Chapter 117 T Lymphocytes, B Lymphocytes, and Natural Killer Cells
Lymphopoiesis in the Fetus
Origin of the Lymphoid System
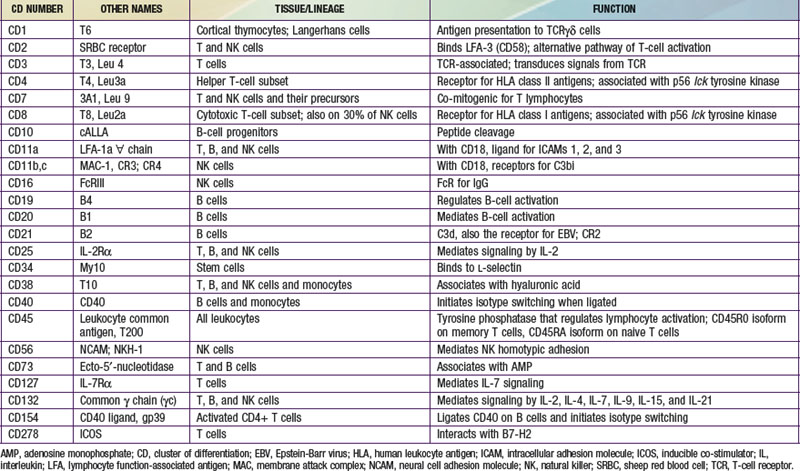
The human immune system arises in the embryo from gut-associated tissue. Pluripotential hematopoietic stem cells 1st appear in the yolk sac at 2.5-3 wk of gestational age, migrate to the fetal liver at 5 wk of gestation, and later reside in the bone marrow, where they remain throughout life (Fig. 117-1). Lymphoid stem cells develop from such precursor cells and differentiate into T, B, or NK cells, depending on the organs or tissues to which the stem cells traffic. Development of the primary lymphoid organs—thymus and bone marrow—begins during the middle of the 1st trimester of gestation and proceeds rapidly. Development of the secondary lymphoid organs—spleen, lymph nodes, tonsils, Peyer patches, and lamina propria—soon follows. These organs serve as sites of differentiation of T, B, and NK lymphocytes from stem cells throughout life. Both the initial organogenesis and the continued cell differentiation occur as a consequence of the interaction of a vast array of lymphocytic and microenvironmental cell surface molecules and proteins secreted by the involved cells. The complexity and number of lymphoid cell surface molecules led to the development of an international nomenclature for clusters of differentiation (CD) (Table 117-1).
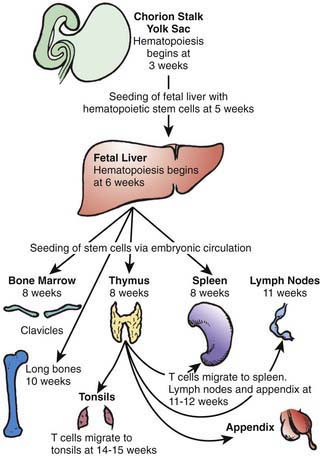
Figure 117-1 Migration patterns of hematopoietic stem cells and mature lymphocytes during human fetal development.
(From Haynes BF, Denning SM: Lymphopoiesis. In Stamatoyannopoulis G, Nienhuis A, Majerus P, editors: Molecular basis of blood diseases, ed 2, Philadelphia, 1994, WB Saunders.)
T and B lymphocytes are the only components of the immune system that have antigen-specific recognition capabilities and are responsible for adaptive immunity. NK cells are lymphocytes that are also derived from hematopoietic stem cells and are thought to have a role in host defense against viral infections, tumor surveillance, and immune regulation, but they do not have antigen receptors. Nonantibody proteins synthesized and secreted by T, B, and NK cells, and by the cells with which they interact, act as intercellular mediators and are referred to as cytokines or interleukins (ILs) (Table 117-2). Cytokines have the ability to act in an autocrine, paracrine, or endocrine manner to promote and facilitate differentiation and proliferation of the cells of the immune system.
Table 117-2 FUNCTIONAL CLASSIFICATION OF CYTOKINES*
GM-CSF, granulocyte macrophage colony stimulating factor; IFN, interferon; IL, interleukin; MHC, major histocompatibility complex; MCP-1, monocyte chemotactic protein; NK, natural killer; RANTES, regulated on activation normal t cell expressed and secreted; SCF, stem cell factor; TGF, transforming growth factor; TNF, tumor necrosis factor.
* This is not an exhaustive list.
Modified from Whiteside TL: Cytokine measurements and interpretation of cytokine assays in human disease, J Clin Immunol 14:327–339, 1994.
B-Cell Development and Differentiation
In parallel with T-cell differentiation, B-cell development begins in the fetal liver before 7 wk of gestation. Fetal liver CD34 stem cells are seeded to the bone marrow of the clavicles by 8 wk of embryonic life and to that of the long bones by 10 wk (see Fig. 117-1). Antigen-independent stages of B-cell development have been defined according to immunoglobulin gene rearrangement patterns and the surface proteins the cells bear. The pro–B cell is the 1st descendent of the pluripotential stem cell committed to B-lineage development that is detected by the presence of both CD34 and CD10 on its surface, although the immunoglobulin genes remain germ line (Fig. 117-2). The next stage is the pre-pre–B cell, during which immunoglobulin genes are rearranged but there is no cytoplasmic expression of µ heavy chains or surface IgM (sIgM). These cells are further characterized by the co-expression of membrane CD34, CD10, CD19, and CD40, and somewhat later by the additional presence of CD73, CD22, CD24, and CD38. The pre–B cell follows and is distinguished by the expression of cytoplasmic µ heavy chains but no sIgM, because no immunoglobulin light chains are produced yet. These cells also continue to express all CD antigens seen at the pre-pre–B-cell stage except CD34 and CD10, which are lost; in addition, they express CD21. Next is the immature B-cell stage, during which sIgM but not sIgD is expressed. The light-chain genes have been rearranged, and CD38 is lost but all other pre–B-cell CD antigens persist. The last stage of antigen-independent B-cell development is the mature or virgin B cell, which co-expresses both sIgM and sIgD; CD23 is also acquired at this stage, and all of the other CD antigens present on immature B cells persist. Pre–B cells can be found in fetal liver at 7 wk gestation, sIgM+ and sIgG+ B cells at between 7 and 11 wk, and sIgD+ and sIgA+ B cells by 12-13 wk. By 14 wk of embryonic life, the percentage of circulating lymphocytes bearing sIgM and sIgD is the same as in cord blood and slightly higher than in the blood of adults.
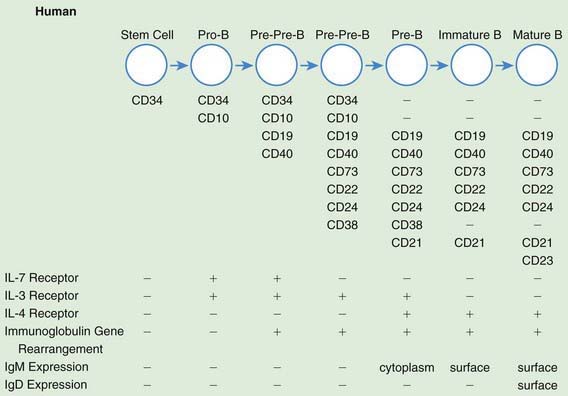
Figure 117-2 Antigen-independent human B-cell development. CD, cluster of differentiation; Ig, immunoglobulin; IL, interleukin.
(From Haynes BF, Denning SM: Lymphopoiesis. In Stamatoyannopoulis G, Nienhuis A, Majerus P, editors: Molecular basis of blood diseases, ed 2, Philadelphia, 1994, WB Saunders.)
Natural Killer (NK)-Cell Development
Unlike T and B cells, NK cells do not rearrange antigen receptor genes during their development but are defined by their functional capacity to mediate non–antigen-specific cytotoxicity. NK cells have killer inhibitory receptors (KIRs) that recognize certain MHC antigens and inhibit the killing of normal allogeneic cells in four specific patterns of reactivity. The genetic loci controlling these receptors are different from MHC alloantigenic loci, and have been mapped to chromosome 19. Virtually all NK cells express CD56, and >90% bear CD16 (FcγRIII) on the cell surface. Other CD antigens found on NK cells include CD57 (50-60%), CD7 and CD2 (70-90%), and CD8 (30-40%) (see Table 117-1). Although NK cells share surface antigens with T and myeloid cells, the lineage relationship of NK cells to the latter is still unclear. Some humans with autosomal recessive SCID who have profound deficiencies in T and B cells have abundant NK cells, whereas those with X-linked and Jak3-deficient SCID have no T or NK cells.
Immune Cell Interactions
In the primary antibody response, native antigen is carried to a lymph node draining the site, taken up by specialized cells called follicular dendritic cells (FDCs), and expressed on their surfaces. Virgin B cells bearing sIg specific for that antigen then bind to the antigen on the surfaces of the FDCs. If the affinity of the B-cell sIg antibody for the antigen present on the FDCs is sufficient, and if other signals are provided by activated T-helper cells, the B cell develops into an antibody-producing plasma cell. If the affinity is not high enough or if T-cell signals are not received, the B cell dies through apoptosis. The signals from activated TH cells include several cytokines (IL-4, IL-5, IL-6, IL-10, IL-13, and IL-21) that they secrete (see Table 117-2) and a surface T-cell molecule, CD154, which, on contact of the T cell with the B cell, binds to CD40 on the B-cell surface. CD40 is a type I integral membrane glycoprotein expressed on B cells, monocytes, some carcinomas, and a few other types of cells. It belongs to the tumor necrosis factor (TNF)/nerve growth factor receptor family. Cross linking of CD40 on B cells by CD154 on T cells in the presence of certain cytokines causes the B cells to undergo proliferation and to initiate immunoglobulin synthesis. In the primary immune response, only IgM antibody is usually made, and most of it is of relatively low affinity. Some B cells become memory B cells during the primary immune response. These cells switch their immunoglobulin genes so that IgG, IgA, and/or IgE antibodies of higher affinity are formed on a secondary exposure to the same antigen. The secondary antibody response occurs when these memory B cells again encounter that antigen. Plasma cells form, just as in the primary response; however, many more cells are rapidly generated, and IgG, IgA, and IgE antibodies are made. In addition, genetic changes in immunoglobulin genes (somatic hypermutation [SHM]) lead to increased affinity of those antibodies. A lack of SHM is seen in deficiency of AID or UNG. The exact pattern of isotype response to antigen in normal individuals varies, depending on the type of antigen and the cytokines present in the microenvironment.
Postnatal Lymphopoiesis
T Cells and T-Cell Subsets
Although the percentage of CD3 T cells in cord blood is somewhat less than in the peripheral blood of children and adults, T cells are actually present in higher number because of a higher absolute lymphocyte count in normal infants. An additional distinction is that the ratio of CD4 to CD8 T cells is usually higher (3.5-4 : 1) in cord blood than in blood of children and adults (1.5-2 : 1). Virtually all T cells in cord blood bear the CD45RA (naive) isoform, and a dominance of CD45RA over CD45RO T cells persists during the 1st 2-3 yr of life, after which time the numbers of cells bearing these 2 isoforms gradually equalize. TH cells can be further subdivided according to the cytokines they produce when activated. TH1 cells produce IL-2 and IFN-γ, which promote cytotoxic T-cell or delayed hypersensitivity types of responses, whereas TH2 cells produce IL-4, IL-5, IL-6, IL-13, and IL-21 (see Table 117-2), which promote B-cell responses and allergic sensitization. There are important additional subsets of T cells that have regulatory functions. These include CD25 high + T cells (Treg cells), considered to be important in the prevention of autoimmune diseases, and T cells that have phenotypic characteristics of NK cells (NK-T cells). Cord blood T cells have the capacity to respond normally to T-cell mitogens (PHA, Con A, and PWM) and are capable of mounting a normal mixed leukocyte response. Normal newborn infants also have the capacity to develop antigen-specific T cell responses at birth, as evidenced by vigorous tuberculin reactivity a few weeks after BCG vaccination on day 1 of life. Because patients in the 1st few months of life may have unrecognized severe T-cell defects, most hospitals now routinely irradiate all blood products given young infants. T-cell defects can readily be detected even at birth by calculating the absolute lymphocyte count because T cells normally constitute 70% of circulating lymphocytes and their absence results in striking lymphopenia (see Fig. 116-2 and Chapter 708).
Inheritance of Abnormalities In T-, B-, and NK-Cell Development
More than 150 immunodeficiency syndromes have been described (see Table 116-8). Specific molecular defects have been identified in approximately 80% of these diseases. Most are recessive traits, several of which are caused by mutations in genes on the X chromosome and others by mutations on autosomal chromosomes. The molecular bases of 7 X-linked immunodeficiency disorders affecting T, B, and/or NK cells are known (Chapters 118 to 120): X-linked immunodeficiency with hyper IgM, X-linked lymphoproliferative syndrome, XIAD, X-linked agammaglobulinemia, X-linked SCID, the Wiskott-Aldrich syndrome, and nuclear factor kappa B essential modulator (NEMO). Autosomal defects for which the molecular basis is known include (1) combined immunodeficiencies due to abnormalities of purine salvage pathway enzymes, either adenosine deaminase (ADA, encoded by a gene on chromosome 20q13-ter) or purine nucleoside phosphorylase (PNP, encoded by a gene on chromosome 14q13.1); (2) combined immunodeficiencies due to mutations in the gene encoding ZAP-70 (localized to chromosome 2q12), a non-src family protein tyrosine kinase important in T-cell signaling; (3) SCID due to mutations in the gene on chromosome 19p13.1 encoding Janus kinase 3 (Jak3), the primary signal transducer from the common cytokine receptor γ chain (γc); (4) mutations in genes on chromosome 11 that encode components of the T-cell receptor, that is, CD3 γ, δ and ε; (5) SCID due to mutations in recombinase activating genes (RAG1 and RAG2); and (6) SCID due to mutations in the gene on chromosome 5p13 that encodes the α chain of the IL-7 receptor. These are only a few of the conditions for which the mutated genes have been discovered and the number is steadily growing.
Prenatal Diagnosis and Carrier Detection
Intrauterine diagnosis of ADA and PNP deficiencies can be established by enzyme analyses on amnion cells (fresh or cultured) obtained before 20 wk gestation. Diagnosis of several X-linked defects can be established by direct mutation analysis of the X chromosome of cells obtained by chorionic villus sampling or by amniocentesis from male infants whose mothers have been identified as carriers. Diagnosis of enzyme-normal SCID or other severe T-cell deficiencies, MHC class I and/or II antigen deficiencies, chronic granulomatous disease (CGD), or Wiskott-Aldrich syndrome (by platelet size) can be established by mutation analysis if the mutation is known in the family or, if not known, by appropriate tests of phenotype or function on small samples of blood obtained by fetoscopy at 18-22 wk of gestation. The same diagnostic procedures can be performed on cord blood, but currently, no immunodeficiency disorders are routinely screened for using cord blood of newborns, although pilot studies have been conducted in Wisconsin and in Massachusetts (Chapter 116). Carriers of ADA and PNP deficiency can be detected by quantitative enzyme analyses of blood samples. Carriers of X-linked agammaglobulinemia, X-linked SCID, or the Wiskott-Aldrich syndrome can be identified by direct mutation analysis if the family’s mutation is known.
Borghesi L, Milcarek C. From B cell to plasma cell: regulation of V(D)J recombination and antibody secretion. Immunol Res. 2006;36:27-32.
Chung JW, Yoon SR, Choi I. The regulation of NK cell function and development. Front Biosci. 2008;13:6432-6442.
LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112:1570-1580.
Luci C, Tomasello E. Natural killer cells: detectors of stress. Int J Biochem Cell Bio. 2008;40:2335-2340.
McKenzie BS, Kastelein RA, Cua DJ. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006;27:17-23.
Mingueneau M, Sansoni A, Gregoire C, et al. The proline-rich sequence of CD3 epsilon controls T cell antigen receptor expression on and signaling potency in preselection CD4+CD8+ thymocytes. Nat Immunol. 2008;9:522-532.
Murphy KM, Travers P, Walport M. Janeway’s immunobiology, ed 7. New York and London: Garland Science Publishing; 2007.
Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008;8:9-21.
Siggs OM, Makaroff LE, Liston A. The why and how of thymocyte negative selection. Curr Opin Immunol. 2006;18:175-183.





