Chapter 13 Cardiovascular system
COMMON CLINICAL PROBLEMS FROM CARDIOVASCULAR DISEASE
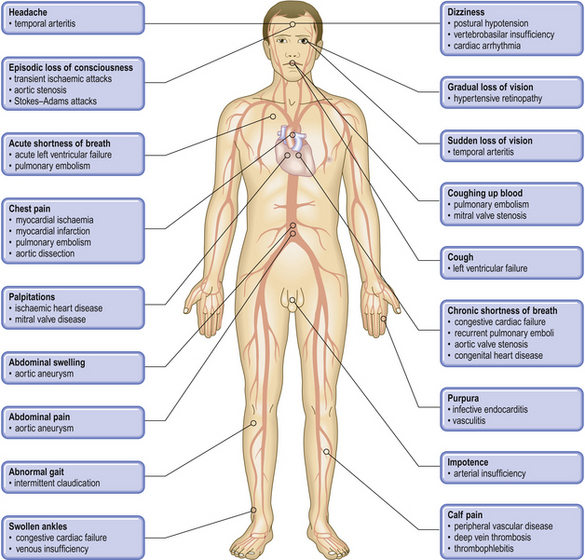
Pathological basis of cardiovascular signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Angina | Myocardial ischaemia due to spasm, atheroma or thrombosis of coronary arteries |
| Abnormal blood pressure | |
Either ‘essential’ (primary, idiopathic) due to as yet undefined genetic and environmental factors, or secondary to a disease resulting in increased levels of hormones with hypertensive effects
Reduction of actual or effective circulating blood volumeAbnormal heart sounds
Turbulence of blood flow through stenotic or incompetent valves
Pericarditis
Pericardial effusionAbnormal ECG
Disturbed myocardial depolarisation/ repolarisation commonly due to ischaemia or infarction
Disturbed conduction of electrical activity due to, for example, disease affecting conducting tissue or causing appearance of foci of ectopic electrical activityAbnormal pulseDisordered heart rhythm or arterial flowRaised jugular venous pressureIncreased central venous pressure due to right or congestive cardiac failureOedemaIf due to vascular disease, attributable to raised venous pressure (e.g. in cardiac failure or venous thrombosis) exceeding plasma oncotic pressureDyspnoeaPulmonary oedema due to left ventricular failure or mitral stenosisCyanosisPartial bypass of pulmonary circulation or acquired impairment of circulation or oxygenationRaised serum troponin or creatinine phosphokinaseRelease of cardiac enzymes into blood due to myocardial infarctionJoint painsSynovial inflammation in rheumatic feverSkin lesions
Impaired arterial or venous flow
Interruption of arterial supply
Microemboli from infective endocarditis
Microhaemorrhages in skin due to vasculitisHemiplegiaCerebral haemorrhage or cerebral artery occlusion by thrombus or embolusVisual impairmentCranial (giant cell) arteritis Hypertensive retinopathySudden collapseVaso-vagal syncope Severe dysrhythmia (e.g. ventricular fibrillation) due to myocardial infarction
DISEASES OF THE ARTERIES AND OTHER VESSELS
Cardiovascular disorders are now the leading cause of death in most Western societies (Ch. 2). In England and Wales ischaemic heart disease currently accounts for 27%, and cerebral vascular disorders for 13%, of all deaths. Atherosclerosis is the commonest and most important vascular disease, but many other vascular disorders are recognised.
Normal arterial structure
In all parts of the arterial system, three anatomical layers can be distinguished. The innermost, the intima, is composed of a single layer of endothelium with a thin supporting framework of connective tissue. The internal elastic lamina separates the intima from the middle layer, the media (Fig. 13.1). The aortic media is particularly rich in elastic tissue, but in most medium-sized arteries, such as the coronary arteries, smooth muscle predominates. The outermost layer, the adventitia, is fibrous connective tissue. Small blood vessels, the vasa vasorum, enter from the adventitial aspect and supply much of the media. The intima and innermost media receive nutrients by direct diffusion from the vascular lumen.
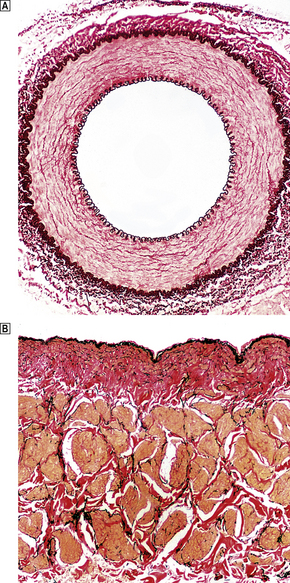
Fig. 13.1 Structure of blood vessels.  Muscular artery from a young child. The intima is extremely thin.
Muscular artery from a young child. The intima is extremely thin.  Renal vein from a 72-year-old man. Elastic lamellae are indistinct and there is some intimal fibrosis (red coloration). The underlying muscle bundles (pale yellow) are not arranged as regularly as in arteries.
Renal vein from a 72-year-old man. Elastic lamellae are indistinct and there is some intimal fibrosis (red coloration). The underlying muscle bundles (pale yellow) are not arranged as regularly as in arteries.
AGE-RELATED VASCULAR CHANGES
A variety of ageing changes occur in the aorta, arteries and arterioles. Although there is considerable individual variation, changes are usually inconsequential before 40 and most common after 70 years of age. The most important changes are:
The net effect of these changes is to reduce both the strength and the elasticity of the vessel wall. Progressive dilatation is a common ageing phenomenon in both the aorta and the coronary arteries. In the ascending aorta this can lead to stretching of the aortic valve ring and aortic incompetence. Dilatation of the arch and thoracic aorta produces the characteristic ‘unfolding’ seen in chest X-rays (Fig. 13.2).
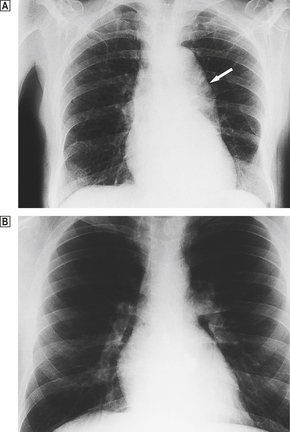
Fig. 13.2 Unfolding of the aorta.  There is a prominent bulge (arrow) caused by dilatation of the arch and descending aorta. If the dilatation involves the aortic valve ring, aortic incompetence may result.
There is a prominent bulge (arrow) caused by dilatation of the arch and descending aorta. If the dilatation involves the aortic valve ring, aortic incompetence may result.  Normal X-ray for comparison.
Normal X-ray for comparison.
The age-related changes that occur in muscular arteries are usually termed arteriosclerosis. Even arterioles can be affected. Characteristic alterations include smooth muscle hypertrophy and the apparent reduplication of the internal elastic laminae by extra layers of collagen. There is often marked intimal fibrosis and this further reduces the diameter of the vessel. Arteriosclerosis contributes to the high frequency of cardiac, cerebral, colonic and renal ischaemia in the elderly population. The clinical effects become most apparent when the cardiovascular system is further stressed by haemorrhage, major surgery, infection or shock.
ATHEROSCLEROSIS





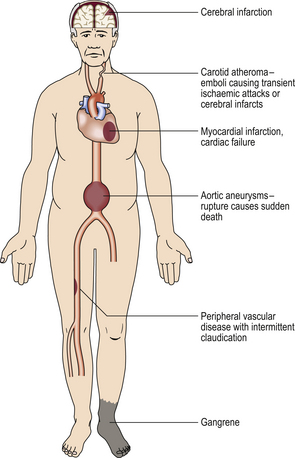
Atherosclerosis is a disease characterised by formation of focal elevated lesions in the intima of large (aorta) and medium-sized arteries (such as coronary arteries)—termed atherosclerotic plaques. Plaques alone are usually benign asymptomatic lesions, even when they are present in large numbers throughout the arterial tree, but life-threatening ischaemic damage of vital organs may occur when an occlusive thrombosis forms on a spontaneously disrupted plaque (atherothrombosis). Such acute obstructions can occur in many different arteries, resulting in a wide range of clinical disorders (Fig. 13.3). The frequency of atherothrombotic complications has increased drastically during the past 50 years, and the condition is now also common in parts of the Middle and Far East, particularly in those countries where a ‘Western style’ of living has been adopted. Coronary atherothrombosis—‘coronary heart disease’—is one of the commonest causes of death in many societies.
Atherosclerotic lesions
The formation of lesions starts in young children, especially in societies with a high dietary fat intake. The earliest significant lesion is called a fatty streak. It is a yellow linear elevation of the intimal lining and is composed of masses of lipid-laden macrophages. These fatty streaks have no clinical significance. They may disappear from the arterial intima, but in patients at risk they progress to atherosclerotic plaques (Fig. 13.4). The fully developed plaque is a lesion with a central lipid core with a cap of fibrous tissue covered by the arterial endothelium (Fig. 13.5). Connective tissues in the cap, mainly collagens, provide the structural strength of the plaque and are produced by smooth muscle cells (SMCs). Inflammatory cells, including macrophages, T-lymphocytes and mast cells, reside in the fibrous cap. They are recruited from the arterial endothelium or, in advanced plaques only, from newly formed microvessels present at the base of, or around, the atheroma.
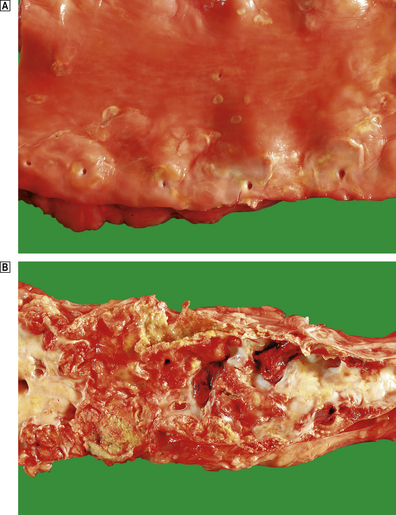
Fig. 13.4 Lesions of atherosclerosis.  Early aortic atherosclerosis. Note the many small fatty streaks. Some larger dot-like lesions are also present. These are common lesions in all racial groups and both genders.
Early aortic atherosclerosis. Note the many small fatty streaks. Some larger dot-like lesions are also present. These are common lesions in all racial groups and both genders.  Advanced complicated atherosclerosis in the abdominal aorta. Many of the lesions have ruptured and become thrombosed.
Advanced complicated atherosclerosis in the abdominal aorta. Many of the lesions have ruptured and become thrombosed.
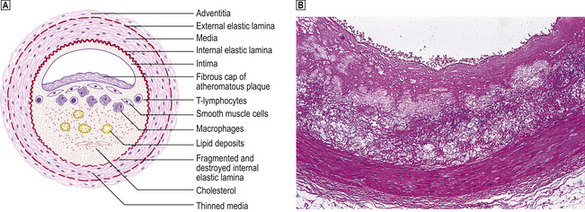
Fig. 13.5 Atheromatous plaque.  Diagram of an atheromatous plaque. Some of the features can be seen in the photomicrograph
Diagram of an atheromatous plaque. Some of the features can be seen in the photomicrograph  from the coronary artery of a 72-year-old.
from the coronary artery of a 72-year-old.
The atheroma is rich in cellular lipids and cellular debris derived from macrophages that have died inside the plaque. It is soft (semi-fluid), highly thrombogenic and often bordered by a rim of so-called foam cells. The foam cell results from uptake of oxidised lipoproteins via a specialised membrane-bound scavenger receptor. This is one of the most distinctive pathological processes in plaque formation. Dystrophic calcification of the plaque can be extensive and occurs late in the process of plaque development. It may serve as a marker for atherosclerotic vessel disease in angiograms or in CT images. Plaques have a tendency to form at arterial branching points and bifurcations. This illustrates the important role of turbulent blood flow in the pathogenesis of atherosclerosis. In the late stages many individual lesions may become confluent and cover large parts of arteries (Fig. 13.4B).
What causes atherosclerosis?
Hypercholesterolaemia is by far the most important risk factor for atherosclerosis. It can cause plaque formation and growth in the absence of other known risk factors. It has been suggested that if plasma cholesterol levels in a population were below 2.5 mmol/l (such as in the traditional Chinese culture), symptomatic atherosclerotic disease would be almost non-existent. The most compelling evidence for the importance of LDL cholesterol comes from studies of patients and animals that have a genetically determined lack of cell membrane receptors for LDL (Fig. 13.6). About 1 in 500 Caucasians is heterozygous for this type of mutation, and has reduced numbers of functional receptors on their cell surfaces and elevated plasma LDL-cholesterol levels (over 8 mmol/l). Such individuals often develop coronary heart disease in their forties or fifties. The rare patients who are homozygous for one of these mutations (approximately 1 per million) have much higher cholesterol levels and usually die from coronary atheroma in infancy or the teens.
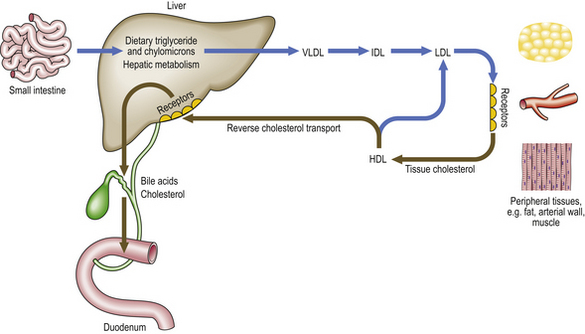
Fig. 13.6 Major pathways of lipoprotein metabolism. This is a much simplified outline of lipid metabolism. Note that LDL uptake in peripheral tissues is receptor-mediated. HDL apoprotein accepts cholesterol from tissues. This can then be absorbed by specific receptors in the liver (reverse cholesterol transport) or recycled into LDL. (HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low-density lipoprotein.)
The importance of other risk factors beyond hypercholesterolaemia is illustrated by the huge variation in expression of severity of disease among groups of patients with the same cholesterol levels. Major risk factors are smoking, hypertension, diabetes, male gender and increasing age. They appear to accelerate the process of plaque formation driven by lipids. Less strong risk factors include obesity, a sedentary lifestyle, low socio-economic status and low birth weight. At present there is also increasing interest in the role of micro-organisms in the evolution of atherosclerotic disease. The cumulative effect of several, often innocent or subclinical, infections with common bacteria such as Chlamydia pneumoniae, cytomegalovirus, influenza and dental pathogens are thought to increase the risk of atherosclerosis by switching on evolutionarily conserved pathways of inflammation. There is also recent evidence that high-fat diets and obesity may promote translocation of commensal-derived endotoxin from the gut into the general circulation and there induce inflammation, insulin resistance and atherosclerosis.
How do lesions develop?
Generally, the development of atherosclerosis is a two-step process. The first step is injury to the endothelium of the arterial wall and the second is a tissue response of the vascular wall to the injurious agents. Chronic or episodic exposure of the arterial wall to these processes leads over many years to formation of plaques. This concept, initially introduced by Ross and Glomset in 1972, is now convincingly supported by carefully designed postmortem studies of patients of different ages and racial origin and from studies in animals that develop atherosclerosis either spontaneously or following high-fat or cholesterol-supplemented diets.
Injured endothelial cells at sites of lesion formation undergo profound functional alterations which include an enhanced expression of cell adhesion molecules, including ICAM-1 and E-selectin, a high permeability for macromolecules such as LDL, and increased thrombogenicity. This allows inflammatory cells and lipids to enter the intimal layer and form plaques. In more advanced stages of plaque formation large amounts of macrophages and T-cells accumulate in the plaque tissue. Lipid-laden macrophages (foam cells) die through apoptosis, spilling their lipid into an ever-enlarging lipid core. In this respect the response to injury in atherosclerosis has all the features of a chronic inflammatory process.
As in all chronic inflammatory diseases the inflammatory reaction is followed by a process of tissue repair. Growth factors, particularly platelet-derived growth factor (PDGF), stimulate the proliferation of intimal smooth muscle cells (myointimal cells) and the subsequent synthesis of collagen, elastin and mucopolysaccharide by smooth muscle cells. A fibrous cap encloses the lipid-rich core (Fig. 13.5). Growth factors are secreted by platelets, injured endothelium, macrophages and smooth muscle cells themselves.
Another important mechanism of plaque growth is initiated by small areas of endothelial loss, especially in fully developed plaques. Microthrombi are formed at the denuded areas of the plaque surface. These become organised by the same repair process of smooth muscle cell invasion and collagen deposition. Repeated cycles of this process gradually increase the plaque volume.
Clinical manifestations of atherosclerosis
Over a lifetime many plaques may develop in a given patient, the great majority of which will remain clinically unnoticed. Clinical disease is usually provoked by only one out of many plaques, and ranges in severity from relatively benign symptoms to life-threatening diseases. The more serious conditions often follow acute changes in the plaques.
Plaque morphology and the vulnerable plaque concept
Autopsy studies on large series of patients who died from myocardial infarction have shown that the atherosclerotic plaques that develop a plaque rupture and subsequent thrombus have distinct morphological features. This has led to the recognition of so-called vulnerable plaques: plaques with a high risk of developing thrombotic complications (Fig. 13.7). Typically vulnerable plaques have a thin fibrous cap, a large lipid core and prominent inflammation. It is thought that pronounced inflammatory activity contributes to degradation and weakening of the plaque that increases the risk of rupture. Secretion of proteolytic enzymes, cytokines and reactive oxygen species by the plaque inflammatory cells orchestrates this process. On the other hand, the plaques that gradually progress to highly stenotic lesions (as, for example, in stable angina pectoris) often have a large fibrocalcific component with little inflammatory activity.
Preventive and therapeutic approaches to atherosclerosis and atherothrombosis
Smoking cessation, control of blood pressure, weight reduction, regular exercise and dietary modifications are all of benefit and are now widely promoted. In Mediterranean communities, a much lower proportion of energy is obtained from saturated fat, and coronary heart disease death rates are much lower. Diets rich in polyunsaturated fat are associated with low coronary heart disease rates. This is the logic behind the advice that we should all eat five portions of fruit or vegetables each day. Fatty acids found in fish have cardioprotective effects. The American Heart Association now recommends at least two servings of fish, especially oily fish, per week.
Secondary prevention of atherosclerotic complications
There is good evidence from many different trials that treatment with cholesterol-lowering drugs reduces cardiac events both in patients with a history of coronary heart disease and in asymptomatic subjects with hypercholesterolaemia. At present ‘statins’ are the most widely used compounds. They act as specific inhibitors of HMG CoA reductase, an enzyme that has a rate-limiting action in hepatic cholesterol synthesis. Besides their cholesterol-lowering effect, they probably reduce inflammation within atheromatous lesions and promote plaque stability (conversion of a lipid-rich inflamed plaque into a fibrous plaque).
Another approach is to minimise the risk of thrombus formation on established atheromatous lesions. The earliest changes in thrombus formation include platelet activation following interaction with thrombogenic plaque components. Low doses of aspirin, which inhibits aggregation of platelets, are given to many patients with clinical evidence of atheromatous disease and have undoubted beneficial effects. The United Kingdom National Service Framework for Coronary Heart Disease also recommends that patients with established coronary heart disease should receive beta-blockers and angiotensin converting enzyme inhibitors or angiotensin receptor antagonists.
Surgical and percutaneous interventions
Several invasive techniques have been developed to reduce the size of lesions, to remove a thrombus or to bypass a severely narrowed or occluded artery. Endarterectomy is a technique by which the atheromatous intima is ‘cored out’ from the underlying media. Embolism of atheromatous debris from the carotid bifurcation is a common cause of transient ischaemic attacks and completed strokes. Controlled trials have shown that carotid endarterectomy reduces the risk of further neurological events. Percutaneous angioplasty is used to ‘crack open’ atheromatous plaques with an inflatable balloon. A metallic expandable stent is usually inserted to maintain the patency of the vessel. These techniques are used in both coronary and lower limb arteries. Surgical bypass procedures use segments of saphenous vein or fabric grafts to divert blood past obstructed segments of lower limb arteries. An atheromatous aneurysm of the distal aorta may be replaced with a Y-shaped fabric graft. Coronary artery stenoses are bypassed with segments of saphenous veins sewn into the proximal aorta or by dissecting the internal mammary artery from the chest wall and anastomosing its distal end to an artery on the anterior surface of the heart, usually the anterior descending branch of the left coronary artery.
ANEURYSMS


 Dissecting. Usually occur in the thoracic aorta; dissection along the media causes vascular occlusion and haemopericardium
Dissecting. Usually occur in the thoracic aorta; dissection along the media causes vascular occlusion and haemopericardium



An aneurysm is a localised permanent dilatation of part of the vascular tree. Permanent dilatation implies that the vessel wall has been weakened. In contrast, a false aneurysm is a blood-filled space that forms around a blood vessel, usually after traumatic rupture or a perforating injury. A haematoma forms and is contained by the adventitial fibrous tissue. A common cause of false aneurysm formation is femoral artery puncture during arteriography or percutaneous angioplasty. The clinical and pathological features of aneurysms are summarised in Table 13.1.
Table 13.1 Clinical effects of aneurysms
| Type of aneurysm | Site | Clinical effects |
|---|---|---|
| Atherosclerotic | Lower abdominal aorta and iliac arteries |
Aortic dissectionAorta and major branches
BerryCircle of WillisSubarachnoid haemorrhageMicro-aneurysms (Charcot–Bouchard)Intracerebral capillariesIntracerebral haemorrhage, associated with hypertensionSyphiliticAscending and arch of aortaAortic incompetenceMycotic (infective)
Thrombosis or rupture, causing cerebral infarction or haemorrhage
Atherosclerotic aortic aneurysms
Atherosclerotic abdominal aortic aneurysms commonly develop in elderly patients (Fig. 13.8). They can be detected by ultrasound examination and the value of screening for these aneurysms is under study. They may impair blood flow to the lower limbs and contribute to the development of peripheral vascular disease. Most importantly, they may rupture into the retroperitoneal space. Elective repair of these aneurysms is comparatively safe but repair after rupture has a high mortality. Some are now managed by percutaneous insertion of supportive stents and this form of treatment may become more common in the future. Aneurysms of the proximal and thoracic aorta are much less common. As with abdominal aneurysms, atherosclerosis is the commonest cause. In atherosclerotic aneurysms there is usually a pronounced loss of elastic tissue and fibrosis of the media. This is due to ischaemia of the aortic media, and release of macrophage enzymes causing fragmentation of elastic fibres. There is evidence that some aortic aneurysms are familial, and inherited defects in collagen have been postulated as the underlying cause.
Aortic dissection (dissecting aneurysms)
In aortic dissection, blood is forced through a tear in the aortic intima to create a blood-filled space in the aortic media (Fig. 13.9). This can track back into the pericardial cavity, causing a fatal haemopericardium, or can rupture through the aortic adventitia. In occasional cases the track re-enters the main lumen to create a ‘double-barrelled’ aorta. The intimal tear and the anatomical features of the aorta can be demonstrated in life by CT or MRI scanning. The underlying pathology is poorly understood. In some, but by no means all, cases there is pronounced degeneration of the aortic media. This is the so-called cystic medial necrosis and is characterised by mucoid degeneration and elastic fibre fragmentation. An exaggerated form of this change is seen in Marfan’s syndrome, a congenital disorder of the expression of a glycoprotein, fibrillin, closely associated with elastin fibres. The strongest risk factor for dissecting aneurysm is systemic hypertension. In some cases the intimal ‘entry’ tears are around atheromatous plaques, but in most cases they involve disease-free parts of the aorta. Without treatment, the mortality from dissecting aneurysm is at least 50% at 48 hours, and 90% within 1 week. The immediate aim of treatment is to contain the propagating haematoma by reducing arterial pressure. Surgical repair is feasible in some patients, especially if the process affects the proximal aorta.
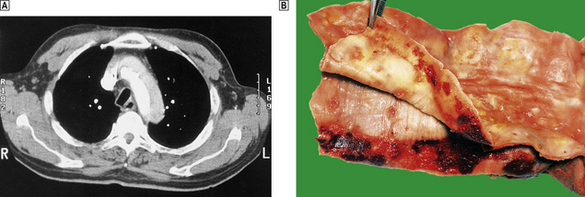
Fig. 13.9 Aortic dissection.  A CT scan of a patient with an acute dissection of the ascending aorta. There are two patterns of contrast enhancement in the aorta. The whiter is the main lumen and the greyer the false lumen.
A CT scan of a patient with an acute dissection of the ascending aorta. There are two patterns of contrast enhancement in the aorta. The whiter is the main lumen and the greyer the false lumen.  The innermost portion of the aortic wall has been peeled away to reveal the underlying haemorrhagic tract.
The innermost portion of the aortic wall has been peeled away to reveal the underlying haemorrhagic tract.
‘Berry’ aneurysms
In the so-called ‘berry’ aneurysms in the circle of Willis, the normal muscular arterial wall is replaced by fibrous tissue. The lesions arise at points of branching on the circle of Willis, and are more common in young hypertensive patients. The most important complication is subarachnoid haemorrhage (Ch. 26).
Capillary micro-aneurysms
Capillary micro-aneurysms (Charcot–Bouchard aneurysms) are associated with both hypertension and diabetic vascular disease (p. 284). In hypertension, they are common in branches of the middle cerebral artery, particularly the lenticulo-striate. They are thought to be the precursors of primary hypertensive intracerebral haemorrhage, which characteristically occurs in the basal ganglia, cerebellum or brainstem.
Syphilitic aneurysms
Tertiary syphilis is now rare in the developed world but was previously a common cause of proximal aortic aneurysms. They rarely rupture but frequently produce aortic incompetence. The aneurysm is due to ischaemic damage to the media, causing fibrosis and loss of elasticity, secondary to inflammation and narrowing of the vasa vasorum.
Mycotic aneurysms
Mycotic aneurysms are the result of weakening of the arterial wall, secondary to bacterial or fungal infection. The organisms are thought to reach the arterial wall via the blood stream and enter the media via the vasa vasorum. Lesions are commonest in the cerebral arteries (Fig. 13.10) but almost any area can be affected. Bacterial endocarditis is the commonest underlying infection.

HYPERTENSION
 Classified aetiologically into essential (primary) hypertension, in which there is no evident cause, and secondary hypertension
Classified aetiologically into essential (primary) hypertension, in which there is no evident cause, and secondary hypertension

Definition
Hypertension is the commonest cause of cardiac failure in many societies and a major risk factor for atherosclerosis. Furthermore, it is a major risk factor for cerebral haemorrhage, another leading cause of death worldwide. There is no universally agreed definition of hypertension, but most authorities would accept that a sustained resting blood pressure of more than 160/95 mmHg is definite hypertension. Furthermore, this would be categorised as:
Borderline hypertension encompasses the range 140/90 to 160/95 mmHg. In the past, less emphasis was placed on high systolic pressure readings if the diastolic pressure was normal or nearly normal. This is now known to be incorrect practice. Guidelines for the diagnosis and treatment of hypertension are altering as new information becomes available. In the future, treatment is likely to be started at lower blood pressure levels, especially in diabetics.
The diagnosis of an individual patient as hypertensive can be fraught with difficulties. Single blood pressure readings are often spuriously high and many patients have ‘ambulatory’ blood pressure monitoring over a 24-hour period. Care must be taken to ensure that the blood pressure is accurately recorded with an inflatable cuff of appropriate size and shape.
Epidemiology
Hypertension is a serious cause of morbidity and mortality. The incidence of hypertension varies markedly in different countries. In most, but not all, communities, blood pressure tends to rise with age. There is good evidence that high blood pressure is heritable. The precise genetic pattern is not known, but the pattern is polygenic. Blood pressures of parents and their natural children are correlated, whereas those of parents and adopted children are not. The correlation of blood pressures in monozygotic twins is higher than in dizygotic twins. Many black communities, both in western Africa and North America, have a high incidence of hypertension, whereas values tend to be lower on the Indian subcontinent. In certain parts of Africa and the South Pacific, average blood pressures are unusually low. Many epidemiological studies have confirmed a positive correlation between body weight and both systolic and diastolic blood pressure. This association is strongest in the young and middle-aged, but is less predictable in the elderly. Hypertensive patients who lose weight can reduce their blood pressure.
Aetiological classification
Hypertension can be classified aetiologically according to whether the cause is unknown—essential (primary or idiopathic) hypertension—or is known—secondary hypertension. Most cases of hypertension are classified as ‘essential’, but the possibility of an underlying cause should always be considered.
Essential hypertension
Up to 90% of patients who present with elevated blood pressure will have no obvious cause for their hypertension and are therefore said to have essential or primary hypertension (Table 13.2).
Table 13.2 Pathogenesis of systemic hypertension
| Aetiological classification | Causes |
|---|---|
| Essential (primary) hypertension | Unknown, but probably multifactorial involving: |
Secondary hypertensionRenal disease
Endocrine causes
Coarctation of aorta Drugs, e.g. corticosteroids, oral contraceptives
Ultimately it is the kidneys that are responsible for the control of blood volume and blood pressure, largely through the handling of sodium in the renal tubules. Factors that influence this include:
The sympathetic nervous system
Blood pressure is a function of total peripheral resistance and cardiac output; both of these are, to some extent, under the control of the sympathetic nervous system. When compared with controls, patients with essential hypertension have higher blood pressures at any given level of circulating plasma catecholamines, suggesting an underlying hypersensitivity to these agents. The circulating levels of catecholamines are highly variable and can be influenced by age, sodium intake, posture, stress and exercise. Nevertheless, young hypertensives tend to have higher resting plasma noradrenaline levels than age-matched, normotensive controls.
The renin–angiotensin–aldosterone system
Renin is released from the juxtaglomerular apparatus of the kidney, diffusing into the blood via the efferent arterioles (Ch. 17). It then acts on a plasma globulin, variously called ‘renin substrate’ or angiotensinogen, to release angiotensin I. This is in turn converted to angiotensin II by angiotensin converting enzyme (ACE). Angiotensin II is a powerful vasoconstrictor and is therefore capable of inducing hypertension. However, only a small proportion of patients with essential hypertension have raised plasma renin levels, and there is no simple correlation between plasma renin activity and the pathogenesis of hypertension. There is some evidence that angiotensin can stimulate the sympathetic nervous system centrally, and many patients with essential hypertension respond to treatment with ACE inhibitors.
Several therapeutic trials have shown that ACE inhibitors given soon after an acute myocardial infarction decrease mortality, perhaps by preventing myocardial dilatation. Recently, variations or mutations in the genes coding for angiotensinogen, ACE and some of the receptors for angiotensin II have been linked with hypertension.
Dietary sodium and potassium
The role of dietary factors in hypertension is controversial. Hypertension is almost unknown in populations with dietary intakes of sodium of less than 50 mmol/day. In most Western societies daily sodium intakes are above 100 mmol daily, but there is no predictable relationship between intake and blood pressure. Studies in hypertensive patients have shown that a 50-mmol/day reduction in sodium intake reduces systolic blood pressure by 4 mmHg. Human kidneys are efficient at conserving sodium and excreting potassium. This was ideal in prehistoric populations where diets were high in potassium and low in sodium—the converse of the modern Western diets. Fruit and vegetables are rich in potassium as well as polyunsaturated fats.
Secondary hypertension
Hypertension may result from several underlying conditions:
Renal hypertension
Some forms of acute, and all forms of chronic, renal disease can be associated with hypertension. The two chief mechanisms involved are:
The possibility of renal disease should be considered in all patients with hypertension. In a few cases, a focal stenosis of one renal artery, as a result of atheroma or fibromuscular dysplasia of the renal artery, is responsible for unilateral renal ischaemia and hyper-reninism. Surgical treatment can be curative in selected patients. Patients in terminal renal failure are extremely sensitive to changes in salt and water balance. Hypertension in these patients can often be managed by restriction of salt and water intake and by careful dialysis.
Endocrine causes
The hypersecretion of corticosteroids in Cushing’s syndrome is associated with systemic hypertension. Similarly, adrenal tumours that secrete aldosterone (Conn’s syndrome) or catecholamines (phaeochromocytoma) can cause hypertension. However, these are found in less than 1% of all hypertensive patients.
Coarctation of the aorta
Systemic hypertension is one of the commonest features in coarctation. Raised blood pressure will be detected in either arm, but not in the legs. The femoral pulse is often delayed relative to the radial. Death usually results from cardiac failure, hypertensive cerebral haemorrhage or dissecting aneurysm (see Fig. 13.43).
Pathological classification
Hypertension is classified also according to the clinicopathological consequences of the blood pressure elevation. Benign or essential hypertension is often asymptomatic and discovered only during a routine medical examination. Malignant hypertension is a serious condition necessitating prompt treatment to minimise organ damage or the risk of sudden death from cerebral haemorrhage.
Benign (essential) hypertension
The increased peripheral vascular resistance and cardiac workload associated with hypertension produce left ventricular hypertrophy. During life this can be detected electrocardiographically, and at postmortem there is often substantial concentric thickening of the left ventricle. With the development of congestive cardiac failure, the hypertrophy can be obscured by left ventricular dilatation. Some patients with hypertension also have coronary arterial atherosclerosis and evidence of consequent ischaemic heart disease.
Longstanding hypertension produces generalised disease of arterioles and small arteries, in addition to enhancing the development of atherosclerosis. The changes are most easily appreciated in the retina during life, and in the kidneys at autopsy. Medium-sized renal arteries and renal arterioles show marked intimal proliferation and hyalinisation of the muscular media. This produces focal areas of ischaemia with scarring, loss of tubules and periglomerular fibrosis. The cortical surfaces are finely granular.
Malignant hypertension
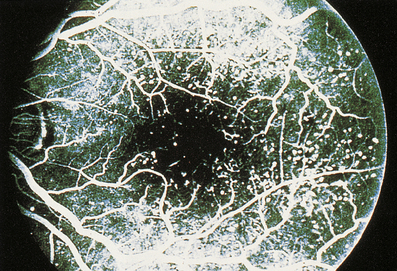
Malignant hypertension is a clinical and pathological syndrome. The characteristic features are a markedly raised diastolic blood pressure, usually over 130–140 mmHg, and progressive renal disease. Renal vascular changes are prominent, and there is usually evidence of acute haemorrhage and papilloedema (Fig. 13.11). Malignant hypertension can occur in previously fit individuals, often black males in their third or fourth decade. However, most cases occur in patients with evidence of previous benign hypertension; this is sometimes termed accelerated hypertension.
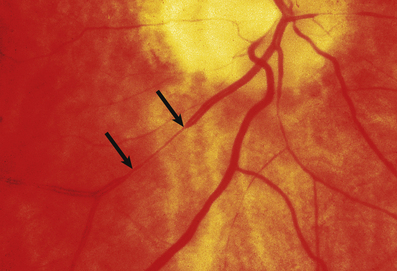
Fig. 13.11 Hypertensive fundus. Ocular fundus from a patient with hypertension. The outline of the blood vessels is caused by the reflection of light from the column of blood (the light reflex). Because the wall of the arteriole is thickened in hypertension, the lumen of the vessel is narrowed and the light reflex is reduced (between the arrows).
The consequences of malignant hypertension are:
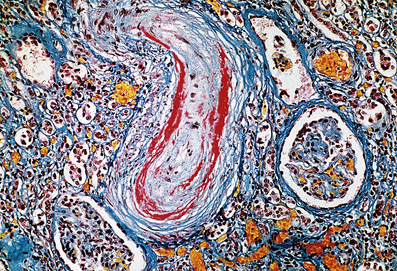
The characteristic histological lesion of malignant hypertension is fibrinoid necrosis of small arteries and arterioles (Fig. 13.12). The kidney is frequently affected and some degree of renal dysfunction is inevitable. Occasionally there is massive proteinuria, and renal failure develops. Acute left ventricular failure can occur.
Pulmonary hypertension
The pathophysiological mechanisms associated with pulmonary hypertension are summarised in Table 13.3.
Table 13.3 Pathological causes and physiological changes in pulmonary hypertension
| Cause | Pathophysiology |
|---|---|
| Acute or chronic left ventricular failure | Raised left ventricular pressure → raised venous pressure |
| Mitral stenosis | Raised left atrial pressure → raised pulmonary venous pressure |
| Chronic bronchitis and emphysema | Hypoxia → pulmonary vasoconstriction → raised pulmonary venous pressure |
| Emphysema | Loss of pulmonary tissue → reduced vascular bed |
| Recurrent pulmonary emboli | Reduction in pulmonary vascular bed available for perfusion |
| Primary pulmonary hypertension | Cause of raised pulmonary pressure unknown |
When pulmonary hypertension develops rapidly (following acute left ventricular failure, for example), there is massive transudation of fluid from the pulmonary capillaries into the pulmonary interstitial space and alveoli. This causes the characteristic clinical picture of acute and distressing shortness of breath and expectoration of lightly bloodstained, watery fluid. In chronic pulmonary hypertension, the pulmonary arteries develop a progressive series of reactive changes. These include muscular hypertrophy, intimal fibrosis and dilatation. There are repeated episodes of haemorrhage into the alveolar spaces, which contain haemosiderin (iron pigment)-laden macrophages.
Vascular and systemic effects
Vascular changes
Hypertension accelerates atherosclerosis, but the lesions have the same histological appearances and distribution as in normotensive subjects. However, hypertension also causes thickening of the media of muscular arteries. This is the result of hyperplasia of smooth muscle cells and collagen deposition close to the internal elastic laminae. In contrast to atherosclerosis, which affects larger arteries, it is the smaller arteries and arterioles that are especially affected in hypertension (Fig. 13.11).
Hypertension increases the normal flow of protein into the vessel wall and the amount of high molecular weight protein, such as fibrinogen, that passes through the junctions between endothelial cells, resulting in protein deposition. These deposits are called hyaline in benign and fibrinoid in malignant hypertension. Hyaline change is a common degenerative feature of many ageing arteries, and refers to the homogeneous appearance of the vessel wall, due to the insudation of plasma proteins. Fibrinoid change is a combination of fibrin with necrosis of the vessel wall. There is no evidence that an immunological reaction is involved in hypertensive vascular disease.
Heart
Hypertension accelerates atherosclerosis, thus ischaemic heart disease is a frequent complication. A large, ongoing longitudinal population study in Framingham, Massachusetts, has shown hypertension to be a major cause of cardiac failure in previously fit subjects. The left ventricle undergoes hypertrophy and may ‘outgrow’ its blood supply, particularly if there is associated coronary atherosclerosis. Patients with hypertensive left ventricular hypertrophy are more liable to spontaneous arrhythmias than normal subjects. The decreased prevalence of systemic hypertension and left ventricular hypertrophy in Western populations has been attributed to the increasing use of antihypertensive medications.
DIABETIC VASCULAR DISEASE



Patients with diabetes, particularly juvenile-onset insulin-dependent diabetes, may develop three forms of vascular disease.
Atherosclerosis
Both males and females develop premature, and sometimes severe, atherosclerosis. Even diabetic pre-menopausal females can develop substantial atheroma.
Hypertensive vascular disease
This is a frequent complication, especially when there is diabetic renal disease (Ch. 21).
Capillary microangiopathy
This is the most important and characteristic change in diabetes. The alterations are found throughout the systemic circulation and can be viewed directly in the retina (Fig. 13.13). Small arterioles and capillaries are affected and the principal clinical effects are diabetic retinopathy, diabetic glomerulosclerosis and peripheral neuropathy. The biochemical changes are complex and include abnormal glycosylation of proteins within the vessel wall. Although thickened, the basement membranes are unusually permeable, and there is increased passive transudation of protein. Small vessels dilate, forming capillary micro-aneurysms. In the eye, protein leakage stimulates a fibrous and vascular response, which damages the complex neural network of the retina. Capillary thrombosis causes retinal ischaemia. This is a stimulus to the ingrowth of new capillaries, which causes further retinal damage. Some degree of diabetic retinal disease is inevitable in longstanding diabetes, but only a minority of patients become blind. Intimal thickening of renal arterioles and micro-aneurysm formation in the glomerular capillaries are the underlying causes of diabetic renal disease. The excretion of small amounts of protein in the urine (micro-albuminuria) is the first evidence of this. Peripheral neuropathy results from disease of small vessels supplying nerves. Multicentre trials have shown that the rate of progression of major complications such as diabetic retinopathy and nephropathy can be reduced by careful control of blood sugar levels and prompt treatment of hypertension.
VASCULITIS
 Multisystem disorders but with a predilection for highly vascular tissues such as skin, renal glomerulus, upper respiratory and gastrointestinal tract
Multisystem disorders but with a predilection for highly vascular tissues such as skin, renal glomerulus, upper respiratory and gastrointestinal tract

Pathogenesis
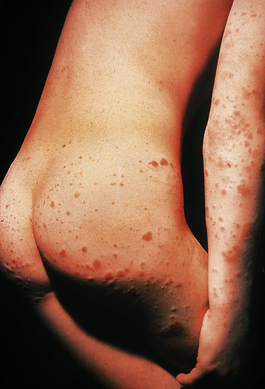
Vasculitis is the name given to inflammatory diseases of blood vessels. The cause of most forms of vasculitis is unknown but clinical and experimental studies suggest that in some cases the underlying pathology is a deposition of complexes of antigen and antibody in the vessel wall. Immune complexes are not inherently harmful, but if they lodge in tissues and activate complement they incite an acute inflammatory reaction and trigger the coagulation system. Repeated minor trauma may be the reason that the lesions of some vascular disorders develop on the extensor surfaces of the arms and on the buttocks (Fig. 13.14). Venous stasis may account for the fact that some examples of vasculitis are particularly prominent in the lower leg.
Clinicopathological features
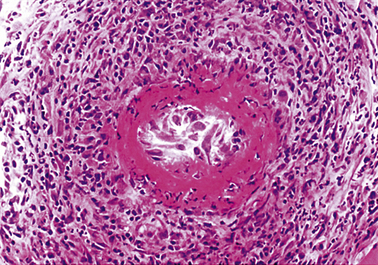
In most forms of vasculitis the pathological changes in the vessels are broadly similar. A dense infiltrate of acute and chronic inflammatory cells is usually present and in some instances immunological techniques may demonstrate abnormal deposits of immunoglobulin and complement in the intima and media. The patterns of disease in vasculitis are largely a result of the organ and the size of the vessel affected. Because of this, systemic vasculitis can present with a wide variety of different signs and symptoms. Pathologists prefer to classify vasculitis according to the type and size of vessel that is chiefly involved. The major clinical and pathological features of some common immunological vascular disorders are summarised in Table 13.4.
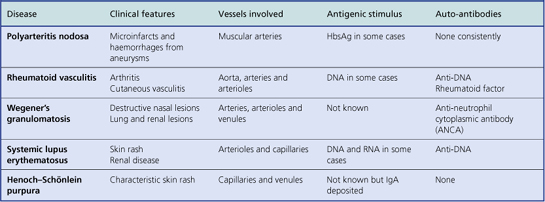
A skin rash is one of the commonest presenting features of acute vasculitis, and is sometimes closely related to treatment with a particular drug. In these circumstances the foreign substance probably acts as a hapten in the induction of an immunological reaction. Many forms of vasculitis are self-limiting, but their clinical course can sometimes be shortened by anti-inflammatory drugs. When the disease affects several different organs and systems the term systemic vasculitis is used. Examples include polyarteritis nodosa (Fig. 13.15), rheumatoid vasculitis and Wegener’s granulomatosis. Without treatment the prognosis in systemic vasculitis is poor. The possibility of systemic vasculitis must be considered in any patient with a multisystem pattern of illness, especially if the respiratory tract is involved. Anti-inflammatory and cytotoxic drugs, such as steroids and cyclophosphamide, may induce clinical remissions in 75% of patients.
Complications
Complications of vasculitis include thrombosis with resulting ischaemia or infarction. Multiple sites are usually involved simultaneously. Tissues with a high blood flow, such as the skin and the glomeruli, are preferentially affected.
In many patients with these disorders there are increased levels of circulating immune complexes, and the complement concentrations may be low, particularly when the disease is active. Auto-antibodies are present in some patients (Table 13.4). Some are directed against components of vascular endothelial cells (anti-neutrophil cytoplasmic antibodies—ANCA) and are useful in supporting a clinical diagnosis.
VASCULAR DISEASES WITH SPECIFIC CLINICAL FEATURES
Some vascular disorders have characteristic clinical and histological features. Very little is known about their cause but, as some show prominent inflammation, they are classified as forms of vasculitis.
Scleroderma (systemic sclerosis)
The vascular changes of scleroderma (systemic sclerosis) are similar to those of benign or malignant hypertension, but only 25% of the patients are hypertensive. Muscular arteries and arterioles are narrowed by newly formed layers of collagen and mucopolysaccharide (Ch. 25). The cause of this curious disorder is unknown. There may be an underlying abnormality of collagen synthesis which affects not only arteries but also the subcutaneous tissues of the extremities and gastrointestinal tract. The most characteristic clinical feature is progressive subcutaneous fibrosis, which leads to marked tightening of the skin of the arms and hands. There is no effective treatment.
Cranial (giant cell) arteritis
Cranial (giant cell) arteritis was first recognised by Sir Jonathan Hutchinson, who in 1890 described an 80-year-old retired hospital porter who was prevented from wearing his hat by tender and inflamed temporal arteries. The arteries of the head and neck, including the aorta, are most frequently involved. If the disease affects the ophthalmic or posterior ciliary arteries, blindness can result. It is the most common form of large vessel vasculitis.
In florid clinical cases the superficial temporal artery is hard, tender and pulseless, and the patient complains of a severe headache. The ESR is usually high, generally above 50 mm/h. Microscopically there is marked intimal thickening and oedema, and a dense, sometimes granulomatous, chronic inflammatory and giant cell reaction with phagocytosis of fragmented elastic fibres (Fig. 13.16). The clinical diagnosis can often be confirmed by biopsy, but this is not always positive; focal involvement of the superficial temporal artery is the probable reason for these negative biopsies.
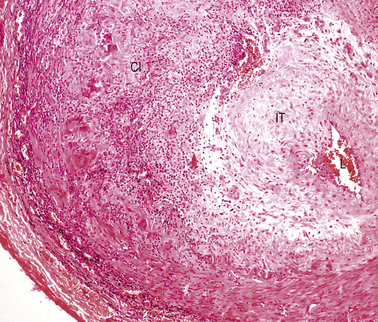
Fig. 13.16 Cranial arteritis. This biopsy of a superficial temporal artery shows marked intimal thickening (IT) and a dense mononuclear and giant cell infiltrate (CI). The lumen is restricted to a tiny slit (L).
The cause of cranial arteritis is unknown. There is little evidence that it is immunological in origin. In up to 50% of cases it is associated with a polymyalgia rheumatica-like illness. Almost all cases of cranial arteritis respond well to steroid therapy and, in severe cases, prompt treatment can prevent blindness.
Pulseless (Takayasu’s) disease
Pulseless, or Takayasu’s, disease is a rare inflammatory disorder of the aorta and its proximal branches. Most patients are young or middle-aged females, who present with hypertension or ischaemic symptoms in the arms. Renal arterial involvement can cause hypertension. Characteristically, there is a severe necrotising inflammation with some similarities to cranial arteritis. Unfortunately, only a proportion of these patients respond to treatment, with either steroids or other agents, and the outlook is much worse than in cranial arteritis. Few patients make a complete recovery, and the associated hypertension may be difficult to control.
Buerger’s disease
Buerger’s disease (thrombo-angiitis obliterans) is a rare disease more strongly associated with smoking than any other vascular disorder. Most patients are male, and Jews are affected twice as commonly as non-Jews. The clinical picture is very distinctive. Peripheral gangrene develops in the fingers and toes, but the changes are progressive and serial amputations are often required (Fig. 13.17).
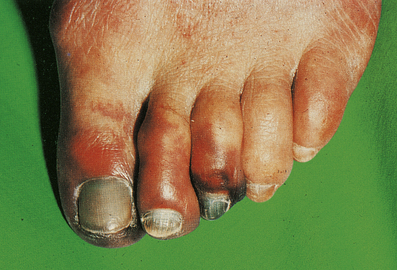
Fig. 13.17 Buerger’s disease. The toes are gangrenous. After 9 months this patient required a below-knee amputation.
Pathological alterations are less specific. Small arteries in the arms and lower leg are mainly involved and show marked intimal fibrosis, thrombus formation with evidence of recanalisation, and peri-arteritis with adventitial tissue changes affecting adjacent veins and nerves. Apart from the striking association with heavy smoking, little is known of its cause.
RADIATION VASCULAR DISEASE
Some pathological change is almost inevitable in any tissue that has been irradiated. The most prominent chronic reactions in vascular tissue are intimal thickening of arteries and arterioles, and dilatation of capillaries and venules. Following radiation there is a substantial reduction of the capillary vascular bed, and this inevitably produces ischaemia and subsequent fibrosis. Strictures in the large and small intestines sometimes follow radiotherapy for carcinoma of the cervix. Patients with lymphoma or other tumours who receive mediastinal radiotherapy frequently develop pericarditis, but damage to small myocardial vessels may produce patchy interstitial fibrosis, and proximal coronary arteries occasionally develop premature atherosclerosis.
DISEASES OF VEINS
Normal venous structure
Like arteries, veins have an intima, media and adventitia (Fig. 13.1B). There is no definite internal elastic lamina and, as in arteries, the thickness of the intima increases with age (phlebosclerosis). Small veins have only a thin muscular wall but in larger channels, such as the saphenous vein and the inferior vena cava, there are coarse bundles of irregular muscle, partially organised into longitudinal and circular layers.
Venous thrombosis
Any condition that impedes normal venous return predisposes to thrombosis (Ch. 8). Common predisposing causes include:
The veins of the lower abdomen, pelvis and legs are most frequently affected. Thrombi often form in the deep veins of the leg when patients are immobilised in bed, for example after a fracture or surgical operation (where the risk is exacerbated by a rise in coagulation factors and platelets) or during a serious illness. There is evidence that anticoagulant drugs reduce the incidence of post-operative deep venous thrombosis, but their beneficial effects must be weighed against the increased danger of post-operative haemorrhage.
In haematological disorders, such as polycythaemia, and in some patients with malignant tumours, the blood is hypercoagulable and venous thrombosis is common. In the distinctive clinical syndrome of thrombophlebitis migrans, superficial venous thrombi form and resolve in different subcutaneous sites.
Inherited disorders enhancing coagulation are of increasing clinical interest (Ch. 23). The most important of these is a specific point mutation in the gene coding for coagulation factor V (factor V Leiden). This abnormal form of factor V is resistant to degradation by activated protein C. In Europe about 5% of subjects are heterozygous for this mutation and have at least a three-fold increased risk of venous thrombosis and pulmonary embolism.
Varicosities
Tortuous and distended (‘varicose’) veins or varices are a common clinical problem. There is often associated ulceration, usually on the medial aspect of the ankle and lower leg (Fig. 13.18). There are both superficial and deep venous plexuses in the lower limb, connected by perforating veins. The return of blood from the deep veins is aided by the normal contraction of the calf and thigh muscles. If the valves in the perforating veins become incompetent, blood can be forced from the deep to the superficial venous plexuses; this is a major factor in the development of varicosities. The exact cause of varicose ulceration is uncertain, but impaired venous return with resulting stasis, lower limb oedema and fibrin deposition around small capillaries and veins has been implicated. In many cases, there has been a previous deep leg vein thrombosis. Some ulcers heal after surgical treatment of varicose veins.
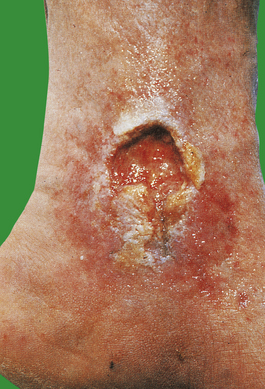
Fig. 13.18 Venous ulceration of the ankle. These ulcers are the result of poor venous drainage from the lower leg. Immobility, previous deep vein thrombosis and varicose veins all contribute to their development.
Varices frequently develop close to the oesophagogastric junction in portal hypertension due to, for example, hepatic cirrhosis (Ch. 16).
COMPLICATIONS IN VESSELS USED AS ARTERIAL BYPASSES
Surgical operations in which atheromatous coronary or lower limb arteries are bypassed with the internal mammary artery or a length of saphenous vein are now common. Although these procedures are often successful in restoring distal blood flow, complications can occur. The two most important are:
Prosthetic vessels made of various types of cloth can be used to repair lower abdominal aortic aneurysms or to bypass iliac or femoral arteries. Although these grafts do not develop true atherosclerosis, a fibrous pseudo-intima develops. Thrombosis may also occur.
DISEASES OF LYMPHATICS
Normal lymphatic structure
The largest lymphatic vessels, such as the thoracic duct, resemble veins. They are lined by endothelium and have a well-defined muscular wall. In contrast, the most peripheral lymphatics begin as closed sacs, lined by a single layer of endothelium and supported by thin strands of collagen. They have valves that give them a beaded appearance.
Lymphatic involvement in disease
Frequently lymphatics provide the channels by which malignant tumours can spread from the primary site to the regional lymph nodes (Ch. 11). In acute inflammation the flow of lymph is markedly increased and, occasionally, lymphatic vessels draining such an area become secondarily inflamed. In infestations with filarial parasites, lymphatic channels are obstructed and marked swelling results; the skin becomes thickened and boggy (‘elephantiasis’).
TUMOURS OF BLOOD VESSELS
Benign tumours
Haemangiomas are common benign tumours of small capillaries (Fig. 13.19). They are particularly common on the face and scalp area of infants, and frequently regress. In adults they can occur on almost any part of the skin. On the lips and fingers they are frequently inflamed and are usually known as pyogenic granulomas; they are probably reactive lesions rather than true neoplasms.
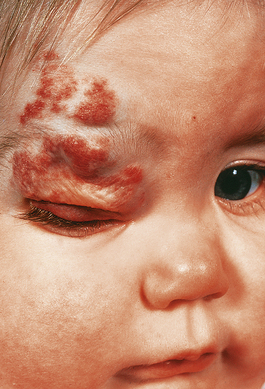
Fig. 13.19 Haemangioma in a child. Although these lesions in this 18-month-old child are unsightly, they are benign and often regress.
A glomus tumour is a distinctive, benign, but sometimes exquisitely painful, blood vessel neoplasm that generally arises in the finger or nail bed. It may develop from some component of the arteriovenous anastomosis that is particularly common in these sites.
Arteriovenous malformations are not strictly true tumours. They are most common in the cerebral and cerebellar hemispheres and in the lungs. The possibility of an arteriovenous malformation should be considered in any young person who presents with cerebral haemorrhage.
Malignant tumours
Angiosarcoma is rare, but has a notoriously aggressive behaviour. The lesion is composed of masses of interconnecting vascular channels lined by a pleomorphic endothelium. Lesions most commonly develop in the soft tissues of the lower limbs, and the head and neck of elderly individuals.
Kaposi’s sarcoma, originally described by a Hungarian dermatologist, is a common malignant tumour in black Africans. Its precise cell of origin is uncertain but may well be lymphatic endothelium. In both blacks and whites, Kaposi’s sarcoma is one of the tumours that develops in patients with acquired immune deficiency following HIV infection.
CARDIAC DISEASE
NORMAL STRUCTURE AND FUNCTION OF THE HEART
The heart is a muscular pump divided on each side into two chambers—an atrium and a ventricle—each separated by a valve, tricuspid on the right, mitral on the left. The embryogenesis of these chambers and valves is covered in the section on congenital cardiovascular disease, where it is of immediate relevance. The inner wall of the cardiac chambers and the surface of the valve cusps are lined by a layer of endothelial cells—the endocardium. The bulk of the chamber wall—the myocardium—comprises a network of striated muscle cells, each separated by an intercalated disc. The heart is invested by patches of adipose tissue and a layer of mesothelium—the epicardium. This layer of mesothelium forms the visceral aspect of the pericardial sac which normally contains a small volume of clear fluid to lubricate the surfaces during cardiac contraction.
Venous blood from the systemic circulation drains into the right atrium, which contracts during diastole to force the blood through the tricuspid valve into the right ventricle. During systole the right ventricle contracts, expelling the blood through the pulmonary valve and into the pulmonary circulation. A synchronous sequence of events takes place on the left side: the pulmonary veins drain oxygenated blood into the left atrium; in diastole the blood is forced through the mitral valve; in systole the left ventricle contracts to expel blood through the aortic valve into the aorta. The atria on each side are of similar dimensions, but the myocardium of the left ventricle is much thicker than that of the right ventricle; this is commensurate with the relative systolic blood pressure in the aorta and pulmonary artery trunk.
The regular and co-ordinated contraction of the myocardium is determined by the pacemaker cells in the sino-atrial (SA) and atrio-ventricular (AV) nodes; the action potentials propagate through the bundle of His and Purkinje network. The electrical activity of the heart can be monitored on the skin surface by electrocardiography (ECG); the P wave corresponds to atrial contraction; the QRS complex reflects propagation of the action potential into the ventricles and their subsequent contraction; and the T wave is due to repolarisation of the myocardium.
Myocardial cell contraction and relaxation is brought about by changes in the concentration of cytosolic calcium. The cyclical contraction of the heart is initiated by the spontaneous depolarisation of the pacemaker cells in the SA node during diastole. The contraction rate, however, is modulated by the autonomic nervous system: beta-adrenergic receptors permit the heart rate to be accelerated by sympathetic stimulation; the vagus nerve through its parasympathetic effects, mediated by acetylcholine, slows the heart rate.
The myocardium is supplied by the coronary arteries originating from the root of the aorta just above the aortic valve cusps. The right coronary artery usually supplies the right ventricle, the posterior part of the interventricular septum, and part of the posterior wall of the left ventricle. The left coronary artery, via its principal branches—the anterior descending and the circumflex arteries—supplies the anterior part of the interventricular septum and most of the left ventricular myocardium. It is not unusual, however, to find that one artery is dominant, supplying a larger territory than usual. Blood flow through the coronary arteries is maximal during diastole when the ventricular myocardium is relaxed.
In life, cardiac structure can be assessed by a variety of invasive and non-invasive techniques. Chest X-rays provide a general guide to cardiac and aortic size. Echocardiography, coupled with Doppler techniques, gives a detailed view of individual chambers and in particular the contractile function of the ventricular cavities, the appearances of the individual valves and the direction of blood flow through them. More detailed images are obtained by passing a transducer into the oesophagus or stomach—trans-oesophageal echocardiography. At present the detailed anatomy of the coronary artery tree can be analysed only by injecting radio-opaque contrast medium into the coronary arterial orifices—coronary arteriography. Computed tomography (CT) and nuclear magnetic resonance imaging (MRI) are increasingly used by cardiac radiologists and can provide detailed images of individual chambers and the aortic lumen and wall.
The cardiac myocytes are permanent cells; if some die, as in myocardial infarction, the others cannot regenerate to replace those that are lost and the defect is repaired by fibrosis. Similarly, in either hypertension or narrowing of the ventricular outflow tracts, the myocardium of the appropriate chamber becomes correspondingly thicker due to hypertrophy rather than hyperplasia.
Part of the right atrial wall produces a peptide hormone—atrial natriuretic peptide (ANP)—that acts on renal tubular epithelium to enhance the urinary excretion of sodium.
Although the heart is vulnerable to ischaemia, it is usually unerringly reliable and robust. For example, assuming an average heart rate of 80 per minute, the cardiac pump completes approximately 42 million contraction/relaxation cycles per annum! In response to increased demand, such as exercise, the rate can be increased rapidly by beta-adrenergic effects. The stroke volume, normally about 65 ml, can be increased in response to increased diastolic ventricular filling and myocardial fibre stretching (Starling’s law of the heart). However, with a sustained increase in workload (e.g. hypertension, aortic or pulmonary stenosis), the ventricular myocardium becomes thickened by hypertrophy.
HEART FAILURE
Chronic heart failure is a common condition with a poor prognosis. It is associated with disabling symptoms such as fatigue, poor exercise tolerance and shortness of breath on light exercise. Patients with established heart failure have median survival rates of about 3 years, less than in many forms of cancer. The clinical diagnosis of early, compensated heart failure is very difficult. There is no simple laboratory test that identifies patients with heart failure, and echocardiography is required for accurate diagnosis. Heart failure is the end result of many different forms of heart disease. Wherever possible the underlying causes of heart failure should be identified early in the course of a patient’s illness.
Pathophysiology
In almost all forms of heart failure the cardiac output is reduced and this causes a degree of underperfusion that is called arterial underfilling. The body compensates by retaining fluid and increasing blood volume. Mechanoreceptors in the left ventricle, the aortic arch, the carotid sinus and the renal afferent arterioles recognise this underfilling and generate afferent signals that stimulate cardioregulatory centres in the brain. Most of these are relayed in the glossopharyngeal and vagus nerves. The cerebral response is largely mediated by the sympathetic nervous system. Increased sympathetic tone causes tachycardia, increased myocardial contractility and arterial and venous constriction. Activation of the renal sympathetic system stimulates release of renin and angiotensin and causes renal vasoconstriction. Sympathetic stimulation of the hypothalamus causes direct (non-osmotic) release of vasopressin. The net effect of these changes is an increase in total body sodium and water, in severe cases of up to 25%. The enhanced activity of the renin–angiotensin–aldosterone system (Fig. 13.20) can be modified pharmacologically. The United Kingdom National Service Framework for the treatment of heart failure emphasises that treatment with ACE inhibitors or angiotensin receptor antagonists is first-line therapy. They improve survival in patients with all forms of heart failure. The term ‘remodelling’ is used to describe the changes in size, shape and composition that occur in diseased cardiac ventricles. Alterations in the network of connective tissue that surrounds the cardiac muscle cells, the interstitial fibrous tissue, may well be important. Ventricular dilatation is a poor prognostic feature in heart failure and ACE inhibitors have been shown to reduce this and may prevent increased interstitial fibrosis. Recent studies have shown that low doses of the aldosterone antagonist spironolactone reduce death rates in congestive cardiac failure. Because enhanced sympathetic tone is an important pathophysiological change in cardiac failure, selective and non-selective beta-adrenergic antagonists, such as carvedilol and metoprolol, have been developed. They have a promising role in the treatment of moderate and severe heart failure.
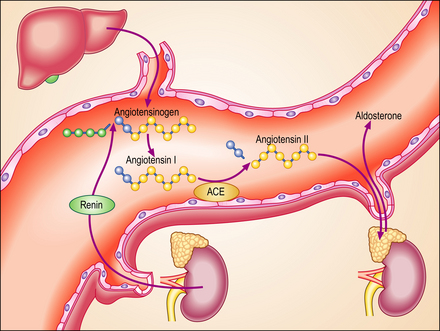
Fig. 13.20 The renin–angiotensin– aldosterone system. At least three hormonal mechanisms are important in the salt and water retention in heart failure. Activation of the renin–angiotensin–aldosterone system is the most important of these. Angiotensin-converting enzyme (ACE) inhibitors or their receptor antagonists are now accepted as the most important therapeutic strategy in heart failure. This diagram summarises the normal pattern of metabolism of angiotensin.
Acute and chronic failure
The clinical features of heart failure depend on the rapidity with which the underlying pathological changes develop. For example, acute failure can occur within minutes of a myocardial infarct. Typically, the patient presents with sudden severe shortness of breath and marked pulmonary oedema. In contrast, valvular defects, such as mitral stenosis and some forms of mitral incompetence, may develop over a period of years and the patient may describe only a very gradual worsening of symptoms. Not infrequently, chronic congestive heart failure develops after an episode of acute failure, for example after a myocardial infarct.
Right and left heart failure
Because the right and left ventricles share an interventricular septum and function together in a closed circuit, it is inevitable that the failure of one ventricular chamber is followed by a failure of the other. Nevertheless, in the early stages of cardiac failure, the clinical signs and symptoms may appear ‘one-sided’. The immediate consequence of left heart failure is pulmonary congestion and oedema. In contrast, right heart failure may produce prominent systemic venous congestion, raised jugular venous pressure and enlargement of the liver. The term cor pulmonale is used to describe right heart failure secondary to lung disease. The commonest cause of cor pulmonale is chronic obstructive airway disease (chronic bronchitis and emphysema). As lung tissue is destroyed, the pulmonary capillary bed is progressively reduced. Effectively the normal cardiac output is pumped into a smaller number of vessels and pulmonary pressure rises. Hypoxia causes reflex pulmonary vasoconstriction and further elevates pulmonary vascular resistance. Just as the left ventricle may fail in systemic hypertension, the right ventricle fails when pulmonary pressures are persistently elevated. Another cause of increased pulmonary pressure and right heart failure is mitral stenosis. In congestive cardiac failure, there is both right and left ventricular failure and a full combination of systemic and pulmonary signs.
Other terminologies in heart failure
In most patients with heart failure, cardiac output fails to increase, or may even decline during exercise, and eventually it is decreased even at rest. This is termed low output failure and is the direct consequence of the inability of the heart to pump normally. In contrast, a few patients may develop pulmonary congestion and oedema when the total cardiac output and ejection fraction of the left ventricle is normal or even increased. This is termed high output failure. The causes of this include an increase in blood volume, for example during pregnancy or from accumulation of excess salt and water due to salt-retaining steroids. It is also associated with an abnormally increased venous return and/or decreased peripheral resistance, for example in hyperthyroidism, cirrhosis, renal failure and severe anaemia.
Because echocardiography is the most sensitive and specific method of diagnosing heart failure, an increasing number of patients have a transthoracic ‘echo’. In the majority of established cases the proportion of blood ejected at each heart beat (the ejection fraction) is reduced. This is termed systolic failure. In some patients systolic function is more normal but there is impaired diastolic filling of the ventricles. This is now called diastolic failure and is increasingly recognised in elderly hypertensive patients.
Causes of heart failure
The principal causes of heart failure in adults are:
Ischaemic heart disease, systemic hypertension and valvular heart disease, either singly or in combination, are responsible for the vast majority of clinical cases of cardiac failure. Only when these have been excluded should other less common causes be considered. Ischaemia, hypertension and most valvular defects initially present with signs and symptoms such as shortness of breath, fatigue and pulmonary oedema, indicating left heart failure. In many patients, right heart failure follows as an inevitable consequence of failure of the opposite ventricle. In about 15% of cases the initial presentation is with pure right-sided failure. When this is secondary to diseases of the lung, such as chronic bronchitis and emphysema, it is termed cor pulmonale. Mitral stenosis is another cause of right ventricular failure.
Clinicopathological features
The major symptoms and signs of heart failure—shortness of breath, pulmonary oedema, systemic venous congestion and oedema—have a clear pathological basis. Not all patients will have all of these changes and it is important to recognise that symptoms in children and the very elderly may be slightly different. At present there is no routine laboratory test that is helpful in the diagnosis of heart failure and echocardiography is the gold standard for diagnosis. There is growing evidence that blood levels of natriuretic peptides, especially brain natriuretic peptide (BNP), are increased in early heart failure and this is likely to become a routine test in clinical laboratories.
Dyspnoea
Except after exercise, breathing is normally automatic and effortless. Dyspnoea is the subjective symptom of shortness of breath or difficulty in breathing and is usually the first symptom of heart failure. Because it is such an important and distressing symptom it has been studied in great detail. Despite this, its exact cause is not understood. In advanced heart failure there is an increase in both the blood and water content of the lungs, and this must be at the expense of the air volume. Intense dyspnoea follows acute left ventricular failure, for example after acute myocardial infarction. The abrupt rise in pulmonary venous pressure causes massive transudation of fluid from the capillaries into the interstitial tissues of the lung, the alveoli and terminal alveoli. Pink frothy fluid pours out from the mouth and nose, and crackles can be heard with a stethoscope, especially in the dependent parts of the lungs.
Shortness of breath while lying flat (orthopnoea) and paroxysmal nocturnal dyspnoea are characteristic signs of left ventricular failure. The basis of orthopnoea is the increased venous return from the legs and gastrointestinal veins to the lungs that results from lying flat. In paroxysmal nocturnal dyspnoea there is a sudden and urgent shortness of breath during sleep, probably because of progressive pulmonary venous congestion.
Increased pulmonary venous pressure and chronic heart failure cause recurrent episodes of alveolar haemorrhage. Some patients in heart failure cough up rusty brown or obviously bloodstained sputum. The rusty colour is the result of haemosiderin-laden macrophages.
Systemic venous congestion and oedema
Fluid retention by the kidney is a compensatory mechanism in cardiac failure. This produces an increased venous return, an increase in ventricular preload and ‘volume overloading’ of the ventricles. Veins are the reservoir for an increased blood volume, and in established heart failure there is widespread congestion of the systemic veins. Distension of the superficial jugular vein in the semi-erect position is one of the earliest signs of this. In congestive heart failure, the liver is enlarged as a direct consequence of engorgement of the centrilobular veins and hepatic sinusoids. The associated ischaemia causes fatty change in hepatocytes (Fig. 13.21). In prolonged cardiac failure, liver function tests can be abnormal, and slight increases in serum bilirubin and transaminases are not uncommon. Dilatation of the left ventricle may be a prominent radiological and echocardiographic feature (Fig. 13.22).

Fig. 13.21 Liver in heart disease. Liver from a patient who died with severe congestive heart failure. Alternating zones of pale fatty change and dark congestion produce a ‘nutmeg’ liver.
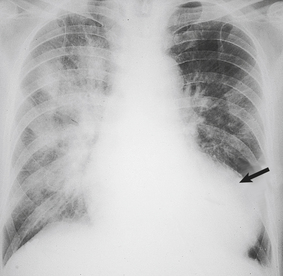
Fig. 13.22 Chest X-ray in congestive heart failure. Note the marked dilatation of the heart (arrow), as compared with the normal chest X-ray in Figure 13.2B, and the increased opacity of the lungs due to congestion and oedema.
A variety of factors contribute to the accumulation of fluid in subcutaneous tissues and in the pleural, pericardial and peritoneal cavities. A hydrothorax is a pleural effusion composed of transudated fluid of low protein content. It is a common feature of congestive cardiac failure, but is rare in uncomplicated left heart failure. High systemic venous pressure is not only responsible for the increased transudation of fluid from pleural capillaries, but also impairs drainage from the lymphatics and the thoracic duct. Large pleural effusions also contribute to the dyspnoea of heart failure. Pericardial and peritoneal effusions (ascites) are features of severe congestive failure.
Other pathophysiological changes
ISCHAEMIC HEART DISEASE




Pathophysiology
Under normal conditions, the blood flow in coronary arteries is closely matched to the metabolic demands of cardiac muscle. Ischaemic heart disease results when the blood supply becomes insufficient, because:
Coronary blood flow is normally independent of aortic pressure. An efficient autoregulatory mechanism exists to control the blood flow through the coronary vascular bed. When an obstruction develops in a major coronary artery, usually because of atherosclerosis, coronary blood flow is initially preserved, because peripheral resistance distal to the obstruction is reduced. When the vessel lumen is more than 75% occluded, ischaemia develops, particularly if the coronary collateral circulation is poorly developed. Cardiac muscle is extremely active metabolically, and mitochondria constitute over 30% of the volume of individual fibres. Aerobic metabolism is essential, as there are very poor reserves of high-energy phosphates. Cardiac muscle death occurs when tissue adenosine triphosphate (ATP) levels are very low and when anaerobic glycolysis has virtually ceased. As with other tissues, the precise cause of death is uncertain, but fatal cardiac muscle injuries are associated with membrane damage and the sudden entry of calcium into the cell cytoplasm. After brief periods of ischaemia, cardiac blood flow can be re-established. However, after a critical interval ‘reperfusion’ is impossible, probably as a result of swelling of capillary endothelial cells.
The sub-endocardial layers of the myocardium are at particular risk from ischaemia. Even though there is a well-developed sub-endocardial plexus of blood vessels, flow in this part of the myocardium is restricted to diastole. Blood vessels are collapsible tubes and are susceptible to compression when tension within the myocardial wall increases. This tension is greatest when the ventricles are dilated, especially in the sub-endocardial layer.
Atherosclerosis accounts for the vast majority of coronary artery disease and is most marked in the proximal (epicardial) parts of the coronary arteries. The intramural branches may show slight intimal thickening, but are generally free of true atherosclerosis. Ischaemia is produced by:
Some cases of ischaemic heart disease result from atherosclerotic narrowing of the openings (ostia) of coronary arteries. In the past this was usually the result of syphilis, but is now usually atherosclerotic in origin. Occasionally emboli lodge in coronary arteries, usually as a result of infective endocarditis or calcific disease of the aortic valve. Other coronary artery diseases are extremely rare.
Ischaemic heart disease can also result from low coronary arterial perfusion. Shock, especially as a result of haemorrhage, is a frequent cause of this. Severe aortic valve disease, either stenosis or incompetence, can also impair coronary blood flow. Some patients with severe anaemia can develop symptoms of ischaemic heart disease.
Acute coronary syndromes




A myocardial infarct is an area of necrosis of heart muscle resulting from a sudden, absolute or relative reduction in the coronary blood supply (Fig. 13.23). The commonest precipitating cause is thrombosis superimposed on, or haemorrhage within, an atheromatous plaque in an epicardial coronary artery.
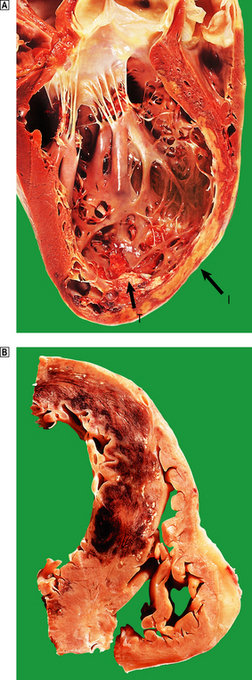
Fig. 13.23 Myocardial infarction.  Note the pale and focally haemorrhagic appearance of the infarcted muscle (I). There is adherent mural thrombus (T).
Note the pale and focally haemorrhagic appearance of the infarcted muscle (I). There is adherent mural thrombus (T).  This patient died 3 days after an acute anteroseptal infarct. Note the extensive haemorrhage into the infarct.
This patient died 3 days after an acute anteroseptal infarct. Note the extensive haemorrhage into the infarct.
Clinical features
The most frequent symptom of acute myocardial infarction is severe chest pain. This often develops suddenly but may build up gradually, and generally lasts for several hours. Pain is usually accompanied by profuse sweating, nausea and vomiting. Many patients give a previous history of angina or of non-specific chest pain in the weeks before the acute event. In at least 10% of patients, myocardial infarction is painless or ‘silent’; this is particularly true in the elderly.
Patients who are admitted with acute chest pain have a variety of underlying pathological changes. These include chronic coronary artery narrowing, rupture or erosion of coronary plaques, acute coronary thrombosis and acute myocardial infarction. The ECG gives a good guide as to which coronary artery is narrowed and the extent of myocardial damage. Acute infarcts are now classified according to the presence or absence of ST-segment elevation. Infarcts with ST elevation on the initial ECG (STEMIs) require emergency treatment to reopen the obstructed coronary artery. This may be achieved with drugs that activate the plasminogen system. Increasingly these patients are treated with emergency angioplasty to reopen the obstructed artery physically. Patients with chest pain but no evidence of ST-segment elevation require emergency estimation of troponin, a cardiac muscle protein that is released into the circulation after cardiac muscle cell death. Significantly increased levels indicate a so-called non-STEMI infarct. In these patients it is thought that the infarct is limited largely to the subendocardial zone of the myocardium. However, detailed clinicopathological studies linking the precise pattern of infarction with ECG changes are difficult to complete in the era of declining autopsy rates. Patients with severe chest pain but no ECG or biochemical evidence of infarction are said to have unstable angina. The underlying pathological change is thought to be rupture or erosion of a plaque of coronary artery atheroma.
Morphology
The location and size of the infarct depend on:
In clinical practice the ECG changes give a good guide to the area of myocardium that is infarcted (Fig. 13.24).
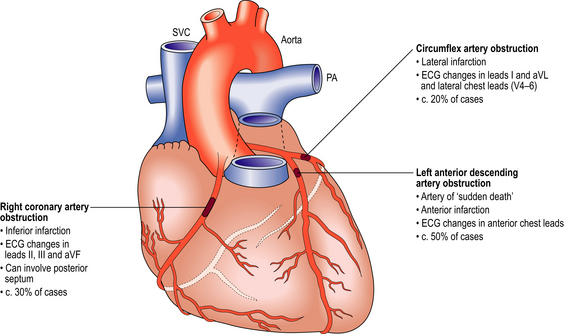
Fig. 13.24 Myocardial infarction. Obstruction of each major coronary artery results in infarction of specific areas of the myocardium.
When coronary angiography is performed in patients with typical symptoms and signs of acute myocardial infarction, a complete obstruction of a major coronary artery can be demonstrated in up to 90% of cases. The immediate objectives of treatment are to relieve pain with opiate analgesia and to restore blood flow in the occluded artery. Activation of the plasminogen system with recombinant forms of human plasminogen activator will partially lyse thrombi and improve blood flow in the occluded artery in at least 50% of patients. Emergency coronary angioplasty is also effective at breaking down thrombi and restoring blood flow. It is used as a primary treatment for coronary thrombosis in most centres worldwide.
The macroscopic and microscopic changes of myocardial infarcts follow a predictable sequence (Table 13.5). The chief features are necrosis, inflammatory cell infiltration and, as cardiac muscle cannot regenerate, repair by fibrous tissue. The extensive necrosis of cardiac muscle is associated with the release of cardiac enzymes and proteins into the circulation. A rapid bedside test for the diagnosis of acute infarction would allow more rational use of thrombolytic drugs or emergency angioplasty. Assays for the blood level of the cardiac muscle protein troponin are the most reliable early biochemical indicator of acute myocardial infarction but may not be increased for some hours after the onset of pain. Raised serum levels of creatine kinase also suggest acute myocardial infarction. Neither of these markers is entirely specific for myocardial infarction. Most patients show a transient leukocytosis in the first 1–3 days, but the value rarely exceeds 15 × 109/l. In clinical practice this is of limited value, largely because of the many other causes of a transient leucocytosis.
Table 13.5 Macroscopic and microscopic features of myocardial infarcts
| Time after onset of clinical symptoms | Macroscopic changes | Microscopic changes |
|---|---|---|
| Up to 18 hours | None | None |
| 24–48 hours | Pale oedematous muscle | Oedema, acute inflammaory cell infiltration, necrosis of myocytes |
| 3–4 days | Yellow rubbery centre with haemorrhagic border | Obvious necrosis and inflammation; early granulation tissue |
| 1–3 weeks | Infarcted area paler and thinner than unaffected ventricle | Granulation tissue, then progressive fibrosis |
| 3–6 weeks | Silvery scar becoming tough and white | Dense fibrosis |
Complications
The complications of myocardial infarction are listed in Table 13.6, and see Figures 13.25 and 13.26.
Table 13.6 Complications of myocardial infarcts
| Complication | Interval | Mechanism |
|---|---|---|
| Sudden death | Usually within hours | Often ventricular fibrillation |
| Arrhythmias | First few days | |
| Persistent pain | 12 hours–few days | Progressive myocardial necrosis (extension of infarct) |
| Angina | Immediate or delayed (weeks) | Ischaemia of non-infarcted cardiac muscle |
| Cardiac failure | Variable | Ventricular dysfunction following muscle necrosis Arrhythmias |
| Mitral incompetence | First few days | Papillary muscle dysfunction, necrosis or rupture |
| Pericarditis | 2–4 days | Transmural infarct with inflammation of pericardium |
| Cardiac rupture (ventricular wall, septum or papillary muscle) | 3–5 days | Weakening of wall following muscle necrosis and acute inflammation |
| Mural thrombosis | 1 week or more | Abnormal endothelial surface following infarction |
| Ventricular aneurysm | 4 weeks or more | Stretching of newly formed collagenous scar tissue |
| Dressler’s syndrome (chest pain, fever, effusions) | Weeks–few months | Autoimmune |
| Pulmonary emboli | 1 week or more | Deep venous thrombosis in lower limbs |
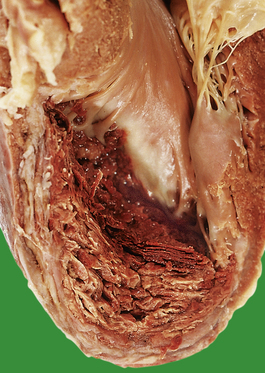
Fig. 13.25 Mural thrombus. Many layers of thrombus have formed on the infarcted myocardium. This can fragment and embolise.
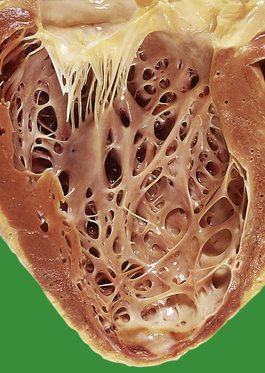
Fig. 13.26 Healed myocardial infarction. Note that the ventricular wall is very thin at the apex of the heart where there is a white fibrous scar.
Early detection and prompt treatment of complications are important in the management of patients with myocardial infarction. Cardiac arrhythmias, sometimes leading to ventricular fibrillation and sudden death, are frequent in the first 24–48 hours after the initial infarct. Cardiac rupture produces electromechanical dissociation (pulseless electrical activity) and rapid death (Fig. 13.27). The incidence of this important complication has been reduced by thrombolytic therapy. Pericarditis, mitral incompetence and cardiac failure are the important complications in the first week after infarction. Later complications include embolism from mural thrombus formation, and the development of ventricular aneurysms.
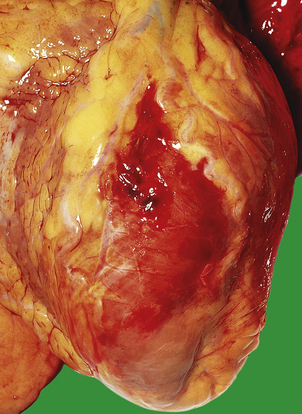
Fig. 13.27 Sudden death following rupture of an acute anterior myocardial infarct. There was a large haemopericardium and the patient died from cardiac tamponade.
As with all patients who are immobilised, there is a substantial risk of deep venous thrombosis and the possibility of subsequent pulmonary embolism. Prolonged bed rest is not essential for cases of uncomplicated myocardial infarction, and early mobilisation has done much to reduce the incidence of post-infarction pulmonary embolism.
Chronic ischaemic heart disease
Clinical features
Angina is one of the commonest clinical features of patients with a long history of ischaemic heart disease. A history of chest pain, induced by exercise and relieved by rest, should be sought in any patient in whom ischaemic heart disease is suspected. Impaired left ventricular function, following one or more previous episodes of myocardial infarction, may result in left ventricular and, ultimately, congestive cardiac failure. Some patients present with severe or rapidly progressive anginal chest pain. This is termed unstable or crescendo angina. It may progress to myocardial infarction and requires emergency management.
Morphology
Most patients with a definite clinical history of angina have extensive coronary arterial atheroma. Typically, two or three of the major coronary arteries have patches of stenosis in which the lumen is reduced to less than 75% of its normal cross-sectional area (Fig. 13.28). Careful postmortem studies in patients who have died after episodes of unstable angina have demonstrated recent rupture of the fibrous cap of large, often eccentric and lipid-rich atheromatous plaques. Paradoxically some patients with a typical clinical history of angina have relatively normal coronary angiograms. It may be that the recurrent episodes of coronary spasm are responsible for pain in these patients; this is sometimes called ‘variant angina’.
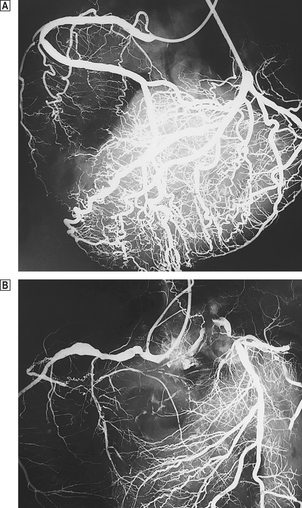
Fig. 13.28 Postmortem coronary angiograms.  Normal; note the widely patent right coronary artery, upper left.
Normal; note the widely patent right coronary artery, upper left.  The contours of the right coronary are irregular and there is a complete obstruction of the circumflex branch. Coronary angiograms made in life do not demonstrate all the small vessels shown above.
The contours of the right coronary are irregular and there is a complete obstruction of the circumflex branch. Coronary angiograms made in life do not demonstrate all the small vessels shown above.
Postmortem examinations on patients with a long history of ischaemic heart disease frequently demonstrate areas of healed myocardial infarction, dilatation of the left ventricle, and other changes related to chronic heart failure such as peripheral oedema, pleural and peritoneal effusions, and pulmonary oedema and congestion.
SUDDEN CARDIAC DEATH
The sudden, unexpected death of a previously fit person is an all too common tragedy in the community. General practitioners, junior hospital doctors and the police are commonly involved, and a medicolegal autopsy may be ordered. In the majority of cases, the cause is directly or indirectly related to the cardiovascular system. Epidemiological studies have demonstrated that there are at least 80 sudden unexpected deaths per 100 000 patients per year in the UK.
Aetiology
Acute cardiac failure as a result of ischaemic heart disease is one of the commonest diagnoses made by pathologists in cases of sudden unexpected death. In many cases, significant narrowing of one or more coronary arteries is identified. When detailed radiological and histological studies are made in patients dying within 6 hours of the onset of ischaemic symptoms, a coronary thrombosis can be found in approximately 55% of cases. At this stage, however, there will be no associated macroscopic or histological evidence of recent myocardial infarction. In the remainder of cases due to ischaemic heart disease there is severe narrowing of one or more coronary arteries, with or without evidence of previous healed myocardial infarction. It is assumed that these patients have died from a ventricular arrhythmia. Pathologists rely on the police evidence that there are no suspicious circumstances. About 50% of patients with established cardiac failure die suddenly, while the remainder deteriorate gradually. Sudden death is also a feature of patients with all forms of cardiomyopathy.
Other common causes of sudden death include ruptured atherosclerotic aneurysms of the abdominal aorta, dissecting aortic aneurysms and pulmonary emboli. Aortic stenosis is a cause of Stokes–Adams (‘drop’) attacks and can also lead to sudden death; acute coronary insufficiency is the probable mechanism (Fig. 13.29). Rupture of a berry aneurysm in the circle of Willis or massive intracerebral haemorrhage from a capillary micro-aneurysm may lead rapidly to death. Despite careful postmortem examination and toxicology, the cause of sudden unexpected death is sometimes not determined. This is increasingly recognised and is sometimes termed sudden adult (or arrhythmic) death syndrome (SADS). In a proportion of these cases biochemical abnormalities in sodium or potassium channels in cardiac muscle have been demonstrated, some of which are due to heritable mutations. It is therefore essential that relatives of sudden cardiac death victims are screened for cardiac abnormalities.
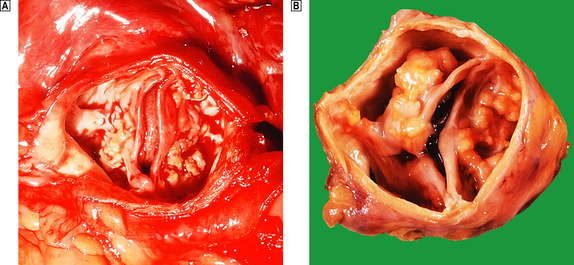
 This 58-year-old male died suddenly. Note that the valve is bicuspid and nodular due to heavy calcification.
This 58-year-old male died suddenly. Note that the valve is bicuspid and nodular due to heavy calcification.  This is the more typical pattern of aortic stenosis in a tricuspid aortic valve. Again there are heavy nodular areas of calcification.
This is the more typical pattern of aortic stenosis in a tricuspid aortic valve. Again there are heavy nodular areas of calcification.Prevention
Ventricular fibrillation is often the immediate cause of death in patients with acute ischaemic heart disease. Many ambulance crews now carry a defibrillator and administer DC shock to appropriate patients en route to hospital. Defibrillators are also installed in public areas such as football grounds and railway stations and are carried in some aircraft. There is good evidence from community studies that prompt cardiopulmonary resuscitation can prevent death in such circumstances. Patients with a history of ventricular arrhythmias can be treated with an implantable defibrillator that senses ventricular fibrillation and automatically delivers a defibrillating shock. Guidelines for the insertion of these devices have been developed and implantation rates are increasing. In some cases cardiac electrophysiologists can ‘map’ the pattern of electrical activity within the ventricle and, using a radiofrequency electrode, ablate the segment of myocardium that is responsible for abnormal electrical impulses.
Pulmonary embolism causes many tragic deaths, sometimes in previously fit patients in the post-operative period. Early mobilisation helps to minimise the risk of deep venous thrombosis. Anticoagulant therapy reduces the incidence of venous thrombosis and subsequent embolism, and patients at high risk of developing venous thrombi are sometimes treated prophylactically.
VALVULAR HEART DISEASE
At least 10% of cases of heart failure are caused by disease of the cardiac valves. The normal function of cardiac valves is to prevent retrograde flow of blood between the atria and ventricles, and between the ventricles and the aorta or pulmonary artery. Valves open noiselessly but heart sounds are produced by the vibration of blood as valves close. The first heart sound is the result of the closure of the mitral valve and, to a lesser extent, the tricuspid valve, early in systole. In the same way, the second heart sound results from aortic and pulmonary valve closure. In many healthy children and adults, the aortic valve closes shortly before the pulmonary valve, leading to a double or ‘split’ second sound. In life the movement of valve leaflets can be studied by echocardiography. Additional heart sounds occur during the filling of an abnormal ventricle. The third heart sound occurs in early diastole because of vibrations produced when blood impacts against an abnormal ventricular wall, for example an immobile area of fibrosis due to ischaemic heart disease. A fourth heart sound is heard later in diastole, just before the first heart sound, and is the result of late diastolic filling of the ventricle in a failing heart.
Pathological problems result from:
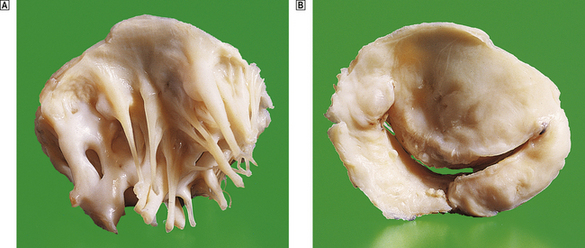
Fig. 13.30 Rheumatic valvular disease. This patient had longstanding mitral stenosis, and a successful mitral valve replacement was performed.  Thickening of chordae tendineae.
Thickening of chordae tendineae.  The atrial aspect shows how narrow the mitral orifice was.
The atrial aspect shows how narrow the mitral orifice was.
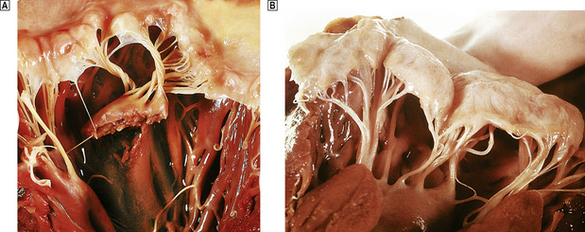
Fig. 13.31 Mitral incompetence.  Acute mitral incompetence. The mitral valve has become acutely incompetent due to infarction and rupture of the capillary muscle.
Acute mitral incompetence. The mitral valve has become acutely incompetent due to infarction and rupture of the capillary muscle.  Mitral valve prolapse (mucoid degeneration of the mitral valve). Note the marked billowing of the cusps in this postmortem specimen.
Mitral valve prolapse (mucoid degeneration of the mitral valve). Note the marked billowing of the cusps in this postmortem specimen.
Clinicopathological features
The most important clinicopathological features of mitral and aortic valve lesions are summarised in Table 13.7. The main pathological causes of diseases in these valves are:
Table 13.7 Pathological causes and clinical features of mitral and aortic valvular lesions
| Valvular lesion | Pathological cause | Clinical features |
|---|---|---|
| Mitral stenosis | Rheumatic fever |
Mitral incompetence
Aortic stenosis
Aortic incompetence
Mitral incompetence
Mitral incompetence is one of the commonest valvular lesions and is sometimes referred to as mitral insufficiency or regurgitation. The commonest cause of mitral incompetence is ischaemic heart disease. Fibrous scarring impairs the normal mobility of the papillary muscle, which leads to tethering of the valve leaflets. Ventricular dilatation may be associated with dilatation of the mitral valve annulus and this in turn worsens mitral incompetence (Fig. 13.32).
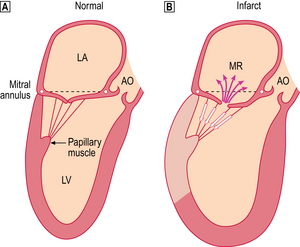
Fig. 13.32 Ischaemic mitral regurgitation. This is now the commonest and most important form of mitral regurgitation (MR).  The papillary muscle complex controls the normal closure of the mitral valve during ventricular systole.
The papillary muscle complex controls the normal closure of the mitral valve during ventricular systole.  A dilated ischaemic left ventricle (LV) with an area of fibrous scarring following previous myocardial infarction (grey colour). The movement of the papillary muscle is impaired and this effectively tethers the mitral valve leaflets away from the mitral valve ring (shown as arrows on the chordae tendinae). In addition, dilatation of the left ventricle increases the diameter of the mitral valve. (AO, aorta; LA, left atrium.)
A dilated ischaemic left ventricle (LV) with an area of fibrous scarring following previous myocardial infarction (grey colour). The movement of the papillary muscle is impaired and this effectively tethers the mitral valve leaflets away from the mitral valve ring (shown as arrows on the chordae tendinae). In addition, dilatation of the left ventricle increases the diameter of the mitral valve. (AO, aorta; LA, left atrium.)
Left atrial pressure is considerably lower than that of the aorta, so blood regurgitates through the mitral valve immediately after the start of ventricular contraction. By the time the aortic valve has opened, as much as a quarter of the stroke volume may already have entered the left atrium. This causes a murmur that begins immediately after the first heart sound and may last throughout systole (‘pansystolic murmur’). Acute mitral incompetence is usually the result of papillary muscle rupture in myocardial infarction (Fig. 13.31A). Most patients go into cardiogenic shock and will die within 48 hours. Surgical replacement of the valve can be life-saving, but the associated mortality is inevitably high.
Mucoid degeneration of the mitral valve is a common finding at postmortem and is seen in at least 15% of patients over the age of 70. The valves have a floppy or billowed appearance (Fig. 13.31B) and prolapse towards the left atrium during ventricular contraction; this is easily seen on echocardiography. Classic clinical signs are a mid-systolic click and a late systolic murmur. In severe disease abnormal stresses on the mitral valve predispose to rupture of chordae tendineae. Excellent results can be obtained by excising segments of the abnormal valve or, if the process is extensive, by mitral valve replacement.
Mitral stenosis
The primary abnormality in mitral stenosis is mechanical obstruction to emptying of the left atrium. The normal cross-sectional area of the mitral valve annulus is about 5 cm2, and signs and symptoms of mitral stenosis result when this is reduced to 1 cm2 or less. In the vast majority of cases mitral stenosis is a long-term result of rheumatic fever. Although this is now a rare disease in the West, a small number of new cases of mitral stenosis present each year. However, in many parts of Africa and the Middle and Far East rheumatic fever is common and there are many cases of rheumatic valve disease. Typically, rheumatic fever develops 2–3 weeks after a streptococcal upper respiratory tract infection. Recurrent attacks are typical, usually in children between 5 and 15 years of age. Although an arthritis and a skin rash (erythema marginatum) are often the presenting signs, involvement of the heart is the most important feature. There may be inflammation in all layers with pericarditis, myocarditis and endocarditis (‘pancarditis’). Heart murmurs are usually heard and heart failure may develop.
Little is known about the exact pathogenesis of rheumatic fever. Bacterial cultures of the heart, joints and other tissues are sterile, but there is serological evidence of a streptococcal infection. Antibodies to streptococcal polysaccharides are substantially elevated (antistreptolysin O titre, ASOT). The chief pathological features are oedema and fibrinoid necrosis of collagen, small aggregates of lymphocytes and macrophages (Aschoff bodies) and fibrosis.
It is even more difficult to explain why some patients go on to develop rheumatic valvular disease. Although rheumatic fever is more common in boys, it is young women who typically present with mitral stenosis, sometimes precipitated by the haemodynamic changes of pregnancy. Pathologically there is scarring of the valve cusps and shortening and thickening of chordae tendineae. Cardiac surgical centres in the East have vast experience of rheumatic valve disease and are especially skilled in its treatment.
Mitral stenosis causes poor emptying of the left atrium, increased pulmonary venous pressure, pulmonary hypertension and right ventricular hypertrophy, dilatation and failure. Atrial fibrillation often complicates mitral stenosis due to rheumatic valvulitis. Other causes of atrial fibrillation include ischaemic heart disease, thyrotoxicosis, hypertension, alcohol abuse and cardiac surgery. Ineffective atrial contraction leads to stasis and thrombus formation within the atrial appendages; these are a potential source of systemic thrombo-emboli.
Aortic stenosis
Calcific aortic valve disease is increasing in importance as the incidence of rheumatic heart disease declines, at least in Western countries. Severe calcific disease produces rigid cusps and results in aortic stenosis. This causes progressive and substantial left ventricular hypertrophy. Coronary blood flow may become inadequate, particularly if there is associated coronary atheroma. Most elderly patients with calcific aortic valve disease have pure aortic stenosis, whereas in rheumatic heart disease there is sometimes aortic incompetence and the mitral valve is also usually involved. The pathological processes responsible for calcification of the aortic valve, largely a disorder of the elderly, are unknown. Approximately 1% of the population have a bicuspid, rather than a tricuspid, aortic valve; these valves are particularly liable to calcification, sometimes at a relatively young age (Fig. 13.29). Unsuspected aortic stenosis is a frequent postmortem finding. The lesion can be present for many years and produce few, if any, clinical symptoms. The major features of aortic stenosis are syncope (abrupt episodes of faintness), angina and left ventricular failure. The systolic murmur typical of aortic stenosis begins well after the first heart sound, and ends before the second. It reaches a peak of intensity in mid or late systole. Older patients should be carefully screened for aortic stenosis. Aortic valve replacement is a successful surgical procedure, even in the very elderly.
Aortic incompetence
In aortic incompetence, blood flows back from the aorta into the left ventricle, producing an increased end diastolic volume. This causes an increased stroke volume, systolic hypertension and a wide pulse pressure, producing the typical ‘collapsing’ or ‘water-hammer’ pulse. A diastolic murmur is characteristic, but systolic ejection murmurs can result from the large stroke volume, and mitral diastolic murmurs from impairment of normal mitral opening by the regurgitant aortic stream. Left ventricular failure is a feature of severe aortic incompetence. Mild aortic incompetence is sometimes detected in healthy subjects, some of whom have bicuspid valves, and in some rheumatological disorders (Ch. 25).
Tricuspid and pulmonary valve disease
Disorders of the aortic and mitral valves produce far more substantial symptoms than do disorders affecting the valves of the right side of the heart.
Many patients with cardiac failure develop tricuspid incompetence, but this may not produce clinical symptoms. The absence of an effective tricuspid valve alters the pattern of the jugular venous pulse during systole. The pulmonary valve is seldom affected by acquired disease, but pulmonary stenosis can occur as an isolated congenital lesion or as part of a complex of malformations such as Fallot’s tetralogy. Pulmonary stenosis can be treated surgically or by percutaneous dilatation with a balloon catheter.
Infective endocarditis
Infective endocarditis is a serious disease resulting from infection of a focal area of the endocardium. The incidence has recently been estimated as between 2 and 6 per 100 000 population per year, and if anything is increasing. The incidence increases with age, and males are affected twice as commonly as females. The median in-hospital mortality rate is 16% (range 11–26%).
A heart valve is usually involved, but the process may affect the mural endocardium of the atrium or ventricle, or a congenital defect such as a patent ductus arteriosus or coarctation of the aorta.
General risk factors for infective endocarditis include poor dental hygiene, systemic sepsis, diabetes mellitus, long-term haemodialysis, immunosuppression and recent surgery or non-surgical invasive procedures. In the past, rheumatic valvular disease was the usual underlying lesion but this has now been replaced by degenerative processes such as aortic valve calcification or mucoid degeneration of the mitral valve. Intravenous drug misuse, previous valve replacement and vascular procedures, such as pacemaker implantation, are now of increasing importance. Infective endocarditis acquired in hospital (noscomial infection) accounts for about 20% of cases. Intravenous and urinary catheters, and recent surgery are particular risk factors. The heart is structurally normal in up to 50% of these cases.
Aetiology
Many different organisms can cause endocarditis. Most of these originate from the normal flora of the body surfaces, liberated into the blood stream in a variety of different ways. In order to survive in the blood stream, they must be resistant to the killing action of antibody and complement. For this reason Gram-positive bacteria, which have a thick layer of rigid mucopeptide protecting the cell membrane, are the usual causes of endocarditis. The proportion of cases caused by staphylococci is increasing. These infections often progress rapidly and may be difficult to treat. Streptococci, especially Streptococcus viridans, form a major part of the normal microbial flora of the oropharynx. Dental procedures, including descaling of teeth and minor fillings, instrumentation of the upper respiratory tract or even aggressive chewing, release small showers of organisms into the blood stream. Various staphylococci and yeasts such as Candida are normally present on skin surfaces. These can be introduced into the blood stream by insertion of cannulae or simple venepuncture. Sometimes, they are directly implanted from the surgeon’s skin if a glove is punctured during an operation. Str. faecalis is normally present in the large intestine and can cause urinary tract infections. During cystoscopy or prostatectomy, organisms may be disseminated into the blood stream and initiate endocarditis (sometimes called enterococcal endocarditis).
Morphology
The characteristic lesion of infective endocarditis is the vegetation. This can vary in size from a small nodule to a large friable mass that can all but occlude the valve orifice (Fig. 13.33). Almost all vegetations occur on valve leaflets or chordae tendineae. Occasionally, congenital defects such as patent ductus arteriosus, coarctation or an arteriovenous fistula can be involved. Surprisingly, vegetations never occur on atheromatous plaques in the aorta; the marked surface irregularities of the latter should make these ideal sites for a vegetation to develop, and why this does not occur is a mystery. Experimental work suggests that vegetations form in areas where there is flow across a high pressure gradient, as in an incompetent valve.
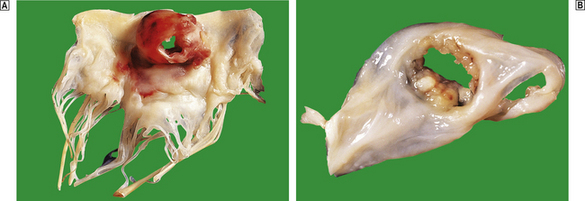
Fig. 13.33 Infective endocarditis.  A surgical resection specimen of a mitral valve. Note the large vegetation, which has perforated.
A surgical resection specimen of a mitral valve. Note the large vegetation, which has perforated.  Another surgical resection specimen. This aortic valve cusp has also perforated.
Another surgical resection specimen. This aortic valve cusp has also perforated.
In the course of septicaemia, virulent bacteria, such as Staphylococcus aureus, are thought to invade normal endocardial tissue. However, less virulent organisms, such as some streptococci, can infect the endocardium only at the sites of pre-existing damage. Binding to endothelial surfaces is facilitated by adhesive processes termed ‘pili’.
Important diseases that predispose to endocarditis include:
The probable sequence of events in the formation of a vegetation is shown in Figure 13.34.
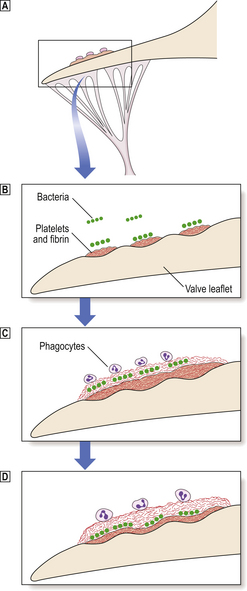
Fig. 13.34 Formation of vegetation on a valve leaflet in infective endocarditis.  A focal area of abnormality on the endocardium of a valve leaflet is covered with tiny deposits of platelets and fibrin; this is in effect the beginnings of a thrombus.
A focal area of abnormality on the endocardium of a valve leaflet is covered with tiny deposits of platelets and fibrin; this is in effect the beginnings of a thrombus.  Circulating micro-organisms (released into the blood stream under any of the circumstances described in the text) colonise the platelet thrombus.
Circulating micro-organisms (released into the blood stream under any of the circumstances described in the text) colonise the platelet thrombus.  and
and  When sufficient bacteria have settled, further blankets of platelets and fibrin are laid down. The bacteria proliferate slowly to form colonies occupying a relatively superficial position in the vegetation. They are separated from the blood stream by a thin layer of fibrinous material. This layer prevents the phagocytes reaching the bacteria, but is not a significant barrier to the diffusion of nutrients from the blood stream.
When sufficient bacteria have settled, further blankets of platelets and fibrin are laid down. The bacteria proliferate slowly to form colonies occupying a relatively superficial position in the vegetation. They are separated from the blood stream by a thin layer of fibrinous material. This layer prevents the phagocytes reaching the bacteria, but is not a significant barrier to the diffusion of nutrients from the blood stream.
Endocarditis in unusual hosts
Patients with prosthetic heart valves
Up to 2–3% of patients with artificial heart valves develop endocarditis (Fig. 13.35). Staphylococci account for at least 50% of cases. Many of these are coagulase-negative.
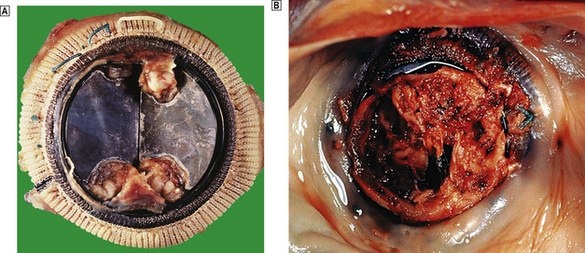
Fig. 13.35 Prosthetic valve endocarditis.  This patient had a mitral valve replacement 5 months previously but developed prosthetic valve endocarditis. Note the large vegetations forming at the margins of the tilting discs. Fortunately this patient recovered.
This patient had a mitral valve replacement 5 months previously but developed prosthetic valve endocarditis. Note the large vegetations forming at the margins of the tilting discs. Fortunately this patient recovered.  This patient underwent mitral valve replacement 7 months before death. Vegetations have virtually obscured the underlying prosthetic valve.
This patient underwent mitral valve replacement 7 months before death. Vegetations have virtually obscured the underlying prosthetic valve.
The commonest initial presenting symptom is post-operative fever. When residual wound sepsis and pulmonary or urinary tract infections have been excluded, prosthetic valve endocarditis should be seriously considered. Typically the process develops about 2 months after valve replacement. Echocardiography and repeated blood cultures are essential for diagnosis. Despite medical and surgical treatment, the mortality rate can be as high as 70%. In some patients, prosthetic valve endocarditis develops many months or years after the operation, and this possibility should always be considered in a pyrexial patient who has had a previous valve replacement.
The elderly
Endocarditis is increasing in incidence in elderly patients. Calcific valve disease is the most frequent pathology. Predisposing factors include genito-urinary infection, diabetes, tooth extraction, pressure sores and surgical procedures. It appears that virulent organisms such as Staph. aureus may be more frequent as the infecting organism in elderly patients. Complications also appear to be more common than in younger patients. Presenting signs and symptoms are often atypical because of other co-existing disease processes, such as respiratory tract infection and cardiac failure.
Drug addicts
Infective endocarditis in drug addicts has increased in all Western societies. The skin is the most common source of micro-organisms; most cases are due to Staph. aureus or Staph. epidermidis, and fungi such as Candida. The bacteraemia has various causes:
Very few intravenous drug abusers presenting with their first episode of endocarditis have previously damaged heart valves. Perhaps the repeated intravenous injection of ‘foreign’ material damages the endocardial surfaces, producing abnormal (roughened) areas. These become sites of platelet aggregation and therefore the development of vegetations. This may account for the high incidence of tricuspid valvular involvement in drug addicts, as this valve is closest to the injection site.
Vegetations in drug addicts are often large, particularly in fungal endocarditis. As infection primarily involves the right side of the heart, vegetations can embolise to the lungs. Endocarditis must be suspected in any drug addict presenting with signs or symptoms of pneumonia, pulmonary embolism or infarction.
Complications
The complications of endocarditis are summarised in Figure 13.36.
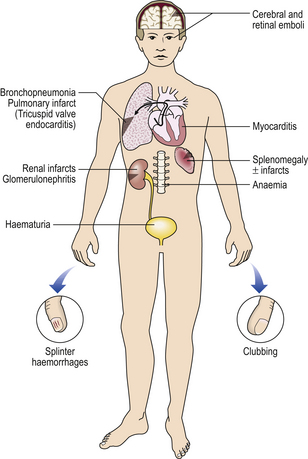
Fig. 13.36 The complications of infective endocarditis. The full range of complications are now rarely encountered in the Western world; for example, clubbing and splenomegaly may not be present in the early stages of the disease. (See text for detail.)
Local effects
All but the smallest vegetations have some effect on valvular function, and many cause valvular incompetence. A heart murmur is one of the most important physical signs of infective endocarditis. As the vegetations enlarge, valve cusps can perforate or chordae tendineae rupture. This is one cause of death in endocarditis and is now one of the indications for valvular replacement. Myocarditis is an important complication of endocarditis; the inflammation probably spreads directly from the valve leaflet to involve the annulus and adjacent myocardium. Vegetations can embolise to coronary arteries but this is extremely uncommon.
Systemic effects
Fever, weight loss, malaise and splenomegaly are the classical findings in infective endocarditis and can be attributed to the persistent bacteraemia. Parts of the vegetations may break away from the heart valves and lodge in many different sites; the spleen, kidney and brain are those most frequently involved. Small emboli produce tiny haemorrhagic lesions, essentially small infarcts, in the skin, mucous membranes and retina. Linear haemorrhages beneath the tips of the nails (splinter haemorrhages, Fig. 13.37A) are frequent in infective endocarditis, but equally can follow everyday trauma. Clubbing (Fig. 13.37B, C) is another clinical feature; its cause is unknown. It is also known as Schamroth’s sign after the physician who described the changes in his own fingers while suffering from endocarditis.
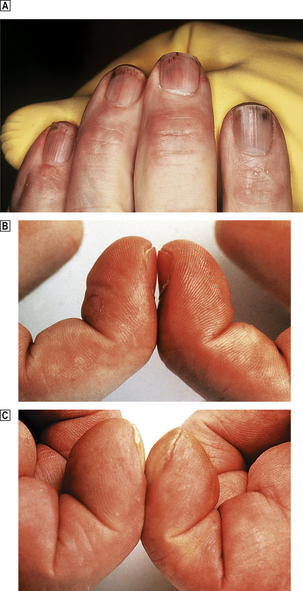
Fig. 13.37 Changes in the hands and nails in infective endocarditis.  Prominent splinter haemorrhages.
Prominent splinter haemorrhages.  Normal nail beds.
Normal nail beds.  Clubbing in infective endocarditis. This is sometimes known as Schamroth’s sign. There are of course many other causes of clubbing, including pulmonary carcinoma, intrathoracic infections and congenital heart disease.
Clubbing in infective endocarditis. This is sometimes known as Schamroth’s sign. There are of course many other causes of clubbing, including pulmonary carcinoma, intrathoracic infections and congenital heart disease.
A focal segmental glomerulonephritis can be seen in infective endocarditis. This is almost certainly the result of immune complex deposition in glomeruli (Ch. 21). The antigen is probably derived from the micro-organism and the antibody produced by the host in response to it. Some other manifestations of infective endocarditis, such as Osler’s nodes in the fingers, may be the result of an immune complex arteritis in the soft tissues.
Diagnosis, treatment and prevention
Investigations
Once a diagnosis of infective endocarditis has been considered, two lines of investigation are essential. The heart valves must be imaged by trans-thoracic or trans-oesophageal echocardiography, and the organism must be cultured from the blood stream. Delay in either of these may have serious consequences and their importance cannot be overemphasised. Echocardiography is valuable in identifying vegetations at initial presentation, for following how their size changes with treatment and for selecting cases for surgical treatment (Fig. 13.38).
Fig. 13.38 Echocardiographic appearance of valve vegetations. In this case the vegetations are on the tricuspid valve (arrowed). The patient was an intravenous drug abuser.
It is important to isolate the causative organism from the blood stream. This not only establishes the diagnosis but indicates which antibiotic or combination of drugs is needed to destroy the infecting organism. In taking blood cultures, it is essential to prevent skin and airborne bacteria contaminating the blood sample. Particular care should be taken to sterilise the skin overlying the vein, using a strong antiseptic such as chlorhexidine in 70% ethanol. Up to one-quarter of blood cultures grow skin bacteria, and this can lead to erroneous diagnoses and inappropriate treatment. Release of bacteria from vegetations is probably episodic and the numbers released may be small. Multiple blood cultures should be taken each day, perhaps for two or even three days. In practice, patients are often so ill that treatment must be started as soon as the diagnosis is suspected clinically, and certainly before the results of blood culture are available. In around 5% of patients with good clinical evidence of endocarditis blood cultures are negative. Failure to recover the causative organism can be due to:
Treatment
Ideally patients should be treated in a cardiac unit with ready access to cardiac surgery. Without adequate antibiotic treatment endocarditis is uniformly fatal. The avascular structure of the vegetation prevents the invasion of large numbers of phagocytes, and because of this it is essential to sterilise the heart valve with antibiotics. The antibiotics chosen must kill the bacteria, not just inhibit their growth. Cardiologists and microbiologists work closely together to select the most appropriate combination and dosages of antibiotics. Serial echocardiography is used to assess the change in size of vegetations. Surgical replacement of valves is increasingly used, especially if vegetations do not reduce in size with antibiotic treatment.
Prevention
Any patient with valvular heart disease is at risk of developing endocarditis as a result of bacteraemia associated with even the most minor surgical or dental procedure. It is therefore essential that the blood contains a high concentration of bactericidal antibiotics immediately before and during these procedures. The aim is to kill bacteria in the blood stream before they settle on the heart valve. Endocarditis still has an appreciable mortality, and the importance of these prophylactic measures cannot be overemphasised. Tragically, cases of endocarditis still occur in previously fit individuals not given appropriate antibiotic cover during minor procedures.
Non-infective endocarditis
Small thrombotic vegetations can occur on the closure lines of valve cusps in debilitated patients, especially those with cancer. These are called marantic vegetations. Thrombotic vegetations develop in some cases of systemic lupus erythematosus (Libman–Sacks endocarditis). In both conditions the thrombotic material can fragment and embolise.
PERICARDITIS AND MYOCARDITIS
Pericarditis
In pericarditis, there is an inflammatory reaction involving the visceral and/or parietal pericardial layers. There are many causes (Table 13.8) but the commonest are acute, non-specific (viral) pericarditis, myocardial infarction and uraemia.
Table 13.8 Clinical causes and pathological forms of pericarditis
| Clinical causes | Pathology |
|---|---|
| Acute non-specific or acute viral pericarditis | Acute fibrinous pericarditis |
| Myocardial infarction | Initially acute fibrinous, and later fibrous, pericardial adhesions |
| Uraemia | Acute fibrinous reaction |
| Carcinomatous pericarditis |
Connective tissue disease (e.g. rheumatic fever or rheumatoid arthritis)Fibrinous pericarditisBacterial pericarditisAcute purulent or fibrinopurulent reactionTuberculosisFibrous or calcific pericarditis, sometimes causing constrictive pericarditisPost-cardiac surgeryAcute fibrinous reactionPost-myocardial infarction (Dressler’s syndrome)Autoimmune infarction
Acute pericarditis
In acute pericarditis there is invariably a fibrinous exudate on the pericardial surfaces, with associated acute inflammation (Fig. 13.39). In many cases, there is an exudate of serous fluid (pericardial effusion) and this may become haemorrhagic. Common viral causes include coxsackievirus A and B, herpes simplex and influenza. Bacterial pericarditis results either from direct spread from an intrathoracic focus or from a blood stream infection.
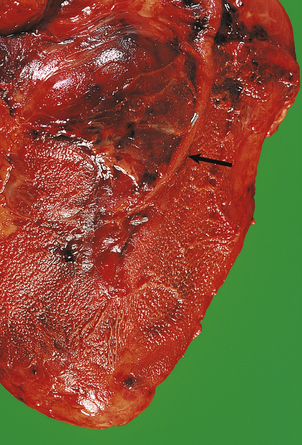
Fig. 13.39 Acute pericarditis. Acute fibrinous pericarditis. Note the granular masses of fibrin on the visceral pericardial surface. Common causes of pericarditis include acute myocardial infarction, uraemia, viral infections and recent cardiac surgery. This patient had a cardiac bypass procedure 7 days before his death. The vein graft is arrowed.
Chronic pericarditis
Chronic pericarditis may have no obvious cause but is a feature of connective tissue diseases such as rheumatoid arthritis, and of tuberculosis. In many cases an effusion develops and there is marked fibrous thickening of the pericardial layers. Many patients with a previous history of myocardial infarction have areas of old pericardial fibrosis, the result of healing of previous acute pericarditis.
Clinicopathological features
Typically, patients with pericarditis complain of chest pain, which may be either sharp or dull and aching. Young patients who present with acute non-specific or viral pericarditis often have severe chest pain which can be confused with acute myocardial infarction. Severe pain is uncommon in other forms of pericarditis. A pericardial friction rub is a characteristic feature of acute fibrinous pericarditis, but it may be transient and variable in its intensity. Pericardial effusions of less than 50 ml are usually undetectable clinically. With large effusions, the area of cardiac dullness to percussion is increased and the heart sound may be diminished. Echocardiography is essential for correct diagnosis. Large effusions may interfere with diastolic filling of the heart and produce cardiac tamponade. The jugular venous pressure is raised, and there is an exaggerated variation in pulse pressure during inspiration and expiration (pulsus paradoxus). Characteristic pressure tracings may be obtained during cardiac catheterisation. Constrictive pericarditis may seriously impair cardiac function, and surgical excision of the pericardium may improve cardiac output. In many cases there is associated calcification of the pericardium, which may be seen on chest X-rays, particularly lateral views.
Myocarditis
Pathogenesis
The chief causes of inflammation of the myocardium are:
Coxsackie B virus is the commonest known infectious cause of pericarditis and myocarditis in western Europe and North America. The diagnosis is suggested by rising titres of specific antibodies in the serum. Many other viruses have been implicated. Diphtheria has not been eradicated from developing countries and cardiac failure is a frequent cause of death; an exotoxin inhibits protein synthesis in cardiac muscle. Myocarditis can complicate infective endocarditis and in some cases myocardial abscesses form around valve rings.
Clinicopathological features
In most patients myocarditis is a self-limiting condition with only mild pleuritic chest pain. Fatalities are relatively uncommon (Fig. 13.40). In some patients cardiac failure develops and coronary angiography may be performed to exclude coronary artery disease. An endomyocardial biopsy may be performed at the same time and may show lymphocytic infiltration and myocyte necrosis. However, even in the most typical clinical cases the proportion of positive biopsies is small. Viral or molecular studies may identify an underlying causative agent. Anti-inflammatory drugs may be used in severe myocarditis but there is no definite evidence that they are effective.
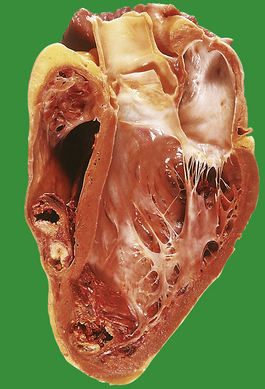
CONGENITAL CARDIOVASCULAR DISEASE




Aetiology
Congenital cardiovascular disease is the result of a structural or functional abnormality of the cardiovascular system at birth. In the vast majority of cases, the structural defects can be attributed to a specific disturbance of normal embryological development.
The incidence of congenital heart disease (CHD) is around 8 per 1000 live births and is much higher if bicuspid aortic valves are included. In about one-third of cases critical illness develops early in life. Associated extracardiac abnormalities occur in about a quarter of infants with CHD. In Down’s syndrome, for example, there is a high incidence of atrial or ventricular septal defect, or a patent ductus arteriosus.
In at least 80% of cases the cause of congenital heart disease is unknown. Environmental factors, such as maternal viral infections (especially rubella), chronic maternal alcohol abuse and drugs such as thalidomide, are all clearly related to CHD. These factors are of greatest importance between the fourth and ninth weeks after conception. During this period, the common atrial and ventricular chambers are divided by septa, the cardiac valves develop and the primitive truncus arteriosus divides into the aorta and pulmonary artery. The incidence of CHD is somewhat increased in the children of mothers with insulin-dependent diabetes or phenylketonuria.
There is a weak but definite family incidence of congenital cardiovascular disorders and the genetic basis of these defects is under investigation. Some congenital heart defects are associated with extracardiac abnormalities and in a small number of these specific chromosomal abnormalities have been detected. The risk of a congenital heart lesion in the siblings of affected individuals varies with the nature of the defect, for example from 2% for coarctation of the aorta to over 4% for ventricular septal defects. When two or more members of a family are affected, the risk appears to be substantially higher and, in these instances, clinical geneticists may be able to provide advice to parents.
Clinicopathological features
Some of the most prominent clinical and pathological features of CHD are:
Most children under 1 year of age who present with cardiac failure have a structural abnormality of the cardiovascular system. The severity of cardiac failure and the presence or absence of additional signs, such as cyanosis, depend on the precise structural abnormalities. Echocardiography and cardiac catheterisation now permit a detailed understanding of disordered anatomy before cardiac surgery.
Individual cardiac disorders
Atrial septal defects (ASD)
Between the fourth and seventh weeks of fetal life two distinct flaps of tissue develop to divide the common cavity into the left and right atria. The first, the septum primum, has two defects but these are normally covered when the second partition, the septum secundum, grows upwards from the atrioventricular ring. The higher of these defects, the ostium secundum, is covered by a flap of the septum secundum. In fetal life this acts as a flap valve, allowing blood entering the right atrium from the systemic veins to bypass the lungs by flowing into the left atrium. When the pulmonary circulation is established, it closes, and in most cases the two layers of the flap fuse together. In many children and some adults a probe can be passed between the layers, the so-called ‘probe patent’ foramen ovale. A defect in this area is the usual form of atrial septal defect (ASD). Less common types of ASD are related to defects low in the interatrial septum, close to the atrioventricular ring. There may be associated abnormalities in the mitral valve, and surgical repair is more complex. Other uncommon types of ASD are associated with defects in the development of the pulmonary veins or the coronary sinus.
Atrial septal defects make up approximately 10% of all congenital abnormalities of the heart. Although they are often asymptomatic, in the past many untreated patients developed signs of right heart failure in the third and fourth decades. A diastolic rumbling murmur, due to increased flow across the tricuspid valve, may be heard and, because of delayed closure of the pulmonary valve, there is often wide splitting of the second heart sound. The right ventricle is compliant and easily dilates to accommodate the increased pulmonary blood flow, but right ventricular hypertrophy and pulmonary hypertension inevitably develop. Closure is therefore indicated.
Ventricular septal defects
Ventricular septal defects account for approximately 25% of all cases of congenital heart disease in infancy. A variety of anatomical forms are recognised and their size and position can be estimated by echocardiography. Small defects in the muscular wall may close spontaneously as the heart grows. Surgical closure is indicated for larger muscular defects and those involving the membranous (fibrous) portion of the septum, close to the atrioventricular ring (Fig. 13.41). As the left ventricular pressure is substantially greater than that in the right, there is always some shunting of blood through the defect. The size and site of the ventricular defect determine the extent of this shunt. In some cases, defects in the membranous septum are also associated with valvular abnormalities, particularly aortic incompetence, and this influences the clinical presentation.
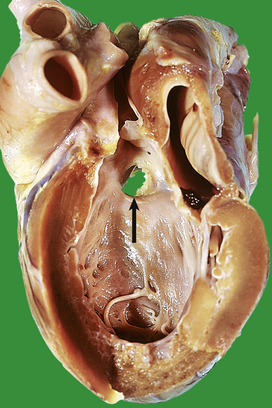
Fig. 13.41 Ventricular septal defect. This child was 4 years old at the time of his death. He had marked left ventricular hypertrophy. This is the heart of a child who died in Africa in the 1970s. Ideally lesions such as these should be treated surgically within the first year of life.
The most prominent physical sign of ventricular septal defect is a loud pansystolic murmur, often with an associated thrill. The most important complication is cardiac failure but there is also a risk of infective endocarditis.
Patent ductus arteriosus
In fetal life, the pulmonary vascular resistance is high and the right heart pressure exceeds that of the left. Consequently there is a flow from the right to the left atrium through the foramen ovale, and from the pulmonary artery to the aorta via the ductus arteriosus. At birth, the pulmonary vascular resistance declines dramatically, and the ductus arteriosus closes within the first few days of life. If the ductus remains open (patent), there is an abnormal shunt of blood from the aorta to the pulmonary artery. This increases both pulmonary arterial and left heart blood flow, but the right atrium and ventricle are virtually unaffected (Fig. 13.42).
Fig. 13.42 Circulation in patent ductus arteriosus. The duct should normally close very soon after birth. (LCC, left common carotid artery; LSC, left subclavian artery; PA, pulmonary artery.)
As in ventricular septal defects the symptoms are proportional to the size of the left-to-right shunt. A continuous ‘machinery’ murmur is characteristic, and is loudest at the time of the second heart sound. If the shunt is large, a left ventricular impulse (‘heave’) is usually present. If a patent ductus does not close spontaneously, surgical treatment is indicated. In all forms of aortic surgery pre-operative echocardiographic and angiographic investigations are used to define the precise anatomy of the aortic arch and its branches.
Coarctation of the aorta
A congenital localised constriction in the diameter of the aorta is known as a ‘coarctation’. This defect accounts for up to 5% of all forms of congenital cardiovascular disease and is substantially more common in males. In the usual form of coarctation the narrowing occurs just distal to the ductus arteriosus, which is usually closed. In a proportion of cases there are associated aortic valve abnormalities, usually a congenitally bicuspid valve.
The signs and symptoms are largely dependent on the degree of constriction (Figs 13.43 and 13.44). If this is severe, symptoms develop soon after birth. If the coarctation is undetected or untreated, a collateral circulation develops to increase blood flow to the lower part of the body. This process involves branches of the intercostal arteries, which become dilated and tortuous. In time, the enlarged vessels may erode portions of the rib, producing ‘notching’ on chest X-ray. The most characteristic clinical finding is hypertension in the upper limbs, with a much lower pressure in vessels distal to the coarctation. The intensity of the femoral pulse is often much reduced. The abnormal blood flow through the coarcted segment may produce a systolic murmur, best heard in the posterior chest.
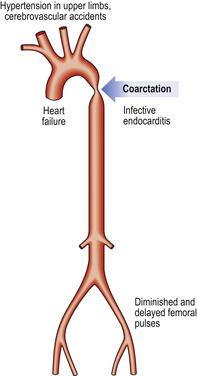
Fig. 13.43 Coarctation of the aorta: clinical features. Echocardiography now detects most cases early in life. The diagnosis should be considered in any child who fails to thrive.
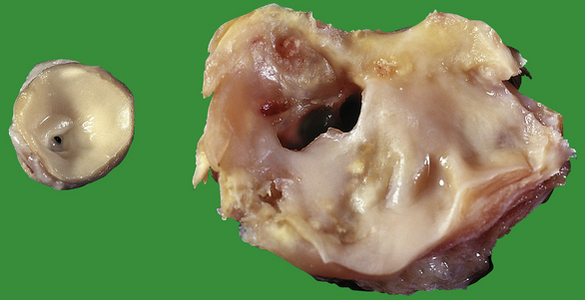
Fig. 13.44 Coarctation of the aorta. Left: A section of an aorta from a 3-day-old child. Note that the lumen of the aorta is less than 2 mm in diameter. Right: A specimen from a 24-year-old male who was found to be hypertensive. The aortic lumen is approximately 5 mm in diameter. Note that atherosclerosis has formed proximal to the coarctation because of turbulent blood flow.
Some patients with coarctation are asymptomatic but most die prematurely, usually as a result of:
In view of these complications, and the shortened life-span of many patients, surgical treatment is usually indicated.
Complex congenital heart disease
These are rare disorders in which individual cardiac chambers are imperfectly developed or incorrectly connected. An exact diagnosis is made by defining the anatomical features using cardiac ultrasonography or other radiological techniques. These disorders usually present soon after birth and a wide range of corrective or palliative surgical techniques have been developed.
The commonest of the complex abnormalities is Fallot’s tetralogy, first described in Marseilles in the 19th century. The four components are:
The clinical features are often characteristic. As the aorta receives both oxygenated blood from the left ventricle and deoxygenated blood from the right, cyanosis develops. Pulmonary stenosis restricts blood flow from the right ventricle into the lungs and, if this is severe, survival is only possible if the ductus arteriosus remains open. The systolic murmurs result from either the ventricular septal defect, or, if severe, the pulmonary stenosis. As in all hypoxic patients, the haemoglobin concentration is increased. Right heart failure is inevitable and bacterial endocarditis can ensue. Dyspnoeic children with Fallot’s sometimes adopt a characteristic squatting posture, with both the knee and hip joint sharply bent, or sit in a ‘knee–chest’ position. This may be an attempt to increase venous return from the lower limbs or, more speculatively, to reduce peripheral arterial perfusion, thereby increasing the flow across the ductus arteriosus or ventricular septal defect to the right side of the circulation. Before the advent of surgical treatment (Fig. 13.45), most patients died well before adult life.
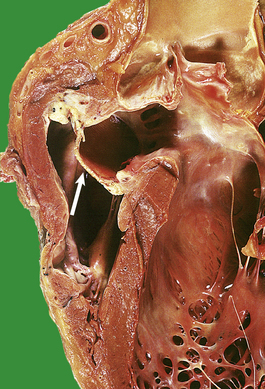
Fig. 13.45 Fallot’s tetralogy. The full features in a classical case are right ventricular hypertrophy, ventricular septal defect, pulmonary hypertension and an overriding aorta. This patient underwent surgical correction as a teenager when pulmonary hypertension was well established. Note the thicknesses of the right and left ventricles are almost equal. An arrow shows a patch that was used to close the ventricular septal defect. This is an old specimen. Nowadays lesions such as these would be corrected within the first year of life.
Another important complex abnormality is transposition of the great arteries. This presents early in life and requires prompt surgery. There are several different forms but in the most important the aorta drains the right ventricle and the pulmonary artery the left. This creates two closed circulations. Post-natal life is only possible if these mix via an atrial or ventricular septal defect or a patent ductus arteriosus. A complete surgical correction is often possible, usually by ‘switching’ the pulmonary artery and aorta at their origin from the heart and re-implanting the coronary arterial ostia. In the best surgical centres excellent results are now obtained, with mortality rates well below 10%.
Congenital valvular abnormalities
The only common congenital abnormality is a bicuspid aortic valve. The vast majority of these are asymptomatic, and the valve is neither incompetent nor stenotic. However, the risk of aortic stenosis in adult life is substantially increased (Fig. 13.29A) and there is a strong association with dissection of the aorta. Small defects (fenestrations) are frequently observed at autopsy, usually in aortic valves, but have no functional significance.
Occasional cases of congenital aortic or pulmonary stenosis do occur. Most tricuspid and mitral abnormalities are part of complex abnormalities rather than isolated lesions.
Coronary arterial abnormalities
There is considerable variation in the normal anatomy of the major coronary vessels and this is usually of no clinical importance. For example, the circumflex branch of the left coronary artery is sometimes small and there is a corresponding increase in the length and distribution of the terminal parts of the right artery. Similarly, the coronary arterial ostia are occasionally malpositioned, but these cause no obvious clinical effects. Many different anomalies have been described. Some are of no clinical importance, while others are associated with an increased risk of sudden death or cardiac disease. For example, one of the coronary arteries may arise from the pulmonary artery, and the myocardium is therefore perfused with deoxygenated blood.
UNUSUAL CARDIAC DISEASES
Most cases of cardiac failure can be attributed to ischaemic heart disease, hypertension, valvular disorders, congenital defects or lung disease. Only when these have been excluded are unusual causes considered. The term cardiomyopathy is often loosely applied to these disorders, but strictly speaking this term should be restricted to disorders of completely unknown cause or clinical association, i.e. idiopathic or primary cardiomyopathy. In practice, expressions such as alcoholic cardiomyopathy, amyloid cardiomyopathy and occasionally ischaemic cardiomyopathy are used to describe cardiac disease in patients with a known systemic disorder that affects the heart.
Unusual disorders of known cause or association
Multisystem diseases
Cardiac changes are often present in association with multisystem disease. In sarcoidosis and rheumatoid disease, for example, granulomatous lesions can develop in the heart, and, if they involve the conduction pathways, arrhythmias or heart block can develop. In some forms of amyloidosis (Ch. 7), the heart is involved. At autopsy, the cardiac muscle has a characteristic glassy brown appearance and, if deposits are extensive, cardiac failure develops. Massive cardiac hypertrophy is a feature of acromegaly, and cardiac failure is the usual cause of death in these patients.
Major cardiac abnormalities are well recognised in both thyrotoxicosis and myxoedema. In severe thyrotoxicosis, the increase in the metabolic rate necessitates an increased cardiac output and peripheral blood flow. Occasionally, this may in itself precipitate ‘high output’ cardiac failure. More frequently, thyrotoxicosis unmasks subclinical coronary or hypertensive heart disease. Atrial fibrillation is particularly common in elderly patients with thyrotoxicosis.
Most patients with myxoedema have an enlarged cardiac outline on chest X-ray. This may be due to left ventricular dilatation or pericardial effusion; these can be distinguished by echocardiography. Characteristically, there is a bradycardia, low voltage ECG and decreased cardiac output. There are usually no specific pathological findings either macroscopically or microscopically. The response to thyroid hormone therapy is often excellent, but angina, and even myocardial infarction, can be precipitated with anything but the smallest doses.
Alcoholism
Cardiac failure is not uncommon in chronic alcoholism. In some cases it can be attributed to common disorders such as coronary artery disease or hypertension. However, in a proportion of patients, no specific cause is determined and ‘alcoholic cardiomyopathy’ is diagnosed. In these patients the macroscopic and microscopic findings are identical to those of other forms of idiopathic cardiomyopathy, and there is some debate as to the exact role of heavy alcohol consumption. Alcohol abuse is associated with an increased risk of sudden death.
Pregnancy
Substantial circulatory changes occur in pregnancy, most notably an increase in circulating blood volume. Cardiac failure may become apparent for the first time during pregnancy, especially in patients with valvular disorders, such as mitral stenosis. Hypertension is one of the cardinal signs of pre-eclampsia but, while cardiac failure and pulmonary oedema can develop in the full syndrome, the disorder is not primarily cardiac in origin (Ch. 19). A characteristic form of cardiomyopathy (discussed below) occasionally develops in the post-partum period.
Iatrogenic disease
Iatrogenic (‘doctor-induced’) cardiac disease is now of some importance because of the increasing use of cytotoxic drugs and of radiotherapy in the treatment of mediastinal tumours. Radiotherapy causes patchy areas of interstitial fibrosis in the myocardium (probably as a result of direct damage to small capillaries) and pericarditis. Some degree of cardiac muscle cell necrosis is a frequent result of treatment with cytotoxic drugs, such as doxorubicin, that interfere with DNA and RNA replication and protein synthesis. This can produce acute cardiac toxicity but may also be associated with an increased long-term incidence of cardiac failure. An increased incidence of heart failure has recently been reported in patients receiving the erb-2 antagonist trastuzumab (Herceptin). It is therefore possible that this agent has a direct cardiotoxic effect.
Cardiomyopathies
A cardiomyopathy is a heart muscle disease of uncertain cause. Consequently the diagnosis of cardiomyopathy should be made only when all other causes of cardiac failure, such as hypertension, ischaemic heart disease, valvular and congenital heart disease, have been excluded. Most patients have either a ‘dilated’ or ‘hypertrophic’ pattern of disease. However, it is now clear that many cases of both forms of cardiomyopathy have a genetic basis. In the future it is likely that individual cases will be classified according to the nature of the underlying mutation in cardiac muscle.
Dilated (congestive) cardiomyopathy (DSCM)
The incidence of this disorder in Europe and North America is about 35 cases per 100 000 per year. The median age at presentation is about 50 years but young adults may be affected. Typically, the coronary arteries are free of significant atheroma but the ventricles are dilated and hypertrophied (Fig. 13.46). There may be adherent mural thrombi and histological evidence of interstitial fibrosis and hypertrophy of muscle fibres. There is clinical evidence of cardiac disease in the relatives of at least 20% of patients. Current studies are demonstrating an increasing range of mutations. The pattern of inheritance can be autosomal dominant or recessive, X-linked or mitochondrial. These mutations involve genes that code for both cytoskeletal and sarcomeric proteins. A variety of other genetic abnormalities have been detected in young patients with cardiomyopathy. Some relate to fatty acid oxidation, mitochondrial oxidative phosphorylation or the cardiac-specific expression of the dystrophin gene.
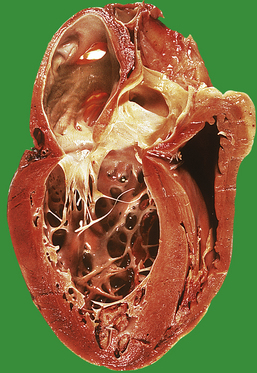
Fig. 13.46 Dilated (congestive) cardiomyopathy. There is marked hypertrophy of both the left and right ventricles. Note the adherent mural thrombus at the apex of the left ventricle. No underlying cause was determined in this patient.
Some cases of viral myocarditis appear to progress to dilated cardiomyopathy but there is no firm evidence of viral infection in the majority of cases. The pathological features of chronic alcoholic heart disease and dilated cardiomyopathy are similar. The outlook in dilated cardiomyopathy is poor and only 50–60% of patients survive 2 years. It is essential to investigate these patients in the hope of identifying a treatable disorder such as coronary artery or aortic valve disease. Young patients with dilated cardiomyopathy are often considered for cardiac transplantation.
Hypertrophic (obstructive) cardiomyopathy (HOCM)
This is a common autosomal dominant disorder that is thought to affect 1 in 500 of the population. The clinical, echocardiographic and pathological features in hypertrophic cardiomyopathy are often characteristic (Fig. 13.47). The chief feature is massive left ventricular hypertrophy, usually most marked in the interventricular septum close to the aortic outflow tract. In most cases the disease becomes apparent after the pubertal growth phase or in early adult life. Patients present with a variety of signs or symptoms but atrial fibrillation, ventricular arrhythmias and sudden death are the most important complications. Histological sections of the left ventricle show fibre hypertrophy, interstitial fibrosis and a characteristic disordered arrangement of muscle fibres termed disarray.
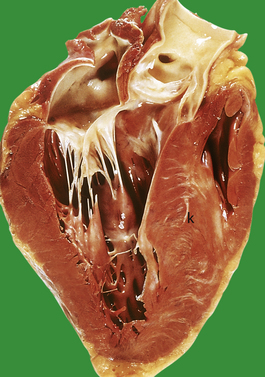
Fig. 13.47 Hypertrophic cardiomyopathy. The patient was a 24-year-old male who died suddenly. Note the marked and asymmetrical enlargement of the interventricular septum (asterisk).
Many cases are familial and chromosomal abnormalities can be identified in up to 50% of patients. Mutations have been described in many different cardiac muscle proteins, and in some the exact nature of the amino acid substitution influences the course of the disease. The products of the genes implicated in hypertrophic cardiomyopathy include the structural proteins troponin T and alpha-tropomyosin and cardiac myosin-binding proteins.
Arrhythmogenic right ventricular cardiomyopathy (ARVC)
Although less common than dilated or hypertrophic cardiomyopathy, this is an important disorder with a strong familial tendency. The exact incidence of this disorder is not known, but after hypertrophic cardiomyopathy it is the commonest cause of unexpected cardiac death in a previously fit young person. The characteristic change is progressive loss of right ventricular myocytes with associated fibrosis, inflammation and adipose tissue replacement. In some cases there is clear evidence of septal or left ventricular disease. A small number of gene mutations have been identified, including proteins involved in cell to cell adhesion, such as desmoplakin. Some patients have unusually curled or woolly hair, and peripheral hyperkeratosis.
In the future it is likely that many forms of cardiomyopathy will be classified and treated on the basis of the exact underlying genetic abnormality.
Other cardiomyopathies
Many other forms of cardiomyopathy have been described, usually in specific clinical settings.
Puerperal cardiomyopathy occurs in the last months of pregnancy, or within 6 months of delivery. There is no history of pre-existing cardiac disease and the clinical outcome is variable. In some cases cardiac failure resolves completely, although recurrence in subsequent pregnancies is likely.
In restrictive cardiomyopathy there is restrictive filling and decreased diastolic volume of one or both ventricles. This is caused by myocardial or endocardial disease that stiffens the heart by infiltration or fibrosis. In some cases there is no obvious cause and the disease is termed primary restrictive cardiomyopathy.
Endomyocardial fibrosis is a curious form of myocardial disease found in the tropics, chiefly in Uganda and the Sudan. The cause is unknown, but there is marked fibrosis of the inner parts of the myocardium, and mural thrombi are common. In some patients with severe cardiac failure, there is a prominent persistent peripheral blood eosinophilia and evidence of multiple systemic emboli (Löffler’s endocarditis). This occurs in both tropical Africa and, sporadically, in the West and is usually fatal. The cause is uncertain.
The most important cause of secondary restrictive cardiomyopathy is infiltration of the myocardium by amyloidosis, of either light chain (AL) or transthyretin-related (ATTR) type. Familial forms of cardiac amyloidosis are caused by mutated TTR.
Newly described forms of cardiac disease
Studies of young patients presenting with cardiac arrhythmias or sudden death have defined a number of important cardiac disorders that have little or no associated cardiac pathology. As discussed in the section on sudden cardiac death (p. 297), a significant number of sudden deaths in young patients are unexplained. Electrophysiological and genetic studies have shown that a proportion of these cases have mutations in myocardial sodium or potassium channels or receptor molecules. These disorders are now termed channelopathies, and some have characteristic alterations in the QT interval. In some patients arrhythmias may be provoked by sudden noise or during swimming. It is likely that many cases of drowning in young subjects were the result of sudden fatal cardiac arrhythmias rather than accidental deaths.
The Brugada syndrome is another potentially fatal disorder with characteristic ECG changes and a normal cardiac structure. Families of these patients require careful assessment in a specialised centre and often contact patient support organisations.
TUMOURS OF THE HEART AND PERICARDIUM
Primary tumours of the heart and pericardium are extremely rare; only a few cases are seen annually in each regional cardiothoracic centre in the UK. The myxoma is the most frequent primary tumour (25%) and usually arises from the endocardium as a polypoid or pedunculated tumour mass (Fig. 13.48). Three-quarters of myxomas occur in the left atrium, and in almost one-half of all cases there are signs and symptoms of mitral valve disease. The tumours are often friable and can fragment and embolise. Myxomas can be present at almost any age, but are most common in adults. The tumours have a characteristic histological appearance and probably arise from undifferentiated connective tissue cells in the sub-endocardial layers of the heart wall. Other primary tumours include lipomas, which are usually found in the interatrial septum. Rhabdomyomas arise from cardiac muscle and are often multiple. Many occur in newborn infants and cause stillbirth, or death within the first days of life. Primary malignant tumours of the myocardium include angiosarcoma and other sarcomas such as leiomyosarcomas.
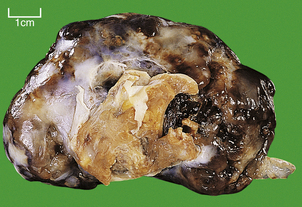
Fig. 13.48 Atrial myxoma. This tumour measured more than 80 mm in diameter and obstructed the mitral valve orifice. The brown tissue in the centre is its point of attachment to the left atrium.
Inevitably, the heart and pericardium are often involved by local extension of primary intrathoracic tumours. Bronchial carcinoma is by far the commonest cause of this and can lead to clinically important pericardial effusions. Malignant mesothelioma, a pleural tumour associated with asbestos exposure, often infiltrates the pericardium. Pericardial effusions may be the result of secondary tumour deposits and require aspiration or drainage. Lung and breast are the commonest primary sites.
Commonly confused conditions and entities relating to cardiovascular pathology
| Commonly confused | Distinction and explanation |
|---|---|
| Atherosclerosis and arteriosclerosis | Atherosclerosis implies hardening (sclerosis) or loss of elasticity of arteries due specifically to atheroma. Arteriosclerosis is hardening or loss of elasticity of arteries from any cause. |
| Angiitis, arteritis and vasculitis | Angiitis is inflammation of any vessel (even a lymphatic, as in lymphangitis). Arteritis is inflammation of an artery or arteriole. Vasculitis is inflammation of any blood vessel (arterial, capillary or venous). |
| Cardiomyopathy and myocarditis | Cardiomyopathy should strictly be reserved for any myocardial disorder of unknown aetiology (once the aetiology is known, the entity is given a more specific name). Myocarditis is inflammation of the myocardium. Both can result in cardiac failure. |
| Phlebothrombosis and thrombophlebitis | Phlebothrombosis is thrombosis in a vein (Greek: phlebos). Thrombophlebitis is an inflammatory reaction to phlebothrombosis. |
| Rheumatic fever and rheumatoid disease | Rheumatic fever is an immunologically mediated post-streptococcal illness affecting the heart and joints. Rheumatoid disease is an autoimmune disorder causing arthritis, completely unrelated to rheumatic fever. |
Adrogué H.J., Madias N.E.. Sodium and potassium in the pathogenesis of hypertension. New England Journal of Medicine. 2007;356:1966-1978.
Andreotti F., Marchese N.. Women and coronary disease. Heart. 2008;94:108-116.
Braunwald E.. Biomarkers in heart failure. New England Journal of Medicine. 2008;358:2148-2159.
Erridge C.. The roles of pathogen-associated molecular patterns in atherosclerosis. Trends in Cardiovascular Medicine. 2008;18:52-56.
Hansson G.K.. Inflammation, atherosclerosis and coronary heart disease. New England Journal of Medicine. 2005;352:1685-1695.
Hughes S.E., McKenna W.J.. New insights into the pathology of inherited cardiomyopathy. Heart. 2005;91:257-264.
Prendergast B.D.. The changing face of infective endocarditis. Heart. 2006;92:879-885.
Roden D.M.. Long QT syndrome. New England Journal of Medicine. 2008;358:167-176.
Shinebourne E.A., Babu-Narayan S.V., Caravalho J.S.. Tetralogy of Fallot; from fetus to adult. Heart. 2006;92:1353-1359.
Van der Wal A.C.. Coronary artery pathology. Heart. 2007;93:1484-1489.
Zipes D.P., Libby P., Bonow R.O., Braunwald E., editors. Braunwald’s heart disease. A textbook of cardiovascular medicine, 7th edn, Philadelphia: Saunders, 2005.




