Chapter 152 Systemic Lupus Erythematosus
Etiology
Pathology
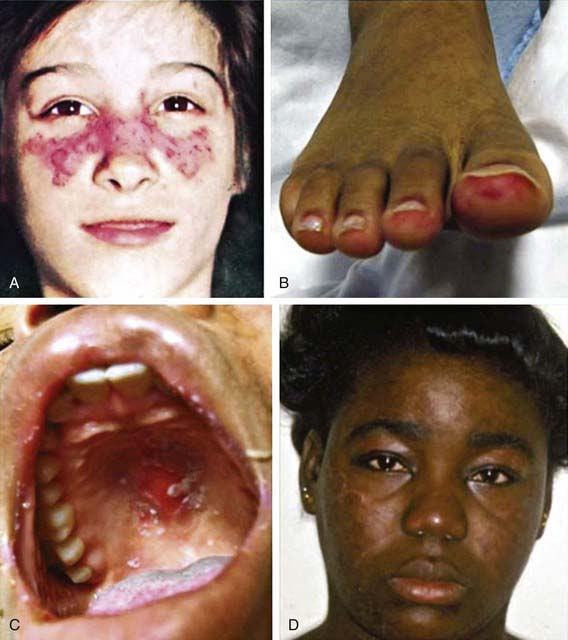
Histologic features most suggestive of SLE include findings in the kidney and skin, especially the discoid rash. Renal manifestations of SLE are classified histologically according to the criteria of the International Society of Nephrology (Chapter 508). The finding of diffuse proliferative glomerulonephritis (class IV) significantly increases risk for renal morbidity. Renal biopsies are very helpful to establish the diagnosis of SLE and to stage disease. Immune complexes are commonly found with “full house” deposition of immunoglobulin and complement. The characteristic discoid rash depicted in Figure 152-1D is characterized on biopsy by hyperkeratosis, follicular plugging, and infiltration of mononuclear cells into the dermal-epidermal junction. The histopathology of photosensitive rashes can be nonspecific, but immunofluorescence examination of both affected and nonaffected skin may reveal deposition of immune complexes within the dermal-epidermal junction. This finding is called the lupus band test, which is specific for SLE.
Clinical Manifestations
Any organ system can be involved in SLE, so the potential clinical manifestations are protean (Table 152-1). The presentation of SLE in childhood or adolescence differs from that in adults. The most common presenting complaints of children with SLE include fever, fatigue, hematologic abnormalities, arthralgia, and arthritis. Renal disease in SLE is often asymptomatic; thus careful monitoring of blood pressure and urinalyses is critical. SLE is often characterized by periods of flare and disease quiescence or may follow a more smoldering disease course. The neuropsychiatric complications of SLE may occur with or without apparently active SLE and are particularly difficult to detect in adolescents, who are already at high risk for mood disorders. Long-term complications of SLE and its therapy, including accelerated atherosclerosis and osteoporosis, become clinically evident in young to middle adulthood. SLE is a disease that evolves over time in each affected individual, and new manifestations may arise even many years after diagnosis.
Table 152-1 POTENTIAL CLINICAL MANIFESTATIONS OF SYSTEMIC LUPUS ERYTHEMATOSUS
| TARGET ORGAN | POTENTIAL CLINICAL MANIFESTATIONS |
|---|---|
| Constitutional | Fatigue, anorexia, weight loss, fever, lymphadenopathy |
| Musculoskeletal | Arthritis, myositis, tendonitis, arthralgias, myalgias, avascular necrosis, osteoporosis |
| Skin | Malar rash, discoid rash, photosensitive rash, cutaneous vasculitis, livedo reticularis, periungual capillary abnormalities, Raynaud’s phenomenon, alopecia, oral and nasal ulcers |
| Renal | Hypertension, proteinuria, hematuria, edema, nephrotic syndrome, renal failure |
| Cardiovascular | Pericarditis, myocarditis, conduction system abnormalities, Libman-Sacks endocarditis |
| Neurologic | Seizures, psychosis, cerebritis, stroke, transverse myelitis, depression, cognitive impairment, headaches, pseudotumor, peripheral neuropathy, chorea, optic neuritis, cranial nerve palsies |
| Pulmonary | Pleuritis, interstitial lung disease, pulmonary hemorrhage, pulmonary hypertension, pulmonary embolism |
| Hematologic | Immune-mediated cytopenias (hemolytic anemia, thrombocytopenia or leukopenia), anemia of chronic inflammation, hypercoaguability, thrombocytopenic thrombotic microangiopathy |
| Gastroenterology | Hepatosplenomegaly, pancreatitis, vasculitis affecting bowel, protein-losing enteropathy |
| Ocular | Retinal vasculitis, scleritis, episcleritis, papilledema |
Diagnosis
The diagnosis of SLE requires a comprehensive clinical and laboratory assessment revealing characteristic multisystem disease and excluding other etiologies, including infection and malignancy. Presence of 4 of the 11 American College of Rheumatology 1997 Revised Classification Criteria for SLE (Table 152-2) simultaneously or cumulatively establishes the diagnosis of SLE. Of note, although a positive antinuclear antibody (ANA) test result is not required for the diagnosis of SLE, ANA-negative lupus is extremely rare. Hypocomplementemia, although common in SLE, is not represented among the classification criteria.
Table 152-2 AMERICAN COLLEGE OF RHEUMATOLOGY 1997 REVISED CLASSIFICATION CRITERIA FOR SYSTEMIC LUPUS ERYTHEMATOSUS*
* The presence of 4/11 criteria establishes the diagnosis of SLE. These criteria were developed for classification in clinical trials and not for clinical diagnosis.
Adapted from Hochberg MC: Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus, Arthritis Rheum 40:1725, 1997.
Differential Diagnosis
Drug-induced lupus refers to the presence of SLE manifestations triggered by exposure to certain medications, including minocycline, many anticonvulsants, sulfonamides, antiarrhythmic agents, and other drugs (Table 152-3). In individuals prone to SLE, these agents may act as a trigger for true SLE. In others, these agents provoke a reversible lupus-like syndrome. Unlike SLE, drug-induced lupus affects males and females equally. An inherited predisposition toward slow acetylation may increase the risk of drug-induced lupus. Circulating antihistone antibodies are often present in drug-induced SLE, and these antibodies are detected in up to 20% of individuals with SLE. Hepatitis, which is rare in SLE, is more common in drug-induced lupus. Individuals with drug-induced lupus are less likely to demonstrate antibodies to double-stranded DNA, hypocomplementemia, and significant renal or neurologic disease. In contrast to SLE, manifestations of drug-induced lupus resolve after withdrawal of the offending medication; complete recovery may take several months to years.
Table 152-3 MEDICATIONS ASSOCIATED WITH DRUG-INDUCED LUPUS
DEFINITE ASSOCIATION
Minocycline, procainamide, hydralazine, isoniazid, penicillamine, diltiazem, interferon-α, methyldopa, chlorpromazine, etanercept, infliximab, adalimumab
PROBABLE ASSOCIATION
Phenytoin, ethosuximide, carbamazepine, sulfasalazine, amiodarone, quinidine, rifampin, nitrofurantoin, beta blockers, lithium, captopril, interferon-gamma, hydrochlorothiazide, glyburide, docetaxel, penicillin, tetracycline, statins, gold, valproate, griseofulvin, gemfibrozil, propylthiouracil
Laboratory Findings
A positive ANA test result is present in 95-99% of individuals with SLE. This test has poor specificity for SLE, as up to 20% of healthy individuals also have a positive ANA test result, making the ANA a poor screening test for SLE. ANA titers are not reflective of disease activity; therefore, repeating ANA titers is not helpful in disease management. Antibodies to double-stranded DNA are more specific for SLE, and in some individuals, anti-dsDNA levels correlate with disease activity, particularly nephritis. Anti-Smith antibody, although found specifically in patients with SLE, does not correlate with disease activity. Serum levels of total hemolytic complement (CH50), C3, and C4 are typically decreased in active disease and often improve with treatment. Table 152-4 lists several autoantibodies found in SLE and their clinical associations. Hypergammaglobulinemia is a common but nonspecific finding. Inflammatory markers, particularly erythrocyte sedimentation rate, are often elevated in active disease. C-reactive protein (CRP) correlates less well with disease activity, and elevated CRP values may reflect infection.
Table 152-4 AUTOANTIBODIES COMMONLY ASSOCIATED WITH SYSTEMIC LUPUS ERYTHEMATOSUS (SLE)
| ANTIBODY | CLINICAL ASSOCIATION |
|---|---|
| Anti–double-stranded DNA | Correlates with disease activity, especially nephritis, in some with SLE |
| Anti-Smith antibody | Specific for the diagnosis of SLE |
| Anti-ribonucleoprotein antibody |
Anti-La antibody (anti-SSB antibody)
Antiphospholipid antibodies, which increase clotting risk, can be found in up to 66% of children and adolescents with SLE. Antiphospholipid antibodies can be detected by several means, and laboratory features that point to the presence of these antibodies include the presence of anticardiolipin antibodies, prolonged phospholipid-dependent coagulation test results (partial thromboplastin time, dilute Russell viper-venom time), and a circulating lupus anticoagulant (which confirms that a prolonged partial thromboplastin time is not corrected with mixing studies). When an arterial or venous clotting event occurs in the presence of an antiphospholipid antibody, antiphospholipid antibody syndrome is diagnosed. Antiphospholipid antibody syndrome can occur in the context of SLE or independent of SLE (Chapter 473).
Complications
Within the first several years of diagnosis, the most common causes of death in individuals with SLE include infection and complications of glomerulonephritis and neuropsychiatric disease (Table 152-5). Over the long term, the most common causes of mortality include complications of atherosclerosis and malignancy. The increased risk of premature atherosclerosis in SLE is not explained by traditional risk factors and is due in part to the chronic immune dysregulation and inflammation associated with SLE. Increased malignancy rates may be caused by immune dysregulation and exposure to medications with carcinogenic potential.
| Renal | Hypertension, dialysis, transplantation |
| Central nervous system | Organic brain syndrome, seizures, psychosis, neurocognitive dysfunction |
| Cardiovascular | Atherosclerosis, myocardial infarction, cardiomyopathy, valvular disease |
| Immune | Recurrent infection, functional asplenia, malignancy |
| Musculoskeletal | Osteopenia, compression fractures, osteonecrosis |
| Ocular | Cataracts, glaucoma |
| Endocrine | Diabetes, obesity, growth failure, infertility, fetal wastage |
From Cassidy JT, Petty RE: Textbook of pediatric rheumatology, ed 5, Philadelphia, 2005, Elsevier/Saunders.
Prognosis
Owing to advances in the diagnosis and treatment of SLE, survival has improved dramatically over the past 50 years. Currently, the 5-year survival rate for pediatric SLE is >90%. However, given their long burden of disease, children and adolescents with SLE face a high risk of future morbidity and mortality from the disease and its complications, especially atherosclerosis and malignancy (see Table 152-5). Given the complex and chronic nature of SLE, it is optimal for children and adolescents with SLE to be treated by pediatric rheumatologists in a multidisciplinary clinic.
Adrianto I, Wen F, Templeton A, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43(3):253-258.
Ardoin SP, Pisetsky DS. Developments in the scientific understanding of lupus. Arthritis Res Ther. 2008;10:218.
Avcin T, Cimaz R, Silverman ED, et al. Pediatric antiphospholipid syndrome: clinical and immunologic features of 121 patients in an international registry. Pediatrics. 2008;122:e1100-e1107.
Berkun Y, Padeh S, Barash J, et al. Antiphospholipid syndrome and recurrent thrombosis in children. Arthritis Rheum. 2006;55:850-855.
Bernatsky S, Boivin JF, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54:2550-2557.
Borchers AT, Keen CL, Gershwin ME. Drug-induced lupus. Ann N Y Acad Sci. 2007;1108:166-182.
Brunner HI, Gladman DD, Ibanez D, et al. Difference in disease features between childhood-onset and adult-onset systemic lupus erythematosus. Arthritis Rheum. 2008;58:556-562.
Buyon JP, Petri MA, Kim MY, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med. 2005;142:953-962.
Chen MJ, Tseng HM, Huang YL, et al. Long-term outcome and short-term survival of patients with systemic lupus erythematosus after bacteraemia episodes: 6-yr follow-up. Rheumatology. 2008;47:1352-1357.
Ginzler EM, Dooley MA, Aranow C, et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N Engl J Med. 2005;353:2219-2228.
Grimaldi CM. Sex and systemic lupus erythematosus: the role of the sex hormones estrogen and prolactin on the regulation of autoreactive B cells. Curr Opin Rheumatol. 2006;18:456-461.
Hiraki LT, Benseler SM, Tyrrell PN, et al. Clinical and laboratory characteristics and long-term outcome of pediatric systemic lupus erythematosus: a longitudinal study. J Pediatr. 2008;152:550-556.
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725.
Izmirly PM, Llanos C, Lee LA, et al. Cutaneous manifestations of neonatal lupus and risk of subsequent congenital heart block. Arth Rheum. 2010;62(4):1153-1157.
Moore TL. Autoimmunity and minocycline. J Pediatr. 2008;153:303-304.
Podolskaya A, Stadermann M, Pilkington C, et al. B cell depletion therapy for 19 patients with refractory systemic lupus erythematosus. Arch Dis Child. 2008;91:401-406.
Ramos-Casals M, Nardi N, Lagrutta M, et al. Vasculitis in systemic lupus erythematosus: prevalence and clinical characteristics in 670 patients. Medicine. 2006;85:95-104.
Sandborg C, Ardoin SP, Schanberg L. Therapy insight: cardiovascular disease in pediatric systemic lupus erythematosus. Nat Clin Pract Rheumatol. 2008;4:258-265.
Siso A, Ramos-Casals M, Bové A, et al. Outcomes in biopsy-proven lupus nephritis. Medicine. 2010;89(5):300-307.
Somers EC, Marder W, Christman GM, et al. Use of a gonadotropin-releasing hormone analog for protection against premature ovarian failure during cyclophosphamide therapy in women with severe lupus. Arthritis Rheum. 2005;52:2761-2767.
Tang X, Huang Y, Deng W, et al. Clinical and serologic correlations and autoantibody clusters in systemic lupus erythematosus. Medicine. 2010;89:62-67.
Tucker LB, Uribe AG, Fernandez M, et al. Adolescent onset of lupus results in more aggressive disease and worse outcomes: results of a nested matched case-control study within LUMINA, a multiethnic US cohort (LUMINA LVII). Lupus. 2008;17:314-322.
Vasoo S. Drug-induced lupus: an update. Lupus. 2006;15:757-761.
Willems M, Haddad E, Niaudet P, et al. Rituximab therapy for childhood-onset systemic lupus erythematosus. J Pediatr. 2006;148:623-627.
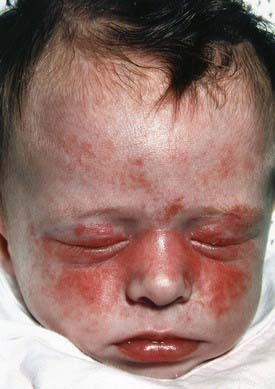
152.1 Neonatal Lupus
Neonatal lupus, an entity distinct from SLE, is one of the few rheumatic disorders manifesting in the neonate. Clinical manifestations of neonatal lupus include a characteristic annular or macular rash typically affecting the face (especially the periorbital area), trunk, and scalp (Fig. 152-2). Infants may also have cytopenias and hepatitis, but the most feared complication is congenital heart block. Conduction system abnormalities range from prolongation of the PR interval to complete heart block, rarely resulting in progressive cardiomyopathy. The noncardiac manifestations of neonatal lupus are usually reversible, but congenital heart block is permanent. The rash typically appears within the first 6 wk of life after exposure to ultraviolet light and lasts 3-4 mo; however, it can be present at birth. Conduction system abnormalities can be detected in utero beginning at 16 wk of gestational age.