Tablets and compaction
Göran Alderborn
Chapter contents
Technical problems during tableting
Tablet production via granulation
Tablet production by direct compaction
Sublingual tablets and buccal tablets
Prolonged-release and pulsatile-release tablets
Uniformity of content of active ingredient
Fundamental aspects of the compression of powders
Mechanisms of compression of particles
Evaluation of compression behaviour
Fundamental aspects of the compaction of powders
Compactability of powders and the strength of tablets
Post-compaction tablet strength changes
Relationships between material properties and tablet strength
Key points
• Tablets of different types represent collectively the dominant type of dosage form.
• Tablets are used for oral administration for both systemic and local drug treatment.
• Several categories of tablets exist that are used in different ways, e.g. swallowed whole or retained in the mouth during the release of the drug.
• Tablets are normally formed by powder compression, i.e. the forcing of particles into close proximity by the application of mechanical force.
• Besides the active ingredient, tablets normally consist of a series of excipients that are included to control biopharmaceutical and other quality attributes, as well as to aid the manufacturing of the tablet.
• The release of the active pharmaceutical ingredient is a key product attribute and can be controlled by the formulation to achieve immediate-release, delayed-release or prolonged- release of the drug.
• In the manufacturing of tablets, a series of unit operations are normally used, including mixing and granulation of active ingredient(s) and excipients.
• In the manufacturing of tablets, a number of technical problems can arise, such as high weight and dose variation, low mechanical strength, capping of the tablets, adhesion and high friction.
• The success of a tableting operation is related to the properties of the powder intended to be formed into tablets and also to the design and conditions of the press and the tooling.
• Important tests of quality attributes of tablets include tablet disintegration and dissolution, tablet friability and tablet fracture resistance.
Introduction
The oral route is the most common way of administering drugs and among the oral dosage forms, tablets of various kinds are the most common type of solid dosage form in contemporary use. The term ‘tablet’ (from Latin tabuletta) is associated with the appearance of the dosage form, i.e. tablets are small disc-like or cylindrical specimens. The Latin name of the dosage form tablet in the European Pharmacopoeia (7th edn, 2011) is compressi, which reflects the fact that the dominating process of tablet fabrication is powder compression in a confined space. Alternative preparation procedures are also in use, such as mouldings and freeze drying. Tablet-like preparations prepared by freeze drying are sometimes referred to as oral lyophilisates. Moulding, i.e. the shaping and hardening of a semi-solid mixture of active substances and excipients, and freeze drying will not be further described in this chapter.
The idea of forming a solid dosage form by powder compression is not new. In 1843 the first patent for a hand-operated device used to form a tablet was granted. The use of tablets as dosage forms became of interest to the growing pharmaceutical industry but within pharmacies, the pill (a dosage form for oral administration formed by hand into spherical particles about 4–6 mm in diameter) remained the most popular solid dosage form for a long time.
A tablet consists of one or more drugs (active pharmaceutical ingredients) as well as a series of other substances (excipients) used in the formulation of a complete preparation. In the European Pharmacopoeia (7th edn, 2011), tablets are defined as ‘solid preparations each containing a single dose of one or more active substances. They are obtained by compressing uniform volumes of particles or by another suitable manufacturing technique, such as extrusion, moulding or freeze-drying (lyophilization). Tablets are intended for oral administration. Some are swallowed whole, some after being chewed, some are dissolved or dispersed in water before being administered and some are retained in the mouth where the active substance is liberated.’
Thus, a variety of tablets exists, and the types of excipients and also the way in which they are incorporated in the tablet vary. Other dosage forms can be prepared in a similar way, but are administered by other routes, such as suppositories.
Tablets are used mainly for systemic drug delivery but may also be used for local drug action. For systemic use, the drug must be released from the tablet, i.e. normally it dissolves in the fluids of the mouth, stomach or intestine, and thereafter the drug is absorbed into the systemic circulation, by which it reaches its site of action. Alternatively, tablets can be formulated for local delivery of drugs in the mouth or gastrointestinal tract, or can be used to increase temporarily the pH of the stomach.
Tablets are popular for several reasons:
• The oral route represents a convenient and safe way of drug administration.
• Compared to liquid dosage forms, tablets (and other solid dosage forms) have general advantages in terms of the chemical, physical and microbiological stability of the dosage form.
• The preparation procedure enables accurate dosing of the drug.
• Tablets are convenient to handle and can be prepared in a versatile way with respect to their use and the delivery of the drug.
• Finally, tablets can be relatively cheaply mass produced, with robust and quality-controlled production procedures giving an elegant preparation of consistent quality.
The main disadvantage of tablets as a dosage form is the problem of poor bioavailability of drugs due to unfavourable drug properties, e.g. poor solubility, poor absorption properties and instability in the gastrointestinal tract. In addition, some drugs may cause local irritant effects or otherwise cause harm to the gastrointestinal mucosa.
Quality attributes of tablets
Like all dosage forms, tablets should fulfil a number of product quality attributes regarding chemical, physical and biological characteristics. Quality issues relating to the final product are worth considering early in the development process (and thus early in this chapter) as they give an indication of the goal to be achieved during the development and manufacture of tablets.
The quality attributes that a tablet must possess can be summarized as follows:
1. The tablet should include the correct dose of the drug.
2. The appearance of the tablet should be elegant, and its weight, size and appearance should be consistent.
3. The drug should be released from the tablet in a controlled and reproducible way.
4. The tablet should be biocompatible, i.e. not include excipients, contaminants and microorganisms that could cause harm to patients.
5. The tablet should be of sufficient mechanical strength to withstand fracture and erosion during handling at all stages of its lifetime.
6. The tablet should be chemically, physically and microbiologically stable during the lifetime of the product.
7. The tablet should be formulated into a product acceptable to the patient.
In order to quantify these quality attributes, tests and specifications must be defined. Some tests and specifications are given in pharmacopoeias, such as dose content, dose uniformity, tablet disintegration, the release of the drug in terms of drug dissolution, and the microbial quality of the preparation. Another important quality attribute of tablets is their resistance to attrition and fracture.
Tablet manufacturing
Stages in tablet formation
The dominating technique of forming tablets (although alternative procedures are in use, as indicated in the definition of tablets in the European Pharmacopoeia, 2011) is by powder compression, i.e. forcing particles into close proximity to each other by confined compression. This enables the particles to cohere into a porous, solid specimen of defined geometry. The compression takes place in a die by the action of two punches, the lower and the upper, by which the compressive force is applied. Powder compression is defined as the reduction in volume of a powder owing to the application of a force. Because of the increased proximity of particle surfaces accomplished during compression, bonds are formed between particles which provide coherence to the powder, i.e. a compact is formed. Compaction is defined as the formation of a solid specimen of defined geometry by powder compression.
The process of tableting can be divided into three stages (sometimes known as the compaction cycle) (Fig. 30.1).
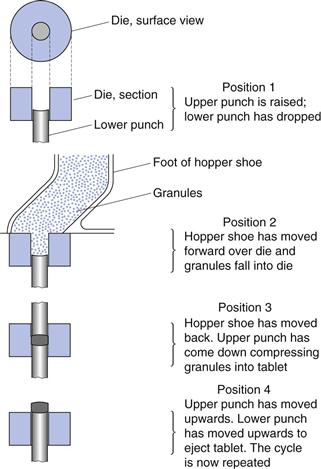
Die filling
This is normally accomplished by gravitational flow of the powder from a hopper via the die table into the die (although presses based on centrifugal die filling are also used). The die is closed at its lower end by the lower punch.
Tablet formation
The upper punch descends and enters the die and the powder is compressed until a tablet is formed. During the compression phase, the lower punch can be stationary or can move upwards in the die. After the maximum applied force is reached, the upper punch leaves the powder, i.e. the decompression phase.
Tablet ejection
During this phase the lower punch rises until its tip reaches the level of the top of the die. The tablet is subsequently removed from the die table by a pushing device.
Tablet presses
There are two types of press in common use during tablet production: the single-punch press and the rotary press. In addition, hydraulic presses are used in research and development work for the initial evaluation of the tableting properties of powders and prediction of the effect of scale-up on the properties of the formed tablets (scale-up refers to the change to a larger apparatus for performing a certain operation on a larger scale).
Single-punch press (eccentric press)
A single-punch press possesses one die and one pair of punches (Fig. 30.2), i.e. a set of tableting tools. The powder is held in a hopper which is connected to a hopper shoe located at the die table. The hopper shoe moves to and fro over the die, by either a rotational or a translational movement. When the hopper shoe is located over the die, the powder is fed into the die by gravitational powder flow. The amount of powder filled into the die is controlled by the position of the lower punch. When the hopper shoe is located beside the die, the upper punch descends and the powder is compressed. The lower punch is stationary during compression and the pressure is thus applied by the upper punch and controlled by the upper punch displacement. After ejection, the tablet is pushed away by the hopper shoe as it moves back to the die for the next tablet.
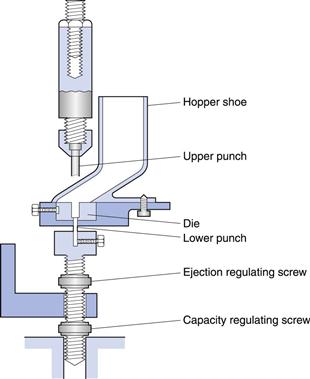
The output of tablets from a single-punch press is up to about 200 tablets per minute. A single-punch press thus has its primary use in the production of small batches of tablets, such as during formulation development and during the production of tablets for clinical trials.
Rotary press
The rotary press (also referred to as a multistation press) was developed to increase the output of tablets. The primary use of this machine is thus during scale-up in the latter part of the formulation work and during large-scale production. Outputs of over 10 000 tablets per minute can be achieved by rotary presses.
A rotary press operates with a number of dies and sets of punches, which can vary considerably from three for small rotary presses, up to 60 or more for large presses. The dies are mounted in a circle in the die table and both the die table and the punches rotate together during operation of the machine, so that one die is always associated with one pair of punches (Figs 30.3 and 30.4). The vertical movement of the punches is controlled by tracks that pass over cams and rollers used to control the volume of powder fed into the die and the pressure applied during compression.
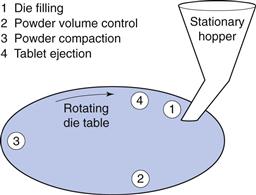
Fig. 30.3 Schematic illustration of the events involved in the formation of tablets with a rotary press.
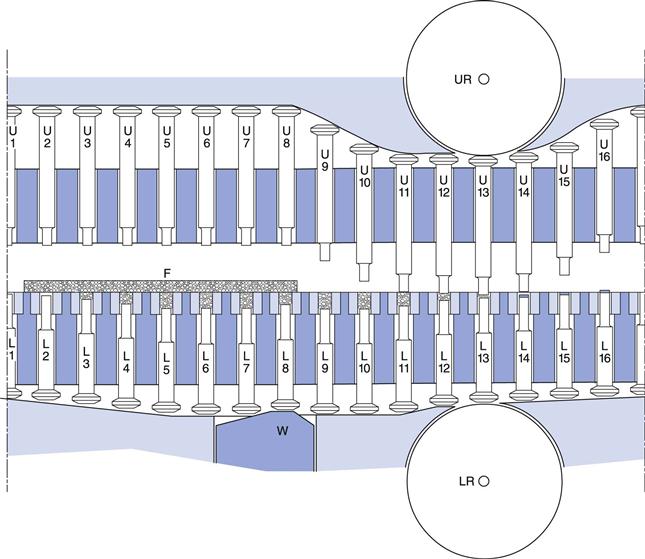
Fig. 30.4 Diagram of punch tracks of a rotary tablet press. UR, upper roller; LR, lower roller; W, powder volume adjuster; F, feed frame with granules. U1 to U8, upper punches in raised position; L1, lower punch at top position, tablet ejected; L2 to L7, lower punches dropping to lowest position and filling die with granules to an overfill at L7; L8, lower punch raised to expel excess granules giving correct volume; U9 to U12, upper punches lowering to enter die at U12; L13 and U13, upper and lower punches pass between rollers and granules are compacted to a tablet; U14 to U16, upper punch rising to top position; L14 to L16, lower punch rising to eject tablet.
The powder is held in a hopper whose lower opening is located just above the die table. The powder flows by gravity onto the die table and is fed into the die by a feed frame. The reproducibility of the die feeding can be improved by a rotating device, referred to as a force-feeding device. During powder compression both punches operate by vertical movement. After tablet ejection, the tablet is knocked away as the die passes the feed frame.
Computerized hydraulic press
For computerized hydraulic presses the movement of the punches can be controlled and varied considerably. Thus, tablets can be prepared under controlled conditions with respect to the loading pattern and loading rate. Possible applications are the investigation of the sensitivity of a drug to such variations, or to mimic the loading pattern of production presses to predict scale-up problems. Because of this latter application, this type of press is also referred to as a ‘compaction simulator’.
Instrumentation of tablet presses
Significant research on the process of tablet preparation was initiated in the 1950s and 1960s, i.e. about 100 years after the introduction of tablets as a dosage form. An important step in the development of such fundamental research was the introduction of instrumented tablet machines. By this instrumentation, the forces involved in the compaction process, i.e. the press forces from the upper and lower punches, and the force transmitted to the die as well as the displacement of the upper and lower punch during the compression and decompression phases, could be recorded.
Instrumented presses are used in research, in development and in the production of tablets. In research and development, instrumented machines are used to provide fundamental information on the mechanical and compaction properties of powders that should be used in tablet formulations. With this application, the work is normally carried out by instrumented single-punch presses or instrumented hydraulic presses (compaction simulators). The two main applications for an instrumented press in research and development are:
1. To prepare tablets under defined conditions, e.g. in terms of applied force during compaction. These tablets are thereafter characterized by different procedures, such as imaging, surface area and tensile strength analysis
2. To describe and analyse the compression properties of materials by studying punch forces and punch displacements during the compression and decompression phases. A series of different procedures exists, involving, for example, the assessment of deformation behaviour of particles during compression and friction properties during ejection. Some of these are described below.
In production, instrumented production machines, i.e. rotary presses, are used to control the tableting operation and to ensure that tablets of consistent quality are produced. Normally, only force signals are used on production machines and the variation in force signal during compression is monitored as it reflects variations in tablet weight.
Force transducers commonly used in the instrumentation of tablet machines are of two types. The most common type is called a strain gauge, which consists of wires through which an electric current is passed. The strain gauge is bonded to a punch or punch holder. During powder compression, a force is applied to the punches and they will temporarily deform. The magnitude of this deformation is dependent on the elastic modulus of the punches and the force applied. When the punch is deformed, the wire of the strain gauge is also deformed and the electrical resistance of the strain gauge will change. This change in electrical resistance can be recorded and calibrated in terms of a force signal. Another, less common type of force transducer employs piezoelectric crystals. These are devices which emit an electrical charge when loaded, the magnitude of which is proportional to the applied force.
Displacement transducers measure the distance which the punches travel during the compression and decompression processes. The most common type of displacement transducer delivers an analogue signal. It consists of a rod and some inductive elements mounted in a tube. When the rod moves within the tube, a signal is obtained which directly reflects the position of the rod. The movable rod is connected to the punch so that they move in parallel, i.e. the signal from the displacement transducer reflects the position of the punch. Digital displacement transducers are also used in instrumented tablet machines. Such transducers are based on differences in signal level depending on the position of an indicator.
Displacement transducers are necessarily mounted some distance from the punch tip. There is therefore a difference in the position given by the transducer and the real position of the punch tip owing to deformation of the punch along the distance between its tip and the connection point of the transducer. This deviation must be determined by a calibration procedure, e.g. by compressing the punch tips against each other, and a correction for this error must be made before the displacement data can be used.
The signals from the force and displacement transducers are normally amplified and sampled into a computer. After conversion into digital form, the signals are transformed into physically relevant units, i.e. N, Pa, mm, etc., and organized as a function of time. To obtain reliable data, the calibration of the signals, the resolution of the measuring systems and the reproducibility of the values must be carefully considered.
Tablet tooling
Tablets are formed in a variety of shapes. The most common tablet shapes are circular, oval and oblong but tablets may also have other shapes, such as triangular or quadratic. From a side-view, tablets may be flat or convex and with or without bevelled edges. Tablets may also bear break marks or symbols and other markings. Break marks (or breaklines) are used to facilitate breaking of tablets in a controlled way to ensure reproducible doses. Markings are used to facilitate identification of a preparation and are of two types: embossed and debossed. Debossed markings are indented into the tablet and embossed markings are raised on the tablet surface. The size, shape and appearance of a tablet formed by powder compaction are controlled by the set of tools and by tooling design; a large variation in tablet size, shape and appearance can thus be obtained.
Punches and dies for rotary presses are designed in a standardized way and standard configurations and terms are in use. The terminology used in describing the punches includes head, neck, barrel, stem and tip and in describing the die includes face, chamfer and bore. The tools are normally fabricated in steel and different steels may be used. Due to the high forces applied to the powder bed, tools may be damaged and under normal use they become worn. The toughness, wear resistance and corrosiveness vary between different types of steels and the choice of steel grade depends on factors such as the tooling configuration, the formulation to be compacted and the cost. In addition, the surface of punches and dies may be coated with a thin layer of another metal, such as chrome, to modify its surface properties, such as hardness and corrosiveness.
Punches and dies are precision tools and they should thus be handled and stored with care. Manufacturing problems may be related to the quality of the compression tooling. Tooling inspection programmes should thus be used in the development and production of tablets.
Technical problems during tableting
A number of technical problems can arise during the tableting procedure, among which the most important are:
• high weight and dose variation of the tablets
• low mechanical strength of the tablets
• capping and lamination of the tablets
Such problems are related to the properties of the powder intended to be formed into tablets and also to the design and conditions of the press and the tooling. They should therefore be avoided by ensuring that the powder possesses adequate technical properties and also that a suitable, well-conditioned tablet press is used, e.g. in terms of the use of forced-feed devices and polished and smooth dies and punches.
Important technical properties of a powder that must be controlled to ensure the success of a tableting operation are:
The technical properties of the powder are controlled by the ingredients of the formulation (i.e. the drug and excipients) and by the way in which the ingredients are combined into a powder during precompaction processing. The precompaction processing often consists of a series of unit operations in sequence. The starting point is normally the drug in a pure, most often crystalline form; the subsequent treatment of the drug particles is sometimes referred to as downstream processing. The unit operations used during this precompaction treatment are mainly particle size reduction, powder mixing, particle size enlargement and powder drying. For further details of these procedures see Chapters 10, 11, 28 and 29, respectively. Granulation of a fine powder is a common means used to preserve the fineness of the drug within larger particles that are suitable for tableting (see below) and granulation procedures are traditionally in common use in preparing a powder for tableting. To save time and energy, precompaction processing without a particle size enlargement operation is chosen if possible. This procedure is called tablet production by direct compression or direct compaction.
Tablet production via granulation
Rationale for granulating powders prior to tableting
Because both granulation and tableting involve the formation of aggregates, tablet production by granulation is based on the combination of two size enlargement processes in sequence. The main rationales for granulating the powder (drug and filler mixture) before tableting are to:
• increase the bulk density of the powder mixture and thus ensure that the required volume of powder can be filled into the die
• improve the flowability of the powder in order to ensure that tablets with a low and acceptable tablet weight variation can be prepared
• improve mixing homogeneity and reduce segregation by mixing small particles which subsequently adhere to each other
• improve the compactability of the powder by adding a solution binder, which is effectively distributed on the particle surfaces
• ensure a homogeneous colour in a tablet by adding the colour in a manner that ensures its effective distribution over the particle surfaces
• affect the dissolution process for hydrophobic, poorly soluble particles by using a fine particulate drug which is thoroughly mixed with a hydrophilic filler and a hydrophilic binder.
Before granulation, the drug might be processed separately in order to obtain a suitable quality in terms of solid-state and particulate properties, such as spray drying and milling. Normally, the drug exists in dry particulate form before granulation. However, it might be suspended or dissolved in a liquid and be added to the filler as a part of the agglomeration liquid.
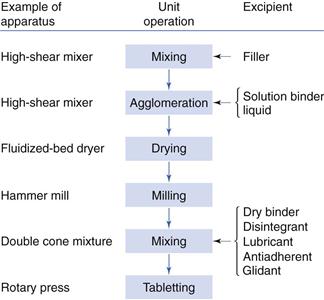
Different procedures may be used for granulation, among which the most important are the use of convective mixers, fluidized-bed driers, spray driers and compaction machines. Chapter 28 discusses granulation in some detail but the process is summarized here in the context of tableting.
Granulation by convective mixing
Agitation of a powder by convection in the presence of a liquid, followed by drying, is the main procedure for the preparation of pharmaceutical granules. This is often considered to be the most effective means in terms of production time and cost to prepare good-quality granules. The process is often referred to as wet granulation.
The ingredients to be granulated in a convective mixer are first dry mixed. The objective is to achieve a good homogeneity. As the components are often cohesive powders, a convective mixer operating at high intensity is normally used (a high-shear mixer). The mixture often consists of the drug and a filler. A disintegrant may also be included (i.e. an intragranular disintegrant) but it is also common to add the disintegrant to the dry granulation (i.e. an extragranular disintegrant). After wet mixing, the wet mass is dried in a separate drier (usually a fluidized-bed dryer; see Chapter 29). Because granulation in a convective mixer is not a very well-controlled operation, large granules (above 1 mm) are often formed which must be broken down into smaller units. This is normally done by milling in a hammer mill or by pressing the granulation through a screen in an oscillating granulator. Granules ranging in size from about 100 to 800 µm are thus obtained.
The prepared granules are finally dry-mixed with the other ingredients, for example in a double-cone mixer, before tableting. Common excipients added in this final mixing operation are disintegrants, lubricants, glidants and colourants. Figure 30.5 summarizes the sequence of unit operations used in the production of tablets with pre-compaction treatment by granulation.
Alternative granulation procedures
A series of alternative granulation procedures may be preferable in certain situations. Granulation in a fluidized-bed apparatus is less common than the use of convective mixers as it is considered to be more time-consuming. However, granules of high quality in terms of homogeneity, flowability and compactability can be prepared by this operation.
By spray drying a suspension of drug particles in a liquid, which can contain a dissolved binder, relatively small spherical granules with uniform size can be prepared. The process is of limited use, except for the preparation of fillers or diluents for direct compaction. The granulation can show a good compactability and this presents the possibility of granulating a drug suspension without a separate drying step for the drug substance.
The formation of granules by compacting the powder into large compacts which are subsequently comminuted into smaller granules is an alternative approach to granulation. The approach can be employed as a means of avoiding exposure of the powder to moisture and heat and is also referred to as dry granulation. In addition, for powders of very low bulk density, compaction can be an effective means to increase markedly their bulk density. The formation of the compacts can be accomplished by powder compression in a die (slugging), giving relatively large tablet-like compacts, or by powder compression between rotating rollers (roller compaction), giving weak compacts with a flake or ribbon-like appearance. Roller compaction is a suitable operation for continuous granulation.
Tablet production by direct compaction
An obvious way to reduce production time and hence cost is to minimize the number of operations involved in the pretreatment of the powder mixture before tableting. Tablet production by direct compaction can be reduced to only two operations in sequence: powder mixing and tableting (Fig. 30.6). The advantage with direct compaction is primarily a reduced production cost. However, in a direct compactable formulation, specially designed fillers and dry binders are normally required, which are usually more expensive than the traditional ones. They may also require a larger number of quality tests before processing. As heat and water are not involved, product stability can be improved. Finally, drug dissolution might be faster from a tablet prepared by direct compaction owing to fast tablet disintegration into primary drug particles.
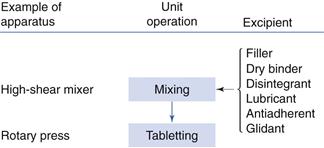
Fig. 30.6 Overview of the sequence of unit operations used in the production of tablets by direct compaction.
The disadvantages of direct compaction are mainly technological. In order to handle a powder of acceptable flowability and bulk density, relatively large particles must be used which may be difficult to mix to a high homogeneity and may be prone to segregation. Moreover, a powder consisting mainly of drug will be difficult to form into tablets if the drug itself has poor compactability. Finally, a uniform colouring of tablets can be difficult to achieve with a colourant in dry particulate form.
Direct compaction may be considered in two common formulation cases; firstly, relatively soluble drugs which can be processed as coarse particles (to ensure good flowability) and secondly, relatively potent drugs which are present in a few milligrams in each tablet and can be mixed with relatively coarse excipient particles (in this latter case the flow and compaction properties of the formulation are mainly controlled by the excipients).
Tablet excipients
In addition to the active ingredient(s), a series of excipients is normally included in a tablet; their role is to ensure that the tableting operation can run satisfactorily and that tablets of specified quality are prepared. Depending on the intended main function, excipients to be used in tablets are subcategorized into different groups. However, one excipient can affect the properties of a powder or the tablet in a series of ways and many substances used in tablet formulations can thus be described as multifunctional. The functions of the most common types of excipients used in tablets are described below. Examples of substances used as excipients in tablets are given in Table 30.1.
Table 30.1
Examples of substances used as excipients in tablet formulation
| Type of excipient | Example of substances |
| Filler | Lactose |
| Sucrose | |
| Glucose | |
| Mannitol | |
| Sorbitol | |
| Calcium phosphate | |
| Calcium carbonate | |
| Cellulose | |
| Disintegrant | Starch |
| Cellulose | |
| Crosslinked polyvinyl pyrrolidone | |
| Sodium starch glycolate | |
| Sodium carboxymethyl cellulose | |
| Solution binder | Gelatin |
| Polyvinyl pyrrolidone | |
| Cellulose derivatives | |
| (e.g. hydroxypropylmethyl cellulose) | |
| Polyethylene glycol | |
| Sucrose | |
| Starch | |
| Dry binder | Cellulose |
| Methyl cellulose | |
| Polyvinyl pyrrolidone | |
| Polyethylene glycol | |
| Glidant | Silica |
| Magnesium stearate | |
| Talc | |
| Lubricant | Magnesium stearate |
| Stearic acid | |
| Polyethylene glycol | |
| Sodium lauryl sulfate | |
| Sodium stearyl fumarate | |
| Liquid paraffin | |
| Antiadherent | Magnesium stearate |
| Talc | |
| Starch | |
| Cellulose |
Filler (or diluent)
In order to form tablets of a size suitable for handling, a lower limit in terms of powder volume and weight is required. Tablets normally weigh at least 50 mg. Therefore, a low dose of a potent drug requires the incorporation of a substance into the formulation to increase the bulk volume of the powder and hence the size of the tablet. This excipient, known as the filler or the diluent, is not necessary if the dose of the drug per tablet is high.
The ideal filler should fulfil a series of requirements, such as:
• possess good biopharmaceutical properties (e.g. water soluble or hydrophilic)
• possess good technical properties (such as compactability and dilution capacity)
As all these requirements cannot be fulfilled by a single substance, different substances have gained use as fillers in tablets, mainly carbohydrates but also some inorganic salts.
Lactose is the most common filler in tablets. It possesses a series of good filler properties, e.g. dissolves readily in water, has a pleasant taste, is non-hygroscopic, is fairly non-reactive and shows good compactability. Its main limitation is that some people have an intolerance to lactose.
Lactose exists in both crystalline and amorphous forms. Crystalline lactose is formed by precipitation and, depending on the crystallization conditions, α-monohydrate or β-lactose (an anhydrous form) can be formed. By thermal treatment of the monohydrate form, crystalline α-anhydrous particles can be prepared. Depending on the crystallization conditions and the use of subsequent size reduction by milling, lactose of different particle sizes is obtained.
Amorphous lactose can be prepared by spray drying a lactose solution (giving nearly completely amorphous particles) or a suspension of crystalline lactose particles in a lactose solution (giving aggregates of crystalline and amorphous lactose). Amorphous lactose dissolves more rapidly than crystalline and shows better compactability. Its main use is therefore in the production of tablets by direct compaction. Amorphous lactose is, however, hygroscopic and physically unstable, i.e. it will spontaneously crystallize if crystallization conditions are met as a result of elevated temperature or high relative humidity (see Chapter 8 for more details).
Other sugars or sugar alcohols, such as glucose, sucrose, sorbitol and mannitol, have been used as alternative fillers to lactose, primarily in lozenges or chewable tablets because of their pleasant taste. Mannitol has a negative heat of solution and imparts a cooling sensation when sucked or chewed.
Apart from the sugars, perhaps the most widely used fillers are powdered celluloses of different types. Celluloses are biocompatible, chemically inert and have good tablet-forming and disintegrating properties. They are therefore also used as dry binders and disintegrants in tablets. They are compatible with many drugs but, owing to their hygroscopicity, may be incompatible with drugs prone to chemical degradation in the solid state.
The most common type of cellulose powder used in tablet formulation is microcrystalline cellulose. The name indicates that the particles have both crystalline and amorphous regions, depending on the relative position of the cellulose chains within the solid. The degree of crystallinity may vary depending on the source of the cellulose and the preparation procedure. The degree of crystallinity will affect the physical and technical properties of the particles, e.g. in terms of hygroscopicity and powder compactability.
Microcrystalline cellulose is prepared by hydrolysis of cellulose followed by spray drying. The particles thus formed are aggregates of smaller cellulose fibres. Depending on the preparation conditions, aggregates of different particle size can be prepared which have different flowabilities.
A final important example of a common filler is an inorganic substance, dicalcium phosphate dihydrate. This is insoluble in water and non-hygroscopic, but is hydrophilic, i.e. easily wetted by water. The substance can be obtained in both a fine particulate form, mainly used in granulation, and an aggregated form. The latter possesses good flowability and is used in tablet production by direct compaction. Calcium phosphate is slightly alkaline and may thus be incompatible with drugs sensitive to alkaline conditions.
Matrix former
In order to affect or control the release of the drug from the tablet, i.e. to speed up or to slow down its release rate, the drug may be dispersed or embedded in a matrix formed by an excipient or a combination of excipients. This type of excipient may thus be referred to as a matrix former. An alternative term is base, a term used in the European Pharmacopoeia (2011) and there defined as ‘the carrier, composed of one or more excipients, for the active substance(s) in semi-solid and solid preparations’.
The matrix former is often a polymer or a lipid and may constitute a significant fraction of the total tablet weight. When the objective is to increase drug dissolution, the matrix former can be a water-soluble substance or a lipid and the drug is dissolved or suspended as fine particles in the matrix. An example of a water-soluble matrix former is poly ethylene glycol (PEG). When the objective is to prolong the drug release, the matrix former can be either an insoluble substance (a polymer or a lipid) or a substance that forms a gel in contact with water. The drug is normally dispersed in particulate form in the matrix (for more details, see below). A common gel-forming substance in tablets is hydroxy propyl methyl cellulose (HPMC). Matrix formers are discussed in more detail in Chapter 31.
Disintegrant
A disintegrant is included in the formulation to ensure that the tablet, when in contact with a liquid, breaks up into small fragments, which promotes rapid drug dissolution. Ideally, the tablet should break up into individual drug particles in order to obtain the largest possible effective surface area during dissolution.
The disintegration process for a tablet occurs in two steps. First, the liquid wets the solid and penetrates the pores of the tablet. Thereafter, the tablet breaks into smaller fragments. The actual fragmentation of the tablet can also occur in steps, i.e. the tablet disintegrates into aggregates of primary particles which subsequently deaggregate into their primary drug particles. A deaggregation directly into primary powder particles will set up conditions for the fastest possible dissolution of the drug. A scheme for the release of the drug from a disintegrating tablet is shown in Figure 30.7.
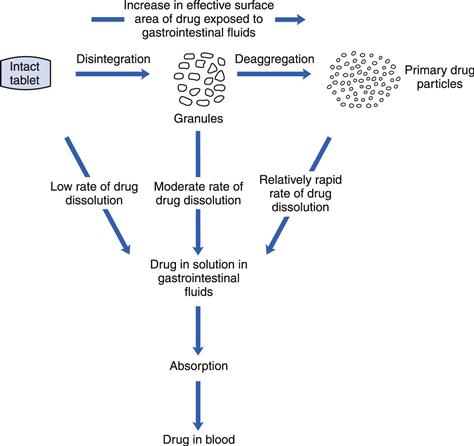
Fig. 30.7 Mechanistic representation of the drug release process from a tablet by disintegration and dissolution. (Courtesy of Wells and Rubinstein, 1976, with permission.)
Several mechanisms of action of disintegrants have been suggested, such as swelling of particles, exothermic wetting reaction, particle repulsion and particle deformation recovery. However, as two main processes are involved in the disintegration event, disintegrants to be used in plain tablets are here classified into two types:
1. Disintegrants that facilitate water uptake. These disintegrants act by facilitating the transport of liquids into the pores of the tablet, with the consequence that the tablet may break into fragments. One obvious type of substance that can promote liquid penetration is surface-active agents. Such substances are used to make the drug particle surfaces more hydrophilic and thus promote the wetting of the solid and the penetration of the liquid into the pores of the tablet. It has also been suggested that other substances can promote the liquid penetration, using capillary forces to suck water into the pores of the tablet.
2. Disintegrants that will rupture the tablet. Rupturing of tablets can be caused by swelling of the disintegrant particles during sorption of water. However, it has also been suggested that non-swelling disintegrants can break the tablet and different mechanisms have been suggested. One such concerns a repulsion of particles in contact with water and another the recovery of deformed particles to their original shape in contact with water, i.e. particles which have been deformed during compaction.
The disintegrant most traditionally used in conventional tablets is starch, among which potato, maize and corn starches are commonly used. The typical concentration range of starch in a tablet formulation is up to 10%. Starch particles swell in contact with water and this swelling can subsequently disrupt the tablet. However, it has also been suggested that starch particles may facilitate disintegration by particle-particle repulsion.
The most common and effective disintegrants act via a swelling mechanism and a series of effective swelling disintegrants has been developed which can swell dramatically during water uptake and thus quickly and effectively break the tablet. These are normally modified starch or modified cellulose. High-swelling disintegrants are included in the formulation at relatively low concentrations, typically 1–5% by weight.
Disintegrants can be mixed with other ingredients prior to granulation and thus be incorporated within the granules (intragranular addition). It is also common for the disintegrant to be mixed with the dry granules before the complete powder mix is compacted (extragranular addition). The latter procedure will contribute to an effective disintegration of the tablet into smaller fragments.
A third group of disintegrants functions by producing gas, normally carbon dioxide, when in contact with water. Such disintegrants are used in effervescent tablets and normally not in tablets that should be swallowed as a solid. The liberation of carbon dioxide is obtained by the decomposition of bicarbonate or carbonate salts in contact with acidic water. The acidic pH is accomplished by the incorporation of a weak acid in the formulation, such as citric acid and tartaric acid.
Dissolution enhancer
For drugs of low aqueous solubility, the dissolution of the drug may be the rate-limiting step in the overall drug release and absorption processes. Agents, other than matrix formers, may therefore sometimes be found in the composition of a tablet with the role to speed up the drug dissolution process by temporarily increasing the solubility of the drug during drug dissolution. An important example of a dissolution enhancer is the incorporation into the formulation of a substance that forms a salt with the drug during dissolution, e.g. increasing the dissolution rate of aspirin by using magnesium oxide in the formulation.
Absorption enhancer
For drugs with poor absorption properties, the absorption can be affected (see Chapter 20) by using substances in the formulation that affect the permeability of the intestinal cell membrane and thus increase the rate at which the drug passes though the intestinal membrane. An additive that modulates the permeability of the intestine is often referred to as an absorption enhancer.
Binder
A binder (also sometimes called an adhesive) is added to a drug-filler mixture to ensure that granules and tablets can be formed with the required mechanical strength. Binders can be added to a powder in different ways:
• As a dry powder which is mixed with the other ingredients before wet agglomeration. During the agglomeration procedure, the binder might thus dissolve partly or completely in the agglomeration liquid
• As a solution which is used as agglomeration liquid during wet agglomeration. The binder is here often referred to as a solution binder
• As a dry powder which is mixed with the other ingredients before compaction (slugging or tableting). The binder is here often referred to as a dry binder.
Both solution binders and dry binders are included in the formulation at relatively low concentrations, typically 2–10% by weight. Common traditional solution binders are starch, sucrose and gelatin. More commonly used binders today, with improved adhesive properties, are polymers such as polyvinylpyrrolidone and cellulose derivatives (in particular hydroxypropyl methylcellulose). Important examples of dry binders are microcrystalline cellulose and crosslinked polyvinylpyrrolidone.
Solution binders are generally considered the most effective and this is therefore the most common way of incorporating a binder into granules; the granules thus formed are often referred to as binder–substrate granules. It is not uncommon, however, for a dry binder to be added to the dry binder–substrate granules before tableting in order to further improve the compactability of the granulation.
Glidant
The role of the glidant is to improve the flowability of the powder. Glidants are used in formulations for direct compaction but are often also added to granules before tableting to ensure that sufficient flowability of the tablet mass is achieved for high production speeds.
Traditionally, talc has been used as a glidant in tablet formulations, in concentrations of about 1–2% by weight. Today, the most commonly used glidant is probably colloidal silica, added in very low proportions (about 0.2% by weight). Because the silica particles are very small they adhere to the particle surfaces of the other ingredients (i.e. an ordered or structured mixture is formed; see Chapter 11) and improve flow by reducing interparticulate friction. Magnesium stearate, normally used as a lubricant, can also promote powder flow at low concentrations (<1% by weight).
Lubricant
The function of the lubricant is to ensure that tablet formation and ejection can occur with low friction between the solid and the die wall. High friction during tableting can cause a series of problems, including inadequate tablet quality (capping or even fragmentation of tablets during ejection and vertical scratches on tablet edges) and may even stop production. Lubricants are thus included in almost all tablet formulations.
Lubrication is achieved mainly by two mechanisms: fluid lubrication and boundary lubrication (Fig. 30.8). In fluid lubrication a layer of fluid is located between and separates the moving surfaces of the solids from each other and thus reduces the friction. Fluid lubricants are seldom used in tablet formulations. However, liquid paraffin has been used, for instance in formulations for effervescent tablets.
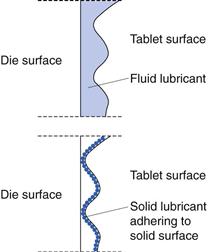
Boundary lubrication is considered as a surface phenomenon, as here the sliding surfaces are separated by only a very thin film of lubricant. The nature of the solid surfaces will therefore affect friction. In boundary lubrication, the friction coefficient and wear of the solids are higher than with fluid lubrication. All substances that can affect the interaction between sliding surfaces can be described as boundary lubricants, including adsorbed gases. The lubricants used in tablet formulations acting by boundary lubrication are fine particulate solids.
A number of mechanisms have been discussed for these boundary lubricants, including that lubricants are substances that show a low resistance towards shearing. The most effective of the boundary lubricants are stearic acid or stearic acid salts, such as magnesium stearate. Magnesium stearate has become the most widely used lubricant owing to its superior lubrication properties. The stearic acid salts are normally used at low concentrations (<1% by weight).
Besides reducing friction, lubricants may cause undesirable changes in the properties of the tablet. The presence of a lubricant in a powder is thought to interfere in a deleterious way with the bonding between the particles during compaction, thus reducing tablet strength (Fig. 30.9). Because many lubricants are hydrophobic, tablet disintegration and dissolution are often retarded by the addition of a lubricant. These negative effects are strongly related to the amount of lubricant present and a minimum amount is normally used in a formulation, i.e. concentrations of 1% or below. In addition, the way in which the lubricant is mixed with the other ingredients should also be considered. It can, for example, be important if the excipients are added sequentially to a granulation rather than simultaneously. The total mixing time and the mixing intensity are also important in this context.
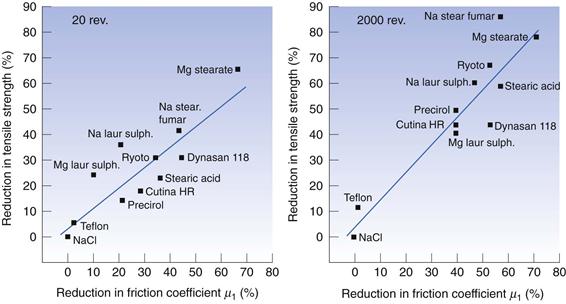
Fig. 30.9 The reduction in tablet tensile strength as a function of the reduction in friction coefficient during tableting of a sodium chloride powder mixed with 0.1% by weight of a series of lubricants admixed at two different mixing intensities. (Courtesy of Hölzer and Sjögren, 1981, with permission.)
The commonly observed retardation of disintegration and dissolution of tablets is related to the hydrophobic character of the most commonly used lubricants. In order to avoid these negative effects, more hydrophilic substances have been suggested as alternatives to the hydrophobic lubricants. Examples are surface-active agents and polyethylene glycol. A combination of hydrophobic and hydrophilic substances might also be used.
The lubricant’s effect on friction and on the changes in tablet properties is related to the tendency of lubricants to adhere to the surface of drugs and fillers during dry mixing. Lubricants are often fine particulate substances which thus are prone to adhere to larger particles. In addition, studies on the mixing behaviour of magnesium stearate have indicated that this substance has the ability to form a film which can cover a fraction of the surface area of the drug or filler particles (the substrate particles). This film can be described as being continuous rather than particulate. A number of factors have been suggested to affect the development of such a lubricant film during mixing and hence also affect friction and changes in tablet properties, such as the shape and surface roughness of the substrate particles; the surface area of the lubricant particles; mixing time and intensity; and the type and size of mixer.
Concerning the tablet strength-reducing effect of a lubricant, apart from the degree of surface coverage of the lubricant film obtained during mixing, the compression behaviour of the substrate particles will also be important. Drugs and fillers can thus be evaluated in terms of their lubricant sensitivity, i.e. the reduction in tablet strength due to the addition of a lubricant compared to a tablet formed from a powder without a lubricant. An important property for this lubricant sensitivity seems to be the degree of fragmentation the substrate particles undergo during compression (see below). It is thus assumed that, during compression, particle surfaces which are not covered with a lubricant film are formed during particle fragmentation and that these clean surfaces will bond differently from the lubricant-covered particle surfaces.
To explain the effect of lubricant film formation on the tensile strength of tablets, a coherent matrix model has been developed. This suggests that when a continuous matrix of lubricant-covered particle surfaces exists in a tablet, along which a fracture plane can be formed, the tablet strength is considerably lower than that of tablets formed from unlubricated powder. However, if the mixing and compression processes do not result in such a coherent lubricant matrix within the tablet, for example due to irregular substrate particles or particle fragmentation, the lubricant sensitivity appears to be lower.
Antiadherent
The function of an antiadherent is to reduce adhesion between the powder and the punch faces and thus prevent particles sticking to the punches. Many powders are prone to adhere to the punches, a phenomenon (known in the industry as sticking or picking) which is affected by the moisture content of the powder. Such adherence is especially prone to happen if the tablet punches have markings or symbols. Adherence can lead to a build-up of a thin layer of powder on the punches, which in turn will lead to an uneven and matt tablet surface with unclear markings or symbols.
Many lubricants, such as magnesium stearate, also have antiadherent properties. However, other substances with limited ability to reduce friction can also act as antiadherents, such as talc and starch.
Sorbent
Sorbents are substances that are capable of sorbing some quantities of fluids in an apparently dry state. Thus, oils or oil-drug solutions can be incorporated into a powder mixture which is granulated and compacted into tablets. Microcrystalline cellulose and silica are examples of sorbing substances used in tablets.
Flavour
Flavouring agents are incorporated into a formulation to give the tablet a more pleasant taste or to mask an unpleasant one. The latter can also be achieved by coating the tablet or the drug particles.
Flavouring agents are often thermolabile and so cannot be added prior to an operation involving heat. They are often mixed with the granules as an alcohol solution.
Colourant
Colourants are added to tablets to aid identification and patient compliance. Colouring is often accomplished during coating (see Chapter 32 for further information) but a colourant can also be included in the formulation prior to compaction. In the latter case, the colourant can be added as an insoluble powder or dissolved in the granulation liquid. The latter procedure may lead to a colour variation in the tablet caused by migration of the soluble dye during the drying stage (see Chapter 29 for more information on the phenomenon of solute migration).
Tablet types
Classification of tablets
Several categories of tablets may be distinguished and the denomination and definitions of different types of tablets can be found in pharmacopoeias. The tablet dosage forms described in the European Pharmacopoeia (2011) appear in the monographs for tablets and for oromucosal preparations (the monograph on oromucosal preparations includes some types of tablets, i.e. compressed lozenges, sublingual tablets and buccal tablets that are intended for administration to the oral cavity and/or the throat to obtain a local or systemic effect).
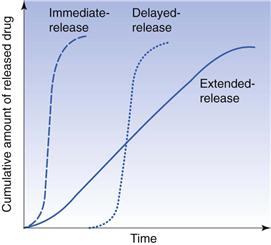
A common means of classifying tablets is based on the drug release pattern from the tablets. The following categories are often used in this context: immediate release, prolonged release, pulsatile release and delayed release. The latter three types are also referred to as modified-release tablets.
For immediate-release tablets, the drug is intended to be released rapidly after administration, or the tablet is dissolved in liquid before intake and thus administered as a solution. Immediate-release tablets are the most common type of tablet and include disintegrating, chewable, effervescent, sublingual and buccal tablets.
Modified-release tablets should normally be swallowed intact. The formulation and thus also the type of excipients used in such tablets might be quite different from those of immediate-release tablets. The term prolonged-release tablet is used to indicate that the drug is released slowly at a nearly constant rate. If the rate of release is constant during a substantial period of time, a zero-order type of release is obtained, i.e. M = kt (where M is the cumulative amount of drug released and t is the release time). This is sometimes described as an ideal type of prolonged-release preparation.
A pulsatile release is another means to increase the time period for drug absorption after a single administration and is accomplished by releasing the drug in two or more pulses.
For delayed-release tablets the drug is liberated from the tablet some time after administration. After this period has elapsed, the release is normally rapid. The most common type of delayed-release tablet is a gastro-resistant (also known as enteric-coated) tablet, for which the drug is released in the upper part of the small intestine after the preparation has passed the stomach. However, a delayed release can also be combined with a prolonged drug release, e.g. for local treatment in the lower part of the intestine or in the colon.
The type of release obtained from immediate-, prolonged- and delayed-release tablets is illustrated in Figure 30.10. In the following sections, the most common types of tablets are described.
Disintegrating tablets
The most common type of tablet is intended to be swallowed and to release the drug in a relatively short time thereafter by disintegration and dissolution, i.e. the goal of the formulation is fast and complete drug release in vivo. Such tablets are often referred to as conventional or plain tablets. A disintegrating tablet includes normally at least the following types of excipients: filler (if the dose of the drug is low), disintegrant, binder, glidant, lubricant and antiadherent.
As discussed above, the drug is released from a disintegrating tablet in a sequence of processes, including tablet disintegration, drug dissolution and drug absorption (see Fig. 30.7). All these processes will affect, and can be rate-limiting steps for, the drug bioavailability. The rate of the processes is affected by both formulation factors and production conditions.
The disintegration time of the tablet is a function of the composition and manufacturing conditions and may thus depend on several factors. Regarding composition, the choice of disintegrant (Fig. 30.11) is of obvious importance but also other excipients, such as the type of filler and lubricant can also be of significant importance for tablet disintegration.
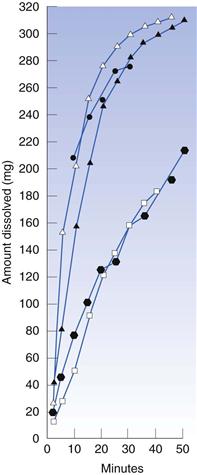
Fig. 30.11 The dissolution rate of salicylic acid, as assessed by an in vitro dissolution method based on agitated baskets, from tablets formed from mixtures of salicylic acid (325 mg) and a series of different types of starches as disintegrant  potato starch,
potato starch,  arrowroot starch,
arrowroot starch,  rice starch, • corn starch, Δ compressible starch. (Courtesy of Underwood and Cadwallader, 1972, with permission.)
rice starch, • corn starch, Δ compressible starch. (Courtesy of Underwood and Cadwallader, 1972, with permission.)
Tablet disintegration may also be affected by production conditions during manufacture. Important examples that may affect tablet disintegration time are the design of the granulation procedure (which will affect the physical properties of the granules), mixing conditions during the addition of lubricants and antiadherents, the applied punch force during tableting and the punch force-time relationship. It has been reported that an increased compaction pressure can either increase or decrease disintegration time, or give complex relationships with maximum or minimum disintegration times.
In many cases, the drug dissolution rate is the rate-limiting step in the drug release process, especially for drugs that are poorly soluble in water but that are readily absorbed in the intestine (i.e. typically class II drugs in the Biopharmaceutics Classification System, see Chapter 21). Since many drugs show very poor solubility, the problem of the rate of drug dissolution is often a critical issue during the development of an immediate release tablet. The most common means to achieve acceptable drug dissolution rate for poorly soluble compounds are:
• control of apparent solubility of the drug.
• control of surface area exposed to the dissolution medium.
The apparent solubility can be increased by changing the solid-state form of the drug, such as a polymorph, using a salt of the drug or using amorphous particles. The surface area of a solid is inversely proportional to particle size and particle size reduction is an obvious means to increase drug dissolution rate. However, a reduced particle size will make a powder more cohesive. A reduction in drug particle size might produce aggregates of particles which are difficult to break up, with the consequence that the drug dissolution rate is not related to the surface area of the primary drug particles. It is thus important to ensure that the tablet is formulated in such a way that it will disintegrate and that the aggregates so formed break up into small drug particles in order that a large surface area of the drug is exposed to the dissolution medium. The preparation and use of small particles, i.e. down to the sub-micrometre range (nano-sized particles), and the use of drug dissolution enhancers and matrix formers in tablet compositions of poorly soluble drugs are important issues in the formulation of disintegrating, immediate-release tablets.
For drugs with poor absorption properties, the absorption can be affected (see Chapter 20) by modifying the drug’s lipophilicity, e.g. by esterification of the drug as well as by adding absorption enhancers.
Single disintegrating tablets can also be prepared in the form of multilayers, i.e. the tablet consists of concentric or parallel layers cohered to each other. Multilayer tablets are prepared by repeated compression of powders and are made primarily to separate incompatible drugs from each other, i.e. incompatible drugs can be incorporated into the same tablet. Although intimate contact exists at the surface between the layers, the reaction between the incompatible drugs is limited. The use of parallel layered tablets where the layers are differently coloured represents an approach to preparing easily identifiable tablets.
Another variation of the disintegrating tablet is coated tablets which are intended to disintegrate and release the drug quickly (in contrast to coated tablets intended for modified release). The rationale for using coated tablets and detailed descriptions of the procedures used for tablet coating are given in Chapter 32.
Chewable tablets
Chewable tablets are chewed and thus are mechanically disintegrated in the mouth. The drug is, however, normally not dissolved in the mouth but swallowed and dissolves in the stomach or intestine. Thus, chewable tablets are used primarily to accomplish a quick and complete disintegration of the tablet – and hence obtain a rapid drug effect – or to facilitate the administration of the tablet. A common example of the former is antacid tablets. In the latter case, the elderly and children in particular have difficulty swallowing tablets and so a chewable tablet is an attractive form of medication. Important examples are vitamin tablets. Another general advantage of a chewable tablet is that this type of medication can be taken when water is not available.
Chewable tablets are similar in composition to conventional tablets except that a disintegrant is normally not included in the composition. Flavouring and colouring agents are common and sorbitol and mannitol are common examples of fillers.
Effervescent tablets
Effervescent tablets are dropped into a glass of water before administration, during which carbon dioxide is liberated. This facilitates tablet disintegration and drug dissolution; the dissolution of the tablet should be complete within a few minutes. As mentioned above, the effervescent carbon dioxide is created by a reaction in water between a carbonate or bicarbonate and a weak acid such as citric or tartaric acids.
Effervescent tablets are used to obtain rapid drug action, for example for analgesic drugs (Fig. 30.12), or to facilitate the intake of the drug, for example for vitamins.
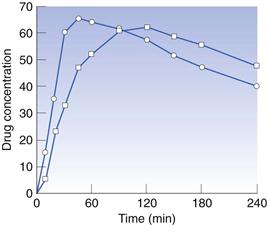
Fig. 30.12 Concentration of salicylates in plasma after administration of acetylsalicylic acid tablets (1 g). Circles, effervescent tablet; squares, conventional tablet. (Courtesy of Ekenved et al, 1975, with permission.)
The amount of sodium bicarbonate in an effervescent tablet is often quite high (about 1 g). After dissolution of such a tablet, a buffered water solution will be obtained which normally temporarily increases the pH of the stomach. The result is a rapid emptying of the stomach and the residence time of the drug in the stomach will thus be short. As drugs are absorbed in the small intestine rather than in the stomach, effervescent tablets can thus show a fast drug absorption, which can be advantageous, for example, for analgesic drugs. Another aspect of the short drug residence time in the stomach is that drug-induced gastric irritation can be avoided, e.g. for aspirin tablets, as the absorption of aspirin in the stomach can cause irritation.
Effervescent tablets also often include a flavour and a colourant. A water-soluble lubricant is preferable in order to avoid a film of a hydrophobic lubricant on the surface of the water after tablet dissolution. A binder is normally not included in the composition.
Effervescent tablets are prepared by both direct compaction and by compaction via granulation. In the latter case, traditional wet granulation is seldom used; instead, granules are formed by the fusion of particles as a result of their partial dissolution during wet massing of a moistened powder.
Effervescent tablets should be packaged in such a way that they are protected against moisture. This is accomplished with waterproof containers, often including a dessicant, or with blister packs or aluminium foils.
Compressed lozenges
Compressed lozenges are tablets that dissolve slowly in the mouth and so release the drug dissolved in the saliva. Lozenges are used for systemic drug uptake or for local medication in the mouth or throat, e.g. with local anaesthetic, antiseptic and antibiotic drugs. The latter type of tablets can thus be described as slow-release tablets for local drug treatment.
Disintegrants are not used in the formulation but otherwise such tablets are similar in composition to conventional tablets. In addition, lozenges are often coloured and include a flavour. The choice of filler and binder is of particular importance in the formulation of lozenges, as these excipients should contribute to a pleasant taste or feeling during tablet dissolution. The filler and binder should therefore be water soluble and have a good taste. Common examples of fillers are glucose, sorbitol and mannitol. A common binder in lozenges is gelatin.
Lozenges are normally prepared by compaction at high applied pressures in order to obtain a tablet of high mechanical strength and low porosity which can dissolve slowly in the mouth.
Sublingual tablets and buccal tablets
Sublingual tablets and buccal tablets are used for drug release in the mouth followed by systemic uptake of the drug. A rapid systemic drug effect can thus be obtained without first-pass liver metabolism. Sublingual tablets are placed under the tongue and buccal tablets are placed in the side of the cheek or high up between the inside of the upper lip and gum.
Sublingual and buccal tablets are often small and porous, the latter facilitating fast disintegration and drug release. Other designs, comprising high molecular weight hydrophilic polymers and/or gums, adhere in place by forming a gel. They remain in position, releasing drug, for 1–2 hours (e.g. prochlorperazine maleate for nausea).
Prolonged-release and pulsatile-release tablets
Classification
In recent years there has been great interest in the development and use of tablets which should be swallowed and thereafter slowly release the drug in the gastrointestinal tract for a period of about 12–24 hours. Such tablets are denominated in various ways, such as slow release, extended release, sustained release and prolonged release. They are often also referred to as controlled-release preparations. This latter term is somewhat misleading, as all tablets, irrespective of their formulation and use, should release the drug in a controlled and reproducible way. The nomenclature for prolonged-release preparations is subject to some debate and no worldwide acceptable system exists. The reader is referred to Chapter 31 for further discussion on this subject.
After release from the tablet, the drug should normally be absorbed into the systemic circulation. The aim is normally to increase the time period during which a therapeutic drug concentration level in the blood is maintained. However, the aim can also be to increase the release time for drugs that can cause local irritation in the stomach or intestine if they are released quickly. Examples of the latter are potassium chloride and iron salts. In addition, drugs for local treatment of diseases in the large intestine are sometimes formulated as prolonged-release tablets.
A prolonged-release preparation can also be categorized as a single-unit or a multiple-unit dosage form. In the first case, the drug dose is incorporated into a single release unit and in the latter is divided into a large number of small release units. A multiple-unit dosage form is often considered to give a more reproducible drug action.
There is a series of rationales behind the increased interest in administering drugs orally for systemic uptake in the form of prolonged-release tablets. However, the drug must fulfil certain criteria in order to render itself suitable for sustained-release medication, otherwise another type of tablet is a more feasible alternative. These rationales and criteria, as well as the pharmacokinetic aspects of prolonged-release drug administration, are described elsewhere in this book (see Chapters 22 and 31). In Chapter 31 the formulation principles used to achieve prolonged drug release are described.
Prolonged-release tablets are often categorized according to the mechanism of drug release. The following are the most common means used to achieve a slow, controlled release of the drug from tablets:
Diffusion-controlled release systems
In diffusion-controlled prolonged-release systems, the transport by diffusion of dissolved drugs in pores filled with gastric or intestinal juice or in a solid (normally polymer) phase is the release-controlling process. Depending on the part of the release unit in which the drug diffusion takes place, diffusion-controlled release systems are divided into matrix systems (also referred to as monolithic systems) and reservoir systems. The release unit can be a tablet or a nearly spherical particle of about 1 mm in diameter (a granule or a millisphere). In both cases the release unit should stay more or less intact during the course of the release process. In matrix systems, diffusion occurs in pores located within the bulk of the release unit and in reservoir systems, diffusion takes place in a thin water-insoluble film or membrane, often about 5–20 µm thick, which surrounds the release unit. Diffusion through the membrane can occur in pores filled with fluid or in the solid phase that forms the membrane.
Drug is released from a diffusion-controlled release unit in two steps:
1. The liquid that surrounds the dosage form penetrates the release unit and dissolves the drug. A concentration gradient of dissolved drug is thus established between the interior and the exterior of the release unit.
2. The dissolved drug will diffuse in the pores of the release unit or the surrounding membrane and thus be released or, alternatively, the dissolved drug will partition into the membrane surrounding the dose unit and diffuse in the membrane.
A dissolution step is thus normally involved in the release process but the diffusion step is the rate-controlling step. The rate at which diffusion will occur depends on three variables: the concentration gradient over the diffusion distance; the area and distance over which diffusion occurs; and the diffusion coefficient of the drug in the diffusion medium. Some of these variables are used to modulate the release rate in the formulation.
Reservoir systems.
In a reservoir system the diffusion occurs in a thin film surrounding the release unit (Fig. 30.13). This film is normally formed from a high molecular weight polymer. The diffusion distance will be constant during the course of the release and, as long as a constant drug concentration gradient is maintained, the release rate will be constant, i.e. a zero-order release (M = kt).
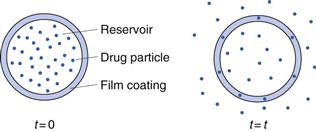
Fig. 30.13 Schematic illustration of the mechanism of drug release from a diffusion-based reservoir tablet (t = time).
One possible process for the release of the drug from a reservoir system involves partition of the drug dissolved inside the release unit to the solid membrane, followed by transport by diffusion of the drug within the membrane. Finally, the drug will partition to the solution surrounding the release unit. The driving force for the release is the concentration gradient of dissolved drug over the membrane. The release rate can be described in a simplified way by the following equation, which also summarizes the formulation factors by which the release rate can be controlled, i.e.:
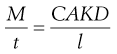 (30.1)
(30.1)
where C is the solubility of the drug in the liquid, A and l are the area and thickness of the membrane, D is the diffusion coefficient of the drug in the membrane, and K the partition coefficient for the drug between the membrane and the liquid at equilibrium.
In practice, the membrane surrounding the release unit often includes a water-soluble component. This can be small particles of a soluble substance, such as sucrose, or a water-soluble polymer, such as a water-soluble cellulose derivative (e.g. hydroxypropyl methylcellulose). In the latter case the polymer is used together with a water-insoluble polymer as the film-forming materials that constitute the coating. In such a membrane, the water-soluble component will dissolve and form pores filled with liquid in which the drug can thereafter diffuse. The area and length of these pores will thus constitute the diffusion area and distance. These factors can be estimated from the porosity of the membrane (e) and the tortuosity (τ) of the pores (the tortuosity refers to the ratio between the actual transport distance in the pores between two positions and the transport distance in a solution). The release rate can thus be described in a simplified way as follows:
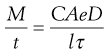 (30.2)
(30.2)
The membrane porosity and pore tortuosity can be affected by the addition of water-soluble components to the membrane.
For oral preparations, the film surrounding the release units is normally based on high molecular weight, water-insoluble polymers, such as certain cellulose derivatives (e.g. ethyl cellulose) and acrylates. The film often also includes a plasticizer. In the case of drug release through liquid-filled pores, a small amount of a water-soluble compound is also added, as described above. Reservoir systems today are normally designed as multiple-unit systems rather than single units.
Matrix systems.
In a matrix system the drug is dispersed as solid particles within a matrix formed of a water-insoluble polymer, such as polyvinyl chloride (Fig. 30.14) or in a matrix forming a gel in contact with water (see below). Initially, drug particles located at the surface of the release unit will be dissolved and the drug released rapidly. Thereafter, drug particles at successively increasing distances from the surface of the release unit will be dissolved and released by diffusion in liquid filled pores of the matrix or in the gel to the exterior of the release unit. Thus, the diffusion distance of dissolved drug will increase as the release process proceeds. The drug release, in terms of the cumulative amount of drug (M) released from a matrix can often be approximated to be proportional to the square root of time, i.e. M = kt1/2.
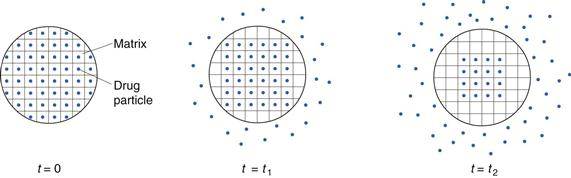
Fig. 30.14 Schematic illustration of the mechanism of drug release from a diffusion-based matrix tablet (t = time).
The main formulation factors by which the release rate from a matrix system can be controlled are the amount of drug in the matrix, the porosity of the release unit, the length of the pores in the release unit (dependent on the size of the release unit and the pore tortuosity) and the solubility of the drug (which regulates the concentration gradient). The characteristics of the pore system can be affected by, for example, the addition of soluble excipients and by the compaction pressure during tableting.
Matrix systems are traditionally designed as single-unit systems, normally tablets, prepared by tableting. However, alternative preparation procedures are also used, especially for release units that are smaller than tablets. Examples of such techniques are extrusion, spray congealing and casting.
Dissolution-controlled release systems
In dissolution-controlled prolonged-release systems, the rate of dissolution of the drug or another ingredient in the gastrointestinal juices is the release-controlling process. It is obvious that a sparingly water-soluble drug can form a preparation of a dissolution-controlled prolonged-release type. Reduced drug solubility can be accomplished by preparing poorly soluble salts or derivatives of the drug. An alternative means of achieving prolonged release based on the dissolution-control principle is to incorporate the drug in a slowly dissolving carrier or by covering drug particles with a slowly dissolving coating. In the latter case, the release of the drug from such units occurs in two steps:
1. The liquid that surrounds the release unit dissolves the coating (rate-limiting dissolution step).
2. The solid drug is exposed to the liquid and subsequently dissolves.
In order to obtain an extended release based on dissolution of a coating, the tablet is formulated to release the drug in a series of consecutive pulses, i.e. a pulsatile release. This can be accomplished by dividing the drug dose into a number of smaller release units, often nearly spherical granules of about 1 mm in diameter, which are coated in such a way that the dissolution time of the coatings will vary (Fig. 30.15). A variation in dissolution time of the coating can be accomplished by varying its thickness or its solubility. Release units with different release times will be mixed and formed into a disintegrating tablet.
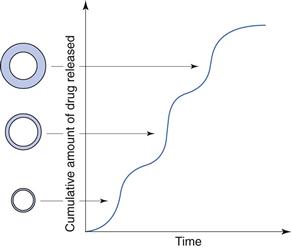
Fig. 30.15 Schematic representation of the cumulative amount of drug released from a dissolution-based (due to differences in coating thickness) pulsatile-release preparation.
Delayed-release tablets are normally formulated as dissolution-controlled preparations. In the case of gastro-resistant tablets, the dissolution is inhibited until the preparation reaches the higher pH of the small intestine, where the drug is released in a relatively short time.
Erosion-controlled release systems
In erosion-controlled prolonged-release systems, the rate of drug release is controlled by the erosion of a matrix in which the drug is dispersed. The matrix is normally formed by a tableting operation and the system can thus be described as a single-unit system. The erosion in its simplest form can be described as a continuous liberation of matrix material (both drug and excipient) from the surface of the tablet, i.e. surface erosion. The consequence will be a continuous reduction in tablet weight during the course of the release process (Fig. 30.16). Drug release from an erosion system can thus be described in two steps:
1. Matrix material, in which the drug is dissolved or dispersed, is liberated from the surface of the tablet.
2. The drug is subsequently exposed to the gastrointestinal fluids and mixed with (if the drug is dissolved in the matrix) or dissolved in (if the drug is suspended in the matrix) the fluid.
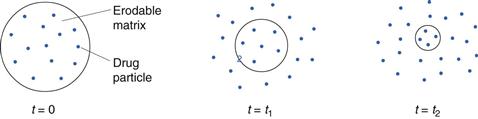
The release rate of a drug from an eroding system can often approximate to zero order for a significant part of the total release time.
The eroding matrix can be formed from different substances. One example is lipids or waxes, in which the drug is dispersed. Another example is polymers that gel when in contact with water (e.g. hydroxyethyl cellulose). The gel will subsequently erode and release the drug dissolved or dispersed in the gel. However, as discussed above, a prolonged-release tablet based on a gel-forming matrix may also be classified as a diffusion-controlled release system. Diffusion may be the dominating release mechanism in some cases.
Osmosis-controlled release systems
In osmosis-controlled prolonged-release systems, the flow of liquid into the release unit, driven by a difference in osmotic pressure between the inside and the outside of the release unit, is used as the release-controlling process. Osmosis can be defined as the flow of a solvent from a compartment with a low concentration of solute to a compartment with a high concentration. The two compartments are separated by a semi-permeable membrane, which allows flow of solvent but not of solute.
In the most simple type of osmosis-controlled drug release, the following sequence of steps is involved in the release process:
1. Osmotic transport of liquid into the release unit.
2. Dissolution of drug within the release unit.
3. Convective transport of a saturated drug solution by pumping of the solution through a single orifice or through pores in the semi-permeable membrane.
The pumping of the drug solution can be accomplished in different ways. One example is a tablet which includes an expansion layer, i.e. a layer of a substance that swells in contact with water, the expansion of which will press out the drug solution from the release unit. Alternatively, the increased volume of fluid inside the release unit will increase the internal pressure and the drug solution will thus be pumped out.
If the flow rate of incoming liquid to the release unit is the rate-controlling process, the drug release rate can be described as:
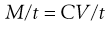 (30.3)
(30.3)
where V is the volume of incoming liquid. The flow rate of incoming liquid under steady-state conditions is a zero-order process and the release rate of the drug will therefore also be a zero-order process. The water flow is not affected by the flow and pH of the dissolution medium. However, the water flow rate and hence drug release rate can be affected by a number of formulation factors, such as the osmotic pressure of the drug solution within the release unit, the drug solubility, and the permeability and mechanical properties of the membrane.
Osmosis-controlled release systems can be designed as single-unit or multiple-unit tablets. In the first case, the drug solution can be forced out from the tablet through a single orifice (Fig. 30.17) formed in the membrane by boring with a laser beam. Alternatively, the drug solution can flow through a number of pores formed during the uptake of water. Such pores can be formed by the dissolution of water-soluble substances present in the membrane, or by straining of the membrane owing to the increased internal pressure in the release unit. In the case of multiple-unit release tablets, the transport occurs in formed pores.
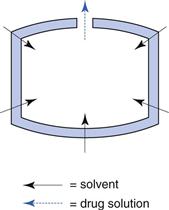
Tablet testing
Test methods
In tablet formulation development and during manufacturing of tablets, a number of procedures are used to assess the quality of the tablets. Some test methods are described in pharmacopoeias and these tests are traditionally concerned with the content and the in vitro release of the active ingredient. Test methods not described in pharmacopoeias are sometimes referred to as non-compendial and concern a variety of quality attributes that need to be evaluated, such as the porosity of tablets. Below, some of the tests used in the quality evaluation of tablets are briefly described.
Uniformity of content of active ingredient
A fundamental quality attribute for all pharmaceutical preparations is the requirement for a constant dose of drug between individual tablets. In practice, small variations between individual preparations are accepted and the limits for this variation appear as standards in pharmacopoeias. Traditionally, uniformity of dose or dose variation between tablets is tested in two separate tests: uniformity of weight (mass) and uniformity of active ingredient.
The test for uniformity of weight is carried out by collecting a sample of tablets from a batch and determining their individual weights. The average weight of the tablets is then calculated. The sample complies with the standard if the individual weights do not deviate from the mean more than is permitted in terms of percentage.
If the drug substance forms the greater part of the tablet mass, any weight variation obviously indicates a variation in the content of active ingredient. Compliance with the standard thus helps to ensure that uniformity of dosage is achieved. However, in the case of potent drugs which are administered in low doses, the excipients form the greater part of the tablet weight and the correlation between tablet weight and amount of active ingredient can be poor (Fig. 30.18). Thus, the test for weight variation must be combined with a test for variation in content of the drug substance. Nevertheless, the test for uniformity of weight is a simple way to assess variation in drug dose, which makes the test useful as a quality control procedure during tablet production.
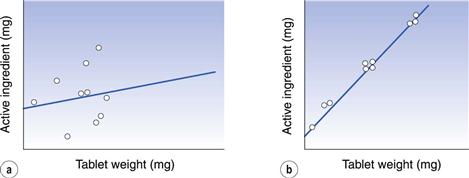
Fig. 30.18 Correlation between amount of active ingredient and tablet weight for (a) a low-dose (drug content 23% of tablet weight) and (b) a high-dose (drug content 90% of tablet weight) tablet. (Courtesy of Airth et al, 1967, with permission.)
The test for uniformity of drug content is carried out by collecting a sample of tablets followed by a determination of the amount of drug in each. The average drug content is calculated and the content of the individual tablets should fall within specified limits.
Disintegration
As discussed above, the drug release process from tablets often includes a step at which the tablet disintegrates into smaller fragments. In order to assess this, disintegration test methods have been developed and examples of these can be found in pharmacopoeias.
The test is carried out by agitating a given number of tablets in an aqueous medium at a defined temperature and the time to reach the endpoint of the test is recorded. The preparation complies with the test if the time to reach this endpoint is below a given limit. The endpoint of the test is the point at which all visible parts of the tablets have been eliminated from a set of tubes in which the tablets have been held during agitation. The tubes are closed at the lower end by a screen and the tablet fragments formed during the disintegration are eliminated from the tubes by passing the screen openings, i.e. disintegration is considered to be achieved when no tablet fragments remain on the screen (fragments of coating may however remain).
A disintegration instrument (Fig. 30.19) consists normally of six chambers, i.e. tubes open at the upper end and closed by a screen at the lower. Before disintegration testing, one tablet is placed in each tube and normally a plastic disc is placed over the tablet. The tubes are placed in a water bath and raised and lowered at a constant frequency in the water in such a way that at the highest position of the tubes, the screen (and thus the tablet held down by the plastic disc) remains below the surface of the water.
Tests for disintegration do not normally seek to establish a correlation with in vivo behaviour. Thus, compliance with the specification is no guarantee of an acceptable release and uptake of the drug in vivo and hence an acceptable clinical effect. However, it is reasonable to assume that a preparation which fails to comply with the test is unlikely to be efficacious. Disintegration tests are, however, useful for assessing the potential importance of formulation and process variables on the biopharmaceutical properties of the tablet, and as a control procedure to evaluate the quality reproducibility of the tablet during production.
Dissolution
Dissolution testing is the most important way to study, under in vitro conditions, the release of a drug from a solid dosage form and thus represents an important tool to assess factors that affect the bioavailability of a drug from a solid preparation. During a dissolution test, the cumulative amount of drug that passes into solution is measured as a function of time. The test thus describes the overall rate of all the processes involved in the release of the drug into a bioavailable form. The reasons for and the procedures to enable the dissolution testing of solid dosage forms is discussed in detail in Chapter 35 but will be covered here briefly in the context of this present chapter.
Dissolution is accomplished by locating the tablet in a chamber containing a flowing dissolution medium. So that the method is reproducible, all factors that can affect the dissolution process are standardized. This includes factors that affect the solubility of the substance (i.e. the composition and temperature of the dissolution medium) and others that affect the dissolution process (such as the concentration of dissolved substance in, and the flow conditions of, the fluid in the dissolution chamber).
A number of compendial and non-compendial methods exist for dissolution testing, which can be applied to both drug substances and formulated preparations. With respect to preparations, the main test methods are based on forced convection of the dissolution medium and can be classified into two groups: stirred-vessel methods and continuous-flow methods.
The composition of the dissolution medium might vary between different test situations. Pure water or water mixed with acids or bases to adjust pH are frequently used. In addition, liquids showing a closer resemblance to physiological conditions are also used. These are often referred to as simulated gastric or intestinal fluids or bio-relevant fluids (see Chapter 35). Also, other dissolution media might be used, such as solvent mixtures, if the water solubility of the drug is very low. Finally, dissolution studies may be carried out in water-ethanol solutions in order to assess the potential effect of intake of alcohol drinks close to the administration of a tablet.
Stirred-vessel methods
The most important stirred-vessel methods are the rotating-basket method (Fig. 30.20) and the paddle method (Fig. 30.21). Details of these can be found in monographs in the European or US pharmacopoeias and are further discussed in Chapter 35. Both use the same type of vessel, which is filled with a dissolution medium of controlled volume and temperature. In the paddle method, the tablet is placed in the vessel and the dissolution medium is agitated by a rotating paddle. In the rotating-basket method, the tablet is placed in a small basket formed from a screen. This is then immersed in the dissolution medium and rotated at a given speed.
Fig. 30.20 Diagram of a dissolution instrument based on the rotating-basket method for the testing of tablet dissolution rate. (Courtesy of Banakar, 1992, with permission.)
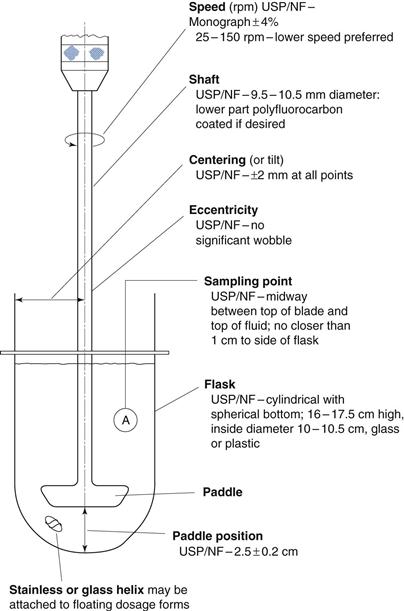
Fig. 30.21 Diagram of a dissolution instrument based on the rotating paddle method for the testing of tablet dissolution rate. (Courtesy of Banakar, 1992, with permission.)
Continuous-flow methods
In the continuous-flow method (e.g. USP Apparatus 4, Chapter 35), the preparation is held within a flow cell, through which the dissolution medium is pumped at a controlled rate from a large reservoir. The liquid which has passed the flow cell is collected for analysis of drug content. The continuous-flow cell method may have advantages over stirred-vessel methods, e.g. it maintains sink conditions throughout the experiment and avoids floating of the preparation.
The amount of drug dissolved is normally analysed more or less continuously as the concentration in the vessel as a function of time. However, sometimes a single measurement can be performed if required in the pharmacopoeial or product specification, i.e. the amount of drug dissolved at a certain time period is determined.
Mechanical strength
The mechanical strength of a tablet is associated with the resistance of the solid specimen to fracturing and attrition. An acceptable tablet must remain intact during handling at all stages, i.e. during production, packaging, warehousing, distribution, dispensing and administration by the patient. Thus, an integral part of the formulation and production of tablets is the determination of their mechanical strength. Such testing is carried out to:
• assess the importance of formulation and production variables for the resistance of a tablet to fracturing and attrition during formulation work, process design and scaling up
• control the quality of tablets during production (in-process and batch control)
• characterize the fundamental mechanical properties of materials used in tablet formulation.
A number of methods is available for measuring mechanical strength and they give different results. Also, the hardness of a tablet is sometimes measured by indentation testing. The hardness of a specimen is associated with its resistance to local permanent deformation and is thus not a measure of the resistance of the tablet to fracturing.
The most commonly used methods for strength testing can be subcategorized into two main groups: attrition resistance methods (friability tests) and fracture resistance methods.
Attrition resistance methods
The idea behind attrition resistance methods is to mimic the kind of forces to which a tablet is subjected during handling between its production and its administration. These are also referred to as friability tests; a friable tablet is one that is liable to erode mechanically during handling. During handling, tablets are subjected to stresses from collisions and tablets sliding towards one another and other solid surfaces, which can result in the removal of particles or particle clusters from the tablet surface. The result will be a progressive reduction in tablet weight and a change in its appearance. Such attrition can occur even though the stresses are not high enough to break or fracture the tablet into smaller pieces. Thus, an important property of a tablet is its ability to resist attrition so as to ensure that the correct amount of drug is administered and that the appearance of the tablet does not change during handling. Another application of a friability method is to detect incipient capping, as tablets with no visible defects can cap or laminate when stressed by an attrition method.
The most common experimental procedure for determining attrition resistance involves the rotation of tablets in a cylinder, followed by the determination of weight loss after a given number of rotations. Another approach is to shake tablets intensively in a jar of similar dimensions to a pack-jar. Normally, weight loss of less than 1% during a friability test is required. In addition, the tablets should not show capping or cracking during such testing.
Fracture resistance methods
Analysis of the fracture resistance of tablets involves the application of a load on the tablet and the determination of the force needed to fracture or break the specimen along its diameter or other axis.
In order to obtain a controlled loading during the test, care must be taken to ensure that the load is applied under defined and reproducible conditions in terms of the type of load applied (compression, pulling, twisting, etc.) and the loading rate.
For compressive loading of tablets, the test is simple and reproducible under controlled conditions and the diametral compression test has therefore a broad use during formulation development and tablet production. In such compression testing, the tablet is placed against a platen (a flat metal plate) and the load is applied along its diameter by a movable platen. The tablet fails ideally along its diameter, i.e. parallel to the compression load, in a single fracture into two pieces of similar size (Fig. 30.22) and the fracture force is recorded. This mode of failure is actually a tensile failure even though it is accomplished here by compressive loading. The force needed to fracture the tablet by diametral compression is often somewhat unfortunately referred to as the crushing strength of the tablet. The term ‘hardness’ is also used in the literature to denote the failure force, which is in this context incorrect as hardness is a deformation property of a solid.
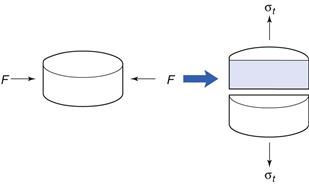
The force needed to fracture a tablet depends on the tablet’s dimensions. An ideal test, however, should allow comparison between tablets of different sizes or even shapes. This can be accomplished by assessing the strength of the tablet, i.e. the force needed to fracture the tablet per unit fracture area. A strength test requires that the fracture mode (i.e. the method by which the crack is formed) can be controlled and that the stress state along the fracture plane can be estimated. The simplest and most common tensile strength test is the indirect diametral compression test described above. For a cylindrical flat-faced tablet, the tensile strength (σT) can be calculated by Eqn 30.4, provided that the tablet fails in a tensile fracture mode characterized by a single linear fracture across the diameter of the cylindrical specimen.
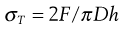 (30.4)
(30.4)
where F is the force needed to fracture the tablet and D and h are the diameter and the height of the cylindrical flat-faced tablet, respectively.
In practice, more complicated failure characteristics than tensile failure are often obtained during diametral compression (Fig. 30.23), which will prevent the strict application of the calculation procedure. It should be pointed out that the tensile strength of convex-faced tablets can also be calculated by using other equations.
Fig. 30.23 Examples of different types of failure induced by diametral compression. (a) Simple tensile failure. (b) Triple cleft failure. (c) Failure due to shear at platen edges. (Courtesy of Davies and Newton, 1996, with permission.)
Alternative procedures to measure the tensile strength of a tablet include breaking the tablet in a bending test or directly pulling the tablet apart until it fractures. The latter may be used to detect weaknesses in the compact in the axial direction, which is an indication of capping or lamination tendencies in the tablet.
Fundamental aspects of the compression of powders
Mechanisms of compression of particles
The compressibility of a powder is defined as its propensity, when held within a confined space, to reduce in volume when subjected to a load. The compression of a powder bed is normally described as a sequence of processes. Initially, the particles in the die are rearranged, resulting in a closer packing structure and reduced porosity. At a certain load the reduced space and the increased interparticulate friction will prevent any further interparticulate movement. The subsequent reduction of the tablet volume is therefore associated with changes in the dimensions of the particles.
Particles, either whole or a part, can change their shape temporarily by elastic deformation and permanently by plastic deformation (Fig. 30.24). Particles can also fracture into a number of smaller, discrete particles, i.e. particle fragmentation. The particle fragments can then find new positions, which will further decrease the volume of the powder bed. When the applied pressure is further increased, the smaller particles formed could again undergo deformation. Thus, one single particle may undergo this cycle of events several times during one compression. As a consequence of compression, particle surfaces are brought into close proximity to each other and particle-particle bonds can be formed.
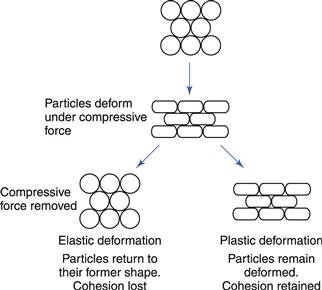
Fig. 30.24 Schematic illustration of particle deformation, elastic and plastic, during compression. (Courtesy of Armstrong, 1982, with permission.)
Elastic and plastic deformations of particles are time-independent processes, i.e. the degree of deformation is related to the applied stress and not the time of loading. However, deformation can also be time-dependent, i.e. the degree of deformation is related to the applied stress and the time of loading. This deformation behaviour is referred to as viscoelastic and viscous deformation of a material. The consequence is that the compression behaviour of a material might depend on the loading conditions during the formation of a tablet in terms of the punch displacement-time relationship. Many pharmaceutical substances seem to have a viscous character, i.e. are strain-rate sensitive, and the properties of the tablet are thus dependent on the punch displacement-time relationship for the compression process.
Elastic deformation can be described as a densification of the particle due to a small movement of the cluster of molecules or ions that forms the particle, e.g. a crystal lattice or a cluster of disordered molecules. Plastic deformation is considered to occur by the sliding of molecules along slip planes within the particles. For real crystals, such slip planes are formed at defects in the crystal lattice, especially dislocations.
The majority of powders handled in pharmaceutical production consist not of non-porous primary particles but rather of granules, i.e. porous secondary particles formed from small dense primary particles. For granules, a larger number of processes is involved in their compression. These can be classified into two groups:
• physical changes in the granules, i.e. the secondary particles
• physical changes in the primary particles from which the granules are formed.
The latter concern changes in the dimensions of the primary particles due to elastic and plastic deformation and fragmentation. Such processes may be significant for the strength of tablets. It is, for example, common for a capping-prone substance, when compacted as dense particles, to also be prone to cap or laminate during compaction in the form of granules, such as substrate–binder granules. However, in terms of the evolution of the tablet structure, the physical changes in the granules that occur during compression are of primary importance.
At low compression forces, the reduction in volume of the bed of granules can occur by a rearrangement within the die. However, granules are normally fairly coarse, which means that they spontaneously form a powder bed of relatively low voidage (i.e. the porosity of the inter-granular spaces). Therefore, this initial rearrangement phase is probably of limited importance with respect to the total change in bed volume of the mass. With increased loading, a further reduction in bed volume therefore requires changes in the structure of the granules. The granules can deform, both elastically and permanently, but also densify, i.e. reduce their intragranular porosity. By these processes, granules can still be described as coherent units but their shape and porosity will change.
Granules can also be broken down into smaller units by two different mechanisms:
• Primary particles might be removed from the surface of granules when they slide against each other or against the die wall. This can be described as erosion or attrition, rather than fracturing. This mechanism occurs primarily for granules with a rough surface texture.
• Granules can fracture into a number of smaller ones, i.e. granule fragmentation.
Studies on the compression properties of granules formed from pharmaceutical substances have indicated that granules are not prone to fracture into smaller units during compression over a normal range of applied pressures. Thus, permanent deformation and densification dominate the compression event although cracking and attrition may occur in parallel, especially for irregular, rough granules.
The dominating compression mechanisms for dense particles and granules are summarized in Table 30.2. The relative occurrence of fragmentation and deformation of solid particles during compression is related to the fundamental mechanical characteristics of the substance, such as its elasticity and plasticity. For granules, both the mechanical properties of the primary particles from which the granules are formed and the physical structure of the granules, such as their porosity and shape, will affect the relative occurrence of each compression mechanism.
Table 30.2
Dominating compression mechanisms for dense particles and granules (porous particles)
| Dense particles | Granules |
| Repositioning of particles | Repositioning of granules |
| Particle deformation | Granule deformation |
| elastic | (is permanent) |
| plastic | Granule densification |
| viscous/viscoelastic | Granule fragmentation/attrition |
| Particle fragmentation | Deformation of primary particles |
Evaluation of compression behaviour
Procedures
The procedures used in research and development work to evaluate the compression behaviour of particles and the mechanisms of compression involved in the volume reduction process are of two types:
• characterization of ejected tablets
• characterization of the compression and decompression events.
Concerning the characterization of ejected tablets, the most important procedures used are inspection and the determination of the pore structure of the tablet, in terms of a mean pore size, pore size distribution and specific surface area. A less common approach is to calculate ratios between the mechanical strengths of tablets measured in different directions.
Concerning characterization of the compression and decompression events, these procedures are based on relationships between parameters that can be derived from the compaction process (Table 30.3). Some of the most common approaches used in this context are described below.
Inspection of tablets
The inspection of tablets, e.g. by scanning electron microscopy and by profilometry, is an important means of studying changes in the physical properties of particles during compression. Such changes include fragmentation into smaller particles, permanent shape changes due to deformation and finally, the formation of cracks within the particles. Such inspection will also give information about the relative positions of particles within the tablet and hence the interparticulate pore structure. The fracture path during strength testing, i.e. failure around or across the particles, can also be estimated from inspection of the tablet fracture surface.
In addition to the inspection of intact tablets, studies of the fragmentation of particles during compression can be obtained by analysing the size and size distribution of particles obtained by deaggregation of a tablet. Such deaggregation can occur spontaneously by disintegration of the tablet in a liquid, or be created mechanically. Studies on such deaggregated tablets have indicated that powder compression can effectively reduce the size of particles and result in a wider particle size distribution of the cohered particles within a tablet.
Pore structure and specific surface area of tablets
One of the most important ways to study the evolution of tablet structure during compression is to measure some characteristic of the pore structure of the tablet. Information on pore size distribution can be obtained by mercury intrusion measurements and by gas adsorption-desorption. However, the most common way to evaluate the pore system of a tablet has been to measure the surface area of the tablet by air permeability or gas adsorption. The former has also been used to derive an indication of the mean pore size in a tablet.
Surface area measurements have been used as a means to assess fragmentation during compaction by measuring the specific surface area of a particulate solid before and after compaction, or measuring changes in tablet surface area with compaction pressure.
The slope of the relationship between tablet surface area and applied pressure represents an indication of the degree of fragmentation that occurs during compression and can be used to classify materials with respect to their fragmentation propensity (Fig. 30.25). It should be pointed out that the calculation of tablet surface area from air permeability measurements may give erroneous values as a result of the assumptions made in the derivation of the calculation procedure. In spite of this, the method has been shown to give useful data in terms of describing the fragmentation propensity of a substance.
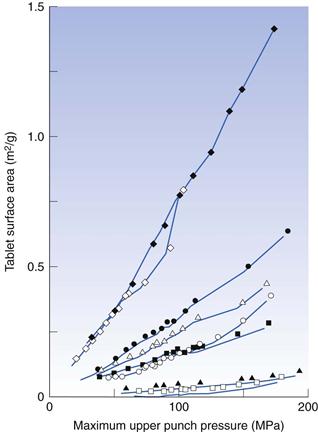
Fig. 30.25 The tablet surface area, measured by air permeametry, as a function of compaction pressure for a series of pharmaceutical substances.  sodium chloride,
sodium chloride, 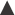 sodium bicarbonate,
sodium bicarbonate,  saccharose,
saccharose,  sodium citrate, Δ ascorbic acid, • lactose, ◊ paracetamol, ♦ Emcompress. (Courtesy of Alderborn et al 1985, with permission.)
sodium citrate, Δ ascorbic acid, • lactose, ◊ paracetamol, ♦ Emcompress. (Courtesy of Alderborn et al 1985, with permission.)
The relationship between tablet surface area and applied pressure is, however, strongly dependent on the original surface area of the powder, i.e. the tablet surface area increases more markedly with applied pressure when the original particle size was smaller. Attempts have been made in the literature to derive an expression similar to those describing the size reduction of particles during milling (see Chapter 10), by which a measure of the propensity of particles to fragment independent of the original powder surface area can be calculated.
It is generally assumed that a change in the size of a particle affects the mechanics of particle deformation, i.e. how a particle responds to an applied load. Such a size-related change in the mechanics of particles can, for example, be attributed to a reduced probability of the presence of flaws in the crystal structure at which a catastrophic failure is initiated. It seems possible, therefore, that at a limiting particle size, fragmentation might cease. Examples of such transitions from a brittle to a plastic behaviour have been reported and the particle size at which this transition takes place is referred to as the critical particle size. This critical size has been suggested to vary markedly between different substances.
Force-displacement profiles
The relationship between upper punch force and upper punch displacement during compression, often referred to as the force-displacement profile, has been used as a means to derive information on the compression behaviour of a powder and to make predictions on its tablet-forming ability. The area under a force-displacement curve represents the work or energy involved in the compression process. Different procedures have been used to analyse the curves.
One suggested approach is based on the division of the force-displacement curve into different regions (denoted E1, E2 and E3 in Fig. 30.26). It has been suggested that the areas of E1 and E3 should be as small as possible for a powder to perform well in a tableting operation and give tablets of a high mechanical strength. An alternative proposed approach is based on mathematical analysis of the force-displacement curve from the compression phase, e.g. in terms of a hyperbolic function.
Fig. 30.26 The relationship between upper punch force and upper punch displacement during compression and decompression of a powder. (Courtesy of Ragnarsson, 1996, with permission.)
Force-displacement curves have some use in pharmaceutical development as an indicator of the tablet-forming ability of powders, including the assessment of the elastic properties of materials from the decompression curve. They can also be used as a means to monitor the compression behaviour of a substance in order to document and evaluate reproducibility between batches.
Force-displacement measurements have also been used in fundamental studies on the energy changes during compaction of powders, i.e. a thermodynamic analysis of the process of compact formation. The energy applied to the powder can be calculated from the area under the force-displacement curve. This compaction energy is used to overcome friction between particles, to deform particles both permanently and reversibly and to create new particle surfaces by fragmentation. The thermal energy released during compaction can be assessed by calorimetry, i.e. the die is constructed as a calorimeter. The heat released during compression is the result of particle deformation, i.e. energy is consumed during deformation and thereafter partly released when the deformation is completed, and the result of the formation of interparticulate bonds.
Data have been reported indicating that the net effect of a compaction process is exothermic, i.e. more thermal energy is released during compaction than is applied to the powder in terms of mechanical energy. The main explanation for this is released bonding energy in the form of heat due to the formation of bonds between particles.
Tablet volume-applied pressure profiles
In both engineering and pharmaceutical sciences, the relationship between volume and applied pressure during compression is the main approach to deriving a mathematical representation of the compression process. A large number of tablet volume-applied pressure relationships exist. In addition to tablet volume and applied pressure parameters, such expressions include some constants which often are defined in physical terms. However, only for a few equations has the physical significance of the constants been generally accepted. Among these, the most recognized expression in pharmaceutical science is the tablet porosity-applied pressure function according to Heckel (see Duberg & Nyström 1986).
Heckel equation.
Tablet porosity can be measured either on an ejected tablet or on a powder column under load, i.e. in a die. The latter approach is more common as it can be performed rapidly with a limited amount of powder. A problem might be that the compression time is different at each pressure, which could affect the profile for materials having pronounced time-dependent compression behaviour.
The compression of a powder can be described in terms of a first-order reaction where the pores are the reactant, and the densification is the product. Based on this assumption, the following expression was derived:
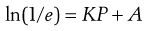 (30.5)
(30.5)
where e is the tablet porosity, P the applied pressure, A is a constant suggested to reflect particle rearrangement and fragmentation, and K the slope of the linear part of the relationship which is suggested to reflect the deformation of particles during compression. The reciprocal of the slope value K is often calculated and considered to represent the yield stress or yield pressure (Py) for the particles, i.e.:
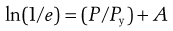 (30.6)
(30.6)
The yield stress is defined as the stress at which plastic deformation of a particle is initiated. To be able to use the Heckel yield pressure parameter to compare different substances, it is important to standardize the experimental conditions, such as tablet dimensions and speed of compaction.
Figure 30.27 shows a typical Heckel profile. The profile often shows an initial curvature (region I) which is associated with particle fragmentation and repositioning. Thereafter, the relationship is often linear over a substantial range of applied pressures (region II) and thus obeys the expression. The linear part is considered to reflect a situation where particle deformation controls the compression process and from the gradient of this linear part, the yield pressure can be calculated. Finally, the profile again deviates from the linear relationship (region III) and this curvature is considered to reflect elastic deformation of the whole tablet.
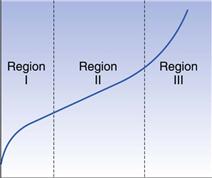
Fig. 30.27 A typical example of a Heckel profile indicating the three regions of powder compression. (Courtesy of Sun and Grant, 2001, with permission.)
Strain rate sensitivity.
Another proposed use of yield pressure values from Heckel profiles is to assess the time-dependent deformation properties of particles during compression by comparing yield pressure values derived under compression at different punch velocities. A term denoted the strain rate sensitivity (SRS) has been proposed (Roberts & Rowe 1985) as a characteristic of the time dependency of a powder:
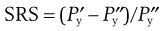 (30.7)
(30.7)
where  is the yield pressure derived at a high punch velocity and
is the yield pressure derived at a high punch velocity and  is that derived at a low punch velocity.
is that derived at a low punch velocity.  is normally higher than
is normally higher than  and the SRS is thus a positive value.
and the SRS is thus a positive value.
The discussion on the use of Heckel profiles to derive a measure of the compression yield pressure is applicable to the compression of powders consisting of solid particles. It should be emphasized that the interpretation of 1/K in terms of a mean yield stress for the particles is under debate. Nevertheless, support has been presented that such an interpretation is valid for solid (non-porous) particles. For porous particles, i.e. granules and pellets, the Heckel procedure is inadequate for the derivation of a measure of deformability or granule strength. The problem of applying the Heckel approach to the compression of porous particles is related to the need to assess the porosity of the reactant pore system. The pore space of interest in relation to the Heckel equation is intergranular and the problem of quantifying this is discussed above.
Kawakita equation.
A promising means of assessing the compression mechanics of granules is to calculate a compression shear strength from the Kawakita equation. This was derived from the assumption that, during powder compression in a confined space, the system is in equilibrium at all stages, so that the product of a pressure term and a volume term is constant. The equation can be written in the following linear form:
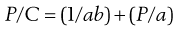 (30.8)
(30.8)
where P is applied pressure, C the degree of volume reduction and a and b are constants. The degree of volume reduction relates the initial height of the powder column (ho) to the height of the powder column (the compact) at an applied pressure P (hp) as follows:
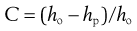 (30.9)
(30.9)
This equation has been applied primarily to powders of solid particles. However, it has been suggested (Adams et al 1994) that the compression parameter 1/b corresponds to the strength of granules in terms of their compression strength. The procedure thus represents a possible means to characterize the mechanical property of granules from a compression experiment.
Evaluation of die wall friction during compression
Friction is a serious problem during tableting. Consequently, a series of procedures has been developed with the aim of assessing the friction between the powder or tablet and the die wall during compression and ejection, which can be used during tablet formulation to evaluate lubricants and formulations. These methods are based mainly on the use of force signals during powder compression or tablet ejection. The type of compression situation most commonly used in this context is a single-punch press with a movable upper punch and a stationary lower punch. In such a rig, the force is applied by the upper punch and transmitted axially to the lower punch and also laterally to the die. The ejection of the tablet involves the application of an ejection force by the lower punch. Typical force profiles during compression in a single-punch press with a stationary lower punch are given in Figure 30.28.
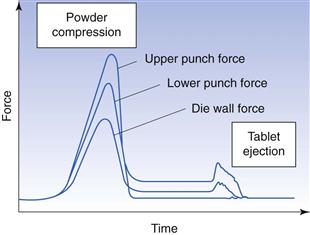
When the descending upper punch establishes contact with the powder bed in the die, the force increases with compression time. The applied force rises to a maximum value and thereafter decreases during the decompression phase to zero. Parallel with the force trace from the upper punch, force traces from the lower punch and the die will be obtained. These can be described as transmitted forces, and the force values are thus generally lower than the applied force. The force transmitted from the upper punch to the lower punch is considered to depend on a number of factors, including the friction between the powder and the die wall. These factors can be summarized in the following expression:
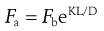 (30.10)
(30.10)
where Fa and Fb are applied and transmitted forces, L and D are the length and diameter of the powder column within the cylindrical die (Fig. 30.29) and K is a constant. The constant K is a function of the friction coefficient between particles and the die wall. Thus, the transmission of force from the upper to the lower punch depends on the friction between the powder and the die wall. Both the difference in transmitted force, i.e. upper punch force-lower punch force, and the ratio between the upper and lower punch force, i.e. lower punch force/upper punch force (often denoted the R value), are used as measures of die wall friction during compression. For a well-lubricated powder, the force transmission corresponds to R > 0.9.
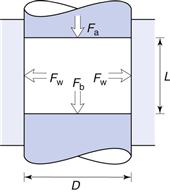
Fig. 30.29 Schematic illustration of punch and die wall forces involved during uniaxial powder compression in a cylindrical die.
After the upper punch has lost contact with the tablet and its force has consequently decreased to zero, the tablet will be positioned in the die in contact with the lower punch and the die wall. In this situation, the tablet will apply a force to both the lower punch and the die wall. The magnitude of these forces is dependent on the mechanical character of the particles formed into the tablet and also on the friction conditions at the interface between tablet and die wall.
The ejection of the tablet will result in an increased force signal from the lower punch, referred to as the ejection force. This is a function of the lateral die wall force but also of the friction condition at the interface between tablet and die wall. The maximum ejection force is thus also used as a measure of friction between tablet and die wall. One approach to assess friction during ejection is to calculate the dimensionless friction coefficient (µ) as the ratio between the ejection force (Fe) and the die wall force (Fw) at the beginning of the ejection phase, i.e.:
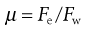 (30.11)
(30.11)
To summarize, the following procedures are mainly used to derive measures of friction between the powder or tablet and the die wall from force signals during tableting in a single-punch press:
The force applied to a powder bed during powder compression is transmitted along columns of particles formed within the powder bed. The consequence is that the applied force is non-uniformly distributed within the powder column, i.e. a stress distribution is obtained within the powder column under loading. The tablet formed will consequently show a distribution in density, i.e. there will be local variations in porosity around an overall mean porosity of the tablet. Stress and density gradients within compacts have been studied experimentally but also by modelling using computational methods. By modelling procedures, density and stress distributions within the compacts can be calculated (Fig. 30.30) and the micro-structure of and local stresses within a compact simulated (Frenning 2010). By such approaches, the importance of the deformation behaviour of particles and friction conditions during compression for the micro-structure of a compact can be studied as well as relationships between micro-structural variations and tablet quality attributes.
Fig. 30.30 Density gradients of a tablet, assessed by experiment (grey image) and by modelling (coloured image) for tablets with high tablet to die wall friction. (Courtesy of Sinka et al., 2003, with permission.)
Fundamental aspects of the compaction of powders
Bonding in tablets
The transformation of a powder into a tablet is fundamentally an interparticulate bonding process, i.e. the increased strength of the assembly of particles is the result of the formation of bonds between them. The nature of these bonds is traditionally subdivided into five types, known as the Rumpf classification:
2. Bonding by liquids (capillary and surface tension forces)
3. Binder bridges (viscous binders and adsorption layers)
In the case of compaction of dry powders, two of the suggested types of bond are often considered to dominate the process of interparticulate bond formation, i.e. bonding due to intermolecular forces and bonding due to the formation of solid bridges. Mechanical interlocking between particles is also considered as a possible but less significant bond type in tablets.
Bonding by intermolecular forces is sometimes also known as adsorption bonding, i.e. the bonds are formed when two solid surfaces are brought into intimate contact and subsequently adsorb to each other. Among the intermolecular forces, dispersion forces are considered to represent the most important bonding mechanism. This force operates in a vacuum and in a gaseous or liquid environment up to a separation distance between the surfaces of approximately 10–100 nm.
The formation of solid bridges, also referred to as the diffusion theory of bonding, occurs when two solids are mixed at their interface and accordingly form a continuous solid phase. Such a mixing process requires that molecules in the solid state are movable, at least temporarily, during compression. An increased molecular mobility can occur due to melting or as a result of a glass-rubber transition of an amorphous solid phase.
Mechanical interlocking is the term used to describe a situation where strength is provided by interparticulate hooking. This phenomenon usually requires that the particles have an atypical shape, such as needle-shaped, or highly irregular and rough particles.
For tablets of a porosity in the range 5–30%, it is normally assumed that bonding by adsorption is the dominant bond type between particles. In tablets formed from amorphous substances or from substances with low melting points, it is possible that solid bridges can be formed across the particle-particle interface. It is also reasonable that if tablets of a very low porosity, i.e. close to zero, are formed, particles can fuse together to a significant degree.
Often granules, i.e. secondary particles formed by the agglomeration of primary particles, are handled in a tableting operation. When granules are compacted, bonds will be formed between adjacent granule surfaces. For granules that do not include a binder, the fusion of adjacent surfaces during compaction is probably not a significant bonding mechanism. Thus, intermolecular bonding forces acting between intergranular surfaces in intimate contact will probably be the dominant bond type in such tablets.
Granules often include a binder. When such binder–substrate granules are compacted it is reasonable to assume that the binder also plays an important role in the formation of intergranular bonds. The binder may fuse together locally and form binder bridges between granule surfaces which cohere the granules to each other. Such bridges may be the result of a softening or melting of binder layers during the compression phase. However, different types of adsorption bonds may be active between granule surfaces. These may be subdivided into three types: binder–binder, binder–substrate and substrate–substrate bonds.
For adsorption bonds between granules in a tablet, the location of the failure during fracturing of the tablet can vary. Fractures occurring predominantly through binder bridges between substrate particles, as well as predominantly at the interface between the binder and the substrate particle, may occur. The location of the failure has been attributed to the relative strength of the cohesive (binder bridge) and adhesive (binder–substrate interface) forces acting within the granules, which can be affected by, for example, the surface geometry of the substrate particles.
The main bond types in tablets formed from dense particles (interparticulate bonds) and from granules (intergranular bonds) are summarized in Table 30.4.
Table 30.4
Suggested predominant bond types in tablets formed from dense particles (interparticulate bonds) and from granules (intergranular bonds)
| Interparticulate bonds | Intergranular bonds |
| Adsorption bonds (intermolecular forces) | Adsorption bonds of three types: binder–binder binder–substrate substrate-substrate |
| Solid bridges | Solid binder bridges |
Compactability of powders and the strength of tablets
The compactability of a powder refers to its propensity to form a coherent tablet and thus represents a critical powder property in successful tableting operations. The ability of a powder to cohere is understood in this context in a broad sense, i.e. a powder with a high compactability forms tablets with a high resistance towards fracturing and without tendencies to cap or laminate (Fig. 30.31). In practice, the most common way to assess powder compactability is to study the effect of compaction pressure on the strength of the resulting tablet, as assessed by the force needed to fracture the formed tablet while loaded diametrically, or the tensile strength of the tablet. Such relationships are often nearly linear (Fig. 30.32) above a lower pressure threshold needed to form a tablet, and up to a pressure corresponding to a tablet of a few per cent porosity. At low porosities the relationship between tablet strength and compaction pressure will often level out. This relationship can thus be described simply in terms of a three-region relationship characterized by lower and upper tablet strength thresholds and an intermediate region in which the tablet strength is pressure-dependent in an almost linear way.
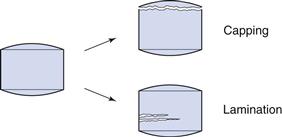
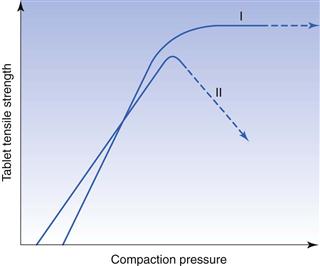
Fig. 30.32 Outline of the relationship between tablet tensile strength and compaction pressure for tablets showing no lamination (I) and for tablets showing lamination or capping (II).
Compactability profiles are sometimes also described by sigmoidal curves that can be divided principally into the three regions described above. At low pressures, the tensile strength of tablets increases as a power function with applied pressure, followed by a nearly linear region that finally levels off.
If cracks are formed in the tablet during tableting, e.g. during the ejection phase, this will often affect the assessed strength. Cracking and capping can often be induced at relatively high compaction pressures. This may be reflected as a drop in the tablet strength-compaction pressure profile.
A series of approaches to quantitatively describe or to model the compactability profile of a powder can be found in the literature. Some modelling approaches aim at describing the microstructure of a tablet in terms of an interparticulate bond structure and are based on the view that bond formation during compaction is significant for the development of coherence, i.e. it is postulated that the tensile strength of a tablet has some proportionality to the interparticulate bonds that act over the fracture area. Such models can thus be described as bond summation approaches and implicit is that all bonds are separated simultaneously during strength assessment. Since this is not consistent with the real mode of failure of a solid, the models are not fundamental approaches to understanding the strength of a tablet (see below).
Examples of equations describing compactability profiles have been given by Leuenberger (1982) and Alderborn and co-workers (Alderborn 2003). In both cases, the bond structure is modelled and related to an endpoint representing the maximum tensile strength (Tmax) that can be obtained for tablets of a specific powder. Leuenberger’s approach is based on the concept of effective numbers of interparticulate bonds in a cross-section of the tablet. It is assumed that over a cross-section of a tablet, a number of bonding and non-bonding sites exists. This number depends on the applied pressure during compression (P) and the tablet relative density (r, which is equivalent to 1 minus the tablet porosity, e). In the derivation of the expression, the term compression susceptibility (γ) was introduced, which described the compressibility of the powder and has the unit 1/pressure. The equation takes the following form:
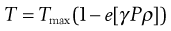 (30.12)
(30.12)
The compactability profile as described by the expression stems from the origin of the tensile strength-compaction pressure axes; the tensile strength will initially increase with compaction pressure and finally level off (compare discussion of compactability profiles above).
Alternatives to tablet strength-compaction pressure relationships for representing the compactability of powders are also used, such as the relationship between tablet strength and tablet porosity and the relationship between tablet strength and the work done by the punches during tablet formation.
Compaction is fundamentally a bonding process, i.e. strength is provided by bonds formed at the interparticulate junctions or contact sites during the compression process. Studies on the structure of fractured tablets indicate that a tablet generally fails by the breakage of interparticulate bonds, i.e. an interparticulate fracture process. However, especially for tablets of low porosity, the tablet can also fracture by breakage of the particles that form the tablet, i.e. a combination of an inter- and an intraparticulate fracture process. In general terms it seems, though, that the interparticulate contacts in a tablet represent the preferred failure path during fracturing. This conclusion is applicable to both tablets formed from solid particles and tablets formed from porous secondary particles (granules and pellets). Consequently, factors that affect the microstructure at the interparticulate junctions have been considered significant for the compactability of a powder.
Our understanding of the mechanical strength of a solid is based on the resistance of a solid body to fracture while loaded. It might seem reasonable that the sum of the bonding forces that cohere the molecules forming the solid will represent the strength of that solid. However, solids fail by a process of crack propagation, i.e. the fracture is initiated at a certain point within the solid and is thereafter propagated across a plane, thereby causing the solid to break. The consequence in terms of the strength of the solid is that the sum of the bonding forces acting over the fracture surface will be higher than the stress required to initiate failure. It is known, for example, that for crystalline solids the theoretical strength due to the summation of intermolecular bonds is much higher than the measured strength of the solid.
In order to understand the strength of solids, the process of fracture has attracted considerable interest in different scientific areas. In this context, important factors associated with the fracturing process and the strength of a specimen are the size of the flaw at which the crack is initiated and the resistance of the solid towards fracturing. The latter property can be described by the critical stress intensity factor, which is an indication of the stress needed to propagate a crack. Another fracture mechanics parameter, which is related to the critical stress intensity factor, is the strain energy release rate, which is a measure of the energy released during crack propagation.
By using the critical stress intensity factor, the tensile strength of the solid (T) is considered to relate to the flaw size (c) and the critical stress intensity factor (KIC) in the following way:
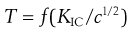 (30.13)
(30.13)
The critical stress intensity factor varies with tablet porosity. It has therefore been suggested that for compacts, such as tablets, factors such as the size of the particles within the tablet and the surface energy of the material will affect the critical stress intensity factor (Kendall 1988). These factors are also considered to control the interparticulate bond structure in a tablet.
Procedures to determine the critical stress intensity factor for a particulate solid have been described. Such a procedure normally involves the formation of a beam-shaped compact in which a notch is formed. When the compact is loaded, the fracture is initiated at the notch. The force needed to fracture the compact is determined and the critical stress intensity factor thereafter calculated. In order to assess a material characteristic, compacts of a series of porosities are formed and the series of values for the critical stress intensity factor subsequently determined is plotted as a function of the compact porosity (Fig. 30.33). The relationship thereafter may sometimes be extrapolated to zero porosity and the value thus derived is sometimes considered a fundamental material characteristic.
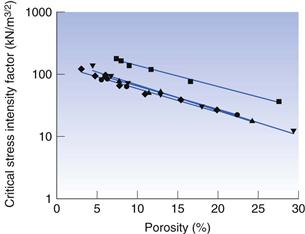
Fig. 30.33 A log-linear relationship between the critical stress intensity factor and the compact porosity for beams formed from polyethylene glycols of different molecular weight. (Courtesy of Al-Nasassrah et al., 1998, with permission.)
In addition to the evaluation of compactability profiles and fracture mechanics studies, indices and expressions have been derived within pharmaceutical science which can be described as indicators of the compactability of a powder. There are several applications of such indicators during pharmaceutical development, such as:
• the evaluation of the compactability of small amounts of particles
• the selection of drug candidates during preformulation based on technical performance
• the detection of batch variations of drugs and excipients
• the selection of excipients and the evaluation of the compactability of formulations.
Examples of such indicators which have found industrial use are the indices of tableting performance derived by Hiestand and co-workers (Hiestand 1996). Hiestand derived three indices of tableting performance, among which the bonding index (BI) and the brittle fracture index (BFI) are suggested to reflect the compactability of the powder. These indices are dimensionless ratios between mechanical properties of compacts formed at some porosity. The BI is proposed to reflect the ability of particles to form a tablet of high tensile strength, whereas the BFI is proposed to reflect the ability of a tablet to resist fracturing and lamination during handling. These indices are defined as follows:
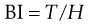 (30.14)
(30.14)
and
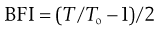 (30.15)
(30.15)
where T is the tensile strength of a normal compact, To is the tensile strength of a compact with a small hole and H is the hardness of the compact.
Post-compaction tablet strength changes
The compactability of a powder is normally understood in terms of the ability of particles to cohere during the compression process and hence to form a porous specimen of defined shape. However, the mechanical strength of tablets can change, increase or decrease, during storage without the application of any external mechanical force. The underlying mechanisms for such changes are often a complex function of the combination of ingredients in the tablet and the storage conditions, such as relative humidity and temperature.
During storage at a fairly high relative humidity, tablets can be softer and their tensile strength reduced. With increased relative humidity, the state of water adsorbed at the solid surface can change from an adsorbed gas to a liquid, i.e. water condenses in the tablet pores. Furthermore, if the solid material is freely soluble in water, it can dissolve. Both the presence of condensed water in the pores and the dissolution of a substance in the condensed water can drastically decrease tablet strength and eventually lead to the collapse of the whole tablet. However, the dissolution of a freely soluble substance in condensed pore water can also give an increase in tablet strength if the water is allowed to evaporate owing to a change in temperature or relative humidity. The result of this evaporation can be crystallization of solid material, with the subsequent formation of solid bridges between particles in the tablet and increased tablet strength.
In addition to the mechanisms involving the presence of condensed pore water, several other mechanisms have been proposed to cause an increase in tablet strength during storage at a relative humidity at which condensation of water is unlikely to occur. One such mechanism is a continuing viscous deformation of particles after the compaction process is completed. This phenomenon is referred to as stress relaxation of tablets. The increase in tablet strength can be significant with no, or minor, detectable changes in its physical structure. However, viscous deformation of small parts of particles might change the microstructure of the tablet in terms of the relative orientation of particle surfaces and the geometry of the interparticle voids and thus affect the resistance to fracturing of a tablet. A characteristic feature for stress relaxation changes is that the tablet strength changes occur for a limited time in connection with the compaction phase. An increase in tablet strength during storage may also be related to a polymorphic transformation, i.e. a change in the crystal structure of particles.
Relationships between material properties and tablet strength
Factors of importance for powder compactability
A number of empirical studies exist in the pharmaceutical literature with the aim of mapping factors that affect the structure of a tablet and its mechanical strength, i.e. tensile strength, resistance towards attrition and capping tendencies. These factors can be classified into three groups: material and formulation factors; processing factors (choice of tablet machine and operation conditions); and environmental factors (relative humidity, etc.).
Of special importance from a formulation perspective are the physical and mechanical properties of the particles used in the formulation, and how these particles are combined in granulation and mixing steps. Relationships for powders consisting of one component; of two components, such as a filler and a lubricant or a dry binder; or of several components have been discussed in this context.
Compaction of solid particles
As discussed above, it is often assumed that the evolution of the interparticulate structure of a tablet, in terms of bonds between particles and the pores between the particles, will be significant for the mechanical strength of the tablet. Thus, the material-related factors that control the evolution of the microstructure of the tablet have been discussed as important factors for the compactability of a powder. In this context, the compression behaviour and the original dimensions of the particles have received special interest.
As already mentioned, the degree of fragmentation and permanent deformation that particles undergo during compression are significant for tablet structure and strength. It has been suggested that both fragmentation and deformation are strength-producing compression mechanisms. The significance of particle fragmentation has been considered to be related to the formation of small particles which constitute the tablet, with the consequence that a large number of contact sites between particles at which bonds can be formed will be developed and the voids between the particles will be reduced in size. The significance of permanent deformation has been explained in terms of an effect on the area of contact of the interparticulate contact sites, with a subsequent increased bonding force. The relative importance of these mechanisms for the bonding between particles in a tablet and the resistance of a tablet towards fracturing has not, however, been fully clarified. Concerning elastic deformation, which is recoverable, this is considered as a disruptive rather than a bond-forming mechanism. Poor compactability, in terms of low tablet strength and capping/lamination, has been attributed to elastic properties of the solid. A summary of proposed advantages and disadvantages of the different particle compression mechanisms for the ability of the particles to form tablets is given in Table 30.5.
Table 30.5
Proposed advantages and disadvantages of the different compression mechanisms in relation to the tablet-forming ability of the powder
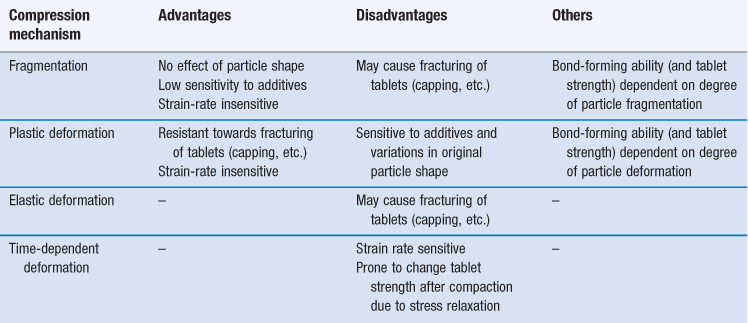
It is sometimes considered that one of the most important properties of particles for the mechanical strength of a tablet is their size before compaction. A number of empirical relationships between particle dimensions before compaction and the mechanical strength of the resultant tablet can thus be found in the literature. As a rule, it is normally assumed that a smaller original particle size increases tablet strength. However, it is also suggested that the effect of original particle size is in relative terms limited for powder compactability, with the possible exception of very small (i.e. micronized) particles. Reported data show, however, that different and sometimes complex relationships between particle size and tablet strength can be obtained, with maximum or minimum tablet strength values. Complex relationships might be associated with a change in the shape, structure (such as the formation of aggregates) or degree of disorder of the particles with particle size. It seems also that increased compaction pressure stresses the relationship between the original particle size and tablet strength in absolute terms.
Some studies have specifically reported on the effect of original particle shape on tablet strength. The results indicate that, for particles that fragment to a limited degree during compression, an increased particle irregularity improved their compactability. However, for particles that fragmented markedly during compression, the original shape of the particles did not affect tablet strength. Moreover, an increased compaction pressure increased the absolute difference in strength of compacts of different original particle shape. Thus, the shape characteristics of particles that fragment markedly during compression seem not to affect the microstructure and the tensile strength of tablets, but the converse applies for particles that showed limited fragmentation.
Compaction of granules
The rationale for granulating a powder mixture before tableting has been discussed above, one reason being to ensure good tableting behaviour, including the compactability. When granules are compacted, the mechanical characteristics of the primary particles and the properties of other excipients will affect the tableting behaviour of the powder. For example, it is a common experience that capping-prone material will show capping tendencies in the granulated form of the substance also. However, the design of the granulation process, such as the method of granulation, will control the physical properties of the granules formed, e.g. in terms of granule size, shape, porosity and strength, which subsequently will affect the evolution in the microstructure of the tablets during compaction. It has, for example, been shown (Fig. 30.34) that tablets formed from granules prepared by roller compaction show a tensile strength that depends on the compaction force used during the dry granulation process. Thus, the evolution of the intergranular tablet microstructure during compression, as well as the evolution in tablet strength, is related to the following two factors:
• the composition of the granules (e.g. choice of filler and binder)
• the physical properties of the granules (e.g. size, porosity and mechanical strength).
During compression of granules, the granules tend to keep their integrity and the formed tablet can in physical terms be described as granules bonded together. When subjected to a load, tablets formed from granules often fail because of breakage of these bonds. Hence, the bonding force of the intergranular bonds and the structure of the intergranular pores will be significant for the tensile strength of the tablets.
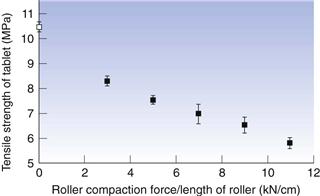
Fig. 30.34 Tensile strength of tablets compacted of a cellulose powder (open symbol) and of cellulose granules formed by roller compaction at different roller-compaction forces (closed symbols). (Courtesy of Herting and Kleinebudde, 2008, with permission.)
In terms of the physical properties of granules, their porosity and compression shear strength are significant properties that influence compactability that can be modulated by the granulation process and by the composition. In general terms, increased porosity and decreased compression shear strength will increase the compactability of the granules. As discussed above, pharmaceutical granules seem to fragment to only a limited degree during compression. The importance of these granule properties for compactability has thus been discussed in terms of a sequential relationship between the original physical character of the granules, the degree of deformation they undergo during compression and the area of contact and the geometry of the intergranular pores of the formed tablet. The formation of large intergranular areas of contact and a closed pore system promote a high tablet strength.
Traditionally, the most important means of controlling the compactability of granules has been to add a binder to the powder to be granulated. This is normally done by adding the binder in a dissolved form, thereby creating binder–substrate granules. An increased amount of binder can correspond to an increased compactability but this is not a general rule. The importance of the presence of a binder for the compactability of such granules can be explained in two ways. First, it has been suggested that intergranular bonds that involve binder-coated granule surfaces can be described as comparatively strong, i.e. difficult to break. Second, binders are often comparatively deformable substances, which can reduce the compression shear strength of the whole granule and thus facilitate the deformation of the granules during compression. An increased degree of granule deformation is sometimes proposed to increase the compactability of the granules. Thus, the binder might have a double role in the compactability of granules, i.e. to increase granule deformation and increase bond strength. Except for the presence of a binder in the granules, the combination of fillers in terms of the hardness and dimensions of the particles can affect the compression shear strength and hence the deformation properties of the granules during compression.
In the preparation of binder–substrate granules, the intention is normally to spread out the binder homogeneously within the granules, i.e. all substrate particles are more or less covered with a layer of binder. However, it is possible that the binder will be concentrated at different regions within the granules, e.g. due to solute migration during drying. The question of the importance of a relatively homogeneous distribution versus a peripheral localization of the binder, i.e. concentration at the granule surface, has been addressed in the literature. It has been argued that a peripheral localization of the binder in the granules before compression should be advantageous, as the binder can thereby be used most effectively for the formation of intergranular bonds. However, the opposite has also been suggested, i.e. a homogeneous binder distribution is advantageous for the compactability of granules. This observation was explained by assuming that, owing to extensive deformation and some attrition of granules during compression, new extragranular surfaces will be formed originating from the interior of the granule. When the binder is distributed homogeneously, such compression-formed surfaces will show a high capacity for bonding.
Figure 30.35 gives an overview of the physical granule properties that may affect the compression behaviour and compactability of granules.
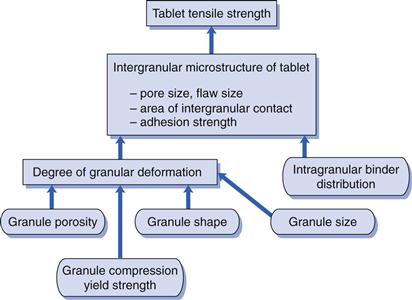
Compaction of binary mixtures
Most of the fundamental work on powder compaction has been carried out on one-component powders. It is, however, of obvious interest to derive knowledge that enables the prediction of the tableting behaviour of mixtures of powders from information on the behaviour of the individual components. In this context, powder mixtures of two components, i.e. binary mixtures, have often been the system of choice in pharmaceutical studies. Binary mixtures can be of two types: simple physical mixtures, i.e. nearly randomized mixtures of particles, and interactive (ordered) mixtures. Most of the studies in this context are empirical, although models for the compaction of binary powder mixtures have been derived.
Concerning simple binary mixtures, the importance of the relative proportions of the ingredients has been studied in relation to the compactability, of or compression parameters for, the respective single component. The mixture can show a linear dependence of the properties of the single powders but deviations from such a simple linear relationship, both positively and negatively, have also been reported. Such non-linear behaviour has been explained in terms of differences between the components in their mechanical and adhesive properties.
Interactive mixtures, especially their compactability after the admixture of lubricants and dry binders, have been the subject of study. The tablet strength-reducing effect of a lubricant mixed with solid particles depends on the surface coverage of the lubricant film obtained during mixing, on the compaction properties of the lubricant per se and on the compression behaviour of the substrate particles. Lubricant sensitivity, also referred to as dilution capacity, seems to be strongly related to the fragmentation propensity of the substrate particles, as discussed earlier.
Concerning the tablet strength-increasing effect of a dry binder mixed with solid particles, similar factors seem to control the compactability of the dry binder mixture as for the lubricant mixture, i.e. the degree of surface coverage of the substrate particle, the binding capacity and deformability of the dry binder and the fragmentation propensity of the substrate particles (Fig. 30.36).
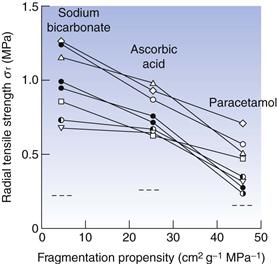
Fig. 30.36 The tensile strength of tablets formed from three substrate substances of different fragmentation propensities in binary mixtures with some fine particulate dry binders, i.e. microcrystalline cellulose, methylcellulose and polyvinylpyrrolidone of different particle size. The dotted lines represent the tensile strength of tablets formed from the single substrate substances. (Courtesy of Nyström and Glazer, 1985, with permission.)
The dilution capacity of interactive mixtures between granules and lubricants or dry binders seems to be related to the degree of deformation the granules undergo during compression, i.e. a high degree of deformation will give a lower sensitivity to a lubricant but also a less positive effect of a dry binder.
References
1. Adams MJ, Mullier MA, Seville JPK. Agglomerate strength measurement using a uniaxial confined compression test. Powder Technology. 1994;78:5–13.
2. Airth JM, Bray DF, Radecka C. Variability of uniformity of weight test as an indicator of the amount of active ingredient in tablets. Journal of Pharmaceutical Sciences. 1967;56:233–235.
3. Alderborn G. A novel approach to derive a compression parameter indicating effective particle deformability. Pharmaceutical Development and Technology. 2003;8:367–377.
4. Alderborn G, Pasanen K, Nyström C. Studies on direct compression of tablets XI Characterization of particle fragmentation during compaction by permeametry measurements of tablets. International Journal of Pharmaceutics. 1985;23:79–86.
5. Al-Nasassrah MA, Podczeck F, Newton JM. The effect of an increase in chain length on the mechanical properties of polyethylene glycols. European Journal of Pharmaceutics and Biopharmaceutics. 1998;46:31–38.
6. Armstrong NA. Causes of tablet compression problems. Manufacturing Chemist. 1982;October:62.
7. Banakar UV. Pharmaceutical Dissolution Testing. New York: Marcel Dekker; 1992.
8. Davies PN, Newton JM. Mechanical strength. In: Alderborn G, Nyström C, eds. Pharmaceutical Powder Compaction Technology. New York: Marcel Dekker; 1996.
9. Duberg M, Nyström C. Studies on direct compression of tablets XVII Porosity-pressure curves for the characterization of volume reduction mechanisms in powder compression. Powder Technology. 1986;46:67–75.
10. Ekenved G, Elofsson R, Sölvell L. Bioavailability studies on a buffered acetylsalicylic acid preparation. Acta Pharmaceutica Suecica. 1975;12:323–332.
11. Frenning G. Compression mechanics of granule beds: A combined finite/discrete element study. Chemical Engineering Science. 2010;65:2464–2471.
12. Herting MG, Kleinebudde P. Studies on the reduction of tensile strength of tablets after roll compaction/dry granulation. European Journal of Pharmaceutics and Biopharmaceutics. 2008;70:372–379.
13. Hiestand EN. Rationale for and the measurement of tableting indices. In: Alderborn G, Nyström C, eds. Pharmaceutical Powder Compaction Technology. New York: Marcel Dekker; 1996.
14. Hölzer AH, Sjögren J. Evaluation of some lubricants by the comparison of friction coefficients and tablet properties. Acta Pharmaceutica Suecica. 1981;18:139–148.
15. Kendall K. Agglomerate strength. Powder Metallurgy. 1988;31:28–31.
16. Leuenberger H. The compressibility and compactability of powder systems. International Journal of Pharmaceutics. 1982;12:41–55.
17. Nyström C, Glazer M. Studies on direct compression of tablets XIII The effect of some dry binders on the tablet strength of compounds with different fragmentation propensity. International Journal of Pharmaceutics. 1985;23:255–263.
18. Ragnarsson G. Force-displacement and network measurements. In: Alderborn G, Nyström C, eds. Pharmaceutical Powder Compaction Technology. New York: Marcel Dekker; 1996.
19. Roberts R, Rowe R. The effect of punch velocity on the compaction of a variety of materials. Journal of Pharmacy and Pharmacology. 1985;37:377–384.
20. Sinka IC, Cunningham JC, Zavaliangos A. The effect of wall friction in the compaction of pharmaceutical tablets with curved faces: a validation study of the Drucker–Prager Cap model. Powder Technology. 2003;133:33–43.
21. Sun C, Grant DJW. Influence of elastic deformation of particles on Heckel analysis. Pharmaceutical Development Technology. 2001;6:193–200.
22. Underwood TW, Cadwallader DE. Influence of various starches on dissolution rate of salicylic acid from tablets. Journal of Pharmaceutical Sciences. 1972;61:239.
23. Wells JI, Rubinstein MH. Pharmaceutical Journal. 1976;217:629.
Bibliography
1. Alderborn G, Nyström C, eds. Pharmaceutical Powder Compaction Technology. New York: Marcel Dekker; 1996.
2. Augsburger LL, Hoag SW. Pharmaceutical Dosage Forms: Tablets. vols 1–3 3rd edn Informa Healthcare: New York; 2008.
3. Chulia D, Deleuil M, Pourcelot Y. Powder Technology and Pharmaceutical Processes. Amsterdam: Elsevier; 1991.
2011. European Pharmacopoeia. 7th edn Council of Europe: Strasbourg; 2011.
5. Levin M. Pharmaceutical Process Scale-Up. New York: Marcel Dekker; 2002.
6. Podczeck F. Particle-particle Adhesion in Pharmaceutical Powder Handling. London: Imperial College Press; 1998.
7. Salman AD, Hounslow MJ, Seville JPK. Granulation Handbook of Powder Technology. vol. 11 Amsterdam: Elsevier; 2007.
8. Sandell E. Industrial Aspects of Pharmaceutics. Stockholm: Swedish Pharmaceutical Press; 1993.