[level-membership-for-pathology-category]
Chapter 17 Endocrine system
COMMON CLINICAL PROBLEMS FROM ENDOCRINE DISEASE

Pathological basis of endocrine signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Signs or symptoms of hormone excess (hyperfunction) |
Signs or symptoms of hormone deficiency (hypofunction)
Diffuse enlargement of gland
Nodular enlargement of glandTumour (benign or malignant)Some organ-specific features
Pituitary tumour
Increased thyroid hormone secretion due to hyperplasia or neoplasia of gland
Autoimmune involvement of retrobulbar connective tissue in Graves’ disease
Adrenocortical hyperplasia or neoplasia
Absolute or relative deficiency of insulin (diabetes mellitus)
NORMAL STRUCTURE AND FUNCTION
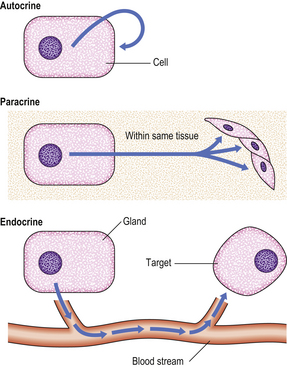
An endocrine gland secretes hormones directly into the blood stream to reach distant ‘target organs’ where the secretory products exert their effects. Endocrine glands are thus distinguished from exocrine glands, whose secretions pass into the gut or respiratory tract, or on to the exterior of the body; examples of exocrine glands include the exocrine pancreas and the bronchial mucous glands. Closely related to the endocrine system is the paracrine (diffuse endocrine) system, consisting of regional distributions of specialised cells producing locally acting hormones, such as those regulating gut motility, and forming part of the neuroendocrine system (Ch. 15); autocrine effects are those acting on the cell producing the hormone (Fig. 17.1).
Hormones exert their effects on the target organs by binding to receptors, protein molecules with high and specific affinity for the hormone. These hormone receptors may be either on the cell surface (for example, thyroid-stimulating hormone receptors on the thyroid epithelium) or intracellular (for example, nuclear receptors for steroid hormones). The binding of a hormone to its cell surface receptor sets off a series of intracellular signals via secondary ‘messenger’ molecules (cyclic nucleotides), which results in changes in metabolic activity, differentiation or mitosis of the stimulated cell.
ENDOCRINE PATHOLOGY
The major disorders of an endocrine gland are:
There are several important general considerations in endocrine pathology. First, disease of one endocrine gland cannot usually be considered in isolation, because it almost always has implications for other endocrine glands:
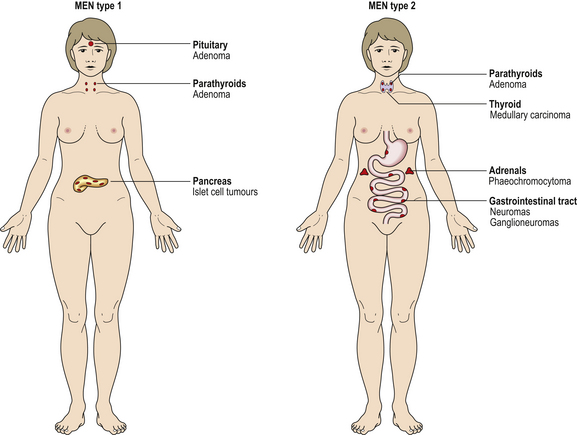
Fig. 17.2 Multiple endocrine neoplasia (MEN) syndromes. MEN syndromes are characterised by the occurrence of tumours in more than one endocrine organ. MEN types 1 and 2 can be distinguished by the organs commonly involved.
Second, one hormone may have many diverse clinical effects, so that malfunction of one endocrine gland may produce numerous clinical features.
Third, the same hormone may be produced in more than one site; thus, ectopic hormone production by tumours of non-endocrine tissues may simulate primary endocrine disease.
PITUITARY
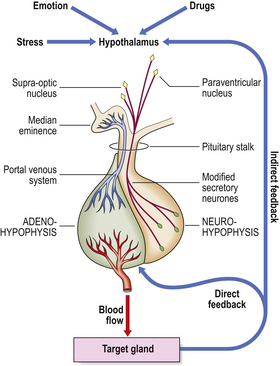
The pituitary is a small gland, weighing only 500–1000 mg. It is situated in the sella turcica of the skull beneath the hypothalamus. Despite its small size, it exerts many essential control functions over the rest of the endocrine system, earning it the title ‘conductor of the endocrine orchestra’. It consists of two parts (Fig. 17.3), each with separate functions. The anterior pituitary, the adenohypophysis, is developed from Rathke’s pouch, an outpouching of the roof of the embryonic oral cavity; it comprises about 75% of the bulk of the gland. The posterior pituitary, the neurohypophysis, is derived from a downgrowth of the hypothalamus.
ADENOHYPOPHYSIS
Classification of cell types
Modern histological classification of the types of hormone-secreting cell is based on immunohistochemistry, a technique in which antibodies raised to a hormone bind to the cells containing that hormone in tissue sections, leading to a coloured stain (Fig. 17.4). This has enabled the true hormone content of the cells to be determined, and has rendered obsolete the traditional classification of the cells into eosinophil, basophil and chromophobe types according to their staining by haematoxylin and eosin (H&E). By electron microscopy, the cells of the adenohypophysis are seen to contain electron-dense granules ranging from 50 to 500 nm in diameter (Fig. 17.5); these contain stored secretory products. The six types of hormone-secreting cell are shown in Table 17.1.
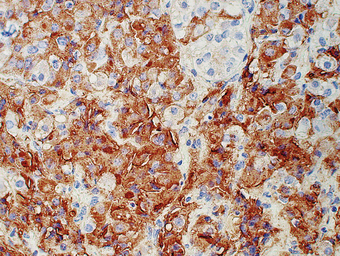
Fig. 17.4 Growth hormone-containing cells in an adenoma of the adenohypophysis. Immunoperoxidase localisation of growth hormone. Cells containing growth hormone are stained brown by this technique.
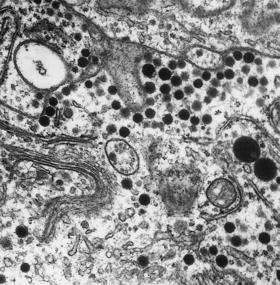
Fig. 17.5 Electron micrograph of a secretory cell of the adenohypophysis. The hormonal products are stored as electron-dense membrane-bound cytoplasmic granules (´ 300 000).
Table 17.1 Hormone-secreting cells of the adenohypophysis
| Cell type | Staining reactionwith H&E | Hormonal product |
|---|---|---|
| Corticotroph | Basophilic | Adenocorticotrophic hormone (ACTH) |
| Thyrotroph | Basophilic | Thyroid-stimulating hormone (TSH) |
| Gonadotroph | Basophilic |
SomatotrophEosinophilicGrowth hormone (GH)LactotrophEosinophilicProlactin (PL)ChromophobePaleUnknown
Control of hormone secretion
Hormonal control factors
The adenohypophysis lacks any direct arterial supply. Blood from the hypothalamus passes down venous portal channels in the pituitary stalk (Fig. 17.3) into sinusoids which ramify within the gland. In this way hormonal control factors produced by neurosecretory cells in the hypothalamus are carried directly to the hormone-producing cells of the adenohypophysis. The known hormonal control factors and their effects are listed in Table 17.2. In general, these factors stimulate the particular secretory cells under their control into activity; the exception is prolactin-inhibiting factor, whose effect on the lactotrophs is inhibitory.
Table 17.2 Hormonal control factors and their effects on the adenohypophysis
| Hormonal control factor | Effect |
|---|---|
| Corticotrophin-releasing factor (CRF) | Corticotrophs release ACTH |
| Thyrotrophin-releasing factor (TRF) | Thyrotrophs release TSH |
| Gonadotrophin-releasing factor (FSH/LH-RF) | Gonadotrophs release FSH/LH |
| Growth hormone-releasing factor (GHRF) | Somatotrophs release GH |
| Prolactin-inhibiting factor (PIF) | Lactotrophs inhibited from releasing PL |
Secretion of these hormonal control factors by the hypothalamus is under two types of control: neural and hormonal. Neural control is via nerves from other parts of the central nervous system, and is important in reactions to stress and in changes during sleep. Hormonal control is a negative feedback mechanism in which the hypothalamus monitors the level of adenohypophysial hormones in the blood and adjusts its output of hormonal control factors accordingly, so as to stabilise the level of each adenohypophysial hormone at the optimum level. This is called the hypothalamic–hypophysial feedback control.
Feedback control
In addition to control via the hypothalamus, a more direct method of control of the adenohypophysis also exists, whereby its cells respond directly to the levels of hormones and metabolites in the blood. Most adenohypophysial hormones stimulate another endocrine gland, termed the ‘target’ gland; for example, ACTH stimulates the adrenal cortex to produce steroid hormones, and TSH stimulates the thyroid to produce thyroxine.
In these examples, the level of hormone from the target gland is monitored for feedback control. However, in the case of growth hormone, which has no single target gland, it is the level of metabolites such as glucose that is monitored. A general scheme of the feedback control mechanisms operating in the regulation of a hypophysial hormone is shown in Figure 17.3.
Adenohypophysial hormones
Adrenocorticotrophic hormone
Adrenocorticotrophic hormone (ACTH), a peptide consisting of 39 amino acids, causes increased cell numbers (hyperplasia) and increased secretory activity in the adrenal cortex. Glucocorticoid output is elevated, but there is no effect on the output of mineralocorticoids, such as aldosterone, that are not under anterior pituitary control. ACTH levels show a marked circadian variation, being highest early in the morning.
Thyroid-stimulating hormone
Thyroid-stimulating hormone (TSH) is a glycoprotein that induces proliferation of the follicular cells of the thyroid, synthesis of thyroxine (T4) and tri-iodothyronine (T3), and secretion of these into the blood. Measurement of TSH levels provides information on the state of the control system of the thyroid and is valuable in the diagnosis of thyroid malfunction.
Gonadotrophic hormones
In the female, follicle-stimulating hormone (FSH) induces growth of Graafian follicles in the ovaries; these secrete oestrogens, which in turn cause endometrial proliferation. After rupture of the follicle at ovulation, luteinising hormone (LH) causes a change in the follicle cells known as luteinisation, whereby their secretory product changes from oestrogens to progesterone which induces secretory changes in the endometrium. Both gonadotrophic hormones are glycoproteins.
The hypothalamus monitors circulating levels of the sex steroids including oestrogens and progesterone, and releases probably a single hormonal control factor, FSH/LH-releasing factor (FSH/LH-RF), to control the adenohypophysial gonadotrophs. Their response to this factor depends on the prevailing levels of sex steroids. Cyclical changes in this feedback loop form the hormonal basis for the menstrual cycle.
In the male, FSH and LH both exist but, in the absence of ovaries as the target organ, their names are inappropriate to their actions. LH stimulates testosterone production by the interstitial cells of Leydig in the testes, while FSH stimulates spermatogenesis.
The circulating levels of FSH and LH vary markedly with age: they increase at puberty and are very high in females after the menopause.
Growth hormone
Growth hormone (GH) is a protein containing 191 amino acids; it binds to receptors on the surface of various cells and thus causes increased protein synthesis, accelerates breakdown of fatty tissue to produce energy, and tends to raise the blood glucose. It is vital for normal growth; deficiency causes dwarfism. Part of its action at tissue level is mediated by a group of peptide growth factors known as somatomedins. The hypothalamic control of GH release from the hypothalamus is complex, there being both a growth hormone-releasing factor (GH-RF) and an inhibitory factor, somatostatin.
Prolactin
Prolactin (PL) is a protein hormone with a structure very similar to that of GH. Although it is present in individuals of both sexes, its function in males remains uncertain. In females, it can produce lactation, provided that the breast has already been prepared during pregnancy by appropriate levels of sex steroids. Prolactin release is a good example of the neural form of hypothalamic control: the sensation of suckling causes reduction in hypothalamic prolactin-inhibiting factor (PIF) release and a consequent rise in PL levels.
Hypofunction



Like other endocrine organs, the adenohypophysis has considerable reserve capacity, and deficiency of its hormones becomes manifest only after extensive destruction; hypofunction is therefore uncommon. Since the pituitary is tightly encased within the sella turcica, any expansile lesion, such as an adenoma, produces compression damage to the adjacent pituitary tissue, in addition to any effect from its own hormonal production. Damage to the hypothalamus or pituitary stalk may also produce adenohypophysial hypofunction through failure of control. Table 17.3 sets out the main causes of hypofunction. These conditions lead to a deficiency of all adenohypophysial hormones, a state known as panhypopituitarism. This is a life-threatening condition, as deficiency of ACTH leads to atrophy of the adrenal cortex and failure of production of vital adrenocorticoids. Diagnosis of hypopituitarism is by measurement of the individual hormones. The commonest causes of pituitary hypofunction are compression by metastatic carcinoma or by an adenoma, but two specific rarer syndromes will be mentioned because they illustrate how congenital and acquired disease may affect the pituitary.
Table 17.3 Causes of adenohypophysial hypofunction
| Site | Lesions |
|---|---|
| Pituitary | Adenoma |
| Metastatic carcinoma | |
| Trauma | |
| Post-partum ischaemic necrosis (Sheehan’s syndrome) | |
| Craniopharyngioma | |
| Infections | |
| Granulomatous diseases | |
| Autoimmunity | |
| Iatrogenic | |
| Hypothalamus | Craniopharyngioma |
| Gliomas |
Pituitary dwarfism
Pituitary dwarfism is due to deficiency of GH, sometimes associated with deficiency of other adenohypophysial hormones. The child fails to grow, although remaining well proportioned. There is a variety of known causes including adenomas, craniopharyngiomas (rare tumours derived from remnants of Rathke’s pouch) and familial forms.
Post-partum ischaemic necrosis
During pregnancy, the pituitary enlarges and becomes highly vascular. Hypotensive shock due to haemorrhage at the time of birth, compounded by the lack of direct arterial supply to the adenohypophysis, may cause ischaemic necrosis. This specific cause of necrosis is known as Sheehan’s syndromeand the effects of the resulting adenohypophysial hypofunction are termed Simmond’s disease. The neurohypophysis is usually spared. The first symptom following delivery is failure of lactation due to PL deficiency; the effects of lack of FSH/LH, TSH and ACTH then follow—loss of sexual function, hypothyroidism, and the diverse effects of glucocorticoid deficiency. Improvements in obstetric management mean that Sheehan’s syndrome is now rare, although hypotensive shock due to trauma may produce similar effects.
Tumours: adenomas




Pituitary tumours account for approximately 10% of primary intracranial neoplasms. They may be derived from any of the hormone-secreting cells and thus may be clinically manifest by virtue of single hormone overproduction, destruction of surrounding normal pituitary and consequent hypofunction, and mechanical effects due to intracranial pressure rise and specific location.
Adenomas are the commonest adenohypophysial tumours; carcinomas are rare. Small adenomas may be asymptomatic and found only at postmortem. Histologically, adenomas consist of nodules containing cells similar to those of the normal adenohypophysis, with many small blood vessels between them. They may produce clinical disease in two ways: excess hormone production and pressure effects.
Excess hormone production
Adenomas may produce any adenohypophysial hormone, depending on their cell of origin (Table 17.4); thus presentation may be via excess production of one of the hormones, for example acromegaly due to excess growth hormone production in an adult (Fig. 17.6), or gigantism if this occurs during childhood.
Table 17.4 Types of adenohypophysial adenoma
| Type | Remarks |
|---|---|
| Prolactinoma (chromophobe) | Commonest type |
| Produces galactorrhoea and menstrual disturbances | |
| GH-secreting (eosinophil) | Produces gigantism in children and acromegaly in adults |
| ACTH-secreting (basophil) | Produces Cushing’s disease |
| Other | Exceptionally rare |
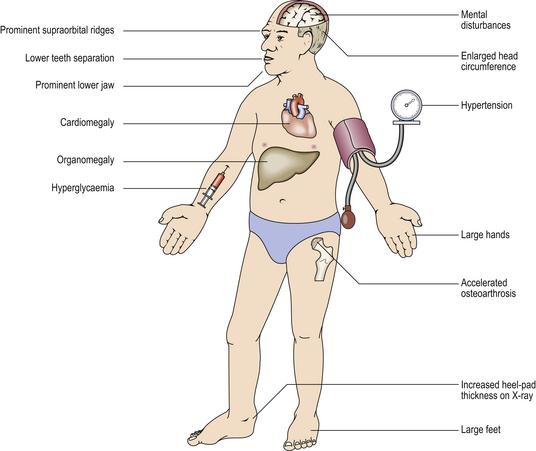
Fig. 17.6 Systemic features of acromegaly. Acromegaly is the clinical syndrome resulting from growth hormone excess in adult life. The chief presenting features are enlargement of the hands, feet and head, but it may also present with secondary diabetes. The cardiovascular effects may be life-threatening.
Pressure effects
These may be either on the surrounding pituitary to produce hypofunction, or on the overlying optic chiasma (Fig. 17.7), producing a characteristic visual field defect called bitemporal hemianopia. Further growth may compress the hypothalamus.
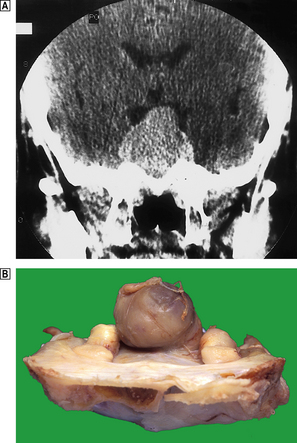
 Coronal plane CT scan of the pituitary fossa showing the sella turcica widened by a pituitary adenoma, which is compressing the optic chiasma and hypothalamus.
Coronal plane CT scan of the pituitary fossa showing the sella turcica widened by a pituitary adenoma, which is compressing the optic chiasma and hypothalamus. Pituitary adenoma revealed at autopsy, protruding above the sella turcica.
Pituitary adenoma revealed at autopsy, protruding above the sella turcica.Types of adenoma
All the following adenomas comprise, histologically, nests and cords of a monotonous single cell type, the islands of cells being supported on a richly vascular sinusoidal framework. Amyloid deposition is not infrequent and calcification may occur.
Chromophobe adenoma
The commonest tumour is one derived from apparently inactive cells; thus hormonal manifestations may be absent but more sensitive biochemical assessments suggest that prolactin may be produced by many of these adenomas. The clinical effects may therefore be limited to infertility and be discovered only because of failed conception in the female.
Eosinophil adenoma
Approximately one-third of lesions are derived from the growth hormone-producing cells and are thus manifest by gigantism in the pre-pubertal patient and acromegaly in the adult.
NEUROHYPOPHYSIS
Neurosecretory cells in the supra-optic and paraventricular nuclei of the hypothalamus give rise to modified nerve fibres which carry the two neurohypophysial hormones—antidiuretic hormone and oxytocin—into the posterior lobe of the pituitary (Fig. 17.3); both hormones are nonapeptides, and are stored until released in response to hypothalamic stimuli.
Antidiuretic hormone
Antidiuretic hormone (ADH) controls plasma osmolarity and body water content by increasing the permeability of the renal collecting ducts; this means that more water is reabsorbed and the urine becomes more concentrated. ADH release is stimulated by increased plasma osmolarity and by hypovolaemia.
Damage to the hypothalamus, for example through trauma or tumours, causes deficiency of ADH, leading to production of large volumes of dilute urine accompanied by compensatory polydipsia (excess drinking). This is called diabetes insipidus, from the days when tasting of the patient’s urine was part of the diagnostic process: the urine is tasteless in this condition, whereas in diabetes mellitus it is sweet due to its high glucose content.
Excess ADH is occasionally produced by the neurohypophysis in response to head injury or meningitis, but most clinical cases of ADH excess are due to its ectopic production by tumours, including bronchial carcinomas. The tumours are almost certainly of neuroendocrine origin and thus equipped for the synthesis of peptide hormones.
The rarity of any neurohypophysial tumour secreting ADH (or oxytocin) is perhaps due to the incapacity of the neurones producing these hormones to undergo mitotic division.
Oxytocin
Oxytocin is an aptly named hormone (it is the Greek word for quick birth) as it stimulates the uterine smooth muscle to contract. Interestingly, it is oxytocin from the fetal pituitary that plays the greater role in initiating parturition, suggesting that the fetus orders its own birth. Oxytocin also causes ejection of milk during lactation. The hormone is present in males, although its function, if any, is unknown.
PINEAL GLAND
The pineal gland is a tiny organ lying above the third ventricle of the brain. Little is known of its function, although its secretory product, melatonin, is thought to be involved in circadian rhythm control and gonadal maturation. The most important tumours of the pineal gland are malignant germ cell tumours (teratomas and seminomas) and pinealoblastomas, resembling neuroblastomas.
ADRENALS
The adrenals consist essentially of two separate endocrine glands within a single anatomical organ. The medulla, of neural crest embryological origin, is part of the sympathetic nervous system; it secretes catecholamines, which are essential in the physiological responses to stress, e.g. infection, shock or injury. The cortex, derived from mesoderm, synthesises a range of steroid hormones with generalised effects on metabolism, the immune system, and water and electrolyte balance.
ADRENAL MEDULLA
Histologically, the adrenal medulla consists of chromaffin cells (so called because they produce brown pigments when fixed in solutions of chrome salts) and sympathetic nerve endings. The adrenal medulla is the main source of adrenaline (epinephrine), as it is produced there from noradrenaline (norepinephrine) by the enzyme phenylethanolamine-N-methyl transferase. Elsewhere in the body, sympathetic nerve endings lack this enzyme and their secretory product is thus noradrenaline. Electron microscopy reveals electron-dense granules in the chromaffin cells (Fig. 17.8), similar to those found in other tissues of the so-called amine precursor uptake and decarboxylation (APUD) system. Islands of similar tissue, known as the organs of Zuckerkandl, are sometimes found in other retroperitoneal sites; these have similar functions and a similar pattern of diseases to that seen in the adrenal medulla. Catecholamines are secreted in states of stress and of hypovolaemic shock, when they are vital in the maintenance of blood pressure by causing vasoconstriction in the skin, gut and skeletal muscles. At tissue level, these hormones bind to cell surface receptors, altering cellular levels of a second messenger, cyclic AMP, which brings about rapid functional changes in the cell.

Fig. 17.8 Electron micrograph of noradrenaline granules in a chromaffin cell. The granules characteristically have eccentric electron-dense cores (× 75 000).
Tumours
Phaeochromocytoma





A phaeochromocytoma is derived from the adrenal medullary chromaffin cells (or from those lying in other sites); it is classified as a paraganglioma. The tumour presents through the effects of its catecholamine secretions: hypertension (which is sometimes intermittent), pallor, headaches, sweating and nervousness. Its presence should be suspected especially in younger hypertensive patients. Although it is a rare cause of hypertension, phaeochromocytoma must not be overlooked as it is one of the few curable causes of elevated blood pressure; other causes include adrenal cortical adenoma, renal artery stenosis and aortic coarctation.
The diagnosis of phaeochromocytoma is usually based on estimating the urinary excretion of vanillylmandelic acid (VMA), a catecholamine metabolite, which is generally at least doubled in the presence of the tumour. Localisation of the tumour is assisted by computed tomography of the abdomen and by radio-isotope scanning with 131I-mIBG, a catecholamine precursor that accumulates in the tumour.
Phaeochromocytoma may be familial, associated with medullary carcinoma of the thyroid or with hyperparathyroidism as part of a multiple endocrine neoplasia (MEN) syndrome. The familial cases are frequently bilateral. Other associations are with neurofibromatosis and the rare von Hippel–Lindau syndrome.
Phaeochromocytomas are brown, solid nodules, usually under 50 mm in diameter, often with areas of haemorrhagic necrosis (Fig. 17.9). Histologically, they consist of groups of polyhedral cells which give the chromaffin reaction, and are highly vascular (Fig. 17.10).
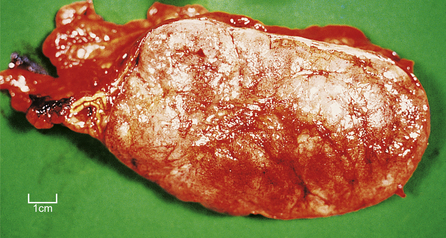
Fig. 17.9 Phaeochromocytoma. The adrenal medulla is expanded by a dark-coloured tumour with areas of degeneration and haemorrhage.
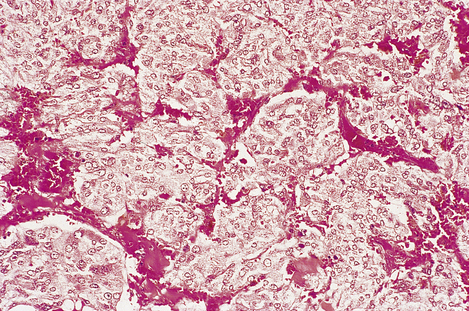
Fig. 17.10 Chromaffin cells in a phaeochromocytoma. There are groups of cells with granular cytoplasm, amidst which there are numerous branching capillaries.
Although most are benign, a few phaeochromocytomas pursue a malignant course. It is not generally possible to predict this behaviour from the histological appearance.
Neuroblastoma
Neuroblastoma is a rare and highly malignant tumour found in infants and children. Derived from sympathetic nerve cells it may, like phaeochromocytoma, secrete catecholamines, and there may be elevated levels of their metabolites in the urine. Neuroblastomas may also originate from parts of the sympathetic chain outside the adrenal medulla. Secondary spread to liver, skin and bones (especially those of the skull) is common. Surprisingly, neuroblastoma may occasionally mature spontaneously to ganglioneuroma, a benign tumour.
ADRENAL CORTEX
Histologically, the adrenal cortex has three zones (Fig. 17.11). Beneath the capsule lies the zona glomerulosa, so called because the cells are grouped into spherical clusters superficially resembling glomeruli. This zone produces mineralocorticoid steroids such as aldosterone. Most of the adrenal cortex comprises the middle and inner zones—zona fasciculata and zona reticularis, respectively. The middle zone is rich in lipid. The inner zone cells convert lipid into corticosteroids, principally glucocorticoids and sex steroids, for secretion.
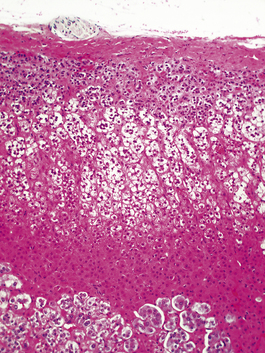
Fig. 17.11 Adrenal cortex. The normal zones are: zona glomerulosa (top), zona fasciculata (middle) and zona reticularis (bottom).
Steroid hormones
Glucocorticoids
The glucocorticoids have important effects on a wide range of tissues and organs. At physiological levels they:
In excess, as a result of therapeutic administration or high levels of endogenous secretion, they cause:
The most important of the hormones is cortisol (hydrocortisone), but other steroid metabolites have similar effects. The synthesis and secretion of glucocorticoids are controlled by ACTH from the pituitary.
Mineralocorticoids
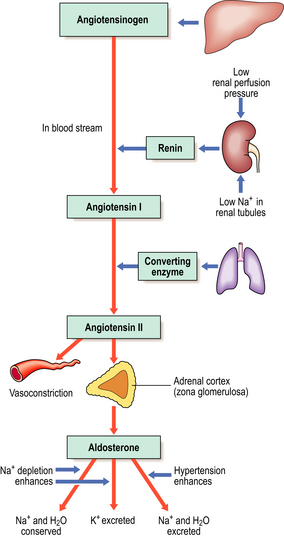
The most important of the mineralocorticoids, aldosterone, acts on the renal tubules to increase reabsorption of sodium and chloride, reducing their loss in urine at the expense of potassium exchange. Unlike the production of glucocorticoids, the synthesis and release of aldosterone is not under pituitary control, but is regulated instead by the renin–angiotensin system (Fig. 17.12). Low perfusion pressure in the kidney stimulates release of renin, an enzyme, from the juxtaglomerular apparatus of the kidney. This converts angiotensinogen into angiotensin I. Angiotensin I is then converted to angiotensin II (an octapeptide) by angiotensin converting enzyme, mainly in the lung. Angiotensin II stimulates secretion of aldosterone from the adrenal cortex. Thus, aldosterone is released to combat fluid depletion.
Sex steroids
The production of sex steroids in the adrenal cortex is low compared with that in the gonads and may not be physiologically important. However, virilising androgens may be produced in conditions such as certain congenital enzyme defects and adrenal cortical tumours, especially if these are malignant.
Hyperfunction
Hyperfunction of the adrenal cortex produces generalised effects, the nature of which depends on whether glucocorticoids, mineralocorticoids or sex steroids are produced in excess.
Cushing’s syndrome



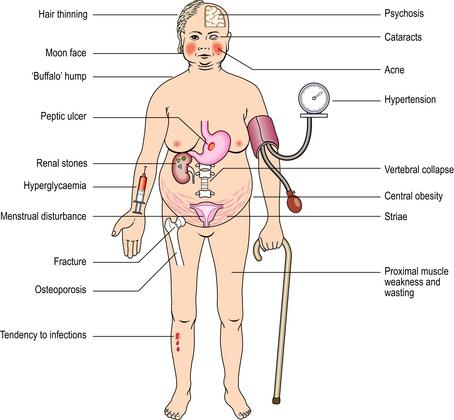
Cushing’s syndrome refers to the constellation of bodily responses to excess glucocorticoids, whatever the underlying cause. Exogenous administration of glucocorticoids or ACTH is a common iatrogenic cause of Cushing’s syndrome. The syndrome occurs most commonly in adult women, and sometimes there is also excess androgen production causing virilisation. The main physical features of the syndrome in an adult are shown in Figures 17.13 and 17.14. In children, there is also growth retardation.
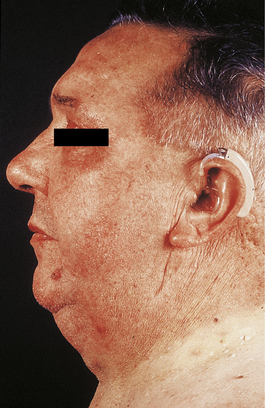
Fig. 17.13 Cushing’s syndrome. There is rounding of the face, acne and central obesity causing double chin.
Diagnosis
Diagnosis is by demonstration of glucocorticoid excess, either as elevated plasma levels of cortisol or as elevated urinary excretion of 17-hydroxysteroids, degradation products of glucocorticoids. Further tests, such as measurement of plasma ACTH levels, are essential to determine the cause of the Cushing’s syndrome (see below).
Pathogenesis
Iatrogenic disease
The therapeutic administration of glucocorticoids to the patient is by far the commonest cause of the features of Cushing’s syndrome.
In addition, three different types of natural disease can cause the syndrome:
Excess ACTH secretion by the adenohypophysis
This was the cause of the syndrome originally described by Harvey Williams Cushing, a Boston neurosurgeon with an interest in the pituitary. Hypersecretion of ACTH by an adenoma of the corticotrophs leads to bilateral adrenal cortical hyperplasia; this combination is termed Cushing’s disease. Histologically, the cells of the adrenal cortex may appear depleted of lipid, indicating that they have discharged their secretions into the blood. Plasma ACTH is raised, and if the dexamethasone suppression test is performed—administration of the synthetic potent steroid, dexamethasone—a fall in cortisol levels will result due to the suppression of pituitary ACTH secretion.
The ideal treatment of this common cause of Cushing’s syndrome is surgical removal of the pituitary adenoma; this not only abolishes the excess ACTH secretion, but also avoids the serious pressure effects that may be produced by a pituitary space-occupying lesion. Removal of the adrenals (once the main form of treatment) is unsatisfactory because the adenohypophysial tumour is left to grow and, in addition to secreting ACTH, may produce a peptide (melanocyte-stimulating hormone) with an amino acid sequence similar to that of the ACTH molecule. In 20% of cases this leads to marked enlargement of the pituitary adenoma (Nelson’s syndrome). Skin pigmentation will occur in most cases.
Adrenal cortical neoplasms
These may secrete cortisol autonomously, independently of ACTH control; low ACTH levels are then found in the presence of elevated cortisol. This is the commonest cause of Cushing’s syndrome in children. The neoplasm is usually an adenoma, but in 5–10% of cases it is a carcinoma, in which case virilising steroid production may be prominent. Treatment is by excision of the neoplasm.
Ectopic ACTH secretion
Certain tumours unrelated to the adenohypophysis may secrete ACTH. Small cell (oat cell) carcinoma of the bronchus (Ch. 14) is the commonest example, although carcinoids, pancreatic islet cell tumours and renal adenocarcinoma (hypernephroma) may occasionally be responsible. Plasma ACTH levels are very high and are not suppressed in the dexamethasone suppression test.
Hyperaldosteronism
Primary hyperaldosteronism (Conn’s syndrome)
This is the autonomous secretion of excess aldosterone. The usual cause is an adenoma of the zona glomerulosa, but generalised hyperplasia of the zona is sometimes responsible. The resulting renal retention of sodium and water leads to hypertension, while potassium loss leads to muscular weakness and cardiac arrhythmias. The hypokalaemia is associated with metabolic alkalosis, causing tetany and paraesthesiae.
Secondary hyperaldosteronism
When renal glomerular perfusion is reduced, for example through a fall in blood volume, the renin–angiotensin system (Fig. 17.12) stimulates aldosterone secretion from the zona glomerulosa in an attempt to correct this. This physiological response is known as secondary hyperaldosteronism, which is by far the commonest type of hyperaldosteronism.
Hypersecretion of sex steroids
Some adrenal cortical adenomas secrete sex steroids, most commonly androgens. In Cushing’s syndrome, quantities of androgens are occasionally secreted along with the glucocorticoids, causing virilisation of females, especially those with adrenocortical carcinomas.
Rarely, congenital enzyme defects of the pathways of steroid synthesis may result in excess production of sex steroids. The least rare example is ‘congenital adrenal hyperplasia’ due to deficiency of the enzyme 21-hydroxylase, needed for the synthesis of both cortisol and aldosterone (Fig. 17.15). Failure of cortisol production leads to increased ACTH secretion, resulting in hyperplasia of the adrenal cortex. The production of androgens occurs before the metabolic block caused by the enzyme deficiency, and their excessive secretion results in masculinisation of females and precocious puberty in males. 21-Hydroxylase deficiency is serious because deficiency of mineralocorticoids causes life-threatening salt loss unless replacement therapy is given.
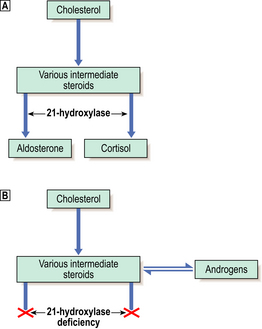
Fig. 17.15 21-Hydroxylase deficiency: the commonest cause of congenital adrenal hyperplasia. Normal metabolism.
Normal metabolism. 21-Hydroxylase deficiency. Failure of aldosterone production leads to salt-wasting, while cortisol lack causes the anterior pituitary to release ACTH, resulting in adrenal cortical hyperplasia. The resulting excess intermediate steroids are converted to androgens, leading to virilisation.
21-Hydroxylase deficiency. Failure of aldosterone production leads to salt-wasting, while cortisol lack causes the anterior pituitary to release ACTH, resulting in adrenal cortical hyperplasia. The resulting excess intermediate steroids are converted to androgens, leading to virilisation.
Tumours
Adenoma
In addition to those ‘functioning’ adrenal cortical adenomas that present by causing Cushing’s or Conn’s syndromes, a clinically unsuspected ‘non-functioning’ adenoma occurs in about 2% of adults. The adenoma is a pale yellow circumscribed nodule, perhaps 20–30 mm in diameter (Fig. 17.16). The cells have clear cytoplasm owing to their high lipid content (Fig. 17.17).
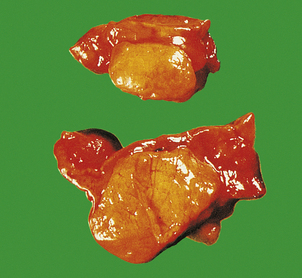
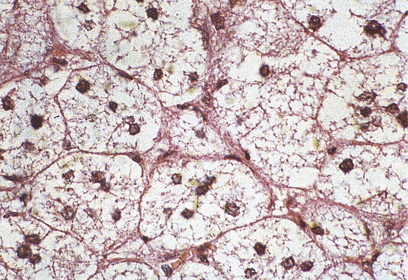
Carcinoma
Adrenal cortical carcinoma is rare; these tumours are usually hormone-secreting, with a tendency to produce androgens. They are commonly large (over 100 g) and exhibit invasive growth. Examination of the adjacent adrenal cortex and that of the opposite gland may give a clue as to the function of the neoplasm; glucocorticoid-secreting tumours will suppress ACTH, resulting in atrophy of the non-neoplastic adrenal cortex.
Adrenal cortical insufficiency



Adrenocortical hypofunction can be primary, due to lesions within the adrenal gland, or secondary, due to failure of ACTH secretion by the adenohypophysis. Acute primary insufficiency is called Waterhouse–Friderichsen syndrome. Causes of chronic primary insufficiency include:
Autoimmune adrenalitis selectively damages and destroys the adrenal cortex, sparing the medulla; tuberculosis destroys the cortex and medulla.
Acute insufficiency
Acute insufficiency (‘adrenal apoplexy’) was first noted in children by Waterhouse and Friderichsen who, in 1911 and 1918 respectively, independently described acute haemorrhagic necrosis of the adrenals in the course of meningococcal septicaemia. Other acute septicaemias, especially those due to Gram-negative bacteria, may cause a similar effect. The adrenal cortices are necrotic and the medullae contain acute haemorrhage (Fig. 17.18). The adrenal necrosis is probably due to disseminated intravascular coagulation (DIC). The symptoms are attributable to lack of mineralocorticoids (salt and water loss with hypovolaemic shock) and of glucocorticoids (failure of gluconeogenesis resulting in hypoglycaemia).
Chronic insufficiency
Thomas Addison first described an association between destruction of the adrenal cortex and the constellation of symptoms caused by the resulting chronic insufficiency of adrenal cortical hormones (Addison’s disease). The effects are due to a combined lack of mineralocorticoids and glucocorticoids:
Patients with chronic adrenocortical insufficiency may develop an acute Addisonian crisis, in which even minor illnesses such as infections may cause vomiting, fluid loss, electrolyte disturbances and circulatory collapse.
The commonest cause of Addison’s disease was once caseous necrosis of the adrenal cortices due to tuberculosis. Autoimmune destruction of the cortex is now a commoner cause; this is associated with other ‘organ-specific’ autoimmune diseases, such as pernicious anaemia (also described by Addison), thyroiditis, insulin-dependent diabetes mellitus and parathyroid failure.
In all cases of Addison’s disease, plasma cortisol levels are low. Estimation of ACTH levels enables a distinction to be made between primary adrenocortical insufficiency (ACTH raised) and secondary insufficiency (ACTH low).
THYROID
The thyroid gland (normal weight 20–30 g) is composed of follicles that are lined by cuboidal epithelial cells (Fig. 17.19) and contain a proteinaceous stored secretion (‘colloid’). The main function of the thyroid epithelial cells is the synthesis of the iodinated amino acids, thyroxine (T4) and tri-iodothyronine (T3).
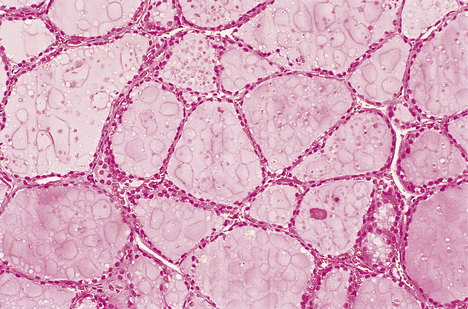
Fig. 17.19 Normal thyroid histology. Colloid-filled follicles are lined by regular cuboidal epithelium.
The secretion of T3 and T4 is under negative feedback control by TSH from the anterior pituitary. For example, a fall in the plasma level of these thyroid hormones causes increased TSH secretion by both direct effects on the adenohypophysis and effects on the hypothalamus.
The thyroid also contains a population of cells known as C-cells; these are sparsely scattered throughout the gland and secrete calcitonin, a peptide hormone involved in calcium metabolism. Medullary carcinoma, a tumour of these cells, is discussed on page 455.
SECRETORY MALFUNCTION
Hyperthyroidism




Thyrotoxicosis is the clinical syndrome resulting from the effect on the tissues of excess circulating T3 and T4; the overall result is an increased metabolic rate. Hyperthyroidism, the commonest cause of thyrotoxicosis, denotes that the source of the high circulating T3 and T4 is a lesion within the thyroid gland. The features are summarised in Figure 17.20. Hyperthyroidism may result from three main pathological lesions:
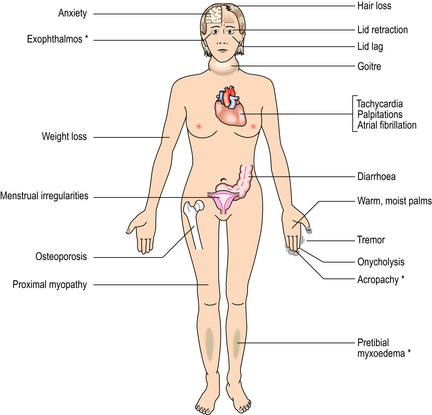
Fig. 17.20 Systemic features of thyrotoxicosis. The features marked ∗ are seen only in thyrotoxicosis due to Graves’ thyroiditis.
More rarely, thyrotoxicosis may result from exogenous thyroid hormones taken by the patient or from ectopic secretion by some rare tumours such as struma ovarii, which is a monophyletic teratoma of the ovary comprising thyroid tissue.
Graves’ thyroiditis
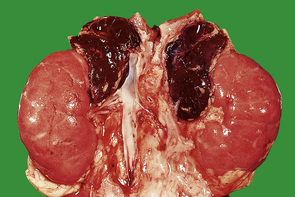
Graves’ thyroiditis is the commonest cause of thyrotoxicosis, usually associated with a diffuse goitre. The thyroid is moderately enlarged, firm and beefy-red due to increased vascularity (Fig. 17.21). Histologically, the gland shows hyperplasia of the acinar epithelium, reduction of stored colloid, and local accumulations of lymphocytes with lymphoid follicle formation (Fig. 17.22). This full spectrum of features is now rarely seen in subtotal thyroidectomy specimens of the condition, because antithyroid drugs are given before surgery.
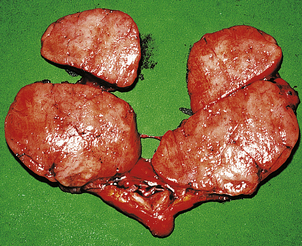
Fig. 17.21 Graves’ thyroiditis. The gland is diffusely enlarged, fleshy and dark-coloured due to increased vascularity.
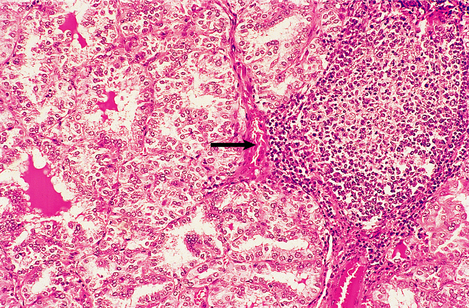
Fig. 17.22 Histological features of Graves’ thyroiditis. There is hyperplasia of the follicular epithelium with nuclear irregularity, depletion of colloid and focal lymphoid aggregates (arrowed).
Graves’ thyroiditis is one of the so-called ‘organ-specific’ autoimmune diseases. The pathogenesis is the production of an auto-antibody of the IgG class which binds to the thyroid epithelial cells and mimics the stimulatory action of TSH. The auto-antibody is known as long-acting thyroid stimulator (LATS) and its effect on the thyroid can be classed as a form of hypersensitivity reaction, ‘stimulatory hypersensitivity’. LATS stimulates the function and growth of thyroid follicular epithelium. In addition to showing the usual features of thyrotoxicosis, patients with Graves’ thyroiditis may also show exophthalmos, pretibial myxoedema (accumulation of mucopolysaccharides in the deep dermis of the skin) and finger-clubbing. The latter two signs are rare effects, but exophthalmos is common. It results from infiltration of the orbital tissues by fat (interestingly, adipocytes have been shown to have cell surface TSH receptors), mucopolysaccharides and lymphocytes, and may be due to an additional auto-antibody reacting with these tissues.
Functioning adenoma
Functioning adenomas of the thyroid may cause thyrotoxicosis, but less than 1% of adenomas show enough secretory activity to do so. Histologically, the tumour is composed of thyroid follicles and is sometimes so small that it is visualised only on an 131I radio-isotope scan. Occasionally it may present as a solitary thyroid mass.
Hypothyroidism



Hypothyroidism (myxoedema) is the clinical syndrome resulting from inadequate levels of circulating T3 and T4. The metabolic rate is lowered and mucopolysaccharides accumulate in the dermal connective tissues to produce the typical myxoedema face (Fig. 17.23). The general features of hypothyroidism are summarised in Figure 17.24. If hypothyroidism is present in the newborn, physical growth and mental development are impaired, sometimes irreversibly; this condition is known as cretinism. Cretinism may be endemic in geographical areas where the diet contains insufficient iodine for thyroid hormone synthesis. Sporadic cases are usually due to a congenital absence of thyroid tissue, or to enzyme defects blocking hormone synthesis.

Fig. 17.23 Myxoedemic face. The skin is coarse and puffy due to accumulation of mucopolysaccharides; the outer third of the eyebrows is lost.
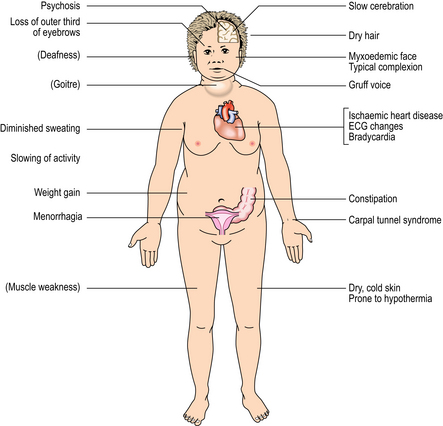
Fig. 17.24 Systemic features of hypothyroidism (myxoedema). The features in brackets are neither common nor essential.
The commonest cause of acquired hypothyroidism in adults is Hashimoto’s thyroiditis (see below), but occasional cases are iatrogenic, for example due to surgical removal of thyroid tissue or to certain drugs that cause unwanted hypothyroidism, such as sulphonylureas, resorcinol, lithium and amiodarone.
Hashimoto’s thyroiditis
Hashimoto’s thyroiditis may initially cause thyroid enlargement, but later there may be atrophy and fibrosis. The gland appears firm, fleshy and pale (Fig. 17.25). Histologically, the gland is densely infiltrated by lymphocytes and plasma cells, with lymphoid follicle formation. Colloid content is reduced, and the thyroid epithelial cells show a characteristic change in which they enlarge and develop eosinophilic granular cytoplasm due to proliferation of mitochondria; they are then termed Askanazy cells, Hürthle cells or oncocytes (Fig. 17.26). In advanced cases there may be fibrosis. Paradoxically, in the early stages of Hashimoto’s thyroiditis, the damage to the thyroid follicles may lead to release of thyroglobulin into the circulation, causing a transient phase of thyrotoxicosis.
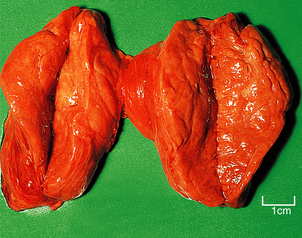
Fig. 17.25 Hashimoto’s thyroiditis. The gland is slightly enlarged and the lobes have been sliced to show the uniformly pale and fleshy cut surface.
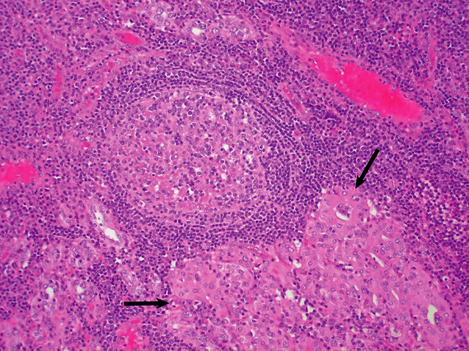
Fig. 17.26 Histological features of Hashimoto’s thyroiditis. There is destruction of follicles by a dense lymphocytic infiltrate with germinal centre formation. Some of the surviving epithelial cells show Hürthle cell change (arrowed).
Like Graves’ thyroiditis, Hashimoto’s thyroiditis is one of the ‘organ-specific’ autoimmune diseases (Ch. 9). Two auto-antibodies can be detected in the serum of most patients with Hashimoto’s thyroiditis, one reacting with thyroid peroxidase and the other reacting with thyroglobulin. These auto-antibodies are probably formed locally by the plasma cells infiltrating the thyroid, and are possibly the result of a loss of specific suppressor T-lymphocytes. In common with other organ-specific autoimmune diseases, there is a female preponderance, and certain HLA antigens (Ch. 3) are commonly found in affected individuals—especially HLA-B8 and -DR5.
GOITRE (ENLARGEMENT OF THE WHOLE GLAND)
The term goitre denotes an enlargement of the thyroid without hyperthyroidism.
Simple goitre
A spectrum of pathological changes may occur, ranging from parenchymatous goitre to colloid goitre.
In parenchymatous goitre there is at first hyperplasia of the thyroid epithelium with loss of stored colloid, but eventually less active areas appear and are compressed by the hyperplastic areas. Tracts of fibrosis may separate these areas, resulting in multinodular goitre (Fig. 17.27). The multiple nodules of this type of goitre can usually be palpated clinically, but occasionally one large nodule may be noted and give rise to suspicion of neoplasia.
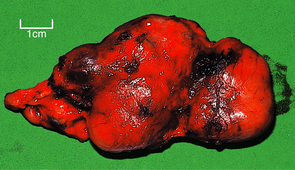
Fig. 17.27 Multinodular goitre. Thyroid lobectomy tissue showing irregular nodular enlargement due to hyperplasia, fibrosis and focally excessive colloid.
In colloid goitre there is no epithelial hyperplasia, but follicles accumulate large volumes of colloid (Fig. 17.28), and coalesce to form colloid-filled cysts. There may be areas of haemorrhage, fibrosis and dystrophic calcification. The thyroid may be diffusely enlarged or multinodular. A complication of this condition is haemorrhage into a cyst, giving rise to rapid enlargement of the cyst, which may cause tracheal compression and stridor.
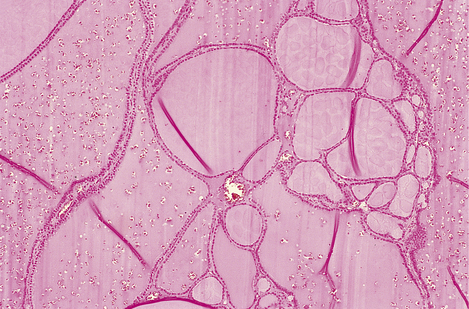
Fig. 17.28 Colloid goitre. The follicles are distended by accumulated colloid. (The folds in the colloid are a common artefact.)
Aetiology
The aetiology of simple goitre is thought to involve a phase of relative lack of T3 and T4 so that TSH rises and causes hyperplasia of the thyroid epithelium. This lack of T3 and T4 can be brought about in three main ways:
Endemic goitre was formerly common in areas remote from the sea, where the soil contains little iodine, for example in the Derbyshire hills (UK), parts of Switzerland and mountainous regions. The addition of iodine to the diet by iodination of table salt has reduced the incidence of goitre in some areas.
Rare causes of goitre
Giant cell thyroiditis
Giant cell thyroiditis (de Quervain’s thyroiditis) is a distinctive form of slight thyroid swelling with tenderness on palpation, and fever, usually with fairly abrupt onset. Histologically, the gland is infiltrated by a mixture of neutrophil polymorphs and lymphocytes, with a focal giant cell reaction possibly due to epithelial cell fusion. The disease is thought to be induced by viral infections such as mumps, but the reason why only a few individuals develop this rare disease is not known.
Riedel’s thyroiditis
Riedel’s thyroiditis is an exceptionally rare cause of thyroid enlargement with dense fibrosis which may involve adjacent muscles; this renders the thyroid firm and immobile on palpation, thus mimicking carcinoma. Histologically, there is dense fibrous replacement of the gland and, characteristically, occlusion of thyroid veins by fibrosis. The condition is of unknown aetiology, but may be associated with retroperitoneal fibrosis.
SOLITARY MASSES
The patient with a solitary mass in the thyroid presents a common clinical problem. The investigation of such a patient, following clinical examination, first involves checking the thyroid secretory status (serum T3, T4 and TSH).
Diagnostic imaging of the thyroid gland may be performed with 99mTc, which localises to the gland in a similar distribution to iodine (Fig. 17.29). ‘Cold’ lesions (which do not take up the radio-isotope) may be cysts or solid tumours; these can be distinguished by ultrasonography, which is gradually superseding 99mTc scanning (Fig. 17.30), or by fine-needle aspiration cytology. Cytology enables a pre-operative diagnosis of thyroid neoplasia to be made and has revolutionised the management of thyroid nodules.

Fig. 17.29 99m Technetium thyroid scans.  Normal thyroid showing symmetrical uptake of the isotope by both lobes.
Normal thyroid showing symmetrical uptake of the isotope by both lobes. Multiple ‘hot’ nodules taking up the isotope, consistent with a multinodular goitre.
Multiple ‘hot’ nodules taking up the isotope, consistent with a multinodular goitre. Solitary ‘cold’ nodule (arrowed); this may be a cyst or a solid tumour.
Solitary ‘cold’ nodule (arrowed); this may be a cyst or a solid tumour.
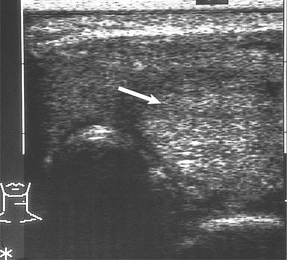
Fig. 17.30 Ultrasound scan of thyroid. There is a solid solitary nodule (arrowed) which may be neoplastic.
Many apparently solitary nodules turn out to be merely one large nodule in an otherwise multinodular goitre. Others, however, are neoplastic.
Tumours

 Malignant forms include carcinomas and lymphoma:
Malignant forms include carcinomas and lymphoma:
Tumours of the thyroid are generally benign. Carcinomas are rare at this site, and lymphomas rarer still. Those tumours that are malignant have a variable behaviour that dictates the clinical management. Histological classification is, therefore, of vital importance.
Benign tumours
Follicular adenoma is a common cause of a solitary thyroid nodule. It usually consists of a solid mass within a fibrous capsule, compressing the adjacent gland (Fig. 17.31), but the centre may show areas of haemorrhage and cystic changes. Microscopically, a range of appearances may be seen, but the commonest type consists of very compact follicles, lined by epithelial cells with slight nuclear hyperchromatism, containing little colloid. There is a surrounding fibrous capsule which is not breached by the tumour.
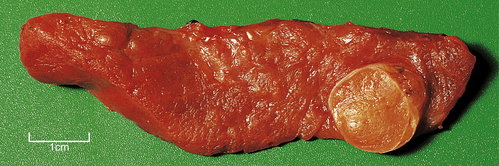
Rarely, follicular adenomas may synthesise excess T3 and T4, appearing ‘hot’ on a radio-isotope scan, and sometimes causing thyrotoxicosis.
Malignant tumours
Carcinoma of the thyroid is not a common tumour and, because the majority of these tumours are well-differentiated types with a good prognosis, it accounts for less than 1% of cancer deaths. It is one of the malignancies known to be associated with radiation exposure, whether as X-rays to the neck or as nuclear fall-out, that contains radio-isotopes of iodine which are selectively trapped by the gland. The main features of the four types of thyroid carcinoma are summarised in Table 17.5. In addition, some of the thyroid tumours once classified as anaplastic carcinoma are now known to be lymphomas.

Papillary adenocarcinoma
Papillary adenocarcinoma is a well-differentiated form of adenocarcinoma most commonly found in younger (less than 45 years old) patients. It presents as a non-encapsulated infiltrative mass (Fig. 17.32) which may be firm and white due to fibrosis. Histologically, it consists of epithelial papillary projections (Fig. 17.33) between which calcified spherules (psammoma bodies) may be present. The epithelial cell nuclei are characteristically large with central clear areas; for this reason, they are sometimes termed ‘Orphan Annie’ nuclei (Fig. 17.34).
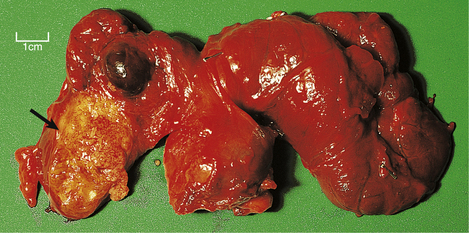
Fig. 17.32 Papillary adenocarcinoma and multinodular goitre. At the lower pole of the right lobe there is an infiltrating white mass (arrowed) which histologically was found to be a papillary adenocarcinoma. The gland also shows multinodular goitre.
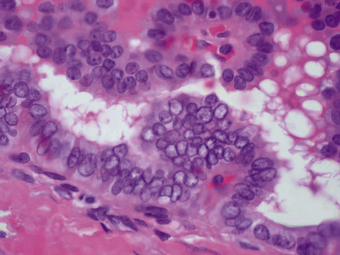
Fig. 17.33 Papillary adenocarcinoma of the thyroid. High-power photomicrograph showing atypical epithelial cells with large vesicular (‘Orphan Annie’) nuclei forming papillae.

Fig. 17.34 Orphan Annie.This cartoon-strip character first appeared in the New York News in 1926. Her forlorn-looking eyes inspired the name ‘Orphan Annie’ for the clear nuclei seen in papillary adenocarcinoma.
Papillary adenocarcinoma metastasises via the lymphatics within the thyroid gland, which may give a multifocal appearance, and to the cervical lymph nodes. However, it is a slow-growing tumour which may even regress; thus the prognosis is excellent.
Follicular adenocarcinoma
Follicular adenocarcinomas usually present in a similar way to follicular adenomas and on naked-eye inspection are often round encapsulated nodules. However, histology reveals invasion of the capsule, blood vessels (Fig. 17.35) or the surrounding gland.
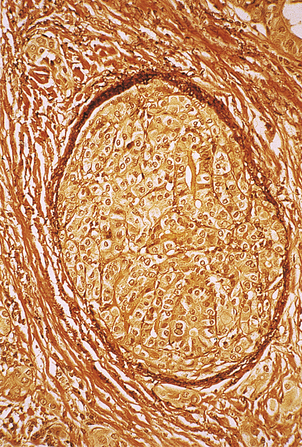
Fig. 17.35 Vascular invasion by follicular carcinoma of the thyroid. Medium-power photomicrograph of a section stained for elastin which appears dark brown in the wall of the large vein (centre). The vein is plugged by infiltrating follicular carcinoma.
Metastasis characteristically occurs via the blood stream, with the bones and lungs the commonest sites of secondary spread. However, many of the tumours retain the ability to take up 131I, which may be used as a highly effective targeted form of radiotherapy. The prognosis is therefore good.
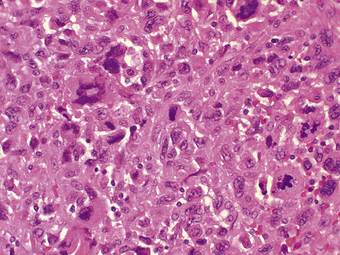
Anaplastic carcinoma
Anaplastic carcinomas (undifferentiated) usually present in the elderly as diffusely infiltrative masses. There are various histological appearances, but spindle cell and giant cell types are common (Fig. 17.36). There is evidence that anaplastic carcinomas are poorly differentiated adenocarcinomas derived from thyroid epithelium. The prognosis is very poor, due to rapid local invasion of structures such as the trachea, producing respiratory obstruction.
Medullary carcinoma
Medullary carcinoma is derived from the thyroid C-cells and commonly both synthesises and secretes calcitonin. Histologically, the tumour is composed of sheets of neoplastic cells with, between them, a hyaline stroma with the staining reactions of amyloid; this is due to polymerisation of calcitonin into a beta-pleated sheet (Fig. 17.37).
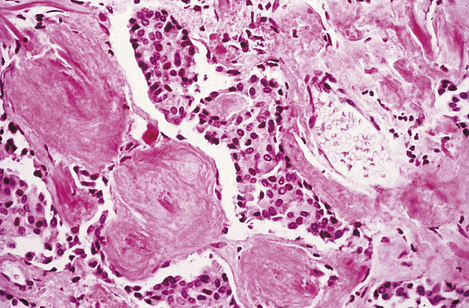
Fig. 17.37 Medullary carcinoma of the thyroid. High-power photomicrograph showing small spherical tumour cells adjacent to masses of amorphous hyaline material; the latter is amyloid.
Although the patient often has very high circulatory levels of calcitonin, this produces no clinical effects. Being a tumour of neuroendocrine (also termed APUD) cells (Ch. 15), medullary carcinoma may produce other secretory products, such as 5-HT, and may lead to symptoms of the carcinoid syndrome (Ch. 15).
Some cases, especially those presenting in young patients, are familial and may be part of one of the so-called multiple endocrine neoplasia (MEN) syndromes. These syndromes are rare, but it is important to recognise them in order to suspect, and diagnose early, associated endocrine neoplasms.
The tumour usually pursues a rather indolent course, but some familial cases are aggressive.
Lymphoma
Some thyroid tumours that previously would have been classified as anaplastic carcinoma are now known, largely on the basis of immunohistochemistry, to be mostly non-Hodgkin’s lymphomas (Fig. 17.38). There is an increased incidence of these types of lymphoma originating in the thyroid in Hashimoto’s thyroiditis. Most thyroid lymphomas are regarded as neoplasms of mucosa-associated lymphoid tissue (see Ch. 22).
PARATHYROIDS
NORMAL STRUCTURE AND FUNCTION
About 90% of individuals have four parathyroid glands, the remainder having three or five. The upper pair are derived from the endoderm of the fourth pharyngeal pouch and lie close to the upper posterior surface of the thyroid. The lower pair, derived from endoderm of the third pharyngeal pouch, are variable in position, lying anywhere from the lower pole of the thyroid to the upper mediastinum. Three types of cell can be recognised—chief cells, water-clear cells and oxyphil cells—but these may represent different functional states of the same cell lineage (Fig. 17.39). Despite having a combined weight of only 120 mg, these tiny glands play a major role in the control of calcium homeostasis through their secretion of parathyroid hormone (PTH).
Calcium homeostasis
Parathyroid hormone, a polypeptide of 84 amino acids, plays a major part in the regulation of bone metabolism and plasma calcium levels. Plasma calcium levels are normally stabilised to within a very narrow range because differences in calcium concentrations across cell membranes are essential in excitable tissues such as muscle. The chief actions of PTH are shown in Table 17.6. The net effect of an increase in PTH secretion is to raise plasma calcium and reduce plasma phosphate levels. The actions of PTH cannot be considered in isolation because of its close relationship with vitamin D and calcitonin in the control of plasma calcium levels.
Table 17.6 The chief actions of PTH
| Site | Action |
|---|---|
| Bone |
Kidney
Vitamin D is acquired in two ways: either by synthesis from 7-dihydrocholesterol in the skin in the presence of sunlight, or from dietary sources such as eggs, butter and fortified margarine. The chief circulating form is 25-hydroxyvitamin D produced in the liver. Under the influence of PTH, the kidney hydroxylates this further to 1,25-dihydroxyvitamin D, the most active form. The actions of vitamin D are shown in Table 17.7.
Table 17.7 The actions of vitamin D
| Site | Action |
|---|---|
| Intestine | Increases calcium absorption |
| Bone | In conjunction with PTH, releases calcium into the circulation |
| Is essential for normal mineralisation of osteoid |
Calcitonin is secreted by the C-cells of the thyroid in response to a rise in plasma calcium. It inhibits resorption of bone by osteoclasts and increases renal phosphate excretion. This tends to lower plasma calcium; however, hypersecretion of the hormone does not cause significant hypocalcaemia in otherwise normal individuals.
DISEASES OF THE PARATHYROIDS
The most important diseases of the parathyroids are hyperparathyroidism, hypoparathyroidism and tumours.
Hyperparathyroidism




Hyperparathyroidism is classified into primary, secondary and tertiary types according to the circumstances in which it occurs (Table 17.8). Primary and tertiary hyperparathyroidism are pathological states with inappropriate excess PTH secretion for the prevailing plasma calcium levels. Secondary hyperparathyroidism, however, is an appropriate physiological response to hypocalcaemia, for example in renal failure.
Table 17.8 Classification of hyperparathyroidism
| Hyperparathyroidism | Cause |
|---|---|
| Primary | Hypersecretion of PTH by an adenoma or hyperplasia of the gland |
| Secondary | Physiological increase in PTH secretions in response to hypocalcaemia of any cause |
| Tertiary | Development of an autonomous hypersecreting adenoma in longstanding secondary hyperparathyroidism |
Primary hyperparathyroidism
Primary hyperparathyroidism is a fairly common condition, occurring in almost 0.1% of the population, most frequently in post-menopausal females. It presents through the symptoms of hypercalcaemia:
In rare cases the effects of hyperparathyroidism on bone—osteitis fibrosa and brown tumour (Ch. 25)—are also apparent clinically.
Hyperparathyroidism is only one of several important possible causes of hypercalcaemia. The commonest causes of hypercalcaemia are:
In the investigation of a patient with hypercalcaemia, the results that point to hyperparathyroidism as the cause are:
In about 80% of cases, primary hyperparathyroidism is due to a secretory adenoma of one of the parathyroid glands; this consists of a neoplastic mass of functioning parathyroid cells surrounded by a compressed rim of inactive parathyroid tissue (Fig. 17.40). The remaining of cases are usually due to hyperplasia of all the parathyroid glands, especially when hyperparathyroidism forms part of one of the MEN syndromes (Ch. 11).
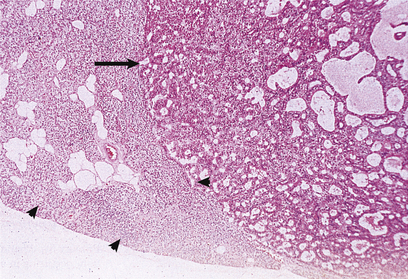
Fig. 17.40 Parathyroid adenoma from a patient with primary hyperparathyroidism. The adenomatous nodule (arrowed) is surrounded by a compressed rim of parathyroid tissue (arrowheads).
The management of hyperparathyroidism usually consists of operative inspection of all four parathyroid glands wherever possible, followed by removal of any suspected adenoma which is then submitted for intra-operative diagnosis by rapid frozen section.
Hypoparathyroidism
Hypoparathyroidism results in a fall in plasma calcium levels accompanied by elevated plasma phosphate levels. The patient presents with the clinical features of hypocalcaemia:
Diagnosis is confirmed by low or absent plasma PTH levels in the presence of hypocalcaemia.
In addition to hypoparathyroidism, other important causes of hypocalcaemia include:
Even when the total plasma calcium levels are normal, symptoms of hypocalcaemia, such as tetany, may be produced by alkalosis; this lowers the proportion of plasma calcium in the ionised state, an important factor in the control of muscle excitability.
The leading causes of hypoparathyroidism are:
Iatrogenic disease, such as accidental removal of the parathyroid glands during thyroidectomy, remains a common cause. Idiopathic hypoparathyroidism is now known to be due to destruction of the parathyroid cells by an auto-antibody. It is associated with other ‘organ-specific’ autoimmune diseases (Ch. 9).
Tumours
The commonest tumours, adenomas, are benign neoplasms of one of the three types of parathyroid cell. They are usually small (less than 50 mm in diameter) and only become clinically apparent through hypersecretion of PTH. Very rarely, they may occur in more than one parathyroid gland. Adenocarcinoma of the parathyroid glands is rare.
ENDOCRINE PANCREAS
The pancreas consists of two functionally distinct components:
Numbering about a million, the islets of Langerhans are derived from endoderm bordering the pancreatic ductal system. Although they comprise only 1–1.5 g of the pancreatic tissue (about 1% of its mass), their endocrine secretions have profound metabolic effects and are essential for life.
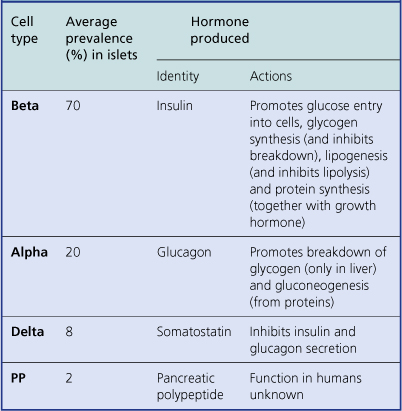
The islets consist of clusters of compact cells interspersed with small blood vessels (Fig. 17.41); they contain at least four distinct cell types, classified according to their hormone content as demonstrated by immunohistochemistry (Fig. 17.42).There are regional differences in hormone content of the islet cells in different parts of the pancreas, but the average hormonal composition is as shown in Table 17.9.
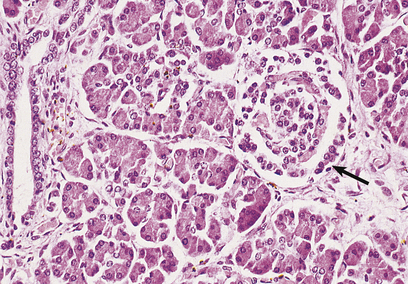
Fig. 17.41 Normal pancreas. An islet of Langerhans (arrowed) is surrounded by exocrine pancreatic acini and a duct.
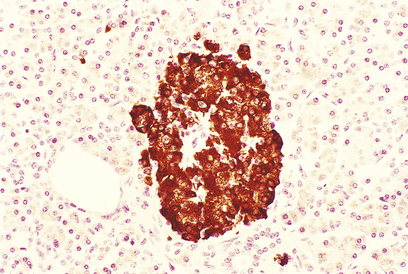
Fig. 17.42 Beta-cells in an islet of Langerhans. This normal pancreas has been stained by the immunoperoxidase technique for insulin. The insulin-containing cells are darkly stained.
On electron microscopy, these endocrine cells contain membrane-bound electron-dense granules, some of which have characteristic shapes revealing their hormone content, and they have the histochemical features of neuroendocrine (APUD) cells (Ch. 15).
The effects of two of the islet hormones, insulin and glucagon, are virtually antagonistic (Table 17.9). It seems that the secretion of pancreatic hormones is controlled locally; for example, a rising blood glucose level appears to stimulate the beta-cells to secrete insulin directly.
The principal diseases of the endocrine pancreas are hypofunction, especially of the beta-cells (diabetes mellitus), and tumours of the islet or other neuroendocrine (APUD) cells, which may produce widespread effects due to hormone hypersecretion.
Diabetes mellitus

 Type 1 (juvenile onset) due to destruction of beta-cells (probably a result of virus infection and genetic factors); insulin-dependent
Type 1 (juvenile onset) due to destruction of beta-cells (probably a result of virus infection and genetic factors); insulin-dependent

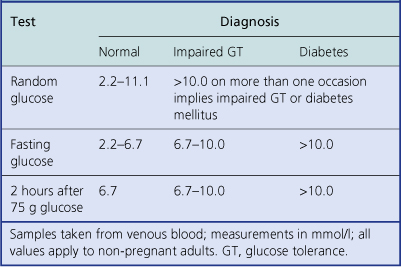
Diabetes mellitus is a disease state rather than a single disease, because it may have several causes. It is defined as an abnormal metabolic state in which there is glucose intolerance due to inadequate insulin action. Diagnosis is based on the clinical demonstration of glucose intolerance (Table 17.10).
Insulin is unique, in that it is the only hormone with a hypoglycaemic effect; there are five hormones that tend to exert a hyperglycaemic effect—glucagon, glucocorticoids, growth hormone, adrenaline (epinephrine) and noradrenaline (norepinephrine). Thus, the hyperglycaemic effects of these hormones cannot be counterbalanced if there is inadequate insulin action.
Pathogenesis
The actions of insulin (Table 17.9) are all anabolic, that is, they promote the laying down of tissue stores from circulating nutrients. The consequences of insulin deficiency are therefore catabolic, that is, there is breakdown of tissue energy stores.
The major features of diabetes mellitus are:
In hyperglycaemia the renal threshold for glucose conservation is exceeded, so that there is osmotic diuresis resulting in polyuria, dehydration and thirst. Lipolysis may also have serious consequences. Free fatty acids are converted in the liver to ketone bodies, such as acetoacetate, acetone and beta-hydroxybutyrate. These dissociate to release hydrogen ions, and a profound metabolic acidosis may ensue.
The combined result of severe ketosis, acidosis, hyperglycaemia, hyperosmolarity and electrolyte disturbance is to impair cerebral function, producing diabetic ketoacidotic coma. This is quite distinct from the hypoglycaemic coma that may also be found in diabetic patients; this is due to insulin overdosage, and has entirely different clinical features.
Classification
The two major types of diabetes mellitus are defined according to the clinical setting in which they occur. Research into pathogenesis of the disease has reinforced this classification, as the two types appear to have distinct pathogeneses. In addition, diabetes sometimes appears as a secondary consequence of other diseases.
Type 1 (juvenile-onset, insulin-dependent diabetes)
Type 1 diabetes mellitus (also called juvenile-onset, or insulin-dependent diabetes) typically presents in childhood. The patient usually shows the catabolic effects described above and is prone to develop ketoacidosis. The central defect is inadequate insulin secretion by the beta-cells of the pancreas, and this can be corrected only by the life-long administration of exogenous insulin.
Postmortem examination of the pancreas in patients who had recently developed type 1 diabetes but died from other causes (e.g. road traffic accident) shows lymphocytic infiltration of the islets with specific destruction of the beta-cells. There are three major theories concerning the aetiology of these changes: autoimmune destruction, genetic factors and viral infection.
Autoimmune destruction
The majority of patients who have recently developed type 1 diabetes have circulatory antibodies to several different types of islet cell. Patients with this type of diabetes are also prone to develop other ‘organ-specific’ autoimmune diseases (Ch. 9).
Genetic factors
As with other ‘organ-specific’ autoimmune diseases, there is an association with certain HLA types (Ch. 3), notably HLA-DR4, especially if HLA-B8 or -DR3 is also present. It seems that environmental factors also play a role, as identical twins show only 40% concordance in development of the disease.
Type 2 (maturity-onset, non-insulin-dependent diabetes)
Type 2 diabetes mellitus (also called maturity-onset, or non-insulin-dependent diabetes) is more common than type 1 and usually presents in middle age, being commonest in the obese. Patients are not prone to ketoacidosis, but occasionally develop a non-ketotic coma in which there is extreme hyperosmolarity of the plasma. Insulin secretion is normal or increased and the central defect may therefore be a reduction in the number of cell surface receptors for insulin.
Genetic factors clearly play an important part in the aetiology of type 2 diabetes, as identical twins show nearly a 100% concordance in development of the disease. No clear Mendelian pattern of inheritance can be recognised. The evidence is against this being an autoimmune disease.
Treatment is usually by weight reduction coupled with orally administered drugs that potentiate the action of insulin.
Secondary diabetes
Hypersecretion of any of the hormones that tend to exert a hyperglycaemic effect may cause glucose intolerance. Thus Cushing’s syndrome, phaeochromocytoma, acromegaly and glucagonomas may cause secondary diabetes. Generalised destruction of the pancreas (Ch. 16) by acute and chronic pancreatitis, haemochromatosis and, occasionally, carcinoma may cause insulin deficiency.
Complications
The major complications of diabetes mellitus are shown in Table 17.11. The commonest complications are seen in blood vessels. Atheroma, often ultimately severe and extensive, develops at an earlier age than in the non-diabetic population. Small blood vessels show basal lamina thickening and endothelial cell proliferation (diabetic microangiopathy), frequently causing retinal and renal damage. About 80% of adult diabetics die from cardiovascular disease, while patients with longstanding diabetes, especially type 1, frequently develop serious renal and retinal disease. Improved metabolic control through modern insulin regimens has only partially reduced the incidence of such serious complications.
Table 17.11 Complications of diabetes
| Situation | Complication |
|---|---|
| Large blood vessels | Accelerated atheroma, leading to: |
Small blood vessels
Peripheral nerveNeuropathy, possibly due to disease of small vessels supplying the nervesNeutrophilsSusceptibility to infectionPregnancy
Skin
Tumours




Adenomas and carcinomas derived from the islet cells are quite rare. They usually present clinically through hypersecretion of their normal hormonal product, producing widespread symptoms; consequently these tumours may be small at the time of presentation. Most consist of cellular nodules within the pancreatic tissue. Histologically, they are composed of cells resembling normal islet cells (Fig. 17.43) and immunohistochemistry may be used to identify the hormonal content of the cells. Like other tumours of APUD tissues (Ch. 11), they contain dense-core secretory granules on electron microscopy. It is usually not possible to predict whether an islet cell tumour will pursue a benign or malignant course on the basis of histological appearance.
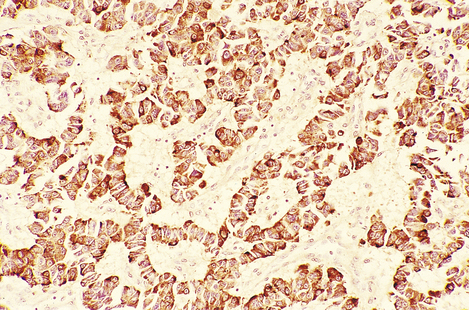
Fig. 17.43 Insulinoma. This tumour has been stained by the immunoperoxidase technique for insulin. Ribbons of brown-stained cells resemble those of the normal islet of Langerhans.
Insulinoma
Insulinoma is the commonest islet cell tumour and produces hypoglycaemia through hypersecretion of insulin. During hypoglycaemic attacks, the patient develops confusion, psychiatric disturbances and possibly coma. Diagnosis is urgent because hypoglycaemia may produce permanent cerebral damage.
Glucagonoma
Glucagonoma is much less common; it leads to hypersecretion of glucagon, producing secondary diabetes and a distinctive skin rash known as necrolytic migratory erythema.
Other islet cell tumours
Other islet cell tumours are very rare, but include somatostatinomas and tumours secreting vasoactive intestinal peptide (VIP), which leads to watery diarrhoea.
Gastrinomas
Although gastrin is usually produced in the G-cells of the stomach, tumours of the G-cells, called gastrinomas, most commonly originate in the pancreas. These APUD tumours lead to intractable hypersecretion of gastric acid due to the action of gastrin, resulting in widespread severe peptic ulceration (Zollinger–Ellison syndrome). Most gastrinomas are malignant.
Islet cell tumours and gastrinomas may occur as part of one of the MEN syndromes (Ch. 11), most commonly MEN type 1.
Commonly confused conditions and entities relating to endocrine pathology
| Commonly confused | Distinction and explanation |
|---|---|
| Gigantism and acromegaly | Both due to excess GH secretion. Gigantism is the result before epiphyseal closure. Acromegaly is the result in adults, with fused epiphyses. |
| Renin and rennin | Renin cleaves angiotensinogen into angiotensin I. Rennin (rennet, chymosin) is a proteinase, usually derived from the mucosa of calf’s stomach, which curdles milk; used in the cheese industry. |
| Addison’s disease and Addisonian pernicious anaemia | Addison’s disease is adrenal failure, usually due to autoimmune destruction. Addison also described pernicious anaemia, also an autoimmune disease, in which the gastric parietal cells are destroyed and hence fail to secrete intrinsic factor, which is required for absorption of vitamin B12. |
| Primary hypertension andsecondary hypertension | Primary hypertension is the commonest (99%) type of hypertension; it has no definite single cause. Secondary hypertension (< 1% of cases) is secondary to other disease (e.g. phaeochromocytoma) and is therefore potentially curable. |
| Graves’ thyroiditis and hyperthyroidism | Graves’ thyroiditis is an organ-specific autoimmune disease; antibodies to TSH receptors cause thyroid hyperfunction. Hyperthyroidism is thyroid hyperfunction from any cause. |
| Hashimoto’s thyroiditis and hypothyroidism | Hashimoto’s thyroiditis is autoimmune thyroid destruction. Hypothyroidism is a generic term for thyroid hypofunction from any cause. |
| Exophthalmos and lid retraction | Exophthalmos is eye protrusion specific to Graves’ thyroiditis, caused by expansion of the retro-orbital tissues by adipose tissue (which also contains TSH receptors). Hyperthyroidism from any cause may also causelid retraction. |
| Myxoedema and pretibial myxoedema | Myxoedema is generalised deposition of mucopolysaccharides in hypothyroidism. Pretibial myxoedema is also mucopolysaccharide deposition, but confined to the pretibial region and specifically associated with Graves’ thyroiditis. |
| Medullary carcinoma of the thyroid and medullary carcinoma of the breast | Medullary carcinoma of the thyroid is derived from the C-cells and is an endocrine neoplasm making calcitonin. Medullary carcinoma of the breast is a type of adenocarcinoma that is soft on gross examination and has no connection with the thyroid tumour. |
| Exocrine and endocrine | Exocrine glands secrete into the gut or onto the surface of the body. Endocrine glands secrete hormones into the blood. |
| Type 2 diabetesand secondary diabetes | Type 2 diabetes is maturity-onset (non-insulin-dependent) diabetes mellitus, so called to distinguish it from type 1 juvenile-onset (insulin-dependent) diabetes mellitus. Secondary diabetes is distinct from both of these: it is the development of glucose intolerance secondary to another disease (e.g. Cushing’s syndrome). |
| Insulin and inulin | Insulin is the hypoglycaemic peptide hormone secreted by the beta-cells of the islets of Langerhans. Inulin is a storage polysaccharide found in Jerusalem artichokes, once used in the investigation of renal clearance, and in some ‘diabetic’ dietary products. |
Al-Shraim M., Asa S.L.. The 2004 World Health Organisation classification of pituitary tumors: what is new? Acta Neuropathologica. 2006;111:1-7.
Asa S.L.. My approach to oncocytic tumours of the thyroid. Journal of Clinical Pathology. 2004;57:225-232.
Balotti S., Chiarelli F., de Martino M.. Autoimmunity: basic mechanisms and implications in endocrine diseases. Hormone Research. 2006;66:132-141.
Bishop A.E., Polak J.M.. Gastrointestinal endocrine tumours. Pathology. Baillière’s Clinical Gastroenterology. 1996;10:555-569.
Boyages S.C., Cheung W.. Goitre in Australia. Medical Journal of Australia. 1995;162:487-489.
Dahia P.L.M.. Evolving concepts in phaeochromocytoma and paraganglioma. Current Opinion in Oncology. 2006;18:1-8.
DeLellis R.A.. Tumors of the parathyroid gland. In: Rosai J., editor. Atlas of tumor pathology, 3rd series. Fascicle 6. Washington, DC: Armed Forces Institute of Pathology, 1993.
Fassnacht M., Kenn W., Allolio B.. Adrenal tumors: how to establish malignancy? Journal of Endocrinological Investigation. 2004;27:387-399.
Fonseca E., Sobrinho-Simões M.. Diagnostic problems in differentiated carcinomas of the thyroid. Pathology. Research and Practice. 1995;191:318-331.
Gepts W., Pepeleers D.. The endocrine pancreas. Functional morphology and histopathology. Acta Medica Scandinavica Supplementum. 1976;601:9-52.
Goldstone A.P., Scott-Coombes D.M., Lynn J.A.. Surgical management of gastrointestinal endocrine tumours. Baillière’s Clinical Gastroenterology. 1996;10:707-736.
Grimelius L., Johansson H.. Pathology of parathyroid tumors. Seminars in Surgical Oncology. 1997;13:142-154.
Julie C., Vieillefond A., Desligneres S., Schaison G., Grunfeld J.P., Franc B.. Hashimoto’s thyroiditis associated with Riedel’s thyroiditis and retroperitoneal fibrosis. Pathology. Research and Practice. 1997;193:573-577.
Kovacs K., Asa S.L.. Functional endocrine pathology, 2nd edn. Oxford: Blackwell Science, 1998.
Lack E.E.. Tumors of the pancreas. In: Rosai J., editor. Atlas of tumor pathology, 3rd series. Fascicle 19. Washington, DC: Armed Forces Institute of Pathology, 1997.
Lahita R.G.. Predisposing factors to autoimmune disease. International Journal of Fertility and Women’s Medicine. 1997;42:115-119.
Lansford C.D., Teknos T.N.. Evaluation of the thyroid nodule. Cancer Control. 2006;13:89-98.
Lewis P.D. The endocrine system symmers textbook of pathology. 1996 Edinburgh: Churchill Livingstone
LiVolsi V.A., Asa S.L.. Endocrine pathology. Edinburgh: Churchill Livingstone, 2002.
Lloyd R.V., Douglas B.R., Young W.F.. Endocrine diseases, 1st series. Fascicle 1. In: In: King D.W., editor. Atlas of nontumor pathology. Washington, DC: Armed Forces Institute of Pathology and American Registry of Pathology, 2000.
Lorini R., Gastaldi R., Taggiai C., Perucchin P.P. Hashimoto’s thyroiditis. Paediatric Endocrinology Reviews 1(Suppl 2). 2003:205-211
McNicol A.M., Nolan C.E., Struthers A.J., Farquharson M.A., Hermans J., Haak H.R.. Expression of p53 in adrenocortical tumours: clinicopathological correlations. Journal of Pathology. 1997;181:146-152.
Rosai J., Carcangiu M.L., DeLellis R.A.. Tumors of the thyroid gland. In: Rosai J., editor. Atlas of tumor pathology, 3rd series. Fascicle 5. Washington, DC: Armed Forces Institute of Pathology, 1992.
Saeger W.. Histopathological classification of adrenal tumours. European Journal of Clinical Investigation. 2000;30(Suppl 3):58-62.
Schott M., Scherbaum W.A., Morgenthaler N.G.. Thyrotropin receptor autoantibodies in Graves’ thyroiditis. Trends in Endocrinology and Metabolism. 2005;16:243-248.
Silver J., Levi R.. Cellular and molecular mechanisms of secondary hyperparathyroidism. Clinical Nephrology. 2005;63:119-126.
Soares P., Sobrinho-Simões M.. Recent advances in cytometry, cytogenetics and molecular genetics of thyroid tumours and tumour-like lesions. Pathology. Research and Practice. 1995;191:304-317.
Solcia E., Capella C., Klöppel G.. Tumors of the pancreas. In: In: Rosai J., editor. Atlas of tumor pathology, 3rd series. Fascicle 20. Washington, DC: Armed Forces Institute of Pathology, 1997.
Stefaneau L., Sasano H., Kovacs K.. Molecular and cellular endocrine pathology. London: Arnold, 2000.
Stephenson T.J.. Criteria for malignancy in endocrine tumours. In: Anthony P.P., MacSween R.N.M., Lowe D.G., editors. Recent advances in histopathology 17. Edinburgh: Churchill Livingstone; 1997:93-111.
Stephenson T.J.. Endocrine pathology. In: Al-Sam S., Davies J.D., Lakhani S.R., editors. A practical atlas of pseudomalignancy. London: Arnold; 1998:1-19.
Stephenson T.J.. Prognostic and predictive factors in endocrine neoplasia. Histopathology. 2006;48:629-643.
Thompson J.D.R.. Endocrine pathology. Foundations in Diagnostic Pathology Series. Edinburgh: Churchill Livingstone, 2006.
van Eeden S., Offerhaus G.J.. Historical, current and future perspectives on gastrointestinal and pancreatic endocrine tumors. Virchows Archives. 2006;448:1-6.
Westermark P.. Islet pathology of non-insulin-dependent diabetes mellitus (NIDDM). Diabetic Medicine. 1996;13(Suppl 6):S46-S48.
Yousem D.M., Scheff A.M.. Thyroid and parathyroid gland pathology. Role of imaging. Otolaryngologic Clinics of North America. 1995;28:621-649.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Chapter 17 Endocrine system
COMMON CLINICAL PROBLEMS FROM ENDOCRINE DISEASE
Pathological basis of endocrine signs and symptoms
| Sign or symptom | Pathological basis |
|---|---|
| Signs or symptoms of hormone excess (hyperfunction) |
Signs or symptoms of hormone deficiency (hypofunction)
Diffuse enlargement of gland
Nodular enlargement of glandTumour (benign or malignant)Some organ-specific features
Pituitary tumour
Increased thyroid hormone secretion due to hyperplasia or neoplasia of gland
Autoimmune involvement of retrobulbar connective tissue in Graves’ disease
Adrenocortical hyperplasia or neoplasia
Absolute or relative deficiency of insulin (diabetes mellitus)
NORMAL STRUCTURE AND FUNCTION
An endocrine gland secretes hormones directly into the blood stream to reach distant ‘target organs’ where the secretory products exert their effects. Endocrine glands are thus distinguished from exocrine glands, whose secretions pass into the gut or respiratory tract, or on to the exterior of the body; examples of exocrine glands include the exocrine pancreas and the bronchial mucous glands. Closely related to the endocrine system is the paracrine (diffuse endocrine) system, consisting of regional distributions of specialised cells producing locally acting hormones, such as those regulating gut motility, and forming part of the neuroendocrine system (Ch. 15); autocrine effects are those acting on the cell producing the hormone (Fig. 17.1).
Hormones exert their effects on the target organs by binding to receptors, protein molecules with high and specific affinity for the hormone. These hormone receptors may be either on the cell surface (for example, thyroid-stimulating hormone receptors on the thyroid epithelium) or intracellular (for example, nuclear receptors for steroid hormones). The binding of a hormone to its cell surface receptor sets off a series of intracellular signals via secondary ‘messenger’ molecules (cyclic nucleotides), which results in changes in metabolic activity, differentiation or mitosis of the stimulated cell.
ENDOCRINE PATHOLOGY
The major disorders of an endocrine gland are:
There are several important general considerations in endocrine pathology. First, disease of one endocrine gland cannot usually be considered in isolation, because it almost always has implications for other endocrine glands:
Fig. 17.2 Multiple endocrine neoplasia (MEN) syndromes. MEN syndromes are characterised by the occurrence of tumours in more than one endocrine organ. MEN types 1 and 2 can be distinguished by the organs commonly involved.
Second, one hormone may have many diverse clinical effects, so that malfunction of one endocrine gland may produce numerous clinical features.
Third, the same hormone may be produced in more than one site; thus, ectopic hormone production by tumours of non-endocrine tissues may simulate primary endocrine disease.
PITUITARY
The pituitary is a small gland, weighing only 500–1000 mg. It is situated in the sella turcica of the skull beneath the hypothalamus. Despite its small size, it exerts many essential control functions over the rest of the endocrine system, earning it the title ‘conductor of the endocrine orchestra’. It consists of two parts (Fig. 17.3), each with separate functions. The anterior pituitary, the adenohypophysis, is developed from Rathke’s pouch, an outpouching of the roof of the embryonic oral cavity; it comprises about 75% of the bulk of the gland. The posterior pituitary, the neurohypophysis, is derived from a downgrowth of the hypothalamus.
ADENOHYPOPHYSIS
Classification of cell types
Modern histological classification of the types of hormone-secreting cell is based on immunohistochemistry, a technique in which antibodies raised to a hormone bind to the cells containing that hormone in tissue sections, leading to a coloured stain (Fig. 17.4). This has enabled the true hormone content of the cells to be determined, and has rendered obsolete the traditional classification of the cells into eosinophil, basophil and chromophobe types according to their staining by haematoxylin and eosin (H&E). By electron microscopy, the cells of the adenohypophysis are seen to contain electron-dense granules ranging from 50 to 500 nm in diameter (Fig. 17.5); these contain stored secretory products. The six types of hormone-secreting cell are shown in Table 17.1.
Fig. 17.4 Growth hormone-containing cells in an adenoma of the adenohypophysis. Immunoperoxidase localisation of growth hormone. Cells containing growth hormone are stained brown by this technique.
Fig. 17.5 Electron micrograph of a secretory cell of the adenohypophysis. The hormonal products are stored as electron-dense membrane-bound cytoplasmic granules (´ 300 000).
Table 17.1 Hormone-secreting cells of the adenohypophysis
| Cell type | Staining reactionwith H&E | Hormonal product |
|---|---|---|
| Corticotroph | Basophilic | Adenocorticotrophic hormone (ACTH) |
| Thyrotroph | Basophilic | Thyroid-stimulating hormone (TSH) |
| Gonadotroph | Basophilic |
SomatotrophEosinophilicGrowth hormone (GH)LactotrophEosinophilicProlactin (PL)ChromophobePaleUnknown
Control of hormone secretion
Hormonal control factors
The adenohypophysis lacks any direct arterial supply. Blood from the hypothalamus passes down venous portal channels in the pituitary stalk (Fig. 17.3) into sinusoids which ramify within the gland. In this way hormonal control factors produced by neurosecretory cells in the hypothalamus are carried directly to the hormone-producing cells of the adenohypophysis. The known hormonal control factors and their effects are listed in Table 17.2. In general, these factors stimulate the particular secretory cells under their control into activity; the exception is prolactin-inhibiting factor, whose effect on the lactotrophs is inhibitory.
Table 17.2 Hormonal control factors and their effects on the adenohypophysis
| Hormonal control factor | Effect |
|---|---|
| Corticotrophin-releasing factor (CRF) | Corticotrophs release ACTH |
| Thyrotrophin-releasing factor (TRF) | Thyrotrophs release TSH |
| Gonadotrophin-releasing factor (FSH/LH-RF) | Gonadotrophs release FSH/LH |
| Growth hormone-releasing factor (GHRF) | Somatotrophs release GH |
| Prolactin-inhibiting factor (PIF) | Lactotrophs inhibited from releasing PL |
Secretion of these hormonal control factors by the hypothalamus is under two types of control: neural and hormonal. Neural control is via nerves from other parts of the central nervous system, and is important in reactions to stress and in changes during sleep. Hormonal control is a negative feedback mechanism in which the hypothalamus monitors the level of adenohypophysial hormones in the blood and adjusts its output of hormonal control factors accordingly, so as to stabilise the level of each adenohypophysial hormone at the optimum level. This is called the hypothalamic–hypophysial feedback control.
Feedback control
In addition to control via the hypothalamus, a more direct method of control of the adenohypophysis also exists, whereby its cells respond directly to the levels of hormones and metabolites in the blood. Most adenohypophysial hormones stimulate another endocrine gland, termed the ‘target’ gland; for example, ACTH stimulates the adrenal cortex to produce steroid hormones, and TSH stimulates the thyroid to produce thyroxine.
In these examples, the level of hormone from the target gland is monitored for feedback control. However, in the case of growth hormone, which has no single target gland, it is the level of metabolites such as glucose that is monitored. A general scheme of the feedback control mechanisms operating in the regulation of a hypophysial hormone is shown in Figure 17.3.
Adenohypophysial hormones
Adrenocorticotrophic hormone
Adrenocorticotrophic hormone (ACTH), a peptide consisting of 39 amino acids, causes increased cell numbers (hyperplasia) and increased secretory activity in the adrenal cortex. Glucocorticoid output is elevated, but there is no effect on the output of mineralocorticoids, such as aldosterone, that are not under anterior pituitary control. ACTH levels show a marked circadian variation, being highest early in the morning.
Thyroid-stimulating hormone
Thyroid-stimulating hormone (TSH) is a glycoprotein that induces proliferation of the follicular cells of the thyroid, synthesis of thyroxine (T4) and tri-iodothyronine (T3), and secretion of these into the blood. Measurement of TSH levels provides information on the state of the control system of the thyroid and is valuable in the diagnosis of thyroid malfunction.
Gonadotrophic hormones
In the female, follicle-stimulating hormone (FSH) induces growth of Graafian follicles in the ovaries; these secrete oestrogens, which in turn cause endometrial proliferation. After rupture of the follicle at ovulation, luteinising hormone (LH) causes a change in the follicle cells known as luteinisation, whereby their secretory product changes from oestrogens to progesterone which induces secretory changes in the endometrium. Both gonadotrophic hormones are glycoproteins.
The hypothalamus monitors circulating levels of the sex steroids including oestrogens and progesterone, and releases probably a single hormonal control factor, FSH/LH-releasing factor (FSH/LH-RF), to control the adenohypophysial gonadotrophs. Their response to this factor depends on the prevailing levels of sex steroids. Cyclical changes in this feedback loop form the hormonal basis for the menstrual cycle.
In the male, FSH and LH both exist but, in the absence of ovaries as the target organ, their names are inappropriate to their actions. LH stimulates testosterone production by the interstitial cells of Leydig in the testes, while FSH stimulates spermatogenesis.
The circulating levels of FSH and LH vary markedly with age: they increase at puberty and are very high in females after the menopause.
Growth hormone
Growth hormone (GH) is a protein containing 191 amino acids; it binds to receptors on the surface of various cells and thus causes increased protein synthesis, accelerates breakdown of fatty tissue to produce energy, and tends to raise the blood glucose. It is vital for normal growth; deficiency causes dwarfism. Part of its action at tissue level is mediated by a group of peptide growth factors known as somatomedins. The hypothalamic control of GH release from the hypothalamus is complex, there being both a growth hormone-releasing factor (GH-RF) and an inhibitory factor, somatostatin.
Prolactin
Prolactin (PL) is a protein hormone with a structure very similar to that of GH. Although it is present in individuals of both sexes, its function in males remains uncertain. In females, it can produce lactation, provided that the breast has already been prepared during pregnancy by appropriate levels of sex steroids. Prolactin release is a good example of the neural form of hypothalamic control: the sensation of suckling causes reduction in hypothalamic prolactin-inhibiting factor (PIF) release and a consequent rise in PL levels.
Hypofunction
Like other endocrine organs, the adenohypophysis has considerable reserve capacity, and deficiency of its hormones becomes manifest only after extensive destruction; hypofunction is therefore uncommon. Since the pituitary is tightly encased within the sella turcica, any expansile lesion, such as an adenoma, produces compression damage to the adjacent pituitary tissue, in addition to any effect from its own hormonal production. Damage to the hypothalamus or pituitary stalk may also produce adenohypophysial hypofunction through failure of control. Table 17.3 sets out the main causes of hypofunction. These conditions lead to a deficiency of all adenohypophysial hormones, a state known as panhypopituitarism. This is a life-threatening condition, as deficiency of ACTH leads to atrophy of the adrenal cortex and failure of production of vital adrenocorticoids. Diagnosis of hypopituitarism is by measurement of the individual hormones. The commonest causes of pituitary hypofunction are compression by metastatic carcinoma or by an adenoma, but two specific rarer syndromes will be mentioned because they illustrate how congenital and acquired disease may affect the pituitary.
Table 17.3 Causes of adenohypophysial hypofunction
| Site | Lesions |
|---|---|
| Pituitary | Adenoma |
| Metastatic carcinoma | |
| Trauma | |
| Post-partum ischaemic necrosis (Sheehan’s syndrome) | |
| Craniopharyngioma | |
| Infections | |
| Granulomatous diseases | |
| Autoimmunity | |
| Iatrogenic | |
| Hypothalamus | Craniopharyngioma |
| Gliomas |
Pituitary dwarfism
Pituitary dwarfism is due to deficiency of GH, sometimes associated with deficiency of other adenohypophysial hormones. The child fails to grow, although remaining well proportioned. There is a variety of known causes including adenomas, craniopharyngiomas (rare tumours derived from remnants of Rathke’s pouch) and familial forms.
Post-partum ischaemic necrosis
During pregnancy, the pituitary enlarges and becomes highly vascular. Hypotensive shock due to haemorrhage at the time of birth, compounded by the lack of direct arterial supply to the adenohypophysis, may cause ischaemic necrosis. This specific cause of necrosis is known as Sheehan’s syndromeand the effects of the resulting adenohypophysial hypofunction are termed Simmond’s disease. The neurohypophysis is usually spared. The first symptom following delivery is failure of lactation due to PL deficiency; the effects of lack of FSH/LH, TSH and ACTH then follow—loss of sexual function, hypothyroidism, and the diverse effects of glucocorticoid deficiency. Improvements in obstetric management mean that Sheehan’s syndrome is now rare, although hypotensive shock due to trauma may produce similar effects.
Tumours: adenomas
Pituitary tumours account for approximately 10% of primary intracranial neoplasms. They may be derived from any of the hormone-secreting cells and thus may be clinically manifest by virtue of single hormone overproduction, destruction of surrounding normal pituitary and consequent hypofunction, and mechanical effects due to intracranial pressure rise and specific location.
Adenomas are the commonest adenohypophysial tumours; carcinomas are rare. Small adenomas may be asymptomatic and found only at postmortem. Histologically, adenomas consist of nodules containing cells similar to those of the normal adenohypophysis, with many small blood vessels between them. They may produce clinical disease in two ways: excess hormone production and pressure effects.
Excess hormone production
Adenomas may produce any adenohypophysial hormone, depending on their cell of origin (Table 17.4); thus presentation may be via excess production of one of the hormones, for example acromegaly due to excess growth hormone production in an adult (Fig. 17.6), or gigantism if this occurs during childhood.
Table 17.4 Types of adenohypophysial adenoma
| Type | Remarks |
|---|---|
| Prolactinoma (chromophobe) | Commonest type |
| Produces galactorrhoea and menstrual disturbances | |
| GH-secreting (eosinophil) | Produces gigantism in children and acromegaly in adults |
| ACTH-secreting (basophil) | Produces Cushing’s disease |
| Other | Exceptionally rare |
Fig. 17.6 Systemic features of acromegaly. Acromegaly is the clinical syndrome resulting from growth hormone excess in adult life. The chief presenting features are enlargement of the hands, feet and head, but it may also present with secondary diabetes. The cardiovascular effects may be life-threatening.
Pressure effects
These may be either on the surrounding pituitary to produce hypofunction, or on the overlying optic chiasma (Fig. 17.7), producing a characteristic visual field defect called bitemporal hemianopia. Further growth may compress the hypothalamus.
Types of adenoma
All the following adenomas comprise, histologically, nests and cords of a monotonous single cell type, the islands of cells being supported on a richly vascular sinusoidal framework. Amyloid deposition is not infrequent and calcification may occur.
Chromophobe adenoma
The commonest tumour is one derived from apparently inactive cells; thus hormonal manifestations may be absent but more sensitive biochemical assessments suggest that prolactin may be produced by many of these adenomas. The clinical effects may therefore be limited to infertility and be discovered only because of failed conception in the female.
Eosinophil adenoma
Approximately one-third of lesions are derived from the growth hormone-producing cells and are thus manifest by gigantism in the pre-pubertal patient and acromegaly in the adult.
NEUROHYPOPHYSIS
Neurosecretory cells in the supra-optic and paraventricular nuclei of the hypothalamus give rise to modified nerve fibres which carry the two neurohypophysial hormones—antidiuretic hormone and oxytocin—into the posterior lobe of the pituitary (Fig. 17.3); both hormones are nonapeptides, and are stored until released in response to hypothalamic stimuli.
Antidiuretic hormone
Antidiuretic hormone (ADH) controls plasma osmolarity and body water content by increasing the permeability of the renal collecting ducts; this means that more water is reabsorbed and the urine becomes more concentrated. ADH release is stimulated by increased plasma osmolarity and by hypovolaemia.
Damage to the hypothalamus, for example through trauma or tumours, causes deficiency of ADH, leading to production of large volumes of dilute urine accompanied by compensatory polydipsia (excess drinking). This is called diabetes insipidus, from the days when tasting of the patient’s urine was part of the diagnostic process: the urine is tasteless in this condition, whereas in diabetes mellitus it is sweet due to its high glucose content.
Excess ADH is occasionally produced by the neurohypophysis in response to head injury or meningitis, but most clinical cases of ADH excess are due to its ectopic production by tumours, including bronchial carcinomas. The tumours are almost certainly of neuroendocrine origin and thus equipped for the synthesis of peptide hormones.
The rarity of any neurohypophysial tumour secreting ADH (or oxytocin) is perhaps due to the incapacity of the neurones producing these hormones to undergo mitotic division.
Oxytocin
Oxytocin is an aptly named hormone (it is the Greek word for quick birth) as it stimulates the uterine smooth muscle to contract. Interestingly, it is oxytocin from the fetal pituitary that plays the greater role in initiating parturition, suggesting that the fetus orders its own birth. Oxytocin also causes ejection of milk during lactation. The hormone is present in males, although its function, if any, is unknown.
PINEAL GLAND
The pineal gland is a tiny organ lying above the third ventricle of the brain. Little is known of its function, although its secretory product, melatonin, is thought to be involved in circadian rhythm control and gonadal maturation. The most important tumours of the pineal gland are malignant germ cell tumours (teratomas and seminomas) and pinealoblastomas, resembling neuroblastomas.
ADRENALS
The adrenals consist essentially of two separate endocrine glands within a single anatomical organ. The medulla, of neural crest embryological origin, is part of the sympathetic nervous system; it secretes catecholamines, which are essential in the physiological responses to stress, e.g. infection, shock or injury. The cortex, derived from mesoderm, synthesises a range of steroid hormones with generalised effects on metabolism, the immune system, and water and electrolyte balance.
ADRENAL MEDULLA
Histologically, the adrenal medulla consists of chromaffin cells (so called because they produce brown pigments when fixed in solutions of chrome salts) and sympathetic nerve endings. The adrenal medulla is the main source of adrenaline (epinephrine), as it is produced there from noradrenaline (norepinephrine) by the enzyme phenylethanolamine-N-methyl transferase. Elsewhere in the body, sympathetic nerve endings lack this enzyme and their secretory product is thus noradrenaline. Electron microscopy reveals electron-dense granules in the chromaffin cells (Fig. 17.8), similar to those found in other tissues of the so-called amine precursor uptake and decarboxylation (APUD) system. Islands of similar tissue, known as the organs of Zuckerkandl, are sometimes found in other retroperitoneal sites; these have similar functions and a similar pattern of diseases to that seen in the adrenal medulla. Catecholamines are secreted in states of stress and of hypovolaemic shock, when they are vital in the maintenance of blood pressure by causing vasoconstriction in the skin, gut and skeletal muscles. At tissue level, these hormones bind to cell surface receptors, altering cellular levels of a second messenger, cyclic AMP, which brings about rapid functional changes in the cell.
Fig. 17.8 Electron micrograph of noradrenaline granules in a chromaffin cell. The granules characteristically have eccentric electron-dense cores (× 75 000).
Tumours
Phaeochromocytoma
A phaeochromocytoma is derived from the adrenal medullary chromaffin cells (or from those lying in other sites); it is classified as a paraganglioma. The tumour presents through the effects of its catecholamine secretions: hypertension (which is sometimes intermittent), pallor, headaches, sweating and nervousness. Its presence should be suspected especially in younger hypertensive patients. Although it is a rare cause of hypertension, phaeochromocytoma must not be overlooked as it is one of the few curable causes of elevated blood pressure; other causes include adrenal cortical adenoma, renal artery stenosis and aortic coarctation.
The diagnosis of phaeochromocytoma is usually based on estimating the urinary excretion of vanillylmandelic acid (VMA), a catecholamine metabolite, which is generally at least doubled in the presence of the tumour. Localisation of the tumour is assisted by computed tomography of the abdomen and by radio-isotope scanning with 131I-mIBG, a catecholamine precursor that accumulates in the tumour.
Phaeochromocytoma may be familial, associated with medullary carcinoma of the thyroid or with hyperparathyroidism as part of a multiple endocrine neoplasia (MEN) syndrome. The familial cases are frequently bilateral. Other associations are with neurofibromatosis and the rare von Hippel–Lindau syndrome.
Phaeochromocytomas are brown, solid nodules, usually under 50 mm in diameter, often with areas of haemorrhagic necrosis (Fig. 17.9). Histologically, they consist of groups of polyhedral cells which give the chromaffin reaction, and are highly vascular (Fig. 17.10).
Fig. 17.9 Phaeochromocytoma. The adrenal medulla is expanded by a dark-coloured tumour with areas of degeneration and haemorrhage.
Fig. 17.10 Chromaffin cells in a phaeochromocytoma. There are groups of cells with granular cytoplasm, amidst which there are numerous branching capillaries.
Although most are benign, a few phaeochromocytomas pursue a malignant course. It is not generally possible to predict this behaviour from the histological appearance.
Neuroblastoma
Neuroblastoma is a rare and highly malignant tumour found in infants and children. Derived from sympathetic nerve cells it may, like phaeochromocytoma, secrete catecholamines, and there may be elevated levels of their metabolites in the urine. Neuroblastomas may also originate from parts of the sympathetic chain outside the adrenal medulla. Secondary spread to liver, skin and bones (especially those of the skull) is common. Surprisingly, neuroblastoma may occasionally mature spontaneously to ganglioneuroma, a benign tumour.
ADRENAL CORTEX
Histologically, the adrenal cortex has three zones (Fig. 17.11). Beneath the capsule lies the zona glomerulosa, so called because the cells are grouped into spherical clusters superficially resembling glomeruli. This zone produces mineralocorticoid steroids such as aldosterone. Most of the adrenal cortex comprises the middle and inner zones—zona fasciculata and zona reticularis, respectively. The middle zone is rich in lipid. The inner zone cells convert lipid into corticosteroids, principally glucocorticoids and sex steroids, for secretion.
Fig. 17.11 Adrenal cortex. The normal zones are: zona glomerulosa (top), zona fasciculata (middle) and zona reticularis (bottom).
Steroid hormones
Glucocorticoids
The glucocorticoids have important effects on a wide range of tissues and organs. At physiological levels they:
[/not-level-membership-for-pathology-category]
