Chapter 303 Syndromes with Oral Manifestations
Many syndromes have distinct or accompanying facial, oral, and dental manifestations (Apert syndrome, Chapter 585; Crouzon disease, Chapter 585; Down syndrome, Chapter 76).
Osteogenesis imperfecta is often accompanied by effects on the teeth, termed dentinogenesis imperfecta (Chapter 299, Fig. 299-2). Depending on the severity of presentation, treatment of the dentition varies from routine preventive and restorative monitoring to covering affected posterior teeth with stainless steel crowns, to prevent further tooth loss and improve appearance. Dentinogenesis imperfecta can also occur in isolation without the bony effects.
Another syndrome, cleidocranial dysplasia, has orofacial variations such as frontal bossing, mandibular prognathism, and a broad nasal base. Tooth eruption is often delayed. The primary teeth can be abnormally retained and the permanent teeth remain unerupted. Supernumerary teeth are common, especially in the premolar area. Although the erupted teeth are usually free of hypoplasia, variations in the size and shape of the teeth are common. Extensive dental rehabilitation therapy may be needed to maintain effective mastication.
Ectodermal dysplasias (Chapter 641) are a heterogeneous group of conditions in which oral manifestations range from little or no involvement (the dentition is completely normal) to cases in which the teeth can be totally or partially absent or malformed. Because alveolar bone does not develop in the absence of teeth, the alveolar processes can be either totally or partially absent, and the resultant overclosure of the mandible causes the lips to protrude. Facial development is otherwise not disturbed. Teeth, when present, can range from normal to small and conical. If aplasia of the buccal and labial salivary glands is present, dryness and irritation of the oral mucosa can occur. People with ectodermal dysplasia might need partial or full dentures, even at a very young age. The vertical height between the jaws is thus restored, improving the position of the lips and facial contours as well as restoring masticatory function.
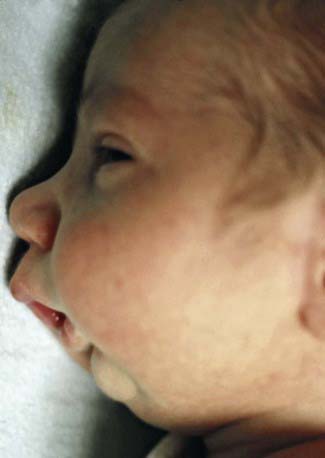
Pierre Robin syndrome consists of micrognathia usually accompanied by a high arched or cleft palate (Fig. 303-1). The tongue is usually of normal size, but the floor of the mouth is foreshortened. The air passages can become obstructed, particularly on inspiration, usually requiring treatment to prevent suffocation. The infant should be maintained in a prone or partially prone position so that the tongue falls forward to relieve respiratory obstruction. Some patients require tracheostomy. Mandibular distraction procedures in the neonate can improve mandibular size, enhance respiration, and facilitate oral feedings.
Sufficient spontaneous mandibular growth can take place within a few months to relieve the potential airway obstruction. Often the growth of the mandible achieves a normal profile in 4-6 yr. The feeding of infants with mandibular hypoplasia requires great care and patience but can usually be accomplished without resorting to gavage. Thirty to 50% of children with Pierre Robin syndrome have Stickler syndrome, an autosomal dominant condition that includes other findings such as prominent joints, arthritis, hypotonia, hyperextensible joints, mitral valve prolapse, and ocular problems (retinal detachment, myopia, cataracts).
In mandibulofacial dysostosis (Treacher Collins syndrome or Franceschetti syndrome), the facial appearance is characterized by downward sloping palpebral fissures, colobomas of the lower eyelids, sunken cheekbones, blind fistulas opening between the angles of the mouth and the ears, deformed pinnae, atypical hair growth extending toward the cheeks, receding chin, and large mouth. Facial clefts, abnormalities of the ears, and deafness are common. The disorder is autosomal dominant, often with incomplete penetrance. The mandible is usually hypoplastic; the ramus may be deficient, and the coronoid and condylar processes are flat or even aplastic. The palatal vault may be either high or cleft. Dental malocclusions are common. The teeth may be widely separated, hypoplastic, or displaced or have an open bite. Orthodontic and routine dental treatments are indicated.
Hemifacial microsomia is usually characterized by unilateral hypoplasia of the mandible and can be associated with partial paralysis of the facial nerve, macrostomia, blind fistulas between the angles of the mouth and the ears, and deformed external ears. Severe facial asymmetry and malocclusion can develop because of the absence or hypoplasia of the mandibular condyle on the affected side. Congenital condylar deformity tends to increase with age. Early craniofacial surgery may be indicated to minimize the deformity. This disorder can be associated with ocular and vertebral anomalies (oculo-auriculo-vertebral spectrum, including Goldenhar syndrome); therefore, radiographs of the vertebrae and ribs should be considered to determine the extent of skeletal involvement.
Nelson Textbook of Pediatrics Expert Consult