[level-membership-for-pediatrics-category]Chapter 57
Systemic Conditions with Lung Involvement
Pulmonary involvement is a frequent component of systemic illness. Pediatric patients often present for chest imaging before the diagnosis of a specific systemic disorder has been made. Therefore, the radiologist plays an important role in directing the diagnostic workup by recognizing that the pulmonary findings reflect an underlying condition. The first imaging study in pediatric patients with respiratory symptoms is usually chest radiography. In some patients such as those with pulmonary edema or sickle cell disease, radiography shows the pulmonary findings adequately to continue appropriate clinical care. In many patients, however, chest radiography is nonspecific, provides initial clues to the diagnosis, or both, prompting further investigation with computed tomography (CT) of the chest. In recent years, the advent of multidetector CT and controlled ventilation techniques (see Chapter 49) has dramatically improved the detailed assessment of lungs in children with systemic disorders.
Specific Systemic Diseases
Vasculitis and Collagen Vascular Disease
Although medium- and large-vessel vasculitides more commonly affect mediastinal vasculature structures and rarely the lung parenchyma, small-vessel vasculitides are the most likely to cause pulmonary parenchymal disease. Pediatric patients with pulmonary vasculitis typically present in their teen years. Of these disorders, Wegener polyangiitis (WP), previously known as granulomatosis, is the most common one in children. Microscopic polyangiitis (MPA) and Churg-Strauss syndrome (CSS) are rarely seen in children. Pulmonary nodules and airspace opacities are typically seen on imaging studies.1–3
Pulmonary involvement, most commonly interstitial changes, may be seen in children with collagen vascular diseases (CVD) such as juvenile arthritis, dermatomyositis, systemic sclerosis (scleroderma), systemic lupus erythematosus (SLE), and mixed connective tissue disease. Pulmonary involvement is less common in children than in adults. Pulmonary disease is seen more frequently in children with systemic sclerosis (59% to 91%) than in the other CVDs, and it is associated with significant morbidity and mortality.4,5 A recent study found that abnormal pulmonary function tests (PFTs) were correlated with the severity of abnormalities on high-resolution CT (HRCT). Thus PFTs provide a monitoring tool to identify children who would benefit from further lung disease evaluation with HRCT.5 Symptomatic lung disease is significantly less common in juvenile rheumatoid arthritis and SLE, reported in only 5% of patients.6
Both the vasculitides and CVDs may cause pulmonary renal syndrome, which is the association of both pulmonary hemorrhage and glomerulonephritis. It is often seen in WP and SLE.1,2 Pulmonary hemorrhage may be seen in both the vasculitides and in CVDs (e-Fig. 57-1) and has a high morbidity and mortality (50% to 90%).1,7
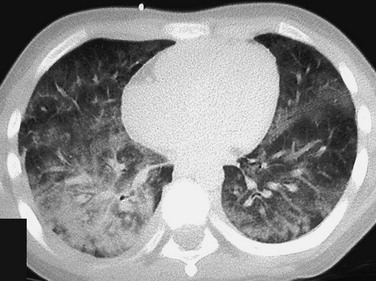
e-Figure 57-1 Pulmonary hemorrhage in a teenage boy with pulmonary vasculitis resulting from collagen vascular disease.
The patient presented with hemoptysis. A computed tomography scan shows diffuse but somewhat patchy ground-glass opacification in both lungs.
Etiology: WP is, by far, the most common of the pulmonary vasculitides seen in children and typically presents with a triad of necrotizing granulomatous lesions in both the upper and lower respiratory tract, as well as glomerulonephritis. MPA is a nongranulomatous necrotizing vasculitis almost always seen with glomerulonephritis. CSS (also known as allergic granulomatosis and angiitis) typically presents with asthma and blood eosinophilia but is rare in children.1 Most of the pulmonary vasculitides are immunologically mediated. WP, MPA, and CSS are associated with antineutrophil cytoplasmic antibodies (ANCA) and are sometimes referred to as ANCA-associated systemic vasculitides.
Imaging: The common imaging findings of pulmonary vasculitis and CVDs are summarized in Table 57-1. The frequent imaging findings of WP are variable sized nodules, followed by ground-glass opacities and air space consolidation; 17% of the nodules show cavitation.8 The nodules are frequently surrounded by a halo of ground-glass opacity, which represents hemorrhage (e-Fig. 57-2 and Fig. 57-3). Airway wall thickening may also be seen, but airway strictures are significantly less common in children (3%) compared with adults (up to 59%).8 Diffuse alveolar hemorrhage (occurring in 44% of pediatric WP) is characterized by lobular or lobar regions of ground-glass opacity or airspace consolidation on CT.9–11 It may also show the “crazy paving” pattern on CT, especially as it evolves.12
Table 57-1
Pulmonary Imaging Findings in Systemic Diseases
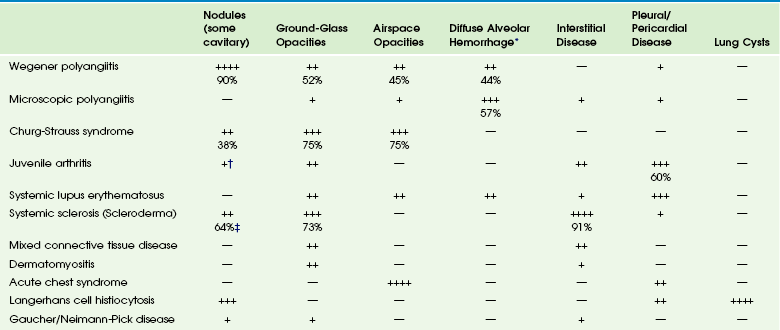
*Specific imaging findings are a spectrum from ground-glass opacities to airspace consolidation.
†Nodules in juvenile arthritis seen in lipoid pneumonia.
‡Seen as subpleural micronodules and were not considered a dominant finding.
Data from references 1, 4, 6, 7, and 11; and Feng RE, Xu WB, Shi JH, et al. Pathological and high resolution CT findings in Churg-Strauss syndrome. Chin Med Sci J. 2011;26(1):1-8.
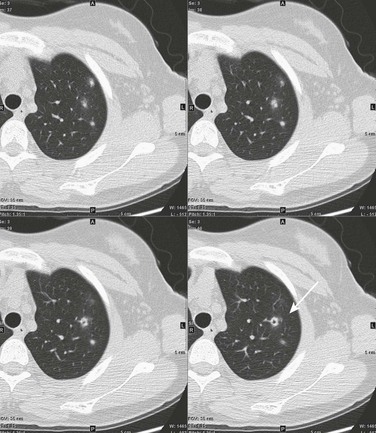
Figure 57-3 Wegener polyangiitis.
This 16-year-old girl had a history of vasculitic skin lesions and headache. Computed tomography (CT) of the sinuses revealed inflammatory changes (not shown). Axial CT images of the chest show left upper lobe cavitary lesion (arrow) with surrounding ground-glass halo as well as several solid pulmonary nodules, also with surrounding halos of ground-glass opacity. Lung biopsy was positive for Wegener polyangiitis.
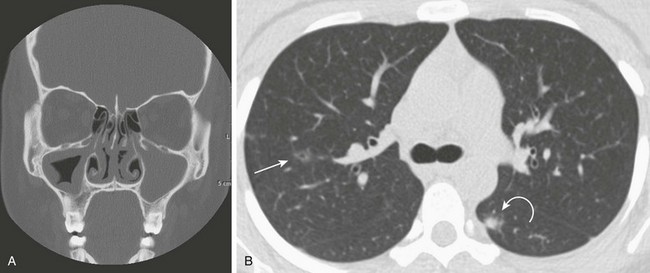
e-Figure 57-2 Wegener polyangiitis.
This 16-year-old girl with a history of chronic sinusitis presented with hematuria, acute renal failure, and sinusitis. She was eventually found to have Wegener polyangiitis. A, A coronal computed tomography (CT) image shows mucosal thickening in the right maxillary sinus and opacified left maxillary sinus. B, An axial CT image in lung windows through the upper lobes show a small thin-walled cavitary lesion in the right upper lobe (arrow). Also shown is a focal nodule with a halo of ground-glass opacity in the superior segment of the left lower lobe (curved arrow).
In the case of CVDs, pleural and pericardial effusions are the most common findings in the chest. Pulmonary findings are significantly less common but follow a similar pattern for most of these disorders and include ground-glass opacity and interstitial septal thickening, which may progress to pulmonary fibrosis. Despite normal chest radiographs or only minimal radiographic abnormalities in children with systemic sclerosis, HRCT demonstrates ground-glass opacities, peripheral areas of pulmonary fibrosis, and subpleural micronodules (Fig. 57-4).4 Some authors have reported occurrence of lipoid pneumonia in children with juvenile idiopathic arthritis, not associated with mineral oil ingestion.12 “Shrinking lung syndrome” has been described in SLE and represents decrease in lung volume that manifests radiographically by an elevated diaphragm.7,12,13
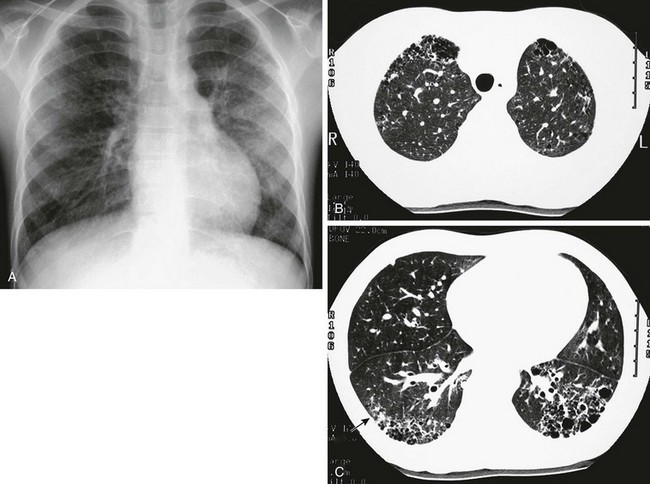
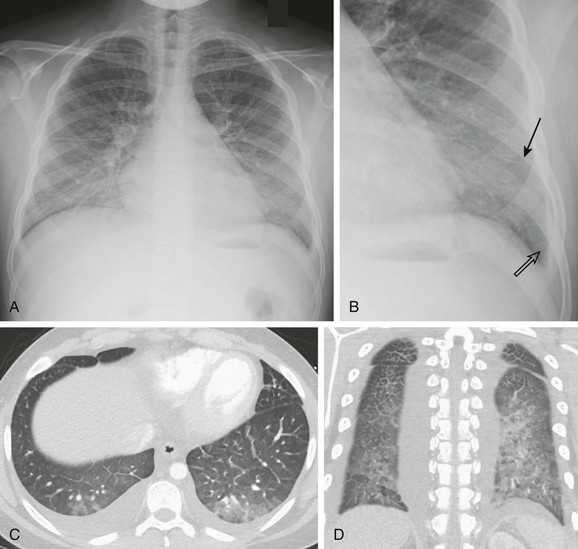
Figure 57-4 An 8-year-old girl with systemic sclerosis (scleroderma) diagnosed at age 5 years.
A, The frontal chest radiograph shows mild increase in perihilar linear opacities. B, High-resolution computed tomography (HRCT) of upper lung zones shows thin-walled, small cystic areas anteriorly in both lungs and a few small scattered subpleural lucencies and opacities more posteriorly. C, HRCT through lower lung zones shows honeycomb pattern dependently in both lungs. More lateral area in right lung shows more ground-glass opacity (arrow) that may represent active disease.
Treatment and Follow-up: Treatment for vasculitides and CVDs is primarily aimed at suppressing the immune response, typically with corticosteroids and chemotherapeutic agents. In patients with vasculitis, CVDs, or both, who experience diffuse alveolar hemorrhage, evolving changes may be seen on CT at follow-up. A more linear and interstitial type of pattern may develop with interlobular septal thickening and the appearance of the “crazy paving” pattern. If episodes of hemorrhage recur, this may also progress to interstitial fibrosis.9 Pulmonary involvement with CVD may progress to interstitial pulmonary fibrosis regardless of treatment, with advanced stages showing honeycombing. Pulmonary artery hypertension may also develop with advanced lung disease, especially in systemic sclerosis.
Sickle Cell Disease
Acute chest syndrome (ACS), a pulmonary illness that occurs in up to 50% of children with sickle cell disease (SCD), is characterized by chest pain, leukocytosis, fever, and a new pulmonary opacity. It is the leading cause of death (25%) and hospitalization in all patients with SCD and usually occurs between 2 and 4 years of age.14 ACS also occurs in patients with other sickle hemoglobinopathies.15 When not fatal, it may lead to chronic lung disease (4%) and pulmonary hypertension.16
Etiology: The etiology of ACS is complex and not always known. In a large multicenter study of 671 episodes, causes were found to be microvascular occlusion and infarction (16%), infection (29%), fat emboli from bone marrow infarcts (9%), and unknown in 46%. Chlamydia, Mycoplasma, and viral agents were the most common pathogens in cases caused by infection. One third of patients presented with symptoms of long bone pain caused by vasoocclusive crisis and developed ACS 2 to 3 days later.17
Imaging: Radiographic findings of ACS are nonspecific but, by definition, include the presence of a pulmonary opacity (e-Fig. 57-5). Opacities were noted in the lower lobes in about 90%, and pleural effusion was noted in 55% of cases in a large multicenter study (see Table 57-1).17
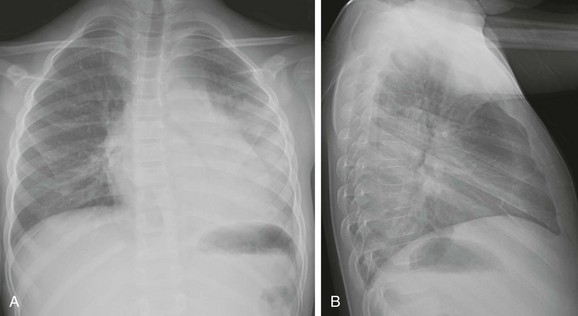
e-Figure 57-5 Sickle cell disease with acute chest syndrome.
An 8-year-old boy with sickle cell disease who presents with fever. A and B, Posteroanterior and lateral chest radiographs show dense airspace consolidation in the left lower lobe with small pleural effusion.
Although CT is not generally used in the setting of ACS, chronic sickle cell lung disease has been studied by using HRCT. Abnormal findings most pronounced at the lung bases include parenchymal bands, interlobular septal thickening, architectural distortion, and traction bronchiectasis. Honeycombing is unusual, in contrast to other types of pulmonary fibrosis.18
Treatment and Follow-up: Therapy for ACS includes hydration, analgesia, respiratory support (including bronchodilators), broad-spectrum antibiotics, transfusion therapy, and sometimes corticosteroids.19 Imaging follow-up is primarily dictated by clinical progression, improvement, or both and may be accomplished by using radiography.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is now the preferred term describing the condition showing a proliferation of a group of histiocytes known as Langerhans cells. Langerhans cells are of myeloid dendritic cell origin and contain characteristic Birbeck bodies on histology.20 The peak age at initial diagnosis is 1 to 3 years, and most patients present with osseous lesions.20 LCH is currently classified according to the extent of involvement, whether it involves a single site (better prognosis) or multiple sites (higher risk of long-term problems). The involvement of “risk organs” (liver, spleen, lung, bone marrow) indicates a poorer prognosis and demands more aggressive treatment.21
Pulmonary involvement is reported to be present in 23% to 50% of children with multisystem LCH.12,20 The mean age of patients with lung disease was 11.9 months in one study. Disease-free survival in these patients was 69%, with 3 out of 4 deaths occurring when “risk organs” other than the lung were involved.22 Although lung disease was traditionally considered a poor prognostic indicator in LCH, recent studies have shown that it does not adversely affect outcome. Primary pulmonary LCH (single site) is rare in children and is typically seen in young adult smokers.21,22
Etiology: The etiology of LCH is not known. Most investigators believe it to be an immune-mediated condition, in which the Langerhans cells accumulate and cause an inflammatory reaction. In the lungs, destructive granulomatous lesions occur in the interstitial tissues, bronchial and bronchiolar epithelium, and subpleural septa that may eventually lead to cystic lesions.20,21 Pulmonary fibrosis occurs later in 10% of children and may demonstrate multiple small cysts giving the lung a honeycomb appearance.23
Imaging: Radiologic findings of LCH vary widely with the extent of the disease and are summarized in Table 57-1. Initial chest radiography may be normal. Small nodules and cysts may be seen, with upper lobe involvement equal to or more extensive than lower lobe involvement (Fig. 57-6). The reticular appearance noted on chest radiography is often from multiple small cysts.20,24 LCH is the most common cause of acquired extensive cystic lung disease in children (e-Fig. 57-7). Spontaneous pneumothorax occurs in about 11% of patients with pulmonary involvement and may be the first indicator of pulmonary disease.25 However, it may be also a manifestation of more advanced disease, as in adolescents who more often have large coalescent cysts (see e-Fig. 57-7).12
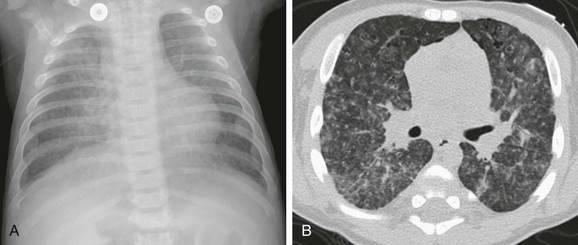
Figure 57-6 Langerhans cell histiocytosis of the lungs in a 2.5-month-old boy who initially presented with a skin rash.
A, An anteroposterior chest radiograph shows a coarse reticulonodular interstitial pattern. B, An axial computed tomography image in a lung window shows the coarse reticulonodular interstitial pattern with some small thin-walled cysts seen in the anterior lung bilaterally.
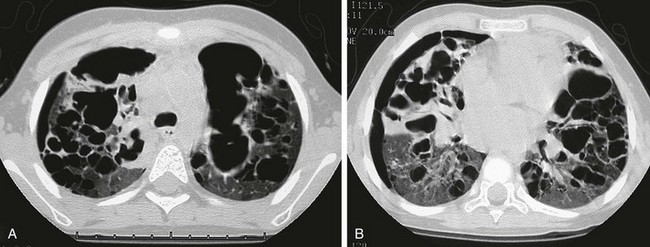
e-Figure 57-7 Progressive advanced changes in Langerhans cell histiocytosis (LCH) of the lungs.
This child first presented at age 2 years with LCH involving both temporal bones and cystic lung disease. The patient was treated with chemotherapy and returned at age 4 years with increasing respiratory distress. The chest radiograph (not shown) showed a marked interval increase in bilateral cystic lung changes and suggested a possible small right pneumothorax. A and B, High-resolution computed tomography shows that multiple cysts of varying sizes have largely replaced much of the lung parenchyma; loculated right pneumothorax was confirmed. Within 3 weeks, the patient developed a large left pneumothorax and died of this cystic lung process 1 week later.
CT is indicated for neonates with LCH and any patient with abnormalities on chest radiography.21 HRCT findings in LCH are characterized by small nodules (with or without cavitation) with upper and middle lobe predominance (e-Fig. 57-8).12,20,26 The pleura may be thickened and sometimes replaced by a thick layer of granulomatous tissue. However, pleural effusions are rare.27 Mediastinal and hilar adenopathy is also rare in children with pulmonary LCH.20,24
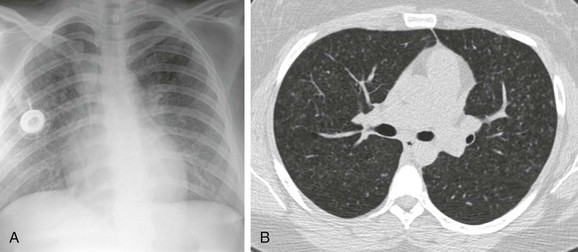
e-Figure 57-8 A patient with known diagnosis of Langerhans cell histiocytosis with relapse of disease with osseous lesion in the scapula (not shown).
A, The chest radiograph shows nodular interstitial abnormality representing pulmonary histiocytosis. B, An axial chest computed tomography (CT) image shows more clearly the small interstitial nodules and tiny thin-walled cysts in the upper lobes, characteristic CT findings of histiocytosis.
Treatment and Follow-up: Treatment of pulmonary LCH depends on the extent of disease and whether “risk” organs are involved. Generally, the first line of treatment is corticosteroids, vinblastine, or both, with duration of therapy being determined by response. Radiation therapy has fallen out of favor except for treating unstable bone lesions.21 Imaging follow-up for pulmonary LCH depends on clinical symptoms and is best performed with CT.
Gaucher Disease and Niemann-Pick Disease
Etiology: Gaucher disease is caused by a deficiency of the enzyme glucocerebrosidase that results in an accumulation of glucocerebroside in phagocytic cells. Pulmonary involvement is most common in patients with neuronopathic types. Neimann-Pick disease is caused by a deficiency of sphingomyelinase, resulting in accumulation of sphingomyelin. Progressive pulmonary fibrosis has rarely been reported in Neimann-Pick disease.28
Imaging: A diffuse reticular lung pattern may be seen on chest radiography with Gaucher disease.29 Niemann-Pick disease may produce similar findings.27 Lipoid pneumonia has been described in patients with Neimann-Pick disease.30 Imaging findings are summarized in Table 57-1.
Treatment and Follow-up: Visceral involvement may improve with enzyme replacement therapy in Gaucher disease, and this therapy may offer an alternative to lung transplantation. The current treatment for Neimann-Pick disease is whole lung lavage.28,30 CT is more sensitive than plain radiography for imaging follow-up, when clinically warranted.
Pulmonary Conditions Occurring with Generalized Systemic Illness
In the pediatric population, cardiogenic edema usually presents in infants less than 6 months of age and is the result of congenital heart disease (CHD) (Figs. 57-9 and 57-10). In an older child, cardiomyopathy may be the cause of cardiogenic edema. Affected infants often present with nonspecific symptoms, including feeding difficulties, grunting, diaphoresis, wheezing, and retractions.31 Rarely, pulmonary edema may be the presenting manifestation of CHD.
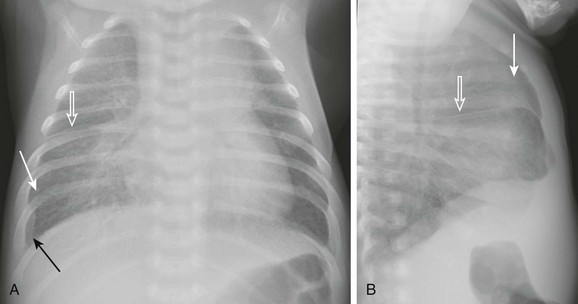
Figure 57-9 Pulmonary edema caused by obstructed pulmonary venous return. This 6-week-old infant presented to the emergency room with tachypnea and feeding difficulties.
A and B, Anteroposterior and lateral chest radiographs show normal heart size and subtle interstitial edema characterized by hyperinflation, septal lines (white arrows), fissural thickening (open arrows), and tiny right pleural effusion (black arrow). Diagnosis of cor triatriatum was made with echocardiography.
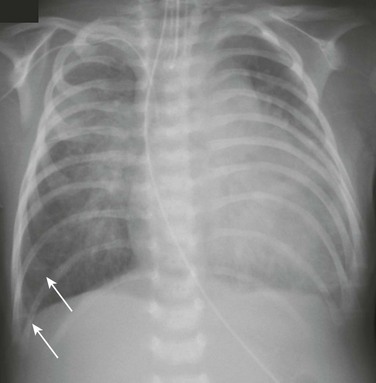
Figure 57-10 Cardiogenic edema.
A 5-week-old male infant with known ventricular septal defect presenting with tachypnea and respiratory distress requiring intensive care unit admission. An anteroposterior chest radiograph shows cardiomegaly, hyperinflation, and pulmonary edema characterized by perihilar airspace opacity, indistinct hilar vessels, and septal lines (arrows).
The frequent causes of noncardiogenic pulmonary edema in children include acute respiratory distress syndrome (ARDS), near-drowning, neurogenic pulmonary edema, renal disease, and upper airway obstruction. Other less common causes of noncardiogenic pulmonary edema include aspiration pneumonia, hydrocarbon pneumonitis, smoke inhalation, and drug reactions.32
Cardiogenic Pulmonary Edema
Etiology: Cardiogenic pulmonary edema occurs significantly less frequently in children than in adults. The common underlying causes for cardiogenic pulmonary edema include left-to-right shunting lesions, left ventricular outlet obstruction, obstruction of pulmonary venous return, or cardiomyopathy (e-Fig. 57-11).31 Cardiogenic pulmonary edema progresses in severity as pulmonary capillary wedge pressure increases.
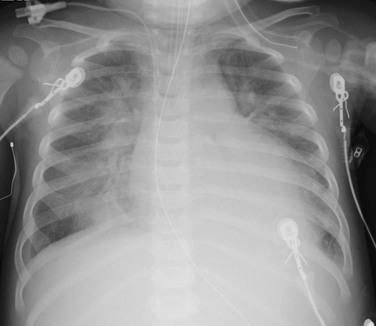
e-Figure 57-11 Dilated cardiomyopathy.
A 14-month-old female presented with 24 hours of increased work of breathing and wheezing. The portable chest radiograph shows characteristic cardiomegaly, perihilar and basilar distribution of airspace opacity caused by pulmonary edema, and a small right pleural effusion.
Imaging: On chest radiography, cardiomegaly is usually found except in instances of pulmonary venous obstruction, in which interstitial edema is present in the absence of cardiac enlargement (see Fig. 57-9).
Early findings in interstitial edema in children with cardiogenic pulmonary edema are indistinct vascular margins and bronchial wall thickening. Interlobular septal thickening (septal lines or Kerley B lines) and interlobar fissural thickening are also often seen. In infants and young children, fissural thickening is often more easily recognized than Kerley lines and is therefore an important clue for diagnosing cardiogenic pulmonary edema in children (see Fig. 57-9). An indirect radiographic finding often seen in children with CHD is hyperinflation. This may be a pitfall for the imager if he or she attributes the findings of peribronchial thickening and hyperinflation to airways disease and does not recognize them as subtle findings of interstitial edema (see Fig. 57-9). Alveolar edema usually occurs after development of interstitial edema and is the most recognizable radiographic finding of pulmonary edema (see Fig. 57-10 and e-Fig. 57-11). The classic appearance of acute alveolar edema is a central or “butterfly” distribution of airspace disease, with central edema and sparing of the lung periphery. Pleural effusions are also usually present (see e-Fig. 57-11).
Although not commonly used to diagnose pulmonary edema, CT is sensitive for the detection of pulmonary edema of any etiology. CT shows peribronchial cuffing, septal lines, ground-glass opacities, and air space consolidation in order of increasing severity (e-Fig. 57-12).33,34
e-Figure 57-12 Glomerulonephritis.
A 13-year-old male with history of asthma presented with 5 days of shortness of breath and was found to be hypoxic. A and B, Posteroanterior chest radiograph and magnified view show pulmonary edema with interstitial septal lines (arrow) and tiny left pleural effusion (open arrow). C and D, Axial and coronal computed tomography images depict typical findings of pulmonary edema showing interlobular septal thickening, dependent ground glass opacity, and bilateral small pleural effusions. The child was diagnosed with postinfectious glomerulonephritis.
Treatment and Follow-up: Treatment for cardiogenic pulmonary edema is generally supportive and includes corrective or palliative treatment for CHD. Diuretics and inotropes may be used for cardiac dysfunction. Ventilator support is provided, if needed. Extracorporeal membrane oxygenation and other types of ventricular assist devices are now being used more frequently in the setting of pediatric cardiac failure.35 Typically, imaging follow-up with chest radiography is adequate, although CT may be used to clarify the vascular anatomy.
Noncardiogenic Pulmonary Edema
Etiology: Many episodes of noncardiogenic pulmonary edema progress to ARDS. ARDS accounts for 1% to 3% of pediatric intensive care admissions, and the mortality in various series ranges from 40% to 60%.32 Sepsis, near-drowning, pneumonia, and smoke inhalation are the most frequent antecedents to pediatric ARDS. In most cases, ARDS progresses through stages of immediate lung injury, exudative alveolitis, fibroproliferative repair, and, in survivors, recovery. Ventilation–perfusion imbalance leads to varying degrees of hypoxemia.32
Neurogenic pulmonary edema may occur with increased intracranial pressure, which, in theory, causes a shift of blood from the systemic to the low-resistance pulmonary circulation. Pulmonary edema caused by renal disease (chronic renal failure, glomerulonephritis, nephrotic syndrome, or all of these) is felt to be multifactorial in mechanism, including factors such as fluid retention and hypoproteinemia (see e-Fig. 57-12). Although uncommon, postobstructive pulmonary edema may also occur in upper airway obstruction from conditions such as croup, epiglottitis, foreign body obstruction, and near-strangulation or when relief of upper airway obstruction occurs. The mechanism is thought to be an abrupt decrease in intrathoracic pressure caused by an attempt to inhale against a closed upper airway, also called negative-pressure pulmonary edema.31
Imaging: The radiographic pattern for ARDS is a multiple stage continuum (Fig. 57-13).36,37 First, the radiograph is reflective of the initial insult, or it may be clear, or both. In 12 to 24 hours, bilateral pulmonary opacities develop, consistent with noncardiogenic pulmonary edema, and opacities become progressively more confluent.37 Subsequent improvement or continued worsening may be seen in the next several days. For patients with progression, the course often is then complicated by air leak phenomenon (pneumothorax, pneumomediastinum, pulmonary interstitial emphysema), with or without ventilator-associated infections. Additionally, further progression to chronic changes of fibrosis may occur.32,36,37
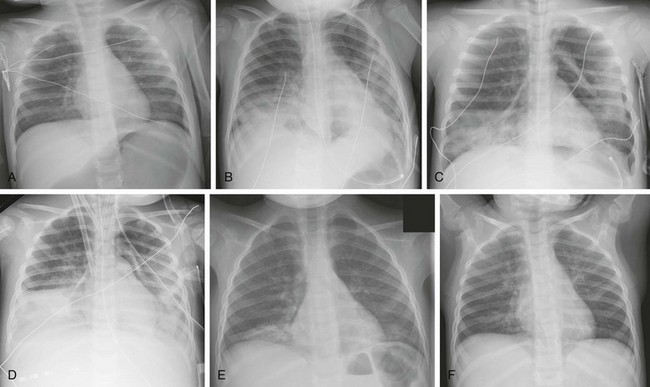
Figure 57-13 A 17-month-old girl who ingested citronella oil from a backyard lamp and developed acute respiratory distress syndrome.
A, The chest radiograph at presentation is near-normal. B, At day 2, bibasilar airspace disease developed. She then deteriorated clinically, with increasing hypoxia that required intubation. C, Shortly thereafter, air leak developed, with pneumomediastinum and left pneumothorax at day 4. Further clinical deterioration occurred, and bacterial cultures from the endotracheal tube were positive consistent with ventilator associated pneumonia. D, She was then placed on arteriovenous extracorporeal membrane oxygenation with cannulas at day 7. Eventually, she clinically improved and was discharged from the hospital. E and F, Follow-up radiographs at 2 and 4 months show gradual clearing of interstitial type opacities in the bilateral lung bases.
The pulmonary findings of noncardiogenic pulmonary edema (e.g., ARDS) are similar to that of cardiogenic pulmonary edema, but the radiographs typically do not show cardiomegaly (see e-Fig. 57-12, Fig. 57-14, and e-Fig. 57-15).37 Pleural effusions may not be a dominant finding, but they are frequently seen, particularly in patients requiring maximum clinical support as a manifestation of anasarca.
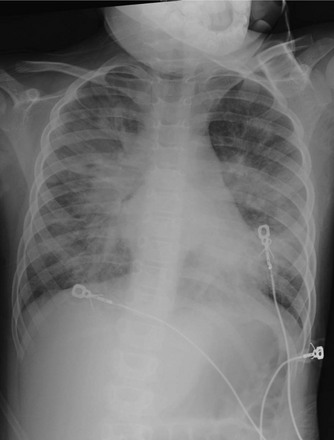
Figure 57-14 A 4-year-old girl was admitted to the intensive care unit for asthma with hypoxia and wheezing.
She had an acute exacerbation one night and was noted to also have obstructive sleep apnea. The following morning this anteroposterior view of the chest showed pulmonary edema pattern caused by upper airway obstruction, which promptly resolved when her symptoms were addressed more aggressively.
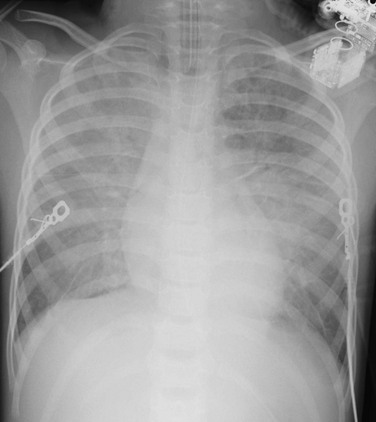
e-Figure 57-15 Neurogenic pulmonary edema.
This 4-year-old girl presented with severe traumatic brain injury of unknown etiology. Portable anteroposterior chest radiograph shows perihilar distribution of airspace disease.
Glomerulonephritis and nephrotic syndrome are often sufficiently characteristic, aiding the radiologist to suggest the correct diagnosis. A child or teenager presents without a known cardiac history with a basilar predominant interstitial edema pattern, often with pleural effusions (see e-Fig. 57-12). Glomerulonephritis may be distinguished from nephrotic syndrome by its tendency to show mild cardiomegaly and increased pulmonary vascularity.38
Treatment and Follow-up: Treatment of noncardiogenic pulmonary edema is directed toward the primary cause, is generally supportive, or both. Ventilatory support is often needed. The use of positive end-expiratory pressure with mechanical ventilation has been shown by CT to improve ventilation of dependent regions of the lung in a setting of ARDS.37,39 Extracorporeal membrane oxygenation is increasingly being used for children with severe respiratory failure. Pulmonary edema from neurogenic causes or upper airway obstruction usually resolves fairly rapidly, often within 12 to 48 hours (see Fig. 57-14).31 Typically, imaging follow-up will be performed with chest radiography, as needed.
Survivors of ARDS may have long-term sequelae, often demonstrable by pulmonary function testing. Follow-up chest radiography most often show a return to normal (80%) but may demonstrate increased interstitial markings, hyperaeration, or both (see Fig. 57-13, E and F).32,37,40
Pulmonary Thromboembolic Disease
Pulmonary thromboembolic disease was traditionally thought to be much less common in children than in adults but is being seen with increasing frequency in children. A 25-year pediatric autopsy series found an incidence of pulmonary embolism (PE) of 3.7% of 3600 autopsies.41 More recent studies have reported a prevalence of 14% to 15% in a population of children undergoing computed tomography pulmonary angiography (CTPA) for clinical suspicion of PE.42,43 Venous thromboembolism (including PE) is associated with an underlying risk factor in 95% of children.42–46 Surprisingly, however, a recent study of pediatric patients undergoing CTPA showed no significant difference in prevalence of risk factors between patients diagnosed with a PE compared with the control group without PE.42 A bimodal age distribution exists for PE in children, which may present in the neonatal time frame or more commonly in late childhood or adolescence.42,43,46
Clinical diagnosis of PE remains a challenge. Many episodes of PE are probably asymptomatic in both children and adults. When signs and symptoms of PE do occur, they are often nonspecific. For example, patients may present with pleuritic chest pain, dyspnea, and tachypnea. Unlike the promising data in adults, d-dimer is not helpful for diagnosis of PE in children. Studies have shown that both pediatric patients at risk for PE as well as patients with a diagnosis of PE had elevated d-dimer.43,46 In addition, no pediatric studies have been done to show that a negative d-dimer may be used to exclude PE in children.
Besides thrombotic emboli, septic embolization is a recognized cause of pulmonary embolic disease in children. Common sites of origin include osteomyelitis, soft tissue infections, infected central venous catheters, endocarditis, and tonsillitis or pharyngitis; the latter may lead to Lemierre syndrome.47,48
Pulmonary fat embolism is characterized clinically by a triad of progressive pulmonary insufficiency, cerebral dysfunction, and petechiae. Fat embolism is a cause of ACS following bone marrow infarcts in sickle cell patients.17 Most other cases of pulmonary fat emboli occur after trauma, especially from fractures of the femora and tibiae. Autopsy studies have shown an incidence of 60% to 97%, but the risk for fat embolism from a single long bone fracture is reported to be 1% to 3%.49 Symptoms usually do not develop until 24 to 48 hours after injury.
Thrombotic Emboli
Etiology: Deep venous thrombosis (DVT) is a common precursor to PE in children. In one study, 56% of PEs in children were found to be associated with thrombosis at another location.46 However, even occurrence of a DVT in a child will usually have a predisposing cause such as the presence of a central venous catheter. Whereas lower extremity DVT is most common in adults, upper extremity DVT is more common in children (accounting for two thirds of DVTs) because of the association with central venous lines.44,46,50 Other factors predisposing to thromboembolism include malignancy, hypercoagulable states (such as SCD, SLE, or coagulation disorders), CHD, trauma, infection, recent surgery, nephrotic syndrome, and use of birth control pills (see Fig. 57-16 and e-Fig. 57-17).46 A recent study found a nearly 2% prevalence of unsuspected PE in pediatric oncology patients undergoing routine thoracic CT studies.51
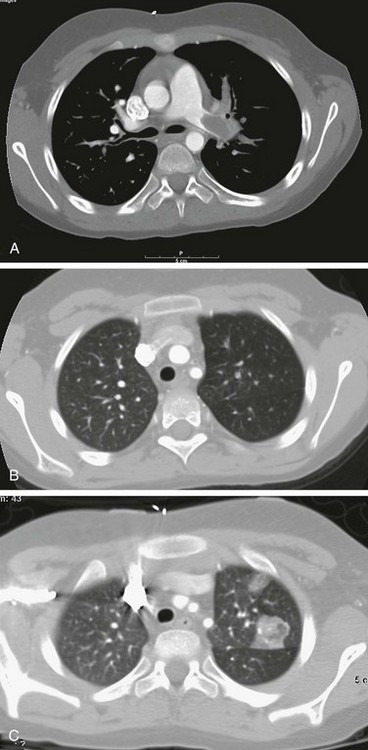
Figure 57-16 Pulmonary embolism found on computed tomography (CT) scan in a 9-year-old patient with a history of nephrotic syndrome and new hypoxia.
A, An axial CT image clearly shows filling defects in the left proximal pulmonary artery and the branch to the left upper lobe. B, Lung window from the initial CT shows paucity of pulmonary vessels in the left upper lobe. C, Follow-up CT image obtained 36 hours later shows interval development of a focal pulmonary infarct in the left upper lobe.
Imaging: Radiographic findings of PE are unfortunately nonspecific. If the embolic episode occurs without infarction, the result of chest radiography is most often normal. If it is abnormal, it may show localized or generalized oligemia (Westermark sign), an enlarged pulmonary artery, or loss of lung volume. PE with associated infarct classically appears as a truncated cone of homogeneous opacity located at the lung periphery and abutting the pleural surface (Hampton hump), but in fact, this pattern is uncommon.
CTPA has a variable but high sensitivity (60% to 100%) and high specificity (81% to 100%) for the diagnosis of PE and provides direct visualization of emboli in central and in smaller, peripheral pulmonary arteries (Fig. 57-16 and e-Fig. 57-17).44 Successful technique requires a multidetector helical scanner and the use of a well-timed contrast bolus, which allows detection of the pulmonary artery filling defects. The interpretation is more challenging if the contrast enhancement is suboptimal or if patient movement limits the image quality. Adjacent hilar lymph nodes may be a pitfall but reformatted images in multiple planes usually clarify their extrinsic location. In addition to the direct vascular findings, CT may also reveal a region of oligemia, peripheral pulmonary consolidation (infarct) with absent or poor enhancement, or both (see Fig. 57-16 and e-Fig. 57-17). Wedge-shaped peripheral consolidation was found to be significantly associated with presence of PE on a review of CTPA studies in children.52
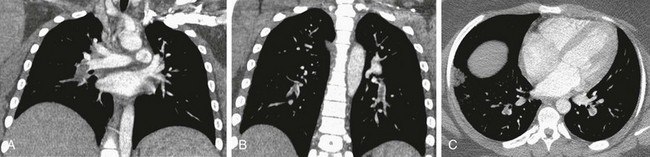
e-Figure 57-17 Pulmonary embolus in a patient with sickle cell disease, shortness of breath, and hypoxia.
A and B, Coronal computed tomographic (CT) images show right and left lower lobe pulmonary artery filling defects. C, An axial CT image in same patient shows the ability of computed tomography to demonstrate the filling defects of a pulmonary embolus even in the distal segmental and subsegmental pulmonary artery branches, as seen in this patient, in both lower lobes. Note also the wedge-shaped airspace opacity at the lateral right lower lobe consistent with an infarct.
Nuclear medicine assessment of pulmonary ventilation and perfusion (V/Q scan) remains an available imaging modality for evaluation of PE in children. A single segmental or larger perfusion defect, with mismatched normal ventilation, signals a high probability of PE. Although some are advocating continued use of V/Q scans in select populations with normal chest radiography results, CTPA is currently preferred in most centers, likely because of the many advantages of CT: it requires minimal patient cooperation, it is rapidly available, and it screens for alternate diagnoses.45,46,53–56
Many studies are underway to determine the utility of contrast-enhanced magnetic resonance angiography (CEMRA) for the diagnosis of PE. This technique would be particularly advantageous in children given the lack of radiation exposure and potential to screen upper extremity veins for thrombosis at the same time as the evaluation for PE.44 PIOPED III (Prospective Investigation of Pulmonary Embolism Diagnosis III) results showed sensitivity of 78% and specificity of 99% in technically adequate CEMRA studies, but unfortunately 25% of studies were not considered technically adequate. Sensitivity was better for the central pulmonary arteries and decreased progressively in the more distal branches.56 The advent and use of new blood pool magnetic resonance imaging contrast agents may improve the capabilities of CEMRA.
Treatment and Follow-up: Anticoagulation therapy is generally used to prevent extension of thrombus and the development of late complications such as recurrences and postthrombotic syndrome. Most children are treated primarily on the basis of recommendations for adults, since large clinical trials of antithrombotic therapy in children are lacking.44,46 Traditionally, the first-line anticoagulant used was unfractionated heparin, but bleeding complications are 2% to 18%, depending on patient population, and it requires intravenous access for administration. The low-molecular-weight heparins being used more frequently have several advantages: lower bleeding complications (0% to 5%), subcutaneous administration, and reduced need for monitoring. These advantages make it ideal for outpatient treatment.44,46
Thrombolytic therapy causes faster resolution of the embolus, but data regarding efficacy and safety in children are lacking. The agent most commonly used is tissue plasminogen activator. A substantially higher risk of bleeding complications exists, with one study reporting bleeding in 68% of patients and transfusion required in 39% of treated children. Given that risk, most feel that thrombolytic therapy should only be considered in pediatric patients with unstable hemodynamic condition, massive PE, or both.44,46
Imaging follow-up for PE is based on clinical factors and is best performed with CTPA.
Septic Embolization
Etiology: In septic emboli, hematogenous spread of infection from the original site occurs, as well as deposition of bacteria in end organs. Osteomyelitis is a common primary site of septic embolic disease (Fig. 57-18 and e-Fig. 57-19). In one study from Taiwan, 8 out of 10 pediatric patients with septic emboli had methicillin-resistant Staphylococcus aureus as the responsible organism, and most patients had primary skin, soft tissue, or bone infections.48 Lemierre syndrome is septic thrombophlebitis of the internal jugular vein secondary to bacterial pharyngitis or tonsillitis, usually from Fusobacterium necrophorum, with subsequent septicemia and systemic spread of infection and lung involvement in 97% of cases (Fig. 57-20).47,57,58
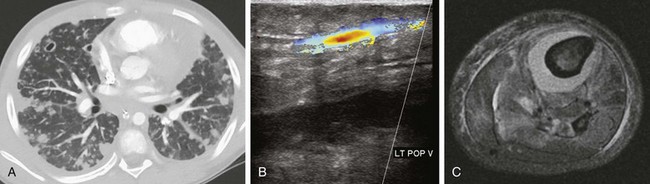
Figure 57-18 Septic emboli.
A 12-year-old male who presented in septic shock with history of left leg swelling. Blood cultures were positive for methicillin-resistant Staphylococcus aureus. A, Initial study was the chest computed tomography showing multiple cavitary and noncavitary pulmonary nodules. B, Lower extremity ultrasonography showed a deep venous thrombosis in the popliteal vein, initially thought to be the source of sepsis. C, A lower leg axial magnetic resonance image (fat-saturated, T2-weighted) obtained 2 days later shows subperiosteal abscess around the tibia. This patient was subsequently found to have osteomyelitis in multiple sites, including right hip and iliac bone, thoracic spine, left sided ribs, left clavicle, and scapula.
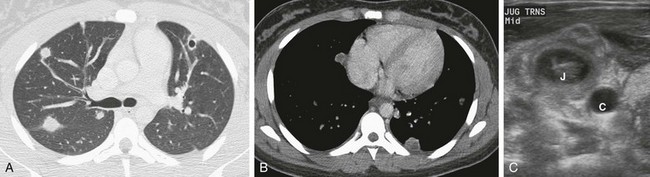
Figure 57-20 Lemierre’s syndrome.
A 16-year-old female seen initially in urgent care for pharyngitis. Ten days later, she presented to the emergency room with persistent fever, night sweats, and cervical lymphadenopathy. Pneumonia reported on radiographs (not shown). A, An axial chest computed tomography (CT) scan showing cavitary and noncavitary nodules. B, An axial chest CT image showing low-attenuation center in multiple nodules. C, A transverse ultrasonographic image through the neck shows thrombus in the right internal jugular vein (J) adjacent to the normal anechoic carotid artery (C).
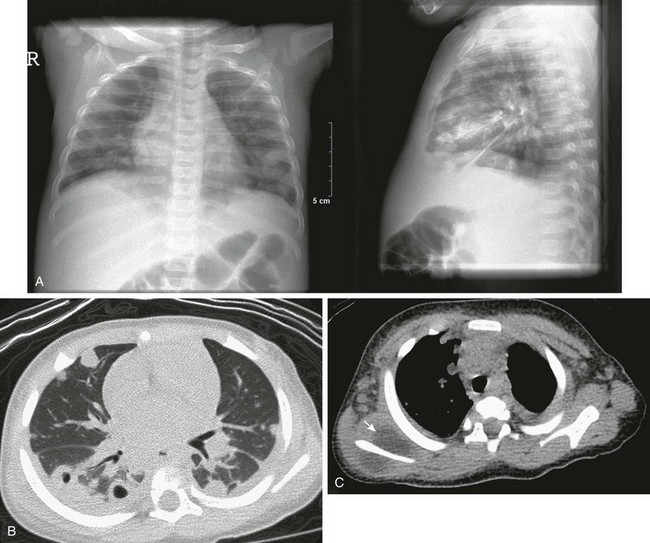
e-Figure 57-19 Septic emboli in the lungs of a patient with sickle cell disease.
An 8-month-old girl with fever, positive blood cultures for Staphylococcus aureus, and shoulder swelling. Shoulder radiographs (not shown) were normal. A, Anteroposterior and lateral chest radiographs show nonspecific patchy areas of airspace disease. B, An axial computed tomography (CT) image in lung window shows multiple pulmonary nodules, several with cavitation, and small bilateral pleural effusions. C, Mediastinal window from the noncontrast chest CT shows low-attenuation collection surrounding the right scapula (arrows). Diagnosis of osteomyelitis with septic emboli was made.
Imaging: The classic radiologic finding is one or more cavitary peripheral nodules, although radiographs more often show nonspecific airspace consolidation (see e-Fig. 57-19). CT is the most sensitive imaging test, more frequently demonstrating cavitation of the nodules (in 85% to 100%) and presence of a “feeding vessel” (in 50% to 71%). Hilar lymph node enlargement and pleural effusions may also be seen.47,48
Treatment and Follow-up: Antibiotics is the mainstay for treating the primary source of infection. If the source is unknown, echocardiography is indicated to look for endocarditis. Drainage of focal abscesses is indicated. Currently, no consensus exists on the use of anticoagulation in the setting of Lemierre syndrome.47 Imaging follow-up may be performed with radiography or CT. Generally, if clinical improvement is prompt, radiography will be adequate. If fever or other symptoms persist, CT may be needed to better assess for persistent sources of infection.
Pulmonary Fat Embolism
Etiology: Fat droplets, typically from the marrow cavity, appear in blood and lodge in the capillaries of the lungs, brain, and other organs. Dyspnea, cyanosis, and tachycardia with cough and fever are the common clinical symptoms, and fat globules may usually be found in blood and urine on laboratory evaluation.
Imaging: Most often, the result of chest radiography is normal. Abnormalities, when present, are usually nonspecific and include airspace consolidation from alveolar hemorrhage and edema, more commonly peripheral and basilar in location. The time lapse between trauma and the development of radiographic findings is usually 1 to 2 days. Such delay may be helpful in differentiating fat embolism from traumatic lung contusion; in the latter, the chest radiograph is abnormal initially.59
Lung Injury Caused by Extrinsic Agents
Drug-Induced Pulmonary Disease
Although a large number of pharmacologic agents may cause or exacerbate pulmonary disease in adults, the incidence and types of pulmonary effects in children are largely unknown.60 Cause and effect are difficult to establish, and the time between drug administration and reaction is highly variable.
Etiology: Chemotherapeutic agents (such as cyclophosphamide, bleomycin, busulfan, cisplatin, nitrosureas, mitomycin-C, and methotrexate) are the most common drugs associated with pulmonary toxicity (occurring in up to 10% of patients).61,62 These medications may cause interstitial pneumonitis or fibrosis, hypersensitivity reaction, ARDS, or bronchiolitis obliterans with organizing pneumonia. Amiodarone may cause interstitial thickening, bronchiolitis obliterans with organizing pneumonia, or both.61
Airway hyperreactivity has been associated with ingestion of aspirin and nonsteroidal antiinflammatory agents. Hypersensitivity reactions may be produced by reactions to methotrexate, sulfonamides, and nitrofurantoin.61 Noncardiogenic pulmonary edema may result from overdoses of heroin, morphine, methadone, and cocaine.61,63 Pulmonary hemorrhage may be seen after administration of anticoagulants, surfactants, and penicillamine.52
Imaging: Radiographic findings of drug-induced reactions are generally nonspecific, consisting of heterogeneous pulmonary opacities, homogeneous pulmonary opacities, or both.60
HRCT is most useful for early diagnosis of pulmonary toxicities. The findings depend on the histologic process and may include interstitial disease, alveolar disease, or both.60,61 Interstitial pneumonitis and fibrosis, typical of bleomycin, produce ground-glass opacities, areas of consolidation, and irregular linear opacities that tend to predominate in the lower lung zones. Methotrexate hypersensitivity resembles hypersensitivity pneumonia, showing ground-glass opacities and poorly defined centrilobular nodules. ARDS results in predominantly dependent airspace consolidation. The least common reaction, a bronchiolitis obliterans with organizing pneumonia-like reaction, causes peribronchial or subpleural areas of consolidation.61
Treatment and Follow-up: Removal of the causative agent is the ideal management for drug-induced pulmonary disease if the toxicity can be recognized and the removal is feasible. Unfortunately, some pulmonary toxicities are often irreversible. If symptoms persist, radiography and CT may help in follow-up management.
Radiation Damage to the Thorax
Lung damage may follow radiation therapy given primarily to the lungs, the mediastinum, the chest wall, or the total body. The incidence of radiation damage to the lung in adult patients treated for lung cancer, mediastinal lymphoma, and breast cancer ranges from 5% to 20%.64 An even larger portion of patients may have changes in pulmonary function tests, although symptoms are present in only a few.65
In pediatric patients, whole-lung irradiation to 12 gray (Gy) was used in the third national Wilms tumor study, and 15 patients (approximately 10%) developed radiation pneumonitis.65 Data from the Childhood Cancer Survivor Study showed a relative risk of lung fibrosis of 4.3 at 5 years or more after diagnosis among children who received chest radiation, total body irradiation, or both. The cumulative incidence of lung fibrosis at 20 years after diagnosis was 3.5%, and the incidence continued to increase from diagnosis up to 25 years. An increased risk for other pulmonary conditions such as supplemental oxygen use, recurrent pneumonia, chronic cough, and pleurisy was also seen in this population.62
Etiology: Important factors in determining whether the lungs are damaged include the volume of tissue irradiated, the dosage and its fractionation, the relative biologic effectiveness of the therapy employed, and the use of concomitant chemotherapy. Radiation-induced lung injury is typically categorized as early pneumonitis (1-6 months) and late pulmonary fibrosis (>6 months).66
Imaging: Radiographic changes, if present, usually appear a few months after the cessation of radiation therapy. The radiographic appearance initially is that of an ill-defined alveolar process. CT typically shows ground-glass opacity in a sharply defined zone not conforming to anatomic boundaries. As the process evolves, it becomes more interstitial in appearance until changes recognized as radiation fibrosis appear (e-Fig. 57-21). CT is more sensitive than chest radiography in showing these findings. If the mediastinum has been irradiated, a central perimediastinal, sharply delineated area of linear increased opacity corresponding to the radiation portal is typically seen (see e-Fig. 57-21).64,66
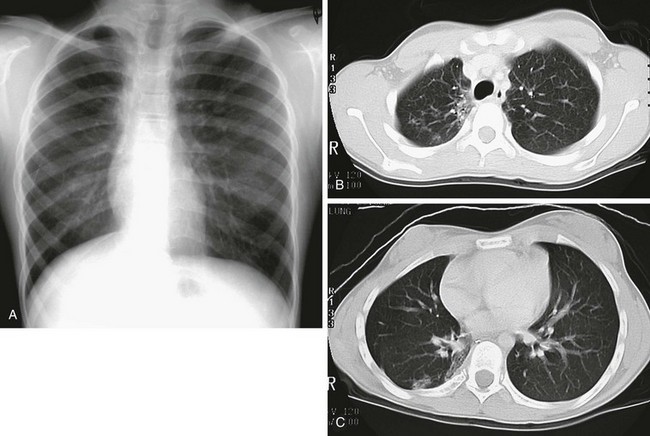
e-Figure 57-21 Effects of radiation therapy in a 12-year-old girl 2 years after intense radiation therapy for a right paraspinal primitive neuroectodermal tumor.
A, A chest radiograph shows a smaller right hemithorax and mild rightward mediastinal shift. B and C, Chest computed tomography scans show a smaller right lung, rather sharp-edged changes of lung opacity along the medial aspect of the right upper lobe, and a few scattered peripheral opacities.
Treatment and Follow-up: Corticosteroids are sometimes used for managing acute radiation-induced pneumonitis, and long-term treatment is aimed at control of symptoms.66 Choice of follow-up imaging is largely determined by clinical factors. CT will be more sensitive to identify subtle findings of fibrosis.
Inhalation Injuries
Children are rarely exposed to the industrial gases, chemicals, and particulates that adult workers may encounter. Pediatric inhalation injury is usually secondary to substances encountered in the home, the most common of which is smoke from house fires. Other causes, far less common, include chlorine gas (pool disinfectant), talcum powder, and household cleaning agents.32
Etiology: Inhalation injuries are rare in pediatric fire victims but should be suspected in those with burns in the head-and-neck area, blackened sputum, and respiratory symptoms. Thermal injury is most likely to produce upper airway edema. More peripheral airway and airspace injuries are usually secondary to toxic chemical effects of the products of combustion. Typically, pulmonary burns progress through three distinct clinical stages: (1) bronchospasm (1 to 12 hours after the burn), (2) pulmonary edema (6 to 72 hours), and (3) bronchopneumonia (after 60 hours) (Fig. 57-22). Bacterial pneumonia is very frequent after the third or fourth day of injury and should be anticipated.32
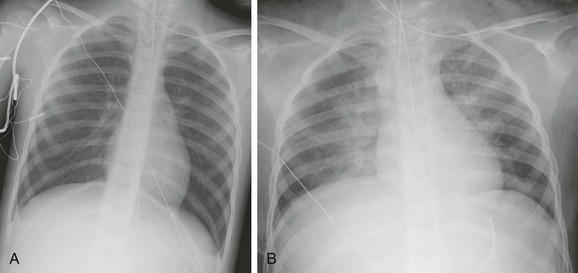
Figure 57-22 A 4-year-old girl rescued from a house fire.
A, Portable anteroposterior chest radiograph at admission shows clear lungs and hyperinflation. B, One day later, bilateral pulmonary edema is developing and lung volumes are lower.
Inhalation injury from chlorine gas inhalation has been reported in children. This irritant gas is found in swimming pool disinfectants and household cleaning agents. It reacts with the water in tissue to produce hydrochloric acid, which leads to mucosal necrosis. Irritation of the upper respiratory tract, cough, and wheezing are early findings.32,67
Another inhalation injury that may occur in infants is caused by inhalation of the small (approximately 5 micrometers) particles of hydrous magnesium silicate present in talcum powder. Inhaled talc produces airway obstruction and impairs ciliary action, which leads to inflammation. Symptoms of cough and respiratory distress are rapid after exposure.68
Passive smoking is associated with an increased rate of respiratory illness in infancy and childhood. The frequency of reactive airways disease is increased in exposed infants.69,70
Imaging: The results of early radiographs are often normal in smoke inhalation, but diffuse or focal patchy opacities or pulmonary edema may be noted (see Fig. 57-22). More diffuse opacities, which usually appear after a latent period of 12 to 48 hours, may clear slowly. Rapid appearance of total or subtotal lung consolidation has been described in up to 10% of burned children and is associated with high early mortality. ARDS is a serious and common complication of inhalation injuries and burns.32
With chlorine gas inhalation, the onset of pulmonary edema is often rapid. The results of chest radiography may be normal or may show hyperinflation or pulmonary edema.67,68 With talcum powder inhalation, chest radiography may show hyperinflation and patchy opacities.68
Treatment and Follow-up: Treatment, as for noncardiogenic pulmonary edema, is primarily supportive. Late sequelae, including severe bronchiectasis, asthma, and bronchiolitis obliterans, may be noted after smoke inhalation.32 Follow-up radiography is adequate if the patient shows clinical improvement. CT may be needed if clinical symptoms persist in a delayed setting.
Hydrocarbon Pneumonitis
Hydrocarbon pneumonitis occurs in 40% of children following ingestion of hydrocarbons such as lamp oil, lighter fluid, cleaning fluid, furniture and floor polishes, kerosene and gasoline.71 These low-viscosity materials are aspirated when ingested. In the United States, such exposures continue to account for 12% to 25% of lethal poisonings in children less than 5 years old.32
Etiology: Aspirated or inhaled material usually results in an intense chemical pneumonitis causing damage to the respiratory epithelium and development of cyanosis through displacement of alveolar gas by the hydrocarbon.32
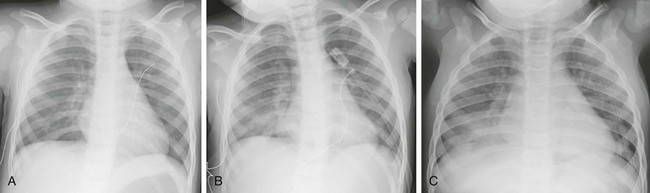
Imaging: Although changes may be evident on chest radiography as early as 30 minutes after aspiration, the findings may be delayed several hours; virtually all affected children with lung disease have abnormal chest radiographs by 12 hours (see Fig. 57-13 and e-Fig. 57-23).32 The typical pattern of hydrocarbon pneumonitis is one of patchy airspace consolidation and alveolar edema involving predominantly the medial basilar portions of the lungs bilaterally. Pneumatoceles are a well-known complication of hydrocarbon aspiration but only develop in 10% of children.71
Lipoid Pneumonia
Oral administration of mineral oil for constipation is the most common cause of exogenous lipoid pneumonia. In a study of affected children in Brazil, 79% of patients were less than 24 months of age.72
Etiology: Lipids aspirated during oral administration of mineral oil may result in an insidious chronic process that produces an intense lung response and may result in severe lung injury. Histologic proof of exposure is gained by finding lipid-laden macrophages and lipid material in alveoli on biopsy. However, the current diagnostic method of choice for diagnosing lipoid pneumonia is by analysis of bronchoalveolar lavage fluid.72
Imaging: The typical radiographic pattern of lipoid pneumonia is perihilar and basilar airspace opacities (Fig. 57-24 and e-Fig. 57-25). CT shows areas of ground-glass opacity or more confluent airspace disease (see Fig. 57-24 and e-Fig. 57-25). On CT, the presence of fat attenuation values of the airspace disease (occurring in 71% of children) should alert the radiologist to the diagnosis of lipoid pneumonia.73 In adults and children with lipoid pneumonia, the crazy-paving pattern has also been noted on thin-section CT.72–74
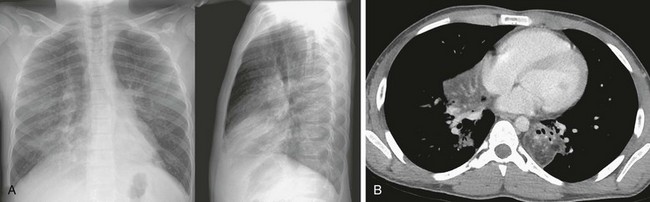
Figure 57-24 Lipoid pneumonia.
This 11-year-old boy presented with chronic cough and drainage, with recent-onset difficulty breathing. He had a history of treatment with oral mineral oil for constipation. A, Bilateral airspace opacities are noted on the initial posteroanterior and lateral chest radiographs, predominantly in the lung bases. B, An axial chest computed tomography image shows fat attenuation and focal consolidation in the left lower lobe and right middle lobe.
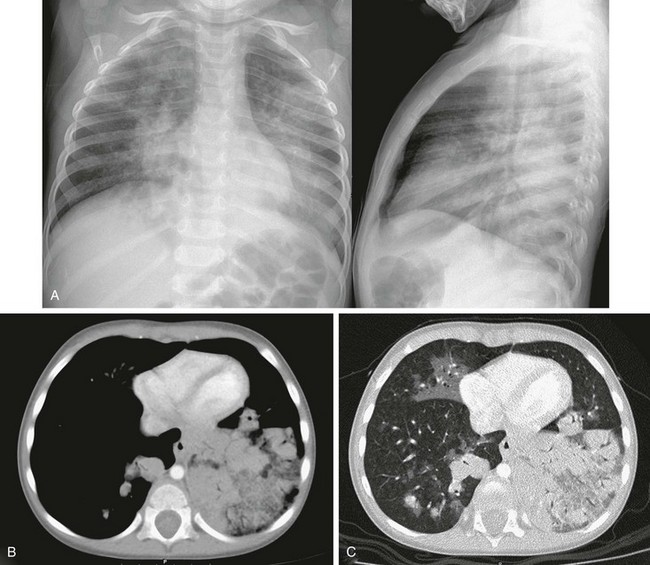
e-Figure 57-25 Lipoid pneumonia.
This 19-month-old boy had persistent fevers despite antibiotic treatment. He had been ingesting mineral oil as a treatment for constipation. A, Frontal and lateral chest radiographs show bilateral patchy areas of airspace disease. B and C, Mediastinal (B) and lung window (C) axial computed tomography images obtained using controlled ventilation technique show low-attenuation airspace disease in the left lower lobe with some degree of the crazy-paving pattern. Patchy areas of airspace disease, some with ground-glass attenuation, are also seen in the right middle lobe and right lower lobe.
Near-Drowning
Drowning is the second most frequent, after motor vehicle accidents, cause of accidental death in children. The most common site for near-drowning is the backyard swimming pool. Accidental submersion injuries demonstrate a bimodal age peak of 6 months to 4 years and 18 to 24 years.32
Etiology: Submersion time, volume of water aspirated, severity of acidosis, and length of delay in cardiopulmonary resuscitation all correlate directly with morbidity and mortality. Most submersion victims aspirate a small amount of water that may contain vomitus or a variety of contaminants. Laryngospasm occurs, which may prevent further aspiration. In about 15% of patients, laryngospasm persists and death is by anoxia (i.e., “dry drowning”).32
Imaging: The radiologic pattern of near-drowning is of pulmonary edema, and the severity depends predominantly on the amount of water aspirated. Opacities are usually bilateral and symmetric, and a significant delay, sometimes as long as 24 to 48 hours after near-drowning, may occur in the appearance of edema (Fig. 57-26).32
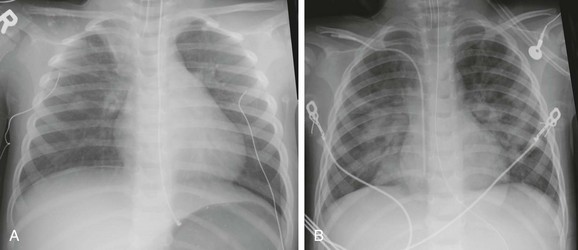
Figure 57-26 A 3-year-old male who suffered a near-drowning episode in a creek.
A, The chest radiograph at presentation is near normal except for atelectasis at the medial right apex. B, The chest radiograph 2 days later shows development of perihilar airspace disease consistent with noncardiogenic edema.
Conditions with Pulmonary Calcifications
Pulmonary Alveolar Microlithiasis
Pulmonary alveolar microlithiasis (PAM) is a rare disease characterized by the presence of innumerable calcium phosphate microliths in the alveoli. Many cases are familial and inherited as an autosomal-recessive trait.75 In one review, 17% of affected patients presented at 12 years of age or younger.76
Etiology: The etiology of PAM is currently unknown. However, the tiny stones in the alveoli consist of concentric calcium rings, which measure from 0.01 to 3 mm in size, are similar to bone histologically, radiographically, and metabolically. The number and size of the calculi and the amount of interstitial reaction increase with the age of the patient and the duration of the disease.75,76
Imaging: On chest radiography, in children with PAM, diffuse ground-glass opacities were more common than nodular calcific densities. HRCT shows ground-glass opacities and calcific densities in the lung, pleura, and interlobar septae.77 The calcified lung lesions demonstrate an avidity for bone scintigraphy agents.76
Miscellaneous Pulmonary Calcifications
Etiology: Focal pulmonary calcifications in childhood are usually the result of granulomatous infection, most commonly tuberculosis or histoplasmosis. Diffuse calcifications are probably dystrophic (calcification caused by tissue damage) and are most commonly seen after varicella infection. Metastatic calcification (calcification in normal tissue) is caused by abnormal calcium and phosphorous metabolism, usually in one of the following clinical situations: chronic renal failure and secondary hyperparathyroidism, acute renal failure, after renal transplantation, and after cardiac surgery.78
Imaging: Initially, plain radiography shows airspace consolidation or nonspecific nodular opacities, which may demonstrate a progressive increase in opacity. These opacities may be mistaken for an infectious process such as pneumonia. CT is more sensitive than chest radiography for the definitive diagnosis of pulmonary calcification.
Dinwiddie, R, Sonnappa, S. Systemic diseases and the lung. Paediatr Respir Rev. 2005;6:181–189.
Garcia-Pena, P, Boixadera, H, Barber, I, et al. Thoracic findings of systemic diseases at high-resolution CT in children. RadioGraphics. 2011;31:465–482.
Patocka, C, Nemeth, J. Pulmonary embolism in pediatrics. J Emerg Med. 2012;42(1):105–116.
Schmidt, S, Eitch, G, Geoffray, A, et al. Extraosseous Langerhans cell histiocytosis in children. RadioGraphics. 2008;28:707–726.
Seely, JM, Effmann, EL. Acute lung injury and acute respiratory distress syndrome in children. Semin Roentgenol. 1998;33:163–173.
References
1. Pesci, A, Manganelli, P. Respiratory system involvement in antineutrophil cytoplasmic-associated systemic vasculitides: clinical, pathological, radiological and therapeutic considerations. Drugs R D. 2007;8(1):25–42.
2. von Vigier, RO, Trummler, SA, Laux-Edn, R, et al. Pulmonary renal syndrome in childhood: A report of twenty-one cases and a review of the literature. Pediatr Pulmonol. 2000;29:382–388.
3. Boyer, D, Vargas, SO, Slattery, S, et al. Churg-Strauss syndrome in children: a clinical and pathologic review. Pediatrics. 2006;118:914–920.
4. Seely, JM, Jones, LT, Wallace, C, et al. Systemic sclerosis: using high-resolution CT to detect lung disease in children. AJR. 1998;170:691–697.
5. Panigada, S, Revelli, A, Silvestri, M, et al. HRCT and pulmonary function tests in monitoring of lung involvement in juvenile systemic sclerosis. Pediatr Pulmonol. 2009;44:1226–1234.
6. Brody, AS. Imaging considerations: interstitial lung disease in children. Radiol Clin North Am. 2005;43:391–403.
7. Dinwiddie, R, Sonnappa, S. Systemic diseases and the lung. Paediatr Respir Rev. 2005;6:181–189.
8. Levine, D, Akikusa, J, Manson, D, et al. Chest CT findings in pediatric Wegener’s granulomatosis. Pediatr Radiol. 2007;37(1):57–62.
9. Marten, K, Schnyder, P, Schirg, E, et al. Pattern-based differential diagnosis in pulmonary vasculitis using volumetric CT. AJR. 2005;184:720–733.
10. Ananthakrishnan, L, Sharma, N, Kanne, JP. Wegener’s granulomatosis in the chest: high-resolution CT findings. AJR. 2009;192:676–682.
11. Akikusa, JD, Schneider, R, Harvey, EA, et al. Clinical features and outcome of pediatric Wegener’s granulomatosis. Arthritis Rheum. 2007;57(5):837–844.
12. Garcia-Pena, P, Boixadera, H, Barber, I, et al. Thoracic findings of systemic diseases at high-resolution CT in children. RadioGraphics. 2011;31:465–482.
13. Delgado, EA, Malleson, PN, Pirie, GE, et al. The pulmonary manifestations of childhood onset systemic lupus erythematosus. Semin Arthritis Rheumatism. 1990;19(5):285–293.
14. Vichinsky, EP, Styles, LA, Colangelo, LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. cooperative study of sickle cell disease. Blood. 1997;89(5):1787–1792.
15. Martin, L, Buonomo, C. Acute chest syndrome of sickle cell disease: radiographic and clinical analysis of 70 cases. Pediatr Radiol. 1997;27:637–641.
16. Lonergan, GJ, Cline, DB, Abbondanzo, SL. Sickle cell anemia. RadioGraphics. 2001;21:971–994.
17. Vichinsky, EP, Neumayr, LD, Earles, AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855–1865.
18. Aquino, SL, Gamsu, G, Fahy, JV, et al. Chronic pulmonary disorders in sickle cell disease: findings at thin-section CT. Radiology. 1994;193:807–811.
19. Heeney, MW, Mahoney, DH, Jr., The acute chest syndrome in children and adolescents with sickle cell disease, Waltham, MA, UpToDate, 2011.
20. Schmidt, S, Eitch, G, Geoffray, A, et al. Extraosseous Langerhans cell histiocytosis in children. RadioGraphics. 2008;28:707–726.
21. McClain, KL, Langerhans cell histiocytosis, Juvenile xanthogranuloma, and Erdheim-Chester disease, Waltham, MA, UpToDate, 2011.
22. Odame, I, Li, P, Lau, L, et al. Pulmonary Langerhans cell histiocytosis: a variable disease in childhood. Pediatr Blood Cancer. 2006;47:889–893.
23. Bernstrand, C, Sandstedt, B, Ahstrom, L, et al. Long-term follow-up of Langerhans cell histiocytosis: 39 years’ experience at a single centre. Acta Paediatr. 2005;94:1073–1084.
24. Smets, A, Mortelé, K, De Praeter, G, et al. Pulmonary and mediastinal lesions in children with Langerhans cell histiocytosis. Pediatr Radiol. 1997;27:873.
25. Ha, SY, Helms, P, Fletcher, M, et al. Lung involvement in Langerhans’ cell histiocytosis: prevalence, clinical features, and outcome. Pediatrics. 1992;89:466.
26. Brauner, MW, Grenier, P, Mouelhi, MM, et al. Pulmonary histiocytosis X: evaluation with high resolution CT. Radiology. 1989;172:255.
27. Matlin, AH, Young, LW, Klemperer, MR. Pleural effusion in two children with histiocytosis X. Chest. 1972;61:33.
28. Klusmann, M, Owens, C. HRCT in paediatric diffuse interstitial lung disease—a review for 2009. Pediatr Radiol. 2009;39(suppl 3):S471–S481.
29. Banjar, H. Pulmonary involvement of Gaucher’s disease in children: a common presentation in Saudi Arabia. Ann Trop Paediatr. 1998;18:55.
30. Nicholson, AC, Wells, AU, Hooper, J, et al. Successful treatment of endogenous lipoid pneumonia due to Niemann-Pick type b disease with whole-lung lavage. Am J Respir Crit Care Med. 2002;165:128–131.
31. Uejima, T. General pediatric emergencies: Acute pulmonary edema. Anesthesiol Clin North Am. 2001;19(2):383–389.
32. Seely, JM, Effmann, EL. Acute lung injury and acute respiratory distress syndrome in children. Semin Roentgenol. 1998;33:163–173.
33. Brasileiro, FC, Vargas, FS, Kavakama, JI, et al. High-resolution CT scan in the evaluation of exercise-induced interstitial pulmonary edema in cardiac patients. Chest. 1997;111:1577–1582.
34. Lewin, S, Goldberg, L, Dec, GW. The spectrum of pulmonary abnormalities on computed chest tomographic imaging in patients with advanced heart failure. Am J Cardiol. 2000;86:98–100.
35. Jefferies, JL, Price, JF, Morales, DLS. Mechanical support in childhood heart failure. Heart Failure Clin. 2010;6:559–573.
36. Terminella, L, Sharma, G. Diagnostic studies in patients with acute respiratory distress syndrome. Semin Thorac Cardiovasc Surg. 2006;18:2–7.
37. Goodman, PC. Radiographic findings in patients with acute respiratory distress syndrome. Clin Chest Med. 2000;21(3):419–433.
38. Kirkpatrick, JA, Jr., Fleisher, DS. The roentgen appearance of the chest in acute glomerulonephritis in children. J Pediatr. 1964;64:492–498.
39. Gattinoni, L, D’Andrea, L, Pelosi, P, et al. Regional effects and mechanism of positive end-expiratory pressure in early adult respiratory distress syndrome. JAMA. 1993;269:2122.
40. Fanconi, S, Kraaemer, R, Weber, J, et al. Long-term sequelae in children surviving adult respiratory distress syndrome. J Pediatr. 1985;106:218.
41. Buck, JR, Connors, RH, Coon, WW, et al. Pulmonary embolism in children. J Pediatr Surg. 1981;16:385–391.
42. Kritsaneepaiboon, S, Lee, EY, Zurakowski, D, et al. MDCT pulmonary angiograph evaluation of pulmonary embolism in children. AJR. 2009;192:1246–1252.
43. Victoria, T, Mong, A, Altes, T, et al. Evaluation of pulmonary embolism in a pediatric population with high clinical suspicion. Pediatr Radiol. 2009;39:35–41.
44. Van Ommen, CH, Peters, M. Acute pulmonary embolism in childhood. Thromb Res. 2005;118:13–25.
45. Babyn, PS, Gahunia, HK, Massicotte, P. Pulmonary thromboembolism in children. Pediatr Radiol. 2005;35:258–274.
46. Patocka, C, Nemeth, J. Pulmonary embolism in pediatrics. J Emerg Med. 2012;42(1):105–116.
47. Cook, RJ, Ashton, RW, Aughenbaugh, GL, et al. Septic pulmonary embolism: presenting features and clinical course of 14 patients. Chest. 2005;128:162–166.
48. Wong, KS, Lin, TY, Huan, YC, et al. Clinical and radiographic spectrum of septic pulmonary embolism. Arch Dis Child. 2002;87:312–315.
49. Johnson, MJ, Lucas, GL. Fat embolism syndrome. Orthopedics. 1996;19:41.
50. Johnson, AS, Bolte, RG. Pulmonary embolism in the pediatric patient. Pediatr Emerg Care. 2004;20(8):555–560.
51. Lee, EY, Kritsaneepaiboon, S, Rios Arellano, CM, et al. Unsuspected pulmonary emboli in pediatric oncology patients: detection with MDCT. AJR. 2010;194:1216–1222.
52. Lee, EY, Zurakowski, D, Diperna, S, et al. Parenchymal and pleural abnormalities in children with and without pulmonary embolism at MDCT pulmonary angiography. Pediatr Radiol. 2010;40:173–181.
53. Kuriakose, J, Patel, S. Acute pulmonary embolism. Radiol Clin North Am. 2010;48:31–50.
54. Lee, EY, Zurakowski, D, Boiselle, PM. Pulmonary embolism in pediatric patients: survey of CT pulmonary angiography practices and policies. Acad Radiol. 2010;17:1543–1549.
55. Lee, EY, Kritsaneepaiboon, S, Zurakowski, D, et al. Beyond the pulmonary arteries: alternative diagnoses in children with MDCT pulmonary angiography negative for pulmonary embolism. AJR. 2009;193:888–894.
56. Stein, PD, Chenevert, TL, Fowler, SE, et al. Gadolinium-enhanced magnetic resonance angiography for pulmonary embolism: a multicenter prospective study (PIOPED III). Ann Intern Med. 2010;152:434–443.
57. Gudinchet, F, Maeder, P, Neveceral, P, et al. Lemierre’s syndrome in children: high-resolution CT and color Doppler sonography patterns. Chest. 1997;112:271–273.
58. Ridgway, JM, Parikh, DA, Wright, R, et al. Lemierre syndrome: a pediatric case series and review of the literature. Am J Otolaryngol. 2010;31(1):38–45.
59. Batra, P. The fat embolism syndrome. J Thorac Imag. 1987;2:12–17.
60. Rossi, SE, Erasmus, JJ, McAdams, HP, et al. Pulmonary drug toxicity: radiologic and pathologic manifestations. RadioGraphics. 2000;20:1245.
61. Ellis, SJ, Cleverley, JR, Müller, NL. Drug-induced lung disease: high resolution CT findings. AJR Am J Roentgenol. 2000;175:1019.
62. Mertens, AC, Yasui, Y, Liu, Y, et al. Pulmonary complications in survivors of childhood and adolescent cancer: a report from the Childhood Cancer Survivor Study. Cancer. 2002;95:2431–2441.
63. McCarroll, KA, Roszler, MH. Lung disorders due to drug abuse. J Thorac Imag. 1991;6:30.
64. Marks, LB, Yu, X, Vujaskovic, Z, et al. Radiation-induced lung injury. Semin Radiat Oncol. 2003;13(3):333–345.
65. Green, DM, Finklestein, JZ, Tefft, ME, et al. Diffuse interstitial pneumonitis after pulmonary irradiation for metastatic Wilms’ tumor: a report from the National Wilms’ Tumor Study. Cancer. 1989;63:450.
66. Davis, SD, Yankelevitz, DF, Henschke, CI. Radiation effects on the lung: clinical features, pathology, and imaging findings. AJR Am J Roentgenol. 1992;159:1157.
67. Ho, M, Yang, C, Cheung, W, et al. Chlorine gas exposure manifesting acute lung injury. J Intern Med Taiwan. 2010;21:210–215.
68. Scalzo, AJ. Inhalation injuries. In: Barkin RM, ed. Pediatric emergency medicine: concepts and clinical practice. 2nd ed. St. Louis, MO: Mosby; 1997:578.
69. Schwartz, J, Timonen, KL, Pekkanen, J. Respiratory effects of environmental tobacco smoke in a panel study of asthmatic and symptomatic children. Am J Respir Crit Care Med. 2000;161:802.
70. Wright, AL, Holberg, C, Martinez, FD, et al. Relationship of parental smoking to wheezing and nonwheezing lower respiratory tract illnesses in infancy. Group Health Medical Associates. J Pediatr. 1991;118:207.
71. Harris, VJ, Brown, R. Pneumatoceles as a complication of chemical pneumonia after hydrocarbon ingestion. AJR Am J Roentgenol. 1975;125(3):531–537.
72. Sias, SM, Ferreira, AS, Daltro, PA, et al. Evolution of exogenous lipoid pneumonia in children: clinical aspects, radiological aspects and the role of bronchoalveolar lavage. J Bras Pneumol. 2009;35(9):839–845.
73. Zanetti, G, Marchiori, E, Gasparetto, TD, et al. Lipoid pneumonia in children following aspiration of mineral oil used in the treatment of constipation: high-resolution CT findings in 17 patients. Pediatr Radiol. 2007;37:1135–1139.
74. Baron, SE, Haramati, LB, Rivera, VT. Radiological and clinical findings in acute and chronic exogenous lipoid pneumonia. J Thorac Imag. 2003;18:217–224.
75. Schmidt, H, Lorcher, U, Kitz, R, et al. Pulmonary alveolar microlithiasis in children. Pediatr Radiol. 1996;26:33.
76. Castellana, G, Lamorgese, V. Pulmonary alveolar microlithiasis. Respiration. 2003;70:549–555.
77. Helbich, TH, Wojnarovsky, C, Wunderbaldinger, P, et al. Pulmonary alveolar microlithiasis in children: radiographic and high-resolution CT findings. AJR Am J Roentgenol. 1997;168:63.
78. Brown, K, Mund, DF, Aberle, DR, et al. Intrathoracic calcifications: radiographic features and differential diagnoses. RadioGraphics. 1994;14:1247.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 57
Systemic Conditions with Lung Involvement
Pulmonary involvement is a frequent component of systemic illness. Pediatric patients often present for chest imaging before the diagnosis of a specific systemic disorder has been made. Therefore, the radiologist plays an important role in directing the diagnostic workup by recognizing that the pulmonary findings reflect an underlying condition. The first imaging study in pediatric patients with respiratory symptoms is usually chest radiography. In some patients such as those with pulmonary edema or sickle cell disease, radiography shows the pulmonary findings adequately to continue appropriate clinical care. In many patients, however, chest radiography is nonspecific, provides initial clues to the diagnosis, or both, prompting further investigation with computed tomography (CT) of the chest. In recent years, the advent of multidetector CT and controlled ventilation techniques (see Chapter 49) has dramatically improved the detailed assessment of lungs in children with systemic disorders.
Specific Systemic Diseases
Vasculitis and Collagen Vascular Disease
Although medium- and large-vessel vasculitides more commonly affect mediastinal vasculature structures and rarely the lung parenchyma, small-vessel vasculitides are the most likely to cause pulmonary parenchymal disease. Pediatric patients with pulmonary vasculitis typically present in their teen years. Of these disorders, Wegener polyangiitis (WP), previously known as granulomatosis, is the most common one in children. Microscopic polyangiitis (MPA) and Churg-Strauss syndrome (CSS) are rarely seen in children. Pulmonary nodules and airspace opacities are typically seen on imaging studies.1–3
Pulmonary involvement, most commonly interstitial changes, may be seen in children with collagen vascular diseases (CVD) such as juvenile arthritis, dermatomyositis, systemic sclerosis (scleroderma), systemic lupus erythematosus (SLE), and mixed connective tissue disease. Pulmonary involvement is less common in children than in adults. Pulmonary disease is seen more frequently in children with systemic sclerosis (59% to 91%) than in the other CVDs, and it is associated with significant morbidity and mortality.4,5 A recent study found that abnormal pulmonary function tests (PFTs) were correlated with the severity of abnormalities on high-resolution CT (HRCT). Thus PFTs provide a monitoring tool to identify children who would benefit from further lung disease evaluation with HRCT.5 Symptomatic lung disease is significantly less common in juvenile rheumatoid arthritis and SLE, reported in only 5% of patients.6
Both the vasculitides and CVDs may cause pulmonary renal syndrome, which is the association of both pulmonary hemorrhage and glomerulonephritis. It is often seen in WP and SLE.1,2 Pulmonary hemorrhage may be seen in both the vasculitides and in CVDs (e-Fig. 57-1) and has a high morbidity and mortality (50% to 90%).1,7
e-Figure 57-1 Pulmonary hemorrhage in a teenage boy with pulmonary vasculitis resulting from collagen vascular disease.
The patient presented with hemoptysis. A computed tomography scan shows diffuse but somewhat patchy ground-glass opacification in both lungs.
Etiology: WP is, by far, the most common of the pulmonary vasculitides seen in children and typically presents with a triad of necrotizing granulomatous lesions in both the upper and lower respiratory tract, as well as glomerulonephritis. MPA is a nongranulomatous necrotizing vasculitis almost always seen with glomerulonephritis. CSS (also known as allergic granulomatosis and angiitis) typically presents with asthma and blood eosinophilia but is rare in children.1 Most of the pulmonary vasculitides are immunologically mediated. WP, MPA, and CSS are associated with antineutrophil cytoplasmic antibodies (ANCA) and are sometimes referred to as ANCA-associated systemic vasculitides.
Imaging: The common imaging findings of pulmonary vasculitis and CVDs are summarized in Table 57-1. The frequent imaging findings of WP are variable sized nodules, followed by ground-glass opacities and air space consolidation; 17% of the nodules show cavitation.8 The nodules are frequently surrounded by a halo of ground-glass opacity, which represents hemorrhage (e-Fig. 57-2 and Fig. 57-3). Airway wall thickening may also be seen, but airway strictures are significantly less common in children (3%) compared with adults (up to 59%).8 Diffuse alveolar hemorrhage (occurring in 44% of pediatric WP) is characterized by lobular or lobar regions of ground-glass opacity or airspace consolidation on CT.9–11 It may also show the “crazy paving” pattern on CT, especially as it evolves.12
Table 57-1
Pulmonary Imaging Findings in Systemic Diseases
*Specific imaging findings are a spectrum from ground-glass opacities to airspace consolidation.
†Nodules in juvenile arthritis seen in lipoid pneumonia.
‡Seen as subpleural micronodules and were not considered a dominant finding.
Data from references 1, 4, 6, 7, and 11; and Feng RE, Xu WB, Shi JH, et al. Pathological and high resolution CT findings in Churg-Strauss syndrome. Chin Med Sci J. 2011;26(1):1-8.
Figure 57-3 Wegener polyangiitis.
This 16-year-old girl had a history of vasculitic skin lesions and headache. Computed tomography (CT) of the sinuses revealed inflammatory changes (not shown). Axial CT images of the chest show left upper lobe cavitary lesion (arrow) with surrounding ground-glass halo as well as several solid pulmonary nodules, also with surrounding halos of ground-glass opacity. Lung biopsy was positive for Wegener polyangiitis.
e-Figure 57-2 Wegener polyangiitis.
This 16-year-old girl with a history of chronic sinusitis presented with hematuria, acute renal failure, and sinusitis. She was eventually found to have Wegener polyangiitis. A, A coronal computed tomography (CT) image shows mucosal thickening in the right maxillary sinus and opacified left maxillary sinus. B, An axial CT image in lung windows through the upper lobes show a small thin-walled cavitary lesion in the right upper lobe (arrow). Also shown is a focal nodule with a halo of ground-glass opacity in the superior segment of the left lower lobe (curved arrow).
In the case of CVDs, pleural and pericardial effusions are the most common findings in the chest. Pulmonary findings are significantly less common but follow a similar pattern for most of these disorders and include ground-glass opacity and interstitial septal thickening, which may progress to pulmonary fibrosis. Despite normal chest radiographs or only minimal radiographic abnormalities in children with systemic sclerosis, HRCT demonstrates ground-glass opacities, peripheral areas of pulmonary fibrosis, and subpleural micronodules (Fig. 57-4).4 Some authors have reported occurrence of lipoid pneumonia in children with juvenile idiopathic arthritis, not associated with mineral oil ingestion.12 “Shrinking lung syndrome” has been described in SLE and represents decrease in lung volume that manifests radiographically by an elevated diaphragm.7,12,13
Figure 57-4 An 8-year-old girl with systemic sclerosis (scleroderma) diagnosed at age 5 years.
A, The frontal chest radiograph shows mild increase in perihilar linear opacities. B, High-resolution computed tomography (HRCT) of upper lung zones shows thin-walled, small cystic areas anteriorly in both lungs and a few small scattered subpleural lucencies and opacities more posteriorly. C, HRCT through lower lung zones shows honeycomb pattern dependently in both lungs. More lateral area in right lung shows more ground-glass opacity (arrow) that may represent active disease.
Treatment and Follow-up: Treatment for vasculitides and CVDs is primarily aimed at suppressing the immune response, typically with corticosteroids and chemotherapeutic agents. In patients with vasculitis, CVDs, or both, who experience diffuse alveolar hemorrhage, evolving changes may be seen on CT at follow-up. A more linear and interstitial type of pattern may develop with interlobular septal thickening and the appearance of the “crazy paving” pattern. If episodes of hemorrhage recur, this may also progress to interstitial fibrosis.9 Pulmonary involvement with CVD may progress to interstitial pulmonary fibrosis regardless of treatment, with advanced stages showing honeycombing. Pulmonary artery hypertension may also develop with advanced lung disease, especially in systemic sclerosis.
Sickle Cell Disease
Acute chest syndrome (ACS), a pulmonary illness that occurs in up to 50% of children with sickle cell disease (SCD), is characterized by chest pain, leukocytosis, fever, and a new pulmonary opacity. It is the leading cause of death (25%) and hospitalization in all patients with SCD and usually occurs between 2 and 4 years of age.14 ACS also occurs in patients with other sickle hemoglobinopathies.15 When not fatal, it may lead to chronic lung disease (4%) and pulmonary hypertension.16
Etiology: The etiology of ACS is complex and not always known. In a large multicenter study of 671 episodes, causes were found to be microvascular occlusion and infarction (16%), infection (29%), fat emboli from bone marrow infarcts (9%), and unknown in 46%. Chlamydia, Mycoplasma, and viral agents were the most common pathogens in cases caused by infection. One third of patients presented with symptoms of long bone pain caused by vasoocclusive crisis and developed ACS 2 to 3 days later.17
Imaging: Radiographic findings of ACS are nonspecific but, by definition, include the presence of a pulmonary opacity (e-Fig. 57-5). Opacities were noted in the lower lobes in about 90%, and pleural effusion was noted in 55% of cases in a large multicenter study (see Table 57-1).17
e-Figure 57-5 Sickle cell disease with acute chest syndrome.
An 8-year-old boy with sickle cell disease who presents with fever. A and B, Posteroanterior and lateral chest radiographs show dense airspace consolidation in the left lower lobe with small pleural effusion.
Although CT is not generally used in the setting of ACS, chronic sickle cell lung disease has been studied by using HRCT. Abnormal findings most pronounced at the lung bases include parenchymal bands, interlobular septal thickening, architectural distortion, and traction bronchiectasis. Honeycombing is unusual, in contrast to other types of pulmonary fibrosis.18
Treatment and Follow-up: Therapy for ACS includes hydration, analgesia, respiratory support (including bronchodilators), broad-spectrum antibiotics, transfusion therapy, and sometimes corticosteroids.19 Imaging follow-up is primarily dictated by clinical progression, improvement, or both and may be accomplished by using radiography.
Langerhans Cell Histiocytosis
Langerhans cell histiocytosis (LCH) is now the preferred term describing the condition showing a proliferation of a group of histiocytes known as Langerhans cells. Langerhans cells are of myeloid dendritic cell origin and contain characteristic Birbeck bodies on histology.20 The peak age at initial diagnosis is 1 to 3 years, and most patients present with osseous lesions.20 LCH is currently classified according to the extent of involvement, whether it involves a single site (better prognosis) or multiple sites (higher risk of long-term problems). The involvement of “risk organs” (liver, spleen, lung, bone marrow) indicates a poorer prognosis and demands more aggressive treatment.21
Pulmonary involvement is reported to be present in 23% to 50% of children with multisystem LCH.12,20 The mean age of patients with lung disease was 11.9 months in one study. Disease-free survival in these patients was 69%, with 3 out of 4 deaths occurring when “risk organs” other than the lung were involved.22 Although lung disease was traditionally considered a poor prognostic indicator in LCH, recent studies have shown that it does not adversely affect outcome. Primary pulmonary LCH (single site) is rare in children and is typically seen in young adult smokers.21,22
Etiology: The etiology of LCH is not known. Most investigators believe it to be an immune-mediated condition, in which the Langerhans cells accumulate and cause an inflammatory reaction. In the lungs, destructive granulomatous lesions occur in the interstitial tissues, bronchial and bronchiolar epithelium, and subpleural septa that may eventually lead to cystic lesions.20,21 Pulmonary fibrosis occurs later in 10% of children and may demonstrate multiple small cysts giving the lung a honeycomb appearance.23
Imaging: Radiologic findings of LCH vary widely with the extent of the disease and are summarized in Table 57-1. Initial chest radiography may be normal. Small nodules and cysts may be seen, with upper lobe involvement equal to or more extensive than lower lobe involvement (Fig. 57-6). The reticular appearance noted on chest radiography is often from multiple small cysts.20,24 LCH is the most common cause of acquired extensive cystic lung disease in children (e-Fig. 57-7). Spontaneous pneumothorax occurs in about 11% of patients with pulmonary involvement and may be the first indicator of pulmonary disease.25 However, it may be also a manifestation of more advanced disease, as in adolescents who more often have large coalescent cysts (see e-Fig. 57-7).12
Figure 57-6 Langerhans cell histiocytosis of the lungs in a 2.5-month-old boy who initially presented with a skin rash.
A, An anteroposterior chest radiograph shows a coarse reticulonodular interstitial pattern. B, An axial computed tomography image in a lung window shows the coarse reticulonodular interstitial pattern with some small thin-walled cysts seen in the anterior lung bilaterally.
e-Figure 57-7 Progressive advanced changes in Langerhans cell histiocytosis (LCH) of the lungs.
This child first presented at age 2 years with LCH involving both temporal bones and cystic lung disease. The patient was treated with chemotherapy and returned at age 4 years with increasing respiratory distress. The chest radiograph (not shown) showed a marked interval increase in bilateral cystic lung changes and suggested a possible small right pneumothorax. A and B, High-resolution computed tomography shows that multiple cysts of varying sizes have largely replaced much of the lung parenchyma; loculated right pneumothorax was confirmed. Within 3 weeks, the patient developed a large left pneumothorax and died of this cystic lung process 1 week later.
CT is indicated for neonates with LCH and any patient with abnormalities on chest radiography.21 HRCT findings in LCH are characterized by small nodules (with or without cavitation) with upper and middle lobe predominance (e-Fig. 57-8).12,20,26 The pleura may be thickened and sometimes replaced by a thick layer of granulomatous tissue. However, pleural effusions are rare.27 Mediastinal and hilar adenopathy is also rare in children with pulmonary LCH.20,24
e-Figure 57-8 A patient with known diagnosis of Langerhans cell histiocytosis with relapse of disease with osseous lesion in the scapula (not shown).
A, The chest radiograph shows nodular interstitial abnormality representing pulmonary histiocytosis. B, An axial chest computed tomography (CT) image shows more clearly the small interstitial nodules and tiny thin-walled cysts in the upper lobes, characteristic CT findings of histiocytosis.
Treatment and Follow-up: Treatment of pulmonary LCH depends on the extent of disease and whether “risk” organs are involved. Generally, the first line of treatment is corticosteroids, vinblastine, or both, with duration of therapy being determined by response. Radiation therapy has fallen out of favor except for treating unstable bone lesions.21 Imaging follow-up for pulmonary LCH depends on clinical symptoms and is best performed with CT.
Gaucher Disease and Niemann-Pick Disease
Etiology: Gaucher disease is caused by a deficiency of the enzyme glucocerebrosidase that results in an accumulation of glucocerebroside in phagocytic cells. Pulmonary involvement is most common in patients with neuronopathic types. Neimann-Pick disease is caused by a deficiency of sphingomyelinase, resulting in accumulation of sphingomyelin. Progressive pulmonary fibrosis has rarely been reported in Neimann-Pick disease.28
Imaging: A diffuse reticular lung pattern may be seen on chest radiography with Gaucher disease.29 Niemann-Pick disease may produce similar findings.27 Lipoid pneumonia has been described in patients with Neimann-Pick disease.30 Imaging findings are summarized in Table 57-1.
Treatment and Follow-up: Visceral involvement may improve with enzyme replacement therapy in Gaucher disease, and this therapy may offer an alternative to lung transplantation. The current treatment for Neimann-Pick disease is whole lung lavage.28,30 CT is more sensitive than plain radiography for imaging follow-up, when clinically warranted.
Pulmonary Conditions Occurring with Generalized Systemic Illness
In the pediatric population, cardiogenic edema usually presents in infants less than 6 months of age and is the result of congenital heart disease (CHD) (Figs. 57-9 and 57-10). In an older child, cardiomyopathy may be the cause of cardiogenic edema. Affected infants often present with nonspecific symptoms, including feeding difficulties, grunting, diaphoresis, wheezing, and retractions.31 Rarely, pulmonary edema may be the presenting manifestation of CHD.
Figure 57-9 Pulmonary edema caused by obstructed pulmonary venous return. This 6-week-old infant presented to the emergency room with tachypnea and feeding difficulties.
A and B, Anteroposterior and lateral chest radiographs show normal heart size and subtle interstitial edema characterized by hyperinflation, septal lines (white arrows), fissural thickening (open arrows), and tiny right pleural effusion (black arrow). Diagnosis of cor triatriatum was made with echocardiography.
Figure 57-10 Cardiogenic edema.
A 5-week-old male infant with known ventricular septal defect presenting with tachypnea and respiratory distress requiring intensive care unit admission. An anteroposterior chest radiograph shows cardiomegaly, hyperinflation, and pulmonary edema characterized by perihilar airspace opacity, indistinct hilar vessels, and septal lines (arrows).
The frequent causes of noncardiogenic pulmonary edema in children include acute respiratory distress syndrome (ARDS), near-drowning, neurogenic pulmonary edema, renal disease, and upper airway obstruction. Other less common causes of noncardiogenic pulmonary edema include aspiration pneumonia, hydrocarbon pneumonitis, smoke inhalation, and drug reactions.32
Cardiogenic Pulmonary Edema
Etiology: Cardiogenic pulmonary edema occurs significantly less frequently in children than in adults. The common underlying causes for cardiogenic pulmonary edema include left-to-right shunting lesions, left ventricular outlet obstruction, obstruction of pulmonary venous return, or cardiomyopathy (e-Fig. 57-11).31 Cardiogenic pulmonary edema progresses in severity as pulmonary capillary wedge pressure increases.
e-Figure 57-11 Dilated cardiomyopathy.
A 14-month-old female presented with 24 hours of increased work of breathing and wheezing. The portable chest radiograph shows characteristic cardiomegaly, perihilar and basilar distribution of airspace opacity caused by pulmonary edema, and a small right pleural effusion.
Imaging: On chest radiography, cardiomegaly is usually found except in instances of pulmonary venous obstruction, in which interstitial edema is present in the absence of cardiac enlargement (see Fig. 57-9).
Early findings in interstitial edema in children with cardiogenic pulmonary edema are indistinct vascular margins and bronchial wall thickening. Interlobular septal thickening (septal lines or Kerley B lines) and interlobar fissural thickening are also often seen. In infants and young children, fissural thickening is often more easily recognized than Kerley lines and is therefore an important clue for diagnosing cardiogenic pulmonary edema in children (see Fig. 57-9). An indirect radiographic finding often seen in children with CHD is hyperinflation. This may be a pitfall for the imager if he or she attributes the findings of peribronchial thickening and hyperinflation to airways disease and does not recognize them as subtle findings of interstitial edema (see Fig. 57-9). Alveolar edema usually occurs after development of interstitial edema and is the most recognizable radiographic finding of pulmonary edema (see Fig. 57-10 and e-Fig. 57-11). The classic appearance of acute alveolar edema is a central or “butterfly” distribution of airspace disease, with central edema and sparing of the lung periphery. Pleural effusions are also usually present (see e-Fig. 57-11).
Although not commonly used to diagnose pulmonary edema, CT is sensitive for the detection of pulmonary edema of any etiology. CT shows peribronchial cuffing, septal lines, ground-glass opacities, and air space consolidation in order of increasing severity (e-Fig. 57-12).33,34
e-Figure 57-12 Glomerulonephritis.
A 13-year-old male with history of asthma presented with 5 days of shortness of breath and was found to be hypoxic. A and B, Posteroanterior chest radiograph and magnified view show pulmonary edema with interstitial septal lines (arrow) and tiny left pleural effusion (open arrow). C and D, Axial and coronal computed tomography images depict typical findings of pulmonary edema showing interlobular septal thickening, dependent ground glass opacity, and bilateral small pleural effusions. The child was diagnosed with postinfectious glomerulonephritis.
Treatment and Follow-up: Treatment for cardiogenic pulmonary edema is generally supportive and includes corrective or palliative treatment for CHD. Diuretics and inotropes may be used for cardiac dysfunction. Ventilator support is provided, if needed. Extracorporeal membrane oxygenation and other types of ventricular assist devices are now being used more frequently in the setting of pediatric cardiac failure.35 Typically, imaging follow-up with chest radiography is adequate, although CT may be used to clarify the vascular anatomy.
Noncardiogenic Pulmonary Edema
Etiology: Many episodes of noncardiogenic pulmonary edema progress to ARDS. ARDS accounts for 1% to 3% of pediatric intensive care admissions, and the mortality in various series ranges from 40% to 60%.32 Sepsis, near-drowning, pneumonia, and smoke inhalation are the most frequent antecedents to pediatric ARDS. In most cases, ARDS progresses through stages of immediate lung injury, exudative alveolitis, fibroproliferative repair, and, in survivors, recovery. Ventilation–perfusion imbalance leads to varying degrees of hypoxemia.32
[/not-level-membership-for-pediatrics-category]
