Endocrine system
A Acromegaly
Definition
Growth hormone (GH) hypersecretion, usually caused by a GH-secreting pituitary adenoma (99% of cases), can produce a highly distinctive syndrome in adults called acromegaly. Acromegaly occurs because of sustained hypersecretion of GH after adolescence. The condition occurs with equal frequency in both sexes. If hypersecretion of GH occurs before puberty—that is, before closure of the growth plates—the individual grows very tall (8–9 feet), a rare condition known as gigantism.
Pathophysiology
The excessive production of GH associated with acromegaly does not induce bone lengthening but rather enhances the growth of periosteal bone. The unrestrained bone growth in patients with acromegaly produces bones that are massive in size and thickness. Bones of the hands and feet (acral) become particularly large. Overgrowth of vertebrae may cause kyphoscoliosis and arthritis.
Soft tissue changes are also prominent with GH hypersecretion. The patient develops coarsened facial features (acromegalic facies), including a large, bulbous nose; a supraorbital ridge overgrowth; dental malocclusion; and a prominent prognathic mandible. The changes in appearance are insidious, and many patients do not seek treatment until the diagnosis is obvious and the disease course is advanced. Overgrowth of the internal organs is less apparent clinically but no less serious. The liver, heart, spleen, and kidneys become enlarged. Lung volumes increase, which may lead to ventilation–perfusion mismatch. Exercise tolerance may be limited because of increased body mass and skeletal muscle weakness.
Cardiomyopathy, hypertension (28% of cases), and accelerated atherosclerosis in patients with acromegaly can lead to symptomatic cardiac disease (congestive heart failure, arrhythmias). Echocardiography often shows left ventricular hypertrophy. Resting electrocardiograms (ECGs) are abnormal in 50% of patients with acromegaly. ST-segment and T-wave depression, conduction defects, and evidence of prior myocardial infarction may be present. The insulin antagonistic effect of GH produces glucose intolerance in up to 50% of patients with acromegaly and frank diabetes mellitus (DM) in 10% to 25% of patients. The insulin antagonistic effect of GH produces glucose intolerance in up to 50% of patients with acromegaly and frank DM in 10% to 25% of patients.
Clinical manifestations
Clinical manifestations resulting from the local effects of the expanding tumor may include headaches (55%), papilledema, and visual field defects (19%), which are caused by compression of the optic nerves and chiasm. Significant increases in intracranial pressure are uncommon. Compression or destruction of normal pituitary tissue by the tumor may lead to panhypopituitarism. Common features of acromegaly are summarized in the box on pg. 58.
Treatment
Treatment for acromegaly is aimed at restoring normal GH levels. The preferred initial therapy for active acromegaly is microsurgical removal of the pituitary tumor with preservation of the gland. The surgical approach to the pituitary tumor most often is via a transsphenoidal route, with the patient in a semi-sitting position. Precautions associated with monitoring of venous air embolism should be part of the anesthesia management plan. Surgical ablation is usually successful in rapidly reducing tumor size, inhibiting GH secretion, and alleviating some symptoms. Administration of octreotide (a long-acting somatostatin analog), pegvisomant (a GH receptor antagonist), and gland irradiation are treatment options for patients who are not surgical candidates.
Anesthetic considerations
Preanesthetic assessment of patients with acromegaly should include a careful examination of the airway. Facial deformities and the large nose may hamper adequate fitting of an anesthesia mask. Endotracheal intubation may be a challenge because these patients have large and thick tongues (macroglossia), enlargement of the thyroid, obstructive teeth, hypertrophy of the epiglottis, and general soft tissue overgrowth in the upper airway. Subglottic narrowing and vocal cord enlargement may dictate the use of a smaller diameter endotracheal tube. Nasotracheal intubation should be approached cautiously because of possible turbinate enlargement.
Preoperative dyspnea, stridor, or hoarseness should alert the anesthetist to airway involvement. Indirect laryngoscopy and neck radiography may be performed for thorough assessment. If difficulties in maintaining an adequate airway are anticipated, a fiberoptic-guided intubation in an awake patient is of proven value. The endotracheal tube should remain in place until the patient is fully awake and has total return of reflexes. The predisposition to airway obstruction in these patients makes assiduous postoperative monitoring of the patient’s respiratory status a wise precaution.
The frequent occurrence of cardiac arrhythmias, coronary artery disease, and hypertension in patients with acromegaly warrants a thorough preanesthetic cardiac evaluation. The increased risk of DM in these patients mandates careful perioperative monitoring of blood glucose and electrolyte levels.
If preoperative assessment reveals impairment of the adrenal or thyroid axis, stress-level glucocorticoid therapy and thyroid replacement should be implemented in the perioperative period.
Entrapment neuropathies, such as carpal tunnel syndrome, are common in patients with acromegaly. An Allen test should be performed before placement of a radial artery catheter because hypertrophy of the carpal ligament may cause inadequate ulnar artery flow.
B Adrenocortical insufficiency
Definition
Primary adrenal insufficiency (Addison’s disease) reflects the absence of cortisol and aldosterone owing to the destruction of the adrenal cortex. In 1855, an English physician, Dr. Thomas Addison, first described a relatively rare clinical syndrome characterized by wasting and skin hyperpigmentation and identified its cause as destruction of the adrenal glands. Primary adrenocortical insufficiency Addison’s disease) becomes apparent when 90% of the gland is destroyed. Tuberculosis is a common cause of primary adrenocortical insufficiency worldwide, but in the United States, most cases are the result of autoimmune dysfunction. Primary adrenocortical insufficiency may also be associated with other autoimmune disorders, such as type 1 diabetes and Hashimoto thyroiditis. Less commonly, primary adrenal insufficiency is congenital or caused by sarcoidosis, human immunodeficiency virus infection, adrenal hemorrhage, malignancy, or trauma.
Clinical manifestations
Clinical symptoms of Addison’s disease reflect destruction of all cortical zones, resulting in adrenal androgen, glucocorticoid, and mineralocorticoid hormone deficiency (see box below).
Weakness and fatigue are cardinal features. Reduced appetite with weight loss, vomiting, abdominal pain, and diarrhea are frequently reported. Hypoglycemia is often present. Volume depletion is a common feature of the disease and may be manifested by orthostatic hypotension. Hyponatremia and hyperkalemia are commonly revealed by laboratory screening.
The adrenal–pituitary axis is intact in primary adrenal insufficiency, and adrenocorticotropic hormone (ACTH) concentrations are elevated as a result of the reduced production of cortisol. Increased melanin formation in the skin and hyperpigmentation of the knuckles of the fingers, toes, knees, elbows, lips, and buccal mucosa may be evident.
Treatment
Treatment for adrenal insufficiency aims to replace both glucocorticoid and mineralocorticoid deficiency. Normal adults secrete 15 to 30 mg of cortisol (hydrocortisone) and 50 to 250 mcg of aldosterone per day. Corticosteroids used for therapy have varying degrees of mineralocorticoid and glucocorticoid effects.
Comparative Pharmacology of Endogenous and Synthetic Corticosteroids
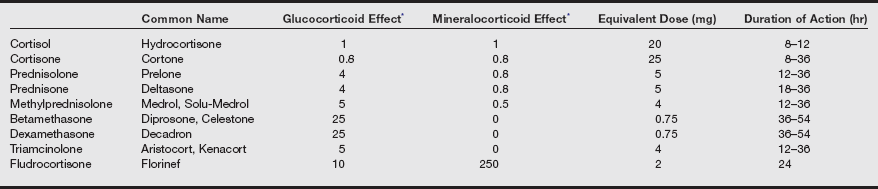
Data from Stoelting RK, Hillier SC. Pharmacology and Physiology in Anesthetic Practice. 4th ed. Philadelphia: Lippincott Williams & Wilkins; 2006:462.
A typical oral replacement dose for Addison’s disease may consist of prednisone, 5 mg in the morning and 2.5 mg in the evening, or hydrocortisone, 20 mg in the morning and 10 mg in the evening. If indicated, mineralocorticoid replacement may consist of 0.05 to 0.2 mg/day of fludrocortisone. Standard glucocorticoid doses should be supplemented during periods of surgical stress.
Anesthetic considerations
Anesthetic management for patients with primary adrenal insufficiency should provide for exogenous corticosteroid supplementation. Etomidate should be avoided because it transiently inhibits synthesis of cortisol in physiologically normal patients. Doses of anesthetic drugs should be minimized because these patients may be sensitive to drug-induced myocardial depression. Invasive monitoring (arterial line and pulmonary artery catheter) is indicated. Because of skeletal muscle weakness, the initial dose of muscle relaxant should be reduced, and further doses should be governed by peripheral nerve stimulator response. Plasma concentrations of glucose and electrolytes should be measured frequently during surgery.
Secondary adrenocortical insufficiency
Definition
Secondary adrenocortical insufficiency is caused by ACTH deficiency from two primary etiologies: (1) hypothalamic–pituitary–adrenal (HPA) axis suppression after exogenous glucocorticoid therapy and (2) ACTH deficiency secondary to hypothalamic or pituitary gland dysfunction (tumor, infection, surgical or radiologic ablation). Long-term treatment with glucocorticoids, for any cause, results in negative feedback to the hypothalamus and pituitary gland, decreased ACTH output, and eventual adrenal cortex atrophy. The longer the duration of glucocorticoid administration, the greater the likelihood of suppression, but the precise dose or duration of therapy that produces adrenal suppression is unknown. Sustained and clinically important adrenal suppression usually does not occur with treatment periods less than 14 days. Treatment periods long enough to provoke signs of Cushing syndrome are usually associated with adrenal suppression of clinically significant.
Clinical manifestations
Clinical manifestations of secondary adrenal insufficiency resemble the primary disease except secondary insufficiency is less likely to be associated with severe hypovolemia, hyperkalemia, or hyponatremia because mineralocorticoid secretion is usually preserved. Hyperpigmentation is absent because ACTH levels are low.
Acute adrenal crisis
Acute adrenal crisis is a sudden exacerbation or onset of severe adrenal insufficiency. It is a rare event associated with high morbidity and mortality if allowed to progress unrecognized. A patient with chronic adrenal insufficiency may deteriorate rapidly into an acute insufficiency state as a result of some superimposed stress, such as infection, acute illness, or sepsis. The stress of surgery or trauma in the patient with inadequate adrenal reserves can precipitate acute adrenal crisis in the perioperative period.
Clinical manifestations
Symptoms of adrenal crisis reflect acute deficiency of corticosteroids and include severe weakness, nausea, hypotension, fever, and decreasing mental status. In the surgical setting, hemodynamic instability or cardiovascular collapse may herald adrenal crisis. The index of suspicion for adrenal crisis should be particularly high if the patient has hyperpigmentation, hyponatremia, or hyperkalemia; a history of autoimmune disease (hypothyroidism, diabetes); or recent prior use of exogenous steroids. The anesthetist should be mindful of the adrenal suppressive effects of etomidate. Even a single dose of etomidate for induction of anesthesia can cause acute adrenocortical insufficiency for up to 24 hours and should be avoided in patients susceptible to adrenal insufficiency.
Treatment
Acute adrenal crisis is a medical emergency requiring aggressive treatment of the steroid insufficiency and associated hypoglycemia, electrolyte imbalance, and volume depletion. Early recognition and intervention are crucial steps in altering the course of acute adrenal insufficiency. Initial therapy begins with rapid intravenous (IV) administration of a glucose-containing isotonic crystalloid solution. If the patient is hemodynamically unstable, advanced hemodynamic monitoring and inotropic support may be necessary. Steroid replacement therapy begins with hydrocortisone, 100 mg IV, followed by hydrocortisone, 100 to 200 mg IV over 24 hours. Mineralocorticoid administration is unnecessary with large doses of steroids (hydrocortisone 100–200 mg) because mineralocorticoid effects are present at these doses.
C Cushing’s disease
Definition
Cushing’s syndrome results in diverse complex symptoms, signs, and biochemical abnormalities caused by excess glucocorticoid hormone.
Pathophysiology
The most common cause of Cushing’s syndrome today is the administration of supraphysiologic doses of glucocorticoids for conditions such as arthritis, asthma, various autoimmune disorders, allergies, and a myriad of other diseases.
Endogenous Cushing’s syndrome is most often the result of one of three distinct pathogenic disorders: pituitary tumor (Cushing’s disease), adrenal tumor, or ectopic hormone production.
Cushing’s disease specifically denotes an anterior pituitary tumor cause of the syndrome. The pituitary tumor produces excessive amounts of ACTH and is associated with bilateral adrenal hyperplasia. Patients often develop skin pigmentation as a result of excess ACTH. Cushing’s disease is the most common cause of endogenous Cushing’s syndrome.
Adrenal Cushing’s syndrome is caused by autonomous corticosteroid production (ACTH independent) by an adrenal tumor, usually unilateral. This form of hyperadrenalism accounts for 20% to 25% of patients with Cushing’s syndrome and is usually associated with suppressed plasma ACTH levels. Adrenal tumors that are malignant are usually large by the time Cushing’s syndrome becomes manifest.
Ectopic Cushing’s syndrome results from autonomous ACTH or corticotropin-releasing hormone (CRH) production by extrapituitary malignancies, producing markedly elevated plasma levels of ACTH. Bronchogenic carcinoma accounts for most of these cases. Carcinoid tumors and malignant tumors of the kidney, ovary, and pancreas also can cause ectopic production of ACTH.
Clinical manifestations
Clinical features reflect cortisol excess, either from overproduction of the adrenal cortex or exogenously administered glucocorticoid. The clinical picture includes central obesity with thin extremities, hypertension, glucose intolerance, plethoric facies, purple striae, muscle weakness, bruising, and osteoporosis. Mineralocorticoid effects include fluid retention and hypokalemic alkalosis. Women manifest a degree of masculinization (hirsutism, hair thinning, acne, amenorrhea), and men manifest a degree of feminization (gynecomastia, impotence) because of the androgenic effects of glucocorticoid excess. The catabolic effects of cortisol result in skin that is thin and atrophic and unable to withstand the stresses of normal activity. Patients with Cushing’s syndrome typically gain weight and develop a characteristic redistribution of fat.
Diagnosis
A widely used test for diagnosis of hyperadrenocorticism is measurement of the plasma cortisol concentration in the morning after a dose of dexamethasone. Dexamethasone suppresses plasma cortisol secretion in normal patients but not in those with endogenous hyperadrenocorticism. Diagnosis of Cushing’s syndrome is also based on elevated levels of plasma and urinary cortisol, plasma ACTH, and urinary 17-hydroxycorticosteroids.
Treatment
Treatment for Cushing’s syndrome depends on the cause. Transsphenoidal hypophysectomy is a primary treatment option for Cushing’s disease. Complications occur in fewer than 5% of patients and include DI (usually transient), cerebrospinal fluid rhinorrhea, and hemorrhage.
Adrenal Cushing’s syndrome may be treated by surgical removal of the adrenal adenoma. Because the contralateral adrenal gland is preoperatively suppressed, glucocorticoid replacement may be necessary for several months after surgery until adrenal function returns. Bilateral adrenalectomy in the patient with Cushing’s syndrome is associated with a high incidence of complications and permanent corticosteroid deficiency.
The treatment of choice for an ectopic ACTH-secreting tumor is surgical removal, but this may not always be feasible because of the nature of the underlying process (e.g., metastatic carcinoma). Metyrapone, an 11-β-hydroxylase inhibitor, and mitotane, an agent that blocks steroidogenesis at several levels, may be used to help normalize cortisol levels.
Anesthetic considerations
Important perioperative considerations for the patient with Cushing’s syndrome include normalizing blood pressure, blood glucose levels, intravascular fluid volume, and electrolyte concentrations. The aldosterone antagonist spironolactone effectively decreases extracellular fluid volume and corrects hypokalemia under these conditions.
Osteopenia is an important consideration in positioning the patient for the operative procedure. Special attention must be given to the patient’s skin, which can easily be abraded by tape or minor trauma. Glucocorticoids are lympholytic and immunosuppressive, placing the patient at increased risk for infection and mandating particular enforcement of aseptic techniques as indicated.
The choice of drugs for induction and maintenance of anesthesia is not specifically influenced by the presence of hyperadrenocorticism. Muscle relaxants may have an exaggerated effect in patients with preexisting myopathy, and a conservative approach to dosing is warranted when significant skeletal muscle weakness is present.
If adrenal resection is planned, glucocorticoids may be indicated after resection and administered at doses equivalent to adrenal output for maximum stress. Hydrocortisone, 100 mg IV, followed by 100 to 200 mg IV over 24 hours can be administered and then reduced over 3 to 6 days postoperatively until a maintenance dose is reached.
D Diabetes insipidus
Definition
Diabetes insipidus (DI) reflects the absence of antidiuretic hormone (ADH) owing to the destruction of the posterior pituitary gland (neurogenic DI) or failure of the renal tubules to respond to ADH (nephrogenic DI).
Pathophysiology
Common causes of neurogenic DI include severe head trauma, neurosurgical procedures (trauma to the median eminence, pituitary surgery), infiltrating pituitary lesions, and brain tumors. DI that develops after pituitary surgery is usually transient and often resolves in 5 to 7 days.
Nephrogenic DI may occur in association with an X-linked genetic mutation, hypercalcemia, hypokalemia, and medication-induced nephrotoxicity. Ethanol, demeclocycline, phenytoin, chlorpromazine, and lithium all inhibit the action of ADH or its release.
Clinical manifestations
The hallmark of DI is polyuria. The inability to produce concentrated urine results in dehydration and hypernatremia. The syndrome is characterized by a urine osmolarity less than 300 mOsm/L, urine specific gravity less than 1.010, and urine volumes greater than 30 mL/kg each day. The tremendous urinary water loss produces serum osmolarities greater than 290 mOsm/L and serum sodium concentrations greater than 145 mEq/L. Neurologic symptoms of hypernatremia and neuronal dehydration may be present and include hyperreflexia, weakness, lethargy, seizures, and coma.
The thirst mechanism assumes a primary role in maintaining water balance in awake patients with DI. Ingestion of large volumes of water prevents hyperosmolarity and life-threatening dehydration.
Treatment
Treatment protocols for DI depend on the degree of ADH deficiency. Most patients have incomplete DI and retain some capacity to concentrate their urine and conserve water. Mild cases (incomplete DI) may be treated with medications that either augment the release of ADH or increase the receptor response to ADH. These drugs may include chlorpropamide (sulfonylurea hypoglycemic agent), carbamazepine (anticonvulsant), and clofibrate (hypolipidemic agent).
Significant deficiency (plasma osmolarity levels >290 mOsm/L) may be treated with various ADH preparations. Aqueous vasopressin is commonly used for short-term therapy, and desmopressin is useful for long-term control. Caution is advised when administering these drugs to patients with coronary artery disease or hypertension because of the arterial constrictive action of ADH. Desmopressin (5 to 10 mcg/day intranasally, or 0.5 to 1 mcg twice daily subcutaneously) is often a preferred agent because it has less pressor activity, a prolonged duration of action (6–24 hours), and enhanced antidiuretic properties.
Anesthetic considerations
Preoperative assessment of the patient with DI includes careful appraisal of plasma electrolytes (especially serum sodium), renal function, and plasma osmolarity. Dehydration makes these patients especially sensitive to the hypotensive effects of anesthesia agents. Intravascular volume should slowly be restored preoperatively over a period of at least 24 to 48 hours.
Perioperative administration of vasopressin is usually not necessary in the patient with partial DI because the stress of surgery causes enhanced ADH release. A surgical patient with a total lack of ADH (complete DI) may be managed with desmopressin (1 mcg subcutaneously) or aqueous vasopressin (an IV bolus of 0.1 units followed by a continuous IV infusion of vasopressin at 0.1–0.2 units/hr). Plasma osmolarity, urine output, and serum sodium concentration should be measured hourly during surgery and in the immediate postoperative period. The surgical patient with DI receiving ADH replacement therapy should be monitored for ECG changes indicative of myocardial ischemia.
Isotonic fluids can generally be administered safely during the intraoperative period. If, however, the plasma osmolarity rises above 290 mOsm/L, hypotonic fluids should be considered and the vasopressin infusion increased above 0.2 units/h.
E Diabetes mellitus
Definition
Diabetes mellitus is a complex metabolic derangement caused by relative or absolute insulin deficiency. Diabetes has been called “starvation in a sea of food.” Glucose is present in abundance, but because of lack of insulin or insulin resistance, it is unable to reach cells for energy provision. Guidelines for diagnosing diabetes include a fasting plasma glucose (FPG) level of 126 mg/dL or greater or a random glucose level greater than 200 mg/dL. The FPG diagnostic level was reduced from a previous value of 140 mg/dL based on findings that patients with an FPG of 126 mg/dL are at risk for diabetes-related complications.
Pathophysiology
The incidence of diabetes has increased dramatically over the past 40 years. Today, it affects nearly 21 million people in the United States (almost 7% of our population). The rise can be attributed to a combination of three factors: (1) an overweight population, (2) more sedentary lifestyles, and (3) a rise in the number of elderly persons. As more of our population advances in age into the decades in which most cases of diabetes occur, the impact of the disease will become even more alarming.
About 5% to 10% of people with DM have type 1 DM. This type of DM was formerly known as insulin-dependent diabetes or juvenile-onset diabetes.
Individuals with type 1 DM have an absolute deficiency of insulin and are therefore entirely dependent on exogenous insulin therapy. In the absence of sufficient exogenous insulin, the disease course may be complicated by periods of ketosis and acidosis.
In most cases, type 1 DM is caused by an unusually vigorous autoimmune destruction of the β cells of the pancreatic islets. Environmental factors, such as infection or exposure to specific antigenic proteins, are cited as possible initiators of the immune assault. Patients with type 1 DM are also more likely to have other autoimmune diseases, such as thyroid disease or Addison’s disease. A genetic predisposition for development of the disease also is involved. Type 1 DM usually develops before the age of 30 years, but it can develop at any age. The classic symptoms of type 1 DM appear only when at least 80% of the β cells are destroyed. The remaining β cells usually are eliminated inexorably over 2 or 3 years. In patients with type 1 DM, daily exogenous insulin therapy is essential for life. Some type 1 DM patients may be candidates for pancreatic transplant. The transplantation of isolated pancreatic islets has been plagued by graft survival and islet isolation setbacks, but it holds out promise for a future cure.
Type 2 diabetes mellitus
About 90% to 95% of patients with diabetes have type 2 DM. Type 2 DM is characterized by impaired insulin secretion, peripheral insulin resistance (a decreased number of insulin receptors or an insulin receptor or postreceptor defect), and excessive hepatic glucose production. This form of diabetes was formerly known as non–insulin-dependent diabetes or maturity-onset diabetes.
Type 2 DM occurs in patients who have some degree of endogenous insulin production but who produce quantities insufficient for sustaining normal carbohydrate homeostasis. Insulin levels may be low, normal, or even elevated, but a relative insulin deficiency exists. The ultimate expression is a hyperglycemic state.
Typically, type 2 DM occurs in patients who are older than 30 years of age, obese (80%), and with a family history of the disease. Type 2 DM has an insidious onset; indeed, it is estimated that half of people who have type 2 DM are not even aware of it. The disease course is rarely associated with ketosis or acidosis, but it may be complicated by a nonketotic, hyperosmolar, hyperglycemic state.
Treatment for this class of diabetes consists primarily of oral hypoglycemic agents, exercise, and diet therapy. Weight reduction in the obese diabetic patient improves tissue responsiveness to endogenous insulin and often restores normoglycemia.
The distinction between insulin-treated DM and insulin-dependent DM is important. Some people with type 2 DM may benefit from exogenously administered insulin, especially during times of illness or stress. Those with type 1 DM, on the other hand, are insulin dependent and require exogenous insulin daily to live.
Diabetes associated with other conditions
Diabetes may result from other conditions such as pancreatectomy, cystic fibrosis, or severe pancreatitis. Certain endocrine conditions, including Cushing’s syndrome, glucagonoma, pheochromocytoma, and acromegaly, may also be associated with a diabetic condition. Steroid-induced diabetes may occur in the patient taking supraphysiologic doses of glucocorticoids. Gestational diabetes occurs in approximately 4% of the pregnancies in the United States. Women who have had gestational DM have a 20% to 50% chance of developing type 2 DM 5 to 10 years postpartum.
Long-term diabetic complications
Diabetics are subject to long-term complications that confer substantial morbidity and premature mortality. These complications include extensive arterial disease, cataracts, sensory and motor neuropathy, infection, and autonomic nervous system dysfunction.
Arterial thrombotic lesions in the diabetic population are widely distributed in the extremities, kidneys, eyes, skeletal muscle, myocardium, and nervous system. As a result of these diffuse lesions, diabetes carries a serious risk for the development of microvascular (nephropathy, retinopathy, neuropathy) and macrovascular (atherosclerosis, stroke, coronary artery disease) complications.
Treatment
Treatment includes a diabetic diet, oral hypoglycemic drugs, and exogenous insulin. Non–insulin-dependent diabetes mellitus (NIDDM) is prevented primarily by avoidance or treatment of obesity. Transplantation of pancreatic tissue may be considered in selected patients.
Anesthetic considerations
Diabetes mellitus is the most common endocrine disorder encountered in surgical patients. Long-standing DM predisposes patients to many diseases that require surgical intervention. Cataract extraction, kidney transplantation, ulcer débridement, and vascular repair are some of the operations frequently performed on patients with DM.
Patients with DM have higher morbidity and mortality rates in the perioperative period compared with nondiabetic patients of similar age. Increased complications are not caused by the disease itself but primarily because of organ damage associated with long-term disease. Ischemic heart disease is the most common cause of perioperative mortality in the patients with DM.
For patients with DM, operations should be scheduled early in the day if possible to minimize disruptions in treatment and nutrition regimens. Day stay for minor surgery may be used for patients with well-controlled DM who are knowledgeable about their disease and treatment and who have proper home support.
Preoperative considerations
Patients with DM may come to the operating room with a spectrum of metabolic aberrations and end-organ complications that warrant careful preanesthetic assessment.
Cardiovascular complications account for most of the surgical deaths in patients with DM. The presence of hypertension, coronary artery disease, or autonomic nervous system dysfunctions can result in a labile cardiovascular course during anesthesia. It is essential that the cardiovascular and volume status of the patient be thoroughly evaluated before surgery.
Preoperative ECG is necessary for all adult patients with DM because of the high incidence of cardiac disease.
Autonomic nervous system dysfunction may result in delayed gastric emptying. It is estimated that gastroparesis occurs in 20% to 30% of all patients with DM. These patients are prone to aspiration, nausea and vomiting, and abdominal distention. Many authorities recommend routine preoperative aspiration prophylaxis with histamine (H2) receptor blockers, metoclopramide, or preinduction antacids for patients with DM. Intubation during general anesthesia is a logical choice for patients with gastroparesis.
Patients with significant autonomic neuropathy may have an impaired respiratory response to hypoxia. These patients are especially sensitive to the respiratory-depressant effects of sedatives and anesthetics and require particular vigilance in the perioperative period.
Peripheral neuropathies (paresthesias, numbness in the hands and feet) should be adequately documented in the preanesthetic evaluation. Their presence may affect the decision to use regional anesthesia.
An estimated 30% to 40% of patients with insulin-dependent diabetes mellitus (IDDM) demonstrate restricted joint mobility. Limited motion of the atlanto-occipital joint can make endotracheal intubation difficult. Demonstration of the “prayer sign,” an inability to approximate the palms of the hands and fingers, may help identify patients with tissue protein glycosylation and potentially difficult airways.
Evidence of kidney disease should be sought, and basic tests of renal function (urinalysis, serum creatinine, blood urea nitrogen) should be performed preoperatively. The presence of renal impairment may influence the choice and dosage of anesthetic agents. The use of potentially nephrotoxic drugs should be avoided.
The anesthetist should examine the patient’s history of glycemic control to ensure preoperative optimization of the patient’s metabolic state. A recommended target blood glucose range for the perioperative period is 80 to 180 mg/dL.
Sustained hyperglycemia, which causes osmotic diuresis, should alert the anesthetist to possible fluid deficits and electrolyte depletion. Preoperative levels of electrolytes should be determined for all patients with DM, and adequate hydration and a good urine output should be maintained. Lactate-containing solutions are generally avoided because lactate conversion to glucose may contribute to hyperglycemia. An important part of the preoperative evaluation is a review of oral hypoglycemic and insulin regimens.
Oral hypoglycemic agents
Oral glucose-lowering agents
Oral glucose-lowering agents and insulin are used as adjuncts to diet therapy and exercise for treating patients with type 2 DM. Currently available oral hypoglycemic agents fall into the following classifications: (1) sulfonylureas, (2) α-glucosidase inhibitors, (3) thiazolidinediones, (4) biguanides, (5) nonsulfonylurea secretagogues, and (6) others. Often, patients take a combination of therapeutic agents. The commonly used medications used to treat type 2 diabetes are listed in the following table.
Oral Agents Available for Treatment of Diabetes Mellitus
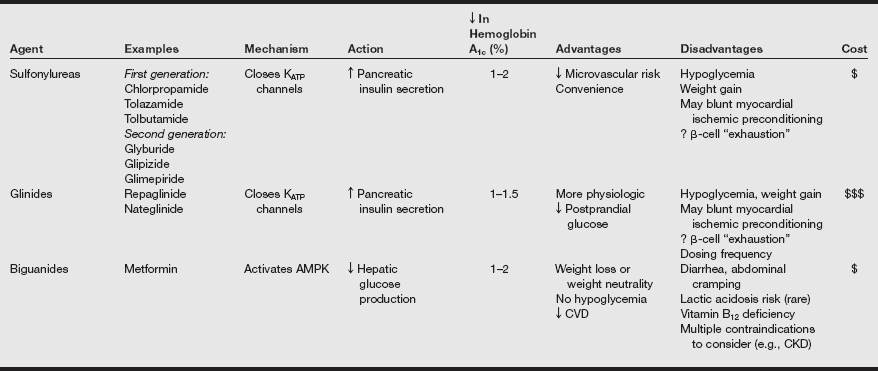
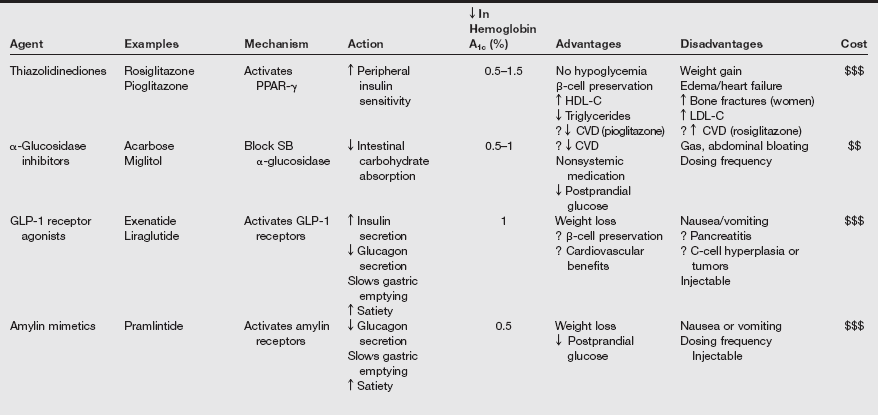
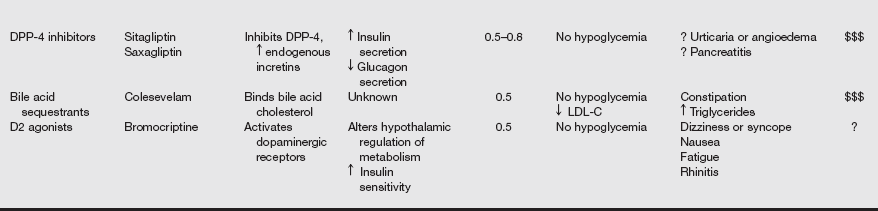
AMPK, Adenosine monophosphate–activated protein kinase; ATP, adenosine triphosphate; CKD, chronic kidney disease; CVD, cardiovascular disease; D2, dopamine-2; DPP, dipeptidyl peptidase; GLP, glucagon-like peptide; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; PPAR, peroxisome proliferator–activated receptor; SB, small bowel.
From Inzucchi SE, Sherwin RS. Type 2 diabetes mellitus. In Goldman L, Schafer AI, et al, eds. Goldman’s Cecil Medicine. 24th ed. Philadelphia: Saunders; 2012:1496.
Insulin preparations
Insulin preparations are generated today by DNA recombinant technology, mimicking the amino acid sequence of human insulin. All insulin formulations in the United States, except for inhaled insulin, are prepared as Unit-100 (100 units/mL).
Insulin preparations differ in onset and duration after subcutaneous administration. In addition to subcutaneous injections, insulin delivery devices (implantable pumps, mechanical syringes) are used to facilitate exogenous administration. The greatest risk with all forms of insulin is hypoglycemia. The major classes of exogenous insulin (regular, rapid acting, inhaled, intermediate acting, and long acting) are listed in the following table.
Pharmacokinetics of Insulin Preparations
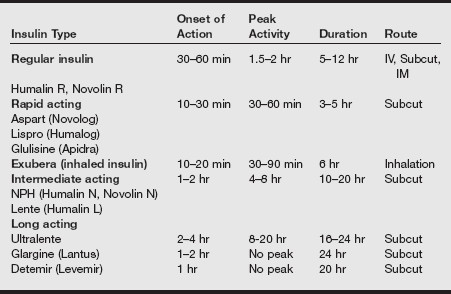
IM, Intramuscular; IV, intravenous; NPH, neutral protamine Hagedorn. Subcut, subcutaneous.
It is imperative to know the surgical patient’s normal insulin dosage regimen and treatment compliance. Some patients are on a fixed regimen that consists of a mixture of rapid- and intermediate-acting insulins taken before breakfast and again at the evening meal. Other patients are on multiple injection regimens designed to provide more physiologic glycemic control. To determine the effectiveness and compliance of antidiabetic therapy, hemoglobin A1c (HbA1c glycated hemoglobin) values provide information about the plasma glucose concentration over time. An HbA1c value of 6.5% or greater is indicative of DM.
Intraoperative management
In patients with DM, operations should be scheduled early in the day, if possible, to minimize disruptions in treatment and nutrition regimens. Surgery produces a catabolic stress response and elevates stress-induced counterregulatory hormones. The hyperglycemic, ketogenic, and lipolytic effects of the counterregulatory hormones in patients with DM compound the state of insulin deficiency. For this reason, perioperative hyperglycemia and other metabolic aberrations are common in the surgical patients with DM.
No specific anesthetic technique is superior overall for patients with DM. Both general anesthesia and regional anesthesia have been used safely. General anesthesia, however, has been shown to induce hormonal changes that accentuate glycogenolysis and gluconeogenesis, compounding the patient’s hyperglycemic state. Regional anesthesia may produce less deleterious changes in glucose homeostasis.
The Certified Registered Nurse Anesthetist (CRNA) must be especially careful in positioning and padding patients with DM on the operating table. Decreased tissue perfusion and peripheral sympathetic neuropathy may contribute to the development of skin breakdown and ulceration.
Patients with DM represent a heterogeneous group requiring individualized perioperative care. The specific approach to metabolic management depends on the type of diabetes (type 1 or 2), the history of glycemic control, and the type of surgery being performed. Frequent blood glucose determinations are an integral part of any diabetic management technique. A glucose meter or other accurate and rapid means of monitoring blood glucose levels should be available. At least hourly intraoperative blood glucose measurement is the prudent course for brittle patients with DM, during long surgical procedures, and for major surgery.
Strict control of even short-term elevations in blood glucose improves perioperative morbidity. Persistent hyperglycemia has been shown to impair wound healing and wound strength. In addition, reports suggest that postoperative infection is more prevalent in patients with DM who have uncontrolled blood sugar levels. Studies also provide evidence that hyperglycemia worsens the neurologic outcome after ischemic brain injury. Avoiding perioperative hyperglycemia is advisable, especially in patients at risk for acute neurologic insult (carotid endarterectomy, intracranial surgery, cardiopulmonary bypass).
Various regimens have been tendered on how to best manage the metabolic changes that occur in surgical patients who have DM. Experts differ on optimal protocols for case management and precisely defined target glucose levels. Current debate centers on the risk-to-benefit ratio of intensive or “tight” blood glucose control versus “nontight” control during surgery. The universal goal with all techniques is to avoid hypoglycemia and to minimize metabolic derangements. Patients under anesthesia are generally maintained with a mild transient hyperglycemia to avoid the potentially catastrophic effects of hypoglycemia. Frequent blood glucose determinations during surgery and in the immediate postoperative period are central to safe practice.
Three different approaches to the metabolic management of adult surgical patients with DM are described below; however, readers should note that there are numerous variations.
Intermediate-acting insulin use
This is a traditional method of managing surgical patients with DM and involves less intensive control of plasma glucose but aims to avoid marked hyperglycemia and dangerous hypoglycemia. Variations of this technique are used for stable patients with DM undergoing elective operative procedures. An example of this regimen follows:
1. On the morning of surgery, fasting blood sugar level is measured.
2. An IV infusion containing 5% dextrose is started at 100 to150 mL/hr.
3. After the IV infusion is started, half of the patient’s normal morning intermediate or long-acting insulin dose is administered subcutaneously.
4. The glucose-containing IV infusion is continued throughout surgery. Additional fluid requirements are met with the administration of a second, glucose-free infusate.
5. Blood glucose levels are checked every 1 to 2 hours during surgery.
6. If the blood glucose level exceeds an established maximum level, commonly 180 mg/dL, regular insulin is administered according to an established “sliding scale.” Insulin sensitivity varies markedly from one patient to the next, but on the average, 1 unit of regular insulin can be expected to decrease the blood glucose level 40 to 50 mg/dL.
This time-tested regimen is easy to implement, and it is usually successful in preventing significant hypo- and hyperglycemia.
The disadvantages of nontight control are as follows:
1. Absorption of preoperatively administered subcutaneous insulin is unpredictable and erratic in surgical patients because of blood pressure, blood flow, and temperature variations that occur with anesthesia.
2. The onset and the peak effect of the preoperative intermediate-acting insulin may not correspond to the time of surgical stress, especially if the operation is delayed or prolonged.
3. The half-life of regular insulin is short, and a “rollercoaster” glucose profile may occur. Plasma glucose levels will vary considerably.
Insulin infusion
An insulin infusion management technique may be used to maintain the blood glucose concentration within relatively narrow boundaries. Intensive perioperative regulation of blood glucose prevents hyperglycemia, but it carries the risk of hypoglycemia and therefore necessitates more frequent blood glucose assays. Regular insulin infusion may range from 0.5 to 5 units/hr depending on the clinical situation and insulin resistance.
Intraoperative insulin infusion may be considered for patients with type 1 DM having major or prolonged surgery, the poorly controlled DM patients, pregnant DM patients, DM patients undergoing coronary artery bypass grafting, and DM patients with serious concurrent illness. An example of this regimen follows:
1. On the morning of surgery, a fasting blood glucose level is measured.
2. An infusion of 5% dextrose is started at a rate of 100 to 150 mL/hr.
3. A regular insulin infusion is begun, piggybacked to the glucose infusion. The insulin infusion rate is set at: insulin (units/hr) = last plasma glucose (mg/dL) ÷ 150. (If the patient is obese, has an infection, or is taking corticosteroids, the divisor is changed to 100.)
4. Blood glucose levels are measured every hour during insulin infusion, and potassium levels are checked after the first hour of the infusion.
5. Additional fluid requirements are met with the administration of a second, glucose-free infusate.
Blood glucose levels less than 80 mg/dL may be treated with D50W and remeasured in 30 minutes. In a 70-kg patient, 15 mL of D50W can be expected to raise the blood glucose concentration by about 30 mg/dL. Surgical patients undergoing renal transplantation or coronary artery bypass graft procedures, obese and septic patients, and patients on steroid therapy usually have higher insulin infusion requirements.
The advantages of tight glucose management in the perioperative period are as follows:
1. The insulin infusion can be finely regulated to correspond to hourly variations in blood glucose levels.
2. Periods of hyperglycemia are less likely. The deleterious effects of hyperglycemia (hyperosmolarity, osmotic diuresis, impaired wound healing, infection) may be prevented.
3. The insulin–glucose infusion can be continued into the postoperative period until the patient is ready to eat, at which time subcutaneous insulin or an oral hypoglycemic agent can be reinstated.
Type 2 diabetes and oral hypoglycemic agents
Patients treated with oral hypoglycemic agents demand the same individualized perioperative management as those with type 1 DM. The duration of action of the patient’s oral agent must be noted. Discontinuing long-acting agents 2 to 3 days before surgery and converting to shorter acting agents or insulin affords better perioperative glucose control. Metformin should be discontinued 2 to 7 days or more before surgery because the surgical risks of hypotension and renal hypoperfusion place patients on this drug at increased risk for lactic acidosis. It is also discontinued before computed tomography (CT) scanning when contrast dye is administered.
For well-controlled surgical patients with type 2 DM who are scheduled for minor to moderate surgery, the patient’s oral hypoglycemic agent may be continued until the evening before surgery. Glucose-containing fluids may be administered intraoperatively to protect against possible residual effects of oral hypoglycemic agents. Other experts adhere to a “no glucose, no insulin” technique for well-controlled type 2 DM patients. Regardless of the technique chosen, plasma glucose should be measured regularly throughout the procedure, and hyperglycemia should be treated with insulin on a “sliding scale.”
Acute derangements in glucose homeostasis
Hypoglycemia is encountered more frequently in patients with DM than in healthy adults, and it can develop insidiously during the perioperative period. Medications (insulin, sulfonylureas, β-adrenergic receptor blocking agents) and toxins (ethanol) are common causes of hypoglycemia. Severe liver disease (impaired hepatic glucose output), the altered physiology associated with gastric bypass surgery, sepsis, or an insulin-secreting tumor of the islets of Langerhans (an insulinoma) are conditions that are often complicated by hypoglycemia.
The blood glucose concentration at which signs and symptoms of hypoglycemia appear varies widely from one person to the next, but blood glucose levels in the range of 45 to 50 mg/dL commonly produce mild symptoms in otherwise healthy patients. Because the brain is the predominant organ of glucose consumption, it is most sensitive to glucose deprivation. Manifestations of impaired cerebral function (confusion, dizziness, headache, weakness) are associated with lack of glucose. As the blood sugar level declines below 50 mg/dL, aberrant behavior, seizures, and loss of consciousness may occur. Other signs of hypoglycemia (tachycardia, diaphoresis, anxiety, tremors, piloerection, pupillary dilation, and vasoconstriction) reflect sympathetic-adrenal hyperactivity. Acute treatment for hypoglycemic surgical patients is the IV administration of 25 to 50 mL of 50% dextrose followed by a continuous infusion of 5% dextrose. Unless prompt glucose therapy is provided, irreversible brain damage may result.
Hypoglycemia is potentially catastrophic during surgery because most of the neural indications of glucose lack are masked by general anesthesia. Signs of sympathetic adrenal discharge may also be blunted by general anesthesia or severe diabetic autonomic neuropathy, making the diagnosis of hypoglycemia extremely difficult. β-Adrenergic receptor blocking agents can reduce the hyperglycemic effects of epinephrine in addition to diminishing the symptomatic warning signs of hypoglycemia. Frequent blood glucose determinations, maintenance of mild hyperglycemia, and diligent monitoring help to avoid this serious complication during anesthesia.
Diabetic ketoacidosis
Diabetic ketoacidosis (DKA) is a medical emergency triggered by a hyperglycemic event, usually in a patient with type 1 DM. Treatment errors, critical illnesses (myocardial infarction, trauma, cerebrovascular accident, burns), and infections are common precipitants of DKA.
Stressful events stimulate the release of hyperglycemic counterregulatory hormones (glucagon, GH, epinephrine, cortisol). People with IDDM are unable to secrete insulin to counterbalance the serum elevations of glucose, free fatty acids, and ketone bodies produced by these stress-induced hormones. Unless exogenous insulin is provided, the glycemic event may progress to severe ketoacidosis, dehydration, and acute metabolic decompensation.
Diabetic ketoacidosis usually develops over 24 hours. Major signs and symptoms include hyperglycemia, volume depletion (average fluid deficit, 5 L), tachycardia, metabolic acidosis, a calculated anion gap greater than 10, electrolyte depletion, hyperosmolarity (>300 mOsm/L), nausea and vomiting, abdominal pain, and lethargy. Blood levels of ketone bodies are elevated, and the patient’s breath may have a fruity odor from excess acetone production. The respiratory center is typically stimulated by the low plasma pH, resulting in rapid, deep breathing (Kussmaul respiration). Acidosis, hyperosmolarity, and dehydration may depress consciousness to the point of coma.
Gangrene and infection of an ischemic lower extremity are common surgical conditions associated with DKA. Preoperative management of surgical patients with DKA requires an aggressive approach to restore intravascular volume, correct electrolyte abnormalities, improve acid–base balance, and reduce blood glucose levels with IV insulin. The airway must be protected in the obtunded patient. After the surgical problem that initiated DKA has resolved, medical management often is more effective.
Hyperglycemic hyperosmolar state
Hyperglycemic hyperosmolar state (HHS) is a life-threatening hyperosmolar condition triggered by a hyperglycemic event. This syndrome commonly occurs in elderly patients with type 2 DM, but it also develops in patients with no history of DM. Patients generally have some endogenous insulin secretion, but the hyperglycemic episode overwhelms the pancreas and produces severe hyperglycemia and glucosuria. The amount of insulin secreted is usually sufficient to prevent lipolysis and ketone production. Therefore, unlike DKA, this syndrome usually is not associated with acidosis or significant ketogenesis. The common features of DKA and HHS can be found on page 77.
Features of Diabetic Ketoacidosis and Hyperglycemic Hyperosmolar Syndrome
| Diabetic Ketoacidosis | Hyperglycemic Hyperosmolar Syndrome | |
| Plasma glucose (mg/dL) | >250 | >600 |
| pH | <7.3 | >7.3 |
| Serum bicarbonate (mmol/L) | <18 | >15 |
| Serum osmolarity | + | ++ |
| Ketonemia | ++ | Normal or slight + |
| Mental obtundation | Variable | Present |
| Hypovolemia | Present | Present |
Common precipitating factors of HHS include infection, sepsis, pneumonia, stroke, and myocardial infarction. A spectrum of symptoms is associated with HHS, culminating in mental confusion, lethargy, and coma. Profound dehydration is present, resulting in hypotension and tachycardia. Laboratory evaluation may reveal a biochemical profile of marked hyperglycemia, normal arterial pH, absent or minimal ketonemia, and hyperosmolarity (>330 mOsm/L). Despite depleted total body potassium stores, the serum potassium levels at presentation may be normal or elevated because of acidosis and insulin lack.
Even with appropriate treatment, the mortality figures for HHS are substantially higher (15%) than those for DKA (<5%), partly because HHS commonly affects an older population group often with accompanying comorbidities.
Treatment goals are similar to those for DKA and include identification and management of the precipitating problem, vigorous isotonic rehydration (average total body water deficit, 9 L), correction of hyperglycemia, and electrolyte replacement. The hazards inherent in aggressive fluid administration in elderly patients dictate central hemodynamic monitoring during treatment.
F Hyperaldosteronism
Definition
Conn’s syndrome, the most common form of primary aldosteronism, results from hypersecretion of aldosterone from an adrenal adenoma independent of stimulus.
Pathophysiology
Primary aldosteronism may also be caused by adrenocortical hyperplasia or rarely carcinoma. An increase in the plasma concentration of aldosterone and an increase in the urinary excretion of potassium with coexisting hypokalemia are pathognomonic of hyperaldosteronism.
Manifestations of the syndrome reflect the exaggerated effects of al-dosterone. Diastolic hypertension and hypernatremia are usually present. Aldosterone’s action of promoting renal excretion of K+ (or H+) in exchange for Na+ results in hypokalemic metabolic alkalosis. Hypertension associated with Conn’s syndrome results from aldosterone-induced sodium retention and subsequent increase in extracellular fluid volume. Primary aldosteronism accounts for approximately 1% of all cases of hypertension.
Primary aldosteronism is associated with low renin levels, a result of the elevated blood pressure’s negative feedback to the juxtaglomerular cells. With secondary hyperaldosteronism, the stimulus of excess aldosterone resides outside of the adrenal gland and is often associated with an increase in circulating renin levels.
Diagnostic and laboratory findings
Diagnosis of hyperaldosteronism is confirmed by increased plasma concentration of aldosterone and increased urinary potassium excretion (>30 mEq/L) despite coexisting hypokalemia. Measurement of plasma renin activity permits classification of the disease as primary (low renin activity) or secondary (increased renin activity).
Treatment
Treatment of primary aldosteronism involves surgical removal of the adenoma or medical management. Surgical intervention is more successful for primary aldosteronism caused by adrenocortical adenoma than for gland hyperplasia because adenomas are almost always unilateral. When the affected adrenal gland is removed, the patient is cured in most cases. For patients with adrenal hyperplasia, medical management has been used successfully to treat primary aldosteronism.
Anesthetic considerations
Preoperative management of the patient with Conn’s syndrome includes correcting electrolyte and blood glucose levels and managing hypertension. Potassium should be replaced slowly to allow for equilibration of intracellular and extracellular potassium stores. Hypokalemia may alter nondepolarizing muscle relaxant responses, making peripheral nerve stimulation monitoring especially valuable. ECG signs of potassium depletion include prominent U waves and arrhythmias. Plasma electrolyte concentrations and acid–base status should be checked often during the perioperative period. Inadvertent hyperventilation may further decrease plasma potassium concentration.
Hypertension may be controlled preoperatively with sodium restriction and aldosterone antagonists such as spironolactone. Spironolactone, 25 to 100 mg every 8 hours, slowly increases potassium levels by inhibiting the action of aldosterone on the distal convoluted tubule. Patients with primary aldosteronism have a higher incidence of left ventricular hypertrophy, albuminuria, and stroke than patients with essential hypertension. Measurement of cardiac filling pressures may be needed to assess fluid volume status in the perioperative period.
Laparoscopic adrenalectomy is currently advocated as the operation of choice for surgically remediable mineralocorticoid excess. Compared with open laparotomy, patients who undergo laparoscopic adrenalectomy have similar improvement in blood pressure control and correction of hypokalemia.
G Hypoaldosteronism
Definition
Hypoaldosteronism causes hyperkalemia in the absence of renal insufficiency. Isolated deficiency of aldosterone secretion may reflect congenital deficiency of aldosterone synthetase or hyporeninemia resulting from a defect in the juxtaglomerular apparatus or treatment with an angiotensin-converting enzyme inhibitor, leading to loss of angiotensin stimulation.
Pathophysiology
Hyporeninemic hypoaldosteronism typically occurs in patients older than 45 years with chronic renal disease, DM, or both. Indomethacin-induced prostaglandin deficiency is a reversible cause of this syndrome.
Clinical manifestations
Symptoms include heart block secondary to hyperkalemia and postural hypotension with or without hyponatremia. Hyperchloremic metabolic acidosis is common.
Treatment
Treatment of hypoaldosteronism includes liberal sodium intake and daily administration of hydrocortisone.
Anesthetic considerations
Begin anesthetic management with preoperative monitoring of the serum potassium level, which should be less than 5.5 mEq/L before elective surgery. ECG monitoring for effects of hyperkalemia (tall, tentlike T waves; heart block) is recommended. Hypoventilation should be avoided to prevent an additional increase in serum potassium. Succinylcholine should be avoided when possible to prevent potassium release. IV fluids should be free of potassium. If hypovolemia is suspected, fluid replacement should be initiated, possibly governed by invasive (i.e., central venous pressure) monitoring.
H Hyperparathyroidism
Definition and incidence
Primary hyperparathyroidism is characterized and diagnosed by the presence of elevated serum parathyroid hormone (PTH) levels despite high serum calcium levels. The incidence of primary hyperparathyroidism in the United States is approximately 0.1% to 0.5%, with a higher occurrence in females and elderly adults. Stimulation of the parathyroid gland during pregnancy or lactation, prior neck irradiation, and a family history of parathyroid disease are predisposing etiologic factors.
Pathophysiology
Primary hyperparathyroidism may result from a parathyroid adenoma, gland hyperplasia, or parathyroid cancer. In approximately 80% of cases, primary hyperparathyroidism is caused by hypersecretion of a single parathyroid adenoma. Hyperplasia of one or more parathyroid glands accounts for about 15% of the cases. Carcinoma of the parathyroid gland is found in fewer than 1% of patients and is associated with particularly high serum calcium levels. Hereditary hyperparathyroidism may exist as part of a multiple endocrine neoplasia (MEN type 1, MEN type 2A).
Clinical manifestations
Sustained overactivity of the parathyroid glands is characterized by high serum calcium levels. Most patients remain asymptomatic until the total serum calcium level rises above 11.5 to 12 mg/dL. Severe hypercalcemia (>14 to 16 mg/dL) may be life threatening and demands immediate attention.
With the development of sensitive laboratory assays for calcium, more than half of patients today with hyperparathyroidism are asymptomatic at diagnosis. Sustained and high levels of PTH over time lead to exaggerated osteoclast activity in bone, resulting in diffuse osteopenia, subperiosteal erosions, and elevated extracellular calcium levels. As osteoblasts attempt to reconstruct the ravaged bone, they secrete large amounts of the enzyme alkaline phosphatase. A heightened serum alkaline phosphatase level, therefore, is a significant diagnostic feature of hyperparathyroidism. Despite an increased mobilization of phosphorus from bone, the serum phosphate concentration usually remains normal or low as a result of increased urinary excretion. The effect of hyperparathyroidism on bone becomes clinically apparent when osteoclastic absorption of bone overwhelms osteoblastic deposition. With severe and protracted disease, the weakened bones become filled with decalcified cavities, making them painful and susceptible to fracture. Because of early diagnosis, the destructive bone disease associated with hyperparathyroidism, osteitis fibrosa cystica, is rare today.
Many of the nonskeletal manifestations of primary hyperparathyroidism are related to the accompanying hypercalcemia. Sustained hypercalcemia may produce calcifications and other deleterious effects in the pancreas (pancreatitis), kidneys (nephrolithiasis, nephrocalcinosis, polyuria), blood vessels (hypertension), heart (shortened ventricular refractory period, bradyarrhythmias, bundle branch block, heart block), and acid-producing areas of the stomach (peptic ulcer). The mnemonic “stones, bones, and groans” summarizes the renal, skeletal, and gastrointestinal features of advanced hyperparathyroidism. Profound muscle weakness, confusion, nausea, vomiting, and lethargy are additional features of the disorder.
Secondary hyperparathyroidism develops in patients with chronically low levels of serum calcium, such as those with chronic renal failure and gastrointestinal malabsorption. A compensatory parathyroid response develops in response to the hypocalcemia. Their clinical course is marked by the same PTH-mediated skeletal assault seen in the primary form of the disorder, but because it is an adaptive response, secondary hyperparathyroidism is seldom associated with hypercalcemia.
Anesthetic considerations
The usual treatment for symptomatic primary hyperparathyroidism is surgical removal of abnormal parathyroid tissue. Surgical treatment for asymptomatic hyperparathyroidism is more controversial. Parathyroidectomy may be performed with the patient under general anesthesia, although minimally invasive neck surgery using cervical plexus block anesthesia is increasingly utilized, especially for excision of a single adenoma.
Parathyroid tissue resembles brown fat, and this can occasionally make it difficult for the surgeon to locate. Furthermore, parathyroid tissue is sometimes footloose and can be found in such ectopic places as the deep recesses of the mediastinum, the carotid sheath, or the thymus gland. Some surgeons use periodic intraoperative determinations of serum PTH and ionized calcium levels to help guide surgical resection.
Blood loss from parathyroid surgery is usually minimal, and advanced monitoring is not required based on the surgical procedure. Serum calcium, magnesium, and phosphorous levels should be monitored in the postoperative period until stable. In most cases, serum calcium levels start to decline within 24 hours and return to normal within 3 to 4 days after successful surgery.
With current methods of detection, most patients with hyperparathyroidism are asymptomatic; however, erosive effects of elevated PTH on bone and the systemic effects of chronic hypercalcemia should be considered in the anesthetic plan for patients with severe untreated disease.
Severe or symptomatic hypercalcemia (>14–16 mg/dL) is treated aggressively. Isotonic saline hydration and loop diuretics (Lasix, 40–80 mg) can rapidly decrease serum calcium levels by hemodilution, increased glomerular filtration, and enhanced excretion. Less frequently, corticosteroids or drugs that inhibit osteoclastic bone resorption (bisphosphonates, pamidronate, plicamycin, calcitonin) are used.
Patients with hypercalcemia may be dehydrated because of anorexia, vomiting, and the impaired ability of the kidneys to concentrate urine. In these patients, hydration with non–calcium-containing solutions should be maintained throughout the perioperative period to dilute serum calcium, maintain adequate glomerular filtration and calcium clearance, and ensure adequate intravascular volume. Vigorous hydration dictates the use of bladder catheterization, central venous pressure monitoring, and frequent determinations of serum electrolytes. Elevated calcium levels may depress the central and peripheral nervous systems. The use of preoperative sedatives in patients with hypercalcemia who appear lethargic or confused should be avoided. General anesthetic requirements may be decreased as well.
Careful review of the patient’s renal status is especially crucial in patients with secondary hyperparathyroidism. Associated complications of renal impairment (volume overload, anemia, electrolyte derangements) may affect anesthetic medication dosages and selection.
Cardiac conduction disturbances such as a shortened QT interval and a prolonged PR interval are observed with hypercalcemia. Dysrhythmias and hypertension may respond to calcium channel antagonists (e.g., verapamil, 5–10 mg IV).
Awareness of the effects of pH on the ionized portion of plasma calcium is important. Alkalosis shifts the ionized calcium to the protein-bound form and decreases serum levels. The response to neuromuscular blockade may be unpredictable. Muscle weakness, hypotonia, and muscle atrophy may increase the patient’s sensitivity to nondepolarizing skeletal muscle relaxants. Careful titration of muscle relaxants with use of a peripheral nerve stimulator is prudent.
Patients with clinically significant bone disease are susceptible to fractures, and care must be exercised in positioning and padding. These patients are prone to postoperative nausea and vomiting; therefore, prophylactic antiemetic medications are advisable.
I Hypoparathyroidism
Definition
Hypoparathyroidism is a disorder characterized by inadequate secretion of PTH or a peripheral resistance to its effect. Patients with hypoparathyroidism typically have low serum calcium levels. The blood phosphate concentration may be elevated because of the decreased renal excretion of phosphate.
Pathophysiology
Inadvertent removal of parathyroid tissue, parathyroid gland injury from irradiation or autoimmune destruction, and chronic severe magnesium deficiency (alcohol abuse, poor nutrition, malabsorption) are possible causes of hypoparathyroidism.
Clinical manifestations
Clinical signs of hypoparathyroidism reflect the degree of hypocalcemia and the rapidity of calcium decline. A sudden drop in ionized calcium usually produces more severe symptoms than a slow decline. Treatment of chronic hypoparathyroidism includes vitamin D and calcium supplementation.
The decreased serum calcium ion concentration accompanying hypoparathyroidism produces hyperexcitability of nerve and muscle cells by lowering the threshold potential of excitable membranes. Cardinal features of the neuromuscular excitability are muscle spasms and hypocalcemic tetany. Symptoms vary in severity and may take the form of muscle cramps, perioral paresthesias, numbness in the feet and toes, or hyperactive deep tendon reflexes. The patient may feel restless or hyperirritable. Life-threatening laryngeal muscle spasm may occur, producing stridor, labored respirations, and asphyxia.
Two classic manifestations of latent hypocalcemic tetany are the Chvostek’s sign and Trousseau’s sign. The Chvostek’s sign is a contracture or twitching of ipsilateral facial muscles produced when the facial nerve is tapped at the angle of the jaw. The Trousseau’s sign is elicited by the inflation of a blood pressure cuff slightly above the systolic level for a few minutes. The resultant ischemia enhances the muscle irritability in hypocalcemic states and causes flexion of the wrist and thumb with extension of the fingers (carpopedal spasm).
Anesthetic considerations
Temporary hypocalcemia often is observed after successful parathyroid surgery for hyperparathyroidism. This may occur within a few hours to a few days after surgery. The transient postoperative hypocalcemia is the result of parathyroid gland suppression (by preoperative hypercalcemia) and rapid bone uptake of calcium (“hungry bone syndrome”). Inadvertent removal of all parathyroid gland tissue induces a substantial decline in the serum calcium concentration, from a normal level to 6 to 7 mg/dL. Even a small amount of remaining parathyroid tissue usually is capable of sufficient hypertrophy to preserve normal calcium–phosphate balance.
Meticulous observation for signs of musculoskeletal irritability and serial measurement of serum calcium, inorganic phosphate, magnesium, and PTH levels should be performed after parathyroid surgery. The threshold for the development of signs of hypocalcemia is variable; however, manifestations of neuromuscular compromise often are observed at serum calcium levels of 6 to 7 mg/dL.
Laryngeal muscles are especially sensitive to tetanic spasm, and laryngospasm may cause life-threatening airway compromise in patients with hypocalcemia. Respiratory distress after parathyroid surgery may be secondary to laryngeal muscle spasm, edema or bleeding in the neck, or bilateral recurrent laryngeal nerve injury. Unilateral recurrent laryngeal nerve injury produces hoarseness and usually requires only close observation. Bilateral recurrent laryngeal nerve injury causes aphonia and requires immediate airway support and intubation.
Hypocalcemia may be apparent on ECG tracings as a prolonged QT interval, reflecting delayed ventricular repolarization. The cardiac rhythm usually remains normal. Decreased cardiac contractility and hypotension may occur, and congestive heart failure, although rare, is a danger.
In addition to parathyroid surgery, circulating levels of ionized calcium can decline from other causes in the perioperative period. Precipitous increases in the circulating levels of anions such as bicarbonate, phosphate, and citrate lower ionized calcium levels. Hyperventilation, the rapid transfusion of citrated blood, or the rapid administration of bicarbonate may induce overt tetany in a previously asymptomatic patient with hypocalcemia. Vigorous diuresis can also augment calcium loss.
Patients with confirmed, symptomatic hypocalcemia require prompt therapy. Those with acute hypocalcemia may be treated with an initial IV bolus of 10 to 20 mL of 10% calcium gluconate administered over 10 minutes followed by 10 mL of 10% calcium gluconate in 500 mL solution over 6 hours. Calcium, magnesium, phosphate, potassium, and creatinine levels should be monitored diligently during calcium replacement. Chronic magnesium deficiency impairs the secretion of PTH and should be corrected.
Common clinical manifestations of hyperparathyroidism and hypoparathyroidism are listed in the table below.
Clinical Features of Hyperparathyroidism and Hypoparathyroidism
| System | Hyperparathyroidism | Hypoparathyroidism |
| Cardiovascular | Hypertension, cardiac conduction disturbances, shortened QT interval | Prolonged QT interval, hypotension, decreased cardiac contractility |
| Musculoskeletal | Bone pain, pathologic fractures, muscle weakness, muscle atrophy | Neuromuscular excitability |
| Neurologic | Somnolence, cognitive impairment, depression, hypotonia | Tetany, paresthesias, numbness in fingers and toes, seizures |
| Gastrointestinal | Anorexia, nausea, vomiting, constipation, abdominal pain, pancreatitis, peptic ulcer | None significant |
| Renal | Tubular absorption defects, diminished renal function, kidney stones, polyuria | None significant |
J Hyperthyroidism
Definition
Hyperthyroidism is defined as thyroid gland hyperactivity. Thyrotoxicosis is more specifically defined as a state of thyroid hormone excess. The most common cause of thyrotoxicosis in the United States is Graves’ disease. Graves’ disease is an autoimmune disease in which thyroid-stimulating hormone (TSH) receptor antibodies bind to and stimulate the thyroid gland, causing excessive production and secretion of thyroxine (T4) and triiodothyronine (T3). The immunoglobulin G (IgG) autoantibodies mimic the action of TSH, but their effects are longer, lasting up to 12 hours compared with 1 hour for normal TSH.
The aberrant immunologic response associated with Graves’ disease targets primarily the thyroid gland but also other tissues, including extraocular muscles and skin. There is a familial tendency and a higher incidence of other autoimmune disorders in patients with Graves’ disease. The disease occurs most often in women (prevalence, 1%–3% in women; 0.1% in men) and between 20 and 50 years of age. Graves’ disease has an unpredictable course marked by relapses and exacerbations.
Pathophysiology
Thyrotoxicosis can also be caused by benign follicular adenomas, which are not believed to have an autoimmune etiology. Exogenous iodine excess (radiocontrast agents or angiography dye) or the administration of thyroid hormones may induce iatrogenic thyrotoxicosis. The antiarrhythmic agent amiodarone is iodine rich and may cause either hypothyroidism or hyperthyroidism. Toxic multinodular goiter, subacute viral thyroiditis, postpartum thyroiditis, TSH-secreting pituitary tumors, and thyroid cancer are less common causes of thyrotoxicosis.
Clinical manifestations
Clinical manifestations associated with thyrotoxicosis reflect the widespread hypermetabolic effects of excess thyroid hormones. Physical signs include tachycardia, tremor, goiter, and muscle weakness. Sleep is often difficult. Weight loss despite increased food consumption, anxiety, fatigue, and heat intolerance are symptoms of thyrotoxicosis.
Signs of ophthalmopathy and dermopathy are associated with Graves’ disease. Thyroid-associated ophthalmopathy may cause proptosis, eye redness, and a gritty sensation in early stages, with diplopia, ocular pain, and (rarely) loss of visual acuity in more advanced stages. Graves’ ophthalmopathy results from cytokine-mediated inflammation and swelling of the periorbital connective tissue and extraocular muscles.
The blood volume increases slightly under the influence of excess thyroid hormone, a result of vasodilation. Mean arterial pressure usually remains unchanged, but the pulse pressure increases. The systolic blood pressure is typically elevated 10 to 15 mmHg, and the diastolic blood pressure is reduced. Blood flow to the skin increases because of the increased need for heat elimination.
The effects of thyrotoxicosis on the heart are pronounced. Palpitations, tachycardia, and cardiac dysrhythmias affect most patients. The cardiac output increases, sometimes to 60% or more above normal. About 10% of thyrotoxic patients have atrial fibrillation. Mitral valve prolapse is more common in patients with Graves’ disease than in the general population. With protracted high thyroid hormone levels, the heart muscle strength may become depressed as a result of protein catabolism. Diagnosis of thyrotoxicosis is more difficult in elderly adults because many of the hyperkinetic manifestations of hyperthyroidism are absent. Elderly patients may initially present with myocardial failure. Hyperthyroid patients may feel a constant fatigue from the exhausting effect of thyroid hormone on the musculature and on the central nervous system.
Diagnosis
The diagnosis of primary disease is biochemically established in most cases by the combined findings of an abnormally high total and unbound serum T3 and T4 assay and depressed TSH levels. With Graves’ disease, the diagnosis may be supported by the presence of stimulatory TSH receptor autoantibodies. An elevated uptake of radioactive iodine by the thyroid gland may be used to confirm gland hyperactivity. Serum alkaline phosphatase and calcium concentrations are mildly elevated in approximately 20% of patients with Graves’ disease.
Other autoimmune diseases such as myasthenia gravis, rheumatoid arthritis, systemic lupus erythematosus, and DM are more common in patients with Graves’ disease.
Treatment
A variety of treatment options are available for patients with Graves’ disease. The three primary treatment options for thyrotoxicosis are radioactive gland ablation, surgery, and antithyroid drug therapy.
Radioactive iodine
A common therapy for Graves’ disease is ablation of the thyroid gland with radioactive iodine. A total of 2 to 4 months is needed to reverse the effects of hyperthyroidism. Hypothyroidism is common after treatment, and it is contraindicated in pregnancy.
Subtotal thyroidectomy
Surgery for treatment of Graves’ disease is an option when antithyroid drugs are ineffective, if radioiodine treatment is refused, in children or pregnant women, or if the thyroid goiter is exceptionally large. Patients should be treated preoperatively with antithyroid medication and rendered euthyroid before surgery. Complications associated with thyroid surgery occur in less than 1% of cases and include damage to the recurrent laryngeal nerve, hypoparathyroidism, and neck hematoma.
Antithyroid drugs and β-adrenergic receptor blockade
The main class of antithyroid medications are the thionamides, which include propylthiouracil (PTU), methimazole, and carbimazole. All thionamides inhibit thyroid hormone synthesis by interfering with the incorporation of iodine into tyrosine residues of thyroglobulin. PTU also inhibits conversion of T4 to T3. A euthyroid state is usually obtained in 6 to 7 weeks. Hepatitis and agranulocytosis are the most serious side effects of these drugs.
About 10 days before surgery, oral potassium iodide (SSKI, Lugol’s solution) is added to the course of therapy to decrease gland vascularity and to block hormone synthesis and release. Propranolol is added to the antithyroid regimen to reduce cardiovascular symptoms and to inhibit the peripheral conversion of T4 to T3.
Anesthetic considerations
Preoperative
The key to successful preoperative preparation of the hyperthyroid surgical patient is a careful assessment of the extent of thyrotoxicosis and the severity of end-organ manifestations. Thyrotoxicosis is associated with increased operative risk, and elective surgery should not proceed until the patient has been rendered euthyroid by medical management. Antithyroid medications should be continued through the morning of surgery.
Hyperthyroid patients have increased blood volume, decreased peripheral resistance, and a wide pulse pressure. The cardiac output, heart rate, and systolic blood pressure may be increased. Appropriate corrections of the patient’s fluid volume and electrolyte status should be accomplished before surgery.
A careful preoperative evaluation of the airway is mandatory in all hyperthyroid patients undergoing surgery. Thyroid gland enlargement can cause tracheal deviation and tracheoesophageal compression. Hoarseness, sore throat, a feeling of pressure in the neck, coughing, or dyspnea suggests tracheal compression that can be caused by thyromegaly. Chest and airway radiographs and CT scans are useful to detect tracheal deviation and compression.
A patient with a large goiter and an obstructed airway poses the same challenge as any other patient in whom airway management is problematic. An awake fiberoptic intubation with topical anesthesia is of proven value under these conditions. Tracheomalacia weakens thyroid cartilage from chronic pressure, and its presence may necessitate a more prolonged intubation and vigilant observation after surgery.
Only life-threatening emergency surgery should be performed in an untreated symptomatic hyperthyroid patient. In an emergency situation, the otherwise healthy patient can be expeditiously prepared for surgery with the oral administration of potassium iodide (3–5 drops every 6 hours) and carefully titrated IV propranolol (1–10 mg) or esmolol (50–300 mcg/kg). Elderly patients who require emergency surgery and have rapid ventricular rates require central pressure monitoring to guide therapy.
Intraoperative
A major goal of perioperative management of hyperthyroid patients is prevention of sympathetic nervous system stimulation. This is accomplished by providing sufficient anesthetic depth and avoiding medications that stimulate the sympathetic nervous system.
A preoperative anxiolytic medication is generally warranted. Atropine should be avoided as an antisialagogue because of its vagolytic effects and its ability to impair sweating.
Induction of anesthesia may be achieved with a number of IV medications. Ketamine should be avoided because it can stimulate the sympathetic nervous system. If the airway is not compromised by an enlarged goiter, administration of a muscle relaxant can facilitate intubation of the trachea. Pancuronium should be avoided because it has the potential to increase the heart rate. Because of the increased incidence of myasthenia gravis and skeletal muscle weakness in hyperthyroid patients, precaution dictates careful titration of muscle relaxant doses with use of a peripheral nerve stimulator.
Isoflurane and sevoflurane are attractive choices for inhalation anesthetics because of their ability to offset sympathetic nervous system responses to surgical stimulation and because they do not sensitize the myocardium to catecholamines. Hyperthyroid patients do not generally require a higher minimum alveolar concentration (MAC) for inhalational anesthesia. The increased cardiac output accompanying hyperthyroidism may accelerate the uptake of an inhaled anesthetic, resulting in the need to increase the delivered concentration, and this may be perceived clinically as an increased anesthetic requirement.
Monitoring of the hyperthyroid patient should focus on early recognition of increased thyroid gland activity, suggesting the onset of thyroid storm. Core body temperature should be monitored closely. The ECG should be assessed for tachycardia or dysrhythmias. Hypotension occurring during surgery is better treated with direct-acting vasopressors than with indirect-acting vasoactive drugs that stimulate the release of catecholamines. Hypercarbia and hypoxia should be stringently avoided because they stimulate the sympathoadrenal axis.
Meticulous care of the eyes is required. The patient with proptosis is at risk for corneal exposure and damage, so special care should be taken to lubricate and protect the eyes perioperatively. The key anesthesia implications for patients with hyperthyroidism are listed below.
A feared complication in hyperthyroid patients is thyroid storm or thyrotoxic crisis. Thyroid storm is a rare event that is caused by acute stress in the previously undiagnosed or incompletely treated hyperthyroid patient. Precipitating events may include trauma, surgery (especially thyroid surgery), the peripartum period, radioiodine treatment, acute illness, and infection.
Thyroid storm is a life-threatening medical emergency that represents a severe exacerbation of hyperthyroid signs and symptoms. The clinical manifestations may include marked tachycardia, hyperthermia, hypertension, atrial fibrillation, sweating, tremor, vomiting, weakness, agitation, shock, and congestive heart failure (see box below). Metabolic acidosis may be present secondary to increased lactate production from the overactive metabolism.
Similarities exist between the clinical features of thyroid storm and those of pheochromocytoma, neuroleptic malignant syndrome, light anesthesia, and malignant hyperthermia, making clinical diagnosis challenging in some cases.
Thyroid storm associated with surgery may occur anytime in the perioperative period but is more likely to occur 6 to 18 hours after surgery. Treatment must be initiated as soon as the diagnosis is made to prevent substantial morbidity and mortality. Mortality rates are as high as 30% even with early diagnosis and management. The high mortality rate associated with thyroid storm underscores the importance of achieving a euthyroid state before surgery.
Management of perioperative thyroid storm includes identifying and treating the precipitating cause, administering antithyroid medications, and providing hemodynamic support. Carefully titrated β-adrenergic receptor blockers, potassium iodide, and antithyroid drugs (PTU or methimazole) block thyroid hormone synthesis and adrenergic manifestations. Supplemental glucocorticoids should be administered because the turnover of endogenous steroids is accelerated by the hypermetabolism of thyrotoxicosis.
Supportive measures include IV hydration with glucose-containing crystalloid solutions, correction of electrolyte and acid base imbalances, and management of hyperthermia. Salicylates may displace T4 from its carrier protein; therefore, acetaminophen is the recommended antipyretic for lowering body temperature. Adequate oxygenation is of paramount importance during thyroid storm. Vasoactive medications and advanced hemodynamic monitoring may be necessary to help manage the labile cardiovascular course.
K Hypothyroidism
Definition and incidence
Hypothyroidism is a state of thyroid gland hypofunction resulting in decreased circulating concentrations of thyroid hormones. Laboratory findings show decreased plasma T3 and T4 concentrations and increased TSH levels in patients with primary hypothyroid disease. The clinical spectrum of thyroid hormone deficiency can range from the asymptomatic patient with no overt physical findings to the classic myxedematous patient with profound symptoms. Hypothyroidism is the most common disorder of thyroid function, occurring in 5% to 10% of women and 0.5% to 2% of men.
Pathophysiology
Primary hypothyroidism accounts for 95% of all cases of hypothyroidism. An autoimmune-mediated destruction of the thyroid gland, known as Hashimoto’s thyroiditis, is the most common form of hypothyroidism in the United States. The disorder most often occurs in women of middle age and is associated with other autoimmune disorders such as myasthenia gravis and adrenal insufficiency.
Primary hypothyroidism may also be the result of severe iodine deficiency, previous thyroid surgery, neck irradiation, or treatment for hyperthyroidism (radioiodine therapy). The antiarrhythmic agent amiodarone is associated with hyper- and hypothyroidism. Lithium inhibits the release of thyroid hormone and causes hypothyroidism in some patients.
Rarely, secondary hypothyroidism is the result of pituitary or hypothalamic disorders. Secondary hypothyroidism is associated with decreased concentrations of both thyroid hormones and TSH. Regardless of the etiology, the clinical manifestations of hypothyroidism are similar.
Clinical manifestations
Most cases of hypothyroidism are subclinical, with laboratory findings of increased plasma TSH but no overt signs. Patients with more significant disease develop signs and symptoms that reflect a slowed metabolism and impaired cellular functions. The thyroid gland usually is enlarged, nontender, and firm. Patients may have dry skin, cold intolerance, paresthesias, slow mental functioning, ataxia, a puffy face, and constipation. Lack of thyroid hormones causes the muscles to become sluggish. Patients with severe hypothyroidism may be hypersomnolent with a decreased ventilatory response to hypoxia and hypercarbia. The hair and nails frequently are brittle.
The accumulation of proteinaceous fluid in serous body cavities is a well-recognized feature of hypothyroidism. The most common sites of effusions associated with hypothyroidism are the pleural, pericardial, and peritoneal cavities. Inappropriate ADH secretion and impaired free water clearance can lead to hyponatremia. Accumulation of mucopolysaccharides and fluid imparts the characteristic edematous appearance, called myxedema.
Cardiovascular complications include sinus bradycardia, dysrhythmias, cardiomegaly, impaired contractility, congestive heart failure, and labile blood pressure. Symptoms of low exercise tolerance and shortness of breath with exertion may be partially the result of decreased cardiac function. Chronic vasoconstriction produces diastolic hypertension and decreases the intravascular fluid volume. The autonomic nervous system response is blunted, and there is a decrease in the sensitivity and number of β-receptors.
Overt hypothyroidism is associated with a number of abnormalities in lipid metabolism that may predispose patients to accelerated coronary artery disease. Hypothyroidism is associated with anemia and decreased erythrocyte production of 2,3-diphosphoglycerate, leading to a leftward shift of the oxyhemoglobin dissociation curve and decreased oxygen delivery to the tissues.
These “classic” clinical features of hypothyroidism are often lacking in elderly hypothyroid patients. In older patients, thyroid status may not always be predicted from clinical signs and symptoms, and diagnosing hypothyroidism is more difficult.
Treatment
Treatment of hypothyroidism requires replacement with thyroid hormone. The agent of choice is synthetic levothyroxine sodium (T4) because of its long half-life (7 days) and its ability to attain physiologic levels of T3. Replacement dosages range from 75 to 150 mcg/day, depending on the underlying autonomous thyroid function.
An area of particular concern during thyroid hormone replacement is the effect on the cardiovascular system. Initiation of thyroid hormone replacement in a patient with coexisting angina pectoris or underlying risk factors for coronary artery disease is potentially hazardous and requires careful monitoring of both cardiovascular and thyroid status. Myocardial oxygen consumption is augmented by thyroid hormone, and a hypothyroid patient with deficient coronary artery circulation may not tolerate full replacement doses.
Anesthetic considerations
Patients with subclinical disease have an overall low risk of complications when undergoing anesthesia and surgery. These patients should receive a careful preoperative evaluation and preoperative continuation of levothyroxine therapy. Patients with severe hypothyroidism are predisposed to multiple complications with anesthesia. Depression of myocardial function, abnormal baroreceptor function, and reduction in plasma volume may be present. Slowed hepatic metabolism and renal clearance of injected drugs may prolong their effects, but MAC is not decreased significantly. Elective surgical procedures should be postponed in the presence of severe or symptomatic hypothyroidism to restore normal thyroid status.
All patients with hypothyroidism should undergo careful preoperative evaluation of the airway. A large goiter may cause airway compromise in the form of tracheal deviation or compression. In some patients with severe hypothyroidism, adequate air exchange may be compromised by an enlarged tongue and myxedematous infiltration of the vocal cords. Depression of the ventilatory responses to hypoxia and hypercarbia must be considered. Preoperative sedation should be avoided in patients with macroglossia or preexisting hypoventilation. The risk of pulmonary aspiration is increased because of associated somatic obesity and delayed gastric emptying.
Patients with hypothyroidism may respond to opioids with increased central nervous system and respiratory depression and to volatile agents with increased hypotension and myocardial depression. Although ketamine has been proposed as the ideal induction agent, even ketamine can produce cardiovascular depression in the absence of a robust sympathetic nervous system.
Intubation of the trachea may be facilitated by the administration of succinylcholine or a nondepolarizing muscle relaxant; however, hypothyroid patients may be more sensitive to standard doses of nondepolarizing muscle relaxants because of coexisting muscle weakness and decreased hepatic metabolism and renal elimination of these drugs. Maintaining muscle paralysis with minimal doses of muscle relaxants is an appropriate goal.
Supplemental perioperative cortisol should be considered in patients with symptomatic disease as a potential for adrenal insufficiency when stress exists. In hypothyroidism, the number of β-receptors is diminished, and responses to inotropic drugs and sympathetic stimulation may be influenced by the altered β-receptor pool.
Body temperature should be monitored closely in hypothyroid patients, and mechanisms for warming the patient should be used during surgery. The box on pg. 91 summarizes the anesthesia implications of hypothyroidism.
Myxedema coma is a rare syndrome that reflects the end stage of untreated hypothyroidism. The presence of coma is a marker of the patient’s clinical deterioration rather than a primary effect of hypothyroidism. A critical insult (infection, surgery, cerebrovascular accident, pneumonia, gastrointestinal bleeding, cold exposure) can precipitate myxedema coma in a patient with hypothyroidism.
Generally, the patient is elderly; has severe clinical features of hypothyroidism; and is hypothermic, hypoventilating, and hyponatremic. The response to hypoxia and hypercapnia is measurably decreased, and mechanical ventilation may be required. The patient is typically lethargic or stuporous. The skin often is pale as a result of cutaneous vasoconstriction. Myxedema coma is a medical emergency. Vigorous therapeutic attention should be paid to body temperature, shock, and ventilatory failure. Treatment consists of hemodynamic and ventilatory support and the IV administration of levothyroxine (300–500 mcg) with continuous ECG monitoring for myocardial ischemia. Supplemental cortisol is appropriate because myxedematous patients may have adrenal atrophy and decreased adrenal reserve. Because these patients may be vulnerable to water intoxication and hyponatremia, meticulous fluid replacement is important. Only lifesaving surgery should proceed in a patient with myxedema coma.
L Pheochromocytoma
Definition
Pheochromocytomas are catecholamine-secreting tumors derived most commonly from adrenomedullary chromaffin cells. The tumors synthesize, store, and secrete catecholamines, mostly norepinephrine and epinephrine. Unlike a normal adrenal medulla, norepinephrine is the predominant catecholamine secreted by most of these tumors. In the majority of cases, however, it is impossible to predict the degree of catecholamine secretion from the clinical features. The tumors are not innervated, and neural stimulation does not stimulate hormone release.
The tumors are sometimes broadly defined as following the “rule of 10s.” Pheochromocytomas involve both adrenal glands in 10% of adult patients with the tumor. Approximately 10% of the tumors arise from extraadrenal chromaffin cells. Approximately 10% of the tumors are in children. Malignant spread of pheochromocytomas occurs in fewer than 10% of cases. Malignant pheochromocytomas are more often extraadrenal and often secrete norepinephrine exclusively. Malignant pheochromocytoma metastasis usually proceeds via venous and lymphatic channels to the liver. See the tables below for the locations of pheochromocytomas.
Locations of Pheochromocytomas

From Landsberg L, Young JB. Catecholamines and the adrenal medulla. In Wilson JD, Foster DW, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia: Saunders; 1998:707.
Locations of Extraadrenal Pheochromocytomas
| Location | % |
| Cervical | 2 |
| Thoracic | 10–20 |
| Intraabdominal | 70–80 |
| Upper abdomen | 40 |
| Organ of Zuckerkandl | 30 |
| Bladder | 15 |
From Landsberg L, Young JB. Catecholamines and the adrenal medulla. In Wilson JD, Foster DW, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia: Saunders; 1998:707.
Diagnostic tests for determining the presence of a catecholamine tumor include measurements of urinary or plasma catecholamines and their metabolites, vanillylmandelic acid, and metanephrines. Free norepinephrine measurement in a 24-hour urine sample is a sensitive index of pheochromocytoma (see the table below). Other methods that can be used to assess for the presence of a pheochromocytoma include a clonidine suppression test, CT scan, and metaiodobenzylguanidine (MIBG) scintigraphy.
Values for Catecholamines and Catecholamine Metabolites
| Hormone or Metabolite | Reference Range |
| Vanillylmandelic acid, urine | 2.0–7.0 mg/24 hr |
| Metanephrines, urine | <1.3 mg/24 hr |
| Norepinephrine, urine | <100 mcg/24 hr |
| Norepinephrine, plasma | 150–450 pg/mL |
| Epinephrine, plasma | <35 pg/mL |
| Catecholamines, free urinary | <110 mcg/24 hr |
Incidence and prevalence
Pheochromocytomas are rare, occurring in approximately 0.1% of hypertensive patients. These tumors may be associated with neurocutaneous syndromes such as von Hippel-Lindau disease, tuberous sclerosis, and Sturge-Weber syndrome. The tumors may also be a component of MEN type 2A or 2B (see the table below). Patients with a family history of MEN syndrome should be regularly screened for pheochromocytoma. Twenty-five percent of pheochromocytomas occur as part of an inherited autosomal dominant trait.
Manifestations of Multiple Endocrine Neoplasia
| Syndrome | Manifestations |
| MEN type 1 (Wermer’s syndrome) | Hyperparathyroidism, pituitary adenomas, pancreatic islet cell tumors |
| MEN type 2A (Sipple’s syndrome) | Medullary thyroid cancer, hyperparathyroidism, pheochromocytoma |
| MEN type 2B (mucosal neuroma syndrome) | Medullary thyroid tumor, pheochromocytoma, neuromas of the oral mucosa, marfanoid habitus |
Pheochromocytomas can occur at any age but usually occur within the third to the fifth decades of life, with equal frequency in both sexes in adults.
Clinical manifestations
Manifestations of a pheochromocytoma reflect massive catecholamine release and include hypertension, diaphoresis, headache, tremors, and palpitations. Hypertension may be paroxysmal or sustained. The combination of paroxysmal diaphoresis, tachycardia, and headache in hypertensive patients is a recognized triad of symptoms for pheochromocytoma.
A catecholamine-mediated paroxysm typically consists of a sudden and alarming increase in blood pressure, a severe throbbing headache, profuse sweating, palpitations, tachycardia, a sense of doom, anxiety, pallor (rarely flushing), and nausea. Orthostatic hypotension may result from plasma volume deficit or a lack of tone in the postural reflexes that defend upright blood pressure caused by the sustained excesses of catecholamines. Paroxysmal symptoms may last several minutes to days and are often followed by physical exhaustion. The frequencies of clinical symptoms associated with pheochromocytoma are outlined in the table below.
Frequency of Symptoms in 100 Patients with Pheochromocytoma
| Symptom | % |
| Headache | 80 |
| Excessive perspiration | 71 |
| Palpitation (with or without tachycardia) | 64 |
| Pallor | 42 |
| Nausea (with or without vomiting) | 42 |
| Tremor or trembling | 31 |
| Weakness or exhaustion | 28 |
| Nervousness or anxiety | 22 |
| Epigastric pain | 22 |
| Chest pain | 19 |
| Dyspnea | 19 |
| Flushing or warmth | 18 |
| Numbness or paresthesia | 11 |
| Blurring of vision | 11 |
| Tightness of throat | 8 |
| Dizziness or faintness | 8 |
| Convulsions | 5 |
| Neck or shoulder pain | 5 |
| Extremity pain | 4 |
| Flank pain | 4 |
From Landsberg L, Young JB. Catecholamines and the adrenal medulla. In Wilson JD, Foster DW, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia: Saunders; 1998:706.
Multiple endocrine neoplasia is a group of rare diseases caused by genetic defects that lead to hyperplasia and hyperfunction of two or more components of the endocrine system.
A paroxysm may be triggered by abdominal palpation, defecation, or any event that provokes pressure on the tumor. Micturition may trigger symptoms if the pheochromocytoma is present in the urinary bladder wall. In some patients, no clearly defined precipitating factor can be found. Mental or psychological stress does not usually initiate a crisis.
Because of the predominance of norepinephrine secretion, the symptoms associated with a pheochromocytoma reflect α-adrenergic activity over β-adrenergic effects. As a result of α-adrenergic inhibition of insulin and enhanced hepatic glucose output, hyperglycemia may be present. The cardiac output and heart rate may be significantly increased. An overall increase in metabolism accelerates oxygen consumption and can cause hyperthermia. Vasoconstriction in the extremities may produce pain, paresthesias, intermittent claudication, or ischemia.
Hypertension is the most common symptom, occurring in more than 90% of patients. Severe paroxysmal hypertension is present in approximately 40% of patients and is a distinctive manifestation of the disease. Sustained hypertension is often resistant to conventional treatment. When pheochromocytomas are predominantly epinephrine secreting, hypertension can alternate with periods of hypotension associated with syncope. Hypotension reflects surges of epinephrine, causing disproportionate β-adrenergic stimulation with vasodilation in the presence of a contracted vascular space.
A catecholamine-induced increase in myocardial oxygen consumption, hypertension, and coronary artery spasm can precipitate myocardial infarction or congestive heart failure even in the absence of coronary artery disease. ECG changes are common. Nonspecific ST-segment and T-wave changes and prominent U waves may be seen. Sinus tachycardia, supraventricular tachycardias, and premature ventricular contractions are commonly noted. Right and left bundle branch blocks and ventricular strain sometimes occur. Ventricular tachycardia has also been reported.
Treatment and anesthetic considerations
Preoperative management
The pharmacologic effects of released catecholamines present major anesthetic challenges. Medical management before tumor excision aims to reverse the effects of excessive adrenergic stimulation. Preoperative antihypertensive therapy and volume replacement has helped to decrease the surgical mortality rate from about 50% to the current 1% to 3%. The preoperative use of α-adrenergic antagonists in concert with reexpansion of the intravascular fluid compartment greatly improves cardiovascular stability intraoperatively. Myocardial infarction, congestive heart failure, cardiac dysrhythmias, and cerebral hemorrhage decrease in frequency when the patient has been treated preoperatively with α-adrenergic receptor antagonists. The drugs used in the management of pheochromocytoma are listed in the following table on pg. 96.
Drugs Used in the Management of Pheochromocytoma
bid, Twice a day; IV, intravenous; PO, oral; tid, three times a day.
Adapted from Schwartz JJ, Rosenbaum S, Graf GJ. Anesthesia and the endocrine system. In Barash PG, Cullen BF, Stoelting RK, eds. Clinical Anesthesia. 4th ed. Philadelphia: Lippincott; 2001:1132.
α-adrenergic receptor blockade
Phenoxybenzamine (Dibenzyline) is the drug of choice for preoperative α-adrenergic blockade and blood pressure stabilization. It is a noncompetitive presynaptic (α2) and postsynaptic (α1) adrenergic receptor antagonist of long duration (24–48 hours). Most patients with a pheochromocytoma require an oral dose between 60 and 250 mg/day. Postural hypotension is a common side effect of treatment.
Typically, patients require 10 to 14 days of α-adrenergic antagonist therapy to stabilize blood pressure, restore fluid volume, and decrease symptoms. Establishment of normotension facilitates reexpansion of the intravascular fluid compartment. Satisfactory α-adrenergic blockade is implied if the hematocrit decreases by 5% during treatment. Half to two-thirds of the normal oral phenoxybenzamine dose may be given the morning of surgery. α-Blockade must be initiated before β-blockade to avoid the potential for a hypertensive crisis.
Prazosin (Minipress), a specific postsynaptic α1-adrenergic receptor antagonist, has also been used successfully to treat the hypertension of pheochromocytoma preoperatively. Labetalol (Trandate, Normodyne), a mixed α- and β-receptor antagonist, has not been as effective as a first-line drug in controlling the blood pressure response, but it may be used as an adjunctive agent.
β-adrenergic receptor blockade
A β-adrenergic receptor antagonist is usually introduced in the preoperative period for control of tachycardia, hypertension, and catecholamine-induced supraventricular dysrhythmias. An important caveat is that β-adrenergic receptor antagonists should not be administered until after α-adrenergic blockade is established. Blocking β-receptor–mediated vasodilation in skeletal muscle without prior α-adrenergic blockade can increase the blood pressure even further in patients with pheochromocytoma.
Other treatment regimens
Calcium channel blockers and magnesium sulfate infusion have been used with variable success as monotherapy for the perioperative management of pheochromocytoma. Some regimens use these agents in conjunction with adrenergic blocking drugs.
α-Methyl tyrosine (Demser) is used for patients requiring long-term therapy such as in patients for whom surgery is contraindicated. The drug competitively inhibits catecholamine formation by blocking tyrosine hydroxylase, the rate-limiting enzyme in catecholamine synthesis.
The following criteria are proposed as end points for the patient awaiting surgery for pheochromocytoma resection:
1. No in-hospital blood pressure reading higher than 160/90 mmHg 24 hours before surgery
2. No blood pressure on standing lower than 80/45 mmHg
3. No ST-segment or T-wave abnormality on the ECG that cannot be attributed to a permanent defect
4. No marked symptoms of catecholamine excess; no more than 1 premature ventricular contraction every 5 minutes
Anesthetic considerations
Effective anesthetic management is based on selecting drugs that do not stimulate catecholamine release and implementing monitoring techniques that facilitate early and appropriate intervention when catecholamine-induced changes in cardiovascular function occur.
Pheochromocytomas are excised by open laparotomy or laparoscopy. Postoperative morbidity is similar with both approaches. During pneumoperitoneum for laparoscopy, significant catecholamine release has been reported.
A number of drugs and conditions can precipitate hypertension in surgical patients with pheochromocytoma. Dopamine antagonists (metoclopramide, droperidol), radiographic contrast media, indirect-acting amines (ephedrine, methyldopa), drugs that block neuronal catecholamine reuptake (tricyclic antidepressants, cocaine), histamine, and glucagon may enhance the physiologic effect of tumor product.
Pheochromocytomas are vascular tumors. Large-bore IV lines and a peripheral arterial catheter should be established preoperatively. A central venous pressure or pulmonary artery catheter should be placed to help guide fluid management and intervention with inotropes or vasoactive drugs. Arterial blood gases, electrolyte concentrations, and blood glucose levels should be assessed regularly during the anesthetic.
Critical intraoperative junctures are: (1) during induction and intubation of the trachea (possible hypertension), (2) during surgical manipulation of the tumor (possible hypertension), and (3) after ligation of the tumor’s venous drainage (possible hypotension).
Anesthesia induction may be accomplished with barbiturates, etomidate, or propofol. Anesthetic depth can be enhanced by mask ventilation of the lungs with a volatile anesthetic prior to laryngoscopy and intubation. Lidocaine (1–2 mg/kg IV) administered 1 minute before intubation may help attenuate the hemodynamic response to laryngoscopy. Rapid-acting vasodilating drugs such as nitroprusside should be readily available to treat hypertension. Short-acting opioids, such as fentanyl or remifentanil, administered before intubation may also help blunt the blood pressure responses to intubation. Morphine sulfate should be avoided because of its propensity for histamine release.
Sevoflurane and short-acting opioids, such as remifentanil, provide cardiovascular stability and possess the ability to rapidly change anesthetic depth, attractive features in the anesthetic management of patients with pheochromocytoma. The tachycardia associated with desflurane makes it a less desirable choice for these cases.
The use of succinylcholine has been questioned because compression of an abdominal tumor by drug-induced skeletal muscle fasciculations may provoke catecholamine release. However, a predictable adverse effect of succinylcholine has not been supported clinically when administered to patients with pheochromocytoma. Skeletal muscle paralysis with a nondepolarizing muscle relaxant devoid of vagolytic or histamine-releasing effects is desirable. Pancuronium should be avoided because of its known chronotropic effect.
The anesthetist should anticipate a labile cardiovascular course during surgery. Hypertension can be treated by increasing the anesthetic depth and administering nitroprusside if necessary. Propranolol, lidocaine, labetalol, or esmolol may be given IV to decrease tachydysrhythmias. β-Adrenergic antagonists must be used cautiously in patients with catecholamine-induced cardiomyopathy because even minimal β-adrenergic blockade can accentuate left ventricular dysfunction. The short half-life of esmolol makes it an advantageous choice for β-adrenergic blockade. Dysrhythmias associated with hypertension may be resolved by lowering an abnormally high blood pressure. Indirect-acting sympathomimetics have an unpredictable pressor effect in these patients and should be avoided.
After surgical ligation of the veins that drain a pheochromocytoma, the rapid decrease in circulating catecholamines and the associated downregulation of adrenergic receptors may precipitate a decrease in blood pressure. During this juncture, close communication with the surgical team is important. Decreasing the inhaled anesthetic agent concentration and increasing the administration of IV crystalloid or colloid solution should adequately increase blood pressure. IV administration of phenylephrine hydrochloride (Neo-Synephrine) or dopamine may be needed until the peripheral vasculature can adapt to the decreased level of endogenous α-stimulation.
Hyperglycemia is common before excision of the pheochromocytoma. With tumor removal, the sudden withdraw of catecholamine stimulation can result in hypoglycemia. Furthermore, β-adrenergic blockade impairs hepatic glucose production. β-Adrenergic blockers may also mask hypoglycemic signs by preventing tachycardia and tremor. Blood glucose levels should be monitored at frequent intervals intraoperatively and postoperatively.
Some pheochromocytomas may first present as a hypermetabolic state during anesthesia for unrelated surgery. The hypertension, tachycardia, hyperthermia, and respiratory acidosis of a pheochromocytoma may mimic light anesthesia, thyroid crisis, malignant hyperthermia, or sepsis.
Postoperative implications
Fluid shifts, pain, hypoxia, hypercapnia, autonomic instability, urinary retention, or residual tumor are all potential causes of postoperative hypertension. Invasive monitoring is indicated during the initial postoperative period to assess blood pressure changes and cardiac status. Fifty percent of patients remain hypertensive during the immediate postanesthesia recovery period despite removal of the pheochromocytoma. Transient hypertension postoperatively usually reflects fluid shifts and autonomic instability. Postoperative catecholamine levels decrease to normal over several days. In 75% of patients, normal blood pressure returns within 14 days after surgery. Relief of postoperative pain can be accomplished with neuraxial opioids and may contribute to early tracheal extubation in otherwise healthy patients.