conduction, and acceleration of the ventricular response has been reported in this situation as well. Nonetheless, the adverse affects previously described after adenosine administration to patients with regular wide QRS complex tachycardia are rare. Multiple consecutive case series have found no greater rate of adverse effects with adenosine than with other antidysrhythmic medications.27,28 Adenosine should be used as a diagnostic and therapeutic agent for regular wide QRS complex tachycardia when the history, physical examination, and ECG findings suggest a supraventricular origin. The EP should ensure that the adenosine is used properly—administered as a 12-mg rapid bolus dose followed immediately by a normal saline bolus flush, with a cardioverter-defibrillator immediately available in the event of destabilization.
A number of ECG findings can be used to diagnose wide QRS complex tachycardia. Possible differentiating characteristics include the heart rate, frontal axis of the QRS deflection, concordance across the precordial leads, QRS duration, morphology of the QRS complex, AV dissociation, and capture or fusion beats. On average, wide QRS complex tachycardia secondary to SVT has a faster heart rate than that secondary to VT.25 This difference has been slightly augmented in the recent past with the use of ICDs. A patient with VT may have already received an ICD, which is preferentially programmed to treat rapid VT, usually greater than 160 to 180 beats/min. It cannot be programmed to treat slower VT because of the risk for inappropriate pacing or shocks for sinus tachycardia. Nevertheless, there is still too much overlap in the heart rate of patients with SVT and VT to use it as a firm differentiating factor. Regardless of the underlying rhythm, higher heart rates are associated with compromised cardiac output because of inadequate diastolic filling and greater electrical instability. Immediate DCCV should be considered for patients with rapid wide QRS tachycardia, regardless of the presumed etiology.
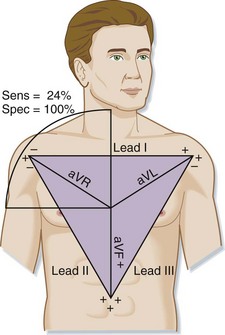
Because of the common ectopic left ventricular origin of the rhythm in VT, abnormal upward and rightward propagation of the excitation wave may be seen. In fact, the QRS axis may point to any of the four quadrants in VT, but an upward and rightward QRS axis is highly unusual for SVT. This finding, defined by a negative QRS deflection in leads I and aVF, is insensitive (24%) but highly specific (100%) for VT (Fig. 59.12).29 An abnormal QRS vector that points toward or away from all of the precordial leads simultaneously is also indicative of VT. A QRS deflection that is primarily positive in all of the precordial leads is defined as positive concordance. The finding of primarily negative QRS deflections in all precordial leads is defined as negative concordance. Each of these findings is also insensitive (10%) but specific (85%) for VT.29
On average, the duration of initial depolarization as recorded by the QRS wave is longer with VT than with wide QRS complex SVT. A QRS duration greater than 140 msec with a right BBB (RBBB) pattern or a QRS duration greater than 160 msec with a left BBB (LBBB) pattern is insensitive but more than 90% specific for VT.29 Brugada et al.30 modified this concept and found that an RS interval greater than 100 msec in the precordial leads has a sensitivity of 66% and a specificity of 98% for VT (Fig. 59.13). Unfortunately, the high specificity of these findings is not applicable if the patient has received a medication that widens the QRS complex, such as a class I antidysrhythmic.
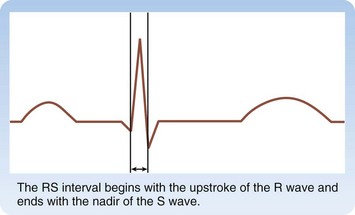
Fig. 59.13 How to measure the RS interval (double arrow).
The RS interval begins with the upstroke of the R wave and ends with the nadir of the S wave.
QRS morphology may be helpful, particularly if an old ECG is available. QRS morphology with tachycardia similar to a previous BBB morphology noted in sinus rhythm may suggest, but does not prove SVT.31 The distinction of RBBB versus LBBB is itself not helpful, but various morphologic variants within these two categories can at least be theoretically helpful.24,25,30
When present, the most definitive evidence of VT is AV dissociation. In SVT an atrial contraction is associated with each ventricular beat. AV dissociation is defined by atrial activity that is separate and independent of ventricular contractions (Fig. 59.14). AV dissociation is present in about half of VT episodes, and it can be seen on the ECG in half of these cases, or about one quarter of all VT episodes. Thus, AV dissociation has a sensitivity of about 25% and specificity approaching 100% for VT. Dissociated P waves should be sought in the V1 rhythm strip, where they are usually most easily seen. The key is to find two candidate P waves. One can then look an equal distance on each side of these two waves for a third deflection. If three consecutive deflections are found that “march out,” they are probably P waves; such identification can be confirmed by marching out more on the ECG. Fusion beats represent fusion of supraventricular excitation mediated by the His-Purkinje system and local ventricular excitation. Capture beats signify complete ventricular depolarization by a supraventricular signal and usually have a narrow QRS complex. Both these phenomena also suggest AV dissociation and VT (Figs. 59.15 and 59.16).
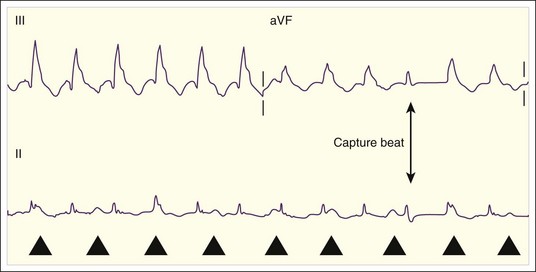
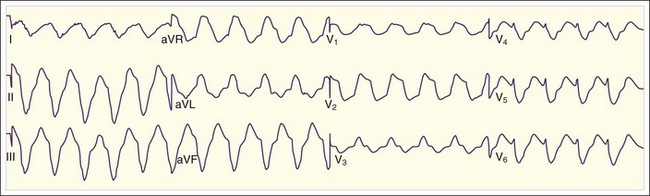
Severe hyperkalemia can also cause wide QRS complex tachycardia. Clues to this diagnosis include a history of renal failure or other medical causes of hyperkalemia and an ECG with very wide and bizarre QRS complexes (Fig. 59.17). Once the condition is diagnosed, it should be confirmed with a serum potassium measurement and treated with intravenous calcium, insulin, glucose, and bicarbonate. Patients who overdose with cyclic antidepressants or other sodium channel blocking agents may have a wide QRS complex tachycardia. An important clue to this diagnosis on the ECG is right axis deviation of the terminal 30 msec of the QRS complex. Patients with suspected overdose should be treated with a continuous intravenous sodium bicarbonate infusion. Patients with acute respiratory distress secondary to pulmonary disease and with an underlying BBB may appear to have a primary tachydysrhythmic condition. Addressing the airway, with intubation if necessary, may slow the heart rate, expose the P waves, and clarify the rhythm.
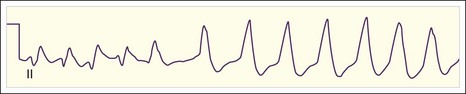
Polymorphic Ventricular Tachycardia
Polymorphic VT is defined as VT with varying QRS wave morphology (Fig. 59.18). A specific type of polymorphic VT characterized by sinusoidal variation of the QRS deflection occurs in patients with a long QT interval during sinus rhythm (Fig. 59.19). This tachydysrhythmia is termed torsades de pointes (TdP), or “twisting of the points” (Fig. 59.20). There are a number of causes of polymorphic VT and TdP, which itself is an important cause of sudden cardiac death. Fortunately, this tachydysrhythmia is rare.
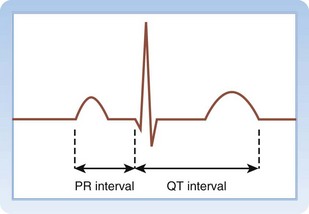

Cellular repolarization and the QT interval vary as a function of the preceding heart rate. Thus, the measured QT interval is corrected for the heart rate, most commonly with the Bazett formula. After puberty, on average, women have a slightly longer QT interval than men do (Table 59.2), and a variety of medications can prolong the QT interval. Patients with a prolonged QT interval are asymptomatic unless TdP or another tachydysrhythmia develops, which may be manifested as syncope or sudden cardiac death.
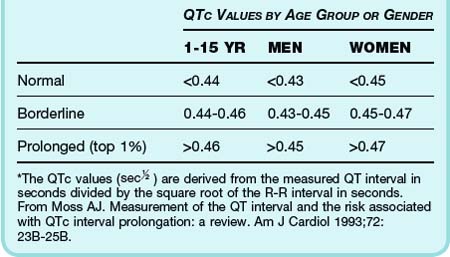
Congenital long QT syndrome with TdP is due to a growing number of known ion channelopathies (Table 59.3). The most common defects lead to deficient potassium channel conduction and augmentation of sodium channel conduction, which cause a prolonged QT interval. Other genetic abnormalities associated with polymorphic VT and sudden cardiac death are rare calcium channel defects, ion channel defects leading to a shortened QT interval, and Brugada syndrome, which is attributed primarily to premature inactivation of sodium channel conduction. As with hemoglobinopathies, there appear to be a wide variety of defects associated with myocardial conducting channels and their supporting proteins, and they are associated with varying risk for tachydysrhythmias and mortality.
See Table 59.3, Known Genetic Tachydysrhythmias, online at www.expertconsult.com
The possibility of congenital long QT interval should be considered in all patients with palpitations, presyncope, syncope, or seizures. What were the circumstances of the event, and has the patient previously had similar events? Did the onset of symptoms occur during rest, sleep, or activity or after the patient was startled (see Table 59.3)? How long did the symptoms last, and were there any associated symptoms of chest pain or shortness of breath? Many of these conditions exhibit autosomal inheritance, although new mutations are common. The family history should be assessed for recurrent syncope and sudden death.
In a patient with sinus rhythm, the physical findings are usually normal. However, analysis of the QT interval and T-wave morphology on the ECG is critical. Different channelopathies tend to be associated with specific T-wave abnormalities. Assess for the morphology of Brugada syndrome, an RBBB pattern in the precordial chest leads with associated downsloping ST-segment elevation (Fig. 59.21). Search for evidence of other cardiologic causes of syncope, including preexcitation and WPW syndrome, ventricular hypertrophy, aortic valvular disease, and other tachydysrhythmias or bradydysrhythmias. Serum electrolytes, including potassium, calcium, and magnesium, should be measured, and treatment should be administered as needed.
Acquired long QT syndrome is due to toxicity from medications that either prolong the QT interval or block the metabolism of QT-prolonging agents (Box 59.4). Medications that prolong the QT interval act by altering ion channel flow, such as by disrupting the outward potassium repolarizing current. Women are more prone to acquired long QT syndrome, perhaps in part because of their longer baseline QT interval. Acute symptoms are similar to those in the congenital form of disease.
See Box 59.4, Causes of Acquired Long QT Syndrome, online at www.expertconsult.com
1 Eisenberg MS, Mengert TJ. Cardiac resuscitation. N Engl J Med. 2001;344:1304–1313.
2 Klein GJ, Bashore TM, Sellers TD, et al. Ventricular fibrillation in the Wolff-Parkinson-White syndrome. N Engl J Med. 1979;301:1080–1085.
3 Schreck DM, Rivera AR, Tricarico VJ. Emergency management of atrial fibrillation and flutter: intravenous diltiazem versus intravenous digoxin. Ann Emerg Med. 1997;29:135–140.
4 Demircan C, Cikriklar HI, Engindeniz Z, et al. Comparison of the effectiveness of intravenous diltiazem and metoprolol in the management of rapid ventricular rate in atrial fibrillation. Emerg Med J. 2005;22:411–414.
5 Chiladakis JA, Stathopoulos C, Davlouros P, et al. Intravenous magnesium sulfate versus diltiazem in paroxysmal atrial fibrillation. Int J Cardiol. 2001;79:287–291.
6 Volgman AS, Carberry PA, Stambler B, et al. Conversion efficacy and safety of intravenous ibutilide compared with intravenous procainamide in patients with atrial flutter or fibrillation. J Am Coll Cardiol. 1998;31:1414–1419.
7 Martinez-Marcos FJ, Garcia-Garmendia JL, Ortega-Carpio A, et al. Comparison of intravenous flecainide, propafenone, and amiodarone for conversion of acute atrial fibrillation to sinus rhythm. Am J Cardiol. 2000;86:950–953.
8 Kalus JS, Spencer AP, Tsikouris JP, et al. Impact of prophylactic i.v. magnesium on the efficacy of ibutilide for conversion of atrial fibrillation or flutter. Am J Health Syst Pharm. 2003;60:2308–2312.
9 Scheuermeyer FX, Grafstein E, Stenstrom R, et al. Thirty-day outcomes of emergency department patients undergoing electrical cardioversion for atrial fibrillation or flutter. Acad Emerg Med. 2010;17:408–415.
10 Scholten MF, Thornton AS, Mekel JM, et al. Anticoagulation in atrial fibrillation and flutter. Europace. 2005;7:492–499.
11 Barrett TW, Martin AR, Storrow AB, et al. A clinical prediction model to estimate risk for 30-day adverse events in emergency department patients with symptomatic atrial fibrillation. Ann Emerg Med. 2011;57:1–12.
12 McCord J, Borzak S. Multifocal atrial tachycardia. Chest. 1998;113:203–209.
13 Lim SH, Anantharaman V, Teo WS, et al. Comparison of treatment of supraventricular tachycardia by Valsalva maneuver and carotid sinus massage. Ann Emerg Med. 1998;31:30–35.
14 DiMarco JP, Miles W, Akhtar M, et al. Adenosine for paroxysmal supraventricular tachycardia: dose ranging and comparison with verapamil. Assessment in placebo-controlled, multicenter trials. Ann Intern Med. 1990;113:104–110.
15 Kirk CR, Gibbs JL, Thomas R, et al. Cardiovascular collapse after verapamil in supraventricular tachycardia. Arch Dis Child. 1987;62:1265–1266.
16 Boahene KA, Klein GJ, Yee R, et al. Termination of acute atrial fibrillation in the Wolff-Parkinson-White syndrome by procainamide and propafenone: importance of atrial fibrillatory cycle length. J Am Coll Cardiol. 1990;16:1408–1414.
17 Boriani G, Biffi M, Frabetti L, et al. Ventricular fibrillation after intravenous amiodarone in Wolff-Parkinson-White syndrome with atrial fibrillation. Am Heart J. 1996;131:1214–1216.
18 Echt DS, Liebson PR, Mitchell LB, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324:781–788.
19 Teo KK, Yusuf S, Furberg CD. Effects of prophylactic antiarrhythmic drug therapy in acute myocardial infarction: an overview of results from randomized controlled trials. JAMA. 1993;270:1589–1595.
20 Ryden L, Ariniego R, Arnman K, et al. A double-blind trial of metoprolol in acute myocardial infarction: effects on ventricular tachyarrhythmias. N Engl J Med. 1983;308:614–618.
21 Scheinman MM, Levine JH, Cannom DS, et al. Dose-ranging study of intravenous amiodarone in patients with life-threatening ventricular tachyarrhythmias. Circulation. 1995;92:3264–3272.
22 Marill KA, deSouza IS, Nishijima DK, et al. Amiodarone is poorly effective for the acute termination of ventricular tachycardia. Ann Emerg Med. 2006;47:217–224.
23 Baerman JM, Morady F, DiCario LA, et al. Differentiation of ventricular tachycardia from supraventricular tachycardia with aberration: value of the clinical history. Ann Emerg Med. 1987;16:40–43.
24 Griffith MJ, de Belder MA, Linker NJ, et al. Multivariate analysis to simplify the differential diagnosis of broad complex tachycardia. Br Heart J. 1991;66:166–174.
25 Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med. 1978;64:27–33.
26 Garratt CJ, Griffith MJ, Young G, et al. Value of physical signs in the diagnosis of ventricular tachycardia. Circulation. 1994;90:3103–3107.
27 Marill KA, Wolfram S, deSouza IS, et al. Adenosine for wide-complex tachycardia: efficacy and safety. Crit Care Med. 2009;37:2512–2518.
28 Herbert ME, Votey SR. Adenosine in wide-complex tachycardia. Ann Emerg Med. 1997;29:172–174.
29 Akhtar M, Shenasa M, Jazayeri M, et al. Wide QRS complex tachycardia. Ann Intern Med. 1988;109:905–912.
30 Brugada P, Brugada J, Mont L, et al. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation. 1991;83:1649–1659.
31 Halperin BD, Kron J, Cutler JE, et al. Misdiagnosing ventricular tachycardia in patients with underlying conduction disease and similar sinus and tachycardia morphologies. West J Med. 1990;152:677–682.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
59 Tachydysrhythmias
• Tachydysrhythmia may be due to intrinsic cardiologic disease or external stimulation.
• Noncardiologic causes, such as hypoxia, inadequate perfusion, metabolic derangements, or toxicity from medications or other agents, should always be considered.
• The higher the tachycardic heart rate, the greater the likelihood of instability.
• Patients with ventricular tachycardia may not necessarily appear unstable.
• If there is uncertainty, wide QRS complex tachycardia should be assumed to be ventricular in origin.
• The most effective treatment of ventricular tachycardia is electrical cardioversion. However, antidysrhythmic medications may also be necessary to prevent recurrence.
• All ostensibly healthy young patients with syncope should be assessed for valvular disease, left ventricular hypertrophy, Wolff-Parkinson-White syndrome, abnormal QT interval or T-wave morphology, and Brugada syndrome.
Epidemiology
Heart rhythms with rapid rates, or tachydysrhythmias, have a range of causes and associated incidences, morbidities, and mortality. Atrial fibrillation (AF) occurs in up to 5% of people older than 65 years. Conversely, sustained ventricular tachycardia (VT) is rare and accounts for less than 0.1% of emergency department (ED) visits. Morbidity ranges from temporary lightheadedness to syncope and deterioration, ventricular fibrillation (VF), or embolization and subsequent permanent disability as a result of ischemic stroke. Mortality from atrial tachydysrhythmias is generally low. Mortality from VT is approximately 5%, and that from VF ranges from 80% to greater than 90%, depending on the clinical circumstances.1 A particular exception to the low mortality associated with atrial tachydysrhythmias is AF or flutter with rapid ventricular excitation via a bypass tract, such as in Wolff-Parkinson-White (WPW) syndrome.2
Pathophysiology and Mechanisms
Reentry refers to a microscopic or macroscopic circular pattern of myocardial depolarization. A wave of excitation passes around the circuit and depolarizes the cells until it arrives at the beginning, and the process repeats itself. Reentry can occur if the circuit is sufficiently long or there is a slow-conducting portion that allows enough time for cells to have recovered from their refractory period before the wave of excitation returns (Fig. 59.1). The reentrant rhythm becomes the cardiac pacemaker if it is sufficiently fast to outpace the sinus node.
Presenting Signs and Symptoms
Treatment
The primary emergency therapies for tachydysrhythmias include medications to slow electrical conduction or to increase the cellular refractory period in the AV node or other groups of cardiac cells (Table 59.1). The goal of therapy is to terminate and suppress the abnormal rhythm or to slow the response in the remainder of the heart. Electrical therapy, including synchronized cardioversion and defibrillation, is used in potentially or frankly unstable patients. Electrical therapy has the advantages of high efficacy and safety, but shocks are painful and do not prevent recurrence of the tachydysrhythmia. Patients with higher heart rates, regardless of the underlying rhythm, have compromised diastolic filling and cardiac output. These patients should be treated more aggressively with early electrical therapy. The emergency physician (EP) should understand and use antidysrhythmic medications but should minimize the use of multiple agents in a single patient. Use of multiple drugs increases the likelihood of drug interactions and compromised cardiac output.
See Table 59.1, Select Tachydysrhythmia Medications, online at www.expertconsult.com
Specific Tachycardias
Primary tachycardiac conditions can be divided most simply into those of supraventricular and ventricular origin (Box 59.1; Fig. 59.2; see also Fig. 59.1). Supraventricular tachycardia (SVT) must involve tissue above the ventricles but may also involve ventricular tissue in a tachycardiac circuit, whereas VT involves only ventricular tissue below the AV node. The remainder of the chapter deals with the diagnosis and treatment of specific tachycardiac conditions.
Supraventricular Tachycardias
Irregular Supraventricular Tachycardias
Atrial Fibrillation
The clinician can usually diagnose AF at the bedside by feeling the pulse, listening to the heart sounds, and observing a single-lead cardiac monitor. The ECG may show fine or coarse irregular undulations of the baseline and an absence of regular P waves. The QRS complex is usually normal; however, if the ventricular response is rapid, the QRS complex may be transiently widened because of intermittent aberrant conduction through the His-Purkinje system (Fig. 59.3). This finding, termed the Ashman phenomenon, can occur when areas of the His-Purkinje tissue have a longer refractory period than the AV node does. Such bursts of wide QRS complex tachycardia can be confused with nonsustained VT, but on close inspection the rhythm remains irregularly irregular.
The most common treatment issue for patients with AF in the emergency setting is control of the ventricular rate. Depending on the function of the patient’s AV node, the ventricular rate may range up to approximately 150 beats/min in adults. Ventricular rates this fast allow insufficient time for diastolic filling of the heart. The rate can be slowed with a variety of agents that act to lengthen the refractory period of the AV nodal tissue, such as calcium channel blockers,3 beta-blockers,4 magnesium,5 digoxin, and amiodarone. Digoxin is less useful in the emergency setting because it takes a few hours to work, but the other agents must be used carefully because they can decrease blood pressure to varying degrees. The calcium channel blocker diltiazem usually provides excellent rate control with minimal loss of blood pressure. Beta-blockers may cause a greater loss of blood pressure and must be used with caution in patients with CHF or pulmonary disease. There is no indication for the administration of adenosine in patients with AF. The diagnosis should be made on the basis of findings from the history, physical examination, and ECG. Furthermore, adenosine, which only transiently blocks AV nodal conduction, would have no lasting therapeutic benefit.
Cardioversion can also be accomplished chemically when circumstances allow. Agents that slow conduction or increase the refractory period of atrial tissue are used to break the reentrant microcircuits within the atria. Vaughn-Williams class III and class I medications that can accomplish this goal include ibutilide, amiodarone, procainamide, propafenone, and flecainide.6,7 These agents can terminate AF alone and increase the likelihood of cardioversion of AF with subsequent DCCV. However, their use entails some risk. There is an approximately 2% to 5% chance of causing torsades de pointes (TdP) after the infusion of ibutilide, amiodarone, and procainamide because these agents variably increase the duration of the depolarized phase and the associated QT interval in the ventricles, as well as the atria. The newly developed agent vernakalant may be less likely to cause TdP because of its action in blocking the ultrarapid potassium channels located primarily in the atria. Hypokalemia and hypomagnesemia raise the risk for TdP. Pretreatment with magnesium may be protective in some circumstances, even in patients with normal serum electrolyte levels.8 Propafenone and flecainide can slow conduction, exacerbate heart failure, and cause ventricular dysrhythmias. Cardioversion also carries a risk for embolic stroke.
In the acute setting, clot may form within 48 hours after the onset of AF. Cardioversion of AF may cause a new clot to embolize into the systemic circulation. If the patient can clearly discern when he or she is in AF, symptoms have begun within the preceding 1 to 2 days, and the patient is not at high risk for stroke, it is generally safe to perform cardioversion without anticoagulation.9 However, if there is any uncertainty about the time of onset of the tachydysrhythmia or if it occurred more than 48 hours ago, the patient should not undergo immediate cardioversion. Instead, the cardiology service should be consulted and transesophageal echocardiography performed to search for atrial clot formation, or cardioversion should be deferred until adequate anticoagulation has been established.10 The only exception would be cardiovascular instability secondary to AF, which requires emergency DCCV. Continuation of anticoagulation should also be considered for 4 weeks after cardioversion because there may be some delay in return of normal atrial contraction.
The disposition of patients with AF depends on a number of factors, including age, comorbid illnesses and social and outpatient medical support, adequacy of control of the ventricular rate, plans for cardioversion, and the state of anticoagulation.11 If the patient requires ongoing adjustment of medications to control a rapid ventricular response, attempted cardioversion is planned, or new anticoagulation is initiated, hospitalization should be considered. The EP should make this decision in concert with the patient and the consulting cardiologist.
Multifocal Atrial Tachycardia
Multifocal AT (MFAT) is a relatively uncommon tachydysrhythmia that primarily affects older patients with chronic lung diseases such as chronic obstructive pulmonary disease, and it appears to be related to the administration of methylxanthine.12 Its mechanism is uncertain, but MFAT is thought to occur as a result of DADs in atrial tissue triggering ectopic beats. The diagnosis is made on the basis of the following ECG criteria: at least three consecutive P waves with different morphologies, variable P-P and PR intervals, and a heart rate greater than 100 beats/min. Treatment of MFAT is centered on treating the underlying pulmonary disease and possible hypoxia. Antidysrhythmic agents are poorly effective in slowing the atrial rate or the ventricular response or in terminating MFAT. Nevertheless, a carefully administered trial of a calcium channel blocker or amiodarone is reasonable. Generally, beta-blockers should be avoided in patients with any evidence of bronchospasm. Magnesium therapy, particularly in combination with potassium replacement, may slow the rate or terminate the tachydysrhythmia. Patients usually require admission to treat the pulmonary disease.
Regular Supraventricular Tachycardias
Atrial Flutter
Atrial flutter can occur as a result of a variety of cardiopulmonary and metabolic derangements, often in association with atrial dilation. Patients with atrial flutter also tend to experience AF, but atrial flutter is less common overall. The mechanism of atrial flutter is a macroreentrant circuit in the right atrium (see Fig. 59.1, A). The most common variant of atrial flutter is termed typical atrial flutter and involves a counterclockwise wave of roughly circular excitation when viewed from a position facing the front of the patient. The flutter pathway is constrained in the inferior portion of the circular loop by anatomic structures, and it travels between the tricuspid annulus anteriorly and the inferior vena cava and coronary sinus posteriorly. This segment of the pathway, the isthmus, contains the relatively slower-conducting tissue in the reentrant loop. Atypical atrial flutter most commonly travels over the same pathway, but in the opposite direction.
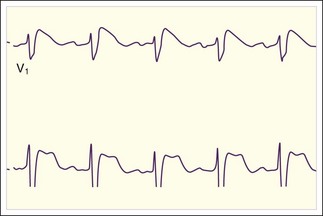
The rate of atrial flutter waves ranges from 250 to 350 beats/min. The diagnosis is made from the ECG tracing, which demonstrates typical sawtooth-shaped flutter waves at the appropriate rate, generally most prominent in the inferior limb leads (II, III, and aVF) and lead V1. The flutter waves tend to be identical and to recur regularly, and there is no isoelectric or flat ECG segment between the waves (Figs. 59.4 and 59.5). As in AF, the refractory period of the AV node usually controls the ventricular response to atrial flutter. The most common response is 2 : 1 AV conduction; however, higher, lower, or variable ratios of conduction are also possible. The ratio of conduction depends on the atrial flutter rate, the health of the AV node, and any modifying physiologic, metabolic, or pharmacologic factors. Because atrial flutter is most commonly manifested as flutter waves at 300 beats/min, 2 : 1 AV conduction, and a ventricular response rate of 150 beats/min, atrial flutter should be the first diagnosis considered in all patients with a regular tachycardia at 150 beats/min. It is important to remember, however, that pharmacologic agents can slow the rate of atrial flutter and decrease or increase AV nodal conduction, thereby altering the usual characteristics.
Cardioversion of atrial flutter is associated with thromboembolism, although the association is probably weaker than that with cardioversion of AF.10 For this reason, cardioversion of atrial flutter should probably not be undertaken in the ED unless the indication is an emergency because of cardiovascular instability, the combined duration of atrial flutter and AF is less than 48 hours, the patient has been adequately anticoagulated before arrival in the ED, or the patient is newly anticoagulated without evidence of atrial thrombi on transesophageal echocardiography. The decision to hospitalize patients with atrial flutter involves weighing issues similar to those described for AF.
Atrial Tachycardia
Regular AT is a relatively uncommon tachydysrhythmia. It may be due to cardiopulmonary disease with dilated atria and a reentrant mechanism; toxicity from digoxin, methylxanthines, or adrenergic agents with increased automaticity; or other causes. AT is distinguished from atrial flutter by separate identifiable P waves with an intervening isoelectric baseline and a rate lower than 200 beats/min (Fig. 59.6). Sometimes the P waves have an abnormal upward or rightward axis, or the onset of tachycardia is abrupt. These characteristics would distinguish the rhythm from sinus tachycardia. Finally, as discussed later, unlike reentrant atrial rhythms that involve the AV node, AT would not be expected to terminate with agents that slow AV nodal conduction. As with atrial flutter, decreasing AV nodal conduction dynamics might slow the ventricular response to the rhythm and facilitate the diagnosis. It would not generally terminate the rhythm because the origin of the tachycardia is contained entirely within the atria.
