Musculoskeletal system
A Ankylosing spondylitis
Definition and incidence
Ankylosing spondylitis (AS), also known as rheumatoid spondylitis and Marie-Strumpell disease, is a chronic inflammatory disorder that primarily affects the spine and sacroiliac joints and produces fusion of the spinal vertebrae and the costovertebral joints. It is a disease of adults younger than 40 years, and it demonstrates a predilection for males (male-to-female ratio is 9:1). The disease is rare in Caucasians.
Pathophysiology
The cause of AS remains unclear. However, it is strongly associated with the histocompatibility antigen HLA-B27, the presence of which is detected in more than 90% of Caucasians with the disease.
Clinical manifestations and diagnosis
Ankylosing spondylitis is diagnosed on the basis of clinical criteria that include: (1) chronic low back pain with limitation of spinal motion (<4 cm as measured by the Schober test), (2) radiographic evidence of bilateral sacroiliitis, and (3) limitation of chest wall expansion (<2.5-cm increase in chest circumference measured at the fourth intercostal space). Extraskeletal manifestations of this disease include iritis, cardiovascular involvement (cardiac conduction defects, aortitis, and aortic insufficiency in 20% of individuals), peripheral arthritis, fever, anemia, fatigue, weight loss, and fibrocavitary (fibrobullous) disease of the apexes of the lungs. The most limiting factors associated with the disease are pain, stiffness, and fatigue.
Complications
Pulmonary complications are reported to occur in 2% to 70% of patients with AS. Apical fibrosis is the most commonly occurring abnormality followed by aspergilloma and pleural effusion with nonspecific pleuritis. In apical fibrosis, the pulmonary lesion begins with apical pleural thickening and patchy consolidation of one or both apexes and often progresses to dense bilateral fibrosis and air space enlargement. Patients with apical fibrosis usually have advanced AS. Impaired thoracic cage excursion caused by AS results in a greater impairment of apical ventilation, and this may be one factor in the pathogenesis of apical fibrosis.
The most common thoracic complication is fixation of the thoracic cage as a result of costovertebral ankylosis, which can lead to pulmonary dysfunction. In patients with this complication, motion of the thoracic cage is restricted because of fusion of the costovertebral joints; this restriction leads to a decrease in thoracic excursion. Respiratory function typically demonstrates a restrictive pattern with mild diminution of total lung capacity (TLC), vital capacity (VC), and carbon monoxide diffusing capacity (Dlco) and normal or slightly increased residual volume (RV) and functional residual capacity (FRC). Pulmonary compliance, diffusion capacity, and arterial blood gas (ABG) values usually are normal. Despite having abnormal pulmonary function, the majority of patients with AS are able to perform normal physical activities without pulmonary symptoms. It has been suggested that patients who exercise regularly and thus improve cardiovascular fitness could maintain a satisfactory work capacity.
Bone ankylosis may occur in the numerous joints around the thorax (the thoracic vertebrae and the costovertebral, costotransverse, sternoclavicular, and sternomanubrial joints), resulting in limitation of chest wall movement. Patients with AS rarely complain of respiratory symptoms or functional impairment unless they have coexisting cardiovascular or respiratory disease. Progressive kyphosis is equivalent to progressive rigidity of the thorax. Increased diaphragmatic function compensates for decreased thoracic motion, allowing lung function to be well preserved. Patients with advanced disease may have an entirely diaphragmatic respiration. Regional lung ventilation in patients with AS is normal unless they have preexisting apical fibrosis.
Cervical spondylosis affects levels C5 to C6 and C6 to C7 most often and less frequently C4 to C5, C7 to T1, and C3 to C4. The degenerative changes may result in nerve root entrapment by foraminal encroachment. The phrenic nerve, which innervates the diaphragm, is supplied primarily by the C4 nerve root and to a lesser extent by the C3 and C5 nerve roots.
Cricoarytenoid involvement may exist and can lead to respiratory dysfunction and upper airway obstruction. Cricoarytenoid dysfunction can manifest as a hoarse, weak voice. Respiratory failure from cricoarytenoid ankylosis has necessitated therapeutic tracheostomy. In all reported cases, laryngeal symptoms were present before cricoarytenoid arthritis caused airway compromise. A case of acute respiratory failure and cor pulmonale resulting from cricoarytenoid arthritis has also been reported in a patient with AS.
Treatment
Medical therapy for adult patients with AS is supportive and preventive. Most patients with AS are asymptomatic. Depending on the severity of disease involvement, management may consist of the use of corticosteroids and nonsteroidal anti-inflammatory drugs (NSAIDs). Patients should refrain from smoking tobacco.
Anesthetic considerations
Patients with AS have specific anesthetic requirements. Management of the upper airway is the priority because of the potential for obstruction. Cervical spine involvement may result in limitation of movement. The ankylosed neck is more susceptible to hyperextension injury, and cervical fracture may occur. Intubation awake with or without the use of a fiberoptic bronchoscope is indicated. In rare situations, tracheostomy must be performed with the patient under local anesthesia before anesthesia can be induced. A regional anesthetic technique may not be feasible because of skeletal involvement that precludes access or because of neurologic complications such as spinal cord compression, cauda equina syndrome, focal epilepsy, vertebral basilar insufficiency, and peripheral nerve lesions. Patients with cardiovascular system involvement may require antibiotics, treatment of heart failure, or insertion of a temporary pacemaker before surgery. Restriction of chest expansion and, rarely, pulmonary fibrosis necessitate performance of a thorough preoperative assessment and immediate postoperative mechanical ventilation. Careful attention to positioning is essential.
B Kyphoscoliosis
Definition and incidence
Kyphosis is a deformity marked by an accentuated posterior curvature. Scoliosis is a lateral curvature of the spine. Kyphoscoliosis results when both kyphosis and scoliosis occur concomitantly, causing a lateral bending and rotation of the vertebral column. Scoliosis alone, despite its severity, does not cause sensory or motor impairment. In contrast, kyphosis and kyphoscoliosis may induce spinal cord damage because of the sharp angulation of the spine. Respiratory dysfunction is associated with scoliosis, significant kyphosis, and severe kyphoscoliosis. Scoliosis is the most common spinal deformity, with an incidence of four persons per 1000.
Scoliosis is classified in five categories: idiopathic, congenital, neuropathic (e.g., poliomyelitis, cerebral palsy, syringomyelia, and Friedreich ataxia), myopathic (e.g., muscular dystrophy and amyotonia), and traumatic. Idiopathic scoliosis is the most common deformity, accounting for 80% of all cases. On the basis of the time of onset, idiopathic scoliosis is divided into the following two categories: (1) the rare infantile form (male-to-female ratio is 6:4) and (2) the common adolescent form (male-to-female ratio is 1:9). The children in the adolescent group are born with straight spines; however, at some point during the growth period, their spines begin to bend and deform, with deformation progressively worsening until growth ends. In general, whereas curves associated with adolescent idiopathic scoliosis are convex and deviated to the right, those related to other disease may be deviated to the left. The presence of cervical scoliosis should alert anesthesia personnel to potential difficulties in airway management. Any significant curvature involving the thoracic spine may alter lung function. Unless the deformity is severe, patients with kyphosis are able to maintain normal pulmonary function; in contrast, even mild forms of scoliosis can result in impaired ventilatory function. Severe thoracic deformity may result in respiratory alterations during sleep. Several types of breathing abnormalities have been documented, including obstructive sleep apnea and hypopnea. The lowest HbO2 saturations occurred during rapid eye movement sleep.
Pathophysiology
Diminution of pulmonary function occurs with curvatures of greater than 60 degrees, and pulmonary symptoms develop with curvatures greater than 70 degrees (as measured by the Cobb technique). Curvatures greater than 100 degrees may be associated with significant gas exchange impairment.
In general, the greater the curvature, the greater the loss of pulmonary function. Because of this, mechanical ventilation becomes inefficient; this inefficiency is the major factor causing respiratory compromise. At the time of diagnosis, it often is possible to document a reduction in lung capacity. The characteristic deformity seen in scoliosis causes one hemithorax to become relatively smaller than the other.
Skeletal chest wall deformity in kyphoscoliosis leads to a reduction in lung volumes and the pulmonary vascular bed. Ventilatory failure associated with severe kyphoscoliosis produces a lung size that is 30% to 65% of normal. As the patient ages, the chest wall becomes less compliant; this increases the work of breathing and leads to hypoventilation and respiratory muscle weakness.
The main features of lung mechanics in the patient with early-stage scoliosis are reduced lung volumes (VC, TLC, FRC, and RV) and reduced chest wall compliance; in the late stages of disease, ventilation/perfusion mismatching with hypoxemia (attributed to alveolar hypoventilation because of a decrease in tidal volume [Vt]), increased pulmonary artery pressure (PAP), hypercapnia, abnormal response to CO2 stimulation, increased work of breathing, and cor pulmonale occur and eventually lead to cardiorespiratory failure. Reduction in VC to 60% to 80% of the predicted value is a typical finding. The ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC) is normal unless other pulmonary diseases are present. Although normocarbia prevails for most of the clinical course, an elevated Paco2 signifies the onset of respiratory failure. The severity of hypercapnia most closely correlates with the patient’s age and inspiratory muscle strength.
Associated conditions
Scoliosis may be associated with several cardiovascular abnormalities, of which mitral valve prolapse is the most common. If mitral regurgitation is present, antibiotic prophylaxis is indicated before surgical manipulation. Other common changes include an increase in pulmonary vascular resistance (PVR) and ensuing pulmonary hypertension (PH), which leads to the development of right ventricular hypertrophy. Several contributing factors are thought to be responsible for the development of increased PVR. First, arterial hypoxemia results in pulmonary vasoconstriction. Second, changes in the pulmonary arterioles consequent to the increased pulmonic pressure may cause narrowing and result in irreversible PH. Third, a compressed chest wall may increase vascular resistance in affected areas. Fourth, development of scoliosis at an early age inhibits growth of the pulmonary vascular bed. Alveolar multiplication is nearly complete by 2 years of age but continues until the age of 8 years. During the first few years, lung growth occurs primarily by enlargement of existing alveoli.
Treatment
The management of scoliosis may include the following: (1) observation of the problem without active medical treatment; (2) treatment by nonoperative methods that include the use of braces or electronic stimulators; and (3) operative methods such as anterior or posterior spinal fusion and instrumentation, such as Harrington rod insertion. The mortality rate among persons with untreated scoliosis is twice that of the normal population, and the rate for those with thoracic curvatures alone was fourfold that of the normal population. Patients with congenital thoracic scoliosis are particularly at risk for cor pulmonale.
Anesthetic considerations
Preoperative evaluation
Before surgery, a thorough review of systems is essential. The severity of scoliosis and of any underlying conditions must be noted. Any reversible pulmonary involvement such as pneumonia should be corrected before elective surgery. Laboratory data should include complete blood count; prothrombin time; partial thromboplastin time; values for electrolytes, blood urea nitrogen, and creatinine; electrocardiography (ECG); chest radiography; and routine pulmonary function test values. ABG analysis may be indicated if the results of the pulmonary function tests reflect significant impairment or if the surgical procedure dictates its need. Because these procedures can potentially involve large blood losses, young, healthy, asymptomatic patients may donate autologous blood. Blood typing and crossmatching also are required.
When sedatives are used in the preoperative area, care must be taken to ensure that respiratory status is not depressed. The need for intraoperative monitoring is dictated by the type of surgery and the physical status of the patient. No specific anesthetic techniques have been shown to be superior in patients with scoliosis; however, N2O may increase PVR by direct vasoconstrictive effects on the pulmonary vasculature. It has been suggested that scoliosis is associated with an increased incidence of malignant hyperthermia (MH). Ventilation should be adjusted so that adequate arterial oxygenation and normocarbia are maintained.
Patients undergoing surgery for correction of the spinal curvature should be informed preoperatively of the possible need for the “wake-up” test; when the patient is able to move both feet on request and surgical correction has been achieved, anesthesia can be quickly reinstituted. The use of somatosensory evoked potentials may require an alteration in anesthetic technique. All anesthetic agents depress somatosensory evoked potentials to a varying degree. Administration of volatile anesthetics should not exceed a minimum alveolar concentration of 1. A continuous infusion opioid technique often is preferred. Communication between the technician and anesthetist is essential.
Intraoperative management
Considerable fluid and blood loss may occur during surgery. The surgeon may request the institution of deliberate hypotension. Deliberate hypotension can be produced with the use of one or more of the following: potent inhalation anesthetics, vasodilators (e.g., sodium nitroprusside, nitroglycerin), or β-adrenergic blocking agents (e.g., propranolol and esmolol). The risks and potential benefits should be weighed against the effects of deliberate hypotension. The mean arterial blood pressure should be maintained at no lower than 60 to 65 mmHg. Cell saver blood is often used. Interventions for prevention of hypothermia, such as use of a hot air warming blanket or heated humidifiers, should be used. Careful positioning is essential.
Postoperative implications
The decision whether to use mechanical ventilation postoperatively is based on the severity of scoliosis and intraoperative events. Most patients with mild to moderate pulmonary dysfunction are able to undergo safe extubation in the operating room. Those with severe deformity or patients who have received massive fluid and blood replacement therapy should be weaned slowly.
C Lambert-eaton myasthenic syndrome
Incidence
Lambert-Eaton myasthenic syndrome (LEMS) is a rare autoimmune disease that classically occurs in patients with malignant disease, particularly small cell carcinoma of the bronchi. One-third to half of patients, however, have no evidence of carcinoma. Most patients with myasthenic syndrome are men between the ages of 50 and 70 years.
Pathophysiology
The basic defect associated with LEMS appears to be an autoantibody-mediated derangement in presynaptic Ca2+ channels leading to a reduction in Ca2+-mediated exocytosis of acetylcholine (ACh) at neuromuscular and autonomic nerve terminals. The decreased release of ACh quanta from the cholinergic nerve endings produces a reduced postjunctional response. Unlike in myasthenia gravis, the number and the quality of postjunctional AChRs remain unaltered, and the end plate sensitivity is normal. The neuromuscular junction abnormality of LEMS is similar in location to that of Mg2+ intoxication or botulism poisoning, in which the release of presynaptic ACh is attenuated.
Clinical manifestations
Muscle weakness, fatigue, hyporeflexia, and proximal limb muscle aches are the dominant features of LEMS. The diaphragm and other respiratory muscles are also involved. Autonomic nervous system dysfunction is often present and is manifested as impaired gastric motility, orthostatic hypotension, and urinary retention.
Patients with LEMS experience a brief increase in muscle strength with voluntary contraction, distinguishing it from myasthenia gravis. Tetanic stimulation results in a progressive augmentation in muscle strength as the frequency of the stimulation is increased. Post-tetanic potentiation is also enhanced.
Treatment
There is no cure for LEMS. Treatment is aimed at removal of the small cell carcinoma if present improving muscle strength and reversing autonomic deficits. 3,4-Diaminopyridine improves muscle strength in some patients by promoting presynaptic Ca2+ influx and increasing the number of ACh quanta that are liberated by a single nerve action potential. Anticholinesterase agents, plasmapheresis, corticosteroids, intravenous immunoglobulin, and immunosuppressive drugs provide improvement for some patients with LEMS.
Anesthetic considerations
An index of suspicion for LEMS should be maintained in surgical patients with a history of muscle weakness and suspected or diagnosed carcinoma of the lung. Patients with LEMS are extremely sensitive to the relaxant effects of both depolarizing and nondepolarizing muscle relaxants. Inhalational anesthetics alone may provide adequate relaxation, but if muscle relaxants are required, their dosages should be reduced and the neuromuscular blockade closely monitored. Neuromuscular reversal with an anticholinesterase agent may be used. Prolonged ventilatory assistance may be required postoperatively.
D Malignant hyperthermia
Definition and incidence
Malignant hyperthermia is an uncommon, life-threatening hypermetabolic disorder of skeletal muscle triggered in susceptible individuals by potent inhalation agents, including sevoflurane, desflurane, isoflurane, and halothane and the depolarizing muscle relaxant succinylcholine. About 52% of cases occur in patients younger than age 15 years, with a mean age of 18.3 years. The exact incidence of MH is unknown, but the rate of occurrence has been estimated to be one in 50,000 in adults and one in 15,000 in children.
Pathophysiology
Although the cause of MH is not yet known with certainty, it is generally agreed that MH is an inherited disorder of skeletal muscle in which a defect in calcium regulation is expressed by exposure to triggering anesthetic agents; intracellular hypercalcemia results. The ryanodine receptor is the major calcium release channel of the sarcoplasmic reticulum, and much attention has been focused on this receptor as the site of the MH defect. The defect involves skeletal muscle, and there is no evidence for a primary defect in cardiac or smooth muscle cells.
Malignant hyperthermia is initiated when specific triggering agents induce increased concentrations of calcium in the muscle cells of MH-susceptible (MHS) patients. Actomyosin cross-bridging, sustained muscle contraction, and rigidity result. Energy-dependent reuptake mechanisms attempt to remove excess calcium from the myoplasm, increasing muscle metabolism two- to threefold. The accelerated cellular processes increase oxygen consumption, augment carbon dioxide and heat production, deplete adenosine triphosphate (ATP) stores, and generate lactic acid. Acidosis, hyperthermia, and ATP depletion cause sarcolemma destruction, producing a marked regress of potassium, myoglobin, and creatine kinase (CK) to the extracellular fluid. Skeletal muscle constitutes 40% to 50% of our body mass, so relatively small changes in muscle metabolism may produce the dramatic systemic biochemical changes observed with MH.
Clinical manifestations
Not all cases of MH are fulminant, but rather there is a spectrum or continuum of severity, ranging from an insidious onset with mild complications to an explosive response with pronounced rigidity, temperature rise, arrhythmias, and death. Although MH may present in several ways, a typical MH episode begins while the patient is under general anesthesia with a volatile anesthetic. Use of succinylcholine may or may not precede the MH episode. The onset of MH symptoms may occur immediately after induction of anesthesia or several hours into the surgery. Desflurane is a weaker MH trigger and has been associated with delayed onset of MH, as long as 6 hours after induction of anesthesia. Succinylcholine appears to accelerate the onset and increase the severity of the MH episode. The presentation of MH may follow a dose-dependent response, with lower concentrations of volatile anesthetics resulting in a more protracted onset of hypermetabolic symptoms. Rarely, MH occurs in the recovery room, usually within 1 hour after general anesthesia.
The clinical features of MH reflect increased intracellular muscle Ca2+ concentration and greatly increased body metabolism and are listed in the following section. Common signs of MH include tachycardia, tachypnea, skin mottling, cyanosis, and total body or jaw muscle rigidity. Muscle rigidity is clinically apparent in 75% of cases. The most sensitive indicator of MH is an unanticipated increase in end-tidal carbon dioxide (ETCO2) levels out of proportion to minute ventilation. The increased ETCO2 may be abrupt, or it may rise gradually over the course of the anesthetic. Hyperthermia, which may climb at a rate of 1° to 2° C every 5 minutes and exceed 43.3° C (110° F), is often a late but confirming sign of MH.
Clinical events
Laboratory results
• ABG analysis: Paco2 >60 mmHg, base excess more negative than –8 mEq/L, pH <7.25
The combination of acidosis, hyperkalemia, and hyperthermia leads to cardiac irritability, a labile blood pressure, and arrhythmias that can rapidly progress to cardiac arrest. Laboratory findings mirror the muscle breakdown and include myoglobinuria and increased serum potassium and CK. Serum CK levels peak 12 to 24 hours after the onset of MH. Myoglobin appears in the plasma within minutes of the hypermetabolic muscle response. Arterial and venous blood gas analysis reveals decreased oxygen tension and mixed metabolic and respiratory acidosis. Late complications may include cerebral edema, myoglobinuric renal failure, disseminated intravascular coagulopathy, hepatic dysfunction, and pulmonary edema.
The variable time course and the nonspecific clinical features and laboratory findings can make the diagnosis of MH difficult. Insufficient anesthetic depth, hypoxia, neuroleptic malignant syndrome, propofol infusion syndrome, thyrotoxicosis, pheochromocytoma, and sepsis can share several characteristics with MH, making the clinical picture ambiguous and the differential diagnosis challenging to even the most experienced practitioner. Surgical procedures performed of necessity in a darkened operating room can further compromise the practitioner’s diagnostic acumen.
In addition to being a trigger of MH, succinylcholine may also induce hyperkalemic-mediated cardiac arrest in children with occult myopathies. Because of this concern, most anesthetists use nondepolarizing muscle relaxants for elective intubation in children and reserve the use of succinylcholine for treatment of laryngospasm or emergency airway management.
Preoperative assessment and prevention
Malignant hyperthermia–susceptible patients may be otherwise healthy and completely unaware of their risk until exposed to a triggering anesthetic. Furthermore, not everyone who has the MH gene develops an MH episode upon each exposure to triggering anesthetics. It is estimated that about 21% of MHS patients have at least one uneventful anesthetic before having an MH episode. Although MH susceptibility cannot be ruled out by history alone, every surgical patient should be questioned about the following information:
• Family or personal history of muscle disorders
• Family history of unexpected intraoperative complications or deaths
• Family or personal history of muscle rigidity or stiffness or high fever under anesthesia
• Personal history of dark or cola-colored urine after surgery
Because MH is considered an inherited disorder, all members of a family in which MH has occurred must be considered MHS unless proven otherwise. Moreover, the absence of a positive family history does not preclude MH susceptibility.
Certain disorders should alert the anesthetist to an increased possibility of MH susceptibility. A clear genetic association between MH and the inherited myopathy central core disease has been demonstrated. Case reports have also linked MH or an MH-like disorder to DMD and BMD dystrophy and forms of periodic paralysis and myotonia. MH-triggering agents should not be administered to patients with these disorders. This caveat is especially consequential in patients undergoing outpatient procedures, who may have more limited postoperative observation.
All patients given general anesthesia for more than 30 minutes should have core temperature monitoring. Stress, fever, prior exercise, and cocaine and alcohol ingestion have been implicated as causal factors of MH, but it is debated whether these factors cause, exacerbate, or have no effect on MH triggering in humans.
Treatment
Enhanced patient monitoring, earlier diagnosis and treatment, and dantrolene administration are responsible for the dramatic decrease in mortality rates from nearly 80% 20 years ago to less than 10% today.
Dantrolene is a unique muscle relaxant that works by reducing the release of calcium from skeletal muscle sarcoplasmic reticulum, counteracting the abnormal intracellular calcium levels accompanying MH. At clinical concentrations, dantrolene does not render the muscle totally flaccid and without tone, but it may cause significant muscle weakness and respiratory insufficiency, especially in patients with preexisting muscle disease. Dantrolene should not be used with calcium channel blockers because the combination may induce life-threatening myocardial depression.
The Malignant Hyperthermia Association of the United States (MHAUS) provides an “Emergency Therapy for MH” poster that should be posted in every surgical site. The following treatment sequence is recommended for an acute MH episode:
• Call for help and alert the surgeon to conclude the procedure promptly.
• Discontinue the volatile anesthetic and succinylcholine.
• Hyperventilate with 100% oxygen at high flows (at least 10 L/min) to improve tissue oxygenation and eliminate CO2.
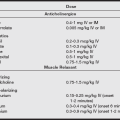
• Administer 2.5 mg/kg of dantrolene intravenous bolus and repeat as necessary until symptoms abate. Occasionally, a total dose greater than 10 mg/kg may be needed, but if greater than 20 mg/kg is given without reversal of symptoms, the diagnosis should be reassessed. The alkaline solution is highly irritating to vessels and should be administered into fast-running large peripheral veins or via central venous catheters.
• Dysrhythmias usually respond to treatment of acidosis or hyperkalemia. Treat persistent or life-threatening arrhythmias with standard antiarrhythmic agents (avoid calcium channel blockers).
• If fever is present, initiate cooling by lavage (orogastric, bladder, open cavities), administration of chilled intravenous normal saline, and surface cooling (hypothermia blanket; ice packs to the groins, axilla, and neck).
• Determine ABGs, serum electrolytes, and blood glucose every 15 minutes until the syndrome stabilizes. Correct severe metabolic acidosis with sodium bicarbonate. Baseline values for coagulation studies, CK, myoglobin, and liver enzymes should be established.
• Treat hyperkalemia with hyperventilation, bicarbonate, and intravenous insulin and glucose.
• Maintain urine output greater than 2 mL/kg/hr with hydration and furosemide (0.5–1.0 mg/kg).
• Large losses of intravascular volume should be anticipated. Consider central venous or pulmonary artery hemodynamic monitoring.
Each vial of dantrolene must be reconstituted with 60 mL of sterile water, and its poor water solubility makes it very time consuming to mix and administer the requisite doses. During an MH emergency, the full-time efforts of additional medical personnel should be enlisted. Warming the diluent fluid to 41° C using an intravenous fluid–warming device expedites dantrolene preparation. Documentation of an MH episode should include patient responses, personnel involved, medications, interventions, and patient outcomes.
Anesthetic considerations
Standard intraoperative monitoring for the MHS surgical patient includes blood pressure, ECG, pulse oximetry, capnography, and continuous measurement of core body temperature. A cooling water mattress should be placed under the MHS patient at the start of the procedure. Dantrolene pretreatment for MHS surgical patients is no longer routine. Inconsistent reports of emotional stress or anxiety predisposing a patient to MH have led to recommendations that anxiolytic agents be included in the premedication.
If the surgical site permits, a regional or local anesthetic technique is preferable for MHS patients. Local anesthetics (both amide and ester) are nontriggering drugs. Nontriggering general anesthetics can also be administered safely in concert with close monitoring of appropriate vital functions. The list of “nontriggering” anesthetic agents is comprehensive enough to meet most anesthetic requirements and are listed below. All volatile inhalation anesthetics and succinylcholine are MH triggers and should not be administered to MHS patients.
Triggering and nontriggering agents
• Barbiturates, propofol, ketamine, etomidate
• Nondepolarizing skeletal muscle relaxants (vecuronium, atracurium, cisatracurium, pancuronium, mivacurium, rocuronium)
Not all drugs have been thoroughly screened as potential MH triggers, but it is clear that the vast majority of prescription and nonprescription drugs are safe, including antibiotics, antihypertensive agents, and drugs used in the treatment of gastrointestinal disorders. Keys to successful perioperative outcome for the MHS patient include the following:
• Avoidance of MH-triggering medications
• Preparation of an anesthesia machine by changing the soda lime and breathing circuits, removing or inactivating vaporizers, and flushing with oxygen or air at 10 L/min for at least 20 minutes or 10 minutes if the fresh gas hose is also replaced
• Assiduous perioperative observation for signs of MH, including continuous intraoperative monitoring of the patient’s ETCO2, arterial oxygen saturation, and core temperature
• A full appreciation of a preestablished treatment protocol by all perioperative medical personnel
• A machine to manufacture ice or the ready availability of ice and the ability to crush it, and a refrigerator containing at least 3000 mL of cold intravenous solution, should be available.
Ambulatory surgery can be safely performed in most MHS patients, provided that appropriate monitoring is used and an adequate supply of dantrolene is available. Patients known or suspected of having MH should be assessed well before their date of outpatient surgery, so that anesthesia records and MH testing center reports (if available) can be collected to corroborate the history.
Outpatient surgical cases for the MHS patient are best scheduled early in the day, allowing for at least 2.5 hours of postanesthesia observation time, including a minimum initial recovery period of 1 hour of monitoring vital signs every 15 minutes. Most MHS patients who experience uneventful outpatient surgery may be discharged on the day of surgery. Some experts recommend conservative management with overnight postoperative hospital admission for patients who have survived a previous fulminant or severe MH episode or when dantrolene prophylaxis is used.
All locations where general anesthesia is administered should contain a fully stocked MH cart with drugs and supplies, including 36 vials of dantrolene. Each minute is critical in an MH emergency, and therefore a dantrolene supply should not be shared with a nearby facility. Dantrolene should be kept in or very close to the operating room so that it is available immediately if MH occurs.
Diagnostic testing
The most accurate and commonly accepted test available for determining MH susceptibility is the caffeine halothane contracture test (CHCT). This test involves taking a biopsy of skeletal muscle from the patient’s thigh and measuring its contractile response to caffeine, halothane, or both. Normal muscle contracts in response to caffeine or halothane, but this is augmented in patients with MH. The test is available at eight medical centers in North America; because it must be completed within hours after muscle biopsy, the patient must travel to the testing site. Patients who have survived an unequivocal episode of MH are considered to have MHS. The CHCT is indicated for family members of an MHS patient or for patients who have had a previous suspicious but undiagnosed reaction to anesthesia. Genetic testing from a blood sample is available, but, at present, this assessment is not highly diagnostic.
Postoperative implications
The patient who has experienced an acute MH episode should be observed in an intensive care unit for at least 24 hours. Intravenous dantrolene should be continued at approximately 1 mg/kg every 6 hours for a minimum of 24 hours after control of the episode. Recrudescence of an intraoperative episode may occur in 20% to 25% of cases. Patients who experience recrudescence are more likely to have a muscular body type and to have had greater than 150 minutes transpire from induction to MH reaction.
For the MHS patient who has undergone an uneventful surgical course, close observation and monitoring should continue into the postanesthesia care unit. MH can first manifest in the recovery room after uneventful surgery and anesthesia.
Masseter muscle rigidity
Masseter muscle rigidity (MMR) or trismus is a sustained and forceful contracture of the masseter muscle. The contracture may be severe enough to make opening the jaw impossible (“jaws of steel”). A mild increase in masseter muscle tone or incomplete jaw relaxation following succinylcholine is fairly common and may be a normal response. However, severe jaw tightness that interferes with intubation may portend an episode of MH. If trismus is further accompanied by generalized body rigidity, MH is highly likely.
Management of trismus in surgical patients is a contentious issue, and authorities are divided on how to proceed after MMR. Some experts recommend cautiously continuing the anesthetic with nontriggering agents after an episode of MMR while monitoring for rhabdomyolysis and signs and symptoms of MH. Others maintain that the safer course is to assume that trismus is a harbinger of MH, discontinue the anesthetic, and cancel elective surgery until results of a muscle biopsy are available.
Because of the likelihood of MH or rhabdomyolysis, the surgical patient should be admitted to the hospital and observed for at least 24 hours after marked jaw rigidity and at least 12 hours after a mild increase in jaw tension. Myoglobinuria may be apparent in the recovery room, and inducing a brisk urine output may lessen the risk of myoglobinuric renal damage. Studies indicate that after MMR, if the CK is greater than 20,000 IU and a concomitant myopathy is not present, the diagnosis of MH is likely. Patients who have experienced MMR should be counseled concerning the possibility that they are MHS and should be referred to a well-informed primary or specialty care physician or genetic counselor for further investigation.
Information resources
The MHAUS is a nonprofit organization that provides educational and technical information to patients and health care providers. Information is available via fax-on-demand at 800-440-9990 or at http://www.mhaus.org. An MH hotline may be accessed for MH emergencies 24 hours a day at 800-MH-HYPER (800-644-9737). Health care providers are encouraged to report MH episodes to the North American MH Registry.
E Muscular dystrophy
Definition
Muscular dystrophy is a heterogeneous set of diseases that includes fascioscapulohumeral dystrophy, limb-girdle dystrophy, Becker muscular dystrophy (BMD), Duchenne muscular dystrophy (DMD), and others. DMD, also known as pseudohypertrophic muscular dystrophy, is the most common and most severe form.
Pathophysiology
Duchenne muscular dystrophy is an inherited, sex-linked recessive disease. The disease presents in early childhood between 2 and 6 years of age. It is clinically evident in boys and has an incidence of one in 3500 live male births. Girls and women are generally unaffected but are carriers of the disorder. Mental retardation, of varying degrees, occurs in about 30% of patients with DMD. Death often occurs in late adolescence or early adulthood and is usually caused by respiratory failure.
Clinical manifestations
Patients with DMD experience an infiltration of fibrous and fatty tissue into the muscle followed by a progressive and painless degeneration and necrosis of muscle fibers. Muscle weakness ends with muscle destruction.
Duchenne muscular dystrophy is characterized by an unremitting weakness and a steady deterioration of the proximal muscle groups of the pelvis and shoulders. The child exhibits a clumsy, waddling gait and falls frequently. Weakness of the pelvic girdle leads to the classic finding of the Gower sign, in which patients use their hands to climb up their legs to arise from the floor. A steady deterioration of muscle strength forces most of these boys to start using wheelchairs by the age of 8 to 12 years.
Skeletal muscle atrophy is usually preceded by fat and fibrous tissue infiltration, resulting in pseudohypertrophy. The infiltrative process is most apparent in the calf muscles, which become particularly enlarged.
Degeneration of respiratory muscles occurs and leads to a restrictive type of ventilatory impairment. Unopposed action by healthy, nondystrophic axial muscles predisposes these patients to kyphoscoliosis, which further decreases the pulmonary reserve. Decreasing muscle strength also results in ineffective cough, impaired swallowing, and an inability to mobilize secretions.
More progressive forms of the disease affect not only skeletal muscle but also smooth muscle of the alimentary tract and cardiac muscle. Alimentary tract involvement can lead to intestinal hypomotility, delayed gastric emptying, and gastric dilation.
Myocardial involvement occurs in almost all patients with progressive disease. Myocardial disease includes fibrotic changes localized primarily to the left ventricle. Echocardiography can effectively evaluate left ventricular function in patients with DMD. Clinical symptoms of heart failure do not usually appear unless the patient is severely stressed or until advanced stages of the disease.
Electrocardiographic changes characteristic of preclinical cardiomyopathy include a large or polyphasic R wave in lead V1, deep Q waves in the lateral precordial leads (V4 to V6), premature beats (atrial and ventricular), and labile sinus or atrial tachycardia.
Although often severe, the compromised cardiac and respiratory conditions may be masked by the limited activity imposed by the patient’s skeletal myopathy. Added stress, such as that produced by surgery and anesthesia, may suddenly increase cardiorespiratory demand and uncover the weakened cardiac and respiratory states.
Laboratory results
Laboratory findings include a serum CK level that is 30 to 300 times normal, even early in the disease, reflecting skeletal muscle necrosis and the increased permeability of skeletal muscle membranes. CK concentration is elevated in approximately 70% of female carriers. Skeletal muscle biopsy early in the course of the disease may demonstrate necrosis and phagocytosis of muscle fibers.
Anesthetic considerations
Patients with DMD are susceptible to untoward anesthesia-related complications. When possible, local or regional anesthesia should be considered.
Generalized muscle weakness, especially in the advanced stages of muscular dystrophy, makes these patients exquisitely sensitive to the respiratory depressant properties of opioids, sedatives, and general anesthetic agents. Preoperative sedation should be minimal, and the smallest possible amounts of anesthetic agents should be used.
Preoperative and postoperative respiratory therapy can help to maximize the patient’s pulmonary condition. In patients with more advanced disease, ABG determinations and preoperative pulmonary function studies may elucidate the extent of respiratory involvement and the amount of respiratory reserve. An FVC of less than 35% of that predicted indicates a risk for postoperative pulmonary complications.
The effects of nondepolarizing muscle relaxants must be scrupulously monitored. There is increased muscle relaxant sensitivity, and recovery may be prolonged by three to six times in patients with DMD. Short-acting nondepolarizing muscle relaxants that are carefully titrated with the use of a nerve stimulator are recommended.
Attention to respiratory function must be continued into the postoperative period. Delayed pulmonary insufficiency, as late as 36 hours after surgery, has been reported. At least 24 hours of observation should be instituted after the patient undergoes anesthesia.
Their decreased cardiac reserve makes these patients sensitive to the myocardial depressant effects of general anesthetic agents, sedatives, and narcotics. Cardiac arrest associated with inhalation anesthetics has been reported. A carefully titrated intravenous “balanced” technique may help to provide a smoother cardiovascular course. Ketamine has been used successfully for anesthesia during diagnostic muscle biopsy in patients with DMD. Judicious administration of intravenous fluids is warranted. The sudden occurrence of tachycardia during anesthesia may indicate acute heart failure.
The potential for delayed gastric emptying, in addition to the presence of weak laryngeal reflexes, dictates that the anesthesia plan of care include measures for guarding against aspiration of stomach contents. Gastrokinetic agents and the prophylactic use of a nasogastric tube are recommended to avoid gastric dilation.
Succinylcholine and the potent inhalational agents should not be used in patients with muscular dystrophy because the altered sarcolemma can lead to rhabdomyolysis with their administration. The resultant massive breakdown of muscle fibers produces a profound hyperkalemia that requires extensive and tenacious treatment with hyperventilation, calcium chloride, sodium bicarbonate, and glucose and insulin. Several cases of ventricular fibrillation or cardiac arrest occurring during anesthetic induction have been associated with succinylcholine or potent inhalational agent administration. Additionally, DMD is included among the myopathies that may be associated with MH. The anesthetist should avoid MH-triggering agents and should vigilantly observe for signs and symptoms of MH when these children undergo surgery. Dantrolene and other treatment modalities for MH should be readily available.
F Myasthenia gravis
Definition
Myasthenia gravis, a chronic disease of the neuromuscular junction, is manifested by increasing skeletal muscle weakness, fatigability on effort, and at least partial restoration of function after rest.
Incidence
In the United States at least one in 7500 people have myasthenia gravis. In individuals younger than 50 years, the ratio of women to men with the disease is 3 to 2; however, in those older than 50 years, the disease is equally distributed between the sexes. Myasthenia gravis can begin spontaneously at any age, but it occurs most frequently between the ages of 30 to 40 years. The onset may be abrupt or insidious, and the course is fluctuating, marked by periods of exacerbation and remission. Spontaneous remissions that do occur sometimes persist for years.
Pathophysiology
Electron microscopic examination of the neuromuscular junction of the patient with myasthenia gravis shows a decrease in the number of functional postsynaptic ACh receptors (AChRs). The AChR lesion appears to be caused by immune-mediated destruction, blockage, or inactivation. The prejunctional ACh pool is normal.
Myasthenia gravis is a prototype autoimmune disease. Circulating antibodies react with myoneural AChR proteins, leading to varying degrees of dysfunction. Anti-AChR antibodies (immunoglobulin G [IgG]) are found in the sera of 85% to 90% of patients with myasthenia gravis, but the antibody level does not necessarily correlate with the severity of the disease. Most patients in clinical remission continue to show elevated serum levels of AChR antibodies.
The initiating stimulus for the production of anti-AChR IgG antibodies is unclear. A genetic cause or induction by microbial antigens has been postulated. The thymus gland seems to play a central role in the pathogenesis.
Pregnancy exacerbates the symptoms of myasthenia gravis in 40% of pregnant women with the disease; however, other patients with the disease experience remission or no change in symptoms during pregnancy. Anti-AChR antibodies that pass across the placenta may produce transitory symptoms of weakness in approximately 10% to 15% of infants born to mothers with myasthenia gravis. Signs of weakness (difficulty with breathing, ptosis, facial weakness) in the affected infant are usually present within the first few hours after birth. The condition lasts as long as 21 days, mirroring the half-life of the IgG antibodies.
Clinical manifestations
The clinical hallmarks of myasthenia gravis include a generalized muscle weakness, which improves with rest, and an inability to sustain or repeat muscular contractions. Enhanced effort produces enhanced weakness. The severity of myasthenia gravis can range from mild (slight ptosis only) to severe (respiratory failure). Environmental, physical, and emotional factors seem to affect the disease process, although unpredictably.
Mouth, eyes, pharynx, proximal limb, and shoulder girdle musculature are most often affected. Visual symptoms (ptosis and diplopia) from extraocular muscle weakness occur in more than 50% of patients with myasthenia gravis. The disease is restricted to the extraocular muscles in 20% of patients. Sensation and cognition are not affected by the disease process.
Thymus gland abnormalities are detectable in about 75% of patients with myasthenia gravis. Other autoimmune disorders, such as thyroid disease, collagen vascular diseases, polymyositis, and RA, occur more frequently in patients with myasthenia gravis. Myocarditis may complicate myasthenia gravis, especially in patients with thymomas. Microscopic lesions of myasthenic cardiac muscles are similar to skeletal muscle lesions, indicating a common pathogenesis. The myocardial inflammation produces dysrhythmias, particularly atrial fibrillation and atrioventricular block.
Treatment
Therapy for patients with myasthenia gravis is directed toward improving neuromuscular transmission and includes cholinesterase inhibitors, corticosteroids and other immunosuppressants, plasmapheresis, intravenous immunoglobulin, and thymectomy.
Treatment with cholinesterase inhibitors can dramatically reduce the symptoms of myasthenia gravis by inhibiting the hydrolysis of ACh and therefore increasing the neurotransmitter’s concentration at the neuromuscular junction. Increasing the synaptic concentration of ACh enhances the possibility of postsynaptic AChR occupation, which is critical for the production of a threshold-reaching end plate potential for muscle contraction. Anticholinesterase treatment is particularly successful in patients with milder disease.
The most commonly used anticholinesterase agent in the United States is oral pyridostigmine. Oral pyridostigmine, 60 mg, lasts 3 to 4 hours and is equivalent to an intramuscular or intravenous dose of pyridostigmine, 2 mg, or neostigmine, 1 mg. Titration of the anticholinesterase dose is challenging. Underdosing does not sufficiently retard the muscle weakness and can result in myasthenic crisis, a severe exacerbation of myasthenic symptoms.
Overmedicating with a cholinesterase inhibitor can produce a surplus of ACh at the myoneural junction, causing a depolarizing-like block and augmenting skeletal muscle weakness. This situation is called cholinergic crisis. Muscarinic side effects (e.g., abdominal cramping, diarrhea, salivation, bradycardia, and miosis) predominate in a cholinergic crisis.
Corticosteroid therapy produces an 80% remission rate in patients with myasthenia gravis, partly by reducing AChR antibody levels. Glucocorticoid therapy is often used in combination with other agents. Use is limited by the side effects (e.g., osteoporosis, gastrointestinal bleeding, suppression of endogenous cortisol release, cataracts, increased susceptibility to acute infections, hypertension, and glucose intolerance) observed with long-term administration.
In patients with more debilitating, widespread disease, immunosuppressive drugs such as azathioprine (Imuran) and cyclosporine may induce remission by interfering with the production of AChR antibodies. Side effects of azathioprine include severe hemopoietic depression and liver dysfunction. Cyclosporine side effects include hypertension and nephrotoxicity.
Excision of the thymus gland is recommended for adults with generalized disease and for patients with thymomas, thymus gland hyperplasia, or drug-resistant myasthenia gravis. Thymectomy effectively arrests or reverses the myasthenic process by removing a major source of antibody production. Clinical improvement of myasthenic symptoms is seen in 75% to 96% of patients within weeks to months after surgery.
Plasmapheresis (plasma exchange) arrests severe refractive myasthenia gravis by reducing the concentration of circulating antibodies. It is used primarily as a short-term treatment because the improvement that it produces in symptoms is generally short lived. Intravenous immunoglobulin may also be used for short-term control of symptoms before surgery.
Anesthetic considerations
Several days before the operation and again immediately before surgery, the surgical candidate with myasthenia gravis should be evaluated for disease control and, if applicable, for stabilization of anticholinesterase dose.
The use of anticholinesterase medication in the immediate preoperative period is controversial. Some experts believe that an awareness of drug mechanisms can enable anticholinesterase therapy to be safely continued into the preoperative period, especially in patients who depend on this therapy for their well-being. Others recommend discontinuing or tapering anticholinesterase medication before surgery to avoid complicating the anesthetic management. Patients with mild myasthenia gravis can usually tolerate the temporary disruption in treatment.
The presence of cholinesterase inhibitors may potentiate vagal responses and complicates both the intraoperative administration of muscle relaxants and the differential diagnosis and treatment of postoperative muscle weakness. Emotional stress and surgery may precipitate or worsen skeletal muscle weakness. Pharyngeal and laryngeal muscle weakness, difficulty in eliminating oral secretions, and the risk of pulmonary aspiration should be considered in the anesthesia plan of care. Swallowing and respiratory muscle dysfunction account for much of the morbidity and potential mortality in patients with myasthenia gravis.
Regional or local anesthesia with careful monitoring are the preferred anesthetic techniques when appropriate. If general anesthesia is indicated, the respiratory depressant effects of barbiturates, sedatives, narcotics, and volatile anesthetic agents, compounded by the presence of an already weakened respiratory system, must be carefully considered.
In many patients, the relaxant effects of a volatile anesthetic in combination with the patient’s preexisting skeletal muscle weakness are sufficient to facilitate intubation of the trachea. Enhanced muscle relaxation may be seen with the administration of all the potent volatile anesthetics.
Small doses of succinylcholine may be used to facilitate tracheal intubation, but the response may be unpredictable. Untreated patients with myasthenia gravis appear to be two to three times more resistant to succinylcholine. Normal dosages of succinylcholine may not effectively depolarize the end plate because of the deficiency of viable AChRs. On the other hand, patients treated with cholinesterase inhibitors exhibit a normal or prolonged response to succinylcholine. Cholinesterase inhibitors block the effects of plasma cholinesterase, as well as those of true cholinesterase; hence, succinylcholine and other medications metabolized by plasma cholinesterase (e.g., ester local anesthetics) may have a delayed hydrolysis and a prolonged duration of action. The ester hydrolysis of atracurium is independent of plasma cholinesterase activity.
The deficient number of functioning AChRs in patients with myasthenia gravis produces an extraordinary sensitivity to nondepolarizing muscle relaxants. Small doses of nondepolarizing agents can produce a profound block with a prolonged effect even in patients being treated with cholinergic drugs. Some patients require no medication at all for surgical muscle relaxation. If muscle relaxation is necessary, cisatracurium offers an advantage of being metabolized by non–organ-dependent mechanisms.
Generally, muscle relaxant requirements are widely variable in patients with myasthenia gravis, a characteristic that makes neuromuscular blockade monitoring an essential and integral part of the anesthetic management. The orbicularis oculi muscle may overestimate the degree of muscle relaxation in patients with myasthenia gravis. This site may be the most ideal site to monitor neuromuscular blockade to avoid the possibility of undetected residual muscle weakness. When needed, the use of smaller doses (half to two-thirds the normal dose) of shorter acting nondepolarizing relaxants is the prudent choice.
Reversal of neuromuscular blockade with an acetylcholinesterase inhibitor should be performed cautiously in patients with myasthenia gravis. Overtreatment with an anticholinesterase agent can precipitate a cholinergic crisis and aggravate rather than reverse the muscle weakness. In many circumstances, the neuromuscular block can be titrated to allow complete spontaneous recovery, avoiding the use of reversal.
Complete, sustained return of muscle strength must be demonstrated before extubation and resumption of spontaneous ventilation. The patient should be informed that postoperative tracheal intubation and ventilatory support may be required. Skeletal muscle strength may appear to be adequate shortly after surgery but may deteriorate a few hours later. Postoperative ventilation is recommended for patients undergoing transsternal thymectomy, who have the disease for longer than 6 years, who take a daily pyridostigmine dose greater than 750 mg, who have chronic obstructive pulmonary disease, or have a preoperative VC less than 2.9 L.
G Myotonic dystrophy
Definition
The myotonias are a group of hereditary degenerative muscle diseases that include myotonic dystrophy, myotonia congenita (Thomsen disease), and paramyotonia congenita. A symptom common to all myotonias is the inability of skeletal muscles to relax after chemical or physical stimulation.
Myotonic dystrophy, also known as Steinert disease, myotonia atrophica, or myotonia dystrophica, is the most common and the most severe form of the myotonias. It is characterized by skeletal muscles that are hypoplastic, dystrophic, and weak yet prone to persistent contraction. Although muscles are primarily affected, myotonic dystrophy is distinguished from nondystrophic myotonias by being a multisystem disease.
Incidence
Myotonic dystrophy is inherited as an autosomal dominant trait. In most cases, an affected person has one affected parent. The onset of symptoms can occur at any age but usually occurs in the second to third decade of life. A slow, progressive deterioration of skeletal, cardiac, and smooth muscle occurs, resulting in death by the sixth decade. An estimated one in 20,000 people worldwide have the disorder, with an equal occurrence in males and females. The severity of clinical symptoms usually increases with transmission to subsequent generations (genetic anticipation). Myotonic dystrophy is the most common and severe inherited muscular dystrophy of adulthood.
Pathophysiology and treatment
Myotonic dystrophy is a disorder of muscle membrane excitability that results in self-sustaining runs of depolarization. Electrophysiologic studies show a lowered resting membrane potential in muscle cells from patients with myotonic dystrophy. Therapeutic agents used to treat the myotonic contractures include quinine, procainamide, and phenytoin. These agents delay the return of membrane excitation by blocking rapid Na+ influx into muscle cells. Regional anesthesia and muscle relaxants do not prevent or relieve the recalcitrant contraction. Dantrolene has also been ineffective in reversing myotonia. Warming the ambient temperature or injecting local anesthetics into the involved muscles may induce relaxation. Steroids and inhalation anesthetic agents may also attenuate the contraction in some patients. No treatment is available for the muscle weakness that develops with myotonic dystrophy.
Clinical manifestations
A wide variety of symptoms are characteristic of myotonic dystrophy. Facial weakness (“expressionless facies”), ptosis, and sternocleidomastoid muscle and distal limb weakness are prominent features of the disease. Frontal balding, cataracts, and testicular atrophy in men form a frequently recognized triad of characteristics. Endocrine abnormalities, such as diabetes mellitus and thyroid disease, occur with a greater frequency in this patient group than in the general population.
Myotonia, an inability to relax a muscle, occurs in most symptomatic patients and may be worsened by pressure, touch, cold, or shivering. Insidious muscle atrophy, particularly of the face, neck, pharynx, and distal limbs, causes severe muscle debility in the later stages of the disease. Myotonic symptoms usually precede the atrophy and weakness.
Cardiac disturbances occur in most patients with myotonic dystrophy, often manifesting as conduction defects and arrhythmias. Conduction defects were present in about 50% of the patients in one series. First-degree atrioventricular block is the most common finding, but greater degrees of heart block are also seen. Arrhythmias include sinus bradycardia, atrial flutter or fibrillation, and ventricular extrasystoles.
Weakening of the thoracic muscles, including the diaphragm, reduces the respiratory reserve and the VC, and a restrictive type of ventilatory impairment develops with progression of the disease. Central sleep apnea and hypersomnolence cause hypoventilation and decreased ventilatory response to carbon dioxide.
Anesthetic considerations
Any drug that has the potential to depolarize skeletal muscle may produce an exaggerated contraction in patients with myotonia dystrophica. Administration of succinylcholine to patients with myotonic dystrophy should be avoided because it can produce an intense generalized myotonic contracture that makes ventilation and intubation difficult or impossible. Agents associated with myoclonus (methohexital, etomidate) have the potential to produce similar effects.
Nondepolarizing muscle relaxants may be used in these patients as long as the degree of muscle wasting and weakness is appreciated. The dose of the nondepolarizer should be reduced according to the degree of muscle impairment, and the neuromuscular block should be monitored closely with a peripheral nerve stimulator.
An abnormal swallowing mechanism resulting from palatal, pharyngeal, and esophageal muscle involvement and gastrointestinal hypomotility renders these patients vulnerable to pulmonary aspiration of gastric contents.
Reversal of neuromuscular blockade with anticholinesterase agents may theoretically precipitate skeletal muscle contraction by producing an ACh-induced depolarizing block. Shorter acting nondepolarizing muscle relaxants have the obvious advantage of being less likely to require reversal. Hypothermia and shivering should be avoided by raising the room temperature, warming inhaled gases and intravenous fluids, and providing a forced-air thermal blanket.
Underestimating the severity of respiratory compromise is not uncommon in these patients. Preoperative ABG determinations and pulmonary function results may serve as useful baselines in the patient with advanced disease. The respiratory depressant effects of barbiturates, opioids, and volatile anesthetics may compromise already weakened respiratory musculature and may lead to unexpected decompensation. Even small doses of short-acting anesthetic agents may be associated with an exaggerated and prolonged anesthetic effect.
Diligent monitoring of cardiovascular parameters should be maintained intraoperatively and postoperatively. Cardiac function that was clinically normal preoperatively may become unacceptably depressed as a result of the administration of general anesthetic agents. The patient should be questioned preoperatively about syncope, and the ECG should be examined closely for advanced conduction blocks to help ascertain the need for cardiac pacing. It may be wise to assume that even asymptomatic patients have some degree of cardiac involvement. Pregnancy may exacerbate the symptoms of myotonia. Uterine atony, postpartum hemorrhage, and retained placenta have accompanied delivery in patients with myotonic dystrophy. Increased progesterone levels are linked to the deleterious effects.
Malignant hyperthermia–triggering agents should be avoided in these patients because associations between some forms of myotonia and MH have been described.
H Pectus deformities
Definition
Pectus excavatum, also referred to as funnel chest, is the most common chest wall deformity, occurring in one in 400 children.
Pathophysiology
Pectus excavatum is a congenital abnormality characterized by depression of the sternum (usually above the xiphisternal junction) and symmetric or asymmetric prominence of the ribs on either side. Its origin is unknown; however, it is thought that excessive diaphragmatic traction on the lower sternum or displacement of the heart into the left hemithorax is largely responsible. Family history of some type of anterior thoracic deformity is present in 37% of patients. If uncorrected, the disease usually worsens at adolescence. Self-limiting deformities are either gone or vastly improved by the age of 3 years.
Clinical manifestations
Clinically, the majority of patients are asymptomatic unless pectus excavatum is extreme. Patients with pectus excavatum have reduced chest cavities and TLC. Pulmonary function often is normal except in severe cases, in which VC, TLC, and maximum breathing capacity may be diminished.
Treatment
The indications for repair of pectus excavatum are the subject of controversy. Conflicting data have been presented regarding whether the repair of pectus excavatum is performed only for cosmetic purposes or whether it actually improves cardiorespiratory function and exercise tolerance. Some clinicians suggest that pectus excavatum should be corrected in childhood—ideally, when patients are between the ages of 4 and 6 years—to relieve the structural compromise of the chest, allow normal growth of the thorax, prevent pulmonary and cardiac dysfunction in teens and adults, and improve cosmetic appearance. Others have found that surgery does not significantly improve pulmonary function and that exercise tolerance and cardiorespiratory function during exercise do not benefit significantly from surgical correction. Patients with Marfan syndrome have a high incidence of chest wall deformities; they usually are seen in their most severe form, often accompanied by scoliosis. Congenital heart disease, mitral valve prolapse, and asthma also occur more frequently in patients with pectus excavatum. Electrocardiographic abnormalities are common and attributable to the abnormal chest wall configuration and to the displacement and rotation of the heart into the left thoracic cavity. A systolic ejection murmur of grades II to III or IV frequently is identified.
Pectus carinatum
Pectus carinatum is characterized by a longitudinal protrusion of the sternum. It is the second most common chest deformity, occurring in one or two persons per 1000. A familial tendency exists, and the disorder is more frequent in males than in females (4:1 ratio).
Pathophysiology
The pathogenesis is unclear, and the disorder may be congenital or acquired. The development of pectus carinatum is thought to result from the overgrowth of the costal cartilages, which results in displacement of the sternum.
Clinical manifestations
The development of pectus carinatum has also been associated with severe childhood asthma and rickets. The physiologic effects are probably related to the restriction of thoracic excursion. Patients with pectus carinatum have an increased incidence of congenital heart disease, including ventricular septal defect, patent ductus arteriosus, atrial septal defects, and mitral valve abnormalities.
Three classifications of pectus carinatum exist. Type I, pigeon breast or keel chest, consists of symmetric protrusion of the sternum and costal cartilages. Type II, pouter pigeon breast or Currarino-Silverman syndrome, is characterized by protrusion of the manubrium of the first two sternal cartilages, backward arching of the sternal body, and anterior displacement of the xiphoid process. Type III, lateral pectus carinatum, is manifested by unilateral protrusion of the anterior chest wall.
I Rheumatoid arthritis
Definition
Rheumatoid arthritis (RA) is a chronic inflammatory polyarthropathy with myriad degrees of systemic involvement. The disease is multifactorial, and the clinical picture varies widely in severity, extent of involvement, and symptoms. The capricious course of the disease may be persistent and debilitating or relapsing and remitting. With each successive exacerbation, new joints may become involved.
Incidence
Rheumatoid arthritis is the most common form of inflammatory arthritis, affecting approximately 0.8% of the U.S. population. The onset of RA can occur at any age, but most cases are diagnosed in patients between the ages of 35 and 50 years. RA is two to three times more likely to develop in women than in men. Patients with RA have a reduced life expectancy ranging from 3 to 7 years.
Pathophysiology
The exact cause of RA remains elusive, but heredity plays some role in increasing a person’s susceptibility. Impaired immunity, stress, and other environmental factors may precipitate or aggravate the disease.
A viral or a bacterial infection that alters the immune system in a genetically susceptible host may play a role in the etiology. The invading microbe may produce a protein similar to those in the body’s own tissue, particularly joint tissue (molecular mimicry). To destroy the antigen, the immune system may mount an autoimmune response and mistakenly direct its attack against its own tissue. Circulating autoantibodies called rheumatoid factors are detectable in 70% to 80% of patients with RA.
Clinical manifestations
Joint involvement
Inflammation and destruction of synovial tissues are responsible for most of the symptoms and chronic disability associated with RA. Joint involvement progresses in three main stages: (1) inflammation of the joint synovial membrane and infiltration by polymorphonuclear leukocytes; (2) rapid division and growth of cells in the joint (synovial proliferation and pannus formation); and (3) liberation of osteolytic enzymes, proteases, and collagenases, which damage small blood vessels, cartilage, ligaments, tendons, and bones. Collapse of normal cortical and medullary architecture leads to erosion and dislocation of bone that is contiguous with the inflammatory cell mass.
The onset of symptoms is most often insidious, evolving over a period of weeks to months. The most common sites of onset are the hands, wrists, and feet. There is often symmetric joint involvement. Swelling, warmth, and pain in the affected joints are caused by the inflammatory process. Morning stiffness, weight loss, and fatigue are noted early in the disease course.
Dissolution of bone and disuse atrophy of bone (osteoporosis) are found in all seriously affected areas. Pain, inflammation, and erosion of bone and tissue may permanently limit the joint’s full range of motion. Later stages of the disease are characterized by severe pain, joint instability, and crippling deformities. Nerve entrapment may occur at any site where peripheral nerves pass near the inflamed joint. Carpal tunnel syndrome is a common peripheral neuropathy.
Synovitis in the temporomandibular joint may limit jaw motion. An estimated 30% to 70% of patients with RA have involvement of the temporomandibular joint. As the disease progresses, flexion contractures and soft tissue swelling may lead to a marked limitation in the patient’s ability to open the jaw.
Although the thoracic and lumbar spine are usually spared, involvement of the cervical spine may be extensive and can lead to limited movement or deformity of the neck and to severe laryngeal deviation.
The most common site of cervical spine synovitis is C1 to C2. Atlantoaxial (C1–C2) instability results from erosion and collapse of bone and from destruction of supporting cervical ligaments. Symptoms occur when excessive motion between C1 and C2 exerts pressure on the spinal cord. Additionally, separation of the atlanto-odontoid articulation may allow the odontoid process of the axis to impinge on the spinal cord, leading to neurologic damage. The atlantoaxial subluxation may also exert pressure and impair blood flow through the vertebral arteries.
Arthritis extends to the cricoarytenoid joint of the larynx in 40% of patients with severe RA. The joint may become swollen, inflamed, and fixed in a position that obstructs air flow. Vocal cord nodules and polyps may also be present. Symptoms of cricoarytenoid arthritis include tenderness over the larynx, hoarseness, pain on swallowing with radiation to the ear, and dyspnea or stridor. Patients with no overt clinical symptoms may also have significant laryngeal disease.
Systemic involvement
Although the effects of RA are most clearly seen in joints, the disease is systemic. The immune-mediated destructive process affects a wide variety of organs, including the heart, lungs, muscle, vasculature, and eyes. The occurrence of extraarticular manifestations is usually associated with more active, erosive articular disease.
Firm, painless subcutaneous nodules occur in approximately 20% to 30% of patients with RA. The nodules usually occur over pressure points, such as the occiput, sacrum, ulna, or Achilles tendon and may be associated with pressure ulcerations. Rheumatoid nodules can also occur in most visceral organs, including the lungs and the heart. Dural nodules can cause spinal cord compression and neurologic complications.
Pericarditis and pericardial effusion may accompany severe progressive RA and impair cardiac performance. Although rare, rheumatoid nodules have been isolated from the cardiac conduction system and may be associated with conduction defects.
Pulmonary involvement manifests as pleural effusion, pneumonitis, pulmonary nodules, or interstitial fibrosis. Decreased lung volume, diffusion capacity, and VC may result from the lung alterations.
Rheumatoid myositis, which is characterized by muscle weakness and eventual muscle necrosis and atrophy, may accompany RA. Inflamed, painful, and underused joints contribute to the skeletal muscle atrophy. Lacrimal duct and salivary gland destruction may result in dryness of the eyes and the mouth (Sjögren syndrome) in about 15% of patients with RA.
Treatment
There is no cure for RA, and all treatment interventions are palliative. Medical therapy is directed toward relief of pain, nonspecific suppression of the inflammatory process, immunosuppression, prevention and correction of deformity, and control of systemic involvement. Most patients, including those with mild to moderate disease, obtain some relief of symptoms with rest, joint immobilization, and use of NSAIDs. NSAIDs relieve joint pain, stiffness, heat, and swelling, partly by blocking cyclooxygenase and inhibiting prostaglandin, thromboxane, and prostacyclin synthesis. Despite their potent antiinflammatory properties, they do not alter the underlying disease process.
Corticosteroids are potent anti-inflammatory drugs that suppress many symptoms of RA. Long-term side effects (osteoporosis, predisposition to infection, suppression of endogenous cortisol release, cataracts, gastrointestinal bleeding, hypertension, and hyperglycemia), however, limit their use to isolated flares of the disease or to adjunctive, rather than primary treatment.
Disease-modifying antirheumatic drugs (DMARDs) can slow the progressive damage and arrest the underlying disease process. Agents such as the anticytokines etanercept (Enbrel), adalimumab (Humira), and infliximab (Remicade) work by interfering with the proinflammatory cytokine, tumor necrosis factor. Notably, these drugs increase the risk of developing serious infection. Biologic agents such as interleukin-1 receptor antagonists offer the potential for more effective treatment of RA. Leflunomide (Arava) is a DMARD that inhibits the proliferation of T lymphocytes and slows disease progression. The major side effect of leflunomide is liver enzyme elevation and liver disease.
The antimetabolite methotrexate (Rheumatrex) is widely used as an effective DMARD for patients with aggressive RA. Bone marrow suppression, oral ulcerations, pneumonitis, and hepatic damage are potential side effects of methotrexate. Gold salts, sulfasalazine, antimalarial drugs, and D-penicillamine are effective DMARDs used when more conservative measures fail to retard symptoms. Immunosuppressive drugs such as cyclophosphamide (Cytoxan) and cyclosporine (Sandimmune) and antimetabolites such as azathioprine (Imuran) are effective agents, generally reserved for more refractory cases. Surgical interventions for relief of pain or for correction or prevention of deformities include total joint replacement, synovectomy, and tenolysis.
Anesthetic considerations
Overall, no individual anesthetic agent or mode of anesthesia is substantially safer than another for patients with RA. Preoperative examination of an individual patient’s disease course and medication history are likely to reveal specific features that affect the anesthesia or surgical course.
NSAID ingestion may result in platelet dysfunction. Mild anemia, a common finding in patients with RA, may be secondary to the disease process or to drug therapy. Long-term NSAID therapy may exert harmful effects on the liver or kidney, exacerbate allergic rhinitis or asthma; and cause gastrointestinal bleeding; these effects may influence the choice of anesthesia.
Patients receiving long-term corticosteroid therapy may develop hypophyseal–pituitary axis suppression, which may require perioperative steroid supplementation. Long-term administration of corticosteroids may increase the patient’s susceptibility to infection by inhibiting normal host defense mechanisms. The newer tumor necrosis factor inhibitors are also associated with serious infections, mandating close attention to sterile techniques.
A thorough preoperative assessment of the airway is essential. Particular attention should be directed to the temporomandibular joints (TMJs), the cervical spine, and the cricoarytenoid joints. Range of motion of the TMJ must be assessed before anesthesia is induced. Patients with severe TMJ involvement may be able to open their mouths only 1 to 2 cm. In such cases, the use of the flexible fiberoptic bronchoscope for tracheal intubation is of proven value.
A thorough neurologic assessment and a radiographic evaluation of the cervical spine should be performed, especially for patients with advanced disease. Some patients with significant radiographic evidence of atlantoaxial or subaxial instability may be entirely asymptomatic. Neck pain is an early symptom of cervical spine instability. Paresthesias that extend into the shoulders and arms, muscle weakness, paresis, and bowel or bladder dysfunction are some of the clinical manifestations of spinal cord compression secondary to atlantoaxial or subaxial subluxation. Compression on the vertebral arteries, with interruption of vertebral artery blood flow, may lead to symptoms such as nausea, vomiting, dysarthria, dysphagia, blurred vision, or transient loss of consciousness.
Altered cervical spine anatomy or laryngeal deviation can make intubation of the trachea an extreme challenge. Deviation of the larynx can frequently be detected preoperatively by palpating the location of the larynx in relation to the sternal notch. Flexion, extension, and rotation of the neck must be avoided in the presence of cervical instability. Such circumstances dictate fiberoptic-guided intubation of the trachea in an awake patient.
Hoarseness in a patient with RA should alert the anesthetist to possible cricoarytenoid joint involvement. A smaller endotracheal tube may be necessary because of narrowing of the glottic opening. Laryngoscopy can assess normal cord motion and glottic patency. The patient should be observed closely for signs of airway obstruction after extubation.
Generalized demineralization of bone may increase the risk of fractures in patients with RA. Glucocorticoid therapy may aggravate the osteopenia. Proper patient positioning and padding of pressure points prevent nerve palsies, skin ulcerations, and further structural damage to the joints.