[level-membership-for-pediatrics-category]Chapter 79
Syndromes and Chromosomal Anomalies
A large number of syndromes, dysplasias, and chromosomal anomalies are associated with congenital or acquired cardiac and vascular disease. This chapter discusses the cardiovascular features of some commonly encountered lesions. A more extensive list is provided in Tables 79-1 to 79-3.
Table 79-1
Syndromes, Dysplasias, and Their Associated Cardiovascular Anomalies
| Syndrome | Cardiovascular Anomalies |
| Achondrogenesis | PDA, ASD, VSD, COA |
| Alagille syndrome (arteriohepatic dysplasia) (see e-Fig. 79-13) |
PS, PPS, ASD, VSD, TOF, PDA, PAT, PAPVR, dysplastic AV valves, COA |
| Apert syndrome (acrocephalosyndactyly) | ASD, PDA, VSD, PS, TOF, EFE, DEXTRO, COA |
| Arthrogryposis | CMY |
| Beckwith-Wiedemann syndrome (EMG syndrome) | CM, CMY, ASD, PDA, TOF, HPLH, subvalvular AS, cardiac fibromas |
| Cardioauditory syndromes | LVH, RVH |
| Cantrell syndrome (pentalogy of Cantrell) (see e-Fig. 79-14) | Combined sternal, pericardial, intracardiac, diaphragmatic, and anterior abdominal wall defects Radiographic findings: sternal defect, ectopia cordis, CHD, ASD, VSD, PS, TOF, APVR, DEXTRO, ventricular diverticulum, intrapericardial herniation of abdominal organs; associated with Turner, trisomy 18, sirenomelia, and amniotic band syndromes |
| Cardiosplenic (heterotaxy) syndromes (see Figs. 79-1 and 79-2) | |
| Right isomerism (see Fig. 79-1) | TAPVR, AVSD, pulmonary outflow obstruction or PAT, DORV, TGA, single atrium (R), single common ventricle, DEXTRO, TAT, TRU (rare), AO-IVC juxtaposition, bilateral right PAs, bilateral SVC, interrupted IVC |
| Left isomerism (see Fig. 79-2) | Cardiac malposition, single atrium (L), single ventricle, VSD, AVSD, DORV, APVR, interrupted IVC, bilateral left PAs, bilateral SVC |
| Cayler syndrome (cardiofacial syndrome) | ASD, PDA, VSD, AVSD, TOF, RAA, COA |
| CHARGE association | ASD, VSD, conotruncal malformations, PDA, TOF, parachute mitral valve |
| Degos syndrome (malignant atrophic papulosis) | MI, pericarditis, constrictive pericarditis, myocardial fibrosis, and renal, cerebral, coronary, visceral, and peripheral arteriopathy |
| Diamond-Blackfan syndrome (congenital red cell aplasia) | VSD, ASD, mitral valve dysplasia |
| Ehlers-Danlos syndrome (see Fig. 79-3) | MVP, dilated AO root, coronary and aortic aneurysms, dissection or rupture, AS, AR, TR, PS, ASD, VSD, TOF, DEXTRO, LV rupture, arteriovenous fistula |
| Ellis–van Creveld syndrome (chondroectodermal dysplasia) | Common atrium, ASD, AVSD |
| Fryns syndrome | Septal defects, arch anomalies, TOF, cystic hygroma |
| Hallermann-Streiff syndrome (oculomandibulofacial syndrome) | PS, TOF, ASD, VSD |
| Holt-Oram syndrome (heart-hand syndrome) (see e-Fig. 79-10) | ASD, VSD, MVP, PDA, HPLH, TAPVR, TRU, conduction disorder, hypoplastic peripheral vessels |
| Jeune syndrome (asphyxiating thoracic dystrophy) | CHF |
| Kartagener syndrome (primary ciliary dyskinesia) (see e-Fig. 79-11) | DEXTRO, CHD |
| Klippel-Trénaunay-Weber syndrome (angioosteohypertrophy syndrome) | CHF, pericardial effusion, superficial varices, telangiectatic nevi, organ hemangiomas, lymphatic obstruction |
| LEOPARD syndrome (cardiomyopathic lentiginosis) (see e-Fig 79-8) | CMY, conduction defect, PS, sub-AS |
| Loeys-Dietz syndrome (see Fig. 79-5) | Congenital heart defects include patent ductus arteriosus, bicuspid aortic valve, bicuspid pulmonary valve, mitral valve prolapse, and atrial septal defect; arterial tortuosity, stenoses, aneurysms, dissection, diffuse arterial involvement; spontaneous rupture of viscera |
| Marfan syndrome (see Fig. 79-4) | MVP, MR, dilation of AO root, AR, CHF, aneurysms (AO, pulmonary, ductus), AO dissection, MI, arrhythmia, TR, ASD, TOF |
| MELAS syndrome | CMY, CHF, conduction abnormalities |
| Mucolipidosis III | AR |
| Mucopolysaccharidoses | |
| IH (Hurler syndrome) | Acute CMY associated with EFE, AR, MR, MS, arteriopathy (coronary, renal, AO, mesenteric) |
| IS (Scheie syndrome) | AS, MS |
| II (Hunter syndrome) | AR, CHF, valve thickening, CMY |
| III (Sanfilippo syndrome) | CMY, MR, AR |
| IV (Morquio syndrome) | AR, CMY, AS, MR, CAD |
| VI (Maroteaux-Lamy syndrome) | AS, MS, CMY |
| Neurofibromatosis type 1 (see Fig. 79-6) | PS, COA, ASD, VSD, CMY, MVP, AS, TOF, PDA, vasculopathy (coronary, pulmonary, renal, systemic), cardiac neurofibroma, arteriovenous fistula, lymphatic abnormality |
| Noonan syndrome (Turner phenotype with normal karyotype) | PS, dysplastic pulmonic valve, hypertrophic CMY, lymphatic abnormalities, PDA, ASD, COA, mitral valve abnormalities, AS, pericarditis, APVR, coronary anomalies |
| Oculoauriculovertebral dysplasia (Goldenhar syndrome) | TOF, VSD, DORV, PAT, TAPVR, RAA, COA, asplenia |
| PHACES | Arch atresia, aberrant subclavian origins, hypoplasia of the descending thoracic aorta, double aortic arch, COA, stenosis and aneurysm formation of the aorta and the cervical arteries, stroke |
| Progeria (Hutchinson-Gilford syndrome) | Accelerated atherosclerosis, CM, MI, CHF, stroke |
| Proteus syndrome | CHD, CMY, myocardial mass, conduction abnormality, venous dilation, hemangioma, lymphangioma |
| Ravitch syndrome (thoracoabdominal wall defect) | Ectopia cordis, pentalogy of Cantrell, TGA, PDA, ASD, VSD, PS, TOF |
| Robinow syndrome (fetal face syndrome) | CHD (right heart lesions) |
| Rubinstein-Taybi syndrome | ASD, VSD, PDA, COA, PS, bicuspid AO valve |
| Silver-Russell syndrome | CHD |
| Smith-Lemli-Opitz syndrome | ASD, complex cardiac anomalies |
| Thrombocytopenia–absent radius (TAR) syndrome | COA, ASD, VSD, PDA, AVSD, TOF |
| Tuberous sclerosis (Bourneville-Pringle syndrome) (see Fig. 79-7) | Rhabdomyoma, hamartoma, CHF, CMY, COA, arrhythmia, arterial aneurysm and stenosis (AO, cerebral, renal, peripheral) |
| VATER/VACTERL association (see Fig. 79-9) | VSD, ASD, PDA, TOF, TGA, single ventricle |
| Velocardiofacial syndrome (Shprintzen syndrome) | TOF, TRU, PA, VSD, absent pulmonary valve, TGA, AS, interrupted AO arch, RAA |
| Williams syndrome (e-Fig. 79-12) | Supravalvular AS, PPS, MR, ASD, VSD, TOF, MI, COA, interrupted arch, hypoplastic AO, aneurysm or stenosis (AO, systemic, renal, cerebral arteries) |
| Zellweger syndrome (cerebrohepatorenal syndrome) | CHD, PDA, VSD, DiGeorge |
Table 79-2
Syndromes that Predominantly Affect the Cardiovascular System
| Syndrome | Cardiovascular Defects |
| Absent pulmonary valve leaflet syndrome (see e-Fig. 79-15) | Maldeveloped nodular myxoid pulmonary valve cusps with aneurysmal dilation of central PAs associated with TOF, airway compression, lobar emphysema and abnormal PA branching, CM, RAA, ASD, VSD, PDA, DORV, AVSD, Marfan syndrome, 18q deletion |
| Berry syndrome | Distal AP window with AO origin of the right PA and arch interruption |
| Bland-White-Garland syndrome | Anomalous origin of left coronary artery from PA |
| Congenital cardiomyopathy: hypertrophic cardiomyopathy | Asymmetric septal hypertrophy, systolic anterior motion of mitral valve, LVOT obstruction, myocardial scar arrhythmias |
| Arrhythmogenic right ventricular dysplasia | Fibrofatty infiltration of right ventricular myocardium, RV dyskinesia/aneurysms, arrhythmias |
| Eisenmenger syndrome | Pulmonary hypertension with bidirectional or reversed shunt at atrial, ventricular, or AP level; cyanosis; dyspnea; sudden death; peripartum CMY radiographic findings: dilated central PAs with tapering, PA calcification |
| Floppy valve syndrome | MVP, prolapse of other valves, CAD, congestive or hypertrophic CMY, ASD, MR, AR, papillary muscle or chordae tendineae rupture |
| Hypoplastic left heart syndrome | Combined mitral and AO obstruction (stenosis or atresia), underdeveloped LA and LV, hypoplastic ascending AO ± COA or AO interruption; may be associated with right diaphragmatic hernia, omphalocele, brain anomalies; radiographic findings: CM and pulmonary edema |
| Lutembacher syndrome | ASD associated with MS |
| Postmyocardial infarction syndrome (Dressler syndrome) | Chest pain, fever, polyserositis—several weeks postinfarction; radiographic findings: pericardial or pleural effusion, noncardiogenic edema |
| Postpericardiotomy syndrome | Chest pain, fever, joint pain—weeks or months after closed or open heart surgery; radiographic findings: pericardial or pleural effusion, noncardiogenic pulmonary edema, constrictive pericarditis |
| Romano-Ward syndrome | Familial Q-T prolongation, arrhythmias, syncope |
| Shone syndrome (or complex) (see e-Fig. 79-16) | Complex of multiple left-sided obstructions, parachute mitral valve, supravalvular ring of LA, sub-AS, COA |
| Sick sinus syndrome | Arrhythmias |
| Tetralogy of Fallot (see Fig. 79-9 and e-Fig. 79-15) | Combination of VSD, overriding AO, RVH, RV outflow obstruction; may be PS and PPS |
| Trilogy of Fallot | PS, ASD (or PFO), right-to-left shunting |
| Uhl syndrome (anomaly) | Congenital aplasia of RV myocardium, RV CMY |
| Wolff-Parkinson-White syndrome | Aberrant intracardiac ECG pathway producing arrhythmias; associated with Ebstein anomaly, IHSS, levo-TGA, giant RA diverticulum |
Table 79-3
Chromosomal Anomalies and Their Associated Cardiovascular Defects
| Chromosomal Anomaly | Cardiovascular Defects |
| Fragile X | MVP, MR, AR, TR, dilated AO root, COA |
| Trisomy 13 (Patau syndrome) | PDA, VSD, ASD, DEXTRO, capillary hemangioma, cervical cystic hygroma |
| Trisomy 18 (Edwards syndrome) | VSD, polyvalvular heart disease (pulmonary and AO valves), ASD, PDA, COA, TOF, TGA, HPLH, VACTERL, pentalogy of Cantrell |
| Trisomy 21 (Down syndrome) (see Fig. 79-18) | AVSD, VSD, ASD, TOF, PDA, PS, MVP, aberrant right SCA, intimal arterial fibrodysplasia, lymphatic abnormality, upper airway obstruction and CHF |
| Cat-eye syndrome (trisomy or tetrasomy 22) | TAPVR, TOF |
| Monosomy X, XO (Turner syndrome) (see Fig. 79-19) | COA, bicuspid AO valve, AO dissection, septal defects, abnormal mitral valve, sub-AS, PS, APVR, pentalogy of Cantrell, DEXTRO, RAA, hemangioma, lymphangiectasia, venous anomalies |
| XXY (Klinefelter syndrome) | MVP, Takayasu arteritis, cerebral aneurysms, varicose veins |
| Deletion Syndromes | |
| Monosomy 1p36 syndrome | Dilated CMY, PDA |
| 22q11 (predominantly DiGeorge syndrome (CATCH 22), also velocardiofacial syndrome) (see Fig. 79-17) | Type B interrupted arch, RAA, VSD, TOF, TRU, COA, aberrant right SCA, isolated SCA |
| 5p: Cri du chat syndrome | CHD |
| 4p: Wolf-Hirschhorn syndrome | ASD, VSD, valve anomalies, complex CHD, persistent left SVC |
| 17p: Miller-Dieker syndrome (lissencephaly type 1) | ASD, CHD, conduction abnormalities |
| 18q syndrome | Absent pulmonary valve, PDA, AS, dilated ascending AO |
Syndromes
Situs and Cardiosplenic (Heterotaxy) Syndromes
Overview: The heterotaxy syndromes, that is, right and left isomerism, feature abnormalities of the visceral situs. These syndromes have an estimated incidence of 1 in 6000 to 1 in 20,000 live births and account for 1% of all congenital heart defects. Although heterotaxy usually occurs sporadically, familial cases have been described.
Although visceral and atrial situs do not always correspond, body situs (from the Latin word meaning location) generally is divided into three types: solitus, inversus, and ambiguus. Situs solitus is the normal arrangement of the viscera in the body (see Chapter 63). Situs inversus is the mirror image of normal; it is seen in 0.01% of the population and is associated with a slightly higher incidence of congenital heart disease (3% to 5%) compared with the solitus population (0.6% to 0.8%). The most common cardiac abnormalities seen in patients with situs inversus are a right-sided aortic arch, atrioventricular discordance, and transposition of the great vessels. Situs ambiguus, or heterotaxy, encompasses all other visceroatrial arrangements. By definition, in situs ambiguus, visceral malposition and dysmorphism associated with an indeterminate atrial arrangement are present.1
Heterotaxy syndrome with right isomerism or bilateral right-sidedness is usually, but not invariably, accompanied by asplenia.1 The condition is more common in males and is characterized by bilateral systemic atria with broad trabeculated appendages (Fig. 79-1), bilateral trilobed lungs with bilateral minor fissures and short eparterial bronchi, a central horizontal liver, bowel malrotation, and the stomach in an indeterminate position (see Fig. 79-1). The abdominal aorta and inferior vena cava often are located on the same side of the spine, frequently in a posterior-anterior orientation. Other occasional anomalies include tracheoesophageal fistula, imperforate anus, absent gallbladder, pancreatic anomalies, fused adrenal glands, and genitourinary abnormalities.2 The prognosis for right isomerism is poor because of an abnormal immune status (asplenia) and the typically complex cardiac anomalies.
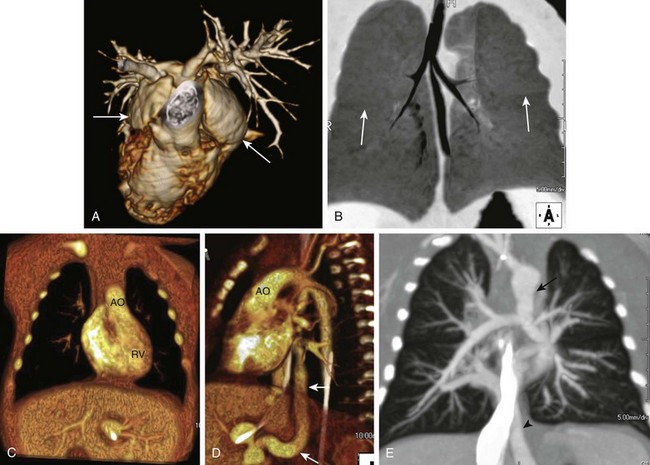
Cardiac anomalies are almost invariable and cause the most common presenting symptoms: cyanosis and severe respiratory distress. The “right isomerism heart” often consists of a common atrioventricular canal, a single ventricle, a large ventricular septal defect (VSD), and a double-outlet right ventricle and/or transposition of the great vessels, along with pulmonary outflow obstruction or atresia and total anomalous pulmonary venous drainage (frequently obstructed) (see Fig. 79-1). The spectrum of cardiovascular anomalies also can include cardiac malposition (dextrocardia or mesocardia), tricuspid atresia, truncus arteriosus, right aortic arch, anomalous systemic venous return, and bilateral superior vena cavae.2,3
Heterotaxy syndrome with left isomerism or bilateral left-sidedness most often accompanies polysplenia. It is characterized by bilateral pulmonary atria with narrow fingerlike appendages, bilateral bilobed lungs, bilateral long hyparterial bronchi, a centrally located liver, the stomach in an indeterminate position, bowel malrotation, and multiclefted or multiple spleens (either right sided or left sided) (Fig. 79-2). An interrupted inferior vena cava with azygos continuation is the most consistent abdominal finding in left isomerism (see Fig. 79-2). Left isomerism is slightly more common in females and generally has a better prognosis than right isomerism with less complex cardiovascular disease. Other associated anomalies include ciliary dyskinesia, biliary atresia, and other gastrointestinal abnormalities including bowel malrotation and pancreatic anomalies, as well as congenital portosystemic shunts (see Fig. 79-2).2,4
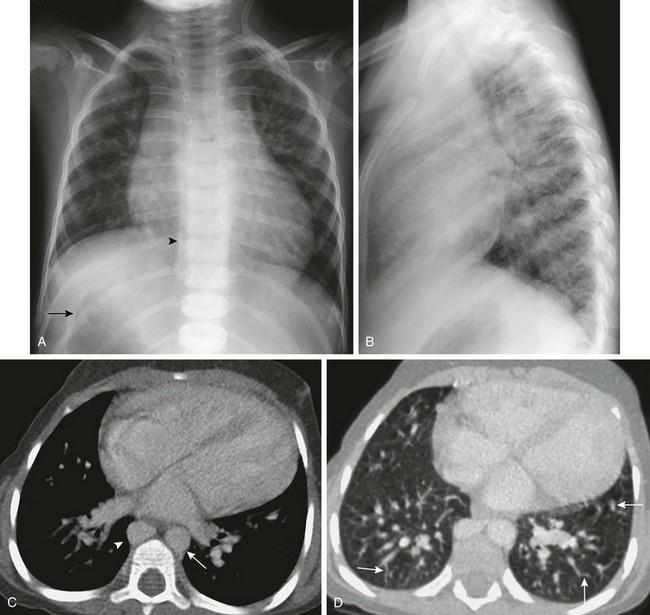
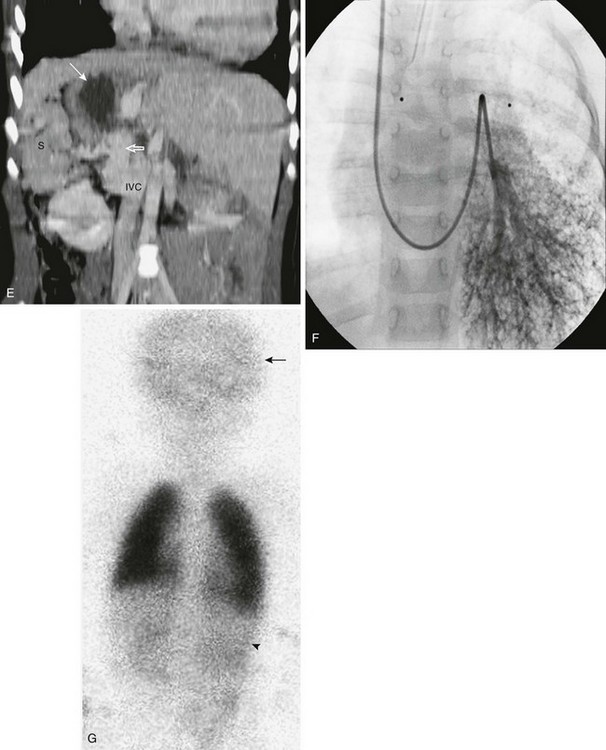
Figure 79-2 This 3-year-old girl, whose mother was diabetic, has polysplenia and Abernethy malformation with hepatopulmonary syndrome.
A, A posteroanterior view of the chest shows heterotaxy with a left-sided cardiac apex, right-sided stomach (arrow), and left-sided liver. Cardiomegaly and increased pulmonary vascularity (a small atrial septal defect and pulmonary arteriovenous shunting) are present. It is difficult to see the bilateral left-sided bronchi. A prominent right paraspinal line is present as a result of an enlarged azygos vein (arrowhead). B, The lateral view of the chest is notable for the absence of the inferior vena cava shadow. C, Contrast-enhanced computed tomography (CT) scan of the chest shows cardiomegaly, a left-sided aorta (arrow), prominent pulmonary veins, and an enlarged azygos vein (arrowhead). D, A contrast-enhanced CT scan of the chest displayed in the lung window shows the presence of multiple dilated peripheral pulmonary vessels (arrows). E, Coronal reconstruction from a contrast-enhanced CT scan of the upper abdomen shows right-sided polysplenia (S), a right-sided stomach (white arrow), and a left-sided liver. A large right-sided portosystemic shunt (splenic vein to renal vein) is present (open arrow). Superiorly, there is interruption of the inferior vena cava (IVC) with azygos continuation (not shown). F, A left pulmonary artery angiogram from a right jugular approach shows dilated peripheral pulmonary arteries and micro arteriovenous connections, consistent with hepatopulmonary syndrome as a result of the congenital portosystemic shunt. This child does not have liver disease, and the intrahepatic portal veins are present but small (Abernethy malformation type II). G, Perfusion portion of a ventilation-perfusion scan using technetium-99m macroaggregated albumin performed after closure of the patient’s atrial septal defect shows normal perfusion of the lungs. There is abnormal radiotracer uptake within the brain (arrow) and kidneys (arrowhead). The percentage of systemic tracer uptake was calculated at 44.7%. These findings are typical of a right-to-left arteriovenous shunt and confirm the presence of hepatopulmonary syndrome.
Cardiac anomalies are seen in more than 50% of patients who have heterotaxy syndrome with left isomerism. The common cardiovascular anomalies include abnormalities of the inferior and superior vena cavae, cardiac malposition, atrial septal defect (ASD), VSD, common atrioventricular canal, double-outlet right ventricle, and anomalous pulmonary venous return (usually partial).2,3
Imaging: Eight major structures are appraised to evaluate situs in heterotaxy syndromes1:
Decreased pulmonary vascularity related to pulmonary outflow obstruction or pulmonary edema as a result of pulmonary venous obstruction is the most typical pattern seen on chest radiographs in persons with right isomerism. Cardiomegaly and increased vascularity on chest radiographs as a result of left-to-right shunting are the most common appearance in persons with left isomerism (see Fig. 79-2). Other chest radiographic features of the heterotaxy syndromes are related to duplicated structures, such as fissures and bronchi, as well as abnormal cardiac and visceral situs.
Echocardiography is the main imaging modality used to evaluate disordered intracardiac anatomy associated with heterotaxy. Both magnetic resonance imaging (MRI) and computed tomography (CT) have a role in fully defining this often complex anatomy. MRI, magnetic resonance angiography (MRA), and functional magnetic resonance (MR) assessment have partially replaced catheter-based angiography in evaluating these anomalies, especially the extracardiac vascular, airway, and visceral abnormalities (see Figs. 79-1 and 79-2).1
Ultrasound often is the first step in evaluating the abdominal anatomy because of its lack of ionizing radiation and ease of use. An upper gastrointestinal study should be performed to evaluate for malrotation. A small bowel follow through in conjunction with the upper gastrointestinal study is useful to determine the relative locations of the small and large bowel and to separate nonrotation from malrotation. Ultrasound, CT, MRI, or liver-spleen scans using technetium-99m sulfur colloid or p-isopropylacetanilidoimidodiacetic acid are used to evaluate the presence and location of splenic tissue, to assess other organs/anomalies, and to help diagnose biliary atresia (associated with polysplenia). Multimodality imaging may be needed to elucidate other less common anomalies associated with heterotaxy such as a congenital portosystemic shunt and a hypoplastic or absent portal vein (Abernethy malformation) with consequent hyperammonemic encephalopathy or hepatopulmonary syndrome (associated with left isomerism; see Fig. 79-2).4
Ehlers-Danlos Syndrome
Overview: Ehlers-Danlos syndrome (EDS) is an inherited group of disorders of connective tissue affecting the skin, ligaments, joints, vasculature, and visceral organs. It is characterized by joint and skin hyperextensibility, excessive bruisability, blood vessel fragility, and poor wound healing.5 It occurs in as many as 1 in 5000 individuals. Although EDS is incompletely understood, six distinct varieties of EDS have been recognized based on genetics, biochemical structure, and clinical presentation.5,6 Mitral valve prolapse (MVP) is the most common anomaly. Other cardiovascular anomalies include a dilated aortic root, aneurysm, and aortic dissection or rupture (Fig. 79-3).7 Papillary muscle dysfunction and left ventricular rupture also can occur.6
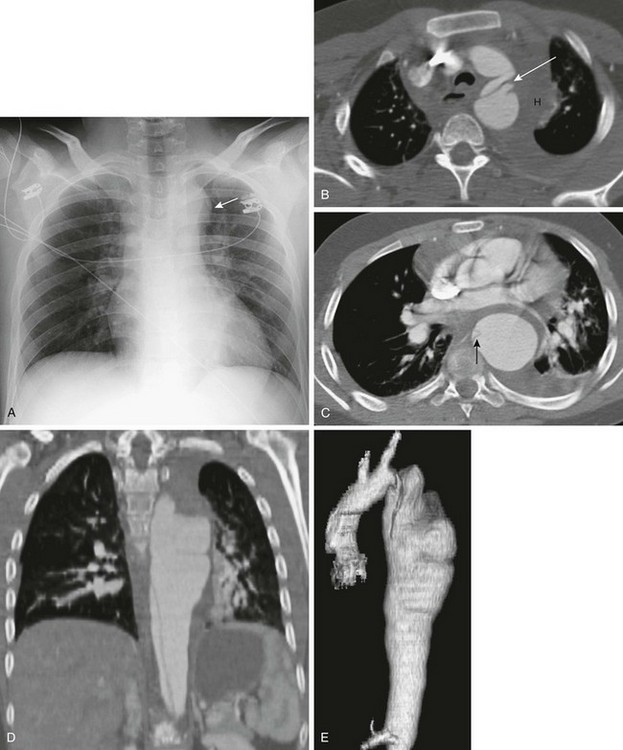
Figure 79-3 Ehlers-Danlos syndrome.
This 14-year-old boy presented to the emergency department with chest pain and a family history of dissecting aortic aneurysm. A, Posteroanterior view of the chest shows a widened mediastinum and prominent aortic knob (arrow). B, Computed tomographic angiography (CTA) of the upper chest shows an intimal flap within the aortic arch (arrow), representing a dissection. Other findings include a mediastinal hematoma (H) and a left-sided pleural effusion. C, CTA of the chest at the level of the descending aorta shows a dilated descending aorta with a small intimal flap (arrow), a moderate amount of periaortic hematoma, and a small left pleural effusion. Coronal reconstruction (D) and three-dimensional reconstruction from the CTA (E) show a dissecting aortic aneurysm and irregular dilated aorta.
The vascular or arterial ecchymotic type of EDS is the most clinically severe form. It is an autosomal dominant disorder of the COL3A1 gene on chromosome 2, which codes for type III procollagen. Patients are subject to spontaneous rupture of the bowel and other viscera. Vascular complications occur either spontaneously or after minor trauma and include arteriovenous fistulas and aneurysms of the aorta and medium-sized arteries, with degeneration and subsequent rupture or dissection (see Fig. 79-3). The thoracic and abdominal arteries are involved in half of the cases, with extremity and head and neck vessels each contributing 25%. Common neurovascular complications include carotid-cavernous fistula formation, carotid dissection, aneurysm formation, and rupture.5–8
Imaging, Treatment, and Follow-up: Echocardiography is the mainstay for evaluation of the heart and valvular function. Ultrasound, CT, or MR vascular imaging is geared toward defining areas of aneurysm, dissection, and rupture. The size and the rate of growth of each aneurysm should be quantified. Because the neurovascular system may be involved, imaging of the cerebral circulation and the carotid arteries may be necessary.
The pattern of aneurysm distribution in persons with EDS differs from that in persons with Marfan syndrome. Whereas Marfan syndrome typically results in dilation of the aortic root and the sinuses of Valsalva, EDS produces discrete, fusiform aneurysms at different locations of the aorta, the iliac arteries, the cervical arteries, and their branches. Dissection in persons with EDS often has a complex morphology (see Fig. 79-3). The abnormal vessels usually are well characterized by noninvasive computed tomographic angiography (CTA) and MRA.8 Catheter-based angiography carries significant hazards in these patients with a reported complication rate of 67%.8
Because of the rarity of the vascular form of EDS, data are lacking about the proper management of these patients. The substrate of the vascular wall is abnormal, and thus surgical repair often is complicated by the formation of new aneurysm, rupture, or dissection. As a result, surgery is a last resort.8
Marfan Syndrome
Overview: Marfan syndrome is an autosomal dominant disorder of connective tissue caused by mutations in the gene that codes for the extracellular matrix protein fibrillin 1.9,10 Marfan syndrome is present in 1 in 3000 to 1 in 5000 persons and is associated with skeletal, ocular, and cardiovascular manifestations.10 The diagnosis of Marfan syndrome is based on clinical criteria—the 2011 revised Ghent nosology.11
Common cardiovascular features of Marfan syndrome include MVP, mitral regurgitation, aortic regurgitation, aortic root dilation, and ascending aortic aneurysm (Fig. 79-4). Aortic dissection and aortic rupture are the most life-threatening complications of Marfan syndrome.
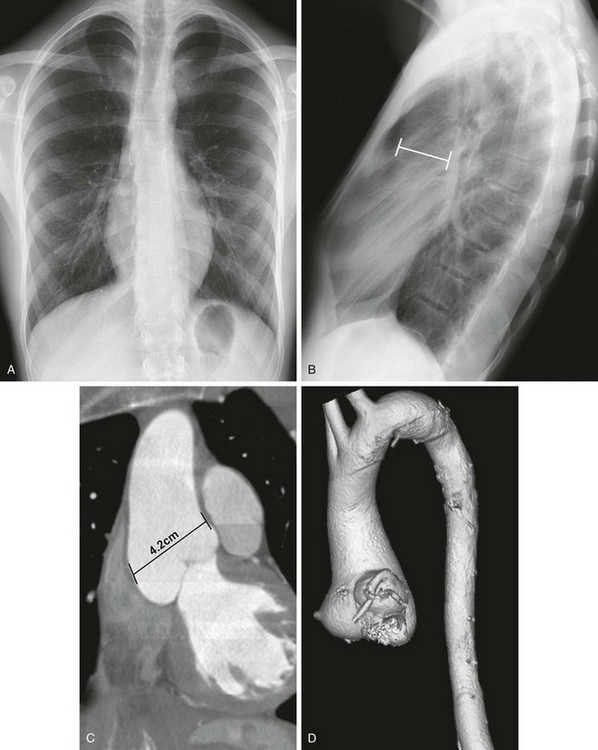
Figure 79-4 An 18-year-old man with a history of Marfan syndrome.
A, A posteroanterior chest radiograph demonstrates a marfanoid appearance, with an elongated chest and little subcutaneous tissue. The anterior ribs are vertically oriented, suggesting a pectus deformity. B, A lateral view of the chest confirms the pectus excavatum and shows a dilated ascending aorta (line). Coronal reconstruction (C) and oblique sagittal three-dimensional reconstruction (D) from computed tomographic angiography of the chest demonstrates the dilated aortic root, which is largest at the level of the sinuses of Valsalva (measurement in C).
MVP affects 35% to 100% of patients with Marfan syndrome and is more common in women and children.12 More than 80% of children with Marfan syndrome have aortic root dilation, MVP, or both before age 18 years; aneurysms can be present even in young children.12,13 Symptoms of heart failure, usually related to valvular insufficiency, can develop in childhood or young adulthood.
Imaging: On chest radiographs, a pectus deformity, kyphoscoliosis, and occasionally cystic lung disease may be present in addition to cardiomegaly and a dilated ascending aorta (see Fig. 79-4). Both MRI and transthoracic echocardiography are used for initial evaluation of the heart and aorta. Both have the advantage of being relatively noninvasive without exposing the patient to ionizing radiation. In Marfan syndrome, the dilated aorta/aneurysm typically is pear shaped because the most marked area of dilatation is at the level of the sinuses of Valsalva (see Fig. 79-4).
Long-term surveillance of the aortic root is performed via MRA or CTA, which have the advantage of providing a global view of the entire aorta. Reformatted and three-dimensional techniques provide excellent visualization of the anatomy in any plane and provide accurate long-term measurements (see Fig. 79-4).14
Treatment and Follow-up: Measurement of the aortic root and assessment of aortic regurgitation are diagnostic criteria for Marfan syndrome. Affected individuals require long-term follow-up to identify progressive aortic root dilation and the appropriate timing for surgical intervention. In older children and adolescents, the incidence of complications such as dissection or rupture increases once the aortic root is larger than 5 cm in diameter.12,15 Cystic medial necrosis is the underlying pathologic abnormality of the aortic wall. Currently, the Bentall procedure, or composite aortic valve graft replacement, is the standard surgical treatment with demonstrated marked improvement in survival. In some centers, the Bentall procedure has been superseded by the Tirone David procedure, which uses an aortic graft with a surgically resuspended native aortic valve, thus avoiding the need for anticoagulation.
It is now recognized that altered regulation of transforming growth factor-β (TGF-β), cytokines that affect cell performance, causes many of the manifestations of Marfan syndrome. Therapy using TGF-β antagonists appears to offer great promise.9,15
Loeys-Dietz Syndrome
Overview: Loeys-Dietz syndrome (LDS) is a rare condition associated with the dysregulation of TGF-β caused by mutations in the TGF-β receptor gene.16 LDS is divided into two types; patients with type 1 LDS have both craniofacial anomalies (e.g., bifid uvula, cleft palate, hypertelorism, craniosynostosis, and cervical spine instability) and widespread vascular anomalies (Fig. 79-5 and Videos 79-1 and 79-2). Patients with type 2 LDS generally have less severe manifestations. They may have a bifid uvula but usually do not have other craniofacial anomalies.16,17
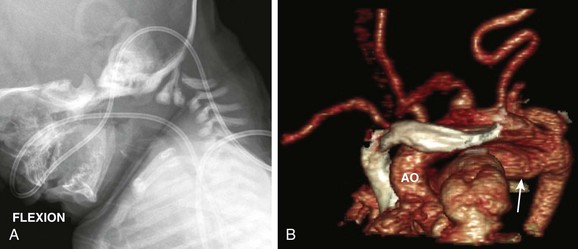
Figure 79-5 An infant with Loeys-Dietz syndrome.
A, A flexion lateral view of the cervical spine demonstrates instability with marked subluxation, posterior hypoplasia, and kyphosis at C2-C3. B, Oblique coronal volumetric reconstruction of computed tomographic angiography. Note a very tortuous aortic (AO) arch and all cervical branches as well as an aneurysmal patent ductus arteriosus (arrow). See Video 79-1 for markedly tortuous aorta and iliac arteries with multiple stenoses and Video 79-2 for diffuse arterial tortuosity and multiple stenoses. (Courtesy Ron Cohen, Oakland Children’s Hospital.)
Vascular manifestations of LDS include elongated and tortuous large arteries, aneurysm formation, and stenoses (see Fig. 79-5 and Videos 79-1 and 79-2).18 Abnormalities may be manifested in infancy. Dissection and aneurysm rupture occur commonly in persons with LDS at an earlier age and are associated with smaller sized aneurysms than are typical for persons with Marfan syndrome.16,17 Although spontaneous visceral perforation may occur in patients with LDS (similar to patients with the vascular form of EDS), both the vascular and visceral lesions tend to do quite well with surgical intervention (unlike in patients with EDS).17
Neurocutaneous Syndromes
Overview: Neurofibromatosis type 1 (NF1) is an autosomal-dominant disorder with multisystem involvement. It is seen in 1 in 3500 newborns. The classic clinical presentation includes café au lait spots, axillary freckling, dermal and plexiform neurofibromas, and learning disabilities.
Congenital heart defects are present in 2% to 4.3% of persons with NF1. The most common defect is pulmonary stenosis.19 Other anomalies include tetralogy of Fallot (TOF), aortic stenosis, aortic coarctation, ASD, VSD, patent ductus arteriosus (PDA), and MVP.2,19,20 Hypertrophic cardiomyopathy has been reported in a small number of patients with NF1. Neurofibromas occasionally can develop within the heart, obstruct cardiovascular blood flow by compression or invasion, or erode a vessel and cause hemorrhage.
Vascular manifestations include stenosis, aneurysms, fistulas, arteriovenous malformations, and spontaneous rupture of systemic arteries and veins (Fig. 79-6). Renovascular stenosis and hypertension are particularly common. Another vascular manifestation known as middle aortic syndrome is caused by the narrowing of the distal thoracic or abdominal aorta, leading to renal vascular hypertension and ischemia of the visceral organs and lower extremities (see Fig. 79-6).21
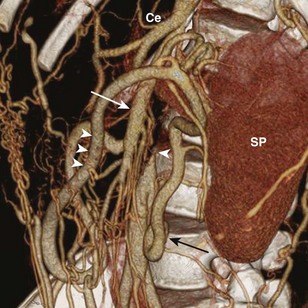
Figure 79-6 Aortic obstruction from neurofibromatosis type 1 in a 14-year-old boy.
Volume rendering of an abdominal computed tomography angiogram shows a complete obstruction of the aorta beyond the origins of the celiac trunk (Ce) and the superior mesenteric artery (white arrow). The distal aorta (single arrowhead) is reconstituted through the superior mesenteric artery, the marginal artery of Drummond (three arrowheads), and the inferior mesenteric artery (black arrow). SP, Spleen.
Imaging and Treatment: In addition to cardiovascular findings, chest imaging in persons with NF1 can show rib deformities (“penciling” and “twisted ribbon”), enlargement of the neural foramen; intercostal, mediastinal, pleural, and soft tissue neurofibromas; and fibrosing alveolitis.
Typically, patients with NF1 are screened with frequent blood pressure measurements and undergo arterial imaging (i.e., CTA or MRA) if hypertension develops.21 Vascular lesions may be treated with drug therapy, angioplasty, or surgery. Development of an aneurysm has been reported after stent or stent graft placement, consistent with an underlying disorder of the vascular wall.
Tuberous Sclerosis
Overview: Tuberous sclerosis is a disease characterized by hamartomas of multiple organs, cortical tubers, subependymal giant cell astrocytomas, and renal angiomyolipomas.22 It is inherited in an autosomal-dominant pattern and occurs in 1 in 6000 live births. The classic clinical triad is seizures, mental retardation, and skin lesions. The presentation and manifestations of tuberous sclerosis are quite varied, and the diagnosis can be missed for a prolonged period.22
Cardiac rhabdomyomas are the most common pediatric cardiac tumors, and 51% to 86% of all cardiac rhabdomyomas are associated with tuberous sclerosis (Fig. 79-7) (see Chapter 81).23
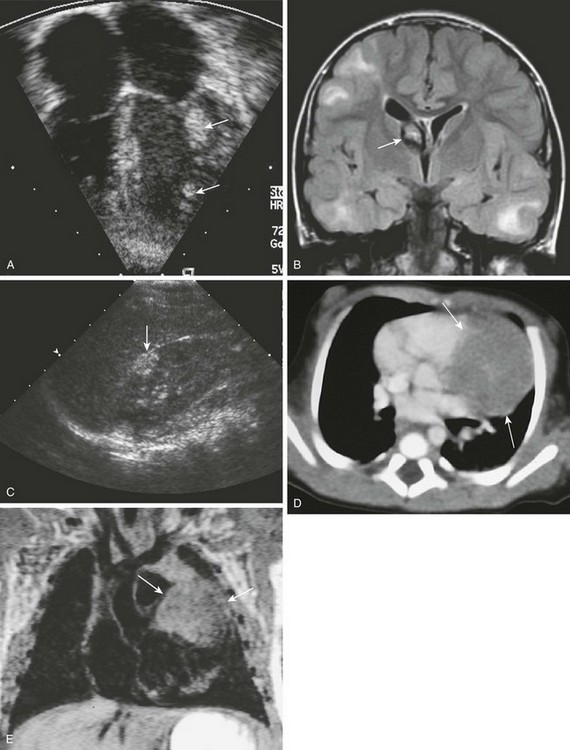
Figure 79-7 Tuberous sclerosis.
A, Four-chamber view from an echocardiogram shows multiple echogenic masses within the free wall of the left ventricle (arrows). The cardiac tumors, representing rhabdomyomas, resolved spontaneously. B, Coronal fluid-attenuated inversion recovery magnetic resonance imaging of the brain from the same child 2 years later shows multiple hyperintense subcortical tubers and a mixed-signal giant cell astrocytoma (arrow) at the foramen of Monro. C, Sagittal ultrasonography of the right kidney shows a hyperechoic lesion in the anterior mid kidney (arrow) that is consistent with an angiomyolipoma. D, Contrast-enhanced computed tomography of a different 3-day-old boy with an abnormal chest radiograph demonstrates a large, hypodense, soft-tissue density mass of the left ventricular wall (arrows). E, A coronal T1-weighted magnetic resonance image demonstrates the large, isointense-to-muscle rhabdomyoma in the left ventricular free wall (arrows). (A, Courtesy Lizabeth Lanford, MD, Children’s Hospital, Pittsburgh, PA.)
Other cardiovascular manifestations of tuberous sclerosis can include central and peripheral arterial aneurysms, aortic coarctation, and vascular stenotic–occlusive disease, including renal artery stenosis.24 Aneurysms in the abdomen appear at an earlier age than do those in the thorax25; the pathologic process is medial atrophy and disruption.
Imaging, Treatment, and Follow-up: Rhabdomyoma is the earliest clinical sign of tuberous sclerosis in utero and can be diagnosed with prenatal or neonatal ultrasound. The presence of multiple cardiac tumors in utero is sufficient to establish a diagnosis of tuberous sclerosis.26 On ultrasound, rhabdomyomas appear as rounded, homogeneous, hyperechoic areas within the myocardium (see Fig. 79-7).
Cardiomegaly with normal or decreased pulmonary vascular markings is the most common finding on chest radiographs after birth. The signal characteristics of rhabdomyomas on MRI are variable. They often are isointense to minimally hyperintense to the myocardium on T1-weighted images (see Fig. 79-7) and hyperintense on T2-weighted images. Rhabdomyomas may be hypointense to the myocardium after administration of gadolinium. They usually appear as low-density masses on contrast-enhanced CT (see Fig. 79-7).
Rhabdomyomas are not considered true neoplasms. They tend to increase in size until 32 weeks of gestation and then progressively regress, especially during the first year of life.20,23 Children can be monitored with serial echocardiograms; complete regression is observed in most patients by age 6 years.
Screening for aortic aneurysms (via ultrasound, MRA, or CTA) is recommended both at the time of diagnosis of the underlying disease and at frequent regular intervals thereafter.25 Open elective repair of an identified aneurysm is important because one third of patients with tuberous sclerosis complex and aneurysms present with rupture.25
PHACES Syndrome
Overview: PHACES syndrome, consisting of posterior fossa malformations, hemangiomas, arterial anomalies, cardiac defects, eye abnormalities, and sternal clefting or a supraumbilical raphe, is a recently described neurocutaneous complex with a strong female predominance.27–30 Features overlap with Sturge-Weber syndrome, leading to confusion as to the nature of the disease and associated vascular lesions.29,30
More than 30% of patients with PHACES syndrome have coarctation of the aorta or other congenital aortic abnormalities such as arch atresia, aberrant subclavian origins, hypoplasia of the descending thoracic aorta, and double aortic arch.28,29 Progressive abnormalities such as stenosis of and aneurysm formation in the aorta and the cervical arteries are of particular concern. Patients with PHACES syndrome are at an unusually high risk of stroke (as a result of a progressive arterial vasculopathy) and cerebrovascular abnormalities.27,29 Currently the imaging strategy is geared toward identification of vascular lesions, treatment planning, and monitoring of disease progression.
Noonan Syndrome
Overview: Noonan syndrome is an autosomal-dominant disorder that occurs in both male and female infants with an incidence of 1 in 1000 to 1 in 2000. It also is known as pseudo–Turner syndrome because it shares several phenotypic similarities with Turner syndrome. Noonan syndrome is typified by characteristic facies and a webbed neck, short stature, cardiac anomalies, deafness, motor delays, and mental retardation. According to Marino and colleagues,31 Noonan syndrome is the second most frequent genetic anomaly associated with congenital heart disease after Down syndrome.
The most commonly reported heart defects in Noonan syndrome are pulmonary stenosis with or without a dysplastic pulmonary valve and hypertrophic cardiomyopathy with or without left ventricular outflow tract obstruction32; these defects are seen in 38.9% and 9.5% of individuals, respectively. Other abnormalities include ASD, VSD, PDA, TOF, anomalous pulmonary venous return, aortic coarctation, partial atrioventricular septal defect, mitral valve abnormalities, and coronary anomalies.31 Lymphatic system abnormalities are particularly common and include lymphedema, lymphangiectasia, cystic hygroma, and chylothorax.32
The term “neurofibromatosis–Noonan syndrome” has been used for cases of NF1 with Noonan features such as pectus, broad neck, and congenital heart disease.2 Noonan syndrome is allelic to, and may be associated with, LEOPARD syndrome (lentigines, electrocardiogram abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, and deafness) (e-Fig. 79-8 and Video 79-3).2
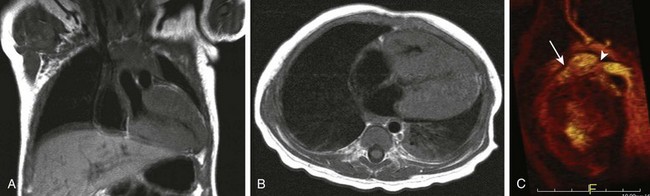
e-Figure 79-8 A 6-month-old girl with a hypertrophic cardiomyopathy and features of LEOPARD syndrome.
LEOPARD syndrome consists of lentigenes, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, and deafness. It is allelic to and may be associated with Noonan syndrome. Coronal (A) and axial T1-weighted magnetic resonance images (B) demonstrate marked thickening of the right ventricle and left ventricle myocardium with small ventricular cavities. See also Video 79-3. C, Oblique sagittal three-dimensional reconstruction from a magnetic resonance angiogram demonstrates moderate pulmonic stenosis with poststenotic dilatation of the pulmonary artery (arrow) and mild stenosis at the origin of the left pulmonary artery (arrowhead).
VATER/VACTERL Association
Overview: The VATER (vertebral defects, VSD, imperforate anus, tracheo-esophageal fistula, radial and renal dysplasia) sequence or VACTERL (vertebral anomaly, anorectal atresia, cardiac lesion, tracheoesophageal fistula, renal anomaly, limb defect) association are nonrandom co-occurrences of birth defects. The VATER/VACTERL association occurs in between 2 and 13 per 10,000 births and is more frequent in infants of diabetic mothers and in persons with trisomy 18.33 A child is diagnosed with the association when he or she has at least two of the characteristics.34
Large, population-based studies have examined the association of defects within this complex and suggested that the association of cardiac defects with other VATER components is no more frequent than with any other birth defect.34 The cardiac anomalies described with the VACTERL association include VSD, ASD, PDA, dextrocardia, TOF, aortic coarctation, a single ventricle, and transposition of the great arteries (see Chapters 74, 75, and 76).
Bronchopulmonary foregut malformations other than the tracheoesophageal fistula complex also can be associated with a spectrum of anomalies similar to VACTERL. These malformations include tracheal agenesis, pulmonary hypoplasia and agenesis, pulmonary sling, scimitar syndrome, congenital lobar hyperinflation, congenital pulmonary airway malformation, bronchial atresia, and pulmonary sequestration. The exact incidence and frequency of anomalies vary somewhat. For example, children with tracheal agenesis tend to have more complex cardiac anomalies than do those who have a tracheoesophageal fistula.35 Imperforate anus occurs much more frequently with a tracheoesophageal fistula than with tracheal agenesis.35
Imaging and Follow-up: Prenatal diagnosis can be made with ultrasound. Prenatal findings include radial atresia or abnormality, vertebral anomalies, polyhydramnios, a single umbilical artery, intrauterine growth restriction, and renal and cardiac anomalies.36 After delivery, echocardiography, plain films, fluoroscopy, CT, and MRI can supplement the clinical findings and confirm the diagnosis (Fig. 79-9). Part of the importance of recognizing this association of anomalies is that when one or more anomalies occur, others should be sought. Therefore infants with esophageal atresia, imperforate anus, or vertebral anomalies should undergo renal and cardiac ultrasound screening. Spinal ultrasound should be performed in the case of imperforate anus to look for occult dysraphism and cord tethering.
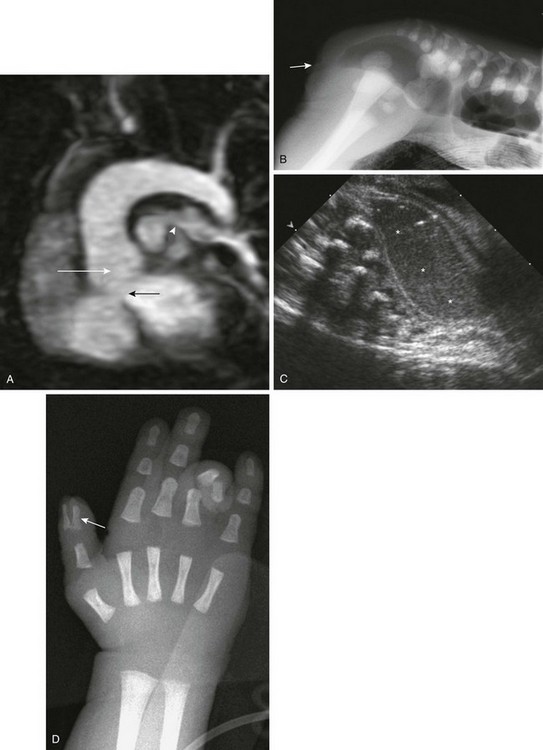
Figure 79-9 A 10-day-old boy with VACTERL (vertebral, anal atresia, cardiac, tracheal, esophageal, renal and rib, limb) association.
A, Oblique, sagittal, thin, maximal intensity projection magnetic resonance angiogram through the left ventricle shows features of tetralogy of Fallot, with a high ventricular septal defect (black arrow) and an overriding aorta (white arrow). Stenosis of the proximal left pulmonary artery is present (arrowhead). A prone, cross-table lateral radiograph (B; arrow on anal dimple) and sagittal transabdominal ultrasonography (C) show an imperforate anus with a dilated, meconium-filled, distal sigmoid colon and rectum. D, A frontal radiograph of the left hand shows a duplicated distal first phalanx of the thumb (arrow). The child’s ring finger is flexed.
The prognosis of an infant diagnosed with VATER/VACTERL is variable and depends on the severity of disease. A study by Tongsong et al36 described a neonatal mortality of 28% in infants with three or more anomalies.36 Because of the variable prognosis, early diagnosis and clear identification of all anomalies are important.
Other Syndromes
Other less common syndromes and their associated cardiovascular components are listed in Table 79-1 (see e-Fig. 79-8, e-Figs. 79-10 through 79-14, and Video 79-4). The most common associated cardiac lesions are listed first. Many other rare syndromes have associated cardiac malformations; however, these syndromes are too numerous to mention specifically. Table 79-2 outlines syndromes that have cardiovascular manifestations almost exclusively (e-Figs. 79-15 and 79-16). This list is not exhaustive but includes the more common, important, and interesting entities.
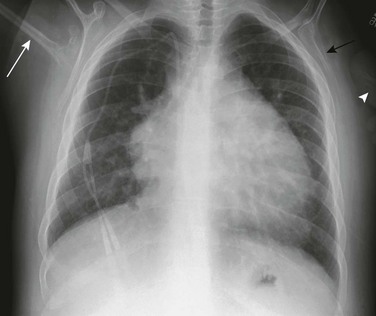
e-Figure 79-10 A 35-year-old man with Holt-Oram syndrome and a history of atrial and ventricular septal defect repairs, as well as pulmonary arterial hypertension.
Chest radiograph shows cardiomegaly, prominent pulmonary vessels, hypoplastic scapulae (left scapula, black arrow), a hypoplastic right humerus (white arrow), and an absent left humerus with a rudimentary digit (arrowhead).
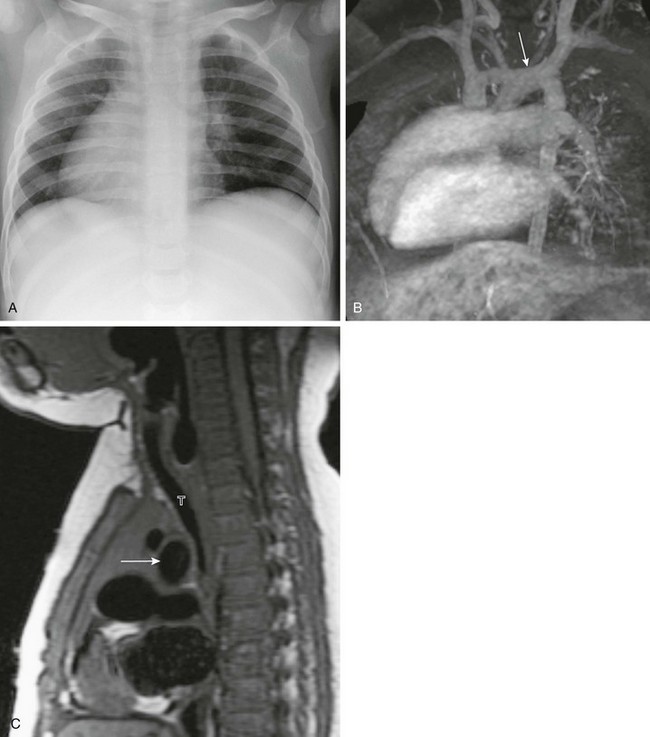
e-Figure 79-11 A 1-year-old boy with Kartagener syndrome and stridor.
A, A chest radiograph shows dextrocardia. The left lung is mildly hyperlucent. The abdominal situs was normal. B, A coronal, three-dimensional, maximum intensity projection magnetic resonance angiogram shows dextrocardia and a right-to-left course of the aorta, with a left-sided aortic arch (arrow). C, A sagittal, T1-weighted magnetic resonance image shows compression of the distal trachea (T) by the crossing aortic arch (arrow).
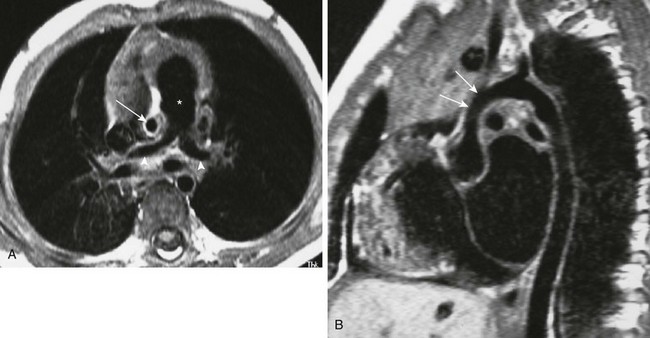
e-Figure 79-12 A 3-year-old boy with Williams syndrome.
A, An axial, T1-weighted magnetic resonance (MR) image of the chest shows an enlarged main pulmonary artery (asterisk), with narrowing of the branch pulmonary arteries (arrowheads). Note the very small ascending aorta (arrow). B, Oblique, sagittal, T1-weighted MR image shows hypoplasia of the supravalvular aorta (arrows).
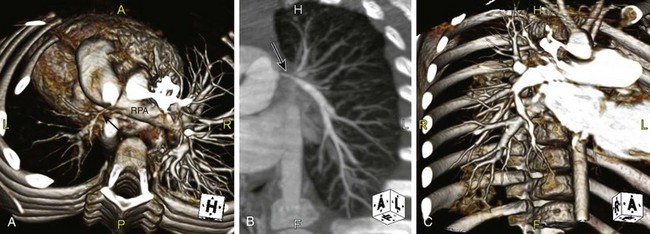
e-Figure 79-13 A 3-year-old child with Alagille syndrome (arteriohepatic dysplasia).
A, Volumetric reconstruction from a computed tomography angiogram (viewed from above) demonstrates marked stenosis and hypoplasia of the left pulmonary artery and branches (arrow). The central right pulmonary artery (RPA) is patent, but numerous peripheral branch stenoses are present (see C). On a nuclear perfusion scan, the distribution of pulmonary flow was 86% right and 14% left. B, Coronal maximum intensity projection reconstruction demonstrates the very stenotic central left pulmonary artery (arrow) with much improved caliber more peripherally. Note additional peripheral stenosis of the origin of the left upper lobe branch. C, Coronal volume rendered reconstruction. Multiple peripheral RPA stenoses are present.
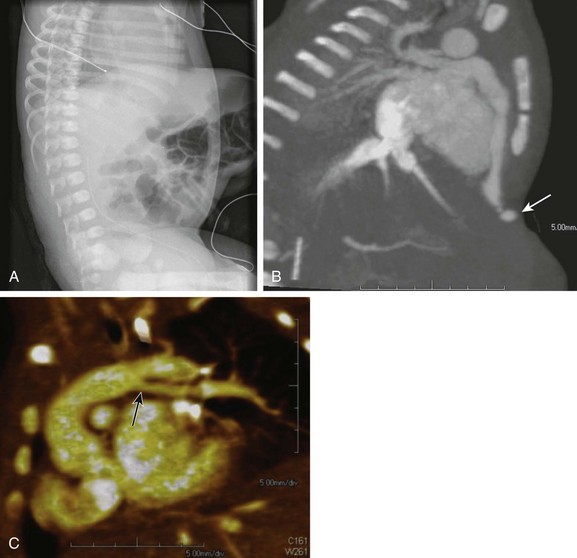
e-Figure 79-14 A 2-day-old girl with Cantrell syndrome (pentalogy of Cantrell), which consists of abnormalities of the anterior abdominal wall, diaphragm sternum, pericardium, and heart.
A, A lateral chest radiograph showing a large, high omphalocele. B, Sagittal maximum intensity projection from a computed tomographic angiogram showing an absent lower sternum and hypoplastic right ventricle with an aneurysm of the right ventricular apex (arrow) extending into the omphalocele, probably through pericardial and diaphragmatic defects. C, An oblique three-dimensional volumetric reconstruction shows the left pulmonary artery (arrow) originating from the proximal portion of the patent ductus arteriosus. The main and right pulmonary arteries were normal (not shown). Also see Video 79-4.
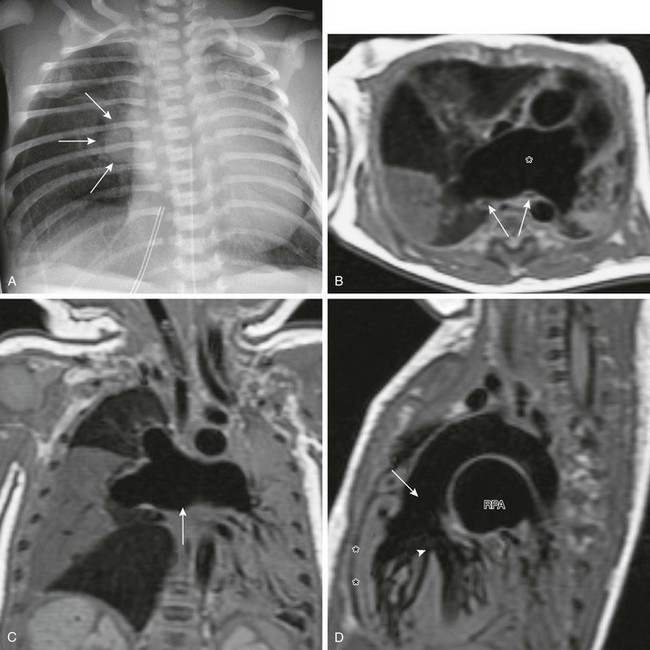
e-Figure 79-15 A 3-day-old boy with respiratory distress who, based on magnetic resonance imaging (MRI), was diagnosed with tetralogy of Fallot–absent pulmonary valve syndrome.
A, A chest radiograph shows a right perihilar “mass” (arrows) and a hyperinflated right lung with associated leftward mediastinal shift and atelectasis. B, An axial T1-weighted MRI obtained to evaluate the perihilar mass shows markedly dilated central pulmonary arteries (asterisk) compressing the bilateral bronchi (arrows) and causing atelectasis. C, Coronal T1-weighted MRI shows markedly dilated central pulmonary arteries (arrow) with aberrant branching. There is complete atelectasis of the right middle lobe and much of the left lung. D, Sagittal T1-weighted MRI shows features of tetralogy of Fallot with right ventricular hypertrophy (asterisks), a ventricular septal defect (arrowhead), and an overriding aorta (arrow). The right pulmonary artery (RPA) is markedly dilated.
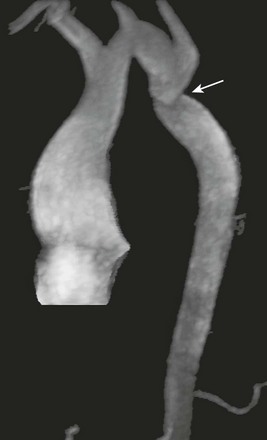
e-Figure 79-16 A 15-year-old boy with a history of Shone syndrome with mitral and aortic valve stenosis and prior coarctation repair.
An oblique, sagittal, volumetric, three-dimensional magnetic resonance angiogram shows a dilated ascending aorta, tortuous cervical aortic arch, and residual aortic coarctation (arrow).
Chromosomal Anomalies
22Q11 Deletion—digeorge Syndrome
Overview: The 22q11 deletion is the most common chromosome deletion in DiGeorge syndrome and the velocardiofacial syndrome.4 DiGeorge syndrome often is remembered by the acronym CATCH 22, which refers to the major manifestations: Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia (absent parathyroid) and 22q11 deletions. The defects seen in the DiGeorge syndrome are field defects of the third and fourth pharyngeal pouches. The most commonly associated cardiac anomalies are truncus arteriosus (Fig. 79-17) and interruption of the aortic arch (usually type B) (see Fig. 79-17), as well as TOF and aberrant or isolated subclavian artery.37
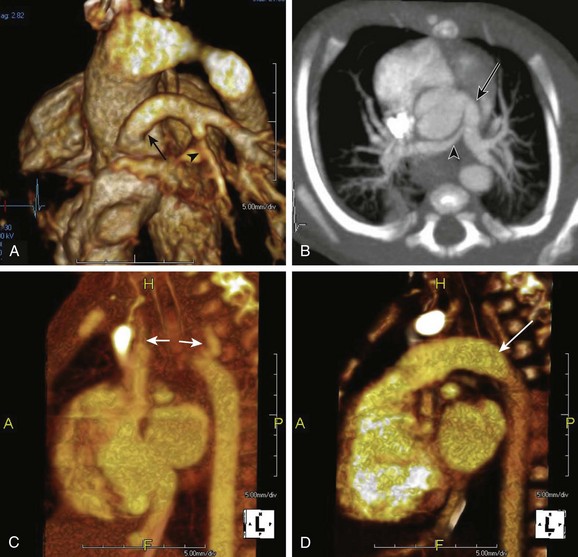
Figure 79-17 Two newborn infants with 22q11 deletion (DiGeorge syndrome).
A and B, Truncus arteriosus type 1. A volume rendered, three-dimensional reconstruction, posterior oblique view (A) and axial thin maximum intensity projection (B) demonstrate a single great artery arising from the heart. The main pulmonary artery (arrow) arises from this vessel, which continues as the ascending aorta. Note branch pulmonary arteries with narrowing of the right pulmonary artery (arrowhead). C and D, Sagittal three-dimensional volumetric reconstructions show interruption of the aortic arch (type B). A marked gap exists between the ascending and descending aorta, with interruption between the left common carotid and left subclavian arteries (arrows in C). A large patent ductus arteriosus extends from the pulmonary artery centrally and supplies the descending aorta (arrow in D).
DiGeorge syndrome is associated with other syndromes including fetal alcohol syndrome, Noonan syndrome, and Zellweger syndrome. It also is associated with infants of diabetic mothers.2
Imaging and management depends on the severity of cardiac and other anomalies. Increased susceptibility to infections, especially respiratory infections, including unusual organisms seen in immunocompromised hosts (such as acid fast bacilli and pneumocystis), are a significant management concern.2
Fragile X Syndrome
Overview: Fragile X syndrome is the most common inherited form of mental retardation. Approximately 1 in 850 people carry the gene for this X-linked disorder; however, about 20% of males with the gene are unaffected. Affected individuals may have large heads and facial abnormalities along with learning disabilities and behavioral problems. Associated cardiac abnormalities include MVP, mitral regurgitation, aortic regurgitation, tricuspid regurgitation, a dilated aortic root, and coarctation.2
Trisomy 21 (Down Syndrome)
Overview: Down syndrome, the most common chromosomal anomaly, is seen in 1 in 600 to 1 in 700 live births. Down syndrome is the most frequent genetic anomaly associated with congenital heart disease. Approximately 40% to 70% of patients with Down syndrome have congenital cardiac malformations, which represent the leading cause of death in the first 2 years of life.38
The most characteristic abnormality in Down syndrome is an ASD with a common atrioventricular junction, also known as a common atrioventricular canal, atrioventricular septal defect, or endocardial cushion defect (Fig. 79-18) (see Chapter 73). Almost 70% of endocardial cushion defects are associated with Down syndrome. Other common cardiac defects include VSD, PDA, TOF, and pulmonary stenosis.2,38
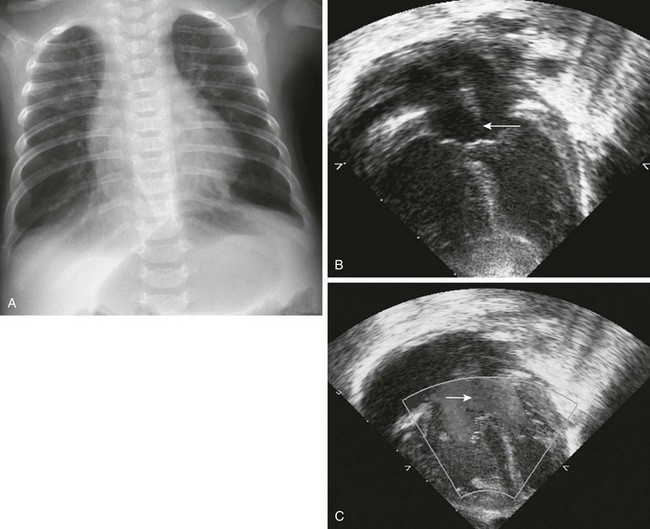
Figure 79-18 A 3-month-old girl with Down syndrome and a known atrioventricular septal defect.
A, A chest radiograph shows increased vascularity, mild congestion, and accompanying hyperinflation of the lungs. Eleven ribs are present bilaterally. B, A four-chamber view from an echocardiogram shows a large atrioventricular septal defect (arrow). C, Left-to-right color Doppler flow is shown across the atrial defect (arrow). (B and C, Courtesy Lizabeth Lanford, MD, Children’s Hospital, Pittsburgh, PA.)
Arterial and lymphatic abnormalities also occur, the latter especially in utero.2 Because persons with Down syndrome have decreased muscular tone, a relatively large tongue, a small pharynx, and laryngotracheomalacia, chronic upper airway obstruction may be a significant clinical problem and can result in congestive heart failure.
Trisomy 13 (Patau Syndrome)
Overview: Patau syndrome is the least common and the most severe of the viable trisomies. It occurs in 1 in 29,000 live births. Eighty-two percent of patients die within the first month of life. Congenital heart defects are seen in approximately 80% of patients. The most common defects include ASD, VSD, PDA, dextrocardia, and a double-outlet right ventricle.2
Trisomy 18 (Edwards Syndrome)
Overview: Trisomy 18 is the second most common autosomal trisomy, with prevalence at birth of about 1 in 7000. It is characterized by severe psychomotor and growth retardation, microcephaly, microphthalmia, micrognathia or retrognathia, microstomia, malformed ears, distinctively clenched fingers, and other congenital malformations.
Cardiac anomalies are seen in 90% to 100% of cases. The most common cardiac anomalies are VSD and valvular heart disease.2 The pulmonary and aortic valves usually are affected. Other cardiac anomalies include ASD, PDA, aortic coarctation, TOF, transposition of the great arteries, and a hypoplastic left heart. Anomalies of the VACTERL spectrum also are associated with trisomy 18.
Turner Syndrome (XO, Monosomy X)
Overview: Turner syndrome is one of the most frequent chromosomal aberrations in females. It affects approximately 1 in 2000 live female births, but it has been estimated that only 1% of fetuses survive to term and that as many as 10% of spontaneous miscarriages have the 45,XO karyotype.39 The characteristic clinical findings include a webbed neck, a shield chest with widely spaced nipples, short stature, gonadal insufficiency, and infertility.40
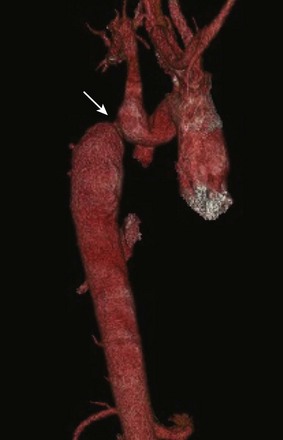
A congenital heart defect occurs in 20% to 40% of patients with Turner syndrome. The most common cardiac anomalies include coarctation of the aorta (30%) (Fig. 79-19) and bicuspid aortic valve (30% to 50%). Dilation of the aortic root is not as common (3% to 8%) but can have devastating consequences such as dissection and rupture.39 Other less common malformations include MVP, aortic stenosis, aortic regurgitation, partial anomalous pulmonary venous drainage, and hypoplastic left heart syndrome. Lymphangiectasia, lymphedema, hemangiomas, and systemic venous anomalies also can occur.40
Klinefelter Syndrome (XXY)
Overview: Klinefelter syndrome is present in 1 in 500 to 1 in 1000 live male births.41 The clinical findings of Klinefelter syndrome include infertility, gynecomastia, hypogonadotropic hypogonadism, cognitive impairment, and predisposition to malignancy.41
MVP, the most common cardiac anomaly, occurs in 55% of patients. Other uncommon defects include TOF, ASD, VSD, tricuspid atresia, and aortic coarctation. Vascular abnormalities include cerebral aneurysms, varicose veins, venous thromboemboli, and arteritis.42
Araoz, PA, Eklund, HE, Welch, TJ, et al. CT and MR imaging of primary cardiac malignancies. Radiographics. 1999;19:1421–1434.
Berdon, WE, Willi, U. Situs inversus, bronchiectasis, and sinusitis and its relation to immotile cilia: history of the diseases and their discoverers—Manes Kartagener and Bjorn Afzelius. Pediatr Radiol. 2004;34:3842.
Cyran, SE, Martinez, R, Daniels, S, et al. Spectrum of congenital heart disease in CHARGE association. J Pediatr. 1987;110:576–578.
Elliott, M, Bayly, R, Cole, T, et al. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin Genet. 1994;46:168–174.
Greenhalgh, KL, Howell, RT, Bottani, A, et al. Thrombocytopenia–absent radius syndrome: a clinical genetic study. J Med Genet. 2002;39:876–881.
Morrison, PJ, Mulholland, HC, Craig, BG, et al. Cardiovascular abnormalities in the oculo-auriculo-vertebral spectrum (Goldenhar syndrome). Am J Med Genet. 1992;44:425–428.
Sletten, LJ, Pierpont, ME. Variation in severity of cardiac disease in Holt-Oram syndrome. Am J Med Genet. 1996;65:128–132.
Stevens, CA, Bhakta, MG. Cardiac abnormalities in the Rubinstein-Taybi syndrome. Am J Med Genet. 1995;59:346–348.
Wippermann, CF, Beck, M, Schranz, D, et al. Mitral and aortic regurgitation in 84 patients with mucopolysaccharidoses. Eur J Pediatr. 1995;154:98–101.
Duarte, AC, Menezes, AIC, Devens, ES, et al. Patau syndrome with a long survival: a case report. Genet Mol Res. 2004;3:288–292.
Versacci, P, Digilio, MC, Sauer, U, et al. Absent pulmonary valve with intact ventricular septum and patent ductus arteriosus: a specific cardiac phenotype associated with deletion 18q syndrome. Am J Med Genet A. 2005;138:185–186.
References
1. Applegate, KE, Goske, MJ, Pierce, G, et al. Situs revisited: imaging of the heterotaxy syndrome. Radiographics. 1999;19:837–852. [discussion 853-834].
2. Taybi, H, Lachman, RS. Taybi and Lachman’s radiology of syndromes, metabolic disorders, and skeletal dysplasias, 5th ed. St Louis: Mosby; 2006.
3. Bartram, U, Wirbelauer, J, Speer, CP. Heterotaxy syndrome—asplenia and polysplenia as indicators of visceral malposition and complex congenital heart disease. Biol Neonate. 2005;88:278–290.
4. Newman, B, Feinstein, JA, Cohen, RA, et al. Congenital extrahepatic portosystemic shunt associated with heterotaxy and polysplenia. Pediatr Radiol. 2010;40:1222–1230.
5. Mao, JR, Bristow, J. The Ehlers-Danlos syndrome: on beyond collagens. J Clin Invest. 2001;107:1063–1069.
6. Beighton, P, De Paepe, A, Steinmann, B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77:31–37.
7. Wenstrup, RJ, Meyer, RA, Lyle, JS, et al. Prevalence of aortic root dilation in the Ehlers-Danlos syndrome. Genet Med. 2002;4:112–117.
8. Germain, DP. The vascular Ehlers-Danlos syndrome. Curr Treat Options Cardiovasc Med. 2006;8:121–127.
9. Judge, DP, Dietz, HC. Marfan’s syndrome. Lancet. 2005;366:1965–1976.
10. Milewicz, DM, Dietz, HC, Miller, DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005;111:e150–e157.
11. Radonic, T, de Witte, P, Groenink, M, et al. Critical appraisal of the revised Ghent criteria for diagnosis of Marfan syndrome. Clin Genet. 2011;80(4):346–353.
12. van Karnebeek, CD, Naeff, MS, Mulder, BJ, et al. Natural history of cardiovascular manifestations in Marfan syndrome. Arch Dis Child. 2001;84:129–137.
13. Knirsch, W, Hillebrand, D, Horke, A, et al. Aortic aneurysm rupture in infantile Marfan’s syndrome. Pediatr Cardiol. 2001;22:156–159.
14. Pemberton, J, Sahn, DJ. Imaging of the aorta. Int J Cardiol. 2004;97(suppl 1):53–60.
15. Ling-Gen, G, Fang, L, Ru-Tai, H, et al. Recent molecular biological progress in Marfan syndrome and Marfan-associated disorders. Ageing Res Rev. 2010;9:363–368.
16. Van Hemelrijk, C, Renard, M, Loeys, B. The Loeys-Dietz syndrome: an update for the clinician. Curr Opin Cardiol. 2010;25:546–551.
17. Johnson, PT, Chen, JK, Loeys, BL, et al. Loeys-Dietz syndrome: MDCT angiography findings. AJR Am J Roentgenol. 2007;189:W29–W35.
18. Kalra, VB, Gilbert, JW, Malhotra, A. Loeys-Dietz syndrome: cardiovascular, neuroradiological and musculoskeletal imaging findings. Pediatr Radiol. 2011;41(12):1495–1504.
19. Lin, AE, Birch, PH, Korf, BR, et al. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet. 2000;95:108–117.
20. Friedman, JM, Arbiser, J, Epstein, JA, et al. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4:105–111.
21. Lama, G, Graziano, L, Calabrese, E, et al. Blood pressure and cardiovascular involvement in children with neurofibromatosis type 1. Pediatr Nephrol. 2004;19:413–418.
22. Staley, BA, Vail, EA, Thiele, EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics. 2011;127:e117–e125.
23. Isaacs, H, Jr. Fetal and neonatal cardiac tumors. Pediatr Cardiol. 2004;25:252–273.
24. Salerno, AE, Marsenic, O, Meyers, KE, et al. Vascular involvement in tuberous sclerosis. Pediatr Nephrol. 2010;25:1555–1561.
25. Jost, CJ, Gloviczki, P, Edwards, WD, et al. Aortic aneurysms in children and young adults with tuberous sclerosis: report of two cases and review of the literature. J Vasc Surg. 2001;33:639–642.
26. Tworetzky, W, McElhinney, DB, Margossian, R, et al. Association between cardiac tumors and tuberous sclerosis in the fetus and neonate. Am J Cardiol. 2003;92:487–489.
27. Drolet, BA, Dohil, M, Golomb, MR, et al. Early stroke and cerebral vasculopathy in children with facial hemangiomas and PHACE association. Pediatrics. 2006;117:959–964.
28. Frieden, IJ, Reese, V, Cohen, D. PHACE syndrome. The association of posterior fossa brain malformations, hemangiomas, arterial anomalies, coarctation of the aorta and cardiac defects, and eye abnormalities. Arch Dermatol. 1996;132:307–311.
29. Metry, DW, Dowd, CF, Barkovich, AJ, et al. The many faces of PHACE syndrome. J Pediatr. 2001;139:117–123.
30. Wendelin, G, Kitzmuller, E, Salzer-Muhar, U. PHACES: a neurocutaneous syndrome with anomalies of the aorta and supraaortic vessels. Cardiol Young. 2004;14:206–209.
31. Marino, B, Digilio, MC, Toscano, A, et al. Congenital heart diseases in children with Noonan syndrome: an expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. 1999;135:703–706.
32. Tartaglia, M, Gelb, BD, Zenker, M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab. 2011;25:161–179.
33. Khoury, MJ, Cordero, JF, Greenberg, F, et al. A population study of the VACTERL association: evidence for its etiologic heterogeneity. Pediatrics. 1983;71:815–820.
34. Rittler, M, Paz, JE, Castilla, EE. VACTERL association, epidemiologic definition and delineation. Am J Med Genet. 1996;63:529–536.
35. Evans, JA, Greenberg, CR, Erdile, L. Tracheal agenesis revisited: analysis of associated anomalies. Am J Med Genet. 1999;82:415–422.
36. Tongsong, T, Wanapirak, C, Piyamongkol, W, et al. Prenatal sonographic diagnosis of VATER association. J Clin Ultrasound. 1999;27:378–384.
37. Chan, FP. Truncus arteriosus. In: Ho H, Reddy GP, eds. Cardiovascular imaging—expert radiology series. St Louis: Mosby, 2011.
38. Vida, VL, Barnoya, J, Larrazabal, LA, et al. Congenital cardiac disease in children with Down’s syndrome in Guatemala. Cardiol Young. 2005;15:286–290.
39. Elsheikh, M, Dunger, DB, Conway, GS, et al. Turner’s syndrome in adulthood. Endocr Rev. 2002;23:120–140.
40. Frias, JL, Davenport, ML. Health supervision for children with Turner syndrome. Pediatrics. 2003;111:692–702.
41. Smyth, CM, Bremner, WJ. Klinefelter syndrome. Arch Intern Med. 1998;158:1309–1314.
42. Wattendorf, DJ, Muenke, M. Klinefelter syndrome. Am Fam Physician. 2005;72:2259–2262.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]Chapter 79
Syndromes and Chromosomal Anomalies
A large number of syndromes, dysplasias, and chromosomal anomalies are associated with congenital or acquired cardiac and vascular disease. This chapter discusses the cardiovascular features of some commonly encountered lesions. A more extensive list is provided in Tables 79-1 to 79-3.
Table 79-1
Syndromes, Dysplasias, and Their Associated Cardiovascular Anomalies
| Syndrome | Cardiovascular Anomalies |
| Achondrogenesis | PDA, ASD, VSD, COA |
| Alagille syndrome (arteriohepatic dysplasia) (see e-Fig. 79-13) |
PS, PPS, ASD, VSD, TOF, PDA, PAT, PAPVR, dysplastic AV valves, COA |
| Apert syndrome (acrocephalosyndactyly) | ASD, PDA, VSD, PS, TOF, EFE, DEXTRO, COA |
| Arthrogryposis | CMY |
| Beckwith-Wiedemann syndrome (EMG syndrome) | CM, CMY, ASD, PDA, TOF, HPLH, subvalvular AS, cardiac fibromas |
| Cardioauditory syndromes | LVH, RVH |
| Cantrell syndrome (pentalogy of Cantrell) (see e-Fig. 79-14) | Combined sternal, pericardial, intracardiac, diaphragmatic, and anterior abdominal wall defects Radiographic findings: sternal defect, ectopia cordis, CHD, ASD, VSD, PS, TOF, APVR, DEXTRO, ventricular diverticulum, intrapericardial herniation of abdominal organs; associated with Turner, trisomy 18, sirenomelia, and amniotic band syndromes |
| Cardiosplenic (heterotaxy) syndromes (see Figs. 79-1 and 79-2) | |
| Right isomerism (see Fig. 79-1) | TAPVR, AVSD, pulmonary outflow obstruction or PAT, DORV, TGA, single atrium (R), single common ventricle, DEXTRO, TAT, TRU (rare), AO-IVC juxtaposition, bilateral right PAs, bilateral SVC, interrupted IVC |
| Left isomerism (see Fig. 79-2) | Cardiac malposition, single atrium (L), single ventricle, VSD, AVSD, DORV, APVR, interrupted IVC, bilateral left PAs, bilateral SVC |
| Cayler syndrome (cardiofacial syndrome) | ASD, PDA, VSD, AVSD, TOF, RAA, COA |
| CHARGE association | ASD, VSD, conotruncal malformations, PDA, TOF, parachute mitral valve |
| Degos syndrome (malignant atrophic papulosis) | MI, pericarditis, constrictive pericarditis, myocardial fibrosis, and renal, cerebral, coronary, visceral, and peripheral arteriopathy |
| Diamond-Blackfan syndrome (congenital red cell aplasia) | VSD, ASD, mitral valve dysplasia |
| Ehlers-Danlos syndrome (see Fig. 79-3) | MVP, dilated AO root, coronary and aortic aneurysms, dissection or rupture, AS, AR, TR, PS, ASD, VSD, TOF, DEXTRO, LV rupture, arteriovenous fistula |
| Ellis–van Creveld syndrome (chondroectodermal dysplasia) | Common atrium, ASD, AVSD |
| Fryns syndrome | Septal defects, arch anomalies, TOF, cystic hygroma |
| Hallermann-Streiff syndrome (oculomandibulofacial syndrome) | PS, TOF, ASD, VSD |
| Holt-Oram syndrome (heart-hand syndrome) (see e-Fig. 79-10) | ASD, VSD, MVP, PDA, HPLH, TAPVR, TRU, conduction disorder, hypoplastic peripheral vessels |
| Jeune syndrome (asphyxiating thoracic dystrophy) | CHF |
| Kartagener syndrome (primary ciliary dyskinesia) (see e-Fig. 79-11) | DEXTRO, CHD |
| Klippel-Trénaunay-Weber syndrome (angioosteohypertrophy syndrome) | CHF, pericardial effusion, superficial varices, telangiectatic nevi, organ hemangiomas, lymphatic obstruction |
| LEOPARD syndrome (cardiomyopathic lentiginosis) (see e-Fig 79-8) | CMY, conduction defect, PS, sub-AS |
| Loeys-Dietz syndrome (see Fig. 79-5) | Congenital heart defects include patent ductus arteriosus, bicuspid aortic valve, bicuspid pulmonary valve, mitral valve prolapse, and atrial septal defect; arterial tortuosity, stenoses, aneurysms, dissection, diffuse arterial involvement; spontaneous rupture of viscera |
| Marfan syndrome (see Fig. 79-4) | MVP, MR, dilation of AO root, AR, CHF, aneurysms (AO, pulmonary, ductus), AO dissection, MI, arrhythmia, TR, ASD, TOF |
| MELAS syndrome | CMY, CHF, conduction abnormalities |
| Mucolipidosis III | AR |
| Mucopolysaccharidoses | |
| IH (Hurler syndrome) | Acute CMY associated with EFE, AR, MR, MS, arteriopathy (coronary, renal, AO, mesenteric) |
| IS (Scheie syndrome) | AS, MS |
| II (Hunter syndrome) | AR, CHF, valve thickening, CMY |
| III (Sanfilippo syndrome) | CMY, MR, AR |
| IV (Morquio syndrome) | AR, CMY, AS, MR, CAD |
| VI (Maroteaux-Lamy syndrome) | AS, MS, CMY |
| Neurofibromatosis type 1 (see Fig. 79-6) | PS, COA, ASD, VSD, CMY, MVP, AS, TOF, PDA, vasculopathy (coronary, pulmonary, renal, systemic), cardiac neurofibroma, arteriovenous fistula, lymphatic abnormality |
| Noonan syndrome (Turner phenotype with normal karyotype) | PS, dysplastic pulmonic valve, hypertrophic CMY, lymphatic abnormalities, PDA, ASD, COA, mitral valve abnormalities, AS, pericarditis, APVR, coronary anomalies |
| Oculoauriculovertebral dysplasia (Goldenhar syndrome) | TOF, VSD, DORV, PAT, TAPVR, RAA, COA, asplenia |
| PHACES | Arch atresia, aberrant subclavian origins, hypoplasia of the descending thoracic aorta, double aortic arch, COA, stenosis and aneurysm formation of the aorta and the cervical arteries, stroke |
| Progeria (Hutchinson-Gilford syndrome) | Accelerated atherosclerosis, CM, MI, CHF, stroke |
| Proteus syndrome | CHD, CMY, myocardial mass, conduction abnormality, venous dilation, hemangioma, lymphangioma |
| Ravitch syndrome (thoracoabdominal wall defect) | Ectopia cordis, pentalogy of Cantrell, TGA, PDA, ASD, VSD, PS, TOF |
| Robinow syndrome (fetal face syndrome) | CHD (right heart lesions) |
| Rubinstein-Taybi syndrome | ASD, VSD, PDA, COA, PS, bicuspid AO valve |
| Silver-Russell syndrome | CHD |
| Smith-Lemli-Opitz syndrome | ASD, complex cardiac anomalies |
| Thrombocytopenia–absent radius (TAR) syndrome | COA, ASD, VSD, PDA, AVSD, TOF |
| Tuberous sclerosis (Bourneville-Pringle syndrome) (see Fig. 79-7) | Rhabdomyoma, hamartoma, CHF, CMY, COA, arrhythmia, arterial aneurysm and stenosis (AO, cerebral, renal, peripheral) |
| VATER/VACTERL association (see Fig. 79-9) | VSD, ASD, PDA, TOF, TGA, single ventricle |
| Velocardiofacial syndrome (Shprintzen syndrome) | TOF, TRU, PA, VSD, absent pulmonary valve, TGA, AS, interrupted AO arch, RAA |
| Williams syndrome (e-Fig. 79-12) | Supravalvular AS, PPS, MR, ASD, VSD, TOF, MI, COA, interrupted arch, hypoplastic AO, aneurysm or stenosis (AO, systemic, renal, cerebral arteries) |
| Zellweger syndrome (cerebrohepatorenal syndrome) | CHD, PDA, VSD, DiGeorge |
Table 79-2
Syndromes that Predominantly Affect the Cardiovascular System
| Syndrome | Cardiovascular Defects |
| Absent pulmonary valve leaflet syndrome (see e-Fig. 79-15) | Maldeveloped nodular myxoid pulmonary valve cusps with aneurysmal dilation of central PAs associated with TOF, airway compression, lobar emphysema and abnormal PA branching, CM, RAA, ASD, VSD, PDA, DORV, AVSD, Marfan syndrome, 18q deletion |
| Berry syndrome | Distal AP window with AO origin of the right PA and arch interruption |
| Bland-White-Garland syndrome | Anomalous origin of left coronary artery from PA |
| Congenital cardiomyopathy: hypertrophic cardiomyopathy | Asymmetric septal hypertrophy, systolic anterior motion of mitral valve, LVOT obstruction, myocardial scar arrhythmias |
| Arrhythmogenic right ventricular dysplasia | Fibrofatty infiltration of right ventricular myocardium, RV dyskinesia/aneurysms, arrhythmias |
| Eisenmenger syndrome | Pulmonary hypertension with bidirectional or reversed shunt at atrial, ventricular, or AP level; cyanosis; dyspnea; sudden death; peripartum CMY radiographic findings: dilated central PAs with tapering, PA calcification |
| Floppy valve syndrome | MVP, prolapse of other valves, CAD, congestive or hypertrophic CMY, ASD, MR, AR, papillary muscle or chordae tendineae rupture |
| Hypoplastic left heart syndrome | Combined mitral and AO obstruction (stenosis or atresia), underdeveloped LA and LV, hypoplastic ascending AO ± COA or AO interruption; may be associated with right diaphragmatic hernia, omphalocele, brain anomalies; radiographic findings: CM and pulmonary edema |
| Lutembacher syndrome | ASD associated with MS |
| Postmyocardial infarction syndrome (Dressler syndrome) | Chest pain, fever, polyserositis—several weeks postinfarction; radiographic findings: pericardial or pleural effusion, noncardiogenic edema |
| Postpericardiotomy syndrome | Chest pain, fever, joint pain—weeks or months after closed or open heart surgery; radiographic findings: pericardial or pleural effusion, noncardiogenic pulmonary edema, constrictive pericarditis |
| Romano-Ward syndrome | Familial Q-T prolongation, arrhythmias, syncope |
| Shone syndrome (or complex) (see e-Fig. 79-16) | Complex of multiple left-sided obstructions, parachute mitral valve, supravalvular ring of LA, sub-AS, COA |
| Sick sinus syndrome | Arrhythmias |
| Tetralogy of Fallot (see Fig. 79-9 and e-Fig. 79-15) | Combination of VSD, overriding AO, RVH, RV outflow obstruction; may be PS and PPS |
| Trilogy of Fallot | PS, ASD (or PFO), right-to-left shunting |
| Uhl syndrome (anomaly) | Congenital aplasia of RV myocardium, RV CMY |
| Wolff-Parkinson-White syndrome | Aberrant intracardiac ECG pathway producing arrhythmias; associated with Ebstein anomaly, IHSS, levo-TGA, giant RA diverticulum |
Table 79-3
Chromosomal Anomalies and Their Associated Cardiovascular Defects
| Chromosomal Anomaly | Cardiovascular Defects |
| Fragile X | MVP, MR, AR, TR, dilated AO root, COA |
| Trisomy 13 (Patau syndrome) | PDA, VSD, ASD, DEXTRO, capillary hemangioma, cervical cystic hygroma |
| Trisomy 18 (Edwards syndrome) | VSD, polyvalvular heart disease (pulmonary and AO valves), ASD, PDA, COA, TOF, TGA, HPLH, VACTERL, pentalogy of Cantrell |
| Trisomy 21 (Down syndrome) (see Fig. 79-18) | AVSD, VSD, ASD, TOF, PDA, PS, MVP, aberrant right SCA, intimal arterial fibrodysplasia, lymphatic abnormality, upper airway obstruction and CHF |
| Cat-eye syndrome (trisomy or tetrasomy 22) | TAPVR, TOF |
| Monosomy X, XO (Turner syndrome) (see Fig. 79-19) | COA, bicuspid AO valve, AO dissection, septal defects, abnormal mitral valve, sub-AS, PS, APVR, pentalogy of Cantrell, DEXTRO, RAA, hemangioma, lymphangiectasia, venous anomalies |
| XXY (Klinefelter syndrome) | MVP, Takayasu arteritis, cerebral aneurysms, varicose veins |
| Deletion Syndromes | |
| Monosomy 1p36 syndrome | Dilated CMY, PDA |
| 22q11 (predominantly DiGeorge syndrome (CATCH 22), also velocardiofacial syndrome) (see Fig. 79-17) | Type B interrupted arch, RAA, VSD, TOF, TRU, COA, aberrant right SCA, isolated SCA |
| 5p: Cri du chat syndrome | CHD |
| 4p: Wolf-Hirschhorn syndrome | ASD, VSD, valve anomalies, complex CHD, persistent left SVC |
| 17p: Miller-Dieker syndrome (lissencephaly type 1) | ASD, CHD, conduction abnormalities |
| 18q syndrome | Absent pulmonary valve, PDA, AS, dilated ascending AO |
Syndromes
Situs and Cardiosplenic (Heterotaxy) Syndromes
Overview: The heterotaxy syndromes, that is, right and left isomerism, feature abnormalities of the visceral situs. These syndromes have an estimated incidence of 1 in 6000 to 1 in 20,000 live births and account for 1% of all congenital heart defects. Although heterotaxy usually occurs sporadically, familial cases have been described.
Although visceral and atrial situs do not always correspond, body situs (from the Latin word meaning location) generally is divided into three types: solitus, inversus, and ambiguus. Situs solitus is the normal arrangement of the viscera in the body (see Chapter 63). Situs inversus is the mirror image of normal; it is seen in 0.01% of the population and is associated with a slightly higher incidence of congenital heart disease (3% to 5%) compared with the solitus population (0.6% to 0.8%). The most common cardiac abnormalities seen in patients with situs inversus are a right-sided aortic arch, atrioventricular discordance, and transposition of the great vessels. Situs ambiguus, or heterotaxy, encompasses all other visceroatrial arrangements. By definition, in situs ambiguus, visceral malposition and dysmorphism associated with an indeterminate atrial arrangement are present.1
Heterotaxy syndrome with right isomerism or bilateral right-sidedness is usually, but not invariably, accompanied by asplenia.1 The condition is more common in males and is characterized by bilateral systemic atria with broad trabeculated appendages (Fig. 79-1), bilateral trilobed lungs with bilateral minor fissures and short eparterial bronchi, a central horizontal liver, bowel malrotation, and the stomach in an indeterminate position (see Fig. 79-1). The abdominal aorta and inferior vena cava often are located on the same side of the spine, frequently in a posterior-anterior orientation. Other occasional anomalies include tracheoesophageal fistula, imperforate anus, absent gallbladder, pancreatic anomalies, fused adrenal glands, and genitourinary abnormalities.2 The prognosis for right isomerism is poor because of an abnormal immune status (asplenia) and the typically complex cardiac anomalies.
Cardiac anomalies are almost invariable and cause the most common presenting symptoms: cyanosis and severe respiratory distress. The “right isomerism heart” often consists of a common atrioventricular canal, a single ventricle, a large ventricular septal defect (VSD), and a double-outlet right ventricle and/or transposition of the great vessels, along with pulmonary outflow obstruction or atresia and total anomalous pulmonary venous drainage (frequently obstructed) (see Fig. 79-1). The spectrum of cardiovascular anomalies also can include cardiac malposition (dextrocardia or mesocardia), tricuspid atresia, truncus arteriosus, right aortic arch, anomalous systemic venous return, and bilateral superior vena cavae.2,3
Heterotaxy syndrome with left isomerism or bilateral left-sidedness most often accompanies polysplenia. It is characterized by bilateral pulmonary atria with narrow fingerlike appendages, bilateral bilobed lungs, bilateral long hyparterial bronchi, a centrally located liver, the stomach in an indeterminate position, bowel malrotation, and multiclefted or multiple spleens (either right sided or left sided) (Fig. 79-2). An interrupted inferior vena cava with azygos continuation is the most consistent abdominal finding in left isomerism (see Fig. 79-2). Left isomerism is slightly more common in females and generally has a better prognosis than right isomerism with less complex cardiovascular disease. Other associated anomalies include ciliary dyskinesia, biliary atresia, and other gastrointestinal abnormalities including bowel malrotation and pancreatic anomalies, as well as congenital portosystemic shunts (see Fig. 79-2).2,4
Figure 79-2 This 3-year-old girl, whose mother was diabetic, has polysplenia and Abernethy malformation with hepatopulmonary syndrome.
A, A posteroanterior view of the chest shows heterotaxy with a left-sided cardiac apex, right-sided stomach (arrow), and left-sided liver. Cardiomegaly and increased pulmonary vascularity (a small atrial septal defect and pulmonary arteriovenous shunting) are present. It is difficult to see the bilateral left-sided bronchi. A prominent right paraspinal line is present as a result of an enlarged azygos vein (arrowhead). B, The lateral view of the chest is notable for the absence of the inferior vena cava shadow. C, Contrast-enhanced computed tomography (CT) scan of the chest shows cardiomegaly, a left-sided aorta (arrow), prominent pulmonary veins, and an enlarged azygos vein (arrowhead). D, A contrast-enhanced CT scan of the chest displayed in the lung window shows the presence of multiple dilated peripheral pulmonary vessels (arrows). E, Coronal reconstruction from a contrast-enhanced CT scan of the upper abdomen shows right-sided polysplenia (S), a right-sided stomach (white arrow), and a left-sided liver. A large right-sided portosystemic shunt (splenic vein to renal vein) is present (open arrow). Superiorly, there is interruption of the inferior vena cava (IVC) with azygos continuation (not shown). F, A left pulmonary artery angiogram from a right jugular approach shows dilated peripheral pulmonary arteries and micro arteriovenous connections, consistent with hepatopulmonary syndrome as a result of the congenital portosystemic shunt. This child does not have liver disease, and the intrahepatic portal veins are present but small (Abernethy malformation type II). G, Perfusion portion of a ventilation-perfusion scan using technetium-99m macroaggregated albumin performed after closure of the patient’s atrial septal defect shows normal perfusion of the lungs. There is abnormal radiotracer uptake within the brain (arrow) and kidneys (arrowhead). The percentage of systemic tracer uptake was calculated at 44.7%. These findings are typical of a right-to-left arteriovenous shunt and confirm the presence of hepatopulmonary syndrome.
Cardiac anomalies are seen in more than 50% of patients who have heterotaxy syndrome with left isomerism. The common cardiovascular anomalies include abnormalities of the inferior and superior vena cavae, cardiac malposition, atrial septal defect (ASD), VSD, common atrioventricular canal, double-outlet right ventricle, and anomalous pulmonary venous return (usually partial).2,3
